

Abstract
In systemic lupus erythematosus, nephrotic-range proteinuria typically signals the presence of a proliferative lupus nephritis (class III/IV) and/or membranous lupus nephritis (class V, with or without concomitant class III or IV lesions). However, in rare instances, systemic lupus erythematosus patients with nephrotic syndrome have kidney biopsy findings of normal glomeruli or focal segmental glomerulosclerosis lesions, with or without mesangial proliferation, on light microscopy; the absence of subepithelial or subendothelial deposits on immunofluorescence and electron microscopy; and diffuse foot process effacement on electron microscopy. This pattern, termed lupus podocytopathy, is a unique form of lupus nephritis that mimics minimal change disease or primary focal segmental glomerulosclerosis and represents approximately 1% of lupus nephritis biopsies. Here we review the clinical features, histological manifestations, diagnostic criteria and classification, pathogenesis, treatment, and prognosis of lupus podocytopathy.
Keywords: Systemic lupus erythematosus, Lupus nephritis, Lupus podocytopathy, Minimal change disease, Focal segmental glomerulosclerosis
Lupus podocytopathy is a glomerular lesion in systemic lupus erythematosus (SLE) characterized by diffuse epithelial cell foot process effacement (FPE) without immune complex deposition or with only mesangial immune complex deposition.1–3 A patient with SLE, presenting with nephrotic syndrome and kidney biopsy findings of normal glomeruli or focal segmental glomerulosclerosis (FSGS) lesions (with or without mesangial proliferation) on light microscopy and the absence of subepithelial or subendothelial deposition, should garner a diagnosis of lupus podocytopathy, which is confirmed by electron microscopy showing diffuse FPE.1–3 In SLE, the nephrotic syndrome far more commonly appears as proliferative class III/IV lupus nephritis (LN) and class V membranous LN (with or without concomitant proliferative lesions), related to endocapillary proliferation and immune complex deposition in glomerular capillary walls.4 Nevertheless, in SLE, nephrotic syndrome can also, in rare instances, appear as lupus podocytopathy, mimicking a minimal change disease (MCD) or primary FSGS.
In the 1980s cases of SLE associated with MCD were reported as patients with steroid-dependent nephrotic syndrome.5,6 Subsequently, simultaneous onset of mesangial LN and minimal change nephrotic syndrome7 or minimal change nephrotic syndrome during the course of SLE8 was described. Hickman and colleagues9 reported a case of concurrent SLE and FSGS. Finally, in 2002, Dube and colleagues3 and Hertig and colleagues10 separately described a series of patients (n = 18 across both series) with SLE, nephrotic syndrome, and biopsy findings of MCD or FSGS. Eleven patients had MCD and seven had FSGS lesions. No subepithelial or subendothelial deposits were described, but 44% of patients (8 of 18 cases) had mesangial deposits concurrent with a class I or class II LN (mesangial LN). Kraft and colleagues1 reported in 2005 eight additional cases of patients with SLE, nephrotic syndrome, and kidney biopsy findings of MCD, FSGS, or mesangial proliferative glomerulonephritis. The frequency of these cases and the observation that the onset of the nephrotic syndrome frequently correlates with the onset of clinical systemic features of SLE led to the idea that they are not 2 coexisting diseases but that the podocytopathy is the result of active SLE, creating the term “lupus podocytopathy.”1 In 2016, Hu and colleagues2 described the largest cohort of lupus podocytopathy with 50 cases (13 with normal light microscopy findings and hence MCD-like picture, 9 with FSGS, and 28 with mesangial proliferative changes). Forty-seven of the 50 cases (94%) had confined mesangial immune deposits by immunofluorescence and electron microscopy. Most of the patients with full nephrotic syndrome had more than 70% FPE. These publications have improved our understanding and recognition of lupus podocytopathy as a distinct entity.11
The prevalence of lupus podocytopathy represents approximately 1% of LN biopsies.2 Lupus podocytopathy can be subdivided based on light microscopy and immunofluorescence findings as MCD (normal LM without mesangial proliferation); FSGS, including the morphologic subtypes of FSGS (not-otherwise-specified [NOS], perihilar, cellular, tip, or collapsing variant); and mesangioproliferative LN (class I or II LN with concomitant podocytopathy).11 The clinical features, histological manifestations, diagnostic criteria and classification, pathogenesis, treatment, and prognosis of lupus podocytopathy will be reviewed in this article.
CLINICAL MANIFESTATIONS OF LUPUS PODOCYTOPATHY
In the largest series of lupus podocytopathy (including MCD and FSGS subtypes) from China, the main clinical manifestation was the full nephrotic syndrome. Like other forms of LN, lupus podocytopathy most commonly affects females in the age range of 20–30 years.2 Acute kidney injury (AKI) is uncommon in lupus podocytopathy (34%) but is more common in the FSGS subtype (78%)2 compared to the MCD subtype. Microscopic hematuria and hypertension are also rare in lupus podocytopathy (18%)2 and may help distinguish from other forms of LN. Nephrotic syndrome in lupus podocytopathy frequently presents as the onset symptom of SLE (88%).2 Nephrotic syndrome relapses usually correlate with lupus activity and extrarenal involvement.2,12
Hematological disorders occur frequently in lupus podocytopathy: leukopenia (44%), anemia (26%), and thrombocytopenia (20%).2 The most common extrarenal clinical manifestation of SLE in patients with lupus podocytopathy is malar rash, affecting practically one-half of the cases (46%).2 One-third of the patients can have arthritis (34%); less frequent signs/symptoms include alopecia (26%), serositis (26%), fever (22%), Raynaud phenomenon (18%), sicca syndrome (12%), and mouth ulcers (10%).2 Other studies1,3,6,9,10,13 with lower number of cases have reported similar frequencies, except for the rate of photosensitivity, which was higher (29%) in these series12 than in the large Chinese series. Regarding serological markers of SLE, all patients reported had positive antinuclear antibody.2,12 Anti-double-stranded DNA antibodies were positive in 26% of cases but reported at higher frequency in other series.1,3,6,9,10,13 Positive anti-Smith antibody and positive anticardiolipin antibody are seen at 32% and 26% frequency, respectively. Low C3 is frequently noted (68%) while low C4 is less common (28%).2
DIAGNOSIS AND CLASSIFICATION OF LUPUS PODOCYTOPATHY
There are no formalized guidelines for diagnosis of lupus podocytopathy, but the most commonly used diagnostic criteria of lupus podocytopathy11 are the following: (1) clinical presentation of nephrotic syndrome in a patient with lupus, (2) kidney biopsy findings of diffuse and severe FPE on electron microscopy, and (3) the absence of subendothelial or subepithelial immune deposits on light, immunofluorescence, and electron microscopy11,14,15 (Table 1).
Table 1.
Proposed Criteria for Diagnosis of Lupus Podocytopathy
| Parameter | Features |
|---|---|
| Clinical | Diagnosis of SLE by ACR criteria; full nephrotic syndrome (ie, nephrotic-range proteinuria, hypoalbuminemia, and edema) |
| Light microscopy | Normal glomeruli or FSGS; mesangial proliferation permitted; endocapillary proliferation, necrosis, and/or crescents not permitted |
| Immunofluorescence microscopy | Deposits absent or confined to mesangium |
| Electron microscopy | Diffuse and severe foot process effacement (typically >70%); deposits absent or confined to mesangium |
Abbreviations: ACR, American College of Rheumatology; FSGS, focal segmental glomerulosclerosis; SLE, systemic lupus erythematosus.
Adapted from Bomback and Markowitz.11
Lupus podocytopathy can be subclassified as MCD or FSGS, including the morphologic subtypes of FSGS (NOS, perihilar, cellular, tip, or collapsing variant)11 as shown in Table 2. Mesangial proliferative lesions share similar clinical and histological features to MCD and could be included in MCD subtype, adding mesangial vs nonmesangial proliferation, or adding the additional secondary diagnosis of mesangial LN (LN class I or II).2,11 Beyond morphology, lupus podocytopathy should be divided into MCD or FSGS subtypes because patients with FSGS have worse outcomes, higher rates of hypertension and AKI on clinical presentation, and more severe tubulointerstitial involvement on kidney biopsy compared with MCD/mesangial proliferative lesions.2,11 Additionally, patients with FSGS are less likely to respond to therapy, and remissions occur later than for patients with MCD (with and without mesangial proliferation).2,11 Lupus podocytopathy FSGS with collapsing lesions have even worse outcomes, progressing to end-stage renal disease in more than 50% of the cases.16 Hu and colleagues2 study only included 1 of the 9 cases of FSGS with collapsing variant where most FSGS cases were tip variant. Salvatore and colleagues16 in a retrospective study of 19 patients with collapsing glomerulopathy and SLE reported massive proteinuria in 95% of patients; segmental and/or global collapsing glomerulopathy was seen in 11%–77% of glomeruli, and extensive FPE was seen in 82% of patients. Seven of 13 patients with follow-up data progressed to end-stage renal disease. Lupus podocytopathy FSGS collapsing variant may be an extreme form of LP17 with worse prognosis remarking the importance of distinguishing different variants of FSGS in lupus podocytopathy.
Table 2.
Proposed Criteria for Clinical Classification of Lupus Podocytopathy
| Classification | Subdivision |
|---|---|
| Lupus podocytopathy, minimal change disease variant | With mesangial proliferation lesions and mesangial deposits (additional diagnosis of LN II) |
| Without mesangial proliferation lesions (if deposits present, additional diagnosis of LN I) | |
| Lupus podocytopathy, focal segmental glomerulosclerosis variant | Not otherwise specified |
| Perihilar | |
| Cellular | |
| Tip | |
| Collapsing variant |
Abbreviation: LN, lupus nephritis.
Wang and colleagues18 studied 125 biopsies of LN patients showing mesangial proliferation with mesangial immune deposits and divided these cases into a “podocytopathy group” with 31 patients (defined as podocyte FPE > 50% with nephrotic syndrome) and “mesangial group” with 94 patients (less or equal than <50% FPE or >50% FPE without nephrotic syndrome). The podocytopathy group presented more frequently with renal involvement as the first manifestation of SLE, had less extrarenal involvement, had less hematuria, and had higher incidence of AKI with higher tubular interstitial injury compared to the mesangial group. The significant difference in the degree of proteinuria (5.57 g/24 h in the podocytopathy group compared with 0.69 g/24 h in the mesangial group) supports the notion that lupus podocytopathy should be a separate clinical diagnosis and classified outside minimal mesangial (class I) and mesangial proliferative (class II) LN.11,15 The degree of proteinuria is associated with the extent of FPE instead of mesangial proliferation or immune complex deposits19; therefore, the predominance of a nephrotic course highlights lupus podocytopathy as the primary diagnosis. An additional or secondary diagnosis of mesangial proliferation or LN II can be added.
We propose classifying lupus podocytopathy in a similar dichotomy as primary podocytopathies, with the major distinction being presence or absence of segmental sclerosis on light microscopy. The predominant clinical feature is the nephrotic syndrome with a high rate of recurrence, mimicking MCD or primary FSGS, which is not typical of class I or II LN. Therefore, rather than making a diagnosis of LN class I or LN class II with or without podocytopathy,12 we believe that lupus podocytopathy should be considered separate from classical forms of nephritis (LN I and II) and considered its own distinct entity. Lupus podocytopathy, subdivided as MCD and FSGS forms, highlights the principal clinical feature (recurrent nephrotic syndrome) that distinguishes it as an entity. Most patients included in the MCD subtype have some degree of mesangial proliferation2; we would add this information in classification as an additional histological information that can help clinicians at the moment of diagnosis, although the presence or absence of mesangial proliferation does not significantly influence clinical characteristics.18 The International Society of Nephrology/Renal Pathology Society classification4 does not include lupus podocytopathy. We believe that lupus podocytopathy is an independent type of LN and not a coexisting histological lesion (Table 3).15
Table 3.
The Classification of Lupus Nephritis, Including Lupus Podocytopathy, With Associated Clinical Presentation
| Class | Biopsy Findings | Clinical Features | Patients Presenting with Nephrotic Syndrome, % |
|---|---|---|---|
| Class I: minimal mesangial LN | No LM abnormalities; isolated mesangial IC deposits on IF and/or EM | Normal urine or microscopic hematuria | 0 |
| Class II: mesangial proliferative LN | Mesangial hypercellularity or matrix expansion with mesangial IC deposits on IF and/or EM | Microscopic hematuria and/or low-grade proteinuria | 0 |
| Lupus podocytopathy | Normal glomeruli, FSGS, or mesangial proliferation on LM; IC deposits absent or limited to mesangium on IF and/or EM; diffuse and severe foot process effacement on EM | Nephrotic syndrome | >90 |
| Class III: focal LN | <50% of glomeruli on LM display segmental (<50% of glomerular tuft) or global (>50% of glomerular tuft) endocapillary and/or extracapillary proliferation or sclerosis; mesangial and focal subendothelial IC deposits on IF and EM | Nephritic urine sediment and subnephrotic proteinuria | 30 |
| Class IV: diffuse LN | ≥50% of glomeruli on LM display endocapillary and/or extracapillary proliferation or sclerosis; mesangial and diffuse subendothelial IC deposits on IF and EM | Nephritic and nephrotic syndromes, hypertension, reduced kidney function | 50 |
| Class V: membranous LN | Diffuse thickening of the glomerular capillary walls on LM with subepithelial IC deposits on IF and EM with or without mesangial IC deposits | Nephrotic syndrome | 80 |
| Class VI: advanced sclerosing LN | >90% of glomeruli on LM are globally sclerosed with no residual activity | Advanced CKD | <10 |
Abbreviations: EM, electron microscopy; FSGS, focal segmental glomerulosclerosis; IC, immune complex; IF, immunofluorescence microscopy; LM, light microscopy; LN, lupus nephritis.
Adapted from Bomback.15
HISTOPATHOLOGY OF LUPUS PODOCYTOPATHY
Lupus podocytopathy presents on light microscopy of a kidney biopsy as a pattern of normal glomeruli (ie, minimal change lesions), mesangial proliferation, or FSGS lesions.1–3,5–10 Mesangial proliferation is the most common presentation (56%) followed by minimal change lesions (26%) and FSGS lesions (18%).2 The FSGS lesions can be further subcategorized as NOS, perihilar, cellular, tip, or collapsing forms.20 Pathology transition with morphing lesions can also occur during the course of the disease. In patients with repeated renal biopsies after recurrence transitions from MCD to FSGS or to LN class IV and LN class V have been reported2,3,12 Endocapillary proliferation, necrosis, and/or crescents are not seen in lupus podocytopathy and suggests other forms of glomerulonephritis.11 Acute tubulointerstitial lesions are common and are more frequent and severe with FSGS patterns.2 On immunofluorescence microscopy, lupus podocytopathy presents as glomerular deposition confined to the mesangium or no deposition. Most patients show depositions of both immunoglobulins and complement in glomeruli (86%) while depositions in cytoplasm of renal tubular epithelial cells or renal tubular basement membrane are less frequent.2 On electron microscopy, the lesion that defines lupus podocytopathy is the diffuse epithelial cell FPE, typically more than 70%1–3 (Fig 1). No deposits should be seen in the subendothelium or subepithelium spaces; such findings should lead to other classes of LN or glomerulonephritis, although the tip and collapsing variant of FSGS can rarely be present in the setting of subendothelial or subepithelial deposits characteristic of class III, IV, or V LN. In these instances, we would recommend adding the tip or collapsing lesions as a second-line diagnosis (to the first-line diagnosis of proliferative or membranous LN) and not considering these cases to be classic lupus podocytopathies. Microvilli, exposure of glomerular basement membrane, and detached foot processes have also been described in some cases.1–3,12 Other causes of podocyte injury in SLE should be excluded. Thrombotic microangiopathy is an example of another mechanism that results in renal injury different from classical immune-complex LN.21 Electron microscopy is essential for differential diagnosis in the study of spectrum of LN and, in particular, for establishing the lupus podocytopathy diagnosis.
Figure 1.
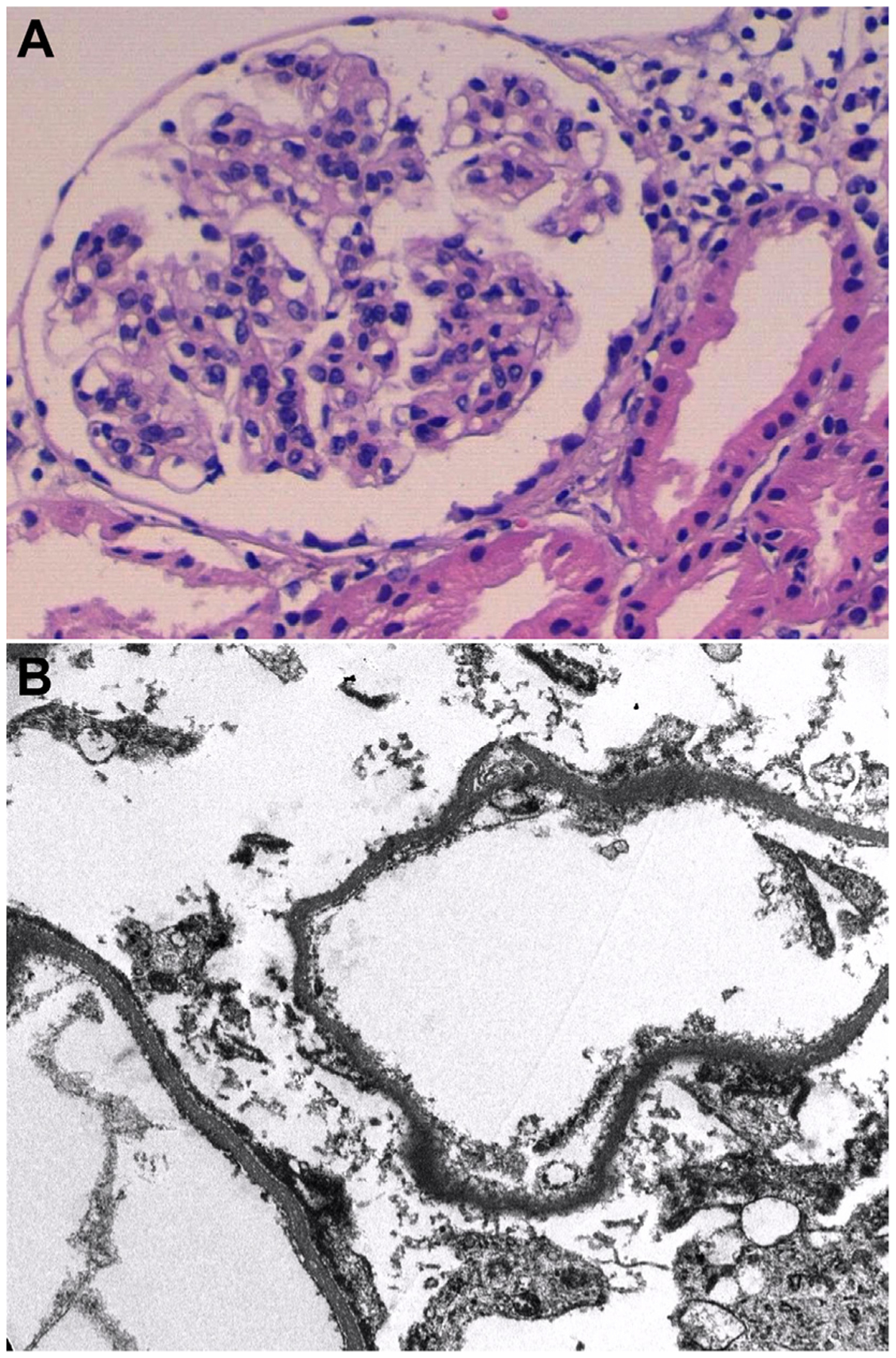
Kidney biopsy with lupus podocytopathy. (A) Light microscopy shows normal glomeruli. Immunofluorescence microscopy, not shown, is without deposits (negative for IgG, IgA, IgM, C3, and C1q). Original magnification. (B) Electron microscopy reveals complete foot process effacement (sample from paraffin with artifact). Original magnification. Courtesy of Teresa Pereda (Department of Pathology of Hospital Costa del Sol Marbella).
PATHOGENESIS OF LUPUS PODOCYTOPATHY
In lupus podocytopathy, the absence of deposits in glomerular capillary walls and the presence of severe FPE imply a mechanism that is independent of the typical immune complex deposition associated with LN. Instead, these findings support a presumably similar pathogenesis for lupus podocytopathy that has been speculated for MCD or primary FSGS, with podocyte injury being mediated by the production of a plasma factor(s) by B and/or T cells. In patients with SLE and mesangial proliferation, the degree of mesangial deposits or mesangial hypercellularity does not correlate with the degree of proteinuria; instead, proteinuria is associated with the extent of FPE suggesting an alternative pathophysiologic pathway.19 Therefore, the pathogenesis of lupus podocytopathy is likely unrelated to immune complex formation and more likely associated with the production of cytokines, lymphokines, or T cell dysfunction that can damage the podocyte (Fig 2).22,23 The T cell dysfunction that seems to play an important role in MCD24 could share pathogenesis with lupus podocytopathy. Other theories could include potential circulating factors, as in primary FSGS with recurrence post-transplantation and response to plasma exchange.25–28 In African-American patients with LN, the presence of APOL1 risk alleles has been associated with SLE collapsing glomerulopathy.29 Lupus podocytopathy FSGS collapsing variant is speculated to have pathogenesis associated with local interferons30 also present in SLE patients. Interferon-α (IFN-α) can be a central regulatory cytokine in SLE and especially in LN and may promote the development of autoreactive plasma cells, helper and memory T cells, and several proinflammatory cytokines.31 A number of cases of lupus podocytopathy have been associated with nonsteroidal anti-inflammatory drug use in patients with SLE.1–3 Pathogenesis of lupus podocytopathy and the underlying biology of SLE in this disease is not well understood and needs future research.
Figure 2.
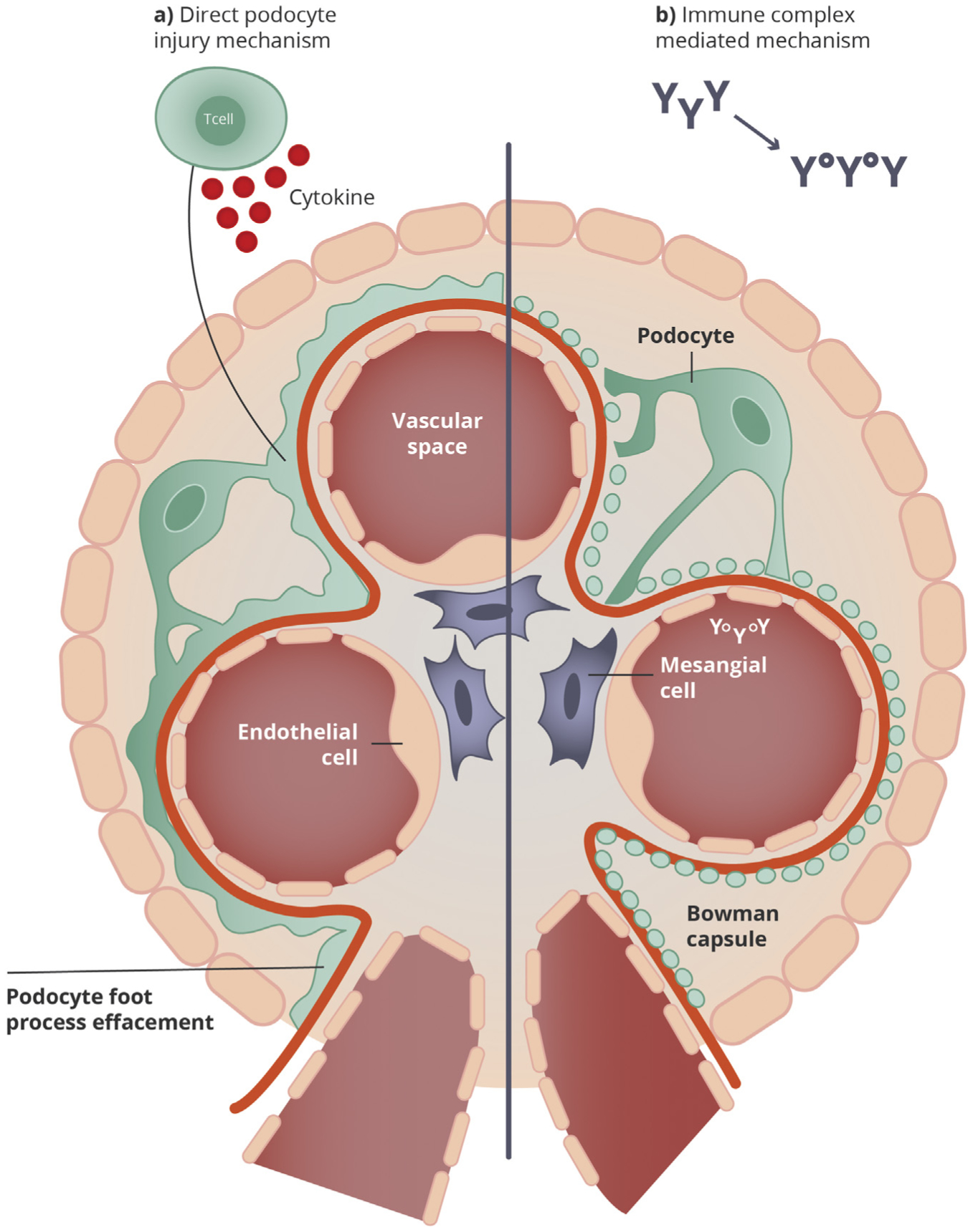
Pathogenesis of lupus podocytopathy. (A) In lupus podocytopathy, the foot process effacement may be explained by a direct injury associated with T cell dysfunction, cytokines, or lymphokines that damage the podocyte, although pathogenesis of lupus podocytopathy is not well understood. (B) In other classes of lupus nephritis, proliferation and hypercellularity, with the infiltration of inflammatory cells and ensuing subendothelial or subepithelial deposits, indicate other mechanisms of injury related to autoantibodies and immune complex depositions. Adapted and modified from Yu et al.22
TREATMENT AND PROGNOSIS
In lupus podocytopathy, as in MCD, patients usually respond to a short course of high-dose glucocorticoids32 and also have a very high rate of relapses.1–3 Lupus podocytopathy therefore is a glucocorticoid-sensitive entity. Hu and colleagues2 demonstrated that steroids alone or combined with another immunosuppressive agent induced remission in 94% of cases, with a median time to remission of 4 weeks. The complete remission rate was 76%. Steroids alone had similar clinical remission rates as induction therapy compared to steroids with another immunosuppressive agent. Nevertheless, within lupus podocytopathy subtypes, FSGS forms have worse response to steroids than MCD (mesangial proliferative cases included). FSGS subtype had a low rate of complete remission (22.2%) and most patients had a partial remission (55.6%) or did not respond (22.2%) adding a longer median time to remission of 8 weeks. Lupus podocytopathy FSGS collapsing variant is a more aggressive form and could require more immunosuppression. African-American patients (APOL1 high-risk variants) with SLE and the collapsing lesions may not respond at all to immunosuppression.16,29
More than one-half of the patients with lupus podocytopathy have relapses of renal disease,2 and these tend to be concurrent with extrarenal manifestations and/or serological activity of SLE.1,2,10 Maintenance treatment with glucocorticoids alone resulted in relapse in 89.5% of the patients, and glucocorticoid in combination with additional immunosuppressive agents showed a markedly reduced relapse rate by more than 50%.33
Therefore, steroids should be the first-line therapy for MCD lupus podocytopathy, and additional immunosuppressive agents may be required to treat relapses. In cases of FSGS lupus podocytopathy, given the more severe phenotype and lower response rates, steroids plus another immunosuppressive drug might be considered as induction therapy and maintenance. As in MCD in adults, following Kidney Disease: Improving Global Outcomes guidelines,34 we suggest prednisone given at a daily single dose of 1 mg/kg (maximum 80 mg) or alternate-day single dose of 2 mg/kg (maximum 120 mg). If tolerated, steroids can be maintained for a minimum period of 4 weeks if complete remission is achieved, and for a maximum period of 16 weeks if complete remission is not achieved. Corticosteroids should be tapered slowly over a total period of up to 6 months after achieving remission.
In the same way as relapsing forms of steroid-sensitive MCD or primary FSGS, lupus podocytopathy can require multiple rounds of immunosuppression for relapses or as steroid-sparing agents. Steroid-sparing agents used in lupus podocytopathy include mycophenolate mofetil, cal-cineurin inhibitors (CNIs), cyclophosphamide, and rituximab.14 In patients with LN with severe podocyte effacement, CNIs can have better remission rates and better long-term renal outcomes than those treated with other regimens,35 suggesting that therapies that facilitate podocyte stability might be beneficial in this subgroup of patients22 and could be considered a second-line therapy agent. In patients with greater foot process width, complete remission rates were significantly higher and the long-term renal outcome was better in the group with CNIs compared with other regimens. In this study, only 9 patients in the CNI group with complete remission and 3 with partial remission continued follow-up to 25 months.35 CNIs can also reduce proteinuria by nonim-munologic mechanisms,36 thus the impact on long-term outcomes is unclear. Rituximab has also been recommended for maintenance treatment in recurrent patients.12 Treatment in lupus podocytopathy is based on observational and retrospective studies. Prospective trials are required to confirm the efficacy of different regimens, although these would be exceedingly difficult to perform in such a rare clinical entity.
CONCLUSIONS
Lupus podocytopathy is a rare cause of nephrotic syndrome in SLE patients with implications for prognosis and treatment.37 This disease is characterized by a full nephrotic syndrome with high rate of relapses and pathological findings of diffuse epithelial cell FPE without immune complex deposition outside the mesangial space. As with other forms of LN, patients with lupus podocytopathy typically present with (1) antinuclear antibody, and depressed complement levels (C3 and/or C4), with less frequent (1) anti-double-stranded DNA testing. Extrarenal clinical manifestations are common. Histologic patterns of FSGS, MCD, and mesangial proliferation can be seen. As these last 2 share clinical features and outcomes, we suggest that lupus podocytopathy can be classified simply as FSGS vs MCD forms, adding additional diagnoses of class I or II LN. Lupus podocytopathy MCD forms respond well to treatment with glucocorticoids as induction therapy, adding a nonglucocorticoid immunosuppressive agent only to treat or, in some cases, avoid relapses. Lupus podocytopathy FSGS subtypes, which are less steroid-responsive, may benefit from initial induction treatment with glucocorticoids and another agent, such as CNIs.
CLINICAL SUMMARY.
The clinical course of lupus podocytopathy is similar to minimal change disease (MCD) and focal segmental glomerulosclerosis (FSGS) and distinct from other forms of lupus nephritis.
The FSGS lesion in lupus podocytopathy suggests lower rates of steroid responsiveness, as is seen in primary forms of FSGS.
In the same fashion as relapsing forms of steroid-sensitive MCD or primary FSGS, lupus podocytopathies can require second- or third-line immunosuppression agents for relapses or as steroid-sparing agents.
ACKNOWLEDGMENTS
This work was supported in part by a grant from the National Institute on Minority Health and Health Disparities (NIMHD) grant number R01-MD009223 (A.S.B.).
Footnotes
Financial Disclosure: The authors declare that they have no relevant financial interests.
REFERENCES
- 1.Kraft SW, Schwartz MM, Korbet SM, Lewis EJ. Glomerular podocytopathy in patients with systemic lupus erythematosus. J Am Soc Nephrol. 2005;16(1):175–179. [DOI] [PubMed] [Google Scholar]
- 2.Hu W, Chen Y, Wang S, et al. Clinical-morphological features and outcomes of lupus podocytopathy. Clin J Am Soc Nephrol. 2016;11(4):585–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dube GK, Markowitz GS, Radhakrishnan J, Appel GB, D’agati VD. Minimal change disease in systemic lupus erythematosus. Clin Nephrol. 2002;57(2):120–126. [DOI] [PubMed] [Google Scholar]
- 4.Weening JJ, D’agati VD, Schwartz MM, et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited. Kidney Int. 2004;65(2):521–530. [DOI] [PubMed] [Google Scholar]
- 5.Abuelo JG, Esparza AR, Garella S. Steroid-dependent nephrotic syndrome in lupus nephritis. Response to chlorambucil. Arch Intern Med. 1984;144(12):2411–2412. [PubMed] [Google Scholar]
- 6.Matsumura N, Dohi K, Shiki H, et al. Three cases presenting with systemic lupus erythematosus and minimal change nephrotic syndrome (in Japanese). Jpn J Nephrol. 1989;31(9):991–999. [PubMed] [Google Scholar]
- 7.Okai T, Soejima A, Suzuki M, et al. A case report of lupus nephritis associated with minimal change nephrotic syndrome: comparison of various histological types of 67 cases with lupus nephritis (in Japanese). Jpn J Nephrol. 1992;34(7):835–840. [PubMed] [Google Scholar]
- 8.Makino H, Haramoto T, Shikata K, Ogura T, Ota Z. Minimal-change nephrotic syndrome associated with systemic lupus erythematosus. Am J Nephrol. 1995;15(5):439–441. [DOI] [PubMed] [Google Scholar]
- 9.Hickman PL, Nolph KD, Jacobs R, Luger AM, Walker SE. Idiopathic focal segmental glomerulosclerosis in a patient with systemic lupus erythematosus: an unusual combination. Am J Kidney Dis. 1994;23(4):582–586. [DOI] [PubMed] [Google Scholar]
- 10.Hertig A, Droz D, Lesavre P, Grünfeld JP, Rieu P. SLE and idiopathic nephrotic syndrome: coincidence or not? Am J Kidney Dis. 2002;40(6):1179–1184. [DOI] [PubMed] [Google Scholar]
- 11.Bomback AS, Markowitz GS. Lupus podocytopathy: a distinct entity. Clin J Am Soc Nephrol. 2016;11(4):547–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen D, Hu W. Lupus podocytopathy: a distinct entity of lupus nephritis. J Nephrol. 2018;31(5):629–634. [DOI] [PubMed] [Google Scholar]
- 13.Wang YT, Chou HH, Chen FF, Chen MJ, Chiou YY. A case of minimal-change nephrotic syndrome in pediatric lupus erythematosus: just a coincidence? Lupus. 2006;15(4):244–247. [DOI] [PubMed] [Google Scholar]
- 14.Bomback AS. Nonproliferative forms of lupus nephritis: an overview. Rheum Dis Clin North Am. 2018;44(4):561–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bomback AS. A pregnant woman with lupus and nephrotic-range proteinuria. Clin J Am Soc Nephrol. 2017;12(9):1534–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Salvatore SP, Barisoni LM, Herzenberg AM, Chander PN, Nickeleit V, Seshan SV. Collapsing glomerulopathy in 19 patients with systemic lupus erythematosus or lupus-like disease. Clin J Am Soc Nephrol. 2012;7(6):914–925. [DOI] [PubMed] [Google Scholar]
- 17.Haas M. Collapsing glomerulopathy in systemic lupus erythematosus: an extreme form of lupus podocytopathy? Clin J Am Soc Nephrol. 2012;7(6):878–880. [DOI] [PubMed] [Google Scholar]
- 18.Wang SF, Chen YH, Chen DQ, et al. Mesangial proliferative lupus nephritis with podocytopathy: a special entity of lupus nephritis. Lupus. 2018;27(2):303–311. [DOI] [PubMed] [Google Scholar]
- 19.Han TS, Schwartz MM, Lewis EJ. Association of glomerular podocytopathy and nephrotic proteinuria in mesangial lupus nephritis. Lupus. 2006;15(2):71–75. [DOI] [PubMed] [Google Scholar]
- 20.D’agati VD, Kaskel FJ, Falk RJ. Focal segmental glomerulosclerosis. N Engl J Med. 2011;365(25):2398–2411. [DOI] [PubMed] [Google Scholar]
- 21.Almaani S, Meara A, Rovin BH. Update on lupus nephritis. Clin J Am Soc Nephrol. 2017;12(5):825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu F, Haas M, Glassock R, Zhao MH. Redefining lupus nephritis: clinical implications of pathophysiologic subtypes. Nat Rev Nephrol. 2017;13(8):483–495. [DOI] [PubMed] [Google Scholar]
- 23.Trivedi S, Zeier M, Reiser J. Role of podocytes in lupus nephritis. Nephrol Dial Transplant. 2009;24(12):3607–3612. [DOI] [PubMed] [Google Scholar]
- 24.Cunard R, Kelly CJ. T cells and minimal change disease. J Am Soc Nephrol. 2002;13(5):1409–1411. [DOI] [PubMed] [Google Scholar]
- 25.Wei C, El hindi S, Li J, et al. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat Med. 2011;17(8):952–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reiser J, Nast CC, Alachkar N. Permeability factors in focal and segmental glomerulosclerosis. Adv Chronic Kidney Dis. 2014;21(5):417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Artero ML, Sharma R, Savin VJ, Vincenti F. Plasmapheresis reduces proteinuria and serum capacity to injure glomeruli in patients with recurrent focal glomerulosclerosis. Am J Kidney Dis. 1994;23(4):574–581. [DOI] [PubMed] [Google Scholar]
- 28.Gohh RY, Yango AF, Morrissey PE, et al. Preemptive plasmapheresis and recurrence of FSGS in high-risk renal transplant recipients. Am J Transplant. 2005;5(12):2907–2912. [DOI] [PubMed] [Google Scholar]
- 29.Larsen CP, Beggs ML, Saeed M, Walker PD. Apolipoprotein L1 risk variants associate with systemic lupus erythematosus-associated collapsing glomerulopathy. J Am Soc Nephrol. 2013;24(5):722–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Markowitz GS, Nasr SH, Stokes MB, D’Agati VD. Treatment with IFN is associated with collapsing focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. 2010;5(4):607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rönnblom L, Alm GV, Eloranta ML. The type I interferon system in the development of lupus. Semin Immunol. 2011;23(2):113–121. [DOI] [PubMed] [Google Scholar]
- 32.Bomback AS, Appel GB. Updates on the treatment of lupus nephritis. J Am Soc Nephrol. 2010;21(12):2028–2035. [DOI] [PubMed] [Google Scholar]
- 33.Hu WX, Chen YH, Bao H, et al. Glucocorticoid with or without additional immunosuppressant therapy for patients with lupus podocytopathy: a retrospective single-center study. Lupus. 2015;24(10):1067–1075. [DOI] [PubMed] [Google Scholar]
- 34.Kidney Disease: Improving Global Outcomes (KDIGO) Glomerulonephritis Work Group. KDIGO clinical practice guideline for glomerulonephritis. Kidney Int Suppl. 2012;2(2):139–274. [Google Scholar]
- 35.Wang Y, Yu F, Song D, Wang SX, Zhao MH. Podocyte involvement in lupus nephritis based on the 2003 ISN/RPS system: a large cohort study from a single centre. Rheumatology (Oxford). 2014;53(7):1235–1244. [DOI] [PubMed] [Google Scholar]
- 36.Rovin BH, Caster DJ, Cattran DC, et al. Management and treatment of glomerular diseases (part 2): conclusions from a Kidney Disease: improving Global Outcomes (KDIGO) controversies conference. Kidney Int. 2019;95(2):281–295. [DOI] [PubMed] [Google Scholar]
- 37.Shea-simonds P, Cairns TD, Roufosse C, Cook T, Vyse TJ. Lupus podocytopathy. Rheumatology (Oxford). 2009;48(12):1616–1618. [DOI] [PubMed] [Google Scholar]