

ABSTRACT
Many integral membrane proteins form oligomeric complexes, but the assembly of these structures is poorly understood. Here, we show that the assembly of OmpC, a trimeric porin that resides in the Escherichia coli outer membrane (OM), can be reconstituted in vitro. Although we observed the insertion of both urea-denatured and in vitro-synthesized OmpC into pure lipid vesicles at physiological pH, the protein assembled only into dead-end dimers. In contrast, in vitro-synthesized OmpC was inserted into proteoliposomes that contained the barrel assembly machinery (Bam) complex, a conserved heterooligomer that catalyzes protein integration into the bacterial OM, and folded into heat-stable trimers by passing through a short-lived dimeric intermediate. Interestingly, complete OmpC assembly was also dependent on the addition of lipopolysaccharide (LPS), a glycolipid located exclusively in the OM. Our results strongly suggest that trimeric porins form through a stepwise process that requires the integration of the protein into the OM in an assembly-competent state. Furthermore, our results provide surprising evidence that interaction with LPS is required not only for trimerization but also for the productive insertion of individual subunits into the lipid bilayer.
KEYWORDS: β barrel, lipopolysaccharide, membrane proteins, outer membrane, protein folding
INTRODUCTION
The great majority of integral membrane proteins in biological membranes contain one or more α-helical membrane-spanning segments and are initially inserted into the eukaryotic endoplasmic reticulum or bacterial cytoplasmic membrane through the Sec61p/SecYEG complex (1). In all three domains of life, up to ∼70% interact to form heterooligomeric or homooligomeric complexes (2). Although significant insights into the insertion process have been reported, the folding of these proteins and their assembly into oligomeric complexes have not been extensively investigated.
In contrast, most of the proteins that reside in the outer membrane (OM) of Gram-negative bacteria (and some of the proteins that reside in the OM of organelles of bacterial origin) do not contain α-helical transmembrane segments but instead contain a unique “β barrel” structure that spans the membrane. β barrels are essentially amphipathic β sheets that range in size from 8 to 36 β strands and fold into a cylindrical structure with a hydrophobic exterior and a hydrophilic interior (3, 4). Like α-helical membrane proteins, many bacterial OM proteins (OMPs) form stable oligomers (5). OMPs are first transported through the SecYEG complex into the periplasm, where they are maintained in an assembly-competent conformation by molecular chaperones, including Skp and SurA (6–9). Subsequently, their insertion into the OM is catalyzed by the conserved barrel assembly machinery (Bam) complex (10, 11), a heterooligomer composed of a single β barrel protein (BamA) and up to four lipoproteins (BamB to BamE in Escherichia coli) that bind to a periplasmic segment (the polypeptide transport-associated, or POTRA, domains) of BamA (11–14). Interestingly, while most β barrels are extremely stable, the BamA β barrel appears to have an unusual ability to open laterally (15). This property of BamA has the potential to perturb the lipid bilayer and has been proposed to be associated with its catalytic function. Indeed, recent biochemical and structural studies provide strong evidence that after undergoing substantial folding in the periplasmic space (16–18), OMPs pass through an assembly intermediate in which they form a hybrid barrel with the open conformation of BamA (19–21). Although the structure of the entire Bam complex has been solved (22–25), the exact mechanism(s) by which it catalyzes the membrane insertion of client proteins is unknown, and it is unclear if it promotes the formation of oligomers either before or after their insertion. The role of membrane lipids and the physical properties of the OM in driving OMP assembly are also poorly understood (26). Furthermore, because the periplasm lacks ATP, the energy source for the insertion reaction has remained enigmatic.
Highly stable homotrimers called porins are by far the best-known oligomeric OMPs. These proteins are widely distributed among Gram-negative bacteria and often represent a large fraction of the total population of OMPs (>105 copies/cell in Enterobacteriaceae) (27). Porins typically function as diffusion pores that promote the uptake of sugars or other specific small molecules or the nonspecific, passive diffusion of hydrophilic compounds that fall below an ∼600-Da cutoff. They are of particular interest because of their role in the emergence of antibiotic resistance. Curiously, each subunit is thought to function independently, and the contribution of trimerization to solute passage is unknown. Furthermore, although porins have been studied extensively for more than 30 years, most studies on their assembly were conducted before the Bam complex was discovered, when OMPs were believed to be inserted into the OM “spontaneously.” Stably folded monomeric forms of the E. coli porins LamB and PhoE and a metastable OmpF dimer were observed in vivo many years ago (8, 28, 29). Although the location of most of these species was not established, available evidence suggests that individual porin subunits can be inserted into the OM before (or in the absence of) trimerization (30). OmpF was also shown to be secreted from spheroplasts as a monomer that forms trimers in the presence of cell membrane preparations (31). In vitro, urea-denatured OmpF has been shown to trimerize in detergent or lipid-detergent mixtures and in short-chain fatty acid bilayers at high pH (32, 33). Likewise, PhoE produced in vitro has been shown to form folded monomers that can be converted to stable trimers in the presence of OM vesicles and 2% Triton X-100 detergent (34). The relevance of these observations to porin assembly in intact cells, however, is unknown.
An especially striking observation that has never been explained is that lipopolysaccharide (LPS), a unique glycolipid that forms the outer leaflet of the OM, greatly facilitates the assembly of at least a subset of porins. LPS is synthesized in the inner membrane and then transported directly to its final destination by the Lpt machinery (35, 36). OmpF synthesized in spheroplasts or in vitro has been shown to form trimers in pure LPS (31, 37), and LPS is required for the above-mentioned folding of PhoE monomers (38). Because LPS binds stably to purified OmpF, it was recently possible to solve the crystal structure of an OmpF-LPS complex and to identify ∼10 specific residues located near the extracellular side of the β barrel that interact with LPS (39). Mutation of these residues reduced LPS binding and impaired OmpF assembly in vivo. Curiously, many of these residues are basic amino acids that face (or are close to) the lipid bilayer. At least in some cases, lipid-facing charged residues have been shown to interfere with OMP insertion (40).
In this study, we used E. coli OmpC, a protein that is closely related to OmpF and PhoE, as a model protein to obtain further insight into the assembly of trimeric porins and the role of the Bam complex and LPS in the assembly process. The basic residues that mediate interactions between OmpF and LPS are all conserved in OmpC (41). Using an in vitro assay in which the purified Bam complex and a chaperone (SurA) are sufficient to reconstitute the assembly of monomeric OMPs into proteoliposomes under relatively physiological conditions (42–44), we found that we could also reconstitute the complete assembly of OmpC trimers, but only if LPS was added to the reaction mixtures. Surprisingly, the LPS appeared to be required at an early stage of assembly because mutations in likely LPS binding sites impaired the integration of the monomer into proteoliposomes. Kinetic studies revealed for the first time that OmpC is assembled through a membrane-embedded dimeric intermediate. Although we observed the assembly of OmpC into a dimer in empty liposomes, these dimers represented a nonnative species that could not form complete trimers. Taken together, our results lead to a new model in which both the Bam complex and LPS play important roles in OMP assembly.
RESULTS
The Bam complex and LPS promote the assembly of OmpC trimers in vitro.
We have demonstrated that a variety of small (≤14-stranded) monomeric β barrel proteins produced by two different methods can be assembled into proteoliposomes that contain the purified Bam complex in the presence of the periplasmic chaperone SurA. In the first method, OMPs are produced in E. coli, purified from inclusion bodies, and solubilized with 8 M urea (43). In the second method, OMPs are synthesized in a coupled transcription-translation system (the PURE system) and detected by the incorporation of a fluorescent tracer (45). In both assays, the assembly of OMPs is observed within minutes at around neutral pH. In this regard, the assays that we developed recapitulate physiological conditions much better than “spontaneous” assembly assays in which the insertion of β barrel proteins into pure lipid vesicles can be observed but often requires long time periods (hours to days) and high pH (33, 46, 47). Furthermore, while the lipid composition of proteoliposomes that contain the Bam complex only slightly affects the efficiency of OMP assembly, spontaneous assembly is strongly influenced by the properties of the lipid bilayer, including surface charge, thickness, and fluidity, and is often abolished by the presence of abundant naturally occurring lipids such as phosphatidylethanolamine (16, 48, 49). Interestingly, OMPs synthesized in vitro are assembled more rapidly than their urea-denatured equivalents and appear to be recognized by fewer periplasmic chaperones (45). These and other results suggest that the mode of production significantly influences the conformational states sampled by OMPs and the time window during which they remain insertion competent.
Based on indications that a wider range of in vitro-synthesized OMPs are assembled into proteoliposomes than urea-denatured OMPs, we first examined the assembly of OmpC produced in the PURE system. We suspected that the assembly of OmpC might be challenging because of both its relatively large size (16 β strands) and its trimeric structure. Indeed, when we used “standard” conditions in which we produced fluorescently labeled OmpC in the presence of SurA and proteoliposomes containing the Bam complex and a nonnative lipid [the short-chain (12:0) lipid 1,2-dilauroyl-sn-glycero-3-phosphocholine (Bam DLPC) or 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (Bam POPC), a longer-chain (16:0, 18:1) lipid that mimics the hydrophobic width and fluidity of E. coli membranes], we observed only OmpC monomers (Fig. 1A and B, lanes 1 and 2). All of the OmpC was sensitive to proteinase K (PK) digestion and therefore was not integrated into the membrane vesicles (Fig. 1A and B, lanes 3 and 4). Given that LPS has been reported to promote the assembly of trimeric porins in permeabilized-cell and spontaneous-assembly assays, we conjectured that the addition of E. coli LPS might facilitate OmpC assembly in our Bam complex-dependent assay. Interestingly, we observed modest amounts of dimeric and trimeric forms of OmpC in the presence of smooth LPS from E. coli strain O55:B5 and Bam DLPC (Fig. 1A, lane 5). Consistent with previous results, the oligomeric forms of OmpC were observed on SDS-PAGE gels only in the absence of heat; heating to 95°C caused denaturation of the protein and its conversion to a monomer (Fig. 1A, lane 6). The resistance of the oligomers to PK digestion and the presence of a significant level of PK-resistant monomer that was presumably derived from the PK-resistant oligomers in heated samples (Fig. 1A, lanes 7 and 8) implied that the dimers and trimers were integrated into the proteoliposomes. No OmpC assembly was observed when smooth LPS was added to reaction mixtures that contained Bam POPC (Fig. 1B, lanes 5 to 8), most likely because thicker membrane vesicles impose a higher kinetic barrier for OMP assembly (16, 50) and place more constraints on the assembly reaction.
FIG 1.

The Bam complex catalyzes OmpC trimerization in vitro in the presence of rough LPS. E. coli OmpC was synthesized in vitro by adding a plasmid carrying ompC lacking its signal peptide under the control of the T7 promoter to the PURExpress coupled transcription-translation system. Reaction mixtures were supplemented with FluoroTect, SurA, the indicated type of LPS (0.5 mg/ml), and proteoliposomes containing the Bam complex and DLPC (A) or POPC (B). After a 30-min incubation at 37°C, half of each sample was treated with PK to digest protein that was not embedded in the proteoliposomes. Half of both treated and untreated samples were heated at 95°C for 5 min, and fluorescently labeled polypeptides were resolved by SDS-PAGE.
Perhaps surprisingly, we next found that rough forms of LPS that are missing parts of the O antigen greatly enhance OmpC assembly. Ra LPS (which has a relatively small polysaccharide deletion) (39, 51) predominantly stimulated the assembly of membrane-embedded, heat-sensitive OmpC trimers into both Bam DLPC and Bam POPC (Fig. 1A and B, lanes 9 to 12). Rc LPS (which has a larger polysaccharide deletion) promoted even more efficient trimerization (Fig. 1A and B, lanes 13 to 16). Curiously, even though Ra and Rc LPSs facilitated a striking increase in trimer assembly, they also facilitated the same low level of dimerization that was promoted by the presence of smooth LPS. As suggested by other experiments (see below), the dimer appears to correspond to an off-pathway species that results from a spontaneous assembly process that fortuitously occurs under a variety of conditions. It seems likely that the size and/or chemical properties (e.g., net hydrophobicity and critical micelle concentration) of the rough forms of LPS account for their enhanced activity in our reconstituted assay system and may have implications for the mechanism by which LPS catalyzes OmpC assembly (see Discussion). In this regard, it is notable that the level of trimerization was highly dependent on the LPS concentration. Maximum trimerization was observed in the presence of 0.5 mg/ml Rc LPS (see Fig. S1A in the supplemental material), and this concentration was used in all subsequent experiments. At this concentration, Rc LPS clearly functioned as a cofactor in the reaction and was insufficient to promote trimerization in the absence of either the Bam complex or SurA (Fig. S1B and Fig. S2). Indeed, the incorporation of the fluorescent tracer at specific positions might interfere with insertion and/or trimerization and thereby limit the efficiency of assembly. Because we were unable to construct functional mixed vesicles containing both lipids and Rc LPS and we did not see significant fusion of Rc LPS micelles with proteoliposomes in dynamic light scattering experiments (data not shown), it appears that the glycolipid promotes OmpC assembly only in solution.
The concentration of Rc LPS affects the efficiency of OmpC trimerization. (A) OmpC was generated in vitro as described in the legend to Fig. 1 using the PURExpress system supplemented with FluoroTect, SurA, the indicated concentration of Rc LPS, and proteoliposomes containing the Bam complex and DLPC (Bam DLPC) or POPC (Bam POPC). After a 30-min incubation at 37°C, half of each sample was heated at 95°C for 5 min, and fluorescently labeled polypeptides were resolved by SDS-PAGE. (B) OmpC was generated in vitro as described in the legend to Fig. 1 using the PURExpress system supplemented with FluoroTect, SurA, and the indicated form of LPS. Proteoliposomes containing the Bam complex and POPC (Bam POPC) were also added to one reaction mixture. After a 30-min incubation at 37°C, half of each sample was heated at 95°C for 5 min, and fluorescently labeled polypeptides were resolved by SDS-PAGE. The results show that LPS alone does not stimulate OmpC folding. Download FIG S1, JPG file, 1.5 MB (1.5MB, jpg) .
{kind=link}
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
SurA is required for OmpC trimerization. OmpC was generated in vitro as described in the legend to Fig. 1 using the PURExpress system supplemented with FluoroTect, Rc LPS (0.5 mg/ml), and proteoliposomes containing the Bam complex and DLPC. SurA was also added as indicated. After a 30-min incubation at 37°C, half of each sample was treated with PK. Half of both treated and untreated samples were heated at 95°C for 5 min, and fluorescently labeled polypeptides were resolved by SDS-PAGE. Download FIG S2, JPG file, 0.3 MB (278.1KB, jpg) .
{kind=link}
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
The presence of the Bam complex and the conformational state of OmpC drive trimerization.
Further experiments strongly suggested that the trimerization of OmpC is promoted by specific activities of the Bam complex. When OmpC was synthesized in vitro in the presence of pure DLPC vesicles, a notable fraction of the protein formed heat-sensitive, PK-resistant dimers (Fig. 2A, top). The addition of Rc LPS did not affect the level of dimerization. Furthermore, only a very low level of dimers was observed when the reaction was performed in the presence of pure POPC vesicles (Fig. 2A, bottom). These results strongly suggest that under highly “permissive” conditions (i.e., in the presence of a low kinetic barrier), OmpC can insert spontaneously into liposomes but forms only a nonphysiological, dead-end dimeric species. This interpretation implies that the Bam complex promotes the insertion of OmpC in one or more assembly-competent conformations and/or that the Bam complex plays a direct role in catalyzing trimerization.
FIG 2.

OmpC forms a dimer in the presence of pure lipid vesicles. (A) OmpC was generated in vitro as described in the legend to Fig. 1 using the PURExpress system supplemented with FluoroTect, SurA, and either Rc LPS or no LPS. Assembly reaction mixtures also contained either pure lipid vesicles (DLPC or POPC) or the corresponding proteoliposomes containing the Bam complex (Bam DLPC or Bam POPC). After a 30-min incubation at 37°C, half of each sample was treated with PK to digest protein that was not embedded in the proteoliposomes. Half of both treated and untreated samples were heated at 95°C for 5 min, and fluorescently labeled polypeptides were resolved by SDS-PAGE. (B) Urea-denatured OmpC was purified from inclusion bodies, fluorescently labeled at residue 71, and added to SurA and either DMPC vesicles or Bam DMPC proteoliposomes in a complete mixture of standard PURExpress reaction components. After a 30-min incubation at 37°C, samples were processed as described above for panel A.
Several observations indicated that the assembly of OmpC is also strongly affected by the conformation of the protein prior to its integration into proteoliposomes. In one set of experiments, we monitored the fate of a cohort of OmpC molecules produced during a short time window (Fig. S3A). After OmpC was synthesized in the PURE system supplemented with Rc LPS at 37°C for 10 min, translation reinitiation (but not the completion of previously initiated nascent chains) was inhibited by the addition of oncocin (Onc112) (45, 52). Reaction mixtures were then returned to 37°C for an additional 20 min. When Bam DLPC or Bam POPC proteoliposomes were added at the start of the reaction, trimers formed efficiently (Fig. S3B). As the proteoliposomes were added at later time points, the level of trimerization was progressively reduced. Because the level of monomers that were PK resistant after heat treatment (which denatures OmpC oligomers) was concomitantly lowered (Fig. S3, lanes 3, 6, and 9), it is likely that the delayed addition of proteoliposomes reduced the insertion of monomers rather than the assembly of trimers. Consistent with the results described above, a small amount of a dimeric form of the protein that was presumably inserted in a Bam complex-independent fashion was observed at all time points. In a second set of experiments, we monitored the assembly of urea-denatured OmpC that was fluorescently labeled. Although a default level of dimerization was observed upon the addition of either pure DLPC vesicles or Bam DLPC, only a small fraction of the protein formed trimers in the presence of the Bam complex and Rc LPS (Fig. 2B). Interestingly, we found that OmpC could trimerize spontaneously during a 16-h incubation into lipid bilayers that contain very short (C10) acyl chains at pH 10 (Fig. S4). Taken together, the results indicate that at near-physiological pH, the trimerization of OmpC is highly dependent on the ability of the Bam complex to recognize specific conformational states of the protein and to maintain its assembly competence following membrane insertion.
Time-dependent loss of insertion competence of OmpC synthesized in vitro. (A) Summary of the experimental strategy. (B) OmpC was generated in vitro as described in the legend to Fig. 1 using the PURExpress system supplemented with FluoroTect, SurA, and Rc LPS (0.5 mg/ml). After 10 min at 37°C (i.e., at t = 0′ in panel A), oncocin (Onc112) was added to halt translation initiation, and incubation was continued for an additional 20 min (until t = 20′). Either Bam DLPC or Bam POPC was added to an aliquot of the reaction mixture at the indicated time points (t = −10′, 0′, or 5′ in panel A) to promote OmpC assembly for the remainder of the incubation. One-half of each sample was treated with PK, and half of the treated samples were heated to 95°C for 5 min. Fluorescently labeled polypeptides were resolved by SDS-PAGE. To compare equal amounts of reaction products, only half of each untreated sample was loaded. Download FIG S3, JPG file, 0.7 MB (738.1KB, jpg) .
{kind=link}
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Trimerization of urea-denatured OmpC at pH 10. Urea-denatured OmpC labeled at residue 71 with BODIPY-FL was assembled into short-chain lipid (diC10PC) vesicles at pH 10 at 37°C for 16 h with and without SurA. The samples were then treated with PK, and half of each sample was heated at 95°C for 5 min. Fluorescently labeled polypeptides were resolved by SDS-PAGE on 4 to 20% minigels. Download FIG S4, JPG file, 0.2 MB (217.6KB, jpg) .
{kind=link}
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
OmpC trimers are assembled through a dimeric intermediate.
Although previous biochemical and structural studies on the mechanism by which the Bam complex catalyzes OMP assembly (e.g., references 19 and 21) strongly suggest that it promotes the membrane insertion of OmpC monomers that subsequently trimerize, our initial results did not rule out the possibility that the Bam complex promotes the insertion of preassembled trimers. To examine the assembly pathway of OmpC in more detail, we next performed a kinetic analysis of OmpC assembly. After OmpC was synthesized in the PURE system in the presence of Rc LPS and Bam POPC for 10 min at 37°C, Onc112 was added to the reaction mixture to halt translation reinitiation. Aliquots were then removed from the reaction mixture at various time points, and half of each sample was treated with PK. Small amounts of PK-resistant monomers and dimers were observed within the first 10 min, which rapidly disappeared; these forms of the protein were concomitantly replaced by increasing amounts of trimers (Fig. 3 and Table S1). The dimer persisted for a longer period of time when the reaction was performed at 30°C, and at the lower incubation temperature it was clearer that this form of the protein was an assembly intermediate that was gradually converted to a trimer (Fig. S5A and Table S1). As expected, a dimeric intermediate was also observed when OmpC was assembled into Bam DLPC proteoliposomes (Fig. 4, right). Interestingly, PK-resistant dimers formed much more rapidly in the presence of Bam DLPC than in the presence of pure DLPC vesicles (Fig. 4, left). These results not only confirm that OmpC trimerizes at a postinsertion stage but also provide further evidence that the terminal dimers that form in the absence of the Bam complex are an off-pathway species.
FIG 3.

The assembly of the OmpC trimer in vitro occurs via a dimeric intermediate. OmpC was generated in vitro as described in the legend to Fig. 1 using the PURExpress system supplemented with FluoroTect, SurA, Rc LPS, and Bam POPC. After 10 min at 37°C, translation reinitiation was halted by the addition of oncocin, and incubation was continued to facilitate further assembly. At the indicated time points, an aliquot was removed, and the reaction was quenched by the addition of RNase A. One-half of each aliquot was left untreated, while the other half was treated with PK. Fluorescently labeled polypeptides were then resolved by SDS-PAGE. The fractions of the total untreated (top) and PK-treated (bottom) proteins that were observed in monomeric, dimeric, and trimeric forms as a function of time in the representative experiment shown on the left are plotted on the right. The numerical data used to generate the graphs are shown in Table S1A in the supplemental material.
FIG 4.

An OmpC dimer forms more rapidly in the presence of Bam DLPC than in the presence of pure DLPC. OmpC was generated in vitro as described in the legend to Fig. 1 using the PURExpress system supplemented with FluoroTect, SurA, Rc LPS, and either DLPC vesicles or Bam POPC proteoliposomes. After 10 min at 30°C, translation reinitiation was halted by the addition of oncocin, and incubation was continued to facilitate further assembly. At the indicated time points, an aliquot was removed, and the reaction was quenched by the addition of RNase A. One-half of each aliquot was left untreated, while the other half was treated with PK. Fluorescently labeled polypeptides were then resolved by SDS-PAGE.
Time course of OmpC assembly at 30°C. (A) The experiment shown in Fig. 3 was repeated except that the incubation temperature was changed to 30°C. The fractions of the total untreated (top) and PK-treated (bottom) proteins that were observed in monomeric, dimeric, and trimeric forms as a function of time in the representative experiment shown on the left are plotted on the right. The numerical data used to generate the graphs are shown in Table S1B in the supplemental material. (B) The experiment shown in panel A was repeated, and the samples were then treated with PK. (Top) Fluorescently labeled polypeptides were resolved by SDS-PAGE and visualized. (Bottom) Proteins were then transferred to PVDF membranes using an overnight wet transfer method, and LPS was detected by Western blotting using an anti-LPS antibody. The results show a time-dependent accumulation of LPS bound to the OmpC trimer. Download FIG S5, JPG file, 1.4 MB (1.4MB, jpg) .
{kind=link}
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Quantitative analysis of the experiments shown in Fig. 3 (A) and Fig. S5A in the supplemental material (B) using ImageJ software. Download Table S1, JPG file, 0.9 MB (901.2KB, jpg) .
{kind=link}
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
A direct interaction between LPS and OmpC is required for trimerization.
We hypothesized that LPS stimulates OmpC trimerization by binding to the protein at a specific stage (or stages) of its biogenesis and thereby driving the protein into an assembly-competent conformation. To test this idea, we first exploited the finding that two groups of lipid-facing basic residues that are located near the extracellular side of E. coli OmpF (designated sites “A” and “B”) serve as LPS binding sites that interact with the phosphate groups of lipid A (39). The mutation of multiple residues within either site strongly impairs LPS binding to purified OmpF. All of these residues are conserved in OmpC. Interestingly, we found that the mutation of three or four residues in site A (K152/K201/R217 or K27/K152/K201/R217) to glutamine or alanine (designated site A-Gln, site A-Ala, site A+-Gln, or site A+-Ala) greatly reduced trimerization (Fig. 5A and B). Likewise, the mutation of three residues in site B (K255/K281/K283) to alanine (designated site B-Ala) or five residues (R246/K255/K281/K283/R328) to glutamine or alanine (designated site B+-Gln or site B+-Ala) almost completely abolished trimerization (Fig. 5A and B). Although the mutation of K255/K281/K283 to glutamine (designated site B-Gln) appeared to only slightly affect trimerization, B-Gln clearly increased the level of PK-resistant monomers observed at early time points (Fig. 5A and Fig. S6). Like the cognate multiple mutations in OmpF, none of the OmpC mutations that we tested affected the folding of the protein into trimers in detergent solution (Fig. S7). Furthermore, consistent with the results described in the above-mentioned study on OmpF, single mutations in either site A or site B did not affect OmpC assembly in our in vitro assay (Fig. S8). Surprisingly, the defect in trimerization associated with multiple mutations correlated with a strong reduction in the level of PK-resistant monomers (Fig. 5A). This observation strongly suggests that mutations that impair the binding of LPS to OmpC inhibit the integration of the monomer into proteoliposomes. As a control to confirm that integrated monomers are protected from PK, we examined the assembly of an OmpC mutant (G19W/R92L) that contains the same mutations as an OmpF mutant that integrates into the OM but does not form trimers in vivo (30). As expected, a significant fraction of the mutant protein was resistant to PK digestion (Fig. 5A).
FIG 5.

Multiple mutations in OmpC LPS binding sites hinder assembly in vitro. (A) Wild-type (WT) OmpC or the indicated site A (K152/K201/R217), site A* (K27/K152/K201/R217), site B (K255/K281/K283), site B* (R246/K255/K281/K283/R328), or G19W/R92L mutants were generated in vitro as described in the legend to Fig. 1 using the PURExpress system supplemented with FluoroTect, SurA, Rc LPS, and Bam POPC proteoliposomes. After a 30-min incubation at 37°C, half of each sample was treated with PK to digest protein that was not embedded in the proteoliposomes. Half of both treated and untreated samples were heated at 95°C for 5 min, and fluorescently labeled polypeptides were resolved by SDS-PAGE. (B) The locations of the site A and site B residues (39) in the E. coli OmpC crystal structure (PDB accession number 2J1N) are shown.
A monomeric intermediate is observed upon reducing the rate of assembly. The experiment shown in Fig. S5 was repeated using the OmpC site B Gln mutant. Download FIG S6, JPG file, 0.5 MB (556.2KB, jpg) .
{kind=link}
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Multiple LPS binding-site mutations do not affect the folding of OmpC in detergent solution. The urea-denatured wild type (WT) or the indicated OmpC mutant was folded into a detergent mixture of 1% dodecyl-β-D-maltoside (DDM)–0.4% DDG in 50 mM Tris (pH 8.0), 1 mM dithiothreitol (DTT), and 0.1 mM EDTA by incubation at 37°C for 24 h. Half of the wild-type sample was heated (lane 1). Proteins were resolved by SDS-PAGE and visualized by colloidal blue staining. Download FIG S7, JPG file, 0.5 MB (475.3KB, jpg) .
{kind=link}
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Single mutations in the LPS binding sites of OmpC do not affect assembly. The experiment described in the legend of Fig. 5A was repeated with the indicated wild-type (WT) and mutant forms of OmpC. Download FIG S8, JPG file, 1.3 MB (1.3MB, jpg) .
{kind=link}
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
The notion that LPS acts at a relatively early stage of OmpC biogenesis in our in vitro assay was supported by the results of site-specific UV cross-linking experiments. To obtain insight into the timing of LPS binding, we introduced amber mutations at two positions (residues 255 and 281) in site B. We then incorporated the photoactivatable amino acid analog benzoyl phenylalanine (Bpa) into OmpC(K255am) and OmpC(K281am) by amber suppression (53) during synthesis in the PURE system in the presence of Rc LPS and Bam POPC proteoliposomes. Half of each reaction mixture was exposed to UV light, and half of both treated and untreated samples were heated. The results showed that the incorporation of Bpa at both positions is compatible with trimer formation (Fig. 6A, top). Interestingly, in the presence of UV light, a polypeptide that migrated slightly slower than the OmpC monomer was observed (Fig. 6A, top, lanes 3, 4, 7, and 8). Western blotting using an anti-LPS antibody confirmed that this polypeptide corresponded to an OmpC monomer-LPS cross-linking product (Fig. 6A, bottom, lanes 3, 4, 7, and 8). The presence of the cross-linking product even in unheated samples suggested that OmpC might interact with LPS prior to its insertion into the proteoliposomes. Consistent with this possibility, the cross-linking product was observed even in reactions that were conducted without Bam POPC proteoliposomes (Fig. 6B, lanes 3 and 4). Only very weak cross-linking was seen when Bpa was introduced at amino acids S71 and E189, residues that are located far away from the LPS binding sites (Fig. S9). Taken together, the results strongly suggest that after OmpC is synthesized in vitro, at least a portion of the protein folds into a near-native insertion-competent conformation in which the LPS binding sites are relatively well formed.
FIG 6.

Residues within the OmpC B site cross-link to LPS even in the absence of the Bam complex. (A) OmpC(K255am) and OmpC(K281am) were generated in vitro as described in the legend to Fig. 1 using a PURExpress system that lacks release factor 1. Reaction mixtures were supplemented with FluoroTect, tRNATAG-BPA, SurA, Rc LPS, and Bam POPC proteoliposomes and incubated at 30°C for 30 min. Half of each reaction mixture was exposed to UV light, and half of both treated and untreated samples were heated to 95°C for 5 min. SDS-PAGE was then conducted to detect fluorescently labeled polypeptides, and an OmpC monomer-LPS cross-linking product was confirmed by Western blotting using an anti-LPS antibody. An unidentified cross-reactive protein is denoted with an asterisk. (B) OmpC(K281am) was generated in vitro as described above for panel A, but the Bam POPC proteoliposomes were omitted from one of two duplicate reactions.
Residues far from an LPS binding site do not interact significantly with LPS. The photo-cross-linking experiment described in the legend of Fig. 6A was repeated with OmpC(S71am), OmpC(E189am), and OmpC(K281am) as a positive control. SDS-PAGE was conducted to detect fluorescently labeled polypeptides, and an OmpC monomer-LPS cross-linking product was confirmed by Western blotting using an anti-LPS antibody. An unidentified cross-reactive protein is denoted with an asterisk. Download FIG S9, JPG file, 0.5 MB (498.9KB, jpg) .
{kind=link}
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Although our analysis provided evidence that the monomeric form of OmpC acquires the ability to interact with LPS at an early stage of assembly, a final set of experiments indicated that the protein remains strongly bound to LPS after trimerization. OmpC was generated in the PURE system supplemented with Rc LPS and Bam DLPC proteoliposomes. Half of each sample was treated with PK, and half of both treated and untreated samples were heated. Polypeptides were then resolved by SDS-PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane. A band that corresponded to the OmpC trimer was not only detected in unheated samples by its fluorescent signal, but was also detected on the same membrane by an anti-LPS antibody (Fig. 7). This observation suggests that after OmpC forms a trimer, LPS remains bound tightly enough to survive both SDS-PAGE and electrophoretic transfer. The finding that the detection of LPS by Western blotting followed the kinetics of trimer formation (Fig. S5B) is consistent with the notion that LPS was bound to the protein prior to trimerization and, at the very least, suggested that binding did not require a lengthy post-assembly conformational transition. Presumably LPS was not detected in association with the OmpC monomer because the interaction does not survive the unfolding of the protein that occurs under SDS-PAGE conditions.
FIG 7.
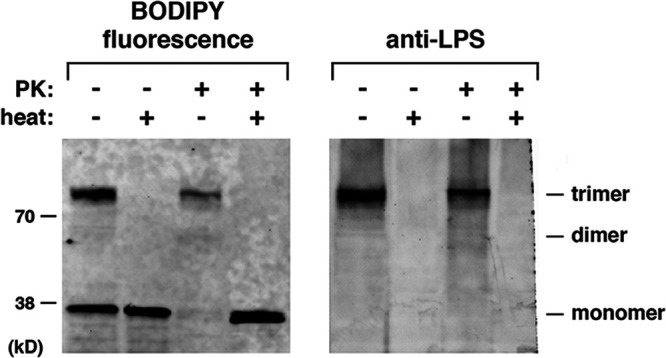
LPS is stably bound to the OmpC trimer. OmpC was generated in vitro as described in the legend to Fig. 1 using the PURExpress system supplemented with FluoroTect, SurA, Rc LPS, and Bam DLPC proteoliposomes. After a 30-min incubation at 37°C, half of each sample was treated with PK. Half of both treated and untreated samples were heated at 95°C for 5 min, and polypeptides were resolved by SDS-PAGE and subsequently transferred to a PVDF membrane using an overnight wet transfer method. Fluorescently labeled OmpC was visualized directly on the membrane (left), and LPS was detected by Western blotting (right) using the same membrane and an anti-LPS antibody.
DISCUSSION
In this study, we describe the complete assembly of an oligomeric OMP in a simple in vitro system. Unlike previous studies that relied on permeabilized whole-cell systems or spontaneous insertion, our study reconstituted the assembly process using only a purified form of the primary factor that catalyzes OMP assembly in vivo (the Bam complex), a periplasmic chaperone that has been implicated in OMP biogenesis (SurA), and membrane lipids (phospholipids and LPS). The pH of the reaction and the kinetics of assembly (trimers were observed within minutes) were near physiological. The native state of the trimeric species that we observed was validated not only by its resistance to PK digestion and SDS denaturation in the absence of heat but also by its ability to remain stably bound to LPS following SDS-PAGE. Curiously, we found that dead-end dimers formed (especially when we used membrane lipids that contained short-chain fatty acids) in the absence of the Bam complex or under conditions in which the preinsertion conformation of OmpC appeared to be altered. Our results clearly indicate, however, that OmpC trimers are assembled in a stepwise fashion through a dimeric intermediate that forms much more rapidly than these terminal dimers. The data suggest that OmpC has a limited ability to insert into lipid vesicles via a Bam complex-independent pathway and even to self-associate but not to form a fully native structure. The finding that the presence of the Bam complex does not increase the level of the dead-end dimers indicates that the two pathways are distinct; once inserted into proteoliposomes, the monomer does not go “off pathway.” While monomers might simply be released into the lipid bilayer and remain in a trimerization-competent conformation in the Bam complex-dependent pathway, it is also possible that the Bam complex catalyzes a postinsertion assembly reaction that remains to be characterized.
It is striking that the assembly of OmpC trimers required LPS in addition to the Bam complex. Several results, most notably those derived from mutagenesis and site-specific cross-linking experiments, suggest that LPS interacts directly with OmpC monomers at a preinsertion stage. At this stage, the protein must be sufficiently well folded to form the site A and B LPS binding sites. Although the concentration of LPS that we used was likely above the critical micelle concentration (∼14 μM, or <0.1 mg/ml, for the short-chain LPS derived from strain O26:B6 [see reference 54]), OmpC might have formed interactions with a small amount of free LPS or LPS that dissociated from micelles. In any case, the finding that the most active form of LPS (Rc LPS) has only a small polysaccharide group suggests that sites A and B form an important interaction with lipid A analogous to the interactions observed in the OmpF-LPS structure (39). Interestingly, OmpC formed a trimerization-competent conformation much more efficiently when it was synthesized in the PURE system than when it was added to proteoliposomes as a fully synthesized urea-denatured protein. Perhaps the de novo-synthesized form of the protein interacts more effectively with LPS, and the time-dependent loss of insertion competence that we observed is due to a gradual conformational change near the LPS binding sites. Regardless, the data are consistent with previous findings that suggest that the acquisition of specific conformational states in solution is critical for the efficient insertion of OMPs into membranes by the Bam complex (45). Based on the observation that mutations in sites A and B inhibit the membrane integration of OmpC monomers, it should be noted that these sites might not only promote LPS binding but might also form key interactions with BamA and/or another Bam complex subunit.
The requirement for LPS in OmpC assembly is especially noteworthy because in a very similar in vitro system in which the folding of a monomeric 10-stranded β barrel protein (OmpT) was reconstituted into proteoliposomes that contain the Bam complex, the addition of LPS (0.25 mg/ml) inhibited assembly by nearly 10-fold (42, 55). Furthermore, several OMPs, including EspP and OmpA, have been assembled in our standard Bam complex-dependent assay system very efficiently (>50%) without LPS (44). It seems likely that LPS is needed to shield the large number of lipid-facing basic residues in OmpC (which are not found in the other OMPs) from membrane lipids or to promote passage through a hydrophobic bilayer environment. Given that charged residues are highly underrepresented on the lipid-facing surface of most OMPs (40) and that membrane partitioning appears to be promoted by a hydrophobic surface (56), it seems likely that there was an evolutionary selection for the conserved network of basic residues found in OmpC and related porins and its interaction with LPS. The basis for this selection is unclear, however, because neither site B nor most of site A is close to the trimerization interface or obviously associated with the uptake of small molecules. Furthermore, the LPS binding sites are not required for folding or trimerization, at least in detergent solution. Perhaps by masking a section of the protein, LPS promotes trimerization on the opposite side of the β barrel in vivo. It is also possible that the tight association of OmpC with LPS is not required for structural purposes per se but rather is required for the packing or even the localization of the protein at specific sites in the OM.
Although we conducted our experiments in an in vitro assay system, the physiological significance of our results is supported by the observation that LPS binding-site mutations (especially site B mutations) inhibit the membrane insertion of OmpC in vivo (J. H. Peterson and H. D. Bernstein, unpublished results). Our data are also consistent with the finding that LPS in mixed micelles induces the formation of folded monomers or trimers of closely related porins (31–38). Nevertheless, our results raise an apparent paradox. In our experiments, we added LPS to proteoliposomes that contain the Bam complex and phospholipids, but there is currently no evidence that LPS exists in solution in the periplasm in vivo. Furthermore, it seems unlikely that the LPS that we add to our assembly reaction mixtures specifically localizes to the inner leaflet of the lipid vesicles (the equivalent of the outer leaflet of the OM). Nevertheless, we believe that our results provide important insight into the assembly of OmpC in living cells. We propose that like other OMPs (17, 18), OmpC undergoes substantial folding at a preintegration stage both in vivo and in vitro. During membrane integration, its N- and C-terminal β strands, like those of other OMPs that have been analyzed, then form a transient hybrid barrel with the open form of the BamA β barrel (19, 21). During the perturbation and thinning of the lipid bilayer that appear to accompany the opening of the BamA β barrel (15), sites A and B, which are located opposite β1 and β16 in the folded structure, are attracted to LPS phosphate groups near the cell surface and thereby effectively pulled into the membrane. In our in vitro system, LPS mimics its function in vivo by binding to OmpC in solution and then inserting readily into the relatively fluid proteoliposomes when the N and C termini of the protein are engaged by BamA. Rc LPS might function more effectively than larger LPS molecules because its smaller polysaccharide moiety poses fewer steric constraints inside the small vesicles. In any case, it should be noted that our model strongly suggests that membrane lipids can play an important role in the membrane integration of membrane proteins.
Finally, our results demonstrate that Bam complex-dependent assembly of oligomeric OMPs can be reconstituted in vitro and provide the first direct evidence that the Bam complex catalyzes the membrane integration of individual subunits of an oligomeric OMP rather than a fully pre-assembled structure. Although a dimeric species of OmpF has been observed in vivo under some conditions (28), its location was not determined. In addition, by showing that the multistage assembly of a trimeric OMP can be reconstituted in a Bam complex-dependent fashion in vitro, we demonstrate the potential utility of our assay to study the assembly of a wide range of OMPs. In principle, our results suggest that it may be possible to use this system to gain insight into the assembly of entire classes of OMPs (e.g., TonB-dependent transporters [TBDTs]) that have not been well studied. It should be of interest to determine, for example, if the single LPS molecule that copurifies with FhuA (57) and that binds to a specific site in the protein is required for its assembly. With respect to trimeric porins, it should also be of interest to determine if proteins that diverge from OmpC (such as the 18-stranded LamB protein) are assembled by a different mechanism.
MATERIALS AND METHODS
OmpC plasmid construction.
The gene that encodes E. coli ompC without a signal peptide was amplified by PCR using genomic DNA from strain MC4100 as a template and the oligonucleotides 5′-ACTTTAAGAAGGAGATATACCATGGCTGAAGTTTACAACAAAGAC-3′ and 5′-GTCGACGGAGCTCGAATTCGGATCCTTAGAACTGGTAAACCAGACC-3′. To clone the gene into pET28b under the control of the T7 promoter, the vector was digested with NcoI and BamHI, added together with a 3-fold excess of the insert to Gibson assembly master mix (New England BioLabs), and incubated at 50°C for 1 h (58). Mutations were introduced into ompC using the QuikChange site-directed mutagenesis kit (Stratagene) as well as Gibson assembly.
Purification of SurA and the Bam complex and production of liposomes/proteoliposomes.
Plasmids pYG120 (pTRC-bamAB2CDE8His) (23) and pSK257 (42) were used to express and purify the E. coli Bam complex and His-C-tagged SurA as previously described (43, 44). Empty liposomes containing DLPC, POPC, or didecanoylphosphatidylcholine (diC10PC) were produced essentially as described previously (44). Dried lipids were hydrated in 20 mM Tris (pH 8.0) at 42°C with occasional vortexing. Following extrusion, sonication was continued for 30 min, and lipids were kept at room temperature before use or stored at 4°C for up to 2 weeks and resonicated before use. The Bam complex was reconstituted into proteoliposomes containing DLPC or POPC and shown to integrate in a right-side-out orientation as previously described (44).
Preparation of LPS.
LPS was prepared by modifying a previously described method (39). LPS from E. coli strains O55:B5, EH-100 (Ra LPS), and J5 (Rc LPS) was obtained from Millipore Sigma and dissolved at 42°C with vortexing (and then further dissolved at 70°C for 5 min) in 20 mM Tris (pH 8) at 10 mg/ml. Samples were then sonicated in a water bath for 5 to 10 min, cycled between 4°C and 70°C (5 min at each temperature) six times, and incubated at 4°C overnight before use. The dissolved LPS was also stored in aliquots at −20°C and sonicated briefly before use in subsequent experiments.
Production of OmpC and assembly assays.
In most experiments, OmpC was synthesized using the PURExpress coupled transcription-translation system (New England BioLabs) according to the manufacturer’s instructions. Typical 10-μl reaction mixtures (used for single-time-point experiments) contained a murine RNase inhibitor (8 U) and 0.4 μl FluoroTect GreenLys, a lysine-charged tRNA labeled with the fluorophore BODIPY-FL at the ε position (Promega) that was used to incorporate fluorescent lysine residues into OmpC. Many reaction mixtures were also supplemented with 2 μM SurA, presonicated empty DLPC or POPC liposomes (1.6 mg/ml) or Bam DLPC or Bam POPC proteoliposomes (0.5 μM Bam complex), and LPS solubilized as described above (0.5 mM Rc LPS unless otherwise noted). After all of the other reaction components were combined, a pET28 plasmid carrying wild-type or mutant ompC was added to a final concentration of 10 ng/μl. Unless otherwise noted, transcription-translation reactions were conducted at 37°C. After a typical 30-min incubation, reactions were stopped by placing the tubes on ice and adding RNase A (2 mg/ml). PK (30 μg/ml) was then added to half of some reaction mixtures. Protease digestions were conducted for 15 min on ice and halted by the addition of 5 mM phenylmethylsulfonyl fluoride (PMSF) and 2× SDS-PAGE sample buffer. To analyze interactions between OmpC and LPS by site-specific photo-cross-linking, the PURExpress ΔRF123 kit was used in place of the PURExpress system. Release factors 2 and 3 were added to the reaction mixtures prior to the synthesis of OmpC amber mutants, and reaction mixtures were supplemented with tRNATAG-BPA (CosmoBio CloverDirect) (16 pmol/μl). Following the addition of RNase A, half of each reaction mixture was exposed to UV light (Spectroline SB-100P super-high-intensity UV lamp) on a cooled plate on ice for 10 min.
To analyze the kinetics of OmpC assembly, reactions were performed as described above, but volumes were scaled up as necessary (up to 80 μl). After transcription-translation reactions were conducted for 8 to 10 min at 37°C or 30°C, translation reinitiation was halted by the addition of 10 μM oncocin (59) (Onc112 [VDKPPYLPRPRPPRrIYNr-NH2], synthesized and high-performance liquid chromatography [HPLC] purified by the Facility for Biotechnology Resources, Center for Biologics Evaluation and Research, FDA), and incubation at the same temperature was continued. At various time points, an 8-μl aliquot was removed, added to RNase A (2 mg/ml), and placed on ice to stop the assembly reaction. Half of each sample was treated with PK as described above.
In some experiments, the assembly of a fluorescently tagged urea-denatured form of OmpC was analyzed. For this purpose, we substituted a cysteine for S71, a nonconserved amino acid that resides at the start of β4, to minimize any effect that a cysteine substitution might have on OmpC folding. Initially, OmpC S71C was produced in vivo, isolated from inclusion bodies, and solubilized in 8 M urea as previously described (44). The purified protein (final concentration, 80 μM) was mixed with 1.6 mM BODIPY-FL maleimide (Thermo Fisher) in a solution containing 20 mM Tris (pH 7.3) and 8 M urea and rotated for 2 h at room temperature. β-mercaptoethanol (3.2 mM) was then added to quench the reaction, and the labeled protein was purified by running it twice over a PD-10 desalting column (GE Healthcare) equilibrated with a solution containing 20 mM Tris (pH 8) and 8 M urea. In one set of experiments, assembly reactions were conducted under the same conditions as those for the PURExpress reactions, but 0.2 μM fluorescently tagged and urea-denatured OmpC S71C was added in place of the pET28-ompC expression plasmid. In a separate set of experiments, the assembly of urea-denatured OmpC into diC10PC vesicles was analyzed essentially as described previously (33). The protein (0.2 μM) was mixed with lipid (160 μM) in a solution containing 10 mM borate (pH 10) and 1 M urea in the presence or absence of 2 μM SurA and incubated at 37°C for 16 h.
Analysis of OmpC folding and interactions with LPS.
Aliquots of PK-treated and untreated assembly reaction mixtures were mixed with SDS-PAGE sample buffer and either heated at 95°C for 5 min or left unheated before proteins were resolved on 8 to 16% NuPAGE minigels (Thermo Fisher Scientific) unless otherwise noted. Either BenchMark fluorescent protein standard (32, 40, 63, and 98 kDa; Thermo Fisher) or Chameleon Vue prestained rainbow ladder (25, 38, 50, 70, 90, and 125 kDa; Li-Cor) molecular weight markers were run on all gels. Assembly was assessed by monitoring the appearance of dimeric and trimeric forms of OmpC in the absence of heat, and membrane integration was assessed by monitoring the resistance of OmpC monomers and oligomers to PK treatment. Fluorescently labeled OmpC was visualized using an Amersham Typhoon scanner at an excitation wavelength of 488 nm. The folded fraction was quantitated using ImageJ and plotted using Igor Pro software as described previously (44). In some experiments, OmpC bound covalently or noncovalently to LPS was detected by Western blotting using a monoclonal antibody against the LPS core (WN-222-5; Hycult). A wet transfer method was used to detect noncovalently linked LPS. In this method, proteins were transferred to a PVDF membrane in an XCell II blot module (Thermo Fisher) in 12 mM Tris base–96 mM glycine containing 20% methanol and 0.02% SDS. Transfers were conducted at 20 V at 4°C overnight.
We slightly modified a previously described method (39) to examine the folding of wild-type and mutant OmpC in detergent solution following purification from inclusion bodies. In our experiments, we incubated the protein for only 24 h at 37°C. In addition, we resuspended ethanol-precipitated protein in a solution containing 20 mM Tris (pH 8) and 0.5% (vol/vol) octylpolyoxyethylene (octyl-POE) and analyzed a portion by SDS-PAGE. Colloidal blue staining was then used to visualize the protein.
ACKNOWLEDGMENTS
We thank Matt Doyle and Thushani Nilaweera for providing helpful comments on the manuscript.
This work was supported by the Intramural Research Program of the National Institutes of Diabetes and Digestive and Kidney Diseases.
S.H. and H.D.B. designed research, S.H. and J.H.P. performed research and analyzed data, and H.D.B. wrote the paper.
We have no competing interests to report.
Footnotes
Citation Hussain S, Peterson JH, Bernstein HD. 2021. Reconstitution of Bam complex-mediated assembly of a trimeric porin into proteoliposomes. mBio 12:e01696-21. https://doi.org/10.1128/mBio.01696-21.
Contributor Information
Harris D. Bernstein, Email: harris_bernstein@nih.gov.
M. Stephen Trent, University of Georgia.
REFERENCES
- 1.Rapoport TA, Li L, Park E. 2017. Structural and mechanistic insights into protein translocation. Annu Rev Cell Dev Biol 33:369–390. doi: 10.1146/annurev-cellbio-100616-060439. [DOI] [PubMed] [Google Scholar]
- 2.Forrest LR. 2015. Structural symmetry in membrane proteins. Annu Rev Biophys 44:311–337. doi: 10.1146/annurev-biophys-051013-023008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fairman JW, Noinaj N, Buchanan SK. 2011. The structural biology of β-barrel membrane proteins: a summary of recent reports. Curr Opin Struct Biol 21:523–531. doi: 10.1016/j.sbi.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lauber F, Deme LC, Lea SM, Berks BC. 2018. Type 9 secretion system structures reveal a new transport mechanism. Nature 564:77–82. doi: 10.1038/s41586-018-0693-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meng G, Fronzes R, Chandran V, Remaut H, Waksman G. 2009. Protein oligomerization in the bacterial outer membrane. Mol Membr Biol 26:136–145. doi: 10.1080/09687680802712422. [DOI] [PubMed] [Google Scholar]
- 6.Chen R, Henning U. 1996. A periplasmic protein (Skp) of Escherichia coli selectively binds a class of outer membrane proteins. Mol Microbiol 19:1287–1294. doi: 10.1111/j.1365-2958.1996.tb02473.x. [DOI] [PubMed] [Google Scholar]
- 7.Lazar SW, Kolter R. 1996. SurA assists the folding of Escherichia coli outer membrane proteins. J Bacteriol 178:1770–1773. doi: 10.1128/jb.178.6.1770-1773.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rouvière PE, Gross CA. 1996. SurA, a periplasmic protein with peptidyl-prolyl isomerase activity, participates in the assembly of outer membrane porins. Genes Dev 10:3170–3182. doi: 10.1101/gad.10.24.3170. [DOI] [PubMed] [Google Scholar]
- 9.Schäfer U, Beck K, Muller M. 1999. Skp, a molecular chaperone of Gram-negative bacteria, is required for the formation of soluble periplasmic intermediates of outer membrane proteins. J Biol Chem 274:24567–24574. doi: 10.1074/jbc.274.35.24567. [DOI] [PubMed] [Google Scholar]
- 10.Voulhoux R, Bos MP, Geurtsen J, Mols M, Tommassen J. 2003. Role of a highly conserved bacterial protein in outer membrane protein assembly. Science 299:262–265. doi: 10.1126/science.1078973. [DOI] [PubMed] [Google Scholar]
- 11.Wu T, Malinverni J, Ruiz N, Kim S, Silhavy TJ, Kahne D. 2005. Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell 121:235–245. doi: 10.1016/j.cell.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 12.Sklar JG, Wu T, Gronenberg LS, Malinverni JC, Kahne D, Silhavy TJ. 2007. Lipoprotein SmpA is a component of the YaeT complex that assembles outer membrane proteins in Escherichia coli. Proc Natl Acad Sci USA 104:6400–6405. doi: 10.1073/pnas.0701579104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim S, Malinverni JC, Sliz P, Silhavy TJ, Harrison SC, Kahne D. 2007. Structure and function of an essential component of the outer membrane protein assembly machine. Science 317:961–964. doi: 10.1126/science.1143993. [DOI] [PubMed] [Google Scholar]
- 14.Webb CT, Heinz E, Lithgow T. 2012. Evolution of the β-barrel assembly machinery. Trends Microbiol 20:612–620. doi: 10.1016/j.tim.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 15.Noinaj N, Kuszak AJ, Gumbart JC, Lukacik P, Chang H, Easley NC, Lithgow T, Buchanan SK. 2013. Structural insight into the biogenesis of β-barrel membrane proteins. Nature 501:385–390. doi: 10.1038/nature12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kleinschmidt JH, Tamm LK. 2002. Secondary and tertiary structure formation of the b-barrel membrane protein OmpA is synchronized and depends on membrane thickness. J Mol Biol 324:319–330. doi: 10.1016/S0022-2836(02)01071-9. [DOI] [PubMed] [Google Scholar]
- 17.Ieva R, Skillman KM, Bernstein HD. 2008. Incorporation of a polypeptide segment into the β-domain pore during the assembly of a bacterial autotransporter. Mol Microbiol 67:188–201. doi: 10.1111/j.1365-2958.2007.06048.x. [DOI] [PubMed] [Google Scholar]
- 18.Sikdar R, Peterson JH, Anderson DE, Bernstein HD. 2017. Folding of a bacterial integral outer membrane protein is initiated in the periplasm. Nat Commun 8:1309. doi: 10.1038/s41467-017-01246-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doyle MT, Bernstein HD. 2019. Bacterial outer membrane proteins assemble via asymmetric interactions with the BamA β-barrel. Nat Commun 10:3358. doi: 10.1038/s41467-019-11230-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee J, Tomasek D, Santos TM, May MD, Meuskens I, Kahne D. 2019. Formation of a β-barrel membrane protein is catalyzed by the interior surface of the assembly machine protein BamA. Elife 8:e49787. doi: 10.7554/eLife.49787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tomasek D, Rawson S, Lee J, Wzorek JS, Harrison SC, Li Z, Kahne D. 2020. Structure of a nascent membrane protein as it folds on the BAM complex. Nature 583:473–476. doi: 10.1038/s41586-020-2370-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bakelar J, Buchanan SK, Noinaj N. 2016. The structure of the β-barrel assembly machinery complex. Science 351:180–186. doi: 10.1126/science.aad3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gu Y, Li H, Dong H, Zeng Y, Zhang Z, Paterson NG, Stansfeld PJ, Wang Z, Zhang Y, Wang W, Dong C. 2016. Structural basis of outer membrane protein insertion by the BAM complex. Nature 531:64–69. doi: 10.1038/nature17199. [DOI] [PubMed] [Google Scholar]
- 24.Han L, Zheng J, Wang Y, Yang X, Liu Y, Sun C, Cao B, Zhou H, Ni D, Lou J, Zhao Y, Huang Y. 2016. Structure of the BAM complex and its implications for biogenesis of outer-membrane proteins. Nat Struct Mol Biol 23:192–196. doi: 10.1038/nsmb.3181. [DOI] [PubMed] [Google Scholar]
- 25.Iadanza MG, Higgins AJ, Schiffrin B, Calabrese AN, Brockwell DJ, Ashcroft AE, Radford SE, Ranson NA. 2016. Lateral opening in the intact β-barrel assembly machinery captured by cryo-EM. Nat Commun 7:12865. doi: 10.1038/ncomms12865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Horne JE, Brockwell DJ, Radford SE. 2020. Role of the lipid bilayer in outer membrane protein folding in Gram-negative bacteria. J Biol Chem 295:10340–10367. doi: 10.1074/jbc.REV120.011473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vergalli J, Bodrenko IV, Masi M, Moynié L, Acosta-Gutiérrez S, Naismith JH, Davin-Regli A, Ceccarelli M, van den Berg B, Winterhalter M, Pagès J-M. 2020. Porins and small-molecule translocation across the outer membrane of Gram-negative bacteria. Nat Rev Microbiol 18:164–176. doi: 10.1038/s41579-019-0294-2. [DOI] [PubMed] [Google Scholar]
- 28.Reid J, Fung H, Gehring K, Klebba PE, Nikaido H. 1988. Targeting of porin to the outer membrane of Escherichia coli: rate of trimer assembly and identification of a dimer intermediate. J Biol Chem 263:7753–7759. doi: 10.1016/S0021-9258(18)68563-1. [DOI] [PubMed] [Google Scholar]
- 29.van Gelder P, Tommassen J. 1996. Demonstration of a folded monomeric form of porin PhoE of Escherichia coli in vivo. J Bacteriol 178:5320–5322. doi: 10.1128/jb.178.17.5320-5322.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naveed H, Jimenez-Morales D, Tian J, Pasupuleti V, Kenney LJ, Liang J. 2012. Engineered oligomerization state of OmpF protein through computational design decouples oligomer dissociation from unfolding. J Mol Biol 419:89–101. doi: 10.1016/j.jmb.2012.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sen K, Nikaido H. 1990. In vitro trimerization of OmpF secreted by spheroplasts of Escherichia coli. Proc Natl Acad Sci USA 87:743–747. doi: 10.1073/pnas.87.2.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Surrey T, Schmid A, Jahnig F. 1996. Folding and membrane insertion of the trimeric β-barrel protein OmpF. Biochemistry 35:2283–2288. doi: 10.1021/bi951216u. [DOI] [PubMed] [Google Scholar]
- 33.Burgess NK, Dao TP, Stanley AM, Fleming KG. 2008. β-Barrel proteins that reside in the Escherichia coli outer membrane in vivo demonstrate varied folding behavior in vitro. J Biol Chem 283:26748–26758. doi: 10.1074/jbc.M802754200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Gelder P, de Cock H, Tommassen J. 1994. Detergent-induced folding of the outer-membrane protein PhoE, a pore protein induced by phosphate limitation. Eur J Biochem 226:783–787. doi: 10.1111/j.1432-1033.1994.00783.x. [DOI] [PubMed] [Google Scholar]
- 35.Okuda S, Sherman DJ, Silhavy TJ, Ruiz N, Kahne D. 2016. Lipopolysaccharide transport and assembly at the outer membrane: the PEZ model. Nat Rev Microbiol 14:337–345. doi: 10.1038/nrmicro.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Owens TW, Taylor RJ, Pahil KS, Bertani BR, Ruiz N, Kruse AC, Kahne D. 2019. Structural basis of unidirectional export of lipopolysaccharide to the cell surface. Nature 567:550–553. doi: 10.1038/s41586-019-1039-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sen K, Nikaido H. 1991. Trimerization of an in vitro synthesized OmpF porin of Escherichia coli outer membrane. J Biol Chem 266:11295–11300. doi: 10.1016/S0021-9258(18)99162-3. [DOI] [PubMed] [Google Scholar]
- 38.de Cock H, Tommassen J. 1996. Lipopolysaccharides and divalent cations are involved in the formation of an assembly-competent intermediate of outer-membrane protein PhoE of E. coli. EMBO J 15:5567–5573. doi: 10.1002/j.1460-2075.1996.tb00941.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arunmanee W, Pathania M, Solovyova AS, Le Brun AP, Ridley H, Baslé A, van den Berg B, Lakey JH. 2016. Gram-negative trimeric porins have specific LPS binding sites that are essential for porin biogenesis. Proc Natl Acad Sci USA 113:E5034–E5043. doi: 10.1073/pnas.1602382113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peterson JH, Plummer AM, Fleming KG, Bernstein HD. 2017. Selective pressure for rapid membrane integration constrains the sequence of bacterial outer membrane proteins. Mol Microbiol 106:777–792. doi: 10.1111/mmi.13845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baslé A, Rummel G, Storici P, Rosenbusch JP, Schirmer T. 2006. Crystal structure of osmoporin OmpC from E. coli at 2.0 Å. J Mol Biol 362:933–942. doi: 10.1016/j.jmb.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 42.Hagan CL, Kim S, Kahne D. 2010. Reconstitution of outer membrane protein assembly from purified components. Science 328:890–892. doi: 10.1126/science.1188919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roman-Hernandez G, Peterson JH, Bernstein HD. 2014. Reconstitution of bacterial autotransporter assembly using purified components. Elife 3:e04234. doi: 10.7554/eLife.04234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hussain S, Bernstein HD. 2018. The Bam complex catalyzes efficient assembly of bacterial outer membrane proteins into membrane vesicles of variable lipid composition. J Biol Chem 293:2959–2973. doi: 10.1074/jbc.RA117.000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hussain S, Peterson JH, Bernstein HD. 2020. Bam complex-mediated assembly of bacterial outer membrane proteins synthesized in an in vitro translation system. Sci Rep 10:4557. doi: 10.1038/s41598-020-61431-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Otzen DE, Andersen KK. 2013. Folding of outer membrane proteins. Arch Biochem Biophys 531:34–43. doi: 10.1016/j.abb.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 47.Fleming KG. 2015. A combined kinetic push and thermodynamic pull as driving forces for outer membrane protein sorting and folding in bacteria. Philos Trans R Soc Lond B Biol Sci 370:20150026. doi: 10.1098/rstb.2015.0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gessmann D, Chung YH, Danoff EJ, Plummer AM, Sandlin CW, Zaccai NR, Fleming KG. 2014. Outer membrane β-barrel protein folding is physically controlled by periplasmic head groups and BamA. Proc Natl Acad Sci USA 111:5878–5883. doi: 10.1073/pnas.1322473111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Danoff EJ, Fleming KG. 2015. Membrane defects accelerate outer membrane β-barrel protein folding. Biochemistry 54:97–99. doi: 10.1021/bi501443p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schiffrin B, Calabrese AN, Higgins AJ, Humes JR, Ashcroft AE, Kalli AC, Brockwell DJ, Radford SE. 2017. Effects of periplasmic chaperones and membrane thickness on BamA-catalyzed outer-membrane protein folding. J Mol Biol 429:3776–3792. doi: 10.1016/j.jmb.2017.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Raetz CR, Whitfield C. 2002. Lipopolysaccharide endotoxins. Annu Rev Biochem 71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seefeldt AC, Nguyen F, Antunes S, Pérébaskine N, Graf M, Arenz S, Inampudi KK, Douat C, Guichard G, Wilson DN, Innis CA. 2015. The proline-rich antimicrobial peptide Onc112 inhibits translation by blocking and destabilizing the initiation complex. Nat Struct Mol Biol 22:470–475. doi: 10.1038/nsmb.3034. [DOI] [PubMed] [Google Scholar]
- 53.Farrell IS, Toroney R, Hazen JL, Mehl RA, Chin JW. 2005. Photo-cross-linking interacting proteins with a genetically encoded benzophenone. Nat Methods 2:377–384. doi: 10.1038/nmeth0505-377. [DOI] [PubMed] [Google Scholar]
- 54.Santos NC, Silva AC, Castanho MARB, Martins-Silva J, Saldanha C. 2003. Evaluation of lipopolysaccharide aggregation by light scattering spectroscopy. Chembiochem 4:96–100. doi: 10.1002/cbic.200390020. [DOI] [PubMed] [Google Scholar]
- 55.Hagan CL, Kahne D. 2011. The reconstituted Escherichia coli Bam complex catalyzes multiple rounds of β-barrel assembly. Biochemistry 50:7444–7446. doi: 10.1021/bi2010784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moon CP, Fleming KG. 2011. Side-chain hydrophobicity scale derived from transmembrane protein folding into lipid bilayers. Proc Natl Acad Sci USA 108:10174–10177. doi: 10.1073/pnas.1103979108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ferguson AD, Hofmann E, Coulton JW, Diederichs K, Welte W. 1998. Siderophore-mediated iron transport: crystal structure of FhuA with bound lipopolysaccharide. Science 282:2215–2220. doi: 10.1126/science.282.5397.2215. [DOI] [PubMed] [Google Scholar]
- 58.Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, III, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 59.Krizsan A, Volke D, Weinert S, Sträter N, Knappe D, Hoffmann R. 2014. Insect-derived proline-rich antimicrobial peptides kill bacteria by inhibiting bacterial protein translation at the 70S ribosome. Angew Chem Int Ed Engl 53:12236–12239. doi: 10.1002/anie.201407145. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The concentration of Rc LPS affects the efficiency of OmpC trimerization. (A) OmpC was generated in vitro as described in the legend to Fig. 1 using the PURExpress system supplemented with FluoroTect, SurA, the indicated concentration of Rc LPS, and proteoliposomes containing the Bam complex and DLPC (Bam DLPC) or POPC (Bam POPC). After a 30-min incubation at 37°C, half of each sample was heated at 95°C for 5 min, and fluorescently labeled polypeptides were resolved by SDS-PAGE. (B) OmpC was generated in vitro as described in the legend to Fig. 1 using the PURExpress system supplemented with FluoroTect, SurA, and the indicated form of LPS. Proteoliposomes containing the Bam complex and POPC (Bam POPC) were also added to one reaction mixture. After a 30-min incubation at 37°C, half of each sample was heated at 95°C for 5 min, and fluorescently labeled polypeptides were resolved by SDS-PAGE. The results show that LPS alone does not stimulate OmpC folding. Download FIG S1, JPG file, 1.5 MB (1.5MB, jpg) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
SurA is required for OmpC trimerization. OmpC was generated in vitro as described in the legend to Fig. 1 using the PURExpress system supplemented with FluoroTect, Rc LPS (0.5 mg/ml), and proteoliposomes containing the Bam complex and DLPC. SurA was also added as indicated. After a 30-min incubation at 37°C, half of each sample was treated with PK. Half of both treated and untreated samples were heated at 95°C for 5 min, and fluorescently labeled polypeptides were resolved by SDS-PAGE. Download FIG S2, JPG file, 0.3 MB (278.1KB, jpg) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Time-dependent loss of insertion competence of OmpC synthesized in vitro. (A) Summary of the experimental strategy. (B) OmpC was generated in vitro as described in the legend to Fig. 1 using the PURExpress system supplemented with FluoroTect, SurA, and Rc LPS (0.5 mg/ml). After 10 min at 37°C (i.e., at t = 0′ in panel A), oncocin (Onc112) was added to halt translation initiation, and incubation was continued for an additional 20 min (until t = 20′). Either Bam DLPC or Bam POPC was added to an aliquot of the reaction mixture at the indicated time points (t = −10′, 0′, or 5′ in panel A) to promote OmpC assembly for the remainder of the incubation. One-half of each sample was treated with PK, and half of the treated samples were heated to 95°C for 5 min. Fluorescently labeled polypeptides were resolved by SDS-PAGE. To compare equal amounts of reaction products, only half of each untreated sample was loaded. Download FIG S3, JPG file, 0.7 MB (738.1KB, jpg) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Trimerization of urea-denatured OmpC at pH 10. Urea-denatured OmpC labeled at residue 71 with BODIPY-FL was assembled into short-chain lipid (diC10PC) vesicles at pH 10 at 37°C for 16 h with and without SurA. The samples were then treated with PK, and half of each sample was heated at 95°C for 5 min. Fluorescently labeled polypeptides were resolved by SDS-PAGE on 4 to 20% minigels. Download FIG S4, JPG file, 0.2 MB (217.6KB, jpg) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Time course of OmpC assembly at 30°C. (A) The experiment shown in Fig. 3 was repeated except that the incubation temperature was changed to 30°C. The fractions of the total untreated (top) and PK-treated (bottom) proteins that were observed in monomeric, dimeric, and trimeric forms as a function of time in the representative experiment shown on the left are plotted on the right. The numerical data used to generate the graphs are shown in Table S1B in the supplemental material. (B) The experiment shown in panel A was repeated, and the samples were then treated with PK. (Top) Fluorescently labeled polypeptides were resolved by SDS-PAGE and visualized. (Bottom) Proteins were then transferred to PVDF membranes using an overnight wet transfer method, and LPS was detected by Western blotting using an anti-LPS antibody. The results show a time-dependent accumulation of LPS bound to the OmpC trimer. Download FIG S5, JPG file, 1.4 MB (1.4MB, jpg) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Quantitative analysis of the experiments shown in Fig. 3 (A) and Fig. S5A in the supplemental material (B) using ImageJ software. Download Table S1, JPG file, 0.9 MB (901.2KB, jpg) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
A monomeric intermediate is observed upon reducing the rate of assembly. The experiment shown in Fig. S5 was repeated using the OmpC site B Gln mutant. Download FIG S6, JPG file, 0.5 MB (556.2KB, jpg) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Multiple LPS binding-site mutations do not affect the folding of OmpC in detergent solution. The urea-denatured wild type (WT) or the indicated OmpC mutant was folded into a detergent mixture of 1% dodecyl-β-D-maltoside (DDM)–0.4% DDG in 50 mM Tris (pH 8.0), 1 mM dithiothreitol (DTT), and 0.1 mM EDTA by incubation at 37°C for 24 h. Half of the wild-type sample was heated (lane 1). Proteins were resolved by SDS-PAGE and visualized by colloidal blue staining. Download FIG S7, JPG file, 0.5 MB (475.3KB, jpg) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Single mutations in the LPS binding sites of OmpC do not affect assembly. The experiment described in the legend of Fig. 5A was repeated with the indicated wild-type (WT) and mutant forms of OmpC. Download FIG S8, JPG file, 1.3 MB (1.3MB, jpg) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Residues far from an LPS binding site do not interact significantly with LPS. The photo-cross-linking experiment described in the legend of Fig. 6A was repeated with OmpC(S71am), OmpC(E189am), and OmpC(K281am) as a positive control. SDS-PAGE was conducted to detect fluorescently labeled polypeptides, and an OmpC monomer-LPS cross-linking product was confirmed by Western blotting using an anti-LPS antibody. An unidentified cross-reactive protein is denoted with an asterisk. Download FIG S9, JPG file, 0.5 MB (498.9KB, jpg) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.