

Abstract
Background:
Reduced fatty acid oxidation (FAO) is a hallmark of metabolic remodeling in heart failure. Enhancing mitochondrial long-chain fatty acid uptake by Acetyl-CoA carboxylase 2 (ACC2) deletion increases FAO and prevents cardiac dysfunction during chronic stresses, but therapeutic efficacy of this approach has not been determined.
Methods:
Male and female ACC2f/f-MCM (ACC2KO) and their respective littermate controls were subjected to chronic pressure overload by TAC surgery. Tamoxifen injection 3 weeks after TAC induced ACC2 deletion and increased FAO in ACC2KO mice with pathological hypertrophy.
Results:
ACC2 deletion in mice with pre-existing cardiac pathology promoted FAO in female and male hearts, but improved cardiac function only in female mice. In males, pressure overload caused a downregulation in the mitochondrial oxidative function. Stimulating FAO by ACC2 deletion caused unproductive acyl-carnitine accumulation, which failed to improve cardiac energetics. In contrast, mitochondrial oxidative capacity was sustained in female pressure overloaded hearts and ACC2 deletion improved myocardial energetics. Mechanistically, we revealed a sex-dependent regulation of PPARα signaling pathway in heart failure, which accounted for the differential response to ACC2 deletion.
Conclusion:
Metabolic remodeling in the failing heart is sex-dependent which could determine the response to metabolic intervention. The findings suggest that both mitochondrial oxidative capacity and substrate preference should be considered for metabolic therapy of heart failure.
Keywords: heart failure, energy metabolism, fatty acid oxidation
Graphical Abstract
Introduction
Cardiac contraction is a high energy consuming process and cardiac metabolism is optimized to supply energy for continuous contraction. Mitochondrial long-chain fatty acid oxidation (FAO) generates the majority of reducing equivalents for ATP production in the heart [1]. A hallmark of heart failure is metabolic remodeling, which presents as one of the earliest changes that precedes structural remodeling, contributing to cardiac deterioration [2].
Changes in cardiac fuel preference are well documented and characterized by a shift from FAO towards increased reliance on glucose [3, 4]. Extensive research has been conducted to determine if the change in substrate metabolism during the cardiac remodeling process is adaptive or maladaptive to maintain myocardial energy production [5]. Evidence from these studies suggest that sustaining a high capacity for ATP synthesis via oxidative metabolism rather than the utilization of specific substrates is critical for maintaining the energy supply to the heart during chronic stress [6–8]. However, cardiac fuel selection directly affects cardiomyocyte growth, as glucose but not fatty acids provides signals and building blocks for structural remodeling of the diseased heart [9–11].
Acetyl-CoA carboxylase 2 (ACC2) catalyzes the production of malonyl-CoA, an inhibitor of long-chain fatty acids (FA) entry into the mitochondria [12]. Mice deficient of ACC2 in the heart have elevated levels of FAO and maintain cardiac energetics during chronic stress, which protects them against cardiac systolic and diastolic dysfunction [6, 13]. Additionally, ACC2 deletion prevents metabolic remodeling and attenuates pathological cardiac growth [9]. These studies suggest that preventing the decline of FAO caused by chronic stress can mitigate the development of heart failure, thus raising the possibility of FAO being a therapeutic target.
An important mechanism for metabolic remodeling in the failing hearts is the downregulation of transcriptional pathways mediated via PGC-1α/PPARα (peroxisome proliferator-activated receptor gamma co-activator 1α/ proliferator-activated receptor α) signaling complex leading to reduced expression and activity of the FAO machinery [14–17]. Moreover, impaired mitochondrial oxidative metabolism in the failing heart impairs efficient energy generation [2]. It remains unclear if stimulating cardiac FAO after the metabolic remodeling has occurred can restore oxidative metabolism and improve the outcome of the diseased heart. Furthermore, despite the distinct susceptibility to heart failure in males and females, how biological sex affects responses to therapeutic interventions has not been assessed.
Thus, in this study, we tested the hypothesis that increasing cardiac FAO after the onset of pathological hypertrophy will attenuate heart failure progression through improved mitochondrial oxidation. For this, we studied both female and male mice and increased FAO during the transition of pathological hypertrophy to heart failure. We find a sex-dependent response to the metabolic intervention, and the outcome is in part determined by the degree of early metabolic remodeling. These findings have significant implications for future development of targeted metabolic therapies.
Materials and Methods
A detailed description of materials and methods is available in the Supplementary Materials and Methods. Additional resources are available from the corresponding author upon reasonable request.
Animal model
All protocols concerning animal use were approved by the Institutional Animal Care and Use Committee at University of Washington.
All mice were housed at 22°C with a 12-hour light, 12-hour dark cycle with free access to water and standard chow. Experiments in C57Bl/6J mice were performed in male and female mice separately. All personnel were blinded during animal experiments (surgery, echocardiography, tissue harvest, perfusion, 31P NMR, 13C NMR).
ACC2-flox/flox (f/f) MerCreMer (MCM) mice were bred on a C57Bl/6J background as described previously [13, 18]. ACC2-f/f-MCM mice were mated to ACC2-f/f to produce both study (ACC2KO) and control (con) littermates. αMHC-MCM Jackson mice were purchased from Jackson (Stock No: 005657) and bred with C57Bl/6J mice (Stock No: 000664) as a separate line. Heterozygous offspring for MCM (genotype MCM/WT) were used for the study (MCM). All mice were confirmed to have Nnt mutant gene expression.
Mice were subjected to TAC surgery at 10 weeks (male) or 13 weeks (females). We waited an additional 3 weeks to perform TAC surgery in female mice in order to achieve a pre-surgery weight in all mice between 20–26g. In our hands, TAC surgery induces similar pressure overload in mice of similar body weight [19]. We find minimal difference in the outcome of wild type mice subjected to TAC at 8–16 weeks [20].
Three weeks later, all mice (ACC2KO, con, MCM) received an intraperitoneal injection of tamoxifen (20 mg/kg) for 5 days, which was sufficient to cause ACC2 deletion in ACC2KO without having side effects in sham mice [13].
Primary endpoints for this study were cardiac function and ventricular remodeling by echocardiography, hypertrophy at 12 weeks after surgery and cardiac energetics (PCr/ ATP ratio) at 6 weeks after surgery.
Transverse aortic constriction (TAC) surgery
TAC or sham surgery was performed as previously described [21]. For detailed information, see Materials and Methods in the Online Data Supplement.
Transthoracic echocardiography
For detailed information, see Materials and Methods in the Online Data Supplement.
Long-chain fatty acid mix preparation
Fatty acid-BSA stock was prepared at a final concentration of ~5 mM fatty acids (FA) with 12% fatty acid-free BSA (molecular ratio 2.2:1) (all purchased from Sigma Aldrich) in glucose/pyruvate-free Krebs-Henseleit Buffer (KHB) with modifications as described previously [9]. For detailed information, see Materials and Methods in the Online Data Supplement.
Isolated Heart Perfusion Experiments and Nuclear Magnetic Resonance (NMR) Spectroscopy
Isolated mouse hearts were perfused in Langendorff-mode at 37°C and a constant pressure as previously described [6, 13]. For detailed information, see Materials and Methods in the Online Data Supplement.
13C NMR Spectroscopy of Tissue Extracts
Hearts were perfused with 13C-labeled substrates (1,6-C glucose and U-13C fatty acids) to determine substrate oxidation as described previously [6]. For detailed information, see Materials and Methods in the Online Data Supplement.
Tissue harvest
Food was removed 4hr before tissue harvest. Hearts were harvested, rinsed briefly in PBS, blotted dry, weighed and freeze clamped. Heart weight was normalized to tibia length to assess changes in hypertrophic growth. Females were not checked for estrus cycle at tissue harvest.
Respiratory complex activities
Frozen cardiac tissue (15 mg) from perfused heart experiments was homogenized in CelLytic™ MT Cell Lysis Reagent (Sigma Aldrich). Respiratory complex activity (Complex I and citrate synthase) were assessed according to previously published methods using a 96 well format plate reader [22]. Activity was calculated by the difference in the change of absorbance in the presence and absence of specific inhibitors and normalized to protein concentration.
Triglyceride Content
Myocardial lipids were extracted from frozen heart tissues (10 mg) with 2:1 chloroform: methanol [23]. The chloroform layer was dried under a nitrogen stream. TG content from lipid fraction was then measured using Triglyceride Colorimetric Assay (Cayman Chemical) according to the manufacturer’s instruction. Values were normalized to frozen tissue weight.
Cardiac acyl-carnitines
Metabolites were extracted from frozen heart tissue (10 mg) by precipitating in 80:20% methanol:water following the addition of 1 ug/ml internal standard (C16 palmitoyl-carnitine, d3, Cambridge Isotope Laboratories, Inc.). After centrifugation, the supernatant was transferred to LC/MS auto sampler vials (Thermo Fisher). The protein pellet was redissolved in 0.1M NaOH and protein concentration measured for normalization.
Acyl-carnitines were analyzed using a Triple TOF – 5600 (AB Sciex) series mass spectrometer in line with a Water Acquity I-class UPLC. For detailed information, see Materials and Methods in the Online Data Supplement.
RNA isolation, real-time PCR and Immunoblots
For detailed information, see Materials and Methods in the Online Data Supplement.
Statistics
The numbers of independent experiments/ animals are specified in the relevant figure legends. Data are expressed as mean + standard error of the mean (SEM). Statistical analysis was performed with Prism 8.0 or 9.0 software (GraphPad). Normal distribution of data was verified by Shapiro-Wilk test. For normal distributed data, statistical comparisons between 2 groups were conducted by unpaired, two-tailed t-test. Statistical comparisons between 3 or more groups were conducted by one-way or two-way ANOVA followed by a Tukey posthoc analysis to determine statistical significance. For comparison over time, Bonferroni posthoc analysis was performed. The value of p < 0.05 was considered statistically significant. Heat maps were generated using MetaboAnalyst 4.0 [24]. Peak areas from LC/MS experiments were normalized as described above and log2 transformed.
Results
ACC2 deletion after TAC improves cardiac function in female but not male mice
To determine the potential of increasing FAO as a therapy for heart failure, we aimed to delete ACC2 to enhance long-chain FA entry into mitochondria in a mouse model of chronic pressure overload (transverse aortic constriction, TAC). We followed separate cohorts of male and female mice for 12 weeks after TAC (Fig. 1A). Since MerCreMer (MCM) toxicity has been reported in tamoxifen-induced genetic mouse models, we used MCM mice as additional control group [25, 26].
Fig. 1: ACC2 deletion in female but not male hearts improves cardiac function after TAC.

A: Experimental strategy to increase fatty acid oxidation (FAO) after TAC. Echocardiographic analysis of cardiac function (fractional shortening, FS) and ventricular remodeling (left ventricular inner diameter, LVID) in (B) male and (C) female mice at different time points after TAC surgery. n = 9–10. D: Representative western blot of ACC2 protein expression 2 weeks after tamoxifen (TAM) injection (6 weeks after surgery) confirming efficient ACC2 deletion in f/f-MCM hearts.
E: Hypertrophy (heart weight to tibia length ratio) in male and female mice 12 weeks after TAC surgery. n = 9–10. F: mRNA expression in male and female mice 12 weeks after TAC surgery. n = 4. Dashed line indicates level of sham. All data are presented as mean+SEM. P values were determined one-way ANOVA (E, F) or two-way ANOVA (C, D) followed by Tukey multiple comparison test. # p < 0.05 ACC2KO-TAC vs. con TAC, $ p < 0.05 ACC2KO-TAC vs. MCM TAC.
Both male and female TAC mice demonstrated preserved contractile function 3 weeks post TAC (Fig. 1B, C, Suppl. Fig. IA, B), when we induced deletion of ACC2 by tamoxifen (TAM) injection in ACC2 f/f-WT (con), ACC2 f/f-MCM (ACC2KO) and WT-MCM (MCM) mice. ACC2 protein was efficiently deleted in ACC2KO-TAC mice two weeks after TAM injection (Fig. 1D), a time point that has previously been shown to increase FAO after ACC2 deletion [13]. This allowed us to target FAO when the heart transitioned from the compensatory phase of pathological hypertrophy to overt dysfunction. Importantly, con and MCM-TAC mice showed a similar decrease in systolic function between three and six weeks after TAC in both female and male mice (Fig. 1B, C, Suppl. Fig. IA, B, Suppl. Table I, II), suggesting that MCM activation after TAC does not cause adverse effects in our study. Despite a similar decline in contractile function by 12 weeks, cardiac hypertrophy and pathological remodeling was less severe in females after TAC compared to males, consistent with prior reports (Fig. 1B–F, Suppl. Fig. IA, B, Suppl. Table I, II) [27–29].
In male mice, ACC2 deletion did not change the course of contractile dysfunction (Fig. 1B, Suppl. Fig. IA, Suppl. Table I) and ACC2KO-TAC males showed a similar degree of left ventricular remodeling as the two control groups at 12 weeks post TAC. In contrast, systolic function was improved and left ventricular dilatation of ACC2KO females was reduced at 12 weeks post TAC (Fig. 1C, Suppl. Fig. IB, Suppl. Table I). However, hypertrophy and molecular remodeling remained similar in con, MCM and ACC2KO mice 12 weeks after TAC (Fig. 1E, F, Suppl. Table II).
Together, this suggests that ACC2 deletion differentially affects cardiac function in female and male mice and points to a sex-specific response. Since cardiac function and pathological remodeling were not different between con and MCM-TAC mice, we only used one control group for subsequent experiments (con TAC).
Mitochondrial and metabolic remodeling after TAC is sex-dependent
Given the limited knowledge about how biological sex affects remodeling of oxidative metabolism and mitochondrial function, we next analyzed the expression of mitochondrial proteins in male and female TAC hearts. Expression of electron transport chain (ETC) subunits were reduced in male TAC hearts, but not in female hearts (Fig. 2A, B). Similarly, mitochondrial membrane and matrix proteins were also reduced in male TAC hearts, but not in females. Similar to the changes in molecular remodeling noted above, mitochondrial remodeling was unaffected by ACC2 deletion in either sex.
Fig. 2: TAC induces adverse mitochondrial remodeling in male but not female mice.

Representative western blot of mitochondrial electron transport chain (ETC) subunits and mitochondrial membrane and matrix proteins in male (A) and female (B) hearts 12 week after surgery. n = 4–5. Dashed line indicates level of sham. All data are presented as mean+SEM. P values were determined one-way ANOVA (A-D) followed by Tukey multiple comparison test. * p < 0.05 vs. con sham, # p < 0.05 vs. con TAC.
Metabolic and mitochondrial remodeling is coordinated by a complex network of transcription factors and co-activators [30, 31]. To determine the upstream signaling responsible for sex-dependent mitochondrial remodeling, we turned our attention to the PGC-1α signaling complex, which is an important player in the downregulation of mitochondrial function and oxidative metabolism in pathological cardiac hypertrophy [15, 31]. PGC-1α expression itself was unaltered in either male or female hearts after TAC, but expression of several interacting transcription factors was differentially affected in females and males (Fig. 3A): Expression of the mitochondrial biogenesis factors Nrf1 (nuclear respiratory factor 1) and Nrf2 and its target genes were downregulated in both male con and ACC2KO-TAC hearts, but unaltered in female TAC hearts (Fig. 3A, B). In line with this, mitochondrial DNA (mtDNA) content was also reduced in male, but not female TAC hearts (Fig. 3C).
Fig. 3: Metabolic signaling is impaired in male but not female mice after TAC.

mRNA levels of metabolic transcription factors, co-regulators (A) and target genes involved in regulation of mitochondrial biogenesis (B) and mitochondrial FAO (D) in male and female hearts 12 week after surgery. n = 3–5. C: mt/nDNA ratio in male and female hearts 12 week after TAC. n = 4–5. Dashed line indicates level of sham. All data are presented as mean+SEM. P values were determined one-way ANOVA (A-D) followed by Tukey multiple comparison test. * p < 0.05 vs. con sham, # p < 0.05 vs. con TAC.
ERRs (estrogen-related receptors) and PPARs (peroxisome proliferator-activated receptor) regulate expression of genes involved in mitochondrial function and FAO, respectively [31]. ERRα expression was unaltered, but PPARα and its interaction partner RxRα (retinoid X receptor) expression were reduced in both male con and ACC2KO-TAC hearts (Fig. 3A). This was accompanied by downregulation of mitochondrial FAO genes in male con-TAC and even further reduced expression in ACC2KO-TAC hearts (Fig. 3D). In contrast, ERRα/PPARα/RxRα expression or their downstream targets were unchanged in female con-TAC (Fig 3A and D). The PPARα and RxRα expression were higher in female ACC2KO-TAC hearts although the expression of downstream FAO genes was unaltered (Fig. 3A and D).
Collectively, these data show that pressure overload induced hypertrophy selectively downregulated pathways for mitochondrial function, biogenesis and FAO in male but not female mouse hearts. These changes were not reverted by increasing the relative oxidation of fatty acids via ACC2 deletion. While these data suggest that the oxidative capacity was preserved in female mice after TAC, it did not explain why ACC2 deletion improved cardiac function.
ACC2 deletion after TAC improves cardiac energetics in female mice
Heart failure is characterized by impaired energy supply, and it has been suggested that this contributes to contractile dysfunction [2]. Considering that FAO could be more effective for supplying energy during chronic increases of demand [5], we next tested if increasing FAO after TAC improved cardiac energetics. In order to determine the early effects of ACC2 deletion, these experiments were performed when cardiac dysfunction was similar in con and ACC2KO-TAC mice (6 weeks post TAC, Fig. 4A). Using 31P NMR spectroscopy of isolated perfused hearts, we measured dynamic changes in cardiac high-energy phosphate content when the hearts were stimulated for higher contractile performance (HWL). This allows to interrogate how changes in substrate supply (increased FAO) affect the energetic status (PCr/ATP) of the heart in response to a higher demand. In male sham hearts, the stimulation resulted in an increase in the rate pressure product (RPP) accompanied by a moderate decrease of PCr/ATP ratio suggesting the heart mobilized its energy reserve to increase contractile performance (Fig. 4B–C). Male TAC hearts, although able to increase left ventricular developed pressure (LVDevP), were unable to maintain the heart rate (HR) during the HWL period (Suppl. Fig. IIA), thus failed to increase RPP. The PCr/ATP ratio at HWL was similar for sham and TAC hearts but the RPP was lower in TAC hearts suggesting depletion of energy reserve prevented the increase of RPP in male TAC (Fig. 4C). There was no improvement in ACC2KO-TAC. In females, RPP increased during HWL conditions in all groups (Fig. 4E, Suppl. Fig. IIB). However, the increase of RPP in female con-TAC caused a progressive decrease in the PCr/ATP ratio, which is prevented in ACC2KO-TAC (Fig. 4F). Of note, citrate synthase and complex I activity were also reduced in male TAC but not female TAC hearts (Suppl. Fig. IIC, D).
Fig. 4: ACC2 deletion improves cardiac energetics in female ACC2KO mice after TAC.

A: Experimental strategy for perfused heart experiments. Cardiac function and energetic parameters at baseline (BL) and under high workload conditions (HWL, 4mM Ca2+) in isolated perfused hearts with mixed substrate buffer assessed by 31P NMR spectroscopy 6 weeks after TAC. Rate pressure product (RPP) at BL and HWL in male (B) and female (E) hearts. n= 6–7. Change of cardiac energetics (PCr/ ATP ratio) during HWL in male (C) and female (F) hearts. n = 6–7. Correlation of RPP and PCr/ATP in male (D) and female (G) hearts during HWL. n = 6–7. All data are presented as mean+SEM. P values were determined two-way ANOVA (A-D) followed by Tukey multiple comparison test (B, D). Bonferroni multiple comparison test was used for comparison over time (BL vs. HWL, A-D). * p < 0.05 con TAC vs. con sham, & p < 0.05 BL vs. HWL for indicated groups, 1 p < 0.05 BL vs. HWL for con TAC, 2 p < 0.05 BL vs. HWL for sham.
These observations suggest a differential response of male and female hearts to cope with increased energy demand for high workload, where male TAC hearts maintained energetic status by limiting cardiac work, while female TAC hearts mobilized energy reserve to increase LV function (Fig. 4D, G). Importantly, we observed a differential response to promoting FAO after TAC in male and female mice: the ability to increase RPP in females TAC hearts was associated with sustained cardiac energetics in ACC2KO, while no improvement of energetic or function was observed in male ACC2KO-TAC. These results suggested that metabolic remodeling during heart failure limited energy supply in TAC hearts, whereas only female hearts benefited from increased fatty acid oxidation.
Deletion of ACC2 in hypertrophic male hearts results in incomplete fatty acid oxidation
Previous studies have demonstrated that ACC2 deletion in the healthy hearts efficiently increases FAO without causing accumulation of lipid intermediates [6, 32]. Cardiac substrate utilization in isolated hearts during the HWL challenge confirmed that ACC2 deletion in the diseased heart elevated the relative oxidation of FA in both male and female hearts after TAC (Fig. 5A, Suppl. Fig. IIIA, B). What was the mechanism for sex-specific difference in myocardial energetics in ACC2KO-TAC hearts?
Fig. 5: ACC2 deletion results in acyl-carnitine accumulation after TAC in male hearts.

A: Contribution of 13C labeled substrates to tricarboxylic acid (TCA) cycle determined by 13C NMR spectroscopy in extracts from isolated perfused hearts from male and female mice under high workload conditions (4mM Ca2+) 6 weeks after TAC. n = 4–5. B: Quantification of acyl-carnitines (sum of medium-, long-chain and hydroxylated acyl-carnitines) in female and male con and ACC2KO mice 12 weeks after TAC. n = 5. C: Heatmap of individual medium-chain (MC), long-chain (LC) and hydroxylated (OH) acyl-carnitines in male and female con and ACC2KO mice 12 weeks after TAC. Values are depicted as log2 fold change normalized to corresponding con TAC. n = 5. D: Bar graphs of statistically significant acyl-carnitines in male ACC2KO-TAC. n = 5. Note that individual acyl-carnitines were not different in female ACC2KO-TAC. E: Regression analysis of ejection fraction (EF) and relative abundance of indicated acyl-carnitines in female and male ACC2KO-TAC hearts. Here, dotted lines indicate 95% confidence intervals. Dashed line indicates level of con TAC of respective sex. All data are presented as mean+SEM. P values were determined two-way ANOVA followed by Tukey multiple comparison test (A), two-tailed t-test (B, D) or linear regression (E). # p < 0.05 vs. con TAC.
We then speculated that increasing FA import into mitochondria by ACC2 deletion in the face of reduced oxidative mitochondrial capacity could lead to incomplete oxidation of FAs in male TAC hearts, thus limiting the effectiveness in energy production. We thus quantified cardiac acyl-carnitines, an intermediate of mitochondrial FAO that serves as a marker for incomplete fatty acid oxidation [33]. We observed a significant increase in total acyl-carnitine levels in male ACC2KO-TAC hearts which was much moderate in female ACC2KO-TAC (Fig. 5B). Specifically, several medium and long-chain acyl-carnitines were increased in male ACC2KO-TAC mice, while hydroxylated acyl-carnitine species were unaltered (Fig. 5C, D). As an important note, acyl-carnitine levels were not different between con and ACC2KO mice of either sex under unstressed conditions (Suppl. Fig. IVA), as previously reported [6]. Thus, it is likely that accumulation of acyl-carnitines in TAC hearts was a consequence of reduced mitochondrial oxidative capacity. Additionally, we found that C16, C18 and C18:1 acyl-carnitines inversely correlated with cardiac function in both male and female ACC2KO-TAC (Fig. 5E). This further supports our conclusion that incomplete FAO caused by impaired mitochondrial oxidative capacity contributed to the differential response to promoting FAO in female and male TAC hearts.
To exclude the possibility that acyl-carnitine accumulation is a consequence of altered cellular fatty acid uptake, we measured triglyceride (TG) concentration and transcript levels of cytosolic fatty acid transporter. Expression of CD36 was unaltered in both male and female hearts after TAC, as were myocardial TG levels (Suppl. Fig. VA, B). This supports the conclusion that the increase in mitochondrial FA uptake due to ACC2 deletion results in specific accumulation of acyl-carnitines, rather than a general increase in FA availability and uptake in ACC2KO-TAC hearts.
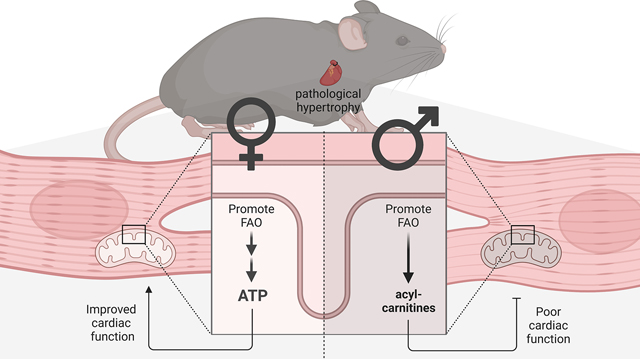
Together, these data allow us to conclude that mitochondrial FAO capacity are reduced in male hearts after TAC. It limits the ability to upregulate FAO flux to match the increased import of FA after ACC2 deletion, creating an oxidative bottleneck and resulting in accumulation of acyl-carnitines. On the other hand, in female mice, oxidative capacity is sustained in TAC hearts. In these hearts, increased FA transport into the mitochondria is adequately oxidized for ATP generation, which ultimately improves myocardial energetics and cardiac contraction (Fig. 6).
Fig. 6: Schematic of study findings.

The healthy heart has high capacity for mitochondrial fatty acid oxidation, resulting in efficient ATP supply. After TAC surgery, FAO is reduced in female and male hearts. Female hearts have sustained oxidative capacity, and increasing FAO results in restored ATP supply. In male hearts, oxidative capacity is reduced and increasing FAO results in accumulation of acyl-carnitines without increasing ATP supply. Created with BioRender.com.
Discussion
In the present study, we sought to test the therapeutic efficacy of augmenting cardiac FAO in heart failure in a clinically relevant setting. Using an inducible gene targeting strategy to increase mitochondrial fatty acid import in hypertrophied hearts, we identified key regulatory mechanisms governing the responses to metabolic therapies in a sex-specific fashion. The most salient finding of our study is that impaired oxidative capacity associated with pathological hypertrophy in male hearts limits the benefit of augmenting fatty acid transport into mitochondria. Nonetheless, female mice demonstrate preserved oxidative capacity, thus benefitting from increased FAO to supply ATP for cardiac contraction. The finding has important translational implication that metabolic therapies for the diseased heart must take the sex-specific metabolic remodeling into consideration.
Female sex is a well-established protective factor in cardiac diseases and female mice develop less severe heart failure under experimental conditions such as pressure overload induced hypertrophy [29]. Estrogen is known to regulate several metabolic processes, including transcription factor expression and genes for substrate utilization [34, 35] As a consequence, female mice demonstrate lesser inactivation of these pathways. However, if this determines the response to therapy remained unclear.
The PGC-1α/PPARα transcriptional cascade is an important regulatory mechanism of mitochondrial oxidation. Coordinated downregulation of the PGC-1α/PPARα signaling complex has been implicated for metabolic remodeling in heart failure and sex-specific remodeling [16, 29, 36]. Corroborating the notion, our study confirms that females are more resistant to the downregulation of mitochondrial oxidative capacity during pathological hypertrophy despite reduced contractile function. The sex-specific changes in PGC-1α/PPARα target genes in mitochondria correlate with the outcome of ACC2 deletion in TAC hearts: While promoting mitochondrial fatty acids import has limited benefit in male failing hearts, it improves myocardial energetics of female hearts in which FAO capacity is preserved.
PPARα has been suggested to play a pivotal role in sex specific remodeling, as a previous study demonstrated that inhibition of PPARα eliminates sex differences in cardiac hypertrophy after chronic Angiotension II infusion [36]. In this light, we found an increase in PPARα/RxRα expression in female ACC2KO-TAC mice. However, this did not translate into increased target gene expression, pointing towards differences in expression and activity of the transcriptional programs. In line with this, our previous work demonstrated that increased FA entry into mitochondria due to ACC2 deletion could reduce the lipid ligands for PPARs resulting in decreased PPAR activity [37]. Thus, it remains to be established how intracellular lipids regulate PPARα activity in females and if this is affected by manipulation of FAO.
As mitochondrial oxidative capacity appears to be the rate-limiting factor for metabolic intervention, one would hypothesize that directly targeting the upstream signaling governing mitochondrial oxidative metabolism would present a more promising approach for heart failure. However, increasing the expression of PPARα or PGC-1α in transgenic animal models has proven unsuccessful to prevent heart failure [20, 38, 39]. This is largely due to cardiomyopathies caused by elevating the transcriptional activities of these signaling cascades to super-physiological level in normal hearts. Alternative approaches that sustain or restore mitochondrial oxidative capacity in the failing hearts are likely more desirable but remain to be developed and tested in clinically relevant model. Future research focusing on treatment rather than prevention will also be necessary for this discovery. Observations in female mice suggest that understanding sex-mediated mechanisms might aid the identification of novel targets for protection against mitochondrial remodeling in heart failure.
Cardiac contraction requires a high volume and continuous supply of energy, primarily from FAO [40]. Accordingly, the mitochondrial capacity is rarely limiting for FAO in the normal heart. Enhancing mitochondrial fatty acid import by deleting ACC2 did not result in accumulation of acyl-carnitines or other evidence of incomplete FAO under physiological conditions and not even during prolonged high fat diet feeding [32, 37]. Accumulation of acyl-carnitines in ACC2KO-TAC hearts observed in the present study suggest that mitochondrial remodeling in pathological hypertrophy has created a bottleneck for oxidative metabolism so that only a limited increase of fatty acids influx can be processed without backup [17].
Elevated level of long-chain acyl-carnitines are associated with adverse outcome of heart failure patients [33, 41], and there is evidence that acyl-carnitines can directly contribute to cardiac dysfunction [42, 43]. Interestingly, pathological remodeling and cardiac dysfunction was not worse in male ACC2KO-TAC mice despite the accumulation of acyl-carnitines. One possibility is that under pathological conditions, acyl-carnitine generation is a part of the compensatory mechanisms that permits mitochondrial efflux of excessive acyl groups to alleviate mitochondrial stress [44]. Alternatively, the benefit of increasing the contribution of fatty acid to ATP production is cancelled by the side effect of acyl-carnitine accumulation in male ACC2KO-TAC mice. This notion appears to be supported by the strong correlation between acyl-carnitine accumulation and cardiac dysfunction in ACC2-KO TAC hearts.
Interestingly, deletion of ACC2 in hypertrophied hearts successfully switched substrate preference to fatty acids, but TAC induced pathological remodeling was not altered at the molecular level. These observations are clearly different from previous reports showing that increasing FAO before the onset of pathological hypertrophy can prevent hypertrophy and remodeling [6, 9]. Previous studies have suggested that the shift in substrate preference is a requirement rather than a driver for molecular remodeling [9, 11]. Thus, metabolic intervention at early stage could blunt the molecular remodeling process and prevent the development of pathological hypertrophy. However, the same intervention is not effective in reverting the established pathological changes as shown here. On the other hand, ACC2 deletion improves cardiac performance of female mice with pathological cardiac hypertrophy in which mitochondrial oxidative capacity is relatively preserved suggesting promoting FAO is advantageous when oxidative capacity permitting. Thus, future metabolic therapy for heart failure should combine the strategies for the preservation of mitochondrial oxidation and manipulation of substrate availability to achieve the maximal benefit. The results reiterate that preventing and treating disease are two distinct entities; it requires significant effort to determine whether successful prevention studies can be translated into rescue approaches.
A limitation of our study is that we did not compare the metabolic remodeling in female and male mice side-by-side, but rather monitored the response to increased FAO after TAC in male and female mice separately. The higher sensitivity to TAC in males could have confounded our results even though we started the intervention before any overt decrease of fractional shortening occurred in either sex. Additionally, the response we observe most likely depends on the timing when FAO was increased by ACC2 deletion. As discussed above, the inducible gene deletion model used here limits our ability to test the effects of ACC2 deletion at very early time points. A pharmacological compound will likely be more suitable for such a purpose. Finally, our study attributes differential changes in PPARα mediated transcription to the sex-specific downregulation of mitochondrial oxidative capacity after TAC, but it does not rule out other pathogenic mechanisms. Further studies and rigorous determination of causal relationships are required in this regard.
In summary, this study reveals a sex-dependent remodeling of cardiac oxidative metabolism in pathological hypertrophy and in response to the metabolic intervention. These findings have important implications for the future development of metabolic therapies for heart failure: First, distinct approaches will be needed for female and male patients. Second, interventions of substrate supply should be combined with strategies directly targeting oxidative capacity. And third, rigorous testing is required when translating successful prevention studies into therapeutic approaches at pre-clinical stage.
Supplementary Material
Highlights.
Pathological hypertrophy induces sex-specific metabolic remodeling in mice
Increasing FAO by ACC2 deletion in mice with pre-existing cardiac pathology improves cardiac function in female but not in male mice
In male mice, stimulation of FAO by ACC2 deletion causes acyl-carnitine accumulation
In female mice, stimulation of FAO by ACC2 deletion improves cardiac energetics
Acknowledgments
We thank the members of the Tian lab for helpful discussion of the study. We thank Bo Zhou and Matthew Walker for technical support with enzyme activity measurements. We also thank Matthew Walker for critical feedback during manuscript revision.
Sources of Funding
This work was supported in part by a postdoctoral fellowship from the German Research Foundation (DFG) RI 2764/1-1 (to JR), a Mitochondria and Metabolism Center Seed Grant (to JR), the U.S. National Institutes of Health (NIH) Grant HL-129510 and HL-142628 (to RT), the American Heart Association Scientist Development Grant 14SDG18590020 (to SK), the American Heart Association Predoctoral Grant 20PRE35120126 (to AC), the Biomedical Research Support Shared Instrumentation Grant S10RR029021 (to 14T High Resolution NMR Core Facility).
Footnotes
Disclosures
The authors have nothing to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Lopaschuk GD, Belke DD, Gamble J, Itoi T, Schonekess BO, Regulation of fatty acid oxidation in the mammalian heart in health and disease, Biochim Biophys Acta 1213(3) (1994) 263–76. [DOI] [PubMed] [Google Scholar]
- [2].Neubauer S, The failing heart--an engine out of fuel, The New England journal of medicine 356(11) (2007) 1140–51. [DOI] [PubMed] [Google Scholar]
- [3].Allard MF, Schonekess BO, Henning SL, English DR, Lopaschuk GD, Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts, The American journal of physiology 267(2 Pt 2) (1994) H742–50. [DOI] [PubMed] [Google Scholar]
- [4].Zhong M, Alonso CE, Taegtmeyer H, Kundu BK, Quantitative PET imaging detects early metabolic remodeling in a mouse model of pressure-overload left ventricular hypertrophy in vivo, J Nucl Med 54(4) (2013) 609–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ritterhoff J, Tian R, Metabolism in cardiomyopathy: every substrate matters, Cardiovascular research 113(4) (2017) 411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kolwicz SC Jr., Olson DP, Marney LC, Garcia-Menendez L, Synovec RE, Tian R, Cardiac-specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy, Circulation research 111(6) (2012) 728–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Horton JL, Davidson MT, Kurishima C, Vega RB, Powers JC, Matsuura TR, Petucci C, Lewandowski ED, Crawford PA, Muoio DM, Recchia FA, Kelly DP, The failing heart utilizes 3-hydroxybutyrate as a metabolic stress defense, JCI Insight 4(4) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Luptak I, Balschi JA, Xing Y, Leone TC, Kelly DP, Tian R, Decreased contractile and metabolic reserve in peroxisome proliferator-activated receptor-alpha-null hearts can be rescued by increasing glucose transport and utilization, Circulation 112(15) (2005) 2339–46. [DOI] [PubMed] [Google Scholar]
- [9].Ritterhoff J, Young S, Villet O, Shao D, Neto FC, Bettcher LF, Hsu YA, Kolwicz SC Jr., Raftery D, Tian R, Metabolic Remodeling Promotes Cardiac Hypertrophy by Directing Glucose to Aspartate Biosynthesis, Circulation research 126(2) (2020) 182–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gibb AA, Lorkiewicz PK, Zheng YT, Zhang X, Bhatnagar A, Jones SP, Hill BG, Integration of flux measurements to resolve changes in anabolic and catabolic metabolism in cardiac myocytes, The Biochemical journal 474(16) (2017) 2785–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Shao D, Villet O, Zhang Z, Choi SW, Yan J, Ritterhoff J, Gu H, Djukovic D, Christodoulou D, Kolwicz SC Jr., Raftery D, Tian R, Glucose promotes cell growth by suppressing branched-chain amino acid degradation, Nat Commun 9(1) (2018) 2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Awan MM, Saggerson ED, Malonyl-CoA metabolism in cardiac myocytes and its relevance to the control of fatty acid oxidation, The Biochemical journal 295 ( Pt 1) (1993) 61–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Choi YS, de Mattos AB, Shao D, Li T, Nabben M, Kim M, Wang W, Tian R, Kolwicz SC Jr., Preservation of myocardial fatty acid oxidation prevents diastolic dysfunction in mice subjected to angiotensin II infusion, Journal of molecular and cellular cardiology 100 (2016) 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC, Myocardial fatty acid metabolism in health and disease, Physiol Rev 90(1) (2010) 207–58. [DOI] [PubMed] [Google Scholar]
- [15].Bugger H, Schwarzer M, Chen D, Schrepper A, Amorim PA, Schoepe M, Nguyen TD, Mohr FW, Khalimonchuk O, Weimer BC, Doenst T, Proteomic remodelling of mitochondrial oxidative pathways in pressure overload-induced heart failure, Cardiovascular research 85(2) (2010) 376–84. [DOI] [PubMed] [Google Scholar]
- [16].Lehman JJ, Kelly DP, Transcriptional activation of energy metabolic switches in the developing and hypertrophied heart, Clinical and experimental pharmacology & physiology 29(4) (2002) 339–45. [DOI] [PubMed] [Google Scholar]
- [17].Lai L, Leone TC, Keller MP, Martin OJ, Broman AT, Nigro J, Kapoor K, Koves TR, Stevens R, Ilkayeva OR, Vega RB, Attie AD, Muoio DM, Kelly DP, Energy metabolic reprogramming in the hypertrophied and early stage failing heart: a multisystems approach, Circulation. Heart failure 7(6) (2014) 1022–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Olson DP, Pulinilkunnil T, Cline GW, Shulman GI, Lowell BB, Gene knockout of Acc2 has little effect on body weight, fat mass, or food intake, Proceedings of the National Academy of Sciences of the United States of America 107(16) (2010) 7598–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Garcia-Menendez L, Karamanlidis G, Kolwicz S, Tian R, Substrain specific response to cardiac pressure overload in C57BL/6 mice, American journal of physiology. Heart and circulatory physiology 305(3) (2013) H397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Karamanlidis G, Garcia-Menendez L, Kolwicz SC Jr., Lee CF, Tian R, Promoting PGC-1alpha-driven mitochondrial biogenesis is detrimental in pressure-overloaded mouse hearts, American journal of physiology. Heart and circulatory physiology 307(9) (2014) H1307–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tarnavski O, McMullen JR, Schinke M, Nie Q, Kong S, Izumo S, Mouse cardiac surgery: comprehensive techniques for the generation of mouse models of human diseases and their application for genomic studies, Physiol Genomics 16(3) (2004) 349–60. [DOI] [PubMed] [Google Scholar]
- [22].Frazier AE, Vincent AE, Turnbull DM, Thorburn DR, Taylor RW, Assessment of mitochondrial respiratory chain enzymes in cells and tissues, Methods in cell biology 155 (2020) 121–156. [DOI] [PubMed] [Google Scholar]
- [23].Folch J, Lees M, Sloane Stanley GH, A simple method for the isolation and purification of total lipides from animal tissues, The Journal of biological chemistry 226(1) (1957) 497–509. [PubMed] [Google Scholar]
- [24].Chong J, Soufan O, Li C, Caraus I, Li S, Bourque G, Wishart DS, Xia J, MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis, Nucleic Acids Res 46(W1) (2018) W486–W494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bersell K, Choudhury S, Mollova M, Polizzotti BD, Ganapathy B, Walsh S, Wadugu B, Arab S, Kuhn B, Moderate and high amounts of tamoxifen in alphaMHC-MerCreMer mice induce a DNA damage response, leading to heart failure and death, Disease models & mechanisms 6(6) (2013) 1459–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Koitabashi N, Bedja D, Zaiman AL, Pinto YM, Zhang M, Gabrielson KL, Takimoto E, Kass DA, Avoidance of transient cardiomyopathy in cardiomyocyte-targeted tamoxifen-induced MerCreMer gene deletion models, Circulation research 105(1) (2009) 12–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Weinberg EO, Mirotsou M, Gannon J, Dzau VJ, Lee RT, Pratt RE, Sex dependence and temporal dependence of the left ventricular genomic response to pressure overload, Physiol Genomics 12(2) (2003) 113–27. [DOI] [PubMed] [Google Scholar]
- [28].Deschepper CF, Llamas B, Hypertensive cardiac remodeling in males and females: from the bench to the bedside, Hypertension 49(3) (2007) 401–7. [DOI] [PubMed] [Google Scholar]
- [29].Murphy E, Amanakis G, Fillmore N, Parks RJ, Sun J, Sex Differences in Metabolic Cardiomyopathy, Cardiovascular research 113(4) (2017) 370–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Barger PM, Brandt JM, Leone TC, Weinheimer CJ, Kelly DP, Deactivation of peroxisome proliferator-activated receptor-alpha during cardiac hypertrophic growth, The Journal of clinical investigation 105(12) (2000) 1723–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Scarpulla RC, Vega RB, Kelly DP, Transcriptional integration of mitochondrial biogenesis, Trends Endocrinol Metab 23(9) (2012) 459–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Shao D, Kolwicz SC Jr., Wang P, Roe ND, Villet O, Nishi K, Hsu YA, Flint GV, Caudal A, Wang W, Regnier M, Tian R, Increasing Fatty Acid Oxidation Prevents High-Fat Diet-Induced Cardiomyopathy Through Regulating Parkin-Mediated Mitophagy, Circulation 142(10) (2020) 983–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ruiz M, Labarthe F, Fortier A, Bouchard B, Thompson Legault J, Bolduc V, Rigal O, Chen J, Ducharme A, Crawford PA, Tardif JC, Des Rosiers C, Circulating acylcarnitine profile in human heart failure: a surrogate of fatty acid metabolic dysregulation in mitochondria and beyond, American journal of physiology. Heart and circulatory physiology 313(4) (2017) H768–H781. [DOI] [PubMed] [Google Scholar]
- [34].Regitz-Zagrosek V, Oertelt-Prigione S, Seeland U, Hetzer R, Sex and gender differences in myocardial hypertrophy and heart failure, Circ J 74(7) (2010) 1265–73. [DOI] [PubMed] [Google Scholar]
- [35].Regitz-Zagrosek V, Seeland U, Sex and gender differences in myocardial hypertrophy and heart failure, Wien Med Wochenschr 161(5–6) (2011) 109–16. [DOI] [PubMed] [Google Scholar]
- [36].Harrington J, Fillmore N, Gao S, Yang Y, Zhang X, Liu P, Stoehr A, Chen Y, Springer D, Zhu J, Wang X, Murphy E, A Systems Biology Approach to Investigating Sex Differences in Cardiac Hypertrophy, Journal of the American Heart Association 6(8) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Liu Z, Ding J, McMillen TS, Villet O, Tian R, Shao D, Enhancing fatty acid oxidation negatively regulates PPARs signaling in the heart, Journal of molecular and cellular cardiology 146 (2020) 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Pereira RO, Wende AR, Crum A, Hunter D, Olsen CD, Rawlings T, Riehle C, Ward WF, Abel ED, Maintaining PGC-1alpha expression following pressure overload-induced cardiac hypertrophy preserves angiogenesis but not contractile or mitochondrial function, FASEB journal : official publication of the Federation of American Societies for Experimental Biology 28(8) (2014) 3691–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, Kelly DP, The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus, The Journal of clinical investigation 109(1) (2002) 121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Murashige D, Jang C, Neinast M, Edwards JJ, Cowan A, Hyman MC, Rabinowitz JD, Frankel DS, Arany Z, Comprehensive quantification of fuel use by the failing and nonfailing human heart, Science 370(6514) (2020) 364–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Martin MA, Gomez MA, Guillen F, Bornstein B, Campos Y, Rubio JC, de la Calzada CS, Arenas J, Myocardial carnitine and carnitine palmitoyltransferase deficiencies in patients with severe heart failure, Biochim Biophys Acta 1502(3) (2000) 330–6. [DOI] [PubMed] [Google Scholar]
- [42].Roussel J, Thireau J, Brenner C, Saint N, Scheuermann V, Lacampagne A, Le Guennec JY, Fauconnier J, Palmitoyl-carnitine increases RyR2 oxidation and sarcoplasmic reticulum Ca2+ leak in cardiomyocytes: Role of adenine nucleotide translocase, Biochim Biophys Acta 1852(5) (2015) 749–58. [DOI] [PubMed] [Google Scholar]
- [43].Seifert EL, Estey C, Xuan JY, Harper ME, Electron transport chain-dependent and -independent mechanisms of mitochondrial H2O2 emission during long-chain fatty acid oxidation, The Journal of biological chemistry 285(8) (2010) 5748–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, Bain J, Stevens R, Dyck JR, Newgard CB, Lopaschuk GD, Muoio DM, Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance, Cell metabolism 7(1) (2008) 45–56. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.