

Abstract
Evidence suggests that species differences exist between rodents and humans in their biological responses to ligand activation of PPARα. Moreover, neonatal/postnatal rodents may be more sensitive to the effects of activating PPARα. Thus, the present studies examined the effects of chronic ligand activation of PPARα initiated during early neonatal development and continued into adulthood on hepatocarcinogenesis in mice. Wild-type, Ppara-null, or PPARA-humanized mice were administered a potent, high-affinity human PPARα agonist GW7647, and cohorts of mice were examined over time. Activation of PPARα with GW7647 increased expression of known PPARα target genes in liver and was associated with hepatomegaly, increased hepatic cytotoxicity and necrosis, increased expression of hepatic MYC, and a high incidence of hepatocarcinogenesis in wild-type mice. These effects did not occur or were largely diminished in Ppara-null and PPARA-humanized mice, although background levels of hepatocarcinogenesis were also noted in both Ppara-null and PPARA-humanized mice. More fatty change (steatosis) was also observed in both Ppara-null and PPARA-humanized mice independent of GW7647 administration. Results from these studies indicate that the mouse PPARα is required to mediate hepatocarcinogenesis induced by GW7647 in mice and that activation of the human PPARα with GW7647 in PPARA-humanized mice are diminished compared with wild-type mice. Ppara-null and PPARA-humanized mice are valuable tools for examining species differences in the mechanisms of PPARα-induced hepatocarcinogenesis, but background levels of liver cancer observed in aged Ppara-null and PPARA-humanized mice must be considered when interpreting results from studies that use these models. These results also demonstrate that early life exposure to a potent human PPARα agonist does not enhance sensitivity to hepatocarcinogenesis.
Keywords: peroxisome proliferator-activated receptors (PPARs), hepatocarcinogenesis, perinatal exposure
Peroxisome proliferator-activated receptors (PPARs) dynamically regulate many physiological and pathological processes including lipid homeostasis, inflammation, differentiation, and carcinogenesis (Corton et al., 2014, 2018; Heikkinen et al., 2007; Peters et al., 2005,2012, 2019). PPARs are best characterized for their critical role in the regulation of target gene expression that modulates cellular function(s) in response to endogenous and exogenous chemicals that act as receptor agonists and/or antagonists. Three different PPAR genes have been identified encoding PPARα, PPARβ/δ, and PPARγ that exhibit different tissue distributions and distinct biological functions (Berger and Moller, 2002). Elucidating the role of PPARs in physiological models has been greatly facilitated by the use of transgenic mouse models (Akiyama et al., 2001, 2002; Lee et al., 1995; Peters et al., 2000). The first PPAR identified was PPARα (Issemann and Green, 1990) that is now recognized as having an essential role in the regulation of genes that modulate many pathways involved in fatty acid transport and catabolism in liver, heart, and kidney. PPARα was subsequently found to mediate the lipid-lowering effects of the fibrate class of drugs that have been used therapeutically in humans since 1963 (Fruchart et al., 1998). The lipid-lowering actions of PPARα is conserved across a number of mammalian species, demonstrating a critical role for PPARα in lipid metabolism.
It is interesting to note that although PPARα has an essential role in regulating lipid homeostasis, prolonged administration of PPARα agonists causes liver cancer in mice and rats (Hays et al., 2005; Peters et al., 1997; Reddy et al., 1980). However, there is a large body of evidence indicating that the hepatocarcinogenic effect of PPARα agonists may be rodent specific (reviewed in Corton et al. [2018], Klaunig et al. [2003], Peters [2008], Peters et al. [2005, 2012]). Epidemiological and prospective studies in humans treated with fibrates do not show any relationship between fibrate administration and an increased incidence of liver cancer, and other key biological responses to fibrates typically observed in rodents are not observed in humans (reviewed in Klaunig et al. [2003]). Differences in PPARα including varying levels of hepatic PPARα expression, the altered binding to DNA response elements of PPARα target genes, and most recently, the function of mouse PPARα as compared with human PPARα have all been postulated to explain the species differences (reviewed in Corton et al. [2018], Klaunig et al. [2003], Peters [2008], Peters et al. [2005], Peters et al. [2012]).
There is evidence that exposure to chemicals during prenatal and perinatal development may alter the developing conceptus differentially as compared with adult, and potentially cause effects that persist/manifest later in life (Prins et al., 2008; Slotkin, 2008; Tremblay and Hamet, 2008; Weinhouse et al., 2014). Although there is widespread use of PPARα agonists and they can be detected in the environment (Barnes et al., 2008; Focazio et al., 2008), little is known about the effects of PPARα agonist administration during perinatal development. This is surprising because neonates may be exposed to chemicals at doses greater than adults (Lyche et al., 2009) and are thought to exhibit more sensitivity than adults to the adverse effects of some drugs and chemicals in general (Allegaert and van den Anker, 2015). Fetal rat liver peroxisomes are found in rat liver as early as postnatal day 15 and their presence/size and enzyme activity increases into the postnatal phase of development without administration of an exogenous PPARα agonist (Tsukada et al., 1968). Ligand activation of PPARα in fetuses achieved by administration of clofibrate to pregnant rats caused clear evidence of peroxisome proliferation but younger fetuses appeared less responsive to this effect as compared with fetuses exposed to clofibrate during later gestation (Stefanini et al., 1989). Other studies also noted that developing fetuses or neonates can exhibit varying levels of peroxisome proliferation and/or induction of hepatic PPARα target genes during perinatal development (Cibelli et al., 1988; Cimini et al., 1994; Dostal et al., 1987; Fahl et al., 1983; Singh and Lazo, 1992; Staubli et al., 1974; Stefanini et al., 1999).
The influence of developmental stage on the hepatocarcinogenic effects of PPARα agonists in rodents has not been examined to date. Additionally, the EC50 for in vitro activation of mouse PPARα by Wy-14,643 is 0.6 µM as compared with the EC50 for in vitro activation of the human PPARα, which is 5.0 µM (reviewed in Shearer and Hoekstra [2003]). Thus, previous studies showing that Ppara-null or PPARα-humanized mice are largely resistant to the hepatocarcinogenic effects of a PPARα agonist using Wy-14,643 are limited because of this differential sensitivity between the mouse and human PPARα to this agonist and because Wy-14,643 was administered for less than a year (Cheung et al., 2004; Hays et al., 2005; Morimura et al., 2006; Peters et al., 1997). Thus, it is possible that differences in the proliferative and hepatocarcinogenic effects of PPARα agonists in these mouse models could be influenced by, developmental stage, ligand affinity for PPARα, and/or the duration of administration of the PPARα agonist. For these reasons, the present study examined the effect of long-term administration of GW7647, a PPARα agonist with high affinity for the human PPARα (EC50 of 6 nM; Brown et al., 2001), on hepatocarcinogenesis using wild-type, Ppara-null, and PPARα-humanized mice with exposure beginning at the age of postnatal day 3.
MATERIALS AND METHODS
Chemical synthesis
2-Methyl-2-[[4-[2-[[(cyclohexylamino)carbonyl](4-cyclohexylbutyl)amino]ethyl]phenyl]thio]-propanoic acid (GW7647) was synthesized as described by others (Brown et al., 2001). Preliminary studies to determine the dietary concentration of GW7647 required to effectively activate PPARα were performed using GW7647 synthesized and purified by the Penn State Cancer Institute Organic Synthesis Shared Resource as briefly described in the companion paper (Foreman et al., 2021). GW7647 used for the other studies was synthesized commercially (Dalton Pharma Services, Toronto, California) and was between 96.6% and 98.4% pure based on HPLC analyses.
Mice and administration of GW7647
Mice were housed in an AAALAC-accredited animal vivarium in a temperature- and light-controlled environment (T = 25°C, 12-h light/12-h dark cycle). The studies were initiated using postnatal day 3 male offspring as determined by anogenital distance, obtained from either wild-type, Ppara-null, or PPARA-humanized homozygous breeding pairs consisting of 1 male and 1 female mouse for each genotype (Figure 1). The experimental mice were randomly assigned to each of the different treatment groups using mice from each litter, to control for potential litter effects. Neonatal mice were housed with their male littermates and the dam until the age of 3 weeks. The 3 congenic lines of mice were all on the 129/Sv genetic background (Akiyama et al., 2001; Cheung et al., 2004). Mouse pups suckled the dams during the neonatal period whereas the maternal diet was a pelleted control diet. Postnatal day 3 neonatal mice were gavaged daily with a 10-µl volume of either corn oil or corn oil containing GW7647 (10 mg GW7647/kg/body weight) until the age of postnatal day 21 when the mice were weaned (Figure 1). The dose of GW7647 delivered by gavage was comparable to the dose achieved by dietary administration of 0.01% GW7647 (based on an average inbred male mouse weight of 25 g with an average food intake of 3–5 g [Bachmanov et al., 2002]). Thus, after the mice were weaned (Figure 1) they were then fed either a pelleted control diet (using Purina 5001 diet ingredients) or one containing 0.01% GW7647 (both diets prepared by Dyets Inc., Bethlehem, Pennsylvania) and provided to mice ad libitum. For mice in the long-term groups, mice that became moribund that required early euthanasia or that died prior to scheduled euthanasia were not included for the calculation/compilation of end points for all groups.
Figure 1.

Schematic of treatments. Postnatal day 3 (PND3) male wild-type, Ppara-null, or PPARA-humanized mouse neonates were gavaged with either corn oil (vehicle control) or GW7647 (10 mg/kg) until weaning. Cohorts of neonatal pups were examined after 1 week of treatment to compare with the adult study. Other cohorts of neonatal pups were weaned on PND21. After weaning, mice were then fed either a control diet or one containing 0.01% GW7647 to provide for examination after a total of 5 weeks, 26 weeks, or long-term administration.
Isolation of samples
Male wild-type, Ppara-null, or PPARA-humanized mice were treated with either vehicle control or GW7647 for either 1 week, 5 weeks, 26 weeks, or long-term administration. The latter treatment group was initially designed for treatment of 104 weeks but the experiment was terminated early due to morbidity and/or mortality. Mice were weighed at the initiation of each experiment and daily from postnatal day 3 through postnatal day 21, but weekly thereafter, and at the time of euthanasia. Mice were euthanized after these 4 different time frames by over exposure to carbon dioxide. Serum was obtained from blood collected after euthanasia and frozen at −80°C until further use. Tissues were weighed and the weights recorded. Gross observations were noted as detected. Representative sections of tissues were snap frozen in liquid nitrogen, stored frozen at −80°C, and used for subsequent molecular/biochemical analyses as described below. Separate sections of representative tissues were also obtained and fixed in 10% phosphate-buffered formalin (PBF) (Fisher Scientific, Fair Lawn, New Jersey) for histopathologic examination as described below.
Pathology
Each liver was examined for the presence of grossly visible lesions at the time of dissection. Representative liver samples were removed and fixed in 10% PBF and embedded in paraffin. Paraffin sections were prepared from these tissue samples, sections were stained with hematoxylin and eosin and examined morphologically for the presence of carcinomas, adenomas, or preneoplastic lesions using established criteria (Thoolen et al., 2010). Histopathological analyses were performed by a pathologist who was blinded to the sample identities. Sample identities were revealed after the histopathological analyses were tabulated.
Target gene analyses of PPARα activation
Quantitative real-time polymerase chain reaction was used to measure the mRNA expression of Cyp4a1, or acyl-CoA oxidase (Acox1) as described previously (Borland et al., 2017; Zhang et al., 2016). Relative expression of each PPARα target gene was normalized to expression of the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (Gapdh) that exhibited no change in expression by any treatment. Each assay included a standard curve with greater than 85% efficiency and a no-template control.
Serum alanine aminotransferase
Serum alanine aminotransferase (ALT) was quantified from representative samples of mice as described in the companion paper (Foreman et al., 2021), using the VetScan MamMalian Liver Profile with the VetScan Chemistry Analyzer (Abaxis, Inc., Union City, California).
Western blot analysis
Liver extracts were prepared from mice-fed control or GW7647 diets as described previously (Koga et al., 2016). Hepatic extracts from mice-fed GW7647 for up to long-term administration were prepared from tissue with no grossly visible tumors. Quantitative Western blot analysis using a radioactive detection method was performed as described previously (Yao et al., 2014). The primary antibodies used were against MYC (catalog no. 9402, Cell Signaling, Danvers, Massachusetts) or lactate dehydrogenase (LDH; catalog no. 200-1173, Rockland, Gilbertsville, Pennsylvania). The expression level of MYC was normalized to LDH and are presented as fold increase compared with controls.
Statistical analysis
The data were subjected to either analysis of variance (ANOVA) followed by Tukey test for post-hoc comparisons (Prism 8.0; GraphPad Software Inc., La Jolla, California). Histopathological and tumor incidence data were analyzed for differences between groups using the Fisher exact test (Prism 8.0; GraphPad Software Inc., La Jolla, California). For all analyses, differences observed are only described when p ≤ .05.
RESULTS
GW7647 Activates Hepatic Mouse and Human PPARα in Mice
After 1 week, 5 weeks, 26 weeks, or long-term administration of GW7647 relative expression of hepatic Cyp4a10 mRNA was higher in wild-type mouse liver as compared with wild-type controls (Figure 2). This did not occur in similarly treated Ppara-null or PPARA-humanized mice (Figure 2). Relative expression of hepatic Acox1 mRNA was higher in wild-type mouse liver as compared with wild-type controls after 1 week, 5 weeks, or 26 weeks of ligand activation of PPARα by GW7647, but this effect was not observed after long-term administration of treatment (Figure 3). Ligand activation of PPARα with GW7647 had no effect on relative expression of Acox1 mRNA in Ppara-null mice (Figure 3). Compared with PPARA-humanized controls, relative expression of Acox1 mRNA was higher in PPARA-humanized mice after 1 week of GW7647 administration but this effect was not seen at later time points (Figure 3).
Figure 2.

Relative hepatic expression of the PPARα target gene cytochrome P450 4A10 (Cyp4a10) in wild-type (Ppara+/+), Ppara-null (Ppara–/–), or PPARA-humanized (PPARA) mice after either 1 week, 5 weeks, 26 weeks, or long-term administration of GW7647 administration initiated on postnatal day 3 of neonatal development. Individual mouse data are presented as circles in the scatter plots, with the mean and standard deviation shown by lines within each scatter plot. Groups with different letters are statistically significant from control at p ≤ .05.
Figure 3.

Relative hepatic expression of the PPARα target gene cytochrome acyl CoA oxidase (Acox1) in wild-type (Ppara+/+), Ppara-null (Ppara–/–), or PPARA-humanized (PPARA) mice after either 1 week, 5 weeks, 26 weeks, or long-term administration of GW7647 administration initiated on postnatal day 3 of neonatal development. Individual mouse data are presented as circles in the scatter plots, with the mean and standard deviation shown by lines within each scatter plot. Groups with different letters are statistically significant from control at p ≤ .05.
Ligand Activation of PPARα Causes Differential Effects in Liver of Wild-Type, Ppara-Null, and PPARA-Humanized Mice following Perinatal Exposure
After 1 week, 5 weeks, 26 weeks, or long-term administration of GW7647, relative liver weight was higher in wild-type mice as compared with wild-type controls and this was not observed in Ppara-null mice at any timepoint following administration of GW7647 (Figure 4). Although relative liver weight was higher in PPARA-humanized mice treated with GW7647 compared with PPARA-humanized controls after 5 weeks, relative liver weight was unchanged in PPARA-humanized mice as compared with PPARA-humanized controls after 1 week, 26 weeks, or long-term administration of GW7647 (Figure 4). Expression of MYC was assessed because it was previously shown to be regulated by PPARα-dependent turnover (Shah et al., 2007). Relative expression of MYC in liver of wild-type mice following ligand activation of PPARα with GW7647 after 5 weeks, 26 weeks, or long-term administration was higher compared with wild-type controls, and this effect was not found in similarly treated Ppara-null mice or PPARA-humanized mice (Figure 5).
Figure 4.

Relative liver weight in wild-type (Ppara+/+), Ppara-null (Ppara–/–), or PPARA-humanized (PPARA) mice after either 1 week, 5 weeks, 26 weeks, or long-term administration of GW7647 administration initiated on postnatal day 3 of neonatal development. Individual mouse data are presented as circles in the scatter plots, with the mean and standard deviation shown by lines within each scatter plot. Groups with different letters are statistically significant from control at p ≤ .05.
Figure 5.

Quantitative Western blot analysis of MYC expression (relative to LDH) in wild-type (Ppara+/+), Ppara-null (Ppara–/–), or PPARA-humanized (PPARA) mice after either 1 week, 5 weeks, 26 weeks, or long-term administration of GW7647 administration initiated on postnatal day 3 of neonatal development. Individual mouse data are presented as circles in the scatter plots, with the mean and standard deviation shown by lines within each scatter plot. Groups with different letters are statistically significant from control at p ≤ .05.
To begin to determine if hepatotoxicity was influenced by activating PPARα, serum levels of ALT and histopathological analyses of liver were performed. Ligand activation of PPARα with GW7647 was not associated with any difference in serum ALT concentration in any genotype of mice after 1 week or 5 weeks of GW7647 administration initiated during the perinatal period (Figure 6). However, average serum ALT was higher in wild-type mice after 26 weeks or long-term administration of GW7647 compared with wild-type controls, and this effect was not observed in similarly treated Ppara-null or PPARA-humanized mice (Figure 6).
Figure 6.

Serum alanine aminotransferase (ALT) in wild-type (Ppara+/+), Ppara-null (Ppara–/–), or PPARA-humanized (PPARA) mice after either 1 week, 5 weeks, 26 weeks, or long-term administration of GW7647 administration initiated on postnatal day 3 of neonatal development. Individual mouse data are presented as circles in the scatter plots, with the mean and standard deviation shown by lines within each scatter plot. Groups with different letters are statistically significant from control at p ≤ .05.
There were more wild-type mice with mild centrilobular hypertrophy after 5 weeks of administration of GW7647 compared with wild-type controls (p ≤ .05), and this effect was not observed in similarly treated Ppara-null or PPARA-humanized mice (Table 1). No differences in hepatocyte necrosis or inflammation were noted after 5 weeks of administration of GW7647 between wild-type, Ppara-null, or PPARA-humanized mice compared with controls (Table 1). Hepatic macrovesicular fatty change was not observed in control wild-type and Ppara-null mice, nor after 5 weeks of administration of GW7647 (Table 1). There was a marked increase in moderate hepatic macrovesicular fatty change in PPARA-humanized mice after 5 weeks of GW7647 administration compared with PPARA-humanized controls (Figure 7 and Table 1, p ≤ .05).
Table 1.
Effect of 5 Weeks of Ligand Activation of PPARα With GW7647 Initiated in Perinatal Development on Liver Histopathology in Wild-Type (Ppara+/+), Ppara-Null (Ppara–/–), or PPARA Humanized Mice (PPARA)
| 5 Weeks |
Ppara
+/+
|
Ppara
–/–
|
PPARA
|
||||
|---|---|---|---|---|---|---|---|
| Control | GW7647 | Control | GW7647 | Control | GW7647 | ||
| Centrilobular hypertrophy | None | 3/5 | 0/5 | 4/5 | 4/7 | 4/5 | 6/6 |
| Mild | 1/5 | 5/5 | 1/5 | 3/7 | 1/5 | 0/6 | |
| Moderate | 1/5 | 0/5 | 0/5 | 0/7 | 0/5 | 0/6 | |
| Severe | 0/5 | 0/5 | 0/5 | 0/7 | 0/5 | 0/6 | |
| Necrosis | None | 5/5 | 5/5 | 5/5 | 6/7 | 5/5 | 6/6 |
| Present | 0/5 | 0/5 | 0/5 | 1/7 | 0/5 | 0/6 | |
| Inflammation | None | 5/5 | 5/5 | 5/5 | 7/7 | 5/5 | 6/6 |
| Acute | 0/5 | 0/5 | 0/5 | 0/7 | 0/5 | 0/6 | |
| Chronic | 0/5 | 0/5 | 0/5 | 0/7 | 0/5 | 0/6 | |
| Macrovesicular fatty change | None | 5/5 | 5/5 | 5/5 | 7/7 | 5/5 | 0/6 |
| Mild | 0/5 | 0/5 | 0/5 | 0/7 | 0/5 | 0/6 | |
| Moderate | 0/5 | 0/5 | 0/5 | 0/7 | 0/5 | 6/6 | |
| Severe | 0/5 | 0/5 | 0/5 | 0/7 | 0/5 | 0/6 | |
Figure 7.
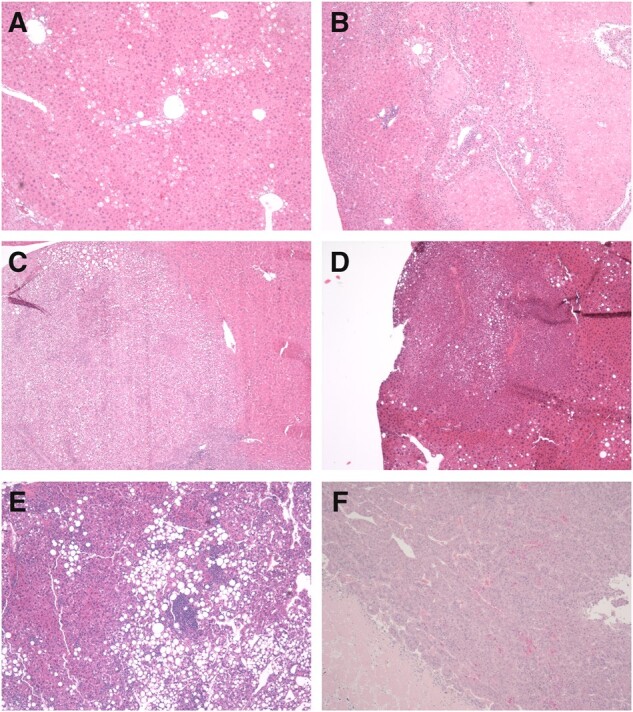
Representative photomicrographs of liver histopathology. A, Hepatic fatty change (steatosis) in a PPARA-humanized mouse liver after 5 weeks of dietary GW7647 administration. B, Hepatic necrosis in a wild-type mouse liver after 26 weeks of GW7647 administration. C, Hepatocellular adenoma in a wild-type mouse liver after long-term dietary GW7647 administration. D, Hepatocellular carcinoma in a control Ppara-null mouse liver after long-term administration of GW7647. E, Hepatocellular carcinoma in a control PPARA-humanized mouse liver. Note fatty change. F, Hepatocellular carcinoma in a PPARA-humanized mouse liver after long-term administration of GW7647. Magnification = 40×.
Fewer wild-type mice exhibited mild to moderate centrilobular hypertrophy after 26 weeks of GW7647 administration compared with wild-type controls and this effect was not observed in Ppara-null mice (Table 2, p ≤ .05). By contrast, centrilobular hypertrophy was essentially absent in both control and GW7647-treated PPARA-humanized mice (Table 2). Hepatic necrosis was markedly higher in wild-type mice administered GW7647 for 26 weeks compared with wild-type controls (Figure 7 and Table 2, p ≤ .05), and this effect was not found in either Ppara-null or PPARA-humanized mice (Table 2). There was no difference in hepatic inflammation between wild-type or PPARA-humanized mice independent of treatment, however chronic hepatic inflammation was observed in Ppara-null mice (both control and GW7647 treated) compared with both wild-type and PPARA-humanized mice (Table 2). Macrovesicular fatty change was higher in wild-type mice administered GW7647 for 26 weeks compared with controls (Table 2, p ≤ .05). Interestingly, macrovesicular fatty change was higher in Ppara-null and PPARA-humanized mice (both control and GW7647 treated) compared with control wild-type mice (Table 2, p ≤ .05).
Table 2.
Effect of 26 Weeks of Ligand Activation of PPARα With GW7647 Initiated in Perinatal Development on Liver Histopathology in Wild-Type (Ppara+/+), Ppara-Null (Ppara–/–), or PPARA Humanized Mice (PPARA)
| 26 Weeks |
Ppara
+/+
|
Ppara
–/–
|
PPARA
|
||||
|---|---|---|---|---|---|---|---|
| Control | GW7647 | Control | GW7647 | Control | GW7647 | ||
| Centrilobular hypertrophy | None | 1/8 | 10/12 | 3/10 | 0/12 | 10/11 | 10/10 |
| Mild | 7/8 | 1/12 | 6/10 | 8/12 | 1/11 | 0/10 | |
| Moderate | 0/8 | 1/12 | 1/10 | 4/12 | 0/11 | 0/10 | |
| Severe | 0/8 | 0/12 | 0/10 | 0/12 | 0/11 | 0/10 | |
| Necrosis | None | 8/8 | 7/12 | 9/10 | 12/12 | 11/11 | 10/10 |
| Present | 0/8 | 5/12 | 1/10 | 0/12 | 0/11 | 0/10 | |
| Inflammation | None | 8/8 | 11/12 | 7/10 | 10/12 | 11/11 | 10/10 |
| Acute | 0/8 | 1/12 | 0/10 | 0/12 | 0/11 | 0/10 | |
| Chronic | 0/8 | 0/12 | 3/10 | 2/12 | 0/11 | 0/10 | |
| Macrovesicular fatty change | None | 8/8 | 0/12 | 3/10 | 0/12 | 2/11 | 2/10 |
| Mild | 0/8 | 5/12 | 6/10 | 8/12 | 5/11 | 6/10 | |
| Moderate | 0/8 | 7/12 | 1/10 | 4/12 | 4/11 | 2/10 | |
| Severe | 0/8 | 0/12 | 0/10 | 0/12 | 0/11 | 0/10 | |
Phenotypic Variance in the Response to Chronic Ligand Activation of Mouse or Human PPARα in a Mouse Model
Average body weight was not different between treatment groups in any genotype in response to long-term administration of GW7647 initiated on postnatal day 3 of perinatal development and extended into adulthood (Figure 8). The incidence of morbidity/mortality in wild-type mice in response to long-term administration of GW7647 was higher compared with wild-type controls, and both control and GW7647 treated Ppara-null and PPARA-humanized mice (Table 3, p ≤ .05). Some mice became moribund and required early euthanasia. The age at time of euthanasia was lower in wild-type mice after long-term administration of GW7647 compared with similarly treated Ppara-null mice or PPARA-humanized mice (Figure 9).
Figure 8.

Average weight gain in wild-type (Ppara+/+), Ppara-null (Ppara–/–), or PPARA-humanized (PPARA) mice during GW7647 administration initiated as adults. Body weight was measured every 4 weeks and values represent the mean ± SD.
Table 3.
Effect of Long-Term Ligand Activation of PPARα With GW7647 Initiated in Perinatal Development on Liver Histopathology (and Overtly Present Liver Lesions) in Wild-Type (Ppara+/+), Ppara-Null (Ppara–/–), or PPARA Humanized Mice (PPARA)
|
Ppara
+/+
|
Ppara
–/–
|
PPARA
|
|||||
|---|---|---|---|---|---|---|---|
| Control | GW7647 | Control | GW7647 | Control | GW7647 | ||
| Centrilobular hypertrophy | None | 6/13 | 6/7 | 6/11 | 12/13 | 14/17 | 0/14 |
| Mild | 6/13 | 1/7 | 5/11 | 1/13 | 3/17 | 8/14 | |
| Moderate | 1/13 | 0/7 | 0/11 | 0/13 | 0/17 | 5/14 | |
| Severe | 0/13 | 0/7 | 0/11 | 0/13 | 0/17 | 1/14 | |
| Necrosis | None | 13/13 | 5/7 | 10/11 | 12/13 | 16/17 | 13/14 |
| Present | 0/13 | 2/7 | 1/11 | 1/13 | 1/17 | 1/14 | |
| Inflammation | None | 5/13 | 0/7 | 4/11 | 4/13 | 6/17 | 3/14 |
| Acute | 0/13 | 0/7 | 0/11 | 0/13 | 0/17 | 0/14 | |
| Chronic | 8/13 | 7/7 | 7/11 | 9/13 | 11/17 | 11/14 | |
| Macrovesicular fatty change | None | 13/13 | 3/7 | 7/11 | 4/13 | 5/17 | 8/14 |
| Mild | 0/13 | 3/7 | 4/11 | 6/13 | 5/17 | 3/14 | |
| Moderate | 0/13 | 1/7 | 0/11 | 3/13 | 7/17 | 3/14 | |
| Severe | 0/13 | 0/7 | 0/11 | 0/13 | 0/17 | 0/14 | |
| Tumora | Total | 1/13 | 6/7 | 1/11 | 1/13 | 1/17 | 4/14 |
| Hepatocellular adenoma | 1/13 | 5/7 | 1/11 | 1/13 | 1/17 | 4/14 | |
| Hepatocellular carcinoma | 0/13 | 1/7 | 0/11 | 0/13 | 0/17 | 1/14 | |
| Gross findingsb | Tumor-like | 0/15 | 14/14 | 2/13 | 4/17 | 3/16 | 5/14 |
| Morbidity/Mortalityc | 1/16 | 7/14 | 2/15 | 0/17 | 1/17 | 2/16 | |
The number of tumors per slide identified histopathologically per group.
The number of mice with gross findings in the liver at the time of necropsy.
Mice that died or were euthanized for health reasons.
Figure 9.

Age at termination in wild-type (Ppara+/+), Ppara-null (Ppara–/–), or PPARA-humanized (PPARA) mice following long-term GW7647 administration initiated as adults. Values represent the mean ± SD. Data with different letters are statistically significant at p ≤ .05.
Consistent with past studies (Maronpot et al., 2010), centrilobular hypertrophy was not noted in any group after long-term GW7647 administration with the exception of PPARA-humanized mice (Table 3). The incidence of hepatocellular necrosis or inflammation was not different between any group or genotype after long-term administration of GW7647 (Table 3). The incidence of hepatic macrovesicular fatty change was higher in wild-type mice after administration of GW7647 compared with wild-type controls (Table 3, p ≤ .05). Additionally, hepatic macrovesicular fatty change was higher in Ppara-null and PPARA-humanized mice with and without administration of GW7647 compared with wild-type controls (Table 3, p ≤ .05).
Livers were examined with a surface light source after euthanasia. The incidence of grossly apparent liver tumors was similar between control wild-type mice compared with control Ppara-null or control PPARA-humanized mice (Table 3). The incidence of gross liver tumors was 86% in wild-type mice treated with GW7647 (6/7) compared with controls with only 1 of 13 mice exhibiting a liver tumor (Table 3, p ≤ .05). The incidence of grossly apparent liver tumors was lower in Ppara-null and PPARA-humanized administered GW7647 as compared with similarly treated wild-type mice (Table 3, p ≤ .05). Histopathological analyses of adenomas and/or carcinomas by light microscopy were consistent with the assessment noted by gross tumor examination between each genotype and treatment group at the time of necropsy (Table 3 and Figure 7). Adenomas and/or carcinomas verified by histopathology were collectively higher in response to long-term administration of GW7647 to wild-type mice compared with wild-type controls (p ≤ .05), and this effect was not found in similarly treated Ppara-null or PPARA-humanized mice (Table 3).
DISCUSSION
There is generally consensus that species differences exist in the response to ligand activation of mouse versus human PPARα (Felter et al., 2018). For example, receptor activities including downstream modulation of gene expression likely explain why rodents develop hepatocarcinogenesis after chronic activation of PPARα, but there are also convincing data indicating that PPARα agonists do not cause liver cancer in humans (Corton et al., 2014, 2018; Klaunig et al., 2003; Peters et al., 2005). PPARα agonist-induced hepatocarcinogenesis is initiated by ligand activation of PPARα, that leads to associative changes in lipid metabolizing enzymes and proteins, and causally related changes in cell cycle signaling leading to increased hepatocellular proliferation and an increased opportunity for spontaneous mutations to contribute to the formation of tumors (Corton et al., 2014, 2018; Klaunig et al., 2003; Peters et al., 2005). However, it was also suggested that this mode of action and the key events induced by ligand activation of PPARα cannot be definitively excluded as a risk to humans for the development of liver cancer (Guyton et al., 2009; Keshava and Caldwell, 2006). For example, studies showing that PPARα is required to mediate liver cancer in mice were approximately 1 year or less (Hays et al., 2005; Morimura et al., 2006; Peters et al., 1997), rather than a typical 2-year cancer bioassay. Additionally, another study revealed that hepatocarcinogenesis was found at low incidence in Ppara-null mice treated with a low affinity PPARα activator (Ito et al., 2007). Differences between these latter observations have recently been discussed in detail Corton et al. (2018).
Results from the present studies are highly impactful because they provide additional evidence to address weaknesses in previous experiments noted by others (Guyton et al., 2009; Keshava and Caldwell, 2006) as well as confirm and provide more evidence that PPARα is required to mediate the hepatocarcinogenic effects of long-term administration of a potent human PPARα agonist in a mouse model. The present studies are significant because of the extended length of time and the inclusion of both the loss of function and the PPARA-humanized mouse models; study design elements that allow for direct comparison between models in examining the functional role of this receptor in PPARα agonist-induced hepatocarcinogenesis. Whereas activating PPARα with GW7647 in wild-type mice causes key events to occur that are part of the mode of action for PPARα agonist-induced hepatocarcinogenesis, Ppara-null mice do not exhibit these changes. This includes increased signaling that promotes cell proliferation (eg, MYC expression) increasing opportunities for cells to ultimately form tumors. Consistent with other studies (Brocker et al., 2017), it is also of interest to note the PPARα-dependent increase in serum ALT, a biomarker of cytotoxicity, a change confirmed by histopathology. The higher incidence of hepatocyte cytotoxicity in wild-type mice treated with GW7647 likely explains the comparable changes in necrosis observed in wild-type mice treated with GW7647 but not similarly treated Ppara-null mice. Cytotoxicity and necrosis induced by ligand activation of PPARα with GW7647 are likely also part of the molecular events that contribute to the mechanisms causing liver cancer by the human PPARα agonist GW7647. These changes occurred very early after ligand activation of PPARα in wild-type mice, present within 5 weeks and after 26 weeks but were not observed in similarly treated Ppara-null or PPARA-humanized mice. This provides new evidence that PPARα is required to increase cytotoxicity by GW7647 and may also underlie the hepatocarcinogenic effects of PPARα agonists. Additionally, this change does not occur by activation of the human PPARα in a mouse model suggesting that a species difference may exist for this phenotype as well.
Results from the present studies also show that the human PPARα is active in a mouse model, consistent with previous studies (Cheung et al., 2004; Morimura et al., 2006). For example, PPARA-humanized mice also exhibit increased expression of target genes involved with lipid metabolism in response to ligand activation of PPARα by the potent human PPARα agonist GW7647, a phenotype previously observed in this model in response to a PPARα agonist with relative greater affinity for the mouse receptor compared with the human receptor (WY-14,643 vs. GW7647; based solely only on in vitro assessment) (Cheung et al., 2004; Morimura et al., 2006). The findings that PPARα agonists activate both the mouse and human receptor supports numerous studies that have shown that PPARα mediates the hypolipidemic effects of drugs that target this receptor. However, there are also differences in the phenotype of the PPARA-humanized mice as compared with mice that express the mouse PPARα that provide new insight into the functional role of mouse versus human PPARα in PPARα agonist-induced hepatocarcinogenesis.
The effect of activating PPARα in PPARA-humanized mice by chronic administration of GW7647 initiated on postnatal day 3 is remarkably similar to the phenotype of Ppara-null mice following GW7647 administration as the incidence of hepatocarcinogenesis was lower in both Ppara-null and PPARA-humanized mice compared with wild-type mice. Ligand activation of PPARα was associated with mild to moderate hepatocyte macrovesicular fatty change in wild-type mice but only after 5 weeks of GW7647 administration. By contrast, mild to moderate hepatocyte macrovesicular fatty change was observed in control Ppara-null and PPARA-humanized mice after 26 weeks and long-term administration of GW7647; an effect that was unchanged in either genotype by activation of PPARα with GW7647. Interestingly, this effect was also noted even earlier in PPARA-humanized mice after only 5 weeks of GW7647 administration. Because fatty change is a form of cytotoxicity (Kanda et al., 2020) and a known risk factor for liver cancer, it remains possible that the increase in hepatic lipids detected in the present studies are mechanistically linked to the relatively low incidence of hepatocarcinogenesis observed in both Ppara-null and PPARA-humanized mice. Higher concentrations of hepatic lipid and/or cytotoxicity could lead to enhanced oxidative stress, a hypothesis currently under investigation.
Because it is known that untreated Ppara-null mice exhibit increased hepatic lipids (Kersten et al., 1999), most likely because of limited ability to catabolize fatty acids for energy due to low constitutive expression of lipid catabolizing enzymes and transport proteins (Aoyama et al., 1998), the reduced constitutive catabolism of fatty acids could cause changes in hepatic lipid accumulation in Ppara-null mice. This might also explain why liver tumors were observed in Ppara-null mice. For example, it was originally shown that relatively older Ppara-null mice develop spontaneous liver tumors with greater incidence compared with control wild-type mice (Howroyd et al., 2004). After chronic administration of another PPARα agonist bezafibrate, a single adenoma was also observed in a Ppara-null mouse (Hays et al., 2005). Others have suggested that a PPARα agonists may cause hepatocarcinogenesis through a mechanism that is not mediated by PPARα (Ito et al., 2007). This was based in part on the observation that Ppara-null mice treated with a weak PPARα agonist (di(2-ethyhexl)phthalate) exhibited a relatively higher incidence of hepatocarcinogenesis compared with similarly treated wild-type mice (Ito et al., 2007). However, the latter study is inconsistent with the present studies and previous studies showing that hepatocarcinogenesis induced by more potent PPARα agonists (GW7647, WY-14,643, bezafibrate) is largely absent or lacking in Ppara-null mice (Hays et al., 2005; Morimura et al., 2006; Peters et al., 1997). There are no known null mutations in humans for PPARA, therefore the phenotype of Ppara-null mice appears to be negatively impacted by the molecular silencing of PPARα expression that elicits hepatic effects (eg, fatty change) resulting in a low-level incidence of hepatocarcinogenesis. Thus, the Ppara-null and PPARA-humanized mice are good models to examine the role of PPARα in liver cancer, but the background incidence of hepatocarcinogenesis must be controlled for in both models.
Another major finding from these studies is the demonstration that exposure to developing perinatal mice did not lead to altered sensitivity to ligand activation of PPARα compared with mice where initiation of ligand activation of PPARα occurs with adults, as suggested by previous work (Cibelli et al., 1988; Cimini et al., 1994; Dostal et al., 1987; Fahl et al., 1983; Singh and Lazo, 1992; Staubli et al., 1974). Indeed, adverse effects of drugs and chemicals could differ as a result of perinatal exposure compared with adult exposure (Allegaert and van den Anker, 2015). Although there are data suggesting that neonatal rodents may exhibit different sensitivity to ligand activation of PPARα (Cibelli et al., 1988; Cimini et al., 1994; Dostal et al., 1987; Fahl et al., 1983; Singh and Lazo, 1992; Staubli et al., 1974), results from the present study indicate that mice do not exhibit either decreased or increased sensitivity to the hepatocarcinogenic effects of a potent human PPARα agonist when exposure is initiated during early postnatal development. Although this is impactful and will be an important reference in the future, further studies with other PPARα agonist using similar dosing paradigms would strengthen these observations.
Lastly, it is worth noting that results from the present studies are consistent with the companion studies that examined identical endpoints using the same experimental approach, with the exception that exposure to GW7647 was initiated in adult mice (Foreman et al., 2021). Both studies indicate that activation of the mouse PPARα with the potent human PPARα agonist GW7647 causes liver cancer in mice by promoting cell proliferation of hepatocytes by PPARα-dependent regulation of MYC and cytotoxicity, and that these effects are absent or largely diminished in similarly treated PPARA-humanized mice. Collectively, these studies provide new rigorous evidence that species differences exist in the responses mediated by either mouse or human PPARα, in particular hepatocarcinogenesis.
ACKNOWLEDGMENTS
The authors gratefully acknowledge the Penn State Cancer Institute Organic Synthesis Shared Resource for the synthesis of some of the GW7647 used for some of these studies. The authors thank Dr Lance Kramer and Mr Leonard Collins of the UNC Biomarker Facility Core for technical assistance with these studies.
FUNDING
This work supported in part by the National Institutes of Health (ES017568, J.E.F.), the USDA National Institute of Food and Federal Appropriations under Project PEN04607 and Accession number 1009993 (J.M.P), the Pennsylvania Department of Health using Tobacco C.U.R.E. Funds (J.M.P). The department specifically disclaims responsibility for any analyses, interpretations, or conclusions.
DECLARATION OF CONFLICTING INTERESTS
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
REFERENCES
- Akiyama T. E., Nicol C. J., Fievet C., Staels B., Ward J. M., Auwerx J., Lee S. S., Gonzalez F. J., Peters J. M. (2001). Peroxisome proliferator-activated receptor-α regulates lipid homeostasis, but is not associated with obesity: Studies with congenic mouse lines. J. Biol. Chem. 276, 39088–39093. [DOI] [PubMed] [Google Scholar]
- Akiyama T. E., Sakai S., Lambert G., Nicol C. J., Matsusue K., Pimprale S., Lee Y. H., Ricote M., Glass C. K., Brewer H. B. Jr., et al. (2002). Conditional disruption of the peroxisome proliferator-activated receptor γ gene in mice results in lowered expression of ABCA1, ABCG1, and apoE in macrophages and reduced cholesterol efflux. Mol. Cell. Biol. 22, 2607–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allegaert K., van den Anker J. N. (2015). Adverse drug reactions in neonates and infants: A population-tailored approach is needed. Br. J. Clin. Pharmacol. 80, 788–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyama T., Peters J. M., Iritani N., Nakajima T., Furihata K., Hashimoto T., Gonzalez F. J. (1998). Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor α (PPARα). J. Biol. Chem. 273, 5678–5684. [DOI] [PubMed] [Google Scholar]
- Bachmanov A. A., Reed D. R., Beauchamp G. K., Tordoff M. G. (2002). Food intake, water intake, and drinking spout side preference of 28 mouse strains. Behav. Genet. 32, 435–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes K. K., Kolpin D. W., Furlong E. T., Zaugg S. D., Meyer M. T., Barber L. B. (2008). A national reconnaissance of pharmaceuticals and other organic wastewater contaminants in the United States–I groundwater. Sci. Total Environ. 402, 192–200. [DOI] [PubMed] [Google Scholar]
- Berger J., Moller D. E. (2002). The mechanisms of action of PPARs. Annu. Rev. Med. 53, 409–435. [DOI] [PubMed] [Google Scholar]
- Borland M. G., Yao P.-L., Kehres E. M., Lee C., Pritzlaff A. M., Ola E., Wagner A. L., Shannon B. E., Albrecht P. P., Zhu B., et al. (2017). Editor's highlight: PPARβ/δ and PPARγ inhibit melanoma tumorigenicity by modulating inflammation and apoptosis. Toxicol. Sci. 159, 436–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocker C. N., Yue J., Kim D., Qu A., Bonzo J. A., Gonzalez F. J. (2017). Hepatocyte-specific PPARA expression exclusively promotes agonist-induced cell proliferation without influence from nonparenchymal cells. Am. J. Physiol. Gastrointest. Liver Physiol. 312, G283–G299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown P. J., Stuart L. W., Hurley K. P., Lewis M. C., Winegar D. A., Wilson J. G., Wilkison W. O., Ittoop O. R., Willson T. M. (2001). Identification of a subtype selective human PPARα agonist through parallel-array synthesis. Bioorg. Med. Chem. Lett. 11, 1225–1227. [DOI] [PubMed] [Google Scholar]
- Cheung C., Akiyama T. E., Ward J. M., Nicol C. J., Feigenbaum L., Vinson C., Gonzalez F. J. (2004). Diminished hepatocellular proliferation in mice humanized for the nuclear receptor peroxisome proliferator-activated receptor-α. Cancer Res. 64, 3849–3854. [DOI] [PubMed] [Google Scholar]
- Cibelli A., Stefanini S., Ceru M. P. (1988). Peroxisomal beta-oxidation and catalase activities in fetal rat liver: effect of maternal treatment with clofibrate. Cell. Mol. Biol. 34, 191–205. [PubMed] [Google Scholar]
- Cimini A. M., Sulli A., Stefanini S., Serafini B., Moreno S., Rossi L., Giorgi M., Ceru M. P. (1994). Effects of Di-(2-ethylhexyl)phthalate on peroxisomes of liver, kidney and brain of lactating rats and their pups. Cell. Mol. Biol. 40, 1063–1076. [PubMed] [Google Scholar]
- Corton J. C., Cunningham M. L., Hummer B. T., Lau C., Meek B., Peters J. M., Popp J. A., Rhomberg L., Seed J., Klaunig J. E. (2014). Mode of action framework analysis for receptor-mediated toxicity: The peroxisome proliferator-activated receptor α (PPARα) as a case study. Crit. Rev. Toxicol. 44, 1–49. [DOI] [PubMed] [Google Scholar]
- Corton J. C., Peters J. M., Klaunig J. E. (2018). The PPARα-dependent rodent liver tumor response is not relevant to humans: addressing misconceptions. Arch. Toxicol. 92, 83–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostal L. A., Jenkins W. L., Schwetz B. A. (1987). Hepatic peroxisome proliferation and hypolipidemic effects of di(2-ethylhexyl)phthalate in neonatal and adult rats. Toxicol. Appl. Pharmacol. 87, 81–90. [DOI] [PubMed] [Google Scholar]
- Fahl W. E., Lalwani N. D., Reddy M. K., Reddy J. K. (1983). Induction of peroxisomal enzymes in livers of neonatal rats exposed to lactating mothers treated with hypolipidaemic drugs. Role of drug metabolite transfer in milk. Biochem. J. 210, 875–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felter S. P., Foreman J. E., Boobis A., Corton J. C., Doi A. M., Flowers L., Goodman J., Haber L. T., Jacobs A., Klaunig J. E., et al. (2018). Human relevance of rodent liver tumors: Key insights from a Toxicology Forum workshop on nongenotoxic modes of action. Regul. Toxicol. Pharmacol. 92, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Focazio M. J., Kolpin D. W., Barnes K. K., Furlong E. T., Meyer M. T., Zaugg S. D., Barber L. B., Thurman M. E. (2008). A national reconnaissance for pharmaceuticals and other organic wastewater contaminants in the United States–II) untreated drinking water sources. Sci. Total Environ. 402, 201–216. [DOI] [PubMed] [Google Scholar]
- Foreman J. E., Koga T., Kosyk O., Kang B.-H., Zhu X., Cohen S. M., Billy L. J., Sharma A., Amin S., Gonzalez F. J., et al. (2021). Diminished hepatocarcinogenesis by a potent, high affinity human PPARα agonist in PPARA-humanized mice. Toxicol. Sci. 183, 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruchart J. C., Brewer H. B. Jr.,, Leitersdorf E. (1998). Consensus for the use of fibrates in the treatment of dyslipoproteinemia and coronary heart disease. Fibrate Consensus Group. Am. J. Cardiol. 81, 912–917. [DOI] [PubMed] [Google Scholar]
- Guyton K. Z., Chiu W. A., Bateson T. F., Jinot J., Scott C. S., Brown R. C., Caldwell J. C. (2009). A reexamination of the PPAR-α activation mode of action as a basis for assessing human cancer risks of environmental contaminants. Environ. Health Perspect. 117, 1664–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hays T., Rusyn I., Burns A. M., Kennett M. J., Ward J. M., Gonzalez F. J., Peters J. M. (2005). Role of peroxisome proliferator-activated receptor-α (PPARα) in bezafibrate-induced hepatocarcinogenesis and cholestasis. Carcinogenesis 26, 219–227. [DOI] [PubMed] [Google Scholar]
- Heikkinen S., Auwerx J., Argmann C. A. (2007). PPARγ in human and mouse physiology. Biochim. Biophys. Acta 1771, 999–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howroyd P., Swanson C., Dunn C., Cattley R. C., Corton J. C. (2004). Decreased longevity and enhancement of age-dependent lesions in mice lacking the nuclear receptor peroxisome proliferator-activated receptor α (PPARα). Toxicol. Pathol. 32, 591–599. [DOI] [PubMed] [Google Scholar]
- Issemann I., Green S. (1990). Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 347, 645–650. [DOI] [PubMed] [Google Scholar]
- Ito Y., Yamanoshita O., Asaeda N., Tagawa Y., Lee C. H., Aoyama T., Ichihara G., Furuhashi K., Kamijima M., Gonzalez F. J., et al. (2007). Di(2-ethylhexyl)phthalate induces hepatic tumorigenesis through a peroxisome proliferator-activated receptor α-independent pathway. J. Occup. Health 49, 172–182. [DOI] [PubMed] [Google Scholar]
- Kanda T., Goto T., Hirotsu Y., Masuzaki R., Moriyama M., Omata M. (2020). Molecular mechanisms: Connections between nonalcoholic fatty liver disease, steatohepatitis and hepatocellular carcinoma. Int. J. Mol. Sci. 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten S., Seydoux J., Peters J. M., Gonzalez F. J., Desvergne B., Wahli W. (1999). Peroxisome proliferator-activated receptor α mediates the adaptive response to fasting. J. Clin. Invest. 103, 1489–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshava N., Caldwell J. C. (2006). Key issues in the role of peroxisome proliferator-activated receptor agonism and cell signaling in trichloroethylene toxicity. Environ. Health Perspect. 114, 1464–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaunig J. E., Babich M. A., Baetcke K. P., Cook J. C., Corton J. C., David R. M., DeLuca J. G., Lai D. Y., McKee R. H., Peters J. M., et al. (2003). PPARα agonist-induced rodent tumors: modes of action and human relevance. Crit. Rev. Toxicol. 33, 655–780. [DOI] [PubMed] [Google Scholar]
- Koga T., Yao P. L., Goudarzi M., Murray I. A., Balandaram G., Gonzalez F. J., Perdew G. H., Fornace A. J. Jr.,, Peters J. M. (2016). Regulation of cytochrome P450 2B10 (CYP2B10) expression in liver by peroxisome proliferator-activated receptor-β/δ modulation of SP1 promoter occupancy. J. Biol. Chem. 291, 25255–25263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. S., Pineau T., Drago J., Lee E. J., Owens J. W., Kroetz D. L., Fernandez-Salguero P. M., Westphal H., Gonzalez F. J. (1995). Targeted disruption of the α isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol. Cell. Biol. 15, 3012–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyche J. L., Gutleb A. C., Bergman A., Eriksen G. S., Murk A. J., Ropstad E., Saunders M., Skaare J. U. (2009). Reproductive and developmental toxicity of phthalates. J. Toxicol. Environ. Health. B Crit. Rev. 12, 225–249. [DOI] [PubMed] [Google Scholar]
- Maronpot R. R., Yoshizawa K., Nyska A., Harada T., Flake G., Mueller G., Singh B., Ward J. M. (2010). Hepatic enzyme induction: histopathology. Toxicol. Pathol. 38, 776–795. [DOI] [PubMed] [Google Scholar]
- Morimura K., Cheung C., Ward J. M., Reddy J. K., Gonzalez F. J. (2006). Differential susceptibility of mice humanized for peroxisome proliferator-activated receptor α to Wy-14,643-induced liver tumorigenesis. Carcinogenesis 27, 1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J. M. (2008). Mechanistic evaluation of PPAR-mediated hepatocarcinogenesis: Are we there yet? Toxicol. Sci. 101, 1–3. [Google Scholar]
- Peters J. M., Cattley R. C., Gonzalez F. J. (1997). Role of PPARα in the mechanism of action of the nongenotoxic carcinogen and peroxisome proliferator Wy-14,643. Carcinogenesis 18, 2029–2033. [DOI] [PubMed] [Google Scholar]
- Peters J. M., Cheung C., Gonzalez F. J. (2005). Peroxisome proliferator-activated receptor-α and liver cancer: where do we stand? J. Mol. Med. 83, 774–785. [DOI] [PubMed] [Google Scholar]
- Peters J. M., Lee S. S., Li W., Ward J. M., Gavrilova O., Everett C., Reitman M. L., Hudson L. D., Gonzalez F. J. (2000). Growth, adipose, brain and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor β(δ). Mol. Cell. Biol. 20, 5119–5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J. M., Shah Y. M., Gonzalez F. J. (2012). The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nat. Rev. Cancer 12, 181–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J. M., Walter V., Patterson A. D., Gonzalez F. J. (2019). Unraveling the role of peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) expression in colon carcinogenesis. NPJ Precis. Oncol. 3, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins G. S., Tang W. Y., Belmonte J., Ho S. M. (2008). Perinatal exposure to oestradiol and bisphenol A alters the prostate epigenome and increases susceptibility to carcinogenesis. Basic Clin. Pharmacol. Toxicol. 102, 134–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy J. K., Azarnoff D. L., Hignite C. E. (1980). Hypolipidaemic hepatic peroxisome proliferators form a novel class of chemical carcinogens. Nature 283, 397–398. [DOI] [PubMed] [Google Scholar]
- Shah Y. M., Morimura K., Yang Q., Tanabe T., Takagi M., Gonzalez F. J. (2007). Peroxisome proliferator-activated receptor α regulates a microRNA-mediated signaling cascade responsible for hepatocellular proliferation. Mol. Cell. Biol. 27, 4238–4247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearer B. G., Hoekstra W. J. (2003). Recent advances in peroxisome proliferator-activated receptor science. Curr. Med. Chem. 10, 267–280. [DOI] [PubMed] [Google Scholar]
- Singh I., Lazo O. (1992). Peroxisomal enzyme activities in brain and liver of pups of lactating mothers treated with ciprofibrate. Neurosci. Lett. 138, 283–286. [DOI] [PubMed] [Google Scholar]
- Slotkin T. A. (2008). If nicotine is a developmental neurotoxicant in animal studies, dare we recommend nicotine replacement therapy in pregnant women and adolescents? Neurotoxicol. Teratol. 30, 1–19. [DOI] [PubMed] [Google Scholar]
- Staubli W., Schweizer W., Suter J., Hess R. (1974). Ultrastructural and biochemical study of the action of benzoctamine and maprotiline on the rat liver. Agents Actions 4, 391–403. [DOI] [PubMed] [Google Scholar]
- Stefanini S., Mauriello A., Farrace M. G., Cibelli A., Ceru M. P. (1989). Proliferative response of foetal liver peroxisomes to clofibrate treatment of pregnant rats. A quantitative evaluation. Biol. Cell 67, 299–305. [PubMed] [Google Scholar]
- Stefanini S., Nardacci R., Farioli-Vecchioli S., Pajalunga D., Sartori C. (1999). Liver and kidney peroxisomes in lactating rats and their pups after treatment with ciprofibrate. Biochemical and morphometric analysis. Cell. Mol. Biol. 45, 815–829. [PubMed] [Google Scholar]
- Thoolen B., Maronpot R. R., Harada T., Nyska A., Rousseaux C., Nolte T., Malarkey D. E., Kaufmann W., Kuttler K., Deschl U., et al. (2010). Proliferative and nonproliferative lesions of the rat and mouse hepatobiliary system. Toxicol. Pathol. 38, 5s–81s. [DOI] [PubMed] [Google Scholar]
- Tremblay J., Hamet P. (2008). Impact of genetic and epigenetic factors from early life to later disease. Metabolism 57(Suppl 2), S27–31. [DOI] [PubMed] [Google Scholar]
- Tsukada H., Mochizuki Y., Konishi T. (1968). Morphogenesis and development of microbodies of hepatocytes of rats during pre- and postnatal growth. J. Cell Biol. 37, 231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinhouse C., Anderson O. S., Bergin I. L., Vandenbergh D. J., Gyekis J. P., Dingman M. A., Yang J., Dolinoy D. C. (2014). Dose-dependent incidence of hepatic tumors in adult mice following perinatal exposure to bisphenol A. Environ. Health Perspect. 122, 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao P. L., Morales J. L., Zhu B., Kang B. H., Gonzalez F. J., Peters J. M. (2014). Activation of peroxisome proliferator-activated receptor-β/δ (PPAR-β/δ) inhibits human breast cancer cell line tumorigenicity. Mol. Cancer Ther. 13, 1008–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Krishnan P., Ehresman D. J., Smith P. B., Dutta M., Bagley B. D., Chang S. C., Butenhoff J. L., Patterson A. D., Peters J. M. (2016). Editor's highlight: Perfluorooctane sulfonate-choline ion pair formation: A potential mechanism modulating hepatic steatosis and oxidative stress in mice. Toxicol. Sci. 153, 186–197. [DOI] [PubMed] [Google Scholar]