

ABSTRACT
Human gut microbes exhibit a spectrum of cooperative and antagonistic interactions with their host and also with other microbes. The major Bacteroides host-targeting virulence factor, Bacteroides fragilis toxin (BFT), is produced as an inactive protoxin by enterotoxigenic B. fragilis strains. BFT is processed by the conserved bacterial cysteine protease fragipain (Fpn), which is also encoded in B. fragilis strains that lack BFT. In this report, we identify a secreted antibacterial protein (fragipain-activated bacteriocin 1 [Fab1]) and its cognate immunity protein (resistance to fragipain-activated bacteriocin 1 [RFab1]) in enterotoxigenic and nontoxigenic strains of B. fragilis. Although BFT and Fab1 share no sequence identity, Fpn also activates the Fab1 protoxin, resulting in its secretion and antibacterial activity. These findings highlight commonalities between host- and bacterium-targeting toxins in intestinal bacteria and suggest that antibacterial antagonism may promote the conservation of pathways that activate host-targeting virulence factors.
KEYWORDS: bacteriocin, Bacteroides, interbacterial interaction, microbiome
INTRODUCTION
The human intestine harbors a complex microbial community that inhabits the length of the gastrointestinal tract, with densities being highest in the colon. In most individuals, the gut microbiome is dominated by representatives of two major phyla (Bacteroidetes and Firmicutes). Although these broad taxonomic groups are ubiquitous across individuals, species- and strain-level differences within these phyla are associated with differences in pathogen susceptibility, metabolism, drug response, and other host phenotypes (1). Notably, gut microbial strains can persist in individuals for years or decades despite continual challenges from the outside environment (2). Multiple mechanisms likely contribute to strain persistence or replacement, including priority effects (3) and nutrient specialization (4). In addition, interbacterial antagonism is increasingly recognized as a factor that determines strain selection and competition in this densely packed ecosystem (5). Antagonistic mechanisms allow bacteria to selectively target closely related and/or physically proximal cells and leave characteristic signatures in gut microbial genomes and metagenomes (6, 7). Understanding these antagonistic interactions can provide insight into the rules of assembly in the gut microbiome and inform future therapeutic manipulation of these microbial communities.
Genetic screens, biochemical isolations, and bioinformatic approaches have identified antimicrobial toxins produced by a wide variety of intestinal bacteria, including Lactobacillus, Bifidobacterium, Enterococcus, Bacteroides, and Escherichia. These studies highlight two general antagonistic strategies: contact-dependent mechanisms include contact-dependent inhibition (CDI) and the type VI secretion system (T6SS) (8, 9); contact-independent antibacterial factors include small-molecule antibiotics and secreted antimicrobial peptides and proteins (e.g., microcins and colicins) (10–13). These antimicrobial factors can mediate competition between bacterial cells across families (broad spectrum) or within strains of the same species (narrow spectrum) (5, 13). Contact-dependent and contact-independent antibacterial factors use a variety of mechanisms of action, including pore formation and inhibition of DNA, RNA, or protein synthesis (13). Both contact-dependent and diffusible toxins can limit the expansion of competing commensals and pathogens in vivo (10, 14, 15).
These systems have been best studied in Proteobacteria; identification and characterization of antimicrobial factors in human gut Bacteroides is constrained by the absence of sequence similarity or protein motifs from previously studied antibacterial effectors. While T6SS-delivered effectors can be identified by genomic context (16), the factors that mediate contact-independent antagonism in human gut Bacteroides have been elusive. Genetic approaches have identified broad-spectrum peptide toxins that target diverse members of the phylum Bacteroidetes (bacteroidetocin A and bacteroidetocin B) (17) and larger proteins that specifically target strains within the same species (18–21). These narrow-spectrum antimicrobial factors share eukaryotic-like features, including membrane attack complex/perforin (MACPF) or ubiquitin-like domains (18–21).
Bacteroides fragilis encodes a diverse repertoire of T6SS-dependent effectors and contact-independent bacteriocins (5). Notably, B. fragilis is implicated in both health and disease (22). This species produces beneficial immunomodulatory factors that mediate host immune system development (23) but can also cause epithelial cell damage, making it the most common anaerobic isolate from abdominal abscesses and bloodstream infections (22). Enterotoxigenic B. fragilis (ETBF) strains are marked by the presence of pathogenicity islands that encode B. fragilis toxin (BFT), which cleaves E-cadherin and causes colonic cell damage and inflammation (24). Fragipain (Fpn), a cysteine protease encoded outside the pathogenicity islands, transforms the 45-kDa full-length BFT protoxin into its 20-kDa active form through cleavage at an arginine-alanine site (25, 26). Interestingly, BFT from an fpn mutant strain is readily activated by host proteases in the gut and efficiently causes epithelial cell damage (25). Fpn is also conserved in nontoxigenic B. fragilis (NTBF) strains that lack BFT, suggesting other roles for Fpn beyond activation of this host-targeting toxin (25, 26). Consistent with this observation, a recent report identifies numerous differences between the secretomes of a wild-type ETBF strain and its isogenic fpn mutant (26).
Here, we report that many NTBF and ETBF B. fragilis strains use Fpn for activation and secretion of a potent, secreted antibacterial toxin. This antibacterial protein, fragipain-activated bacteriocin 1 (Fab1), lacks domains found in previously characterized Bacteroides bacteriocins and directly kills susceptible strains upon activation by Fpn. Transfer of the open reading frame downstream of fab1 into otherwise susceptible strains confers protection from Fab1, suggesting that this downstream gene encodes an immunity protein. Together, these results expand the repertoire of antagonistic activities in human gut microbes, suggest that contact-independent host- and bacteria-targeting toxins can leverage the same machinery for activation and provide an explanation for the maintenance of this machinery in the absence of its best-characterized substrate.
RESULTS
Bacteroides fragilis exhibits potent antibacterial activity independent of type VI secretion.
In the course of screening B. fragilis isolates for contact-dependent antibacterial activity, we observed that the T6SS-positive NTBF strain NCTC9343 (BfN) exhibits potent antagonistic activity toward diverse B. fragilis strains that is independent of a functional T6SS (Fig. 1A). Target strains (B. fragilis HMW160 [BfH610], B. fragilis HMW615 [BfH615], and B. fragilis 638R [BfR]) were selected to represent distinct branches of the B. fragilis phylogeny (14) that can be differentiated from BfN by selective plating (see Table S1 in the supplemental material). By contrast, killing of Bacteroides thetaiotaomicron by BfN was T6SS dependent, as previously reported (14). To distinguish this T6SS-independent activity from T6SS-dependent antagonism, we used deletion mutants in the essential T6SS component tssC (referred to as parental strains) in subsequent experiments. Culture supernatants from the BfN parental strain also possess bactericidal activity toward susceptible B. fragilis strains (Fig. 1B and C) but not B. thetaiotaomicron (Fig. S1). Both heat and proteinase K treatment abolished this T6SS-independent antimicrobial activity, suggesting that one or more secreted protein factor(s) are required (Fig. 1B and C). Notably, BfN lacks homologs of previously reported MACPF (membrane attack complex/perforin) domain-containing antimicrobial proteins and bacteroidetocin peptide toxins (17–19, 21, 27).
FIG 1.

B. fragilis strain NCTC9343 (BfN) secretes antimicrobial protein(s) to target susceptible B. fragilis strains independent of the type VI secretion system. (A). Input and output CFU of target strains after surface cocultivation with the indicated BfN producer strains. Error bars indicate standard deviations (SD) (n = 2; representative of three independent experiments). Strain designations: Bt, B. thetaiotaomicron; BfR, B. fragilis strain 638R; BfH610, B. fragilis strain HMW610; BfH615, B. fragilis strain HMW615. Parental strains carry deletions in tssC which disable the type VI secretion system (T6SS). (B and C). BfN culture supernatants contain antimicrobial activity. CFU of two target strains after exposure to culture supernatant from the BfN parental strain or minimal medium with different treatments are reported. Error bars indicate SD (n = 2; representative of three independent experiments). *, P < 0.05; **, P < 0.01; n.s., not significant.
B. thetaiotaomicron (Bt) is resistant to the antimicrobial protein(s) produced by BfN. B. thetaiotaomicron CFU after exposure to BfN parental culture supernatant or minimal medium with different treatments are reported. Error bars indicate SD (n = 2; representative of three independent experiments). Download FIG S1, EPS file, 1.9 MB (1.9MB, eps) .
Copyright © 2021 Bao et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Bacterial strains, plasmids, and oligonucleotide primers used in this study. Download Table S1, XLSX file, 0.02 MB (19.6KB, xlsx) .
Copyright © 2021 Bao et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Identification of genetic regions required for BfN antagonistic activity.
To identify genetic regions required for antibacterial activity, we constructed a mariner transposon mutant library in the BfN parental background and screened individual clones for loss of antagonism toward BfH610. This representative target strain is susceptible to the T6SS-independent BfN antagonistic activity (Fig. 1A and B) and, unlike BfN, is naturally resistant to tetracycline. As a result, selective plating of the competition assay on tetracycline serves as an indicator of the extent of BfN::TN antagonistic activity (Fig. 2A). From a total of 15,000 mutants screened, three independent clones (carrying transposon insertions in the intergenic region upstream of BF9343_2671, within the BF9343_2671 open reading frame, and within the BF9343_1466 open reading frame) demonstrated significant reductions in bactericidal activity (Fig. 2B). Based on results described below, we designated BF9343_2671 as fpn and BF9343_1466 as fab1. The strain carrying a transposon in the intergenic region upstream of fpn exhibited significantly reduced fpn expression (Fig. S2). For subsequent studies, we used the tetracycline-sensitive, genetically tractable NTBF parent strain 638R (BfR) strain as a representative target strain. Unmarked, in-frame deletions of fpn and fab1 in BfN recapitulate the decreased antibacterial capacity of the transposon mutant strains, and complementation of gene expression in single copy in trans significantly (but not completely) restores antagonistic activity (Fig. 2C and D). Together, these results implicate fpn and fab1 in BfN antagonistic activity.
FIG 2.
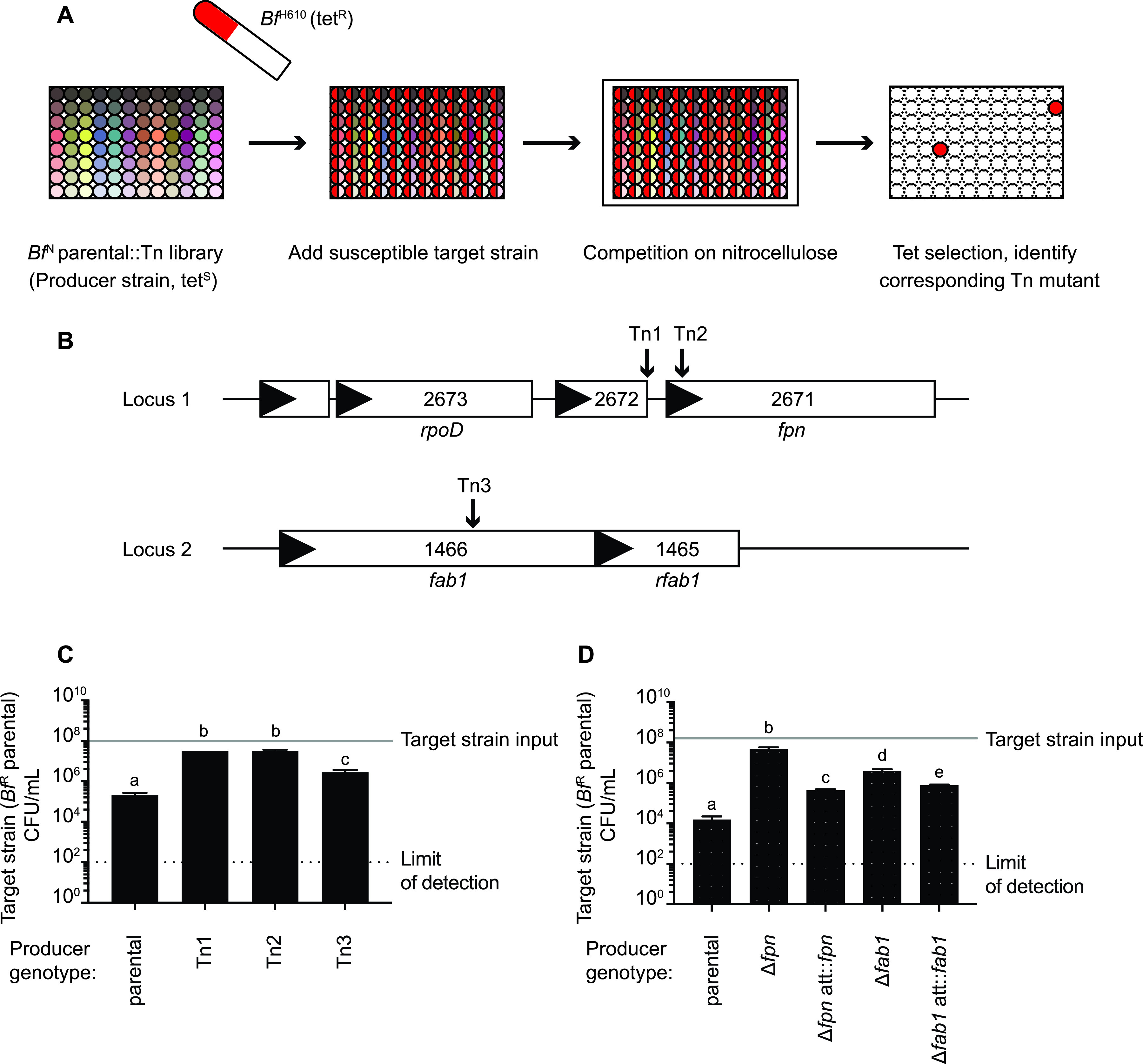
A loss-of-function (LoF) genetic screen identifies genes involved in antimicrobial activity. (A) Workflow of the LoF genetic screen. (B) Genomic locations of BfN transposon insertions that abrogate its capacity to inhibit growth of BfH610. (C) Quantification of killing activity by BfN parental and transposon mutant strains identified in panel B. (D) Quantification of killing activity by BfN parental, isogenic deletion strains, and complemented mutants. Complemented mutants carry the deleted gene in single copy in a neutral locus (att). For panels C and D, mean number of target strain input CFU is indicated with a gray line (mean = 2.2 × 107); line width represents SD (SD = 8.5 × 106). Target strain output CFU are reported. Error bars indicate SD (n = 2; representative of three independent experiments). Different letters represent groups that are statistically significantly different (P < 0.05).
Transposon insertion in the intergenic region between BF9343_2672 and fpn significantly reduces fpn gene expression. Expression of fpn and BF9343_2672 in the mutant strain carrying a transposon insertion in the intergenic region upstream of fpn (Tn1) (Fig. 2B) relative to the BfN parental strain was determined by reverse transcription-qPCR (qRT-PCR). Mean expression levels are calculated from technical triplicates from two independent experiments and normalized to 16S RNA. Error bars indicate SD. Download FIG S2, EPS file, 1.9 MB (1.9MB, eps) .
Copyright © 2021 Bao et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Designation of BF9343_2671 as fpn.
BfN Fpn shares 99.7% identity with a clostripain-related cysteine protease that mediates maturation of the host-targeting toxin BFT in ETBF strains (25). This activity requires a conserved histidine-cysteine dyad, which is common to cysteine proteases (28). We expressed and purified BfN Fpn in Escherichia coli. Although BfN does not encode BFT, this purified Fpn also cleaves purified BFT to form the 20-kDa toxin (Fig. 3A). Cleavage activity is abrogated by heat deactivation of Fpn or by substitution of the predicted active site residues H135 or C180 with alanine (Fig. 3A). The active-site residues required for the BFT cleavage role of Fpn are also required for its contribution to antimicrobial activity (Fig. 3B), suggesting that the cysteine protease activity of this protein is also important for its contribution to antimicrobial antagonism.
FIG 3.
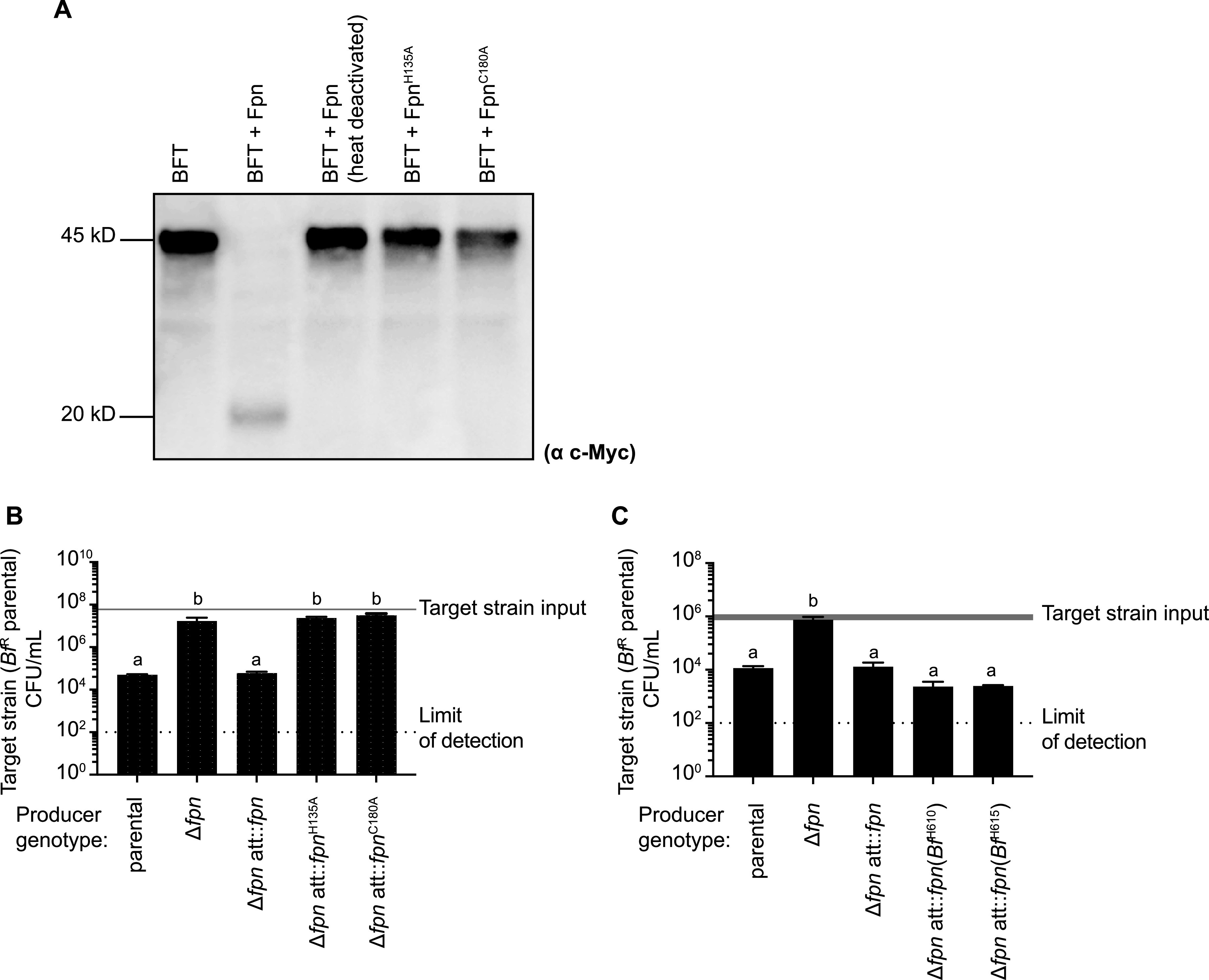
Activity of Fpn homologs from BfN and susceptible target strains. (A) Western blot analysis of BFT products generated following incubation of purified BfN Fpn or its catalytic residue mutants (FpnH135A; FpnC180A) with BFT carrying a C-terminal c-Myc tag. (B) Mutation of Fpn catalytic residues in BfN abrogates its ability to inhibit BfR growth. The mean number of target strain input CFU is indicated with a gray line (mean = 6.0 × 107); line width represents SD (SD = 5.0 × 106). Target strain output CFU are reported. Error bars indicate SD (n = 2; representative of three independent experiments). (C) Expression of fpn genes derived from susceptible strains BfH610 and BfH615 restores killing activity to a BfNΔfpn mutant. The mean number of target strain input CFU is indicated with a gray line (mean = 9.6 × 105); line width represents SD (SD = 2.4 × 105). Target strain output CFU are reported. Error bars indicate SD (n = 2; representative of three independent experiments). For panels B and C, different letters represent groups that are statistically significantly different (P < 0.05).
Surprisingly, BfH610 and BfH615, which are both susceptible to BfN antimicrobial activity (Fig. 1A), also encode Fpn homologs. Expression of each homolog restores antimicrobial activity to the BfN Δfpn deletion strain, indicating that Fpn homologs from these susceptible B. fragilis strains are functional (Fig. 3C). This result also suggests that the antimicrobial capacity of BfN is dependent on Fpn and another factor that is missing in BfH610 and BfH615.
Fpn activates the antimicrobial function of Fab1.
The loss-of-function screen also implicated Fab1, a predicted 50-kDa protein with no known function or recognizable domains, in BfN antimicrobial activity (Fig. 2B). Fab1 homologs are absent in BfH610 and BfH615. C-terminal epitope tagging of the Fab1 open reading frame in BfN followed by Western blotting using an epitope-targeted antibody revealed that the C-terminal end of the protein is almost entirely localized to the secreted (supernatant) fraction as a 28-kDa fragment (Fig. 4A). Untargeted secretome analysis also suggests that Fab1 is abundant in culture supernatants (14). Because both Fpn and Fab1 are required for antimicrobial activity, we next examined the impact of Fpn on the production and secretion of the 28-kDa C-terminal fragment of Fab1. Indeed, the Δfpn deletion strain fails to secrete this 28-kDa fragment of Fab1 and instead accumulates the full-length (50-kDa) protein in the cell pellet (Fig. 4A). To determine whether Fpn acts directly on Fab1, we next incubated purified Fab1 with purified Fpn and examined Fab1 processing by Western blotting. Indeed, Fpn directly cleaves recombinant Fab1 to produce a 28-kDa C-terminal fragment (as observed in the supernatant of the BfN parental strain but not the BfN Δfpn mutant); heat deactivation of Fpn or mutating its active site residues abolishes this activity (Fig. 4B). Using liquid chromatography-mass spectrometry (LC-MS), we identified a cleavage site between amino acid residues R200 and A201 of Fab1 that produces the secreted 28-kDa fragment (Fig. S3). Consistent with this prediction, Fab1R200A is not processed into the 28-kDa form upon incubation with Fpn in vivo (Fig. 4A) or in vitro (Fig. 4B). This arginine-alanine sequence in Fab1 is consistent with the primary cleavage site of BFT (R211-A212) targeted by Fpn (25). Fab1 likely contains an additional cleavage site(s), because Fab1R200A is secreted (Fig. 4A) and migrates at a different size than Fab1 controls in which Fpn is absent (Fig. 4B).
FIG 4.
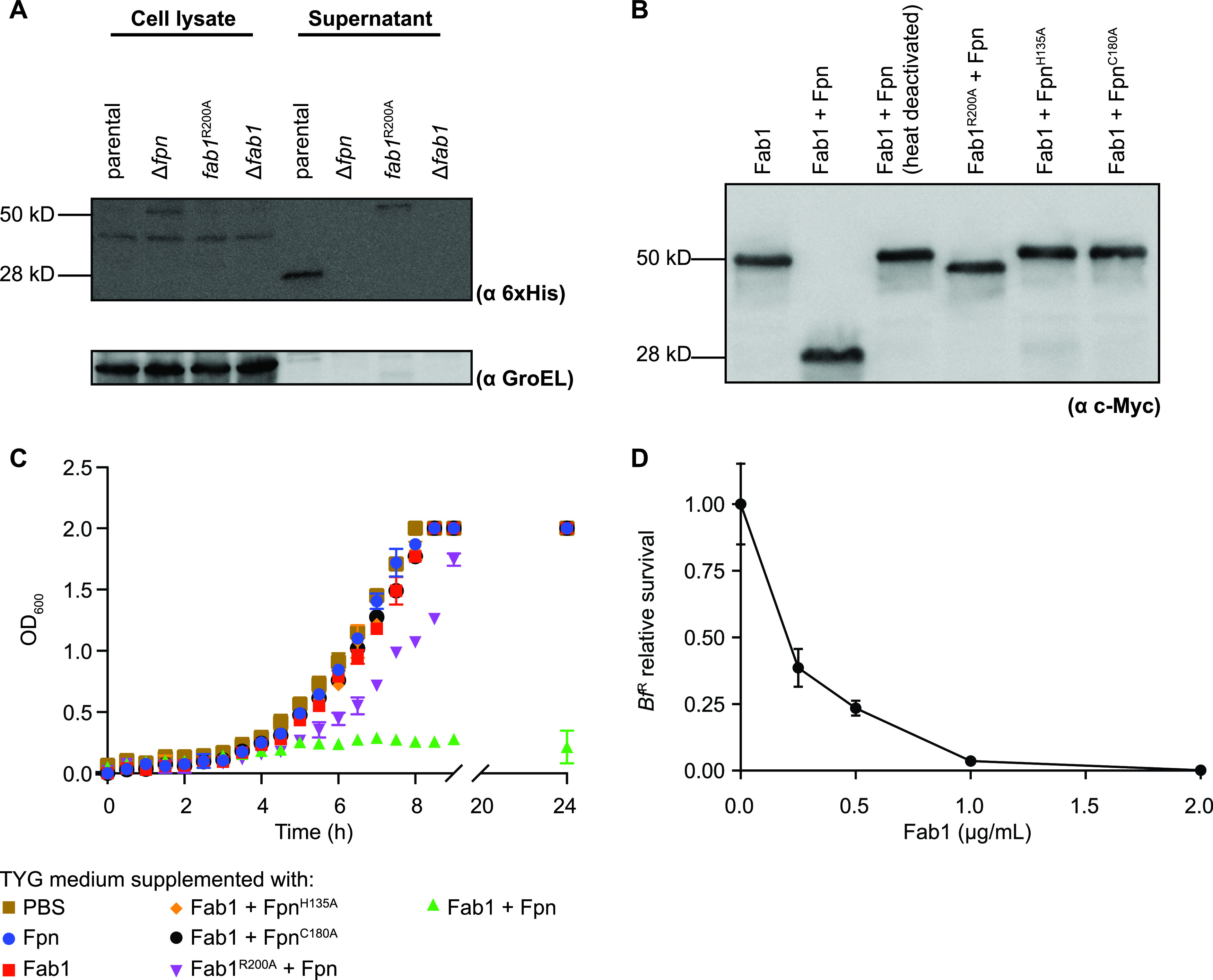
Fpn cleaves Fab1 to activate its antimicrobial function. (A) Western blot analysis of BfN parental and mutant strains carrying Fab1-His6 in the native genomic location and probed with anti-6×His. (B) Western blot analysis of Fab1 products generated following incubation of purified Fpn (wild type or catalytic residue mutants) with purified Fab1 (wild type or cleavage site mutant). Fab1 carries a C-terminal c-Myc tag; Western blots were probed with anti-c-Myc. (C) Growth of the susceptible BfR parental strain in TYG medium supplemented with combinations of purified Fpn (wild type or catalytic residue mutants) and purified Fab1 (wild type or cleavage site mutant). Error bars indicate SD (n = 2; representative of three independent experiments). (D) Concentration dependence of Fab1 activity on BfR viability. Fpn is included at 2 μg/ml under all conditions. Cell viability (CFU/ml) is normalized to the mean viability of cultures incubated in the absence of Fab1. Error bars indicate SD (n = 2; representative of three independent experiments).
Fpn cleaves Fab1 between R200 and A201 to produce the secreted 28-kDa fragment. (A) Fab1 peptides identified by liquid chromatography-mass spectrometry (LC-MS). (B) Comparison of the number of peptide spectrum matches (PSMs) between full-length Fab1 and its Fpn-dependent C-terminal 28-kDa fragment shows that the primary cleavage site of Fab1 is between R200 and A201. Peptide locations A to J are as described in panel A. Download FIG S3, EPS file, 1.7 MB (1.7MB, eps) .
Copyright © 2021 Bao et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
To assess whether Fpn, full-length Fab1, or the Fpn-processed Fab1 is responsible for the observed antibacterial activity, we next grew the susceptible BfR parental strain in the presence of these proteins, either alone or in combination, and determined growth rates of BfR. While neither Fpn nor Fab1 alone inhibited growth, the combination of these proteins exhibited potent antibacterial activity (Fig. 4C). Substitution of Fpn with catalytically inactive mutants or substitution of Fab1 with Fab1R200A blocked toxicity, indicating that processing of Fab1 is required for its antibacterial activity (Fig. 4C). Titrating purified Fab1 indicates an MIC50 of 0.2 μg/ml under these conditions (Fig. 4D). Together, these results suggest that Fab1 is an antibacterial protoxin that is processed by Fpn into a C-terminal 28-kDa form to mediate its secretion and activity. Based on these results, we designated this protein fragipain-activated bacteriocin 1 (Fab1).
Gut microbes use antibacterial antagonism to prevent or delay expansion of invading strains (5). In the genus Bacteroides, invasion is also influenced by the origin of the invading strain: bacterial cultures prepared in vitro have a diminished invasion capacity compared to in vivo-prepared bacteria collected from the feces of monocolonized gnotobiotic mice (3). To examine the role of Fab1 in strain dynamics in the gut environment, we colonized germfree Swiss-Webster (outbred) mice with either the BfN parental or BfNΔfab1 strains and measured the ability of in vivo-prepared BfR parental cells to invade the gastrointestinal tracts of these animals after oral gavage. Invasion was significantly delayed in mice carrying the parental BfN strain compared to mice carrying BfNΔfab1 (Fig. S4A), although the relative abundance of the invading strain in mice carrying BfNΔfab1 varied between 59% and 98% among individual mice by the end of the experiment. This interanimal variability was likely not due to genetic differences in these outbred animals, because repeating these studies in C57BL/6 (inbred) mice did not reduce the observed variability; indeed, the time points at which invading strain abundance was dependent on resident strain genotype differed in separate experiments (Fig. S4B and C). Laboratory-grown BfR exhibited minimal invasion in mice carrying BfN or BfNΔfab1 (Fig. S4D). The BfNΔfab1 mutant does not exhibit a fitness defect in direct competition with its isogenic parental strain in gnotobiotic mice, suggesting that a reduced capacity to delay invasion is not due to generic fitness defect in the gut (Fig. S5).
Impact of Fab1 on invasion of a susceptible strain in gnotobiotic mice. (A to C) Invasion of the BfR parental strain, harvested from monocolonized mice, into gnotobiotic mice carrying parental or Δfab1 mutant BfN strains. (A) Swiss-Webster (outbred) mice; (B to C), C57BL/6 (inbred) mice. (D) Invasion of the BfR parental strain, harvested from laboratory cultures, into gnotobiotic C57BL/6 mice carrying parental or Δfab1 mutant BfN strains. In panels A to D, the abundance of each strain was determined by quantitative PCR using gDNA from fecal samples collected over time. The relative abundance of the BfR parental strain in each group (n = 4 [A], 5 [B and C], and 10 [D]) is shown; error bars indicate SD. *, P < 0.05, **, P < 0.01, ***, P < 0.001, ****, P < 0.0001. Download FIG S4, EPS file, 2.2 MB (2.2MB, eps) .
Copyright © 2021 Bao et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
A fab1 deletion mutant does not have a fitness defect in gnotobiotic mice. BfN parental and Δfab1 mutant strains were introduced into C57BL/6 germfree mice (n = 5). The abundance of each strain was determined by quantitative PCR using gDNA from fecal samples collected over time. Error bars indicate SD. Download FIG S5, EPS file, 2.0 MB (2MB, eps) .
Copyright © 2021 Bao et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Rfab1 protects susceptible strains from Fab1.
Antimicrobial toxins are frequently encoded in tandem with cognate immunity genes that protect against intoxication by sister cells (14, 19, 29). BF9343_1465 (Rfab1), encoded downstream of fab1 (Fig. 2B), has no known function or recognizable domains. Notably, expression of the genetic fragment containing fab1 and rfab1 with their native promoter in the susceptible strain BfR protects this strain from Fpn- and Fab1-mediated killing (Fig. 5). Expression of fab1 alone in BfR fails to confer any protective effect, implicating Rfab1 as an immunity factor. Accordingly, we designated this protein resistance to fragipain-activated bacteriocin 1 (Rfab1).
FIG 5.

Expression of RFab1 protects otherwise susceptible strains from Fab1-mediated antagonism. Expression of the genetic cassette containing fab1 and rfab1, but not fab1 alone, in the susceptible BfR parental strain protects against Fab1-mediated antagonism. Input and output CFU of BfR target strains are reported. Error bars indicate SD (n = 2; representative of three independent experiments). **, P < 0.01; n.s., not significant.
The fab1/rfab1 gene cluster is likely acquired through horizontal gene transfer independent of Fpn.
To assess the potential of diverse B. fragilis strains to utilize this antagonistic pathway, we first searched 92 sequenced B. fragilis genomes (14) for homologs of Fpn. Consistent with its distribution across prominent gut Bacteroides species (30), fpn is conserved in nearly all of these B. fragilis genomes (Fig. 6). These strains (including BfN and the ETBF strain B. fragilis ATCC 43859) generally encode two Fpn homologs, one sharing 70 to 100% identity and the other sharing 30% identity to BfN Fpn. The fpn allele encoded in BfH610 (which has 73% amino acid sequence identity with BfN Fpn) restores antagonistic activity to a BfNΔfpn mutant (Fig. 3C), suggesting that other homologs with 70 to 100% amino acid identity may share this capacity. Because deletion of fpn abolishes Fab1 activity in BfN (Fig. 2D) and BFT activity in Bf ATCC 43859 (25), the second (30% identity) homolog of Fpn likely targets substrates other than Fab1 and BFT (or is nonfunctional); as a result, we did not include it in our analysis.
FIG 6.
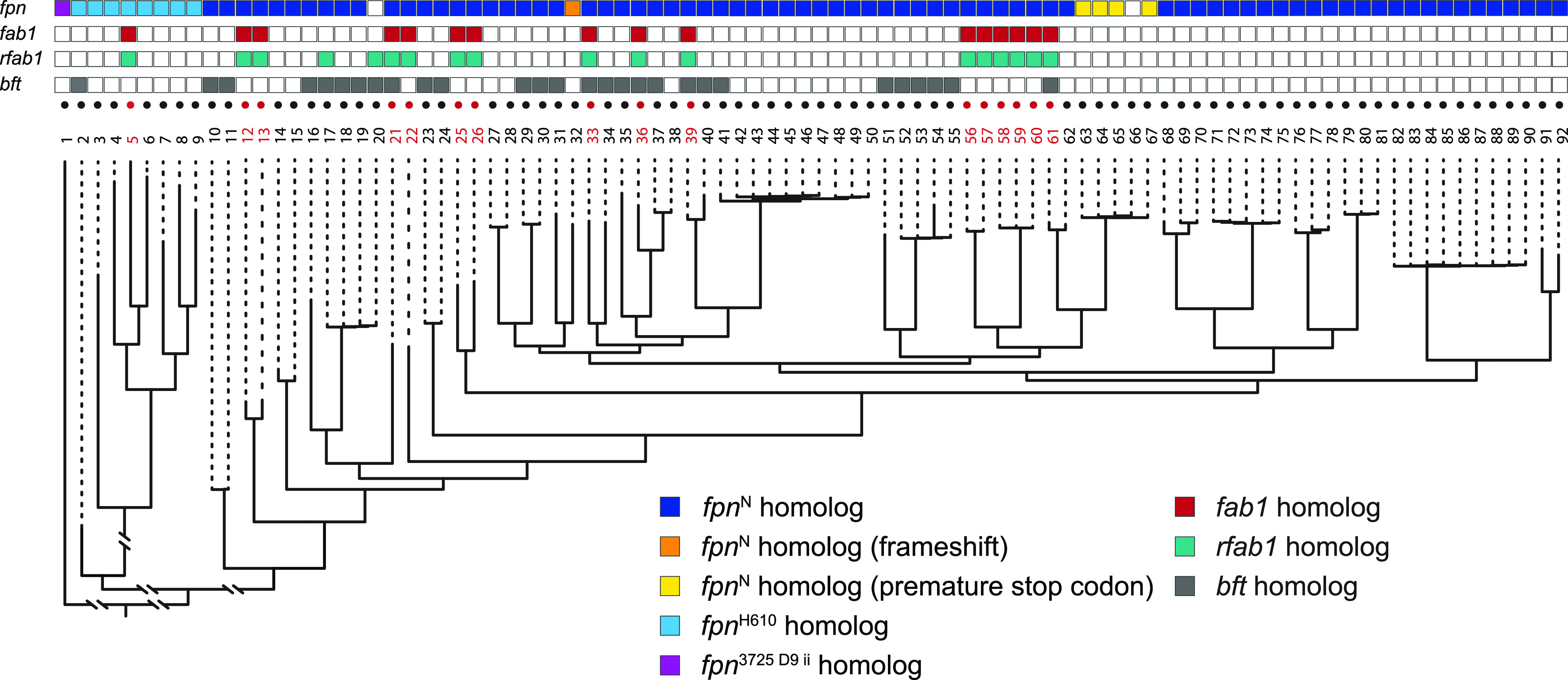
Comparative genomic analysis reveals multiple independent acquisitions of fab1 and rfab1 across B. fragilis strains. B. fragilis strains (numbered 1 to 92; Table S2) that carry a fab1 homolog are in red, and strains with no identified fab1 homolog are in black. (Bottom) Whole-genome phylogeny; (top) presence of fpn (purple, light blue, dark blue, orange, or yellow), fab1 (red), rfab1 (green), or bft (gray). Empty boxes indicate that no homolog was identified.
Ninety-two sequenced strains of B. fragilis analyzed in this study. Download Table S2, XLSX file, 0.01 MB (11.9KB, xlsx) .
Copyright © 2021 Bao et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Analysis of these genomes for fab1 and rfab1 homologs revealed that fab1 is heterogeneously distributed within the B. fragilis phylogeny, consistent with repeated acquisition by horizontal gene transfer. Fab1 is encoded in approximately 20% of analyzed B. fragilis genomes, and its presence is independent of the presence of bft; like bft and many B. fragilis T6SS-dependent effectors, fab1 is rarely identified in genomes outside B. fragilis. Rfab1 is encoded directly downstream of fab1 in all fab1-positive genomes, consistent with a role as a cognate immunity factor for Fab1. Additionally, we identified two strains that encode rfab1 without fab1, suggesting that Rfab1 may also function as an orphan immunity factor that protects these strains from Fab1-mediated killing by other strains. Together, these results suggest that the capacity of a strain to express or resist Fab1-mediated antagonistic activity will not be readily predicted by phylogeny.
DISCUSSION
The gut microbiome harbors enormous bacterial populations (31). These microbes encode a diverse repertoire of contact-dependent and contact-independent mechanisms that determine strain fitness in vitro and in animal models (14, 19). Metagenomic analyses suggest that interbacterial antagonistic mechanisms can also provide a selective advantage in the human gut microbiome (6). However, how contact-independent antibacterial toxins are translocated and released from commensal microbes is not understood.
Human intestinal Bacteroides, specifically certain strains of B. fragilis, also translocate and secrete host-targeting toxins such as BFT, which causes epithelial cell damage in the cecum and colon (22). This predicted lipoprotein is exported to the cell surface; Fpn activity removes the N-terminal BFT prodomain to release the active toxin (26). Notably, ETBF B. fragilis strains also cause BFT-dependent host epithelial damage in the absence of Fpn (25). The observations that Fpn is dispensable for BFT activity in the gut and is encoded in many nontoxigenic strains are consistent with the hypothesis that this protease mediates other functions in human gut Bacteroides. Indeed, a recent secretome analysis of an ETBF strain and its isogenic Δfpn mutant suggests that Fpn is likely involved in releasing many proteins in addition to BFT (26).
In this report, we demonstrate that in addition to its role in activating a host-targeting virulence factor, Fpn also mediates the activation and secretion of the potent antibacterial toxin Fab1. Several B. fragilis strains encode both BFT and Fab1 (Fig. 6), although whether these strains secrete both toxins under the same conditions is unknown. Cleavage by Fpn is important for both the secretion and activity of Fab1 (Fig. 4A and C). Purified Fab1 begins to aggregate after Fpn-mediated cleavage, consistent with a role for the prodomain in folding and stabilization. While the arginine residue at position 200 in Fab1 is required for normal cleavage and antibacterial activity of the protein (Fig. 4A to C), additional cleavage site(s) are likely because Fpn also alters the size of Fab1R200A (Fig. 4B).
Fab1 does not contain any identifiable domains or high-confidence structural predictions, and its molecular target (and which species express this target) is unknown. Rfab1, which also does not contain any identifiable domains, is encoded directly downstream of fab1 in all fab1-carrying strains. This conserved genetic organization and the observation that expression of a fab1-rfab1 cassette (but not fab1 alone) protects an otherwise susceptible strain from Fab1-mediated antagonism suggest that Rfab1 confers immunity against Fab1. Multiple B. fragilis strains that do not carry fab1 do have rfab1 homologs (98% identity with BfN Rfab1) in their genomes, suggesting that this gene has been repeatedly acquired and maintained as an orphan immunity factor to protect against antagonism by Fab1-producing strains. In two independent gnotobiotic experiments using inbred mice and a third experiment using outbred mice, Fab1 expression by BfN significantly delayed the invasion of the Fab1-sensitive strain BfR in the gut. However, the dynamics of the delay were variable between mice (and experiments) and sensitive to the origin of the invading strain. BfN and BfR vary by over 800 genes (32) and BfR exhibits a significant fitness advantage in competition with BfN in vivo (7); it is possible that Fab1 provides a local fitness advantage that is not readily measured in feces or primarily allows BfN to antagonize strains other than BfR.
Notably, the BfNΔfpn deletion mutant exhibits significantly less antibacterial activity than the corresponding Δfab1 deletion mutant (Fig. 2D), suggesting that Fpn may be responsible for the activation of an additional antimicrobial factor(s) in BfN. Because Fab1 lacks sequence similarity to any previously identified bacteriocins, a combination of genetic and proteomic methods may be required to identify additional Fpn-dependent bacteriocins.
Our study adds to the emerging evidence that diverse effector delivery pathways can deliver proteins that target both bacterial and host cells (14, 29, 33–35); selection for pathways that mediate antibacterial antagonism may prepare strains to utilize host-targeting toxins acquired by horizontal gene transfer, and vice versa. Future studies may also resolve whether bacteria- and host-targeting toxins provide a common benefit to the producer, possibly releasing nutrients from susceptible bacteria or host tissues or countering other antagonistic activities from the microbiome or host.
MATERIALS AND METHODS
Bacterial culture conditions.
Bacteroides strains were grown in liquid TYG (tryptone yeast glucose) medium, in liquid minimal medium (14), or on brain heart infusion (BHI; Becton Dickinson) agar supplemented with 50 mg/liter hemin (MP Biomedicals) and 0.5 mg/liter vitamin K3 (MP Biomedicals) in an anaerobic chamber (Coy Laboratory Products) filled with 70% N2, 20% CO2, and 10% H2. Escherichia coli S17-1 lambda pir and BL21 Rosetta strains were grown in LB medium and incubated aerobically at 37°C with shaking at 300 rpm. Antibiotics were added when required at the following concentrations: anhydrotetracycline (aTC), 2 ng/ml; ampicillin, 100 μg/ml; erythromycin, 25 μg/ml; gentamicin, 200 μg/ml; kanamycin, 50 μg/ml; tetracycline, 2 μg/ml; and 5-fluoro-2′-deoxyuridine (FUdR), 200 μg/ml.
Bacteroides genetic manipulations.
All primers used in this study were obtained from the Keck Biotechnology Resource Laboratory (Yale University). DNA amplification was carried out using Kapa HiFi ReadyMix (Kapa Biosystems). The creation, maintenance, and transformation of plasmid constructs were performed using standard molecular cloning procedures. Primer sequences are provided in Table S1. The identity of the BfN and BfR strains used in these studies was confirmed by whole-genome sequencing and comparison to the reference genome sequences (GCA_900445515.1 and GCA_000210835.1).
(i) Deletion of tssC from B. fragilis 638R. The BfRΔtssC strain was constructed using pSIE2 (36). In brief, flanking regions (1,000 to 1,500 bp) of the tssC gene were PCR amplified (Table S1) and assembled with pSIE2 by Gibson cloning to make pSIE2-BF638R_tssC. This plasmid was sequence-verified and transformed into E. coli S17-1 λpir, which was used for conjugation with BfR. Merodiploids were selected by plating on BHI supplemented with gentamicin and erythromycin, and second recombination events were generated by overnight culture in TYG followed by plating on BHI supplemented with aTC as described previously (36). Individual clones were then screened by PCR for deletion of tssC (Table S1).
(ii) Mutant construction in B. fragilis NCTC9343.BfNΔtdk and BfNΔtdkΔtssC mutant strains were previously described (14), and all other BfN mutant strains were constructed using the BfNΔtdkΔtssC parental strain as previously described (37). Briefly, flanking regions (1,000 to 1,500 bp) of genes of interest were PCR amplified (Table S1) and assembled with pExchange-tdk (37) by Gibson assembly. The resulting vectors were sequence verified and cloned into E. coli S17-1 λpir by transformation. Plasmids were then mobilized into BfNΔtdkΔtssC by conjugation. Merodiploids were selected on BHI plates containing gentamicin and erythromycin, grown in liquid TYG to allow generation of second recombination events, and plated onto BHI agar supplemented with FUdR. Gene deletions were verified through PCR (Table S1).
(iii) Genetic complementation. Genes of interest were PCR amplified (Table S1), assembled with pNBU2 vectors (with or without oligonucleotide barcodes) by Gibson assembly, and introduced in single copy into BfN as previously described (38). Fpn and its mutants (FpnH135A and FpnC180A) were cloned downstream of the synthetic promoter P5E4 (39). fab1 and rfab1 were cloned with the predicted endogenous promoter 300 bp upstream of fab1. Integration sites were verified through PCR (Table S1).
Bacterial antagonism studies.
(i) Transposon mutagenesis screen. To create pSAM_BfN, pSAM_Bt (40) was modified by replacing the promoter upstream of the erythromycin resistance gene ermG with the promoter 300 bp upstream of the RpoD (BF9343_2673) gene of BfN. The construct was verified by sequencing and transformed into E. coli S17-1 lambda pir. This strain was used for conjugation with BfN as described elsewhere (40). Clones with transposon insertions were selected on BHI agar with gentamicin and erythromycin and transferred into 96-well plates (termed BfN::TN plates) containing TYG medium with erythromycin using a microbial colony picker (QPix 420; Molecular Devices, San Jose, CA, USA). After 20 h anaerobic incubation at 37°C, an aliquot from each well was individually combined with an equal volume of early-log-phase (optical density at 600 nm [OD600], 0.1) BfH610 culture, and 10 μl of each mixture was spotted in a 96-well format onto nitrocellulose filters on BHI agar plates by robotic liquid handling (epMotion 5075; Eppendorf, Hamburg, Germany). Plates were incubated under anaerobic conditions at 37°C for 20 h, and spots were patched onto BHI agar with gentamicin and tetracycline to select for BfH610. Transposon mutants from corresponding wells in the BfN::TN plates were colony purified on BHI agar containing erythromycin and gentamicin. Colony-purified isolates were then subjected to secondary screening using the procedure described above and transposon insertion site mapping using primers described in Table S1 and a previously described protocol (40).
(ii) Cocultivation assay. The cocultivation assay was conducted largely as described elsewhere (14). Strains were grown on BHI agar plates for 16 to 20 h at 37°C. Bacterial lawns were resuspended from the plates in phosphate-buffered saline (PBS), and cell suspensions were adjusted to an OD600 of 0.1. Cells were mixed at a 1:1 (vol/vol) ratio, and 10 μl of each mixture was spotted onto nitrocellulose squares placed on BHI agar plates. After incubation at 37°C anaerobically for 8 h, viable cells were enumerated by serial dilution and selective plating based on the antibiotic resistance profile of each strain (Table S1). Significant differences were determined by repeated-measures analysis of variance (ANOVA) and post hoc Tukey’s honestly significant difference (HSD) test.
(iii) Supernatant activity assay.(a) Preparation of culture supernatants. To minimize cellular material in supernatant activity assays, supernatant test cultures were initiated from actively growing starter cultures. To this end, supernatant producer strains were inoculated from glycerol stocks into 5 ml minimal medium and grown anaerobically at 37°C for 20 h. These cultures were subcultured (1:1,000), grown as described above to an OD600 of 0.6 to 0.8, and used to initiate test cultures at a starting OD600 of 0.02. These cultures were grown as described above to an OD600 of 0.3 before being pelleted by centrifugation at 3,220 × g for 10 min at 4°C. Supernatants were collected and filtered through a 0.2-μm filter. In certain studies, supernatants were heat treated at 95°C for 40 min or incubated with proteinase K (100 μg/ml) at 37°C for 30 min followed by 65°C for 10 min to inactivate proteinase K. In control samples, untreated supernatants were incubated at 25°C for 40 min.
(b) Preparation of target strain cultures. Target strains were inoculated from glycerol stocks into 5 ml minimal medium and grown anaerobically at 37°C for 20 h. After subculturing (1:1,000), cultures were grown to an OD600 of 0.15 to 0.3 and used to inoculate supernatants or medium controls (prepared as described above) at an OD600 of 0.001. Cultures were incubated at 37°C anaerobically and viable cells enumerated by serial dilution and plating. Significant differences were determined by repeated-measures ANOVA and post hoc Tukey’s HSD test.
(iv) Protein activity assays. For growth inhibition studies, target strains were inoculated from glycerol stocks into 5 ml TYG medium and grown anaerobically at 37°C for 20 h. After 1:1,000 dilution into fresh TYG medium, cultures were grown to an OD600 of 0.15 to 0.3 and used to inoculate TYG medium to a starting OD600 of 0.02. Cultures were supplemented with purified proteins at 20 μg/ml (for controls containing Fpn or Fab1 alone) or Fab1 at 15 μg/ml and Fpn at 5 μg/ml (for test cultures containing both proteins).
For MIC50 determination, the BfR target strain was inoculated from glycerol stocks into 5 ml TYG medium and grown anaerobically at 37°C for 20 h. After 1:250 dilution into fresh TYG medium, cultures were grown to an OD600 of 0.2 to 0.3, and a bacterial pellet corresponding to 1 ml of culture was resuspended in 1× PBS to a final concentration of ∼105 CFU/ml. Recombinant Fab1 was added at final concentrations of 0, 0.25, 0.5, 1, and 2 μg/ml, and Fpn was added at a final concentration of 2 μg/ml at all Fab1 concentrations. The cells were incubated with Fpn and Fab1 at 37°C, anaerobically, for 2 h before plating dilutions on BHI-HK (BHI-hemin-vitamin K) agar to determine CFU/ml.
Protein and molecular assays.
(i) Reverse transcription-quantitative PCR. Cells were harvested at an OD600 of 0.3 with RNA Protect (Qiagen) according to the manufacturer’s instructions. RNA was extracted using the RNeasy kit (Qiagen) and reverse transcribed into cDNA using SuperScript II reverse transcriptase (Thermo Fisher Scientific) with random priming. Quantitative PCR (qPCR) was performed using a CFX96 detection system (Bio-Rad) and SYBR FAST universal master mix (Kapa Biosystems) with primers described in Table S1. Primers were optimized using an Integrated DNA Technologies (IDT) OligoAnalyzer such that primers used together have melting temperatures within ∼2°C. Expression levels were compared by normalizing fpn and BF9343_2672 transcripts to the amount of 16S transcript present in each sample.
(ii) Expression and purification of bacterial proteins.E. coli BL21 Rosetta carrying pET21_NESG expression vectors (38) were used for protein expression and purification. BFT was amplified using published primer sequences (25) and assembled with pET21_NESG using Gibson assembly. For both Fpn and Fab1, N-terminal predicted signal sequences were omitted from the open reading frames and a C-terminal 6×His tag and/or C-terminal c-Myc-tag was added and cloned into pET21_NESG (Table S1).
For protein purification, E. coli expression strains were grown for 20 h, subcultured (1:200), and allowed to grow to an OD600 of 0.4 to 0.6, before induction with IPTG (isopropyl-β-d-thiogalactopyranoside; 0.5 mM) for 4 h. Cells were harvested and lysed using BugBuster reagent (Millipore Sigma, Burlington, MA, USA). Lysates were incubated for 1 h at 4°C with nickel-nitrilotriacetic acid (Ni-NTA) agarose beads (Qiagen, Hilden, Germany), washed with 30 ml of wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole [pH 7.4]), and eluted with 5 ml elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole [pH 7.4]). Fab1 was further purified using a Pierce strong cation exchange column (Thermo Fisher Scientific, Waltham, MA, USA).
(iii) In vitro cleavage of BFT and Fab1. Purified proteins (BFT, Fab1, and Fpn) were dialyzed twice against PBS. Equimolar amounts of wild-type Fpn or mutant variants were incubated with substrates in PBS at 37°C for 30 min followed by deactivation at 95°C for 5 min.
(iv) Immunoblotting. Proteins were separated by SDS-PAGE and transferred onto a polyvinylidene difluoride (PVDF) membrane (Bio-Rad). Membranes were blocked with 5% nonfat milk in PBS with 0.1% Tween 20 (PBST). Primary antibodies, including anti-C-myc mouse monoclonal antibody (Invitrogen) and anti-His6 mouse monoclonal antibody (Invitrogen), were diluted in 5% nonfat milk in PBST at 1:1,000; secondary antibodies were diluted in PBST at 1:10,000.
Gnotobiotic animal studies.
All animal experiments were performed using protocols approved by the Yale University Institutional Animal Care and Use Committee. Male and female germfree 10- to 16-week-old Swiss Webster or C57BL/6 mice were individually caged and maintained in flexible plastic gnotobiotic isolators with a 12-h light/dark cycle. Mice were provided with standard autoclaved mouse chow (5K67 LabDiet; Purina, St. Louis, MO, USA) ad libitum.
(i) BfN competition.BfN parental and BfNΔfab1 strains were stored as single-use aliquots in 10% glycerol at −80°C. After CFU quantification from representative aliquots of each strain, strains were thawed, mixed, and introduced into germfree mice by oral gavage at a starting ratio of 1:1 (5 × 108 CFU:5 × 108 CFU).
(ii) BfR invasion experiments. Invasion experiments were conducted using two previously described protocols (3, 7). In the first approach, germfree mice were monoassociated (day −7) with 109 CFU of either barcoded parental BfN (n = 5) or barcoded BfNΔfab1 (n = 5) by oral gavage. One mouse was monoassociated with 109 CFU of the barcoded BfR parental strain. On day −3, a fecal sample was collected from the BfR monoassociated mouse, and viable cells were enumerated by serial dilution and plating. On day 0, BfN-monoassociated mice were subjected to gavage with fecal material from the BfR-monoassociated mouse (108 CFU, determined based on the CFU assessment from day −3). In the second approach, germfree mice were monoassociated (day −7) with 109 CFU of either barcoded parental BfN (n = 5) or barcoded BfNΔfab1 (n = 5) by oral gavage. Stationary-phase BfR cultures were resuspended in PBS with 20% glycerol and stored in aliquots at −80°C prior to CFU determination. On day 0, BfN-monoassociated mice were subjected to gavage with 108 CFU of these in vitro-grown BfR cultures. For all mouse experiments, fecal samples were collected over time and stored at −20°C before genomic DNA extraction. Total gDNA was extracted, and the relative abundance of each strain was determined by qPCR using a CFX96 detection system (Bio-Rad), SYBR FAST universal master mix (Kapa Biosystems), and oligonucleotide barcode-specific primers (Table S1) (41). Significant (P < 0.05) differences were determined by Student's t test.
Genomic analysis.
(i) Genome phylogeny. The phylogenetic tree of B. fragilis strains (listed in Table S2) is adapted from a previous study (14). In that study, these genomes (many of which are in draft stage) were also queried for the presence of 14 housekeeping genes conserved across all bacteria; for each genome, 14/14 of these genes were successfully identified (14).
Protein homolog identifications were conducted through BLASTp using the PATRIC database (42) as described below.
(ii) Identification of fpn homologs. The presence/absence of fpn was identified by BLASTp search using an E-value cutoff of 9e−43. fpn homologs were manually categorized into groups based on reciprocal BLAST using an identity cutoff of 75% and screened for mutations.
(iii) Identification of bft homologs. The presence/absence of bft was identified by BLASTp search for the 186-residue active fragment (43) of translated bft from strain ATCC 43859 using an E-value cutoff of 2e−35 and identity cutoff of 87%.
(iv) Identification of fab1 homologs. The presence/absence of fab1 was identified by BLASTp search using an E-value cutoff of 0.0 and identity cutoff of 60%.
(v) Identification of rfab1 homologs. The presence/absence of rfab1 was identified by BLASTp search using an E-value cutoff of 4e−18 and identity cutoff of 95%.
ACKNOWLEDGMENTS
We thank Bentley Lim and other members of the Goodman lab for helpful discussions. We also thank Jingyao Qu and Zhifeng Li from State Key Laboratory of Microbial Technology of Shandong University for help and guidance in LC-MS.
This work was supported by NIH grants GM118159, the HHMI Faculty Scholars Program, and the Burroughs Wellcome Fund to A.L.G. and the Taishan Young Scholars Program (tsqn20161005) to X.G.
Footnotes
Citation Bao Y, Verdegaal AA, Anderson BW, Barry NA, He J, Gao X, Goodman AL. 2021. A common pathway for activation of host-targeting and bacteria-targeting toxins in human intestinal bacteria. mBio 12:e00656-21. https://doi.org/10.1128/mBio.00656-21.
Contributor Information
Andrew L. Goodman, Email: andrew.goodman@yale.edu.
Joerg Graf, University of Connecticut.
REFERENCES
- 1.Libertucci J, Young VB. 2019. The role of the microbiota in infectious diseases. Nat Microbiol 4:35–45. doi: 10.1038/s41564-018-0278-4. [DOI] [PubMed] [Google Scholar]
- 2.Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL, Clemente JC, Knight R, Heath AC, Leibel RL, Rosenbaum M, Gordon JI. 2013. The long-term stability of the human gut microbiota. Science 341:1237439. doi: 10.1126/science.1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee SM, Donaldson GP, Mikulski Z, Boyajian S, Ley K, Mazmanian SK. 2013. Bacterial colonization factors control specificity and stability of the gut microbiota. Nature 501:426–429. doi: 10.1038/nature12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shepherd ES, DeLoache WC, Pruss KM, Whitaker WR, Sonnenburg JL. 2018. An exclusive metabolic niche enables strain engraftment in the gut microbiota. Nature 557:434–438. doi: 10.1038/s41586-018-0092-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.García-Bayona L, Comstock LE. 2018. Bacterial antagonism in host-associated microbial communities. Science 361:eaat2456. doi: 10.1126/science.aat2456. [DOI] [PubMed] [Google Scholar]
- 6.Verster AJ, Ross BD, Radey MC, Bao Y, Goodman AL, Mougous JD, Borenstein E. 2017. The landscape of type VI secretion across human gut microbiomes reveals its role in community composition. Cell Host Microbe 22:411–419.E4. doi: 10.1016/j.chom.2017.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ross BD, Verster AJ, Radey MC, Schmidtke DT, Pope CE, Hoffman LR, Hajjar AM, Peterson SB, Borenstein E, Mougous JD. 2019. Human gut bacteria contain acquired interbacterial defence systems. Nature 575:224–228. doi: 10.1038/s41586-019-1708-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aoki SK, Pamma R, Hernday AD, Bickham JE, Braaten BA, Low DA. 2005. Contact-dependent inhibition of growth in Escherichia coli. Science 309:1245–1248. doi: 10.1126/science.1115109. [DOI] [PubMed] [Google Scholar]
- 9.Navarro-Garcia F, Ruiz-Perez F, Cataldi A, Larzábal M. 2019. Type VI secretion system in pathogenic Escherichia coli: structure, role in virulence, and acquisition. Front Microbiol 10:01965. doi: 10.3389/fmicb.2019.01965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sassone-Corsi M, Nuccio S-P, Liu H, Hernandez D, Vu CT, Takahashi AA, Edwards RA, Raffatellu M. 2016. Microcins mediate competition among Enterobacteriaceae in the inflamed gut. Nature 540:280–283. doi: 10.1038/nature20557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cascales E, Buchanan SK, Duché D, Kleanthous C, Lloubès R, Postle K, Riley M, Slatin S, Cavard D. 2007. Colicin biology. Microbiol Mol Biol Rev 71:158–229. doi: 10.1128/MMBR.00036-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corr SC, Li Y, Riedel CU, O'Toole PW, Hill C, Gahan CGM. 2007. Bacteriocin production as a mechanism for the antiinfective activity of Lactobacillus salivarius UCC118. Proc Natl Acad Sci USA 104:7617–7621. doi: 10.1073/pnas.0700440104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cotter PD, Ross RP, Hill C. 2013. Bacteriocins—a viable alternative to antibiotics? Nat Rev Microbiol 11:95–105. doi: 10.1038/nrmicro2937. [DOI] [PubMed] [Google Scholar]
- 14.Wexler AG, Bao Y, Whitney JC, Bobay L-M, Xavier JB, Schofield WB, Barry NA, Russell AB, Tran BQ, Goo YA, Goodlett DR, Ochman H, Mougous JD, Goodman AL. 2016. Human symbionts inject and neutralize antibacterial toxins to persist in the gut. Proc Natl Acad Sci USA 113:3639–3644. doi: 10.1073/pnas.1525637113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hecht AL, Casterline BW, Earley ZM, Goo YA, Goodlett DR, Bubeck Wardenburg J. 2016. Strain competition restricts colonization of an enteric pathogen and prevents colitis. EMBO Rep 17:1281–1291. doi: 10.15252/embr.201642282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coyne MJ, Comstock LE. 2019. Type VI secretion systems and the gut microbiota. Microbiol Spectr 7 doi: 10.1128/microbiolspec.PSIB-0009-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coyne MJ, Béchon N, Matano LM, McEneany VL, Chatzidaki-Livanis M, Comstock LE. 2019. A family of anti-Bacteroidales peptide toxins wide-spread in the human gut microbiota. Nat Commun 10:3460. doi: 10.1038/s41467-019-11494-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chatzidaki-Livanis M, Coyne MJ, Comstock LE. 2014. An antimicrobial protein of the gut symbiont Bacteroides fragilis with a MACPF domain of host immune proteins. Mol Microbiol 94:1361–1374. doi: 10.1111/mmi.12839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roelofs KG, Coyne MJ, Gentyala RR, Chatzidaki-Livanis M, Comstock LE. 2016. Bacteroidales secreted antimicrobial proteins target surface molecules necessary for gut colonization and mediate competition in vivo. mBio 7:e01055-16. doi: 10.1128/mBio.01055-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chatzidaki-Livanis M, et al. 2017. Gut symbiont Bacteroides fragilis secretes a eukaryotic-like ubiquitin protein that mediates intraspecies antagonism. mBio 8:e01902-17. doi: 10.1128/mBio.01902-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shumaker AM, Laclare McEneany V, Coyne MJ, Silver PA, Comstock LE. 2019. Identification of a fifth antibacterial toxin produced by a single Bacteroides fragilis strain. J Bacteriol 201:e00577-18. doi: 10.1128/JB.00577-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sears CL. 2009. Enterotoxigenic Bacteroides fragilis: a rogue among symbiotes. Clin Microbiol Rev 22:349–369. doi: 10.1128/CMR.00053-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. 2005. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 122:107–118. doi: 10.1016/j.cell.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 24.Wu S, Lim KC, Huang J, Saidi RF, Sears CL. 1998. Bacteroides fragilis enterotoxin cleaves the zonula adherens protein, E-cadherin. Proc Natl Acad Sci USA 95:14979–14984. doi: 10.1073/pnas.95.25.14979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi VM, Herrou J, Hecht AL, Teoh WP, Turner JR, Crosson S, Bubeck Wardenburg J. 2016. Activation of Bacteroides fragilis toxin by a novel bacterial protease contributes to anaerobic sepsis in mice. Nat Med 22:563–567. doi: 10.1038/nm.4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pierce JV, Fellows JD, Anderson DE, Bernstein HD. 2020. A clostripain-like protease plays a major role in generating the secretome of enterotoxigenic Bacteroides fragilis. Mol Microbiol doi: 10.1111/mmi.14616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McEneany VL, Coyne MJ, Chatzidaki-Livanis M, Comstock LE. 2018. Acquisition of MACPF domain-encoding genes is the main contributor to LPS glycan diversity in gut Bacteroides species. ISME J 12:2919–2928. doi: 10.1038/s41396-018-0244-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herrou J, Choi VM, Bubeck Wardenburg J, Crosson S. 2016. Activation mechanism of the Bacteroides fragilis cysteine peptidase, fragipain. Biochemistry 55:4077–4084. doi: 10.1021/acs.biochem.6b00546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chatzidaki-Livanis M, Geva-Zatorsky N, Comstock LE. 2016. Bacteroides fragilis type VI secretion systems use novel effector and immunity proteins to antagonize human gut Bacteroidales species. Proc Natl Acad Sci USA 113:3627–3632. doi: 10.1073/pnas.1522510113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roncase EJ, Gonzalez-Paez GE, Wolan DW. 2019. X-ray structures of two Bacteroides thetaiotaomicron C11 proteases in complex with peptide-based inhibitors. Biochemistry 58:1728–1737. doi: 10.1021/acs.biochem.9b00098. [DOI] [PubMed] [Google Scholar]
- 31.Rinninella E, Raoul P, Cintoni M, Franceschi F, Miggiano GAD, Gasbarrini A, Mele MC. 2019. What is the healthy gut microbiota composition? A changing ecosystem across age, environment, diet, and diseases. Microorganisms 7:14. doi: 10.3390/microorganisms7010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Avram O, Rapoport D, Portugez S, Pupko T. 2019. M1CR0B1AL1Z3R—a user-friendly web server for the analysis of large-scale microbial genomics data. Nucleic Acids Res 47:W88–W92. doi: 10.1093/nar/gkz423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mougous JD, Cuff ME, Raunser S, Shen A, Zhou M, Gifford CA, Goodman AL, Joachimiak G, Ordoñez CL, Lory S, Walz T, Joachimiak A, Mekalanos JJ. 2006. A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science 312:1526–1530. doi: 10.1126/science.1128393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pukatzki S, Ma AT, Sturtevant D, Krastins B, Sarracino D, Nelson WC, Heidelberg JF, Mekalanos JJ. 2006. Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc Natl Acad Sci USA 103:1528–1533. doi: 10.1073/pnas.0510322103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bayer-Santos E, Cenens W, Matsuyama BY, Oka GU, Di Sessa G, Mininel IDV, Alves TL, Farah CS. 2019. The opportunistic pathogen Stenotrophomonas maltophilia utilizes a type IV secretion system for interbacterial killing. PLoS Pathog 15:e1007651. doi: 10.1371/journal.ppat.1007651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bencivenga-Barry NA, Lim B, Herrera CM, Trent MS, Goodman AL. 2020. Genetic manipulation of wild human gut Bacteroides. J Bacteriol 202:e00544-19. doi: 10.1128/JB.00544-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koropatkin NM, Martens EC, Gordon JI, Smith TJ. 2008. Starch catabolism by a prominent human gut symbiont is directed by the recognition of amylose helices. Structure 16:1105–1115. doi: 10.1016/j.str.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wexler AG, et al. 2018. Human gut Bacteroides capture vitamin B12 via cell surface-exposed lipoproteins. Elife 7:e37138., doi: 10.7554/eLife.37138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zimmermann M, Zimmermann-Kogadeeva M, Wegmann R, Goodman AL. 2019. Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature 570:462–467. doi: 10.1038/s41586-019-1291-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goodman AL, McNulty NP, Zhao Y, Leip D, Mitra RD, Lozupone CA, Knight R, Gordon JI. 2009. Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe 6:279–289. doi: 10.1016/j.chom.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Degnan PH, Barry NA, Mok KC, Taga ME, Goodman AL. 2014. Human gut microbes use multiple transporters to distinguish vitamin B12 analogs and compete in the gut. Cell Host Microbe 15:47–57. doi: 10.1016/j.chom.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davis JJ, Wattam AR, Aziz RK, Brettin T, Butler R, Butler RM, Chlenski P, Conrad N, Dickerman A, Dietrich EM, Gabbard JL, Gerdes S, Guard A, Kenyon RW, Machi D, Mao C, Murphy-Olson D, Nguyen M, Nordberg EK, Olsen GJ, Olson RD, Overbeek JC, Overbeek R, Parrello B, Pusch GD, Shukla M, Thomas C, VanOeffelen M, Vonstein V, Warren AS, Xia F, Xie D, Yoo H, Stevens R. 2020. The PATRIC Bioinformatics Resource Center: expanding data and analysis capabilities. Nucleic Acids Res 48:D606–D612. doi: 10.1093/nar/gkz943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chung GT, Franco AA, Wu S, Rhie GE, Cheng R, Oh HB, Sears CL. 1999. Identification of a third metalloprotease toxin gene in extraintestinal isolates of Bacteroides fragilis. Infect Immun 67:4945–4949. doi: 10.1128/IAI.67.9.4945-4949.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
B. thetaiotaomicron (Bt) is resistant to the antimicrobial protein(s) produced by BfN. B. thetaiotaomicron CFU after exposure to BfN parental culture supernatant or minimal medium with different treatments are reported. Error bars indicate SD (n = 2; representative of three independent experiments). Download FIG S1, EPS file, 1.9 MB (1.9MB, eps) .
Copyright © 2021 Bao et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Bacterial strains, plasmids, and oligonucleotide primers used in this study. Download Table S1, XLSX file, 0.02 MB (19.6KB, xlsx) .
Copyright © 2021 Bao et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Transposon insertion in the intergenic region between BF9343_2672 and fpn significantly reduces fpn gene expression. Expression of fpn and BF9343_2672 in the mutant strain carrying a transposon insertion in the intergenic region upstream of fpn (Tn1) (Fig. 2B) relative to the BfN parental strain was determined by reverse transcription-qPCR (qRT-PCR). Mean expression levels are calculated from technical triplicates from two independent experiments and normalized to 16S RNA. Error bars indicate SD. Download FIG S2, EPS file, 1.9 MB (1.9MB, eps) .
Copyright © 2021 Bao et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Fpn cleaves Fab1 between R200 and A201 to produce the secreted 28-kDa fragment. (A) Fab1 peptides identified by liquid chromatography-mass spectrometry (LC-MS). (B) Comparison of the number of peptide spectrum matches (PSMs) between full-length Fab1 and its Fpn-dependent C-terminal 28-kDa fragment shows that the primary cleavage site of Fab1 is between R200 and A201. Peptide locations A to J are as described in panel A. Download FIG S3, EPS file, 1.7 MB (1.7MB, eps) .
Copyright © 2021 Bao et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Impact of Fab1 on invasion of a susceptible strain in gnotobiotic mice. (A to C) Invasion of the BfR parental strain, harvested from monocolonized mice, into gnotobiotic mice carrying parental or Δfab1 mutant BfN strains. (A) Swiss-Webster (outbred) mice; (B to C), C57BL/6 (inbred) mice. (D) Invasion of the BfR parental strain, harvested from laboratory cultures, into gnotobiotic C57BL/6 mice carrying parental or Δfab1 mutant BfN strains. In panels A to D, the abundance of each strain was determined by quantitative PCR using gDNA from fecal samples collected over time. The relative abundance of the BfR parental strain in each group (n = 4 [A], 5 [B and C], and 10 [D]) is shown; error bars indicate SD. *, P < 0.05, **, P < 0.01, ***, P < 0.001, ****, P < 0.0001. Download FIG S4, EPS file, 2.2 MB (2.2MB, eps) .
Copyright © 2021 Bao et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
A fab1 deletion mutant does not have a fitness defect in gnotobiotic mice. BfN parental and Δfab1 mutant strains were introduced into C57BL/6 germfree mice (n = 5). The abundance of each strain was determined by quantitative PCR using gDNA from fecal samples collected over time. Error bars indicate SD. Download FIG S5, EPS file, 2.0 MB (2MB, eps) .
Copyright © 2021 Bao et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Ninety-two sequenced strains of B. fragilis analyzed in this study. Download Table S2, XLSX file, 0.01 MB (11.9KB, xlsx) .
Copyright © 2021 Bao et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.