

Abstract
Membrane proteins have historically been recalcitrant to biophysical folding studies. However, recent adaptations of methods from the soluble protein folding field have found success in their applications to transmembrane proteins composed of both α-helical and β-barrel conformations. Avoiding aggregation is critical for the success of these experiments. Altogether these studies are leading to discoveries of folding trajectories, foundational stabilizing forces and better-defined endpoints that enable more accurate interpretation of thermodynamic data. Increased information on membrane protein folding in the cell shows that the emerging biophysical principles are largely recapitulated even in the complex biological environment.
Introduction
Included among the National Academy of Engineering grand challenges for the 21st century are goals to advance health informatics, to engineer better medicines, to reverse engineer the brain, and to engineer the tools of scientific discovery [1]. Achieving these goals will rely on overcoming the contemporary biophysical problem of describing how a polypeptide sequence encodes the structure and function of a protein. Because membrane proteins play key roles in human health, cognitive functions [2], and are thought to bind over half of the therapeutics on the market today, advancing an understanding of how and why membrane proteins attain their native folds will be key to meeting the grand challenges. There are two important perspectives to be addressed: (1) a biophysical description of driving forces underlying how a sequence encodes a structure and (2) a biological description of folding within a complex cellular environment. Here, we review the major advances from the biophysical vantage and comment on how these may be manifested in the cell (see Figure 1).
Figure 1. Membrane protein folding and stability flow chart.
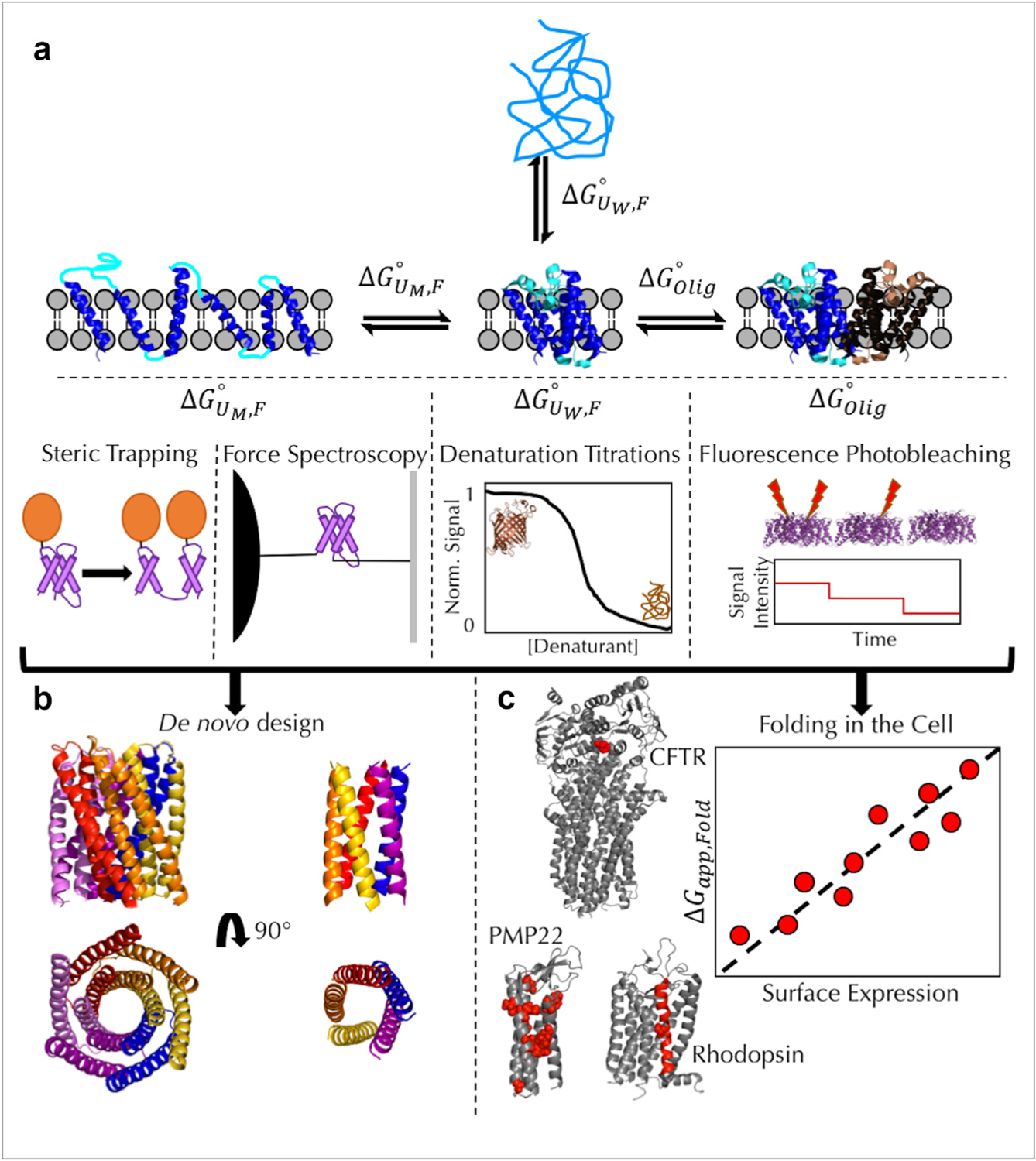
(a) The relevant thermodynamic equilibria describing membrane protein stability and the experimental approaches used to measure each free energy are shown. describes the coupled folding and insertion of an unfolded, water-soluble membrane protein into the bilayer and is calculated from chemical denaturation titrations of β-barrels (PDB: 1QD5). This approach has been used to investigate side-chain transfer free energies [3–5,8,13,53] and folding transition states [3]. describes the association/folding of helices in a membrane unfolded state and has been measured using both steric trapping [20,23] and single-molecule force spectroscopy [29,30]. describes the oligomerization of membrane proteins and is currently measured using single-molecule fluorescence photobleaching (PDB: 3Q17) [25,26]. (b) The growing knowledge of the thermodynamic parameters that define membrane protein folding and structure have led to the successful design of functional membrane proteins (PDBs: 6TMS (left) and 6MCT (right)) [27,35]. (c) In vitro-derived parameters of membrane protein stability (Panel A) have also been applied to membrane protein folding. Model systems for investigating folding in vivo include CFTR (PDB: 5UAK), PMP22 [54], and rhodopsin (PDB: 1L9H). The residues for each protein that have been discussed here are shown with a space-filling representation and are colored red. For rhodopsin, the entire TM7 helix has been investigated using deep mutational scanning [43]. For each system, the general trend is that stability is correlated with the surface expression of each membrane protein.
The value of water-to-bilayer end points
Water-solvated unfolded, UW, and bilayer-embedded folded states, F, represent the two most extreme endpoints of biophysical interest for membrane-protein folding reactions. A deceptively simple parameter — the free energy of folding — captures the population bias at equilibrium, and the free energy change between these end states reveals the maximum energetic contributions of the various atomic interactions responsible for stabilizing a particular folded state over its aqueous-unfolded conformational ensemble. Although the water-soluble unfolded state is not typically observed in a cellular context, these endpoints are nevertheless useful in theoretical considerations that seek to describe the underlying chemical reactions. Taking cues from the water-soluble protein-folding field, a number of groups used chemical denaturation titrations and extensive condition tweaking to measure path-independent equilibrium values for several transmembrane β-barrels [3–6]. These experiments reveal an extremely favorable folding stability for β-barrels, ranging from −18 to −32 kcal mol−1, and the systems have proved useful in addressing the energetic contributions of side-chain partitioning and backbone hydrogen bond formation [4,7–10].
Hydrophobicity energies from water-to-bilayer folding
Statistically, membrane-embedded segments are highly enriched in apolar side chains that favorably interact with the nonpolar core of the bilayer [11]. One key question concerns how much energy is gained by the removal of a nonpolar moiety from water and its placement within the bilayer. The answer is captured by the driving force known as the hydrophobic effect. Historically, water-to-octanol partitioning of peptide segments has been employed to mimic this energetic contribution as manifested through the construct of a hydrophobicity scale [12]. More recently, folding studies using two different transmembrane β-barrels employing a host–guest system strategy and a phospholipid bilayer instead of octanol have enabled a novel hydrophobicity scale [4,5]. These new measurements demonstrate that the magnitudes of the membrane partitioning energies are nearly twice as high as previously concluded from the octanol scale.
Moving the membrane mimic from an organic solvent to an actual bilayer brought an understanding of hydrophobicity closer to the cellular condition; however, the membrane itself is still not a uniform solvent. Rather, the bilayer interface is a chemically complex environment with a steeply changing water concentration. How does this aqueous gradient change the driving force energy of the hydrophobic effect along the bilayer normal? The favorable stability of the transmembrane β-barrel scaffold enabled nonpolar partitioning energies to be assessed at different locations in the bilayer and thus, under widely varying water concentrations [13]. This work reveals a continuously changing nonpolar solvation parameter function that connects the value for the energy of insertion of nonpolar moieties at the interface to that at the center of the bilayer. By relating the energy of this important driving force to chemical parameters of the membrane, and not, for example, the position along a transmembrane α-helix, these results have the potential to be adopted for proteins in any bilayer.
Energetic features of native folds
Since the availability of the earliest membrane-protein structures, it has been observed that most membrane proteins are enriched in either the transmembrane α-helical or the β-sheet (barrel) secondary structure that is formed by regular patterns of backbone hydrogen bonds. Backbone hydrogen bond (bbHB) formation is favored in membrane-embedded regions because there is a larger energetic penalty for the water-to-bilayer partitioning of the nonhydrogen bonded backbone. Recent advances in NMR experimental methodologies have allowed for bbHB strengths to be measured both in α-helical and β-barrel transmembrane proteins using a hydrogen––deuterium exchange [14]. Cao et al. reported that bbHB strengths for the transmembrane α-helical amyloid precursor protein reach −6 kcal mol−1, a value much more favorable than previous estimates using organic solvents and small peptides, or even soluble proteins [10,14]. Lessen et al. performed similar experiments using the transmembrane β-barrel OmpW and found strengths ranging from −3 to −4 kcal mol−1 on average [9]. In contrast to the partitioning free energy changes of nonpolar side chains discussed above, both NMR investigations found bbHB strengths to be relatively insensitive to the position of the membrane. Together, these studies indicate that bbHB energies appear to be affected by neither sequence nor secondary structure. In sum, the unchanging bbHB energy in membrane proteins across the bilayer implicates sidechain partitioning interactions as the main driving force for transmembrane protein insertion into the bilayer.
Side-chain entropy can be another energy source in protein folding. Compared to Uw, in which the polypeptide chain can assume a large and heterogeneous conformational ensemble, the folding of a transmembrane α-helix upon insertion limits the conformational space and perhaps the motions of side chains [15]. In contrast to this assumption, solution NMR relaxation studies suggest that membrane proteins are extraordinarily dynamic with fast internal motions on methyl-bearing side chains [16]. This finding was equally true for the α-helical sensory rhodopsin II as well as the OmpW β-barrel and was independent of the hydrophobic, membrane-mimicking cosolvent. The energetic contribution of side-chain motion to folding will depend on the extent to which it is preferentially enhanced in F as compared to Uw. Crucially, this remains to be tested [16].
Membrane-embedded unfolded-to-folded endpoints dominate α-helical membrane protein measurements
To date, there are no water-to-bilayer stabilities measured for α-helical transmembrane proteins. This is presumably due to the enhanced aggregation propensities of transmembrane α-helical regions that are composed of continuous stretches of nonpolar amino acids. Stability measurements of α-helical membrane proteins have accordingly been tractable only in experimental setups in which unfolded states remain embedded in a membrane or in a membrane mimic, which we term UM, regardless of its secondary structure. In these reactions, the energy derived from the hydrophobic effect is attenuated because the water concentration is not bulk, and a smaller energy difference between UM and F is expected. If the α-helical secondary structure is stable in isolated segments in the unfolded ensemble, for example UM, H, these experiments should report on transmembrane helix–helix interactions, for example UM, H ↔ F.
The classic example of this reaction includes the dimerization of the single-transmembrane domain of glycophorin A, GpATM [17–19]. However, new methods that interrogate helix–helix interactions in more complex multispan proteins show that the lateral interactions are not going to be simple to understand. Local interactions show varied stabilities in the intramembrane rhomboid protease GlpG as assessed using a ‘steric trapping’ strategy [20–23]. In contrast, the ClC-ec1 Cl−/H+ antiporter has a high affinity in bilayers using a promising new single-molecule microscopy technique [24–26]. The method is model-independent and can be carried out in any bilayer of choice using single-molecule fluorescence bleaching steps to quantify the membrane protein oligomer size following equilibration in what is essentially an “infinite” bilayer. In 2:1 POPE:POPG, the authors found that ClC-ec1 forms a high-affinity dimer with a mole fraction equilibrium dissociation constant equal to 4.7 × 10−8 subunits lipid−1. For context, this is only ~1.3 kcal mol−1 less favorable than the GpATM dimer in POPC [19], which was a surprising outcome because the ClC-ec1 dimerization interface is much larger by comparison. Because the CLC-ec1 lacks a so-called GxxxG dimerization motif, future mutational analysis on this protein will be needed to rationalize the distinct physical mechanisms these two proteins employ in subunit recognition. The distinction between these two structural modes for dimerization also begs the question of whether the packing of nonpolar side chains is sufficient to drive protein–protein interactions in lipids, which is an area of high interest in the membrane protein design field [27].
Aspects of the folding trajectory as assessed by force spectroscopy
The folding reaction of ClC-ec1 has also been measured using single-molecule force spectroscopy, a second single-molecule technique that is gaining popularity in its ability to probe folding at infinite dilution [28,29]. In the ClC-ec1 experiments, a force ramp strategy interrogated the unfolding of the monomeric ClC-ec1 protein in a DMPC bilayer wrapped in CHAPSO [29]. This protomer possesses an inverted topological arrangement of structurally similar N- and C-domains connected by a linker. The authors found that the ClC-ec1 N- and C-domains unfolded in separate events suggesting the idea that the protein evolved from gene duplication of subunits that fused together. The work further revealed that aggregation is not the only factor subverting folding: even under these single-molecule conditions, misfolded states of the two ClC-ec1 domains refold slowly and inefficiently and are prone to forming a non-native structure.
Showing its versatility to a wide variety of proteins [28], single-molecule force spectroscopy was recently used to elucidate intrinsic folding pathways for GlpG and the human β-adrenergic receptor β2AR [30]. Of significance is the observation that the β2AR folding occurred N- to C-terminal, which is intriguing because it implies that transmembrane α-helices may have evolved to laterally interact as they are inserted into the bilayer using the translocon.
Designer membrane proteins
Design efforts challenge our current understanding of how a sequence encodes a structure. The driving questions in this area may be summarized by two pithy phrases, What I cannot create, I do not understand [31] and Do I understand what I can create? [32] These two are at odds because design efforts take advantage not only of advances in fundamental thermodynamic principles but also of the ever-increasing structural knowledge base to create novel proteins. Despite the balance of input arguments, engineering efforts have led to some exciting successes that foretell the power of this approach (Figure 1b). The landmark achievement of the Rocker coiled-coil Zn2+/H+ antiporter [33] was followed by Rosetta-driven design of α-helical transmembrane bundles of varying stoichiometries [34], a dodecameric-helix pore that conducts ions with a selectivity of K+ over Na+ [35], and the de novo design of β-barrel transmembrane proteins [36]. Promise has also been demonstrated for the rational control of cellular signaling by the design of single-pass transmembrane domains that may alter receptor signaling through competition for helix–helix interactions in integrins [37]. Complementing these structural achievements is the continued development of energy functions that seek to more explicitly model interactions between the surfaces of transmembrane proteins and the lipidic membrane environment with additions that include differentiable models of multiple membrane compositions, nonpolar energy functions that increase the variety of side chains in design so that they more accurately reflect the biological diversity, and a lipophilicity-based force field for scoring [38,39].
Membrane protein folding in the cell
Recognizing that this entire literature cannot be summarized in a short review, we conclude with some comments on how the biophysical measurements discussed above impact our understanding of folding in the cell. The biophysical experiments are carried out under controlled conditions with purified components and carefully assessed endpoints. In contrast, it is widely appreciated that there is additional complexity within the living biological system. Foremost is the concept that evolution selects for fitness over stability, and it does so within the context of the cellular machinery. For example, there can be coupling between the biological processes of insertion and helix–helix association that can be difficult to disentangle [40]; putative transmembrane α-helices may be sorted by the translocon while simultaneously exploring conformational space in an unanticipated manner [41]; cotranslational forces are increasingly recognized in their ability to influence folding [42]; and the biogenesis process itself may place limitations on allowed mutations [43]. Thus, it is expected that mechanistic adaptations from the biophysically derived principles may arise because of constraints or benefits imparted by the proteostasis networks or cellular trafficking. Even in face of the complex cellular environment, works on the cystic fibrosis transmembrane conductance regulator (CFTR) and peripheral myelin protein 22 (PMP22) proteins involved in cystic fibrosis and Charcot-Marie-Tooth diseases, respectively, demonstrate the protein folding rules gleaned in the test tube are guiding principles largely applicable to the cellular context (Figure 1c).
There is a large body of literature supporting the conclusion that the most commonly occurring mutation in cystic fibrosis, ΔF508, is at its heart a protein folding defect [44]. The mutant protein has a propensity to sample misfolded conformations and is degraded before reaching the plasma membrane. Early in these studies, it was appreciated that ΔF508 is temperature sensitive and could undergo conditional rescue at the permissive temperature [45] Consistent with this observation, the severity of the disease correlates with the fraction of folded CFTR protein that is trafficked to the plasma membrane [46]. This led to the discovery of folding correctors, including an FDA-approved drug (VX-809 [47,48]), and more recently to the demonstration that the peripheral quality control system can rescue the fold by suppression of the CFTR ΔF508 mutant instability in cells [49].
Charcot-Marie-Tooth disease is a second example in which pathogenic severity is related to protein folding. In this case, the connection was directly established by showing that conformational stability and cellular trafficking of 12 variants of the PMP22 protein are linearly correlated [50]. Importantly, the work discovered that motor nerve conduction velocities in affected patients in vivo also tracked with thermodynamic stability of PMP22 assessed by classical protein-folding experiments in vitro [50]. The recent finding that overexpression of PMP22 leads to mistrafficking implies that overwhelming the proteostasis network is deleterious in the cell and is consistent with the very slow folding kinetics observed for PMP22 in vitro [51,52].
Conclusions and future directions
The work reviewed here highlights the creative applications and concomitant expansion of technical approaches that can be used to elucidate fundamental principles governing membrane protein folding. Continued increases in computational power and the advent of more widespread cryoEM structural solutions of recalcitrant membrane protein complexes will significantly add to the knowledge database from which design efforts can be drawn. Library expression of variants coupled with functional assays in vivo and deep mutational scanning methods are already showing promise in shaping the biologically allowed sequence space [43]. As the distinct steps of membrane protein folding are interrogated in the cellular context [41], scientists will gain greater insight into how the biophysical rules are played out within the living cell.
Funding
This work was funded by the National Institutes of Health grants R01 GM079440 and T32 GM008403 and the National Science Foundation grant MCB1412108.
Footnotes
Conflict of interest statement
Nothing declared.
References
Papers of particular interest, published within the period of review, have been highlighted as:
* of special interest
** of outstanding interest
- 1.Engineering NA of: grand challenges for the 21st century. http://www.engineeringchallenges.org/challenges/16091.aspx2019.
- 2.O’Donovan SM, Sullivan CR, McCullumsmith RE: The role of glutamate transporters in the pathophysiology of neuropsychiatric disorders. npj Schizophr 2017, 10.1038/s41537-017-0037-1. [DOI] [PMC free article] [PubMed]
- 3.Huysmans GH, Baldwin SA, Brockwell DJ, Radford SE: The transition state for folding of an outer membrane protein. Proc Natl Acad Sci U S A 2010, 107:4099–4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moon CP, Fleming KG: Side-chain hydrophobicity scale derived from transmembrane protein folding into lipid bilayers. Proc Natl Acad Sci U S A 2011, 108:10174–10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marx DC, Fleming KG: Influence of protein scaffold on side-chain transfer free energies. Biophys J 2017, 113: 597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moon CP, Kwon S, Fleming KG: Overcoming hysteresis to attain reversible equilibrium folding for outer membrane phospholipase A in phospholipid bilayers. J Mol Biol 2011, 413:484–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fleming PJ, Freites JA, Moon CP, Tobias DJ, Fleming KG: Outer membrane phospholipase A in phospholipid bilayers: a model system for concerted computational and experimental investigations of amino acid side chain partitioning into lipid bilayers. Biochim Biophys Acta Biomembr 2012, 1818:126–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McDonald SK, Fleming KG: Aromatic side chain water-to-lipid transfer free energies show a depth dependence across the membrane normal. J Am Chem Soc 2016, 138: 7946–7950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lessen HJ, Majumdar A, Fleming KG: Backbone hydrogen bond energies in membrane proteins are insensitive to large changes in local water concentration. J Am Chem Soc 2020, 142:6227–6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao Z, Hutchison JM, Sanders CR, JU Bowie: Backbone hydrogen bond strengths can vary widely in transmembrane helices. J Am Chem Soc 2017, 139:10742–10749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ulmschneider MB, Sansom MSP: Amino acid distributions in integral membrane protein structures. Biochim Biophys Acta Biomembr 2001, 1512:1–14. [DOI] [PubMed] [Google Scholar]
- 12.Wimley WC, Creamer TP, White SH: Solvation energies of amino acid side chains and backbone in a family of host-guest pentapeptides. Biochemistry 1996, 35:5109–5124. [DOI] [PubMed] [Google Scholar]
- 13.**. Marx DC, Fleming KG: Local bilayer hydrophobicity modulates membrane protein stability. J Am Chem Soc 2021, 143: 764–772. This paper reports the linear relationship between the nonpolar solvation parameter and the local water concentration in the bilayer. This allowed for functions relating side-chain transfer free energy and bilayer z-position to be derived for all nonpolar and aromatic side chains.
- 14.Cao ZJU Bowie: An energetic scale for equilibrium H/D fractionation factors illuminates hydrogen bond free energies in proteins. Protein Sci 2014, 23:566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bower MJ, Cohen FE, Dunbrack RL: Sidechain prediction from a backbone-dependent rotamer library: a new tool for homology modeling. J Mol Biol 1997, 267:1268–1282. [DOI] [PubMed] [Google Scholar]
- 16.*. O’Brien ES, Fuglestad B, Lessen HJ, Stetz MA, Lin DW, Marques BS, Gupta K, Fleming KG, Wand AJ: Membrane proteins have distinct fast internal motion and residual conformational entropy. Angew Chemie - Int Ed 2020, 10.1002/anie.202003527. NMR is used to measure side-chain motion in both α-helical and β-barrel membrane proteins folded in multiple membrane mimetics. Reported dynamics are faster than folded, soluble proteins and contribute to membrane protein stability and function.
- 17.Doura AK, Kobus FJ, Dubrovsky L, Hibbard E, Fleming KG: Sequence context modulates the stability of a GxxxG mediated transmembrane helix-helix dimer. J Mol Biol 2004, 341: 991–998. [DOI] [PubMed] [Google Scholar]
- 18.Duong MT, Jaszewski TM, Fleming KG, MacKenzie KR: Changes in apparent free energy of helix-helix dimerization in a biological membrane due to point mutations. J Mol Biol 2007, 371:422–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hong HJU Bowie: Dramatic destabilization of transmembrane helix interactions by features of natural membrane environments. J Am Chem Soc 2011, 133:11389–11398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gaffney KA, Hong H: The rhomboid protease GlpG has weak interaction energies in its active site hydrogen bond network. J Gen Physiol 2019, 10.1085/jgp.201812047. [DOI] [PMC free article] [PubMed]
- 21.Chang YCJU Bowie: Measuring membrane protein stability under native conditions. Proc Natl Acad Sci U S A 2014, 111: 219–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hong H, Chang YC, JU Bowie: Measuring transmembrane helix interaction strengths in lipid bilayers using steric trapping. Methods Mol Biol 2013, 1063:37–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo R, Gaffney K, Yang Z, Kim M, Sungsuwan S, Huang X, Hubbell WL, Hong H: Steric trapping reveals a cooperativity network in the intramembrane protease GlpG. Nat Chem Biol 2016, 12:353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chadda R, Robertson JL: Measuring membrane protein dimerization equilibrium in lipid bilayers by single-molecule fluorescence microscopy. Methods Enzym 2016, 581:53–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.**. Chadda R, Krishnamani V, Mersch K, Wong J, Brimberry M, Chadda A, Kolmakova-Partensky L, Friedman LJ, Gelles J, Robertson JL: The dimerization equilibrium of a ClC Cl(−)/H(+) antiporter in lipid bilayers. Elife 2016, 5. This paper introduces a model-independent analysis to calculate membrane protein oligomerization energetics using a previously reported single-molecule fluorescence photobleaching assay.
- 26.Chadda R, Cliff L, Brimberry M, Robertson JL: A model-free method for measuring dimerization free energies of CLC-ec1 in lipid bilayers. J Gen Physiol 2018, 10.1085/jgp.201711893. [DOI] [PMC free article] [PubMed]
- 27.Mravic M, Thomaston JL, Tucker M, Solomon PE, Liu L, DeGrado WF: Packing of apolar side chains enables accurate design of highly stable membrane proteins. Science 2019, 80, 10.1126/science.aav7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jefferson RE, Min D, Corin K, Wang JY, Bowie JU: Applications of single-molecule methods to membrane protein folding studies. J Mol Biol 2018, 10.1016/j.jmb.2017.05.021. [DOI] [PMC free article] [PubMed]
- 29.Min D, Jefferson RE, Qi Y, Wang JY, Arbing MA, Im W, Bowie JU: Unfolding of a ClC chloride transporter retains memory of its evolutionary history. Nat Chem Biol 2018, 10.1038/s41589-018-0025-4. [DOI] [PMC free article] [PubMed]
- 30.**. Choi HK, Min D, Kang H, Shon MJ, Rah SH, Kim HC, Jeong H, Choi HJ, JU Bowie, Yoon TY: Watching helical membrane proteins fold reveals a common N-to-C-terminal folding pathway. Science 2019, 80, 10.1126/science.aaw8208. Single-molecule force spectroscopy is used to monitor the membrane-embedded folding ΔGU;M trajectory in both bicelles and vesicles. A concerted N-to-C terminal folding pathway was discovered for two evolutionarily distinct proteins.
- 31.Lupas AN: What I cannot create, I do not understand. Science 2014, 80, 10.1126/science.aaa2721. [DOI] [PubMed] [Google Scholar]
- 32.Schmidt M: Do I understand what I can create?. In Synthetic Biology; 2009.
- 33.Joh NH, Wang T, Bhate MP, Acharya R, Wu Y, Grabe M, Hong M, Grigoryan G, DeGrado WF: De novo design of a transmembrane zn2+-transporting four-helix bundle. Science 2014, 80, 10.1126/science.1261172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu P, Min D, DiMaio F, Wei KY, Vahey MD, Boyken SE, Chen Z, Fallas JA, Ueda G, Sheffler W, et al. : Accurate computational design of multipass transmembrane proteins. Science 2018, 80, 10.1126/science.aaq1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.*. Xu C, Lu P, Gamal El-Din TM, Pei XY, Johnson MC, Uyeda A, Bick MJ, Xu Q, Jiang D, Bai H, et al. : Computational design of transmembrane pores. Nature 2020, 10.1038/s41586-020-2646-5. This paper reports the de novo design of functional membrane protein channels, paving the way for more complex membrane proteins to be created in the future.
- 36.**. Vorobieva AA, White P, Liang B, Horne JE, Bera AK, Chow CM, Gerben S, Marx S, Kang A, Stiving AQ, et al. : De novo design of transmembrane β-barrels. Science 2021, 80:371. This paper reports the de novo design of eight-stranded transmembrane β-barrel proteins. The principles of β-barrel membrane protein assembly elucidated here provide building blocks to study β-barrel folding and assembly in outer membranes in vivo.
- 37.Mravic M, Hu H, Lu Z, Bennett JS, Sanders CR, Orr AW, Degrado WF: De novo designed transmembrane peptides activating the α5b1 integrin. Protein Eng Des Sel 2018, 10.1093/protein/gzy014. [DOI] [PMC free article] [PubMed]
- 38.Alford RF, Fleming PJ, Fleming KG, Gray JJ: Protein structure prediction and design in a biologically realistic implicit membrane. Biophys J 2020, 118:2042–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weinstein JY, Elazar A, Fleishman SJ: A lipophilicity-based energy function for membrane-protein modelling and design. PLoS Comput Biol 2019, 15. [DOI] [PMC free article] [PubMed]
- 40.Elazar A, Weinstein J, Biran I, Fridman Y, Bibi E, Fleishman SJ: Mutational scanning reveals the determinants of protein insertion and association energetics in the plasma membrane. Elife 2016, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guerriero CJ, Gomez YK, Daskivich GJ, Reutter KR, Augustine AA, Weiberth KF, Nakatsukasa K, Grabe M, Brodsky JL: Harmonizing experimental data with modeling to predict membrane protein insertion in yeast. Biophys J 2019, 10.1016/j.bpj.2019.07.013. [DOI] [PMC free article] [PubMed]
- 42.Harrington HR, Zimmer MH, Chamness LM, Nash V, Penn WD, Miller TF, Mukhopadhyay S, Schlebach JP: Cotranslational folding stimulates programmed ribosomal frameshifting in the alphavirus structural polyprotein. J Biol Chem 2020, 10.1074/jbc.RA120.012706. [DOI] [PMC free article] [PubMed]
- 43.*. Penn WD, McKee AG, Kuntz CP, Woods H, Nash V, Gruenhagen TC, Roushar FJ, Chandak M, Hemmerich C, Rusch DB, et al. : Probing biophysical sequence constraints within the transmembrane domains of rhodopsin by deep mutational scanning. Sci Adv 2020, 6:7505. Deep mutational scanning is used to generate a library of over 800 variants of transmembrane helices in rhodopsin. This paper provides a method to couple membrane protein stability, surface expression, and evolution to derive a holistic understanding of membrane protein biogenesis.
- 44.Dechecchi MC, Tamanini A, Cabrini G: Molecular basis of cystic fibrosis: from bench to bedside. Ann Transl Med 2018, 10.21037/atm.2018.06.48. [DOI] [PMC free article] [PubMed]
- 45.Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ: Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 1992, 358:761–764. [DOI] [PubMed] [Google Scholar]
- 46.Bronsveld I, Mekus F, Bijman J, Ballmann M, De Jonge HR, Laabs U, Halley DJ, Ellemunter H, Mastella G, Thomas S, et al. : Chloride conductance and genetic background modulate the cystic fibrosis phenotype of ΔF508 homozygous twins and siblings. J Clin Invest 2001, 10.1172/JCI12108. [DOI] [PMC free article] [PubMed]
- 47.Van Goor F, Hadida S, Grootenhuis PDJ, Burton B, Stack JH, Straley KS, Decker CJ, Miller M, McCartney J, Olson ER, et al. : Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A 2011, 10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed]
- 48.Okiyoneda T, Veit G, Dekkers JF, Bagdany M, Soya N, Xu H, Roldan A, Verkman AS, Kurth M, Simon A, et al. : Mechanism-based corrector combination restores ΔF508-CFTR folding and function. Nat Chem Biol 2013, 10.1038/nchembio.1253. [DOI] [PMC free article] [PubMed]
- 49.Bagdany M, Veit G, Fukuda R, Avramescu RG, Okiyoneda T, Baaklini I, Singh J, Sovak G, Xu H, Apaja PM, et al. : Chaperones rescue the energetic landscape of mutant CFTR at single molecule and in cell. Nat Commun 2017, 10.1038/s41467-017-00444-4. [DOI] [PMC free article] [PubMed]
- 50.Schlebach JP, Narayan M, Alford C, Mittendorf KF, Carter BD, Li J, Sanders CR: Conformational stability and pathogenic misfolding of the integral membrane protein PMP22. J Am Chem Soc 2015, 137:8758–8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Myers JK, Mobley CK, Sanders CR: The peripheral neuropathy-linked Trembler and Trembler-J mutant forms of peripheral myelin protein 22 are folding-destabilized. Biochemistry 2008, 47:10620–10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.**. Marinko JT, Carter BD, Sanders CR: Direct relationship between increased expression and mistrafficking of the Charcot-Marie-Tooth-associated protein PMP22. J Biol Chem 2020, 10.1074/jbc.AC120.014940. This paper assesses the relationship between membrane protein expression levels and surface trafficking using single-cell flow cytometry. The observed nonlinear relationship between expression and trafficking implies that stability is not the only constraint on membrane protein assembly in vivo.
- 53.Moon CP, Zaccai NR, Fleming PJ, Gessman D, Fleming KG: Membrane protein stability may be the energy sink for sorting in the periplasm. Proc Natl Acad Sci U S A 2013, 110: 5285–5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mittendorf KF, Kroncke BM, Meiler J, Sanders CR: The homology model of PMP22 suggests mutations resulting in peripheral neuropathy disrupt transmembrane helix packing. Biochemistry 2014, 10.1021/bi500809t. [DOI] [PMC free article] [PubMed]