

Abstract
Background
In a multinational phase 3 trial (VIALE-C), venetoclax plus low-dose cytarabine prolonged overall survival vs placebo plus low-dose cytarabine in patients with newly diagnosed acute myeloid leukaemia ineligible for intensive chemotherapy, although it was not statistically significant. Herein, we assess the benefit of venetoclax plus low-dose cytarabine in the Japanese subgroup of VIALE-C patients (n = 27).
Methods
VIALE-C, a randomized (2:1), double-blind study (NCT03069352), enrolled untreated patients (≥18 years) with acute myeloid leukaemia. Patients received venetoclax (600 mg days 1–28, 4-day ramp-up in cycle 1) or placebo in 28-day cycles with low-dose cytarabine (20 mg/m2 days 1–10). The primary endpoint was median overall survival.
Results
In the Japanese subgroup, at a 6-month follow-up from the primary analysis, median overall survival for venetoclax (n = 18) and placebo (n = 9), plus low-dose cytarabine, was 4.7 and 8.1 months, respectively (hazard ratio, 0.928, 95% confidence intervals : 0.399, 2.156). The rate of complete remission plus complete remission with incomplete blood count recovery was higher with venetoclax plus low-dose cytarabine (44.4%) vs placebo plus low-dose cytarabine (11.1%). All patients experienced at least 1 adverse event. The most common grade ≥3 adverse events with venetoclax or placebo, plus low-dose cytarabine, were febrile neutropenia (50.0% vs 44.4%, respectively) and thrombocytopenia (27.8% vs 44.4%, respectively). Serious adverse events were reported in 50.0 and 33.3% of patients in the venetoclax and placebo, plus low-dose cytarabine arms, respectively; pneumonia was the most common (22.2% each).
Conclusions
Limited survival benefit in the Japanese subgroup can be attributed to small patient numbers and to baseline imbalances observed between treatment arms, with more patients in the venetoclax plus low-dose cytarabine arm presenting poor prognostic factors. Venetoclax plus low-dose cytarabine was well tolerated in Japanese patients with acute myeloid leukaemia ineligible for intensive chemotherapy.
Keywords: acute myeloid leukaemia, venetoclax, low-dose cytarabine, VIALE-C, Japan
In the Japanese subgroup of VIALE-C patients (n = 27), venetoclax plus low-dose cytarabine (LDAC) was well tolerated and showed high response vs placebo plus low-dose cytarabine in patients with acute myeloid leukaemia ineligible for intensive chemotherapy.
Introduction
Acute myeloid leukaemia (AML), although relatively rare, is the most common adult leukaemia in Japan accounting for ~70% of myeloid leukaemias (1,2). The incidence of AML is age dependent with a median age at diagnosis in Japan of 65 years (3). Survival rates for patients with AML are the lowest amongst all leukaemias. Retrospective population-based studies that included the overall AML population (young adult to elderly) observed relatively low 5-year overall survival (OS) rates of 10–20% (4–7).
Standard first-line therapy for adults with newly diagnosed AML is intensive chemotherapy (1,8). However, many patients are ineligible for intensive therapy because of advanced age or comorbidities (3,9,10). When compared with younger adult patients with AML, elderly patients with AML also have increased frequencies of adverse prognostic factors such as unfavourable risk karyotype and secondary AML (11), leading to a poorer prognosis (12–14). Less intensive treatment options include azacitidine, decitabine or low-dose cytarabine (LDAC) (8) with only LDAC monotherapy approved in Japan (prior to March 2021) for the treatment of patients with AML who are ineligible for intensive chemotherapy (10). However, reported rates of complete remission (CR) or CR with incomplete blood count recovery (CRi) are ≤30% (3,15–17), underscoring the need for additional new treatment options.
Venetoclax, a selective inhibitor of B-cell leukaemia/lymphoma-2 (BCL2), has been evaluated either alone or in combination with other active agents in several hematologic malignancies (18–24). Venetoclax and cytarabine have complementary mechanisms of action that provide a biologic rationale for evaluation in AML. Cancer cell survival is mediated by BCL2 family members, including BCL2, BCL-XL, and MCL1. In AML, BCL2 promotes chemoresistance, the survival of leukemic progenitor and blast cells, and has been associated with poor outcomes (25,26). Resistance to the BCL2 inhibitor venetoclax may be mediated by other pro-survival factors, like MCL1, that sequester endogenous BCL2 homology (BH)3-only proteins released by venetoclax upon binding to BCL2. In preclinical models, cytarabine synergized with venetoclax by enhancing BH3-only activity and/or suppressing MCL1 to promote apoptosis (27,28).
Venetoclax-based therapy in elderly patients with previously untreated AML was assessed in 2 large phase 1b/2 studies (24,29). Combination therapy with venetoclax plus azacitidine or decitabine resulted in a CR plus CRi rate of 67% and a median OS of 17.5 months (29). When combined with LDAC, venetoclax produced a CR plus CRi rate of 54% and median OS of 10.1 months (24). These results prompted the initiation of 2 phase 3 placebo-controlled trials to compare azacitidine (VIALE-A) or LDAC (VIALE-C) with or without venetoclax (30,31). Both studies enrolled patients globally and included patients from Japan. In the VIALE-A study, the addition of venetoclax to azacitidine significantly increased the CR plus CRi rate (66% vs 28%; P < 0.001) and median OS (14.7 vs 9.6 months; hazard ratio [HR]: 0.66; P < 0.001) compared with the control group (30). In the VIALE-C study at a 6-month follow-up analysis, the addition of venetoclax to LDAC significantly increased the CR plus CRi rate (48% vs 13%; P < 0.001) compared with the control group; median OS was 8.4 months vs 4.1 months (HR, 0.70; P = 0.040) (31).
Venetoclax has been approved for use in the USA and several other countries in combination with azacitidine, decitabine, or LDAC in patients with newly diagnosed AML ≥75 years of age who are ineligible for intensive induction chemotherapy; in March 2021, venetoclax in combination with azacitidine or LDAC was approved in Japan. Here we present efficacy and safety outcomes in the subgroup of Japanese patients with AML ineligible for intensive chemotherapy who participated in the VIALE-C study.
Methods
Study design
VIALE-C (NCT03069352) is a phase 3 randomized, double-blind placebo-controlled, multicenter study that assessed the efficacy and safety of venetoclax plus LDAC compared with placebo plus LDAC in treatment-naive patients with AML who were ineligible for intensive chemotherapy (31). The primary endpoint of the study was OS and secondary endpoints included response rates (CR, CR plus CRi, proportion of patients with CR plus CRi by initiation of cycle 2), transfusion independence rates, and event-free survival (EFS). The study was conducted in accordance with the International Council for Harmonization requirements, Good Clinical Practice guidelines, and the Declaration of Helsinki. The protocol was reviewed and approved by an independent ethics committee/institutional review board at each site before initiation. All patients provided written informed consent before participating.
Patients
Full eligibility criteria have been published previously (31). Briefly, eligible patients were adults (≥18 years) with newly diagnosed AML, as defined by the World Health Organization (32). Patients were ineligible for standard induction therapy either due to age (≥75 years) or lack of fitness, defined by the presence of at least 1 of the following: Eastern Cooperative Oncology Group (ECOG) performance status 2 or 3, history of congestive heart failure requiring treatment or ejection fraction ≤50% or chronic stable angina, diffusion capacity of the lung for carbon monoxide ≤65% or forced expiratory volume in 1 second ≤65%, creatinine clearance ≥0.5 to <0.75 ml/s/m2, moderate hepatic impairment with total bilirubin >1.5 to ≤3.0 times the upper limit of normal, or other comorbidities deemed incompatible with standard therapy. Patients with secondary AML could have received prior therapy for myelodysplastic syndromes (MDS). Main exclusion criteria comprised a projected life expectancy <12 weeks, prior therapy for AML (except for hydroxyurea either prior to or during the first cycle of treatment) and previous treatment with cytarabine for any indication.
Randomization and treatment
Patients were randomized 2:1 via interactive response technology to either venetoclax plus LDAC or placebo plus LDAC. Randomization was stratified by AML status (de novo vs secondary), patient age (<75 vs ≥75 years) and region (USA, Europe, China, Japan and rest of world).
Venetoclax was administered orally once daily (QD), and to avoid the risk of tumour lysis syndrome (TLS), dosing began at 100 mg on day 1 of cycle 1 and increased stepwise over 4 days (ramp-up period) to reach the target dose of 600 mg (100 mg day 1, 200 mg day 2, 400 mg day 3, 600 mg days 4–28 of cycle 1). Venetoclax was given at the target dose of 600 mg QD in all subsequent 28-day cycles. During the ramp-up period and until 24 hours after the target dose of venetoclax was reached, all patients were hospitalized to monitor for TLS and received TLS prophylaxis (uric acid-reducing agents and hydration). Patients in the placebo arm received a placebo (identical-looking tablet) in the same manner as venetoclax. All patients received subcutaneous LDAC at a dose of 20 mg/m2 QD on days 1–10 of each 28-day cycle. Treatment was continued until disease progression (PD), unacceptable toxicity or other pre-established treatment discontinuation criteria were met (31).
Assessments
Disease assessments were performed on bone marrow samples collected at screening, end of cycles 1 and 4, and every three cycles thereafter (in the absence of PD) until 2 consecutive samples confirmed stable achievement of CR or CRi. Disease assessments were also performed when relapse was suspected and/or at the final study visit. Clinical responses were defined according to the modified International Working Group criteria for AML (33), and PD was defined as per European LeukemiaNet recommendations (34). Details on the criteria for evaluating disease assessment have been reported previously in the primary publication of this study (31). OS was defined as the time from study randomization to death due to any cause. EFS was defined as the time from study randomization to PD, confirmed relapse from CR or CRi, treatment failure (failure to achieve CR, CRi, partial remission or morphologic leukaemia-free state as assessed by the investigator) or death from any cause. Post-baseline transfusion independence was defined as a period of at least 56 consecutive days without transfusions of either red blood cells (RBCs) or platelets occurring between the first dose of study drug and 30 days after the last dose of study drug.
Safety evaluations were performed throughout the study. Patients were monitored for adverse events (AEs), serious AEs, vital signs, laboratory measures, and clinically significant cardiac, pulmonary or radiologic findings. AEs were defined as those that occurred between the first dose of study drug until 30 days after the last dose of study drug. AEs were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events Version 4.03.
Statistical methods
Efficacy analyses were performed on the full analysis set, consisting of all patients who were randomized, whereas safety analyses were performed on all patients who received at least 1 dose of study drug. The pre-planned sample size for the VIALE-C study was 210 patients (randomized 2:1) to detect a statistically significant reduction in mortality of 45.5%, with 90% power at an alpha level of 0.05. OS, and EFS were analysed using Kaplan–Meier methodology and compared between treatment arms using the log-rank test. The HR was estimated using the Cox proportional-hazards model. Cox proportional-hazard regression models with stepwise variable selection were performed on OS in the Japan region as sensitivity analyses to identify relevant prognostic factors for OS and to better understand the treatment effect on OS when adjusting for these factors. Response rates and transfusion-independence outcomes were compared between treatment arms using the Cochran–Mantel–Haenszel test and 95% confidence intervals (CIs) were determined using the Clopper–Pearson Exact method.
For this analysis, baseline characteristics and study outcomes are described for the Japanese subgroup. The data cutoff for the primary analysis of the study was 15 February 2019; the cutoff for the 6-month follow-up analysis presented herein was 15 August 2019.
Results
Patient demographics and baseline characteristics
Between May 2017 and the data cutoff for the 6-month follow-up analysis of 15 August 2019, 211 patients were enrolled across 21 countries. Of the 211 patients,14 sites in Japan enrolled 27 (12.8%) patients who received venetoclax plus LDAC (n = 18) or placebo plus LDAC (n = 9). The median age was 81 (range: 60–89) years for patients treated with venetoclax plus LDAC and 78 (range: 71–85) years for those treated with placebo plus LDAC. Demographics and baseline characteristics of the Japanese subgroup are shown in Table 1. Treatment arms were balanced in terms of patient age (85% of patients ≥75 years; venetoclax plus LDAC vs placebo plus LDAC: 83.3% vs 88.9%), the proportion of patients with de novo AML, and the prevalence of transfusion dependence. Some imbalances were observed between the venetoclax plus LDAC and placebo plus LDAC arms, including the proportion of patients with AML with myelodysplasia-related changes (venetoclax plus LDAC vs placebo plus LDAC: 55.6% vs 44.4%), bone marrow blast count ≥50% (27.8% vs 11.1%), poor cytogenetic risk (55.6% vs 33.3%), tumour protein 53 (TP53) mutations (43.8% vs 22.2%), fms-like tyrosine kinase 3 (FLT3) mutations (18.8% vs 0%), nucleophosmin (NPM1) mutations (6.3% vs 22.2%) and prior use of hypomethylating agents (HMAs) (16.7% vs 33.3%). More than half of the patients in the venetoclax plus LDAC treatment arm (n = 10 [55.6%]) and n = 4 [44.4%] patients in the placebo plus LDAC arm had ≥2 reasons for ineligibility to receive intensive therapies.
Table 1.
Patient demographics and baseline characteristics
| Characteristic | Placebo + LDAC (n = 9) | Venetoclax + LDAC (n = 18) |
|---|---|---|
| Age | ||
| Median, years (range) | 78 (71–85) | 81 (60–89) |
| ≥75 years, n (%) | 8 (88.9) | 15 (83.3) |
| Male, n (%) | 4 (44.4) | 12 (66.7) |
| ECOG performance status, n (%) | ||
| 0 | 3 (33.3) | 5 (27.8) |
| 1 | 3 (33.3) | 10 (55.6) |
| 2 | 2 (22.2) | 3 (16.7) |
| 3 | 1 (11.1) | 0 |
| AML type, n (%) | ||
| De novo | 6 (66.7) | 13 (72.2) |
| Secondary | 3 (33.3) | 5 (27.8) |
| Secondary AML type, n/N (%) | ||
| Treatment-related AML | 0/3 | 1/5 (20.0) |
| Prior hematologic disorder | 3/3 (100.0) | 4/5 (80.0) |
| Myelodysplasia-related changes, n (%) | 4 (44.4) | 10 (55.6) |
| Prior treatment with HMAs, n (%) | 3 (33.3) | 3 (16.7) |
| Bone marrow blast count, n (%) | ||
| <30% | 4 (44.4) | 7 (38.9) |
| ≥30%–<50% | 4 (44.4) | 6 (33.3) |
| ≥50% | 1 (11.1) | 5 (27.8) |
| Cytogenetic risk, n (%) | ||
| Favourable | 0 | 0 |
| Intermediate | 6 (66.7) | 8 (44.4) |
| Poor | 3 (33.3) | 10 (55.6) |
| Somatic mutations, n/N (%) | ||
| TP53 | 2/9 (22.2) | 7/16 (43.8) |
| FLT3 | 0/9 | 3/16 (18.8) |
| IDH1/2 | 2/9 (22.2) | 3/16 (18.8) |
| NPM1 | 2/9 (22.2) | 1/16 (6.3) |
| Baseline hepatic impairment | 3 (33.3) | 8 (44.4) |
| Baseline renal impairment | 8 (88.9) | 18 (100.0) |
| Transfusion dependenta at baseline, n (%) | ||
| RBC or platelet | 7 (77.8) | 14 (77.8) |
| RBC | 6 (66.7) | 13 (72.2) |
| Platelet | 3 (33.3) | 8 (44.4) |
| Number of reasons for ineligibility to receive intensive therapy, n (%) | ||
| 1 | 5 (55.6) | 8 (44.4) |
| 2 | 4 (44.4) | 9 (50.0) |
| 3 | 0 | 1 (5.6) |
| ≥4 | 0 | 0 |
Transfusion dependence defined as transfusion within 56 days before first dose of study drug.
AML, acute myeloid leukaemia; ECOG, Eastern Cooperative Oncology Group; FLT3, fms-like tyrosine kinase 3; HMA, hypomethylating agent; IDH, isocitrate dehydrogenase; LDAC, low-dose cytarabine; NPM1, nucleophosmin; RBC, red blood cell; TP53, tumour protein 53.
The median treatment duration in the venetoclax plus LDAC and placebo plus LDAC arms was 2.1 (range: 0.2–23.5) months and 1.9 (range: 0.3–14.6) months, respectively. The proportion of patients who received any post-study treatment and intensive chemotherapy as post-study treatment was markedly higher in the placebo plus LDAC arm (77.8 and 55.6%, respectively) than in the venetoclax plus LDAC arm (27.8 and 16.7%, respectively; Table 2). The most common post-study treatments in the placebo plus LDAC arm were cytarabine (66.7%), aclarubicin hydrochloride (33.3%), and hydroxycarbamide (33.3%); azacitidine, cytarabine, daunorubicin and gemtuzumab ozogamicin (11.1% each) were the most common for patients in the venetoclax plus LDAC arm. Individual chemotherapy drugs were noted, but treatment regimens were not.
Table 2.
Summary of post-study treatment
| Treatment, n (%) | Placebo + LDAC (n = 9) | Venetoclax + LDAC (n = 18) |
|---|---|---|
| Any post-study treatment | 7 (77.8) | 5 (27.8) |
| Intensive chemotherapy | 5 (55.6) | 3 (16.7) |
| Aclarubicin/aclarubicin hydrochloride | 4 (44.4) | 1 (5.6) |
| Cytarabine | 4 (44.4) | 2 (11.1) |
| Daunorubicin/daunorubicin hydrochloride | 2 (22.2) | 3 (16.7) |
Treatment regimen was not collected, only individual chemotherapy drug.
LDAC, low-dose cytarabine.
Overall, as of the data cutoff date, 26 patients in the Japanese subgroup (venetoclax plus LDAC, n = 17; placebo plus LDAC, n = 9) had discontinued treatment. The primary reasons for study drug discontinuation were (venetoclax plus LDAC vs placebo plus LDAC): treatment failure (22.2% vs 44.4%), PD (11.1% vs 33.3%), physician decision (16.7% vs 11.1%), AE not related to PD (16.7% vs 0%), withdrawal of consent (11.1% each), morphologic relapse (11.1% vs 0%) and AE related to PD (5.6% vs 0%).
Efficacy
OS outcomes in the Japanese subgroup at the primary analysis and at the 6-month follow-up analysis are shown in Fig. 1A and B, respectively. At both analyses, median OS was 4.7 months in the venetoclax plus LDAC arm, and 8.1 months in the placebo plus LDAC arm. The HR at the 6-month follow-up was 0.928 (95% CI: 0.399–2.156). Considering the observed imbalance in baseline patient characteristics, a stepwise multivariate Cox regression analysis was performed to identify pre-treatment factors associated with OS. Factors included in the analysis were treatment arm, age, sex, AML status, bone marrow blast count, ECOG performance status, cytogenetic risk, prior use of HMAs and mutation status of FLT3, isocitrate dehydrogenase (IDH) and NPM1. To estimate the adjusted treatment effect, inclusion of treatment arm was forced into the model. Based on the stepwise variable selection, cytogenetic risk was identified as being significantly correlated with OS. At the 6-month follow-up, the HR for cytogenetic risk (intermediate vs poor) was 0.264 (95% CI: 0.102–0.685; P = 0.006). The covariate-adjusted HR for treatment arm (venetoclax plus LDAC vs placebo plus LDAC) was 0.800 (95% CI: 0.337–1.898), which was similar to that observed in the primary analysis.
Figure 1.
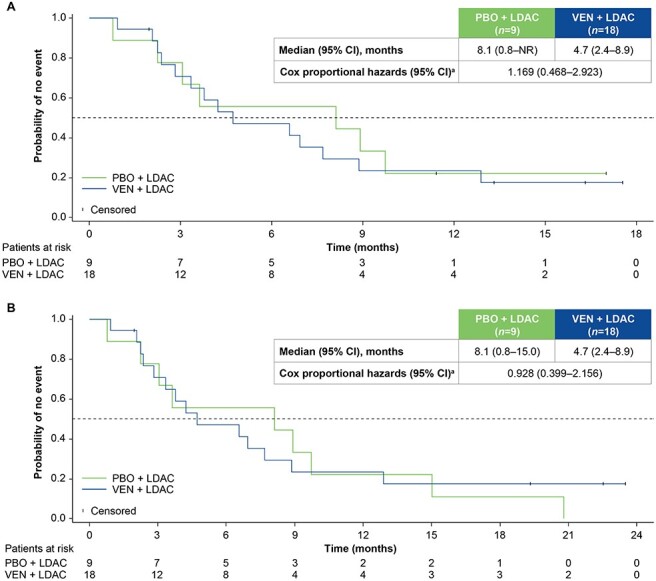
Overall survival (OS) by treatment arm at the primary analysis (A) and 6-month follow-up (B). aUnstratified Cox proportional hazards model. CI, confidence interval; HR, hazard ratio; LDAC, low-dose cytarabine; PBO, placebo; VEN, venetoclax.
Response rates for the Japanese subgroup are summarized in Fig. 2. The rates of CR and CR plus CRi were consistently higher in patients treated with venetoclax plus LDAC (22.2 and 44.4%, respectively) than in patients treated with placebo plus LDAC (11.1% each). The proportion of patients achieving CR plus CRi by initiation of cycle 2 was also higher in the venetoclax plus LDAC arm (44.4%) vs the placebo plus LDAC arm (0%).
Figure 2.
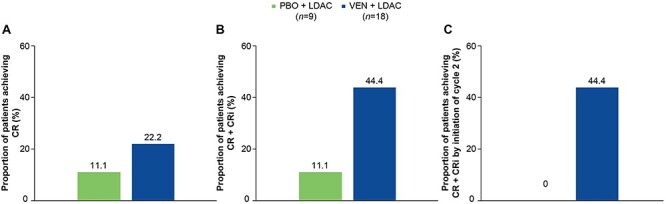
Rates of complete response (CR) (A), CR + CR with incomplete blood count recovery (CRi) (B), and CR + CRi by initiation of cycle 2 (C). LDAC, low-dose cytarabine; PBO, placebo; VEN, venetoclax.
Median EFS was numerically higher in the venetoclax plus LDAC arm vs the placebo plus LDAC arm both at the primary analysis (3.7 vs 2.2 months; HR: 0.565; 95% CI: 0.197–1.624; Fig. 3A) and at the 6-month follow-up (3.7 vs 2.2 months; HR: 0.620; 95% CI: 0.246–1.566; Fig. 3B).
Figure 3.
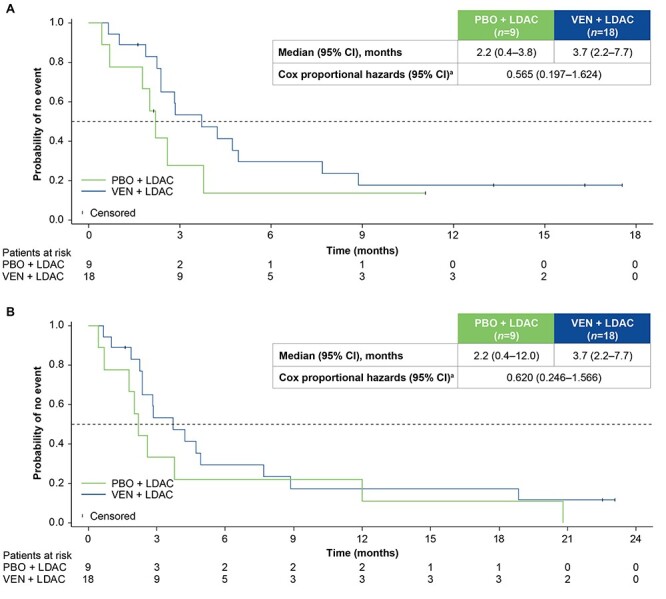
Event-free survival (EFS) by treatment arm at the primary analysis (A) and 6-month follow-up (B). aStratified by AML state (de novo vs secondary) and age (18–74 vs ≥75 years). AML, acute myeloid leukaemia; CI, confidence interval; LDAC, low-dose cytarabine; PBO, placebo; VEN, venetoclax.
The proportion of patients with post-baseline transfusion independence was similar across treatment groups in the Japanese subgroup (Fig. 4A). The median time to transfusion independence (RBC plus platelet) was shorter in the venetoclax plus LDAC arm than in the placebo plus LDAC arm (50 vs 96 days, respectively; Fig. 4B). One of 14 (7.1%) patients who were transfusion-dependent at baseline achieved transfusion independence during treatment with venetoclax plus LDAC, and 1 of 7 (14.3%) transfusion-dependent patients assigned to placebo plus LDAC achieved transfusion independence.
Figure 4.
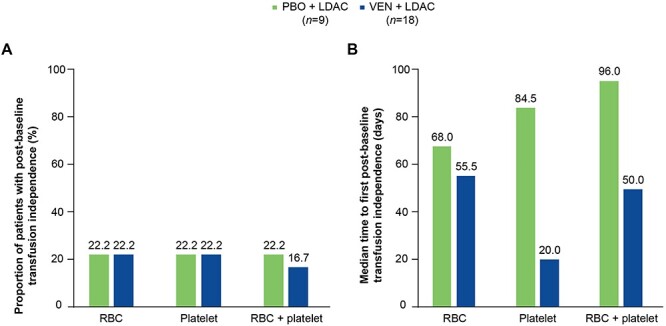
Proportion of patients with post-baseline transfusion independence (A) and median time to post-baseline transfusion independence (B), by treatment arm. Post-baseline transfusion independence was defined as a period of at least 56 consecutive days without transfusions. LDAC, low-dose cytarabine; PBO, placebo; RBC, red blood cell; VEN, venetoclax.
Safety
All patients in the Japanese subgroup experienced at least 1 AE (Table 3). The most frequently reported AEs (≥40% of patients) of any grade for the venetoclax plus LDAC or placebo plus LDAC arms, respectively, were nausea (66.7% vs 22.2%), febrile neutropenia (50.0% vs 44.4%), vomiting (50.0% vs 11.1%), decreased appetite (33.3% vs 44.4%), hypokalemia (33.3% vs 44.4%), thrombocytopenia (27.8% vs 44.4%) and pyrexia (22.2% vs 44.4%). The most frequently reported grade ≥ 3 AEs in the venetoclax plus LDAC or placebo plus LDAC arms, respectively, were hematologic: febrile neutropenia (50.0% vs 44.4%) and thrombocytopenia (27.8% vs 44.4%). Grade ≥ 3 pneumonia was reported in 3/18 (16.7%) patients in the venetoclax plus LDAC arm and in 3/9 (33.3%) in the placebo plus LDAC arm.
Table 3.
Adverse events (AEs) reported in ≥15% of patients in the venetoclax plus low-dose cytarabine (LDAC) arm only
| AE, n (%) | Placebo + LDAC (n = 9) | Venetoclax + LDAC (n = 18) | ||
|---|---|---|---|---|
| Any grade | Grade ≥ 3 | Any grade | Grade ≥ 3 | |
| Any | 9 (100.0) | 8 (88.9) | 18 (100.0) | 17 (94.4) |
| Hematologic | ||||
| Febrile neutropenia | 4 (44.4) | 4 (44.4) | 9 (50.0) | 9 (50.0) |
| Leukopenia | 0 | 0 | 3 (16.7) | 3 (16.7) |
| Neutropenia | 0 | 0 | 3 (16.7) | 3 (16.7) |
| Thrombocytopenia | 4 (44.4) | 4 (44.4) | 5 (27.8) | 5 (27.8) |
| Nonhematologic | ||||
| Back pain | 1 (11.1) | 0 | 3 (16.7) | 0 |
| Constipation | 3 (33.3) | 0 | 5 (27.8) | 0 |
| Decreased appetite | 4 (44.4) | 0 | 6 (33.3) | 1 (5.6) |
| Decreased weight | 0 | 0 | 3 (16.7) | 0 |
| Delirium | 1 (11.1) | 0 | 3 (16.7) | 0 |
| Diarrhoea | 2 (22.2) | 0 | 5 (27.8) | 0 |
| Dry skin | 0 | 0 | 3 (16.7) | 0 |
| Epistaxis | 0 | 0 | 3 (16.7) | 0 |
| Fatigue | 0 | 0 | 3 (16.7) | 1 (5.6) |
| Hypokalemia | 4 (44.4) | 3 (33.3) | 6 (33.3) | 3 (16.7) |
| Increased blood bilirubin | 0 | 0 | 3 (16.7) | 0 |
| Insomnia | 2 (22.2) | 0 | 5 (27.8) | 0 |
| Malaise | 2 (22.2) | 0 | 3 (16.7) | 0 |
| Nausea | 2 (22.2) | 0 | 12 (66.7) | 0 |
| Oropharyngeal pain | 1 (11.1) | 0 | 3 (16.7) | 1 (5.6) |
| Peripheral edema | 3 (33.3) | 0 | 4 (22.2) | 0 |
| Pneumonia | 3 (33.3) | 3 (33.3) | 5 (27.8) | 3 (16.7) |
| Proctalgia | 0 | 0 | 4 (22.2) | 0 |
| Pyrexia | 4 (44.4) | 1 (11.1) | 4 (22.2) | 1 (5.6) |
| Transfusion reaction | 0 | 0 | 4 (22.2) | 0 |
| Upper GI haemorrhage | 0 | 0 | 3 (16.7) | 1 (5.6) |
| Vomiting | 1 (11.1) | 0 | 9 (50.0) | 0 |
GI, gastrointestinal.
Serious AEs were reported in 9/18 (50.0%) and 3/9 (33.3%) patients in the venetoclax plus LDAC and placebo plus LDAC arms, respectively; pneumonia was the most common (22.2% each; Table 4). TLS was not observed in any patients in the Japanese subgroup. Fourteen (77.8%) patients died in the venetoclax plus LDAC arm and 9 (100.0%) in the placebo plus LDAC arm, mainly because of PD (61.1 and 77.8%, respectively). The rate of death within 60 days of initiating study treatment was similar in both treatment group (11.1% each).
Table 4.
Serious adverse events (AEs) reported in all patients
| Serious AE, n (%) | Placebo + LDAC (n = 9) | Venetoclax + LDAC (n = 18) |
|---|---|---|
| Any | 3 (33.3) | 9 (50.0) |
| Pneumonia | 2 (22.2) | 4 (22.2) |
| Multiple organ dysfunction syndrome | 1 (11.1) | 0 |
| Enterococcal infection | 1 (11.1) | 0 |
| Upper GI haemorrhage | 0 | 2 (11.1) |
| Congestive cardiac failure | 0 | 1 (5.6) |
| GI haemorrhage | 0 | 1 (5.6) |
| Intracranial haemorrhage | 0 | 1 (5.6) |
| Acute pancreatitis | 0 | 1 (5.6) |
| Acute cholecystitis | 0 | 1 (5.6) |
| Neutrophil count decreased | 0 | 1 (5.6) |
| WBC count decreased | 0 | 1 (5.6) |
GI, gastrointestinal; LDAC, low-dose cytarabine; WBC, white blood cell.
AEs led to study treatment discontinuation in 6/18 (33.3%) and 0/9 patients treated with venetoclax plus LDAC and placebo plus LDAC, respectively, including pneumonia (11.1% vs 0%), and neutropenia, congestive heart failure, acute pancreatitis, fatigue, decreased appetite, intracranial haemorrhage and organizing pneumonia (5.6% vs 0% for each). Dose interruption and/or reduction occurred in 11/18 (61.1%) patients in the venetoclax plus LDAC arm and 4/9 (44.4%) patients in the placebo plus LDAC arm. The most common AEs (≥10% of patients) leading to dose interruption and/or reduction were (venetoclax plus LDAC vs placebo plus LDAC): febrile neutropenia (16.7% vs 11.1%), thrombocytopenia (11.1% each), decreased neutrophil count (11.1% vs 0%), upper GI haemorrhage (11.1% vs 0%), atrial fibrillation (5.6% vs 11.1%), pneumonia (5.6% vs 11.1%), cellulitis (0% vs 11.1%) and fasciitis (0% vs 11.1%).
Discussion
In this small subgroup of Japanese patients participating in VIALE-C (n = 27), the unadjusted OS appeared comparable though unbalanced covariates may have obscured the observation of treatment effect. At a 6-month follow-up analysis, the addition of venetoclax to LDAC showed a median OS of 4.7 months vs 8.1 months with placebo plus LDAC. In contrast, median OS of the total VIALE-C study population at a 6-month follow-up was 8.4 and 4.1 months for venetoclax plus LDAC and placebo plus LDAC, respectively (31).
Several important factors may have influenced outcomes in the Japanese subgroup. First, the number of patients in the subgroup was small (venetoclax plus LDAC, n = 18; placebo plus LDAC, n = 9). Second, patients in the venetoclax plus LDAC arm were more likely to have high-risk features at baseline than patients in the placebo plus LDAC arm, including a poor-risk cytogenetic profile (55.6% vs 33.3%, respectively), which was shown to correlate with OS. After adjusting for cytogenetic risk in Japanese patients, the HR for the venetoclax plus LDAC treatment arm was 0.800 (95% CI: 0.337–1.898). Last, patients in the Japanese subgroup assigned to the venetoclax plus LDAC arm were less likely to receive post-study treatment than those in the placebo plus LDAC arm (27.8% vs 77.8%, respectively), including intensive chemotherapy (16.7% vs 55.6%, respectively), which may have reduced the ability to detect the effects of study treatment on OS in this subgroup. It is noteworthy that the decision to administer post-study treatment and the choice of regimen to be used was at the investigator’s discretion. The selection of further therapy was dependent upon important intermediate events during the study, such as treatment failure or PD. In the Japanese subgroup of the VIALE-C study, treatment failure and PD were reported as the primary reason for study discontinuation at least twice as often in the placebo plus LDAC arm (33.3 and 44.4%, respectively) compared with venetoclax plus LDAC arm (11.1 and 22.1%, respectively). The increased use of intensive chemotherapy as salvage treatment in Japanese patients compared with the total study population (29.6% vs 12.7%, respectively), and the greater use amongst Japanese patients randomized to the placebo plus LDAC arm compared with the venetoclax plus LDAC arm (n = 5/9 [55.6%] vs 3/18 [16.7%], respectively), may have reduced the ability to detect the effect of study treatment on survival within the Japanese subgroup.
A broad consensus on criteria for selection of the ideal ‘unfit’ patient with AML for inclusion in clinical trials was lacking and remains the subject of scientific debate. The structure for identifying patients who would not be suitable for intensive treatment in the VIALE-C, as well as the VIALE-A study, was based on age ≥ 75 years or age ≥ 18 to 74 years plus at least 1 criterion associated with lack of fitness for intensive induction chemotherapy (e.g. ECOG performance status of 2 or 3, particular defined comorbid conditions) (30,31). It is noteworthy that eligibility criteria for VIALE-C include prior MDS treated with HMAs, whereas VIALE-A excluded pre-treated MDS. Thus, VIALE-C included more patients with refractory MDS than VIALE-A, which may partially explain the difference in median OS between both studies. In a commentary by Lӧwenberg et al., it was noted that by using these inclusion criteria, a considerable proportion of the study population for these trials may not have only been ‘unfit’, but potentially ‘frail’ (e.g. patients had ECOG performance status of 3). Thus, a proportion of enrolled patients might have been too frail to benefit from almost any antileukemic treatment that introduces toxicities (35).
Despite the imbalances between treatment arms in baseline characteristics (e.g. poor cytogenetic risk, bone marrow blast count ≥50%, prior treatment with HMAs, TP53, NPM1, or FLT3 mutation, and AML with myelodysplasia-related changes) and older median age, the addition of venetoclax to LDAC was associated with an increased CR plus CRi rate in Japanese patients (44.4% vs 11.1%), and all responses to treatment with venetoclax plus LDAC were achieved within the first cycle of therapy (vs 0% with placebo plus LDAC). The transfusion-independence rate was similar in the venetoclax plus LDAC and placebo plus LDAC arms likely due to the small patient numbers. In addition, transfusion prescription was not defined within the protocol and was at the investigator’s discretion which could depend on institutional/regional guidelines. However, transfusion independence (RBC plus platelet) was achieved more rapidly with venetoclax plus LDAC compared with placebo plus LDAC (median 50 vs 96 days, respectively).
Of note, the differences in rates of post-study treatment received between treatment arms did not obscure the treatment effect of venetoclax plus LDAC when evaluating EFS as a secondary endpoint. At the 6-month follow-up, the Japanese subgroup reported a median EFS of 2.2 months in the placebo plus LDAC arm compared with 3.7 months in the venetoclax plus LDAC arm (HR: 0.620; 95% CI: 0.246–1.566), which was an opposite trend relative to the OS observation. It is noteworthy that the analysis of treatment arms shows a separation of EFS curves that implies benefit of venetoclax plus LDAC over placebo plus LDAC prior to receipt of subsequent salvage therapy. These data suggest that patients treated with venetoclax plus LDAC derived clinical benefit from therapy.
The safety profile of venetoclax plus LDAC was consistent with previous studies of venetoclax in AML, including the total study population of VIALE-C (24,29–31). AEs consisted mainly of hematologic events, such as febrile neutropenia and thrombocytopenia, and GI AEs (e.g. grade 1 or 2 nausea and vomiting). No cases of TLS were reported in the Japanese subgroup.
Despite the limitations of the current analysis (e.g. small patient numbers, imbalances between treatment arms in baseline characteristics, impact of post-study treatment), the data indicate a tolerable safety profile along with a trend toward beneficial improvements for patients treated with venetoclax plus LDAC in comparison to placebo plus LDAC. Treatment with venetoclax plus LDAC was well tolerated and led to higher CR plus CRi rates in comparison to treatment with placebo plus LDAC. These results support the consideration of venetoclax plus LDAC as a first-line treatment option for Japanese patients with AML ineligible for intensive chemotherapy.
Data sharing and data accessibility statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial-level data (analysis data sets), as well as other information (e.g. protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications.
These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and statistical analysis plan and execution of a data sharing agreement. Data requests can be submitted at any time, and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.
Contributor Information
Takahiro Yamauchi, Department of Hematology and Oncology, University of Fukui Hospital, Fukui, Japan.
Chikashi Yoshida, Department of Hematology, National Hospital Organization, Mito Medical Center, Ibaraki, Japan.
Kensuke Usuki, Department of Hematology, NTT Medical Center Tokyo, Tokyo, Japan.
Satoru Takada, Leukemia Research Center, Saiseikai Maebashi Hospital, Maebashi, Japan.
Itaru Matsumura, Department of Hematology and Rheumatology, Kindai University Hospital, Osaka, Japan.
Nobuaki Dobashi, Division of Clinical Oncology/Hematology, The Jikei University Daisan Hospital, Tokyo, Japan.
Yasushi Miyazaki, Department of Hematology, Atomic Bomb Disease Institute, Nagasaki University, Nagasaki, Japan.
Toshihiro Miyamoto, Medicine and Biosystemic Science, Graduate School of Medical Sciences, Kyushu University, Fukuoka, Japan.
Hiroatsu Iida, Department of Hematology, National Hospital Organization Nagoya Medical Center, Nagoya, Japan.
Norio Asou, Department of Hematology, Saitama Medical University International Medical Center, Hidaka, Japan.
Junya Kuroda, Division of Hematology and Oncology, Department of Medicine, Kyoto Prefectural University of Medicine, Kyoto, Japan.
Satoshi Ichikawa, Department of Hematology, Tohoku University Hospital, Sendai, Japan.
Norio Komatsu, Department of Hematology, Juntendo University School of Medicine, Tokyo, Japan.
Wellington Mendes, AbbVie Inc., North Chicago, IL, USA.
Hideyuki Honda, AbbVie GK, Tokyo, Japan.
Sumiko Okubo, AbbVie GK, Osaka, Japan.
Misaki Kurokawa, AbbVie GK, Tokyo, Japan.
Qi Jiang, AbbVie Inc., North Chicago, IL, USA.
Andrew Wei, Department of Clinical Haematology, The Alfred Hospital and Monash University, Melbourne, VIC, Australia.
Kenichi Ishizawa, Department of Third Internal Medicine, Yamagata University Hospital, Yamagata, Japan.
Author contributions
Takahiro Yamauchi: investigation and writing (reviewing and editing). Chikashi Yoshida: investigation and writing (reviewing and editing). Kensuke Usuki: investigation and writing (reviewing and editing). Satoru Takada: investigation and writing (reviewing and editing). Itaru Matsumura: investigation and writing (reviewing and editing). Nobuaki Dobashi: investigation and writing (reviewing and editing). Yasushi Miyazaki: investigation and writing (reviewing and editing). Toshihiro Miyamoto: investigation and writing (reviewing and editing). Hiroatsu Iida: investigation and writing (reviewing and editing). Norio Asou: investigation and writing (reviewing and editing). Junya Kuroda: investigation and writing (reviewing and editing). Satoshi Ichikawa: investigation and writing (reviewing and editing). Norio Komatsu: investigation and writing (reviewing and editing). Wellington Mendes: supervision, writing (original draft preparation) and writing (reviewing and editing). Hideyuki Honda: formal analysis, data curation and writing (reviewing and editing). Sumiko Okubo: formal analysis, data curation and writing (reviewing and editing). Misaki Kurokawa: formal analysis, data curation and writing (reviewing and editing). Qi Jiang: formal analysis, data curation and writing (reviewing and editing). Andrew Wei: investigation and writing (reviewing and editing). Kenichi Ishizawa: investigation and writing (reviewing and editing).
Acknowledgements
AbbVie and the authors thank all the trial investigators and the patients who participated in this clinical trial as well as Sathej Gopalakrishnan and Jiuhong Zha for their contributions to this publication. Medical writing support was provided by Mary L. Smith, PhD, CMPP from Aptitude Health, Atlanta, GA, USA and funded by AbbVie.
Funding
Venetoclax is being developed in a collaboration between AbbVie and Genentech. AbbVie sponsored the study, contributed to its design, collection, analysis and interpretation of the data, and participated in the writing, review and approval of the manuscript. All authors had access to relevant data and participated in the drafting, review and approval of this manuscript. No honoraria or payments were made for authorship.
Conflict of interest statement
Takahiro Yamauchi has received research support and honoraria from and has served as an advisor for AbbVie, Astellas Pharma, Boehringer Ingelheim, Chugai Pharmaceutical Co., Ltd, Gilead Sciences, Inc., Mundipharma, Jansen (research support only), Ono Pharmaceutical, Otsuka Pharmaceutical Co., Ltd, Pfizer, Solasia Pharma, SymBio, Takeda and Teijin Pharma. Chikashi Yoshida has received honoraria from AbbVie GK, Astellas Pharma, Bristol-Myers Squibb, Daiichi Sankyo Co. Ltd, Janssen Pharmaceutical KK, Meiji Seika Pharma Co., Ltd, Nippon Shinyaku Co., Ltd, Novartis Pharma KK and Otsuka Pharmaceutical Co., Ltd Kensuke Usuki has received research funding from AbbVie, Alexion, Astellas Pharma, Chugai Pharmaceutical Co., Ltd, Daiichi Sankyo Co., Gilead Sciences, Inc., Sumitomo Dainippon Pharma and SymBio, and has received honoraria from Novartis. Satoru Takada has nothing to disclose. Itaru Matsumura has received research funding from Chugai Pharmaceutical Co., Ltd, Eisai, Kyowa Kirin Co. Ltd, Shionogi & Co. Ltd and Sumitomo Dainippon Pharma, and has served on a speakers bureau for Astellas Pharma, Bristol-Myers Squibb, Inc., Daiichi Sankyo Co. Ltd, Janssen Pharmaceutical KK, Novartis Pharma KK, Otsuka Pharmaceutical Co., Ltd and Pfizer Japan Inc. Nobuaki Dobashi from Otsuka Pharmaceutical Co., Ltd. Yasushi Miyazaki has received honoraria from Astellas Pharma, Celgene, Chugai Pharmaceutical Co., Ltd, Kyowa Kirin, Nippon Shinyaku, Novartis, Otsuka Pharmaceutical Co., Ltd and Sumitomo Dainippon Pharma, and has received research funding from Chugai Pharmaceutical Co., Ltd and Sumitomo Dainippon Pharma. Toshihiro Miyamoto has received honoraria from AbbVie, Astellas Pharmaceutical Co., Ltd, Astellas Amgen Pharmaceutical Co., Ltd, Bristol-Myers Squibb, Celgene, Merch Sharp & Dohme, Otsuka Pharmaceutical Co., Ltd and Takeda Pharmaceutical Co. Hiroatsu Iida has received research funding from Chugai Pharmaceutical Co., Ltd, and has received honoraria from Astellas Pharma, Celgene, Janssen and Novartis. Norio Asou has received research funding from AbbVie; Astellas Pharma, Chugai Pharmaceutical Co., Ltd, Eisai and Sumitomo Dainippon Pharma; has served on a speakers bureau for Asahi Kasei, Fuji Pharma, Nippon Shinyaku and Sumitomo Dainippon Pharma; and has received honoraria from Novartis and Nippon Shinyaku. Junya Kuroda has received honoraria from AbbVie, Astellas Pharma, Bristol-Myers Squibb, Celgene, Chugai Pharmaceutical Co., Ltd, Daiichi Sankyo Co., Eisai, Fujimoto Pharmaceutical, Janssen Pharmaceutical KK, Kyowa Kirin, Nippon Shinyaku, Ono Pharmaceutical, Otsuka Pharmaceutical Co., Ltd, Pfizer, Sanofi, Sumitomo Dainippon Pharma and Takeda Pharmaceutical; has received research funding: from AbbVie, Asahi Kasei, Astellas Pharma, Bristol-Myers Squibb, Celgene, Chugai Pharmaceutical Co., Ltd, Daiichi Sankyo Co., Eisai, Fujimoto Pharmaceutical, Kyowa Kirin, MSD, Nippon Shinyaku, Ono Pharmaceutical, Otsuka Pharmaceutical Co., Ltd, Pfizer, Sanofi, Shionogi, Sumitomo Dainippon Pharma, Sysmex, Taiho Pharmaceutical and Takeda Pharmaceutical; and has served as a consultant for AbbVie, Bristol-Myers Squibb, Celgene, Janssen Pharmaceutical KK and Sanofi. Satoshi Ichikawa from Chugai Pharmaceutical Co., Ltd. Norio Komatsu has received research funding from Bristol-Myers Squibb KK, Chugai Pharmaceutical Co., Ltd, Fujifilm Wako Pure Chemical Corporation, Fuso Pharmaceutical Industries, Ltd, Kyowa Hakko Kirin Co., Ltd, Otsuka Pharmaceutical Co., Ltd, Novartis Pharma KK, Pfizer Japan Inc., PharmaEssentia Japan KK, Shire Japan KK, Sumitomo Dainippon Pharma and Takeda Pharmaceutical Co., Ltd; has received advisory fees from AbbVie GK, Celgene KK, Japan Tobacco Inc. (Funding provided was intended only to support the investigator and does not support the manuscript or any work involved in producing the manuscript. The manuscript content is not in any way related to the tobacco industry), Novartis Pharma KK, Otsuka Pharmaceutical Co., Ltd, PharmaEssentia Japan KK and Shire Japan KK; has served on a speakers bureau for Novartis Pharma KK, Shire Japan KK and Takeda Pharmaceutical Co., Ltd; and served as a member of safety assessment committee in the M13–834 clinical trial. Wellington Mendes, Hideyuki Honda, Sumiko Okubo, Misaki Kurokawa and Qi Jiang are employees of AbbVie and may own stock. Andrew Wei has received research funding from AbbVie, Amgen, AstraZeneca, Celgene/BMS, Novartis, Servier and F.Hoffmann-La Roche; has received honoraria from AbbVie, Amgen, Astellas, AstraZeneca, Celgene/Bristol-Myers Squibb, Genentech, Janssen, MacroGenics, Novartis, Pfizer and Servier; has served on advisory boards for AbbVie, Amgen, Astellas, AstraZeneca, Celgene/Bristol-Myers Squibb, Genentech, Janssen, MacroGenics, Novartis, Pfizer and Servier; has served on a speakers bureau for Novartis, AbbVie and Celgene/Bristol-Myers Squibb; has served as a consultant for Servier; is a former employee of the Walter and Eliza Hall Institute of Medical Research, which receives royalties in relation to venetoclax, and is entitled to a fraction of these payments. Kenichi Ishizawa has received research funding from AbbVie, Bayer, Novartis and SymBio, and has served on a speakers bureau for Celgene, Chugai Pharmaceutical Co., Ltd, Eisai, Novartis, Ono Pharmaceutical and Takeda.
Statement of prior presentation
Results of this study have been partially presented at the 43rd Annual Meeting of the Japan Society for Hematopoietic Cell Transplantation Congress, Virtual, 5–7 March 2021.
Statement on originality of the work
The manuscript represents original work and has not been submitted for publication elsewhere nor previously published.
Abbreviations
AE, adverse event; AML, acute myeloid leukaemia; BCL2, B-cell leukaemia/lymphoma-2; BH, BCL2 homology; CI, confidence interval; CR, complete remission; CRi, complete remission with incomplete blood count recovery; ECOG, Eastern Cooperative Oncology Group; EFS, event-free survival; FLT3, fms-like tyrosine kinase 3; GI, gastrointestinal; HMA, hypomethylating agent; HR, hazard ratio; IDH, isocitrate dehydrogenase; LDAC, low-dose cytarabine; MDS, myelodysplastic syndromes; NPM1, nucleophosmin; OS, overall survival; PBO, placebo; QD, once daily; RBC, red blood cell; TLS, tumour lysis syndrome; TP53, tumour protein 53; VEN, venetoclax; WBC, white blood cell.
References
- 1.Miyawaki S. Clinical studies of acute myeloid leukemia in the Japan Adult Leukemia Study Group. Int J Hematol 2012;96:171–7. [DOI] [PubMed] [Google Scholar]
- 2.Niino M, Matsuda T. Type distribution of myeloid leukemia from Cancer Incidence in Five Continents Vol. X. Jpn J Clin Oncol 2016;46:394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tasaki T, Yamauchi T, Matsuda Y, et al. The response to induction therapy is crucial for the treatment outcomes of elderly patients with acute myeloid leukemia: single-institution experience. Anticancer Res 2014;34:5631–6. [PubMed] [Google Scholar]
- 4.Bhayat F, Das-Gupta E, Smith C, McKeever T, Hubbard R. The incidence of and mortality from leukaemias in the UK: a general population-based study. BMC Cancer 2009;9:252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Phekoo KJ, Richards MA, Møller H, Schey SA. South Thames Haematology Specialist Committee. The incidence and outcome of myeloid malignancies in 2,112 adult patients in southeast England. Haematologica 2006;91:1400–4. [PubMed] [Google Scholar]
- 6.Pulte D, Gondos A, Brenner H. Improvements in survival of adults diagnosed with acute myeloblastic leukemia in the early 21st century. Haematologica 2008;93:594–600. [DOI] [PubMed] [Google Scholar]
- 7.Ohnishi H, Imataki O, Kawachi Y, Ide M, Kawakami K, Waki M, et al. Age is an independent adverse prognostic factor for overall survival in acute myeloid leukemia in Japan. World J Hematol 2014;3:105–14. [Google Scholar]
- 8.Heuser M, Ofran Y, Boissel N, et al. Acute myeloid leukaemia in adult patients: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2020;31:697–712. [DOI] [PubMed] [Google Scholar]
- 9.Kantarjian H, Ravandi F, O'Brien S, et al. Intensive chemotherapy does not benefit most older patients (age 70 years or older) with acute myeloid leukemia. Blood 2010;116:4422–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kiyoi H, Yamaguchi H, Maeda Y, Yamauchi T. JSH practical guidelines for hematological malignancies, 2018: I. leukemia-1. Acute myeloid leukemia (AML). Int J Hematol 2020;111:595–613. [DOI] [PubMed] [Google Scholar]
- 11.Grimwade D, Hills RK, Moorman AV, et al. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 2010;116:354–65. [DOI] [PubMed] [Google Scholar]
- 12.Estey E. Acute myeloid leukemia and myelodysplastic syndromes in older patients. J Clin Oncol 2007;25:1908–15. [DOI] [PubMed] [Google Scholar]
- 13.Dombret H, Raffoux E, Gardin C. Acute myeloid leukemia in the elderly. Semin Oncol 2008;35:430–8. [DOI] [PubMed] [Google Scholar]
- 14.Pollyea DA, Kohrt HE, Medeiros BC. Acute myeloid leukaemia in the elderly: a review. Br J Haematol 2011;152:524–42. [DOI] [PubMed] [Google Scholar]
- 15.He P-F, Zhou J-D, Yao D-M, et al. Efficacy and safety of decitabine in treatment of elderly patients with acute myeloid leukemia: a systematic review and meta-analysis. Oncotarget 2017;8:41498–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dombret H, Seymour JF, Butrym A, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015;126:291–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kantarjian HM, Thomas XG, Dmoszynska A, et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol 2012;30:2670–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fischer K, Al-Sawaf O, Fink A-M, et al. Venetoclax and obinutuzumab in chronic lymphocytic leukemia. Blood 2017;129:2702–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar S, Kaufman JL, Gasparetto C, et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood 2017;130:2401–9. [DOI] [PubMed] [Google Scholar]
- 20.Moreau P, Chanan-Khan A, Roberts AW, et al. Promising efficacy and acceptable safety of venetoclax plus bortezomib and dexamethasone in relapsed/refractory MM. Blood 2017;130:2392–400. [DOI] [PubMed] [Google Scholar]
- 21.Roberts AW, Davids MS, Pagel JM, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med 2016;374:311–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seymour JF, Kipps TJ, Eichhorst B, et al. Venetoclax–rituximab in relapsed or refractory chronic lymphocytic leukemia. N Engl J Med 2018;378:1107–20. [DOI] [PubMed] [Google Scholar]
- 23.Stilgenbauer S, Eichhorst B, Schetelig J, et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol 2016;17:768–78. [DOI] [PubMed] [Google Scholar]
- 24.Wei AH, Strickland SA Jr, Hou J-Z, et al. Venetoclax combined with low-dose cytarabine for previously untreated patients with acute myeloid leukemia: results from a phase Ib/II study. J Clin Oncol 2019;37:1277–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Del Poeta G, Venditti A, Del Principe MI, et al. Amount of spontaneous apoptosis detected by Bax/Bcl-2 ratio predicts outcome in acute myeloid leukemia (AML). Blood 2003;101:2125–31. [DOI] [PubMed] [Google Scholar]
- 26.Lagadinou ED, Sach A, Callahan K, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013;12:329–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Teh T-C, Nguyen N-Y, Moujalled DM, et al. Enhancing venetoclax activity in acute myeloid leukemia by co-targeting MCL1. Leukemia 2018;32:303–12. [DOI] [PubMed] [Google Scholar]
- 28.Niu X, Zhao J, Ma J, et al. Binding of released bim to Mcl-1 is a mechanism of intrinsic resistance to ABT-199 which can be overcome by combination with daunorubicin or cytarabine in AML cells. Clin Cancer Res 2016;22:4440–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DiNardo CD, Pratz K, Pullarkat V, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019;133:7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DiNardo C, Jonas B, Pullarkat V, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med 2020;383:617–29. [DOI] [PubMed] [Google Scholar]
- 31.Wei AH, Montesinos P, Ivanov V, et al. Venetoclax plus LDAC for newly diagnosed AML ineligible for intensive chemotherapy: a phase 3 randomized placebo-controlled trial. Blood 2020;135:2137–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391–405. [DOI] [PubMed] [Google Scholar]
- 33.Cheson BD, Bennett JM, Kopecky KJ, et al. Revised recommendations of the International Working Group for Diagnosis, standardization of response criteria, treatment outcomes, and reporting standards for therapeutic trials in acute myeloid leukemia. J Clin Oncol 2003;21:4642–9. [DOI] [PubMed] [Google Scholar]
- 34.Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017;129:424–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Löwenberg B, Huls G. The long road: improving outcome in elderly "unfit" AML? Blood 2020;11;135:2114–5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial-level data (analysis data sets), as well as other information (e.g. protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications.
These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and statistical analysis plan and execution of a data sharing agreement. Data requests can be submitted at any time, and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.