

Abstract
Rationale
COPD and smoking are characterised by pulmonary inflammation. 18F-fluorodeoxyglucose positron emission tomography/computed tomography (FDG PET/CT) imaging may improve knowledge of pulmonary inflammation in COPD patients and aid early development of novel therapies as an imaging biomarker.
Objectives
To evaluate pulmonary inflammation, assessed by FDG uptake, in whole and regional lung in “usual” (smoking-related) COPD patients, alpha-1 antitrypsin deficiency (α1ATD) COPD patients, smokers without COPD and never-smokers using FDG PET/CT. Secondly, to explore cross-sectional associations between FDG PET/CT and systemic inflammatory markers in COPD patients and repeatability of the technique in COPD patients.
Methods
Data from two imaging studies were evaluated. Pulmonary FDG uptake (normalised Ki; nKi) was measured by Patlak graphical analysis in four subject groups: 84 COPD patients, 11 α1ATD-COPD patients, 12 smokers and 10 never-smokers. Within the COPD group, associations between nKi and systemic markers of inflammation were assessed. Repeatability was evaluated in 32 COPD patients comparing nKi values at baseline and at 4-month follow-up.
Results
COPD patients, α1ATD-COPD patients and smokers had increased whole lung FDG uptake (nKi) compared with never-smokers (0.0037±0.001, 0.0040±0.001, 0.0040±0.001 versus 0.0028±0.001 mL·cm−3·min−1, respectively, p<0.05 for all). Similar results were observed in upper and middle lung regions. In COPD participants, plasma fibrinogen was associated with whole lung nKi (β=0.30, p=0.02) in multivariate analysis adjusted for current smoking, forced expiratory volume in 1 s % predicted, systemic neutrophils and C-reactive protein levels. Mean percentage difference in nKi between the baseline and follow-up was 3.2%, and the within subject coefficient of variability was 7.7%.
Conclusions
FDG PET/CT has potential as a noninvasive tool to enable whole lung and regional quantification of FDG uptake to assess smoking- and COPD-related pulmonary inflammation.
Short abstract
FDG PET/CT has potential utility to noninvasively evaluate pulmonary inflammation in COPD. Pulmonary FDG uptake is increased in COPD patients, positively associated with systemic inflammatory markers and shows low inter-occasion variability. https://bit.ly/3dELYAW
Introduction
COPD is a heterogenous condition characterised by persistent respiratory symptoms and airflow limitation due to airway and alveolar pathology [1]. Although the molecular origins of the disease have yet to be fully elucidated, it is known that tobacco smoking remains the main modifiable risk factor for the development of COPD and it is understood that tobacco smoke inhalation (or other toxic air particulates or gases) trigger an abnormal exuberant pulmonary inflammatory response, which may continue even when the noxious stimuli is removed. Persistent inflammation subsequently leads to irreversible tissue destruction and airway remodelling changes [2]. Inflammation is a driver of disease progression in COPD and is characterised by increased lymphocytes, macrophages and neutrophils within the lung [3]. While forced expiratory volume in 1 s (FEV1), assessed by spirometry, is used to diagnose COPD, this measure does not relate well to pulmonary inflammation or indeed the symptoms that patients experience [4]. Moreover, lung damage has often developed and progressed before the spirometric diagnostic threshold of COPD is reached [5]. Current treatment options for COPD remain limited, which may in part be due to the lack of biomarkers reflective of disease phenotypes, progression or severity [6].
18F-fluorodeoxyglucose positron emission tomography paired with computed tomography (FDG PET/CT) is a functional noninvasive imaging modality which enables in vivo regional visualisation and quantification of glucose metabolism to assess pulmonary inflammation [7]. To date, Jones et al. [8] showed that FDG uptake quantified by PET could distinguish COPD patients from healthy never-smokers and asthma sufferers. Subramanian et al. [9] observed that in COPD patients FDG uptake correlated with FEV1 but found no difference in FDG uptake in alpha-1 antitrypsin PiZZ deficiency (α1ATD) patients with COPD (α1ATD-COPD) compared with healthy never-smoking controls.
Despite these encouraging results from small studies, it remains to be elucidated whether FDG PET has potential utility as a research tool to advance pathophysiological understanding and therapeutics development for COPD. Important questions that have not previously been addressed to support FDG PET's utility in this domain, include the impact of smoking on pulmonary inflammation quantified by FDG PET uptake, repeatability of pulmonary FDG PET in COPD patients and the clinical relevance of FDG PET as a biomarker to evaluate pulmonary inflammation in COPD patients in relation to validated peripheral biomarkers of inflammation and CT features of disease [10].
In this study, we hypothesised that usual (smoking-related) COPD patients have increased pulmonary inflammation, as measured by FDG PET uptake, compared with α1ATD-COPD patients, chronic smokers (≥10 pack-years) without COPD and healthy never-smokers. We sought to determine whole lung and regional FDG PET uptake in four subject groups and evaluate the impact of smoking on FDG PET uptake. Within the COPD group, we also assessed the association of pulmonary inflammation assessed by FDG uptake with peripheral markers of inflammation and explored FDG PET uptake stratified by high-resolution computed tomography (HRCT) subtypes in COPD patients [10]. Finally, we evaluated the repeatability of FDG PET/CT to quantify FDG uptake in COPD patients at baseline and at 4 months.
Methods
Study design
Data reported in this manuscript include participants from two parallel FDG PET imaging studies: 1) an observational cross-sectional study (EVOLVE) (REC 13/EE/0165, UK CRN ID 1513), which included COPD patients with low fibrinogen levels (<2.8 g·L−1), α1ATD-COPD, and both smokers and never-smokers without COPD; and 2) the EVOLUTION clinical trial (ClinicalTrials.gov identifier: NCT01541852), a double-blind, placebo-controlled phase 2a trial in COPD patients with baseline fibrinogen ≥2.8 g·L−1 with baseline FDG PET/CT scans before the intervention of Losmapimod (a p38 mitogen-activated protein kinase inhibitor)/placebo with repeat imaging at 4-months follow-up. In this manuscript, baseline data from all trial participants are incorporated into the COPD group to create a large cohort of COPD patients for analysis. The trial's placebo group was used to assess repeatability of FDG PET/CT data from baseline to 4-month follow-up. Methodology and results from both these studies have been published previously [11, 12]. Both studies recruited participants from two UK tertiary centres and received favourable opinions from the Cambridge South Research Ethics Committee (EVOLVE: REC 13/EE/0165, UK CRN ID 1513, EVOLUTION: 12/EE/0135). Medicines and Healthcare products Regulatory Agency approval was obtained for the EVOLUTION clinical trial. Written informed consent was obtained from all participants and the studies were carried out in accordance with institutional guidelines and the Declaration of Helsinki.
Participant groups
There were four participant groups included in the study. 1) Individuals clinically diagnosed with COPD as confirmed by post-bronchodilator spirometry FEV1/forced vital capacity (FVC) <0.70, and a reported smoking history of ≥10 pack-years smoked. 2) α1ATD-COPD patients (PiZZ phenotype), where clinical diagnosis had been confirmed and post-bronchodilator spirometry confirmed FEV1/FVC <0.70. 3) Chronic smokers without COPD, defined as ≥10 pack-years smoking history, who smoked approximately ≥10 cigarettes per day in the 12 months preceding study enrolment, with no clinical diagnosis of COPD, and spirometric values within the normal range (for both FEV1 and FEV1/FVC). 4) Never-smokers, similarly with spirometric values in the normal range.
Both COPD and α1ATD-COPD participants had to be clinically stable, and free of exacerbations in the preceding 4 weeks before enrolment in the study. The PET acquisition protocol required a body mass index (BMI) in the range 17–35 kg·m−2. Age and sex were prospectively matched across the four groups as closely as possible to enable cross-sectional analysis of the different subject groups.
Lung quantitative PET/CT protocol
Scans were performed in Cambridge (PET/CT unit, Addenbrookes Hospital) and London (Imanova Centre, Hammersmith). A General Electric Lightspeed VCT (GE Healthcare, Milwaukee, WI, USA) scanner was used in Cambridge and a Siemens Biograph (Siemens, Munich, Germany) scanner in London. All participants were imaged under a closely matched acquisition protocol.
Participants were required to fast for 6 h prior to the scan and to avoid any strenuous exercise in the preceding 24 h to limit muscle uptake of tracer. Blood glucose levels had to be <11 mmol·L−1 to proceed with the scan. Participants were positioned comfortably on the scanner couch with arms down by sides.
In COPD and α1ATD patients, a HRCT scan was performed at maximum inspiration, dependent on the subject's breath hold capability. In all participants, a non-contrast low-dose CT scan covering one bed position centred on the lungs with the participant breathing freely was performed to enable attenuation-correction and anatomical co-registration of PET data. Following the CT scan, the PET scan was immediately commenced. A dose of approximately 240 MBq 18F-fluorodeoxyglucose (FDG) was injected at the start of the scan, followed by 10 mL flush of normal saline. Dynamic data acquisition by list mode was acquired for 60 min from injection using the standard energy and coincidence timing window settings for the scanner and acquired in 23 dynamic frames.
Lung image analysis
Using Analyze 11.0 (AnalyzeDirect, Inc., Overland Park, KS, USA), the co-registered attenuation-correction CT images were segmented using a semi-automated process into whole right and left lung, any large arteries or obvious airways were excluded. Next, the CT mask was down-sampled to match the PET resolution, and was further refined: 1) to exclude any holes where vessels were removed; 2) a further 1 cm3 rim was removed from the edge of the fused mask to avoid motion artefact; and 3) to remove any areas of the mask proximal to the diaphragm with conspicuous motion artefact. The mask was then automatically divided into three regions of equal volumes along the transverse axis: upper, middle and lower.
The rate of FDG uptake, Ki, was evaluated using the widely established Patlak graphical technique [13] as pre-specified in the trial protocol [11]; Ki was normalised (nKi) as described in previous studies [8] and used as a surrogate marker of pulmonary inflammation. Further information is provided in the online supplementary material. A lung segmentation of the FDG PET/CT scan image of a COPD patient in the study is shown in figure 1.
FIGURE 1.
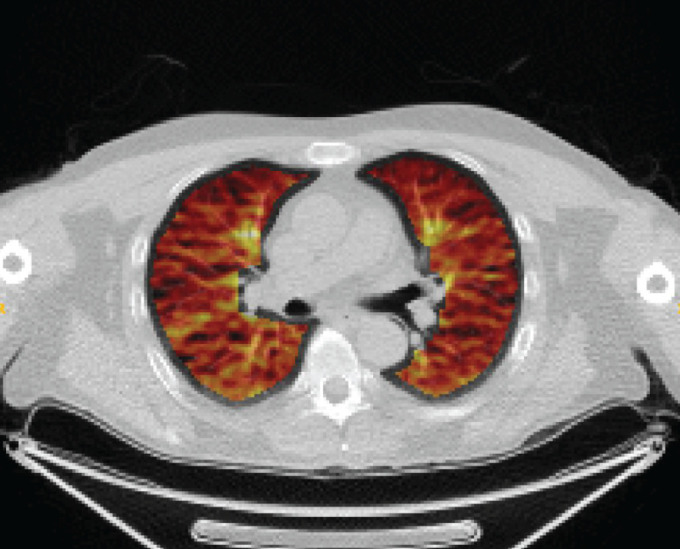
Pulmonary 18F-fluorodeoxyglucose positron emission tomography/computed tomography fused image of participant in the study.
Perc15
In COPD and α1ATD patients, the Perc15 score, calculated from the HRCT scan as the 15th percentile of Hounsfield units (HU) distribution (as described previously [14]), was used as a surrogate of emphysema severity.
COPD CT image subtypes
HRCT scans from usual COPD participants were visually analysed using the HOROS imaging platform to define the dominant CT-definable subtype of COPD according to the Fleischner classification system [10]. The Fleischner classification of COPD CT-definable subtypes includes main patterns of centrilobular emphysema, panlobular, paraseptal emphysema, airway disease and associated features. Scans were assessed for the predominant CT pattern by a radiology trainee with 3 years experience, following a period of initial training by a thoracic radiologist.
Other markers
Blood samples were analysed for total white blood cell count (WCC), neutrophils, plasma fibrinogen (Klauss Method) and high sensitivity C-reactive protein (hsCRP) in National Health Service hospital laboratories using quality-controlled validated assays used in clinical practice. Spirometry was performed in accordance with American Thoracic Society (ATS) guidance [15]. For patients, up to 400 μg of salbutamol inhaler was administered prior to assessment. For volunteers, no bronchodilator reversibility testing was performed.
The 6-min walking distance (6MWD) test was performed in usual COPD participants according to the ATS guidelines with the exception that a practice test was not conducted [16].
Statistical analyses
Data were analysed using SPSS software (version 23; IBM Corp., Armonk, NY, USA) and R version 3.0.0 for Microsoft Windows with R studio version 0.98.953 (www.r-project.org/). For cross-sectional analyses, never-smokers were the control group. Unpaired t-test was used to compare imaging measures between the groups adjusted for multiple testing using the Bonferroni correction. Homogeneity of variance was assessed by Levene's test and Shapiro–Wilks used to check for normality prior to performing statistical tests. For unequal sample sizes, Welch's t-test was used in cases of unequal variance. Pearson's correlation coefficient and multivariate linear regression analysis were used to assess associations between variables, using mean values unless otherwise stated. Data were assessed for normality and log-transformed if necessary. A Bland–Altman plot was used to evaluate the repeatability of FDG PET/CT in the placebo arm of the longitudinal EVOLUTION cohort and paired t-tests used to compare values at baseline and follow-up. p-values <0.05 were considered significant for all statistical analyses. All data are presented as mean±sd, percentages, or with 95% confidence intervals.
Results
The demographics of the groups and main results are shown in table 1. The total number of evaluable cross-sectional scans were 84 COPD patients, 11 α1ATD-COPD patients, 12 chronic smokers without COPD and 10 never-smokers. Four participants were excluded due to excessive movement on the PET/CT scan, which could not be corrected (one COPD patient, one α1ATD-COPD patient and two never-smokers). COPD patients and never-smokers were both older (68±8 years and 69±7 years) and had a higher BMI (25.9±3.9 and 26.6±2.6 kg·m−2) than α1ATD patients (62±8 years, 25.0±3.3 kg·m−2) and smokers (62±6 years, 23.1±2.3 kg·m−2) (table 1). There were a higher proportion of women in the COPD and smokers without COPD groups compared with never-smokers, although this did not reach statistical significance. There was no difference in pack years smoked between COPD patients and smokers (45±25 versus 37±19 pack years, p=0.36), but both groups smoked significantly more than α1ATD-COPD patients (19±11 pack years, p<0.001 for both). 68 (81%) of COPD patients and 10 (91%) of α1ATD patients used a combined long-acting β-agonist/inhaled corticosteroid inhaler.
TABLE 1.
Demographics, spirometry and image data
| COPD | α1ATD-COPD | Smokers | Never-smokers | |
| Subjects n | 84 | 11 | 12 | 10 |
| Demographics | ||||
| Age years | 68±8 | 62±8* | 62±6* | 69±7 |
| Male % | 67 | 73 | 58 | 83 |
| BMI kg·m−2 | 25.9±3.9 | 25.0±3.3 | 23.1±2.3* | 26.6±2.6 |
| Current smoker n (%) | 11 (13)*** | 2 (17)*** | 12 (100)*** | 0 |
| Pack-years smoked | 45±25*** | 19±11*** | 37±19*** | 0 |
| LABA/LAMA/ICS n (%) | 68 (81) | 10 (91) | ||
| Lung function | ||||
| FEV1 L | 1.37±0.6*** | 1.47±0.4*** | 2.84±0.56 | 2.88±0.6 |
| FEV1 % predicted | 51±20*** | 45±16*** | 95±17 | 100±15 |
| FEV1/FVC | 0.45±0.15 | 0.36±0.10 | 0.79±0.08 | 0.77±0.06 |
| Laboratory data | ||||
| Fibrinogen g·L−1 | 3.4±0.7* | 3.1±0.6 | 2.8±0.6 | 2.7±0.5 |
| hsCRP mg·L−1 | 5.2±7.0* | 3.3±2.3* | 2.1±1.4 | 1.2±0.6 |
| White cell count ×109·L−1 | 6.54±1.83 | 7.01±2.72 | 7.28±2.02 | 5.84±1.31 |
| Neutrophils ×109·L−1 | 4.43±3.6 | 4.68±2.47 | 4.53±1.45 | 3.63±1.11 |
| Pulmonary image data | ||||
| Whole lung | ||||
| nKi mL·cm−3·min−1 | 0.0037±0.001* | 0.0040±0.001* | 0.0040±0.001* | 0.0028±0.001 |
| Perc15 HU | −889±54*** | −942±28*** | ||
| Upper lung | ||||
| nKi mL·cm−3·min−1 | 0.0040±0.001** | 0.0038±0.001* | 0.0044±0.00** | 0.0027±0.001 |
| Perc15 HU | −884±61* | −922±34*** | ||
| Middle lung | ||||
| nKi mL·cm−3·min−1 | 0.0037±0.001** | 0.0042±0.001** | 0.0041±0.001** | 0.0027±0.001 |
| Perc15 HU | −889±52* | −943±29*** | ||
| Lower lung | ||||
| nKi mL·cm−3·min−1 | 0.0036±0.001 | 0.0038±0.001 | 0.0040±0.001 | 0.0032±0.001 |
| Perc15 HU | −880±49** | −952±26*** | ||
Data are presented as mean±sd, unless otherwise stated. α1ATD: alpha-1 antitrypsin deficiency; BMI: body mass index; LABA: inhaled long-acting β-agonist; LAMA: long-acting muscarinic antagonist; ICS: inhaled corticosteroid; FEV1: forced expiratory volume in 1 s; FVC: forced vital capacity; hsCRP: high sensitivity C-reactive protein; HU: Hounsfield units. ***: p<0.001; **: p<0.01; *: p<0.05 significant difference compared to never-smokers.
Pulmonary FDG uptake (nKi)
Whole lung, upper and middle lung nKi values were higher in COPD, α1ATD-COPD and smokers without COPD compared with never-smokers (p<0.05) (figure 2, table 1). There were no statistical differences in FDG uptake between COPD, α1ATD-COPD or smokers, or between all groups in the lower lung regions. A within-group comparison of regional nKi values in the upper, middle and lower lung revealed no significant differences in any of the subject groups (table S1). The maximum within group difference observed was 0.49±0.03×10−3 mL·cm−3·min−1, p=0.66, between the upper and lower lung zones in smokers.
FIGURE 2.
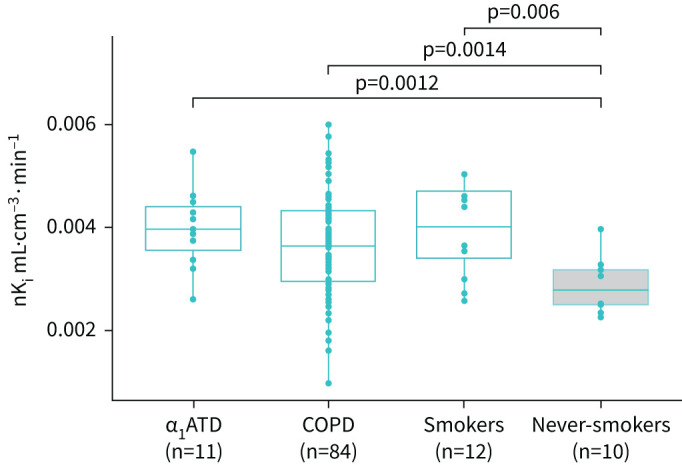
Rate of 18F-fluorodeoxyglucose uptake measured using nKi as a marker of whole lung pulmonary inflammation in COPD groups, smokers and never-smokers. Data are presented as individual values together with median, first and third quartile values (boxes). A statistical difference was observed between COPD patients, alpha-1 antitrypsin deficiency (α1ATD) patients and smokers versus never-smokers as a control group.
Smoking
In the α1ATD-COPD group, there was no difference in whole lung nKi between α1ATD patients who were current smokers (n=2, 4.2±0.1×10−3 mL·cm−3·min−1) versus ex-smokers (n=8, 3.9±0.1×10−3 mL·cm−3·min−1, p=0.66). Grouping usual-COPD patients by smoking status showed that nKi was modestly elevated in COPD current smokers compared with COPD ex-smokers (n=11 and 73; 4.2±0.1×10−3 mL·cm−3·min−1 versus 3.7±0.1×10−3 mL·cm−3·min−1, respectively; p=0.047) despite no significant difference in FEV1 between them (FEV1 % predicted 47±18% versus 50±22%, respectively). There was no difference in nKi in ex-smoker usual-COPD participants (3.7±0.1×10−3 mL·cm−3·min−1) compared with current smokers without COPD (4.0±0.1×10−3 mL·cm−3·min−1, p=0.10). Within the usual-COPD cohort, there was no correlation between the total pack-years smoked and whole lung nKi (r=0.07, p=0.44). Stratifying whole lung nKi by quartiles of total pack-years showed no statistical difference across groups (see online supplementary material). Smoking intensity (average number of cigarettes smoked per day) was also not associated with nKi (r=0.13, p=0.71).
Association with peripheral inflammatory biomarkers and disease severity
In the COPD group, correlation between whole lung nKi and plasma fibrinogen (r=0.40, p<0.001), log10hsCRP (r=0.26, p=0.02), WCC (r=0.24, p=0.03) and neutrophils (r=0.28, p=0.01) were observed (table 2). Upper and middle lung nKi also correlated with systemic inflammatory markers (p<0.05), but not lower lung FDG uptake (table 2). In regression analyses, plasma fibrinogen was associated with whole lung nKi independently of confounders including current smoking status, FEV1 % predicted, neutrophil counts and hsCRP (table 3). A modest inverse correlation was observed between FEV1 (% predicted) (r=−0.22, p=0.04) and whole lung nKi. However, no correlation was found between nKi and 6MWD (−0.26, p=0.15) or whole lung emphysema assessed as Perc15 (r=−0.10, p=0.42) (table 2). The association of whole lung nKi with plasma fibrinogen was also assessed in the whole cohort (r=0.39, p<0.001) (figure 3).
TABLE 2.
Correlations between rate of 18F-fluorodeoxyglucose uptake classified by lung regions in participants with COPD
| Variable (x) | Upper lung | Middle lung | Lower lung | |||
| r | sig | r | sig | r | sig | |
| Fibrinogen g·L−1 | 0.42 | <0.001 | 0.38 | <0.001 | 0.16 | 0.16 |
| Log10hsCRP mg·L−1 | 0.31 | 0.005 | 0.31 | 0.005 | 0.09 | 0.41 |
| White cell count ×109·L−1 | 0.24 | 0.04 | 0.25 | 0.03 | 0.17 | 0.14 |
| Neutrophils ×109·L−1 | 0.31 | 0.005 | 0.29 | 0.009 | 0.19 | 0.10 |
| FEV1 L | −0.33 | 0.003 | −0.18 | 0.10 | 0.02 | 0.89 |
| FEV1 % predicted | −0.26 | 0.02 | −0.16 | 0.17 | −0.04 | 0.74 |
| 6MWD m | 0.28 | 0.01 | −0.25 | 0.03 | 0.08 | 0.51 |
| Perc15 score HU | −0.35 | 0.004 | −0.11 | 0.38 | −0.04 | 0.76 |
Pearson's bivariate test used. Strength of correlation determined by r and significance (sig) by p-value. hsCRP: high sensitivity C-reactive protein, FEV1: forced expiratory volume in 1 s; 6MWD: 6-min walking distance.
TABLE 3.
Variables associated with whole lung pulmonary 18F-fluorodeoxyglucose uptake in participants with COPD assessed by regression analysis
| Variable (x) | Standardised beta coefficient | Coefficient standard error | Significance |
| Fibrinogen g·L−1 | 0.30 | 0.01 | 0.02 |
| Current smoking (yes/no) | 0.20 | 0.00 | 0.07 |
| FEV1 % predicted | −0.12 | 0.00 | 0.37 |
| Neutrophils ×109·L−1 | 0.10 | 0.00 | 0.41 |
| Log10hsCRP mg·L−1 | 0.02 | 0.00 | 0.90 |
Dependent variable=whole lung 18F-fluorodeoxyglucose uptake (nKi, units mL·cm−3·min−1). Multivariable model, R=0.48, R2=0.23, adjusted R2=0.17. hsCRP: high sensitivity C-reactive protein, FEV1: forced expiratory volume in 1 s.
FIGURE 3.

Scatterplot of plasma fibrinogen and whole lung nKi values for the whole cohort. Pearson correlation between 111 cases with both fibrinogen and nKi available, r=0.391, p<0.001. α1ATD: alpha-1 antitrypsin deficiency.
HRCT subtypes
There were 74 evaluable HRCT scans in the COPD cohort. Table 4 summarises clinical information stratified by the visual subtypes defined according to Fleischner classification [10]. The majority of scans were defined as advanced destructive centrilobular emphysema followed by moderate or trace centrilobular emphysema. Three scans were classified as panlobular emphysema but given the small number of scans of this subtype, they were not included in analysis. No scans were characterised as paraseptal emphysema or airways disease as the predominant visual CT pattern or had associated features such as bronchiectasis identified. There were no significant differences in nKi values between the sub-classifications of centrilobular emphysema identified (table 4).
TABLE 4.
Clinical and imaging measurements for COPD subtypes defined by computed tomography#
| ADE | Confluent | Moderate | Mild | Trace | p-value ¶ | |
| Subjects n (%) | 22 (30%) | 9 (12%) | 10 (14%) | 15 (20%) | 15 (20%) | |
| FEV1 L | 0.99±0.30 | 1.05±0.46 | 1.5±0.73 | 1.4±0.46 | 1.8±0.52 | <0.001 |
| FVC L | 3.1±0.78 | 3.1±0.70 | 3.0±1.2 | 2.7±0.75 | 3.1±0.74 | ns |
| Smoker % | 10 | 11 | 20 | 11 | 7 | |
| Smoking years | 42±6 | 44±13 | 39±12 | 35±11 | 35±13 | ns |
| 6MWD m | 343±104 | 335±145 | 375±98 | 377±81 | 478±115 | <0.05 |
| Perc15 HU | −949±41 | −918±18 | −890±35 | −865±24 | −833±32 | <0.001 |
| nKi ×10−3 mL·cm−3·min−1 | 4.0±1.1 | 4.0±1.1 | 3.9±1.1 | 3.5±1.1 | 3.5±0.9 | ns |
Data are presented as mean±sd, unless otherwise stated. ADE: advanced destructive emphysema; FEV1: forced expiratory volume in 1 s; FVC: forced vital capacity; 6MWD: 6-min walking distance; ns: nonsignificant. #: only centrilobular emphysema subtypes are analysed; n=74 high-resolution computed tomography COPD scans analysed, three (4%) scans were panlobular emphysema so were not included in analysis; no scans were identified as paraseptal emphysema, airway disease or associated features noted. ¶: overall comparison across five subtypes.
Repeatability
We assessed repeatability of FDG PET, using data from the placebo arm of the EVOLUTION clinical trial, in COPD patients who had a follow-up scan available (n=32). The mean percentage difference in nKi between the baseline and follow-up was 3.2% and the within subject coefficient of variability was 7.7% (see figure 4 for the Bland–Altman plot), which is similar to previously reported values using FDG [17] and other tracers [18]. For further context of the between group differences observed, the mean percentage difference in nKi between never-smoking controls and COPD patients was +32% (2.8±0.1×10−3 versus 3.7±0.1×10−3 mL·cm−3·min−1). 10 COPD patients who had an exacerbation between the baseline and follow-up scan, exhibited a slight increase in whole lung FDG uptake at the follow-up scan (baseline nKi=3.6±0.1×10−3 mL·cm−3·min−1; follow-up nKi=4.1±0.1×10−3 mL·cm−3·min−1, p=0.05), despite the fact study participants had to be exacerbation free for 1 month prior to the follow-up scan. 22 patients who did not experience an exacerbation had a baseline nKi=3.8±0.1×10−3 mL·cm−3·min−1 and follow-up scan of nKi=3.7±0.1×10−3 mL·cm−3·min−1, p=0.5 (figure 5).
FIGURE 4.
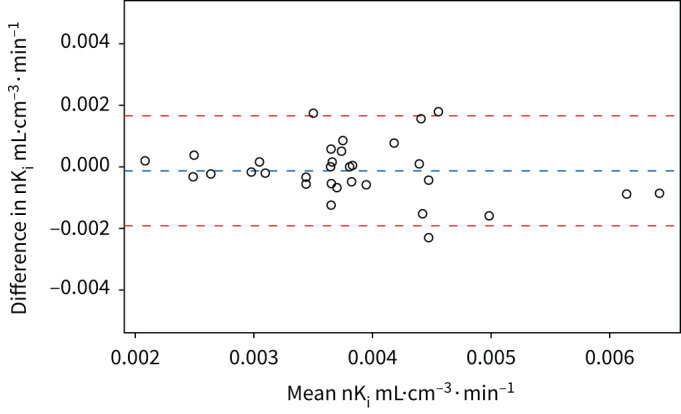
Bland–Altman plot of the rate of 18F-fluorodeoxyglucose (FDG) uptake (nKi). These data were obtained from the baseline and follow-up FDG scans of COPD participants in the placebo arm. The interval between scans was ∼4 months. Horizontal lines show the mean difference and upper and lower limits (±1.96 standard deviation) of the mean difference between paired baseline and 4-month values.
FIGURE 5.

a) nKi values at baseline and 4 months for participants without an exacerbation in between these time-points (n=22). b) nKi values at baseline and 4 months for participants that experienced an exacerbation (n=10).
Discussion
This study sought to extend knowledge from prior small studies to evaluate whether FDG PET/CT is useful as a noninvasive tool to quantify pulmonary inflammation associated with COPD [8, 9]. We found that pulmonary FDG uptake measured by nKi, as a surrogate marker of pulmonary inflammation, was increased in both COPD and α1ATD-COPD individuals and in smokers with unobstructed spirometry when compared with never-smokers, but there was no difference in FDG uptake between COPD, α1ATD-COPD or smokers without COPD. Furthermore, COPD individuals who were current smokers had increased inflammation compared with COPD ex-smokers. Additional findings of this study are the modest associations between pulmonary FDG uptake (especially in the upper lung regions) and peripheral inflammatory markers in COPD individuals and the finding that FDG PET/CT imaging provides a reproducible, stable signal of FDG uptake, measured by nKi, over 4 months follow-up in COPD patients. A crude comparison of +32% percentage mean difference between groups compared with a 3.2% repeatability difference, shows it is of a magnitude 10 times higher. This supports the validity of our between-subject findings. Given these differences in nKi were observed despite the widespread use of inhaled corticosteroids in COPD patients, is also encouraging for the utility of this imaging modality to evaluate pulmonary inflammation.
We observed increased nKi in α1ATD-COPD patients versus never-smokers in this study, in contrast to findings reported by Subramanian et al. [9] who observed no difference between these two groups. Our results are consistent with understanding of the inflammatory pathological pathways underlying α1ATD, and highlight the high inflammatory burden of this condition, besides its well-recognised protease–antiprotease imbalance [19, 20]. Variation in scan acquisition protocol (for example Subramanian et al. [9] administered a lower FDG dose than used in this study) may well account for the differences in data observed between studies.
The current study included current smokers with spirometry in the normal predicted range. As far as we are aware, this is the first prospective pulmonary FDG PET/CT study in current smokers, although animal studies have demonstrated the pulmonary inflammatory effects of smoking using FDG PET/CT [21]. A previous study of PET imaging with 11C-carbon monoxide-labelled erythrocytes, found that smokers had increased pulmonary extravascular tissue density in comparison to never-smokers [22]. A possible explanation suggested by the authors was that chronic pulmonary inflammation in smokers accounted for increased density, and this may outweigh any offsetting tissue loss due to emphysema that may be expected in smokers [20]. This is an important consideration in interpreting tissue density measurements within COPD patients and also in comparison to smokers without COPD and lends support to our study findings. We observed that smokers with normal spirometry values, had similar FDG uptake values compared with COPD patients and COPD individuals who were current smokers had higher FDG uptake than COPD ex-smokers. Although we observed no relationship with total pack-years smoked or smoking intensity, determined by number smoked per day, current smoking seems to be associated with higher FDG uptake determined by nKi. This suggests that smoking induces significant pulmonary inflammation and supports the pathophysiological concept that the initiator of COPD is an inflammatory response to noxious inhaled stimuli [3]. In this study, we evaluated tobacco smoking although the effects of other air pollutants may be predicted to be similar. Further, our study highlights the importance of smoking cessation as an extremely critical public health need, given smokers without COPD had similar pulmonary FDG uptake values to COPD patients.
We noted regional differences in FDG uptake across subject groups, with higher nKi values in upper and middle lung regions in both COPD and α1ATD-COPD patients as well as smokers without COPD compared with these lung regions in never-smokers. This is consistent with previously reported results [9], which showed a upper lung regional predilection for FDG uptake that correlated with COPD disease severity. There was no difference in nKi values in the lower lung across participant groups and we speculate that this was due to the motion of the diaphragm during the free-breathing acquisition of PET data. A further point of consideration is that emphysema predominantly affects the upper lobes; therefore, perhaps the number of patients with lower zone changes related to pulmonary inflammation, evaluated by nKi, was too small to elicit differences across groups. Moreover, we observed no difference in nKi values across lung regions within groups, if any regional differences within the lungs of individuals within a group exist, they may be too subtle to detect with this technique. We also evaluated the effect of gravity on pulmonary FDG uptake in the study by determing nKi values stratified by anterior and posterior distribution (data not shown) and observed no significant differences between them. Similarly, this may be because the technique is not sensitive enough to detect differences in blood flow due to gravity. However, nKi values were averaged across the anterior–posterior direction to limit any potential effect.
We found modest correlations in COPD participants between nKi and peripheral inflammation markers. Given that we previously found no strong associations between systemic inflammation measures and vascular inflammation assessed by FDG PET/CT in the same cohort [7], these data support the hypothesis that increased systemic inflammatory markers in COPD patients likely derive from the lung. Plasma fibrinogen in particular had an independent association with nKi, and interestingly, in vitro studies suggest the pulmonary epithelium can be an extrahepatic source of fibrinogen in response to local inflammatory mediators [23, 24]. These data further support the relevance of plasma fibrinogen as a US Food and Drug Administration and European Medicines Agency qualified drug development tool assessing risk for exacerbation and mortality in COPD [25]. Direct assessment of pulmonary inflammation via bronchoalveolar lavage or tissue biopsies may have yielded stronger correlations with pulmonary FDG uptake, although the invasive nature of these techniques, the patient-related challenges of such invasive techniques to repeatability in an interventional drug study, and probable need for spatial consideration of where to sample from within the lungs are caveats to this consideration. Another interesting finding of our study, which demonstrates the potential of FDG PET as a drug development tool sensitive to a change in COPD disease activity, was increased nKi in participants who had an exacerbation between the baseline and follow-up scan. This was evident despite a minimum 1-month period without exacerbation prior to the follow-up scan. Our data suggest a potential role for FDG PET as a noninvasive tool to demonstrate response to novel anti-inflammatory therapy as proof of concept in COPD experimental studies and in other pulmonary diseases, such as pulmonary fibrosis [26]. In-depth evaluation of FDG PET/CT to evaluation pulmonary inflammation in comparison to other biomarkers is more far-reaching than this discussion permits, and each research technique or biomarker has its own benefits and limitations. However, for context, in a very large cohort of COPD patients sputum eosinophils, a marker of specifically eosinophilic airway inflammation, showed differences in spirometry and some lung regions’ CT parameters between stratified levels of this biomarker, whereas peripheral eosinophils had little association with these markers of COPD severity. As far as we are aware, no study has compared sputum eosinophils in smokers without COPD versus COPD patients [27].
99mTc-DPTA (technetium-99m-diethylenetriaminepentaacetic acid) is an imaging biomarker that has been used to assess epithelial permeability and similarly showed significant differences in COPD patients and smokers without COPD versus controls [28].
Our large dataset of COPD participants with HRCT and FDG PET/CT data also enabled stratification of FDG uptake values by visual subtypes of CT imaging, which has not been evaluated before. The majority of scans had a centrilobular predominant pattern with advanced destructive emphysema and no scans were classified as predominant airway disease. We did not observe any differences in FDG uptake across the CT-defined subtypes of COPD observed in this study, which is probably to be expected given that although a large FDG PET/CT study, the HRCT scans are predominantly of the same visual subtype or phenotype (i.e. centrilobular emphysema). Previous studies have found that CT-defined COPD subtypes exhibit differences in mortality and disease progression [29, 30]. Although there was an indication that severe forms of centrilobular emphysema had higher nKi values than milder forms, the FDG signal may have been affected by differences in air and pulmonary blood volume between these different severities of emphysema. Further study of the relationship between CT classification and FDG uptake in patients with defined phenotypes of airways disease versus emphysema may be helpful. HRCT scans were not performed in smokers without COPD, but this may have enabled exploration of FDG uptake with visual CT subtypes in this group compared with COPD patients.
The strengths of this study include the large COPD group, which enabled robust evaluation of associations with inflammatory markers and the ability to assess the reproducibility of the technique, which is similar to previously reported values using FDG [17] and other tracers [18], and is reassuring for future research studies in COPD patients. Nevertheless, there were limitations. Smoking status was self-reported and α1ATD-COPD patients were studied at one centre only. CT attenuation-correction scans were acquired under free breathing, which may lead to inaccuracies when quantifying PET data. FDG PET scans were analysed using the Patlak graphical approach, which has been shown to be influenced by blood and air volume [31], future work will be required to determine the most accurate PET measure of inflammation. Indeed, standardised uptake value (SUV), which is a measure of FDG uptake often used in clinical practice, is highly influenced by blood and air volume [7]. Normalised Patlak analysis is the most widely established method to measure pulmonary FDG uptake in research studies because it uses dynamic data rather than a single time-point and is therefore generally considered more accurate than SUV. A further point of consideration is that exposure to ionising radiation will limit widespread use of FDG PET/CT imaging in clinical trials requiring measurement of inflammation at multiple time-points.
In summary, patients with COPD, α1ATD-COPD and current smokers without COPD have increased levels of pulmonary FDG uptake quantified by nKi from FDG PET/CT imaging compared with never-smokers. nKi as a surrogate marker of pulmonary inflammation is a stable and reproducible parameter at 4 months in COPD patients, is associated with current smoking, has modest correlations with peripheral systemic inflammatory markers including plasma fibrinogen and may reflect COPD disease activity defined by exacerbations. Further work is needed to elucidate reasons for regional differences in FDG uptake across subject groups and determine if meaningful COPD phenotypes can be defined by FDG PET, although exposure to ionising radiation will limit the study sample size of such studies. This study supports advancing research into pulmonary FDG PET as a noninvasive tool to evaluate visualisation and quantification of pulmonary inflammation in modest size, precision medicine experimental studies.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00699-2020.supplement (200.2KB, pdf)
Acknowledgements
We would like to thank all participants who took part in this study. We would also like to thank the staff at the National Institute for Health Research (NIHR) Respiratory Biomedical Research Unit and NHS laboratory at the Royal Brompton and Harefield NHS Foundation Trust and Imperial College, and the NIHR Comprehensive Biomedical Research Centre and Blood Sciences laboratory at Cambridge University Hospitals NHS Foundation Trust for their help with conducting this work. We would also like to thank the staff at the PET unit at Cambridge University Hospitals NHS Foundation Trust and Imanova Ltd, Hammersmith.
Footnotes
This article has an editorial commentary: https://doi.org/10.1183/23120541.00445-2021
Data availability: Data available from the authors upon request.
Author contributions: I.B. Wilkinson, M.I. Polkey, R. Tal-Singer, J. Cheriyan, J.R. Cockcroft, D.A. Lomas, C.M. McEniery, M. Fisk and D. Mohan contributed to the conception and design of the study. M. Fisk, D. Mohan, J. Cheriyan, J. Fuld, I.B. Wilkinson, M.I. Polkey, N.S. Hopkinson and K.M. Mäki-Petäjä contributed to the acquisition of data. L. Vass, M. Fisk, D. Mohan, J. Forman, J.R. Cockcroft, C.M. McEniery, K.M. Mäki-Petäjä, A. Oseni and A. Devaraj contributed to data analysis. All authors contributed to interpretation of the data of the work, drafting and revising the manuscript and have approved the final version for publication. All authors are accountable for all aspects of the work performed.
Conflict of interest: L. Vass reports a PhD studentship grant from GlaxoSmithKline during the conduct of the study.
Conflict of interest: M. Fisk has nothing to disclose.
Conflict of interest: J. Cheriyan reports grants from GlaxoSmithKline (GSK) during the conduct of the study and as a Cambridge University Hospital National Health Service (NHS) Trust employee was obligated to spend 50% of his NHS time to do clinical trial work for GSK. However, he received no employee benefits from GSK. The payment for this time was made directly by GSK to Cambridge University Hospitals NHS Trust.
Conflict of interest: D. Mohan is an employee of and shareholder in GSK.
Conflict of interest: J. Forman has nothing to disclose.
Conflict of interest: A. Oseni has nothing to disclose.
Conflict of interest: A. Devaraj has nothing to disclose.
Conflict of interest: K.M. Mäki-Petäjä has nothing to disclose.
Conflict of interest: C.M. McEniery has nothing to disclose.
Conflict of interest: J. Fuld has nothing to disclose.
Conflict of interest: N.S. Hopkinson has nothing to disclose.
Conflict of interest: D.A. Lomas reports grants, personal fees and nonfinancial support from GlaxoSmithKline, during the conduct of the study; and personal fees and grants from GlaxoSmithKline, and personal fees from Griffols, outside the submitted work.
Conflict of interest: J.R. Cockcroft received a grant from GSK outside this submitted work.
Conflict of interest: R. Tal-Singer was an employee and shareholder of GSK during the conduct of the study, and has received personal fees from Immunomet, Vocalis Health and Ena Respiratory.
Conflict of interest: M.I. Polkey reports personal fees for an advisory board on this topic from GSK and grants from Innovate UK paid to his institution to conduct this work, during the conduct of the study.
Conflict of interest: I.B. Wilkinson received a grant from Innovate UK and an educational imaging award from GSK during the conduct of the study.
Support statement: This study was jointly sponsored by the University of Cambridge and Cambridge University Hospitals NHS Foundation Trust and was funded by a grant from Innovate UK (UK Technology Strategy Board) as a component study of the ERICA (Evaluation of the Role of Inflammation in Chronic Airways Disease) Collaboration, which includes academic and industry partners. GSK, a consortium partner, made contributions towards study management and provided study medication. This work was partly undertaken at the NIHR Respiratory Biomedical Research Unit at the Royal Brompton and Harefield NHS Foundation Trust and Imperial College. The views expressed in this publication are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care. J. Cheriyan acknowledges funding support from the NIHR Cambridge Biomedical Research Centre (BRC-1215-20014). D.A. Lomas is supported by the NIHR UCLH Biomedical Research Centre and is an NIHR Senior Investigator.
References
- 1.Soriano JB, Polverino F, Cosio BG. What is early COPD and why is it important? Eur Respir J 2018; 52: 1801448. doi: 10.1183/13993003.01448-2018 [DOI] [PubMed] [Google Scholar]
- 2.Wang Y, Xu J, Meng Y, et al. Role of inflammatory cells in airway remodeling in COPD. Int J Chron Obstruct Pulmon Dis 2018; 13: 3341–3348. doi: 10.2147/COPD.S176122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barnes PJ. Cellular and molecular mechanisms of chronic obstructive pulmonary disease. Clin Chest Med 2014; 35: 71–86. doi: 10.1016/j.ccm.2013.10.004 [DOI] [PubMed] [Google Scholar]
- 4.Jones PW, Agusti AGN. Outcomes and markers in the assessment of chronic obstructive pulmonary disease. Eur Respir J 2006; 27: 822–832. doi: 10.1183/09031936.06.00145104 [DOI] [PubMed] [Google Scholar]
- 5.Hoesterey D, Das N, Janssens W, et al. Spirometric indices of early airflow impairment in individuals at risk of developing COPD: spirometry beyond FEV1/FVC. Respir Med 2019; 156: 58–68. doi: 10.1016/j.rmed.2019.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinez FJ, Han MK, Allinson JP, et al. At the root: defining and halting progression of early chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2018; 197: 1540–1551. doi: 10.1164/rccm.201710-2028PP [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen DL, Cheriyan J, Chilvers ER, et al. Quantification of lung PET images: challenges and opportunities. J Nucl Med 2017; 58: 201–207. doi: 10.2967/jnumed.116.184796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones HA, Marino PS, Shakur BH, et al. In vivo assessment of lung inflammatory cell activity in patients with COPD and asthma. Eur Respir J 2003; 21: 567–573. doi: 10.1183/09031936.03.00048502 [DOI] [PubMed] [Google Scholar]
- 9.Subramanian DR, Jenkins L, Edgar R, et al. Assessment of pulmonary neutrophilic inflammation in emphysema by quantitative positron emission tomography. Am J Respir Crit Care Med 2012; 186: 1125–1132. doi: 10.1164/rccm.201201-0051OC [DOI] [PubMed] [Google Scholar]
- 10.Lynch DA, Austin JH, Hogg JC, et al. CT-definable subtypes of chronic obstructive pulmonary disease: a statement of the Fleischner Society. Radiology 2015; 277: 192–205. doi: 10.1148/radiol.2015141579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fisk M, Mohan D, Cheriyan J, et al. Evaluation of losmapimod in patients with chronic obstructive pulmonary disease (COPD) with systemic inflammation stratified using fibrinogen (‘EVOLUTION’): rationale and protocol. Artery Res 2014; 8: 24–34. doi: 10.1016/j.artres.2013.10.380 [DOI] [Google Scholar]
- 12.Fisk M, Cheriyan J, Mohan D, et al. Vascular inflammation and aortic stiffness: potential mechanisms of increased vascular risk in chronic obstructive pulmonary disease. Respir Res 2018; 19: 100. doi: 10.1186/s12931-018-0792-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patlak CS, Blasberg RG, Fenstermacher JD. Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. J Cereb Blood Flow Metab 1983; 3: 1–7. doi: 10.1038/jcbfm.1983.1 [DOI] [PubMed] [Google Scholar]
- 14.Newell JD, Hogg JC, Snider GL. Report of a workshop: quantitative computed tomography scanning in longitudinal studies of emphysema. Eur Respir J 2004; 23: 769–775. doi: 10.1183/09031936.04.00026504 [DOI] [PubMed] [Google Scholar]
- 15.Wanger J, Clausen JL, Coates A, et al. Standardisation of the measurement of lung volumes. Eur Respir J 2005; 26: 511–522. doi: 10.1183/09031936.05.00035005 [DOI] [PubMed] [Google Scholar]
- 16.ATS statement: guidelines for the six-minute walk test. Am J Respir Crit Care Med 2002; 166: 111–117. doi: 10.1164/ajrccm.166.1.at1102 [DOI] [PubMed] [Google Scholar]
- 17.Fraum TJ, Fowler KJ, Crandall JP, et al. Measurement repeatability of 18F-FDG PET/CT versus 18F-FDG PET/MRI in solid tumors of the pelvis. J Nucl Med 2019; 60: 1080–1086. doi: 10.2967/jnumed.118.218735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin C, Bradshaw T, Perk T, et al. Repeatability of quantitative 18F-NaF PET: a multicenter study. J Nucl Med 2016; 57: 1872–1879. doi: 10.2967/jnumed.116.177295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baraldo S, Turato G, Lunardi F, et al. Immune activation in α1-antitrypsin-deficiency emphysema. Beyond the protease–antiprotease paradigm. Am J Respir Crit Care Med 2015; 191: 402–409. doi: 10.1164/rccm.201403-0529OC [DOI] [PubMed] [Google Scholar]
- 20.Cosio MG, Bazzan E, Rigobello C, et al. Alpha-1 antitrypsin deficiency: beyond the protease/antiprotease paradigm. Ann Am Thorac Soc 2016; 13: Suppl. 4, S305–S310. doi: 10.1513/AnnalsATS.201510-671KV [DOI] [PubMed] [Google Scholar]
- 21.Schroeder T, Vidal Melo MF, Musch G, et al. PET imaging of regional 18F-FDG uptake and lung function after cigarette smoke inhalation. J Nucl Med 2007; 48: 413–419. [PubMed] [Google Scholar]
- 22.Brudin LH, Rhodes CG, Valind SO, et al. Regional lung density and blood volume in nonsmoking and smoking subjects measured by PET. J Appl Physiol 1987; 63: 1324–1334. doi: 10.1152/jappl.1987.63.4.1324 [DOI] [PubMed] [Google Scholar]
- 23.Haidaris PJS. Induction of fibrinogen biosynthesis and secretion from cultured pulmonary epithelial cells. Blood 1997; 89: 873–882. doi: 10.1182/blood.V89.3.873 [DOI] [PubMed] [Google Scholar]
- 24.Lawrence SO, Simpson-Haidaris PJ. Regulated de novo biosynthesis of fibrinogen in extrahepatic epithelial cells in response to inflammation. Thromb Haemost 2004; 92: 234–243. doi: 10.1160/TH04-01-0024 [DOI] [PubMed] [Google Scholar]
- 25.Miller BE, Tal-Singer R, Rennard SI, et al. Plasma fibrinogen qualification as a drug development tool in chronic obstructive pulmonary disease. Perspective of the chronic obstructive pulmonary disease biomarker qualification consortium. Am J Respir Crit Care Med 2016; 193: 607–613. doi: 10.1164/rccm.201509-1722PP [DOI] [PubMed] [Google Scholar]
- 26.Win T, Screaton NJ, Porter JC, et al. Pulmonary 18F-FDG uptake helps refine current risk stratification in idiopathic pulmonary fibrosis (IPF). Eur J Nucl Med Mol Imaging 2018; 45: 806–815. doi: 10.1007/s00259-017-3917-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hastie AT, Martinez FJ, Curtis JL, et al. Association of sputum and blood eosinophil concentrations with clinical measures of COPD severity: an analysis of the SPIROMICS cohort. Lancet Respir Med 2017; 5: 956–967. doi: 10.1016/S2213-2600(17)30432-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maini CL, Bonetti MG, Giordano A, et al. Scintigrafia polmonare dinamica con 99mTc radioaerosol per la valutazione della permeabilità della barriera alveolo-capillare [Dynamic pulmonary scintigraphy with Tc99m radioaerosol for the evaluation of the permeability of the alveolo-capillary barrier]. Radiol Med 1987; 74: 520–524. [PubMed] [Google Scholar]
- 29.Lynch DA, Moore CM, Wilson C, et al. CT-based visual classification of emphysema: association with mortality in the COPDGene study. Radiology 2018; 288: 859–866. doi: 10.1148/radiol.2018172294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park J, Hobbs BD, Crapo JD, et al. Subtyping COPD by using visual and quantitative CT imaging features. Chest 2020; 157: 47–60. doi: 10.1016/j.chest.2019.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coello C, Fisk M, Mohan D, et al. Quantitative analysis of dynamic 18F-FDG PET/CT for measurement of lung inflammation. EJNMMI Res 2017; 7: 47. doi: 10.1186/s13550-017-0291-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00699-2020.supplement (200.2KB, pdf)