

Abstract
Fusobacterium nucleatum (Fn) is a Gram‐negative oral commensal, prevalent in various human diseases. It is unknown how this common commensal converts to a rampant pathogen. We report that Fn secretes an adhesin (FadA) with amyloid properties via a Fap2‐like autotransporter to enhance its virulence. The extracellular FadA binds Congo Red, Thioflavin‐T, and antibodies raised against human amyloid β42. Fn produces amyloid‐like FadA under stress and disease conditions, but not in healthy sites or tissues. It functions as a scaffold for biofilm formation, confers acid tolerance, and mediates Fn binding to host cells. Furthermore, amyloid‐like FadA induces periodontal bone loss and promotes CRC progression in mice, with virulence attenuated by amyloid‐binding compounds. The uncleaved signal peptide of FadA is required for the formation and stability of mature amyloid FadA fibrils. We propose a model in which hydrophobic signal peptides serve as “hooks” to crosslink neighboring FadA filaments to form a stable amyloid‐like structure. Our study provides a potential mechanistic link between periodontal disease and CRC and suggests anti‐amyloid therapies as possible interventions for Fn‐mediated disease processes.
Keywords: amyloid, colorectal cancer, Fusobacterium nucleatum, FadA, periodontal disease
Subject Categories: Cancer; Microbiology, Virology & Host Pathogen Interaction; Molecular Biology of Disease
Fusobacterium nucleatum, an oral commensal anaerobe, secretes amyloid‐like FadA to enhance its pathogenicity. Intact pre‐FadA is a critical component of amyloid‐like FadA, which is secreted via the autotransporter Fap2.

Introduction
Fusobacterium nucleatum (Fn) is a filamentous Gram‐negative anaerobe ubiquitous in the oral cavity. As an opportunistic commensal, it is the most predominant core component in the subgingival microbiome in both health and disease. Outside the oral cavity, Fn is absent or infrequently detected under healthy conditions (Segata et al, 2012). Under disease conditions, however, Fn is one of the most prevalent species involved in organ abscesses, atherosclerosis, pregnancy complications, rheumatoid arthritis, respiratory tract infections, and GI disorders, e.g., appendicitis, inflammatory bowel disease, esophageal, gastric, pancreatic, and colorectal cancers (CRC) (Han et al, 2009; Ortiz et al, 2009; Han et al, 2010; Strauss et al, 2011; Swidsinski et al, 2011; Castellarin et al, 2012; Kostic et al, 2012; Wang et al, 2013; Mitsuhashi et al, 2015; Hsieh et al, 2018; Liu et al, 2019; Thomas et al, 2019). It is not known how Fn acts both as a common commensal and a rampant pathogen. Understanding the molecular signal and mechanism is critical for controlling Fn pathogenesis.
Several lines of evidence implicate that the FadA adhesin (for Fusobacterium adhesin A) plays a critical role in the pathogenicity of Fn. FadA is conserved among Fn, Fusobacterium periodonticum and Fusobacterium necrophorum, but absent in most other Fusobacteria species (Han et al, 2005; Umana et al, 2019). In human colonic tissues, the fadA gene levels increase stepwise from normal to adenoma and from adenoma to carcinoma (Rubinstein et al, 2013). Meta‐analysis of diverse populations showed that fadA is also consistently enriched in the fecal microbiome of CRC patients (Wirbel et al, 2019). The pathogenesis mechanisms of FadA in CRC and pregnancy complications have been reported previously (Ikegami et al, 2009; Rubinstein et al, 2013; Rubinstein et al, 2019). FadA mediates Fn binding and invasion of epithelial and endothelial cells and colonization in the murine placenta (Han et al, 2005; Xu et al, 2007; Ikegami et al, 2009; Fardini et al, 2011). It binds VE‐cadherin on endothelial cells, loosening of the cell–cell junctions enabling systemic bacterial dissemination (Ikegami et al, 2009; Fardini et al, 2011). It also enables Fn to preferentially bind CRC cells expressing Annexin A1, a β‐catenin modulator required for CRC cell growth. Upon binding to CRC cells, FadA further elevates Annexin A1 expression. This positive feedback loop between FadA and Annexin A1 exacerbates CRC progression (Rubinstein et al, 2019). While the implication of FadA in various pathologies is well documented, how this virulence factor functions in health and disease is not known. In this study, we report that FadA undergoes dramatic biochemical changes to become “amyloid‐like” to enhance Fn virulence.
Amyloids are fibrous protein aggregates that are implicated in numerous human diseases such as Alzheimer’s, Parkinson’s, and prion diseases. A related, but biochemically distinct, group of protein aggregates is known as ‘amyloid‐like’. This class shares a subset of biochemical properties of the disease‐related amyloids including fiber formation, stable cross‐beta sheets, and/or resistance to ionic detergents (Berchowitz et al, 2015; Boke et al, 2016). FadA readily forms fibers in vitro with a diameter similar to well‐studied amyloids (Nithianantham et al, 2009). These preliminary data led us to speculate that FadA forms functional amyloid‐like assemblies and that the formation of these structures is critical for Fn pathogenesis.
Results
Fn produces amyloid‐like FadA regulated by growth phase
A distinctive property of amyloid and amyloid‐like assemblies is their ability to bind amyloidophilic compounds such as Congo Red and Thioflavin‐T (Tukel et al, 2009; Evans et al, 2018). To determine whether Fn expresses amyloid‐like FadA, we assessed whether Fn and its mutants bind Congo Red. Wild‐type Fn 12230 clearly bound Congo Red, depleting it from the solution in a density‐dependent manner, while the fadA‐deletion mutant US1 (ΔfadA) was defective (Fig 1A). In order to compare with the well‐characterized bacterial amyloid‐like adhesin curli (Evans et al, 2018), wild‐type curli‐producing E. coli and curli mutants were tested in the same assay (Fig 1A). The similarities observed between Fn and E. coli support that FadA is an amyloid‐like adhesin.
Figure 1. Fn produces amyloid‐like FadA in stationary phase.

-
ACongo Red depletion assay. Fn 12230 (black), the fadA‐deletion mutant US1 (ΔfadA) (orange), and the spontaneous lam mutant (blue) (top panel) were grown to OD600 > 0.8. E. coli MC4100, Δcurli, and ΔcsgA (bottom panel) were grown on TSA‐blood agar plate at 26°C for 48 h. The bacteria were suspended in PBS to OD600 of 0.5, 1.0, 1.5, and 2.0, followed by incubation in 10 μg/ml Congo Red (CR) for 10 min. After centrifugation, the supernatants were measured at OD500. The results shown are the average of five independent experiments, each performed in duplicate. The error bars indicate SD. *P < 0.05, **P < 0.01, ***P < 0.001 (compared to wild type, t‐test).
-
BKinetics of FadA production in Fn. Fn 12230 and US1 were inoculated to OD600 = 0.1. 10 OD600 units of bacteria were harvested by centrifugation at 12‐h intervals. Bacteria were lysed with 2 mg/ml lysozyme, followed by incubation in 1% sarkosyl at 4°C for 20 min, which became the ‘total’ sample. Lysates were then centrifuged at 100,000 g for 20 min, and the supernatants and pellets were collected. An aliquot of 5 μl is loaded onto each lane, followed by immunoblot using anti‐FadA monoclonal antibody (mAb) 7H7 at 1:4,000 dilution to detect FadA protein in total, supernatant, and pellet fractions. Ponceau staining of lysozyme is shown as a loading control. Pre‐FadA and mFadA are pointed by arrows. Note pre‐FadA migrates faster than mFadA on SDS–PAGE as previously reported (Xu et al, 2007).
-
CAnalysis of detergent‐resistant FadA polymers in wild‐type Fn and its mutants by semi‐denaturing detergent agarose gel electrophoresis (SDD–AGE) and Western blot analysis using mAb 7H7. Fn12230 (wt), fadA‐deletion mutant US1 (ΔfadA) and spontaneous mutant lam (lam) were grown to log (OD600 0.3–0.4) or stationary phase (OD600 > 0.8), E. coli MC4100, Δcurli, and ΔcsgA were grown to stationary phase. The bacteria were harvested by centrifugation. Following sequential incubation in lysis buffer containing 2 mg/ml lysozyme and 1% sarkosyl, the insoluble pellets were obtained by centrifugation. An aliquot of 100 μg of each pellet was loaded onto 1.7% agarose gel followed by overnight electrophoresis in 0.5xTAE and 0.1% SDS. Following transfer to nitrocellulose membrane, Western blot was performed using anti‐FadA mAb 7H7 at 1:4,000 dilution or anti‐CsgA antibody at 1:15,000 dilution. The large heterogeneous detergent‐resistant FadA polymers were detected in wild‐type Fn in the stationary phase, defective in US1 and lam, absent in log phase.
-
DImmunohistochemical analysis of FadA in log and stationary phases. Fn12230 (Fn), fadA‐deletion mutant US1 (ΔfadA), and spontaneous mutant lam in log and stationary phase were fixed and incubated with mAb 7H7 at 1:800 dilution, followed by incubation of HRP‐conjugated anti‐mouse IgG and developed by DAB. The large FadA aggregates were detected specifically in wild‐type Fn 12230 in the stationary but not in log phase. FadA was detected on lam, which was defective in secreted aggregates. No FadA was detected in US1. The images were taken using a 40× objective. Scale bar equals 20 μm.
-
EDouble immunofluorescent staining of Fn12230, fadA‐deletion mutant US1 (ΔfadA) and spontaneous mutant lam in log and stationary phases using mAb 7H7 at 1:800 dilution and polyclonal anti‐human β‐amyloid antibody A11 at 1:500 dilution, followed by incubation with Alexa Fluor 680‐conjugated donkey anti‐rabbit and Alexa Fluor 555‐conjugated goat anti‐mouse, both at 1:1,000 dilution. Co‐staining of FadA and A11 was observed in Fn in stationary phase, not in log phase, or in US1 or lam. The images were taken using a 60X objective. Scale bar equals 5 μm.
-
FScanning electron microscopy of Fn 12230, US1 (ΔfadA) and lam in log and stationary phase at 10,000× magnification. Note the fibrous structure coating Fn in stationary phase, pointed by the clear arrows, but not in log phase. Scale bar equals 1 μm.
Source data are available online for this figure.
Because Congo Red was depleted in a bacterial density‐dependent manner, we conducted a kinetic analysis to examine production of amyloid‐like FadA over the course of growth. Fn cultures were harvested and lysed at increasing time points following subculture and incubated with 1% sarkosyl. Resistance to ionic detergent is a characteristic of amyloid‐like proteins (Sondheimer & Lindquist, 2000). Sarkosyl‐resistant (i.e., amyloid‐like) FadA (pellet) increased over time, and by 48 h (late stationary phase), FadA was only detected in the pellet but not in supernatant, indicating the existence of FadA as predominantly insoluble aggregates (Fig 1B). [Note that on SDS–PAGE, pre‐FadA migrates faster than mFadA (Xu et al, 2007).] This result indicates that although FadA is constitutively expressed in Fn, its molecular characteristics change during growth.
To examine the production of amyloid‐like FadA aggregates, we employed semi‐denaturing detergent agarose gel electrophoresis (SDD–AGE), followed by Western blot analysis using anti‐FadA monoclonal antibody (mAb) 7H7. Amyloid‐like (as opposed to amorphous or globular) assemblies are SDS‐resistant, and SDD‐AGE allows for the resolution of these structures based on their size and resistance to the ionic detergent SDS. Fn produced massive and heterogeneously‐sized amyloid‐like FadA assemblies in stationary phase, similar as the curli‐producing E. coli, but not in log phase (Fig 1C). This observation suggests FadA assembly could be regulated by nutrient depletion, a feature observed with other amyloid‐like proteins (Berchowitz et al, 2015).
Image analyses were then conducted to visually examine the FadA aggregates. Increased secretion of FadA aggregates by wild‐type Fn was detected by immunohistochemistry (IHC) in stationary phase compared to log phase (Fig 1D). To further examine the amyloid‐like properties of extracellular FadA aggregates, double‐immunofluorescent staining was performed using mAb 7H7 and polyclonal anti‐human β‐amyloid antibody A11, which specifically recognizes amyloid oligomers independent of amino‐acid (aa) sequence. Co‐staining between A11 and FadA was observed in stationary phase but not in log phase of wild‐type Fn (Fig 1E). Scanning electron microscopy (SEM) confirmed the existence of extracellular “plaque‐like” fibrils coating wild‐type Fn in stationary phase, but not in log phase, which were significantly reduced in US1 (ΔfadA) (Fig 1F). Together, these data confirm that Fn produces amyloid‐like FadA as the bacterium enters stationary phase, possibly in response to nutrient deprivation.
Pre‐FadA is a key component of amyloid‐like FadA secreted by Fap2‐like Type V autotransporter
FadA is an unusual adhesin in that it exists in two forms: a full‐length peptide consisting of 129 aa residues (15.5 kDa), termed pre‐FadA, and a cleaved “mature” form consisting of 111 aa residues (13.6 kDa) without the signal peptide, termed mFadA (Fig 2A). Crystallographic analysis of mFadA reveals a filamentous structure, with predominantly α‐helical monomers linked in a head‐to‐tail pattern through a “leucine chain” motif (Nithianantham et al, 2009). The mFadA alone exhibits little virulence, and pre‐FadA by itself is insoluble under neutral pH. Together, these two forms constitute a heterogeneous complex termed FadAc (Xu et al, 2007). Previous studies have demonstrated that recombinant FadAc, but not mFadA, is the active form for binding and invading host cells and stimulating CRC growth (Xu et al, 2007; Fardini et al, 2011; Rubinstein et al, 2013; Rubinstein et al, 2019). Pre‐FadA plays a critical role in the size, heterogeneity and function of FadAc. With increasing pre‐FadA, the multimeric complex becomes more heterogeneous, and the complex size and cell‐binding activities also increase (Xu et al, 2007; Temoin et al, 2012a).
Figure 2. Pre‐FadA is a key component of amyloid‐like FadA.

-
ASingle‐letter amino‐acid (aa) sequence of FadA. The intact pre‐FadA consists of 129 aa, with the first 18‐aa (top row) constitute the signal peptide. The remaining 111 aa constitute mFadA. The residues that are replaced in the mutant proteins used in this study are shown in red.
-
BAnalysis of detergent‐resistant recombinant FadA polymers expressed in E. coli. An aliquot of 50 μg detergent‐resistant pellets prepared from E. coli BL21(DE3) carrying the cloning vector pET21(b), pYWH417‐6 (p_FadAc), or pYWH418 (p_mFadA) were analyzed by SDD‐AGE, followed by Western blot analysis. FadA polymers were only detected in E. coli expressing FadAc, but not in mFadA or the vector control.
-
CReactivity of recombinant FadA and its variants with anti‐amyloid fibril antibody OC. A total of 10 μg each of recombinant protein mFadA, FadAc, FadA‐L‐9A, FadA‐L14A, FadA‐S71A, and FadA‐L76A purified from E. coli was slot‐blotted onto nitrocellulose membrane and incubated with polyclonal OC antibody at 1:5,000 dilution. Amyloid‐like recombinant Rim4 (Berchowitz et al, 2015) was used as a positive control and histone H1 was used as a negative control. Ponceau stain (pink color) is shown as a loading control.
-
DThioflavin‐T binding assay. FadA proteins at indicated concentrations were incubated with 10 μM Thioflavin‐T at room temperature for 10 min. The fluorescent intensity was measured at excitation wavelength of 440 nm and emission wavelength of 500 nm. The experiment was performed in duplicate and repeated three times. The error bars indicate SD. *P < 0.05, **P < 0.01, ***P < 0.001 (compared to BSA, t‐test).
-
EThioflavin‐T binding assay in the presence of 0.1% SDS. FadA proteins (0.8 mg/ml) were incubated with 10 μM Thioflavin‐T as described above. The experiment was performed in duplicate and repeated three times. The error bars indicate SD. *P < 0.05, **P < 0.01 (compared to BSA, t‐test).
-
FWestern blot analysis of Fn12230 (Fn), fadA‐deletion mutant US1 (ΔfadA), and spontaneous mutant lam following SDS–PAGE. A total of 400 μl of each culture grown to OD600 of 1.0 was pelleted and loaded onto 12% SDS–PAGE. FadA protein was detected using anti‐FadA mAb 7H7 at 1:4,000 dilution. Compared to the wild type, lam produced significantly less pre‐FadA, but not mFadA.
-
GAnalysis of amyloid‐like FadA produced by Fn ATCC 23726 and its Δfap2 mutant. An aliquot of 50 μg detergent‐resistant pellets was loaded onto each lane, followed by SDD‐AGE and Western blot analysis as described above. The mutant produced significantly less amyloid‐like polymers than the wild type.
Source data are available online for this figure.
Using SDD‐AGE and Western blot analysis, we found that FadAc expressed in E. coli also formed detergent‐resistant polymers, although the sizes were smaller than those produced by Fn. In contrast, mFadA did not form polymers (Fig 2B). These results indicate that while the full‐size amyloid‐like FadA polymers likely involve additional factors and processes that are specific to Fn, FadAc has intrinsic capability to assemble into amyloid‐like structure and that pre‐FadA is a key component for amyloid formation.
To evaluate whether, and to what degree, FadA and its variants self‐assemble into amyloid‐like aggregates, we assessed binding of recombinant FadA proteins to two different antibodies that recognize amyloid structures: polyclonal anti‐human amyloid‐β antibodies OC and A11. The difference between these two antibodies is that A11 recognizes pre‐fibril oligomers, while OC recognizes mature amyloid fibrils (Kayed et al, 2007). Only A11 is suitable for immunofluorescence staining, but both can be used for the analysis of purified proteins by slot blot. Among the recombinant FadA mutants tested, L14A and L76A did not produce pre‐FadA (Fig EV1A) and exhibited no filamentous structure or cell‐binding function (Xu et al, 2007). These mutant proteins did not react with either OC or A11, similar as the negative control protein histone H1 (Figs 2C and EV1B). In contrast, FadAc, L‐9A (carrying Leu‐Ala mutation at position −9 in the signal peptide) and S71A reacted with both OC and A11, similar as the positive control protein Rim4, which readily forms β‐sheet‐rich amyloid‐like aggregates (Figs 2C and EV1B) (Berchowitz et al, 2015). Previous study showed these latter group all retained filamentous structure and were functional (Temoin et al, 2012a). Interestingly, they all express both forms of FadA (Fig EV1A). Notably, mFadA reacted weakly with the anti‐oligomer A11 but not with the anti‐mature fibril OC, indicating that mFadA weakly forms oligomers, but not mature amyloid fibrils (compare Figs 2C and EV1A). This is consistent with the crystal structure of mFadA, in which mFadA monomers link to one another in a head‐to‐tail manner to form a filament (Nithianantham et al, 2009). However, the mFadA filament is not resistant to SDS, as shown by the Thioflavin‐T binding assay (Fig 2D and E).
Figure EV1. Reactivity of recombinant FadA with anti‐amyloid antibody A11 and detection of FadA in Fn 23726 and its Δfap2 mutant.

-
ASDS–PAGE analysis of recombinant FadAc and its variants. The proteins were purified as previously described (Xu et al, 2007). Approximately 2.5 μg of each purified protein was loaded onto 12% SDS–PAGE. Following electrophoresis, the gel was stained with Coomassie Blue G‐250. M, protein size markers.
-
BReactivities of recombinant FadA and its variants with anti‐amyloid oligomer antibody A11. A total of 10 μg each of recombinant protein FadAc, mFadA, FadA‐S71A, FadA‐L76A, and FadA‐L14A purified from E. coli was slot‐blotted onto nitrocellulose membrane and incubated with polyclonal anti‐amyloid oligomer antibody A11 at 1:5,000 dilution. Amyloid‐like recombinant Rim4 (Berchowitz et al, 2015) was used as a positive control and histone H1 was used as a negative control. Ponceau stain (pink color) is shown as a loading control.
-
CWestern blot analysis of wild‐type Fn 23726 (1) and its mutant Δfap2 (2) using anti‐FadA mAb 7H7. The bottom panel is a Coomassie blue stain as loading control.
Source data are available online for this figure.
FadA proteins that reacted with A11 or OC bound the amyloid‐specific dye Thioflavin‐T in a dose‐dependent manner. Without SDS, mFadA bound more Thioflavin‐T than FadAc at the same protein concentrations tested, possibly because more filaments were present (Fig 2D). However, mFadA did not bind Thioflavin‐T in the presence of 0.1% SDS, while FadAc, L‐9A, and S71A, all of which contained pre‐FadA, bound Thioflavin‐T. These results indicate that mFadA only forms the less stable oligomers but not fibrils and that the signal peptide plays a critical role in the assembly of mature amyloid fibrils (Fig 2E), consistent with the results of SDD‐AGE (Fig 2B). L14A, which does not form filamentous structure (Nithianantham et al, 2009) or react with A11 or OC (Figs 2C and EV1B), did not bind Thioflavin‐T (Fig 2D). The FadA proteins were also found to contain varying levels of β‐sheets by circular dichroism (Table EV1), a characteristic of amyloid proteins. Taken together, these data indicate that FadA proteins are capable to self‐assemble into amyloid‐like oligomers or fibrils.
To further investigate the role of pre‐FadA in amyloid formation, we utilized a previously isolated spontaneous mutant, lam (for less adhesive mutant), which was defective in Fn autoaggregation, binding to host cells, and induction of IL 8 (Han et al, 2000). The lam mutant was defective in producing pre‐FadA but not mFadA (Fig 2F). Consequently, it was severely defective in binding Congo Red (Fig 1A), or producing large amyloid‐like assemblies (Fig 1C). Furthermore, IHC analysis showed that lam was defective in secreting FadA aggregates, but not bacterial surface FadA (Fig 1D). It did not react with anti‐amyloid antibody A11 by immunofluorescence staining (Fig 1E) and lacked extracellular fibrils as determined by SEM (Fig 1F). Their observations support that pre‐FadA is a critical component of amyloid‐like FadA and is likely secreted.
DNA sequencing analysis of wild‐type Fn 12230 and lam revealed two variations: (i) a 6‐aa in‐frame deletion in an open reading frame (ORF) sharing 62.3% identity to the Fap2 protein from Fn ATCC 23726 and (ii) a missense mutation (N284I) in a glycerol kinase homolog (Table EV2). Fap2 is a Type V autotransporter required for inter‐species adherence and for Fn to interact with the immune cells, as well as colonization in colorectal tumors (Kaplan et al, 2009; Kaplan et al, 2010; Coppenhagen‐Glazer et al, 2015; Abed et al, 2016). We analyzed Fn 23726 and its fap2 mutant in stationary phase by SDD‐AGE and Western blot analysis. Similar to Fn 12230, Fn 23726 formed highly heterogeneous SDS‐resistant FadA polymers while the fap2 mutant was almost completely defective (Fig 2G). SDS–PAGE analysis showed the fap2 mutant was defective in producing pre‐FadA, although mFadA was unaffected (Fig EV1C). These results demonstrate that the Fap2 autotransporter is required for producing pre‐FadA, which is a critical component of the amyloid fibrils.
Amyloid‐like FadA is detected in periodontal disease and CRC
Our data indicate that not only does FadA assemble into an amyloid‐like structure, but the formation of this structure is a regulated process. We hypothesize that the transition of FadA from monomers to amyloid‐like fibrils substantiates Fn pathogenicity. Therefore, we examined the presence of amyloid‐like FadA in periodontal disease and CRC. Previous studies have reported Fn and fadA gene levels are significantly increased in the colonic tissues and fecal microbiome of CRC patients, compared to the controls (Kostic et al, 2013; Rubinstein et al, 2013; Wirbel et al, 2019). Similarly, Fn 16S rRNA and fadA genes are also increased in periodontal diseased sites, compared to healthy subjects or healthy sites of periodontitis patients (Fig EV2). The increase is not only quantitative, but also qualitative, as FadA aggregates were detected in periodontal diseased sites but not in the healthy sites from the same patients (Fig 3A). Furthermore, amyloid‐like FadA was detected in colorectal carcinoma tissues by fluorescent staining, but not in the paired normal tissues (Fig 3B). It should be noted that since OC is not suitable for immunofluorescent staining, A11 was used, which was less ideal because it captures the oligomeric seeds rather than the mature fibrils. This could partially explain the incomplete overlap of staining of A11 and anti‐FadA 7H7. Nonetheless, the overlap staining was only observed in the carcinoma tissues, but not in the matched normal controls. These observations indicate that amyloid‐like FadA is associated with disease and may play an important role in pathogenesis. The potential pathogenicity of amyloid‐like FadA was then assessed in a series of in vitro and in vivo tests.
Figure EV2. Quantification of Fn and fadA DNA levels in subgingival plaque samples.

DNA was extracted from subgingival bacterial plaque samples from two groups of participants: periodontal healthy subjects (n = 20) and patients with periodontitis (n = 20). For the latter group, subgingival plaques were collected from healthy sites (probing depth ≤ 3 mm) and diseased sites (probing depth ≥ 7 mm). Gene copy numbers of total 16S rRNA, Fn 16S rRNA, and fadA genes were measured by qPCR and determined using the standard curves. The percentages of Fn 16S rRNA (left panel) and fadA gene (right panel) levels relative to total 16S RNA gene levels of each patient were calculated. The horizontal bars represent the mean values. *P < 0.05, **P < 0.01 (one‐way ANOVA).
Figure 3. Amyloid‐like FadA is detected in periodontal disease and CRC.
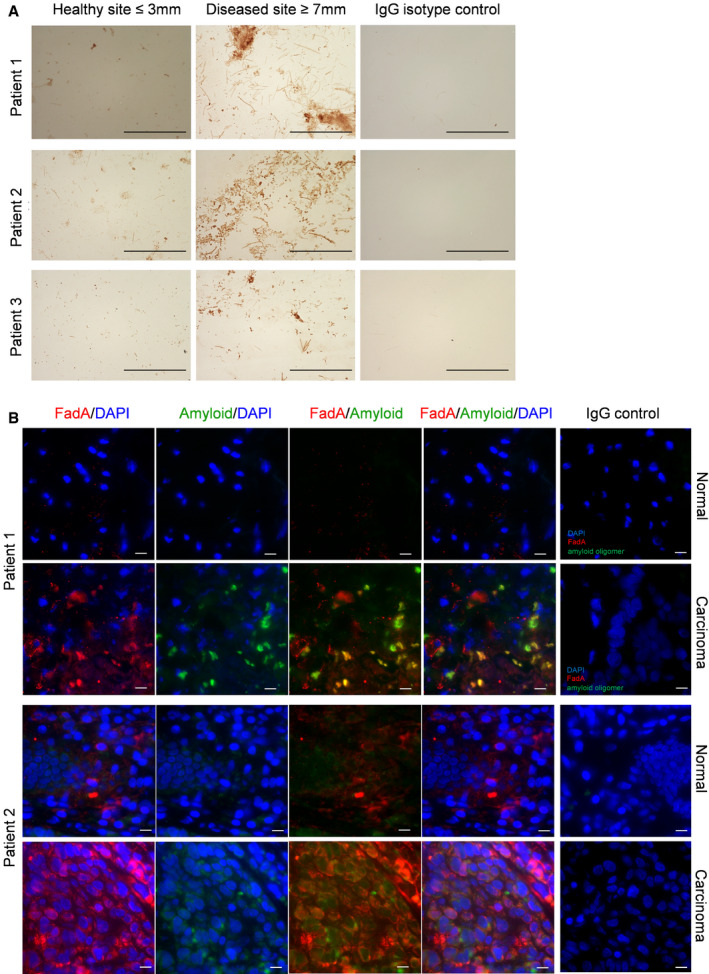
-
ADetection of FadA in subgingival plaque samples by IHC. Three patients with periodontitis are shown here who provided plaque samples from their healthy sites (probing depth ≤ 3 mm, left panels) and periodontal diseased sites (probing depth ≥ 7 mm, middle panels). IHC was performed as described using mAb 7H7 at 1:800 dilution. Anti‐mouse IgG isotype control of the diseased site is shown (right panels). The images were taken using a 100X objective. Scale bar equals 50 μm.
-
BDouble immunofluorescent staining of paired normal and carcinoma tissues from two CRC patients. The frozen tissue sections were incubated with mAb 7H7 at 1:50 dilution and anti‐amyloid antibody A11 at 1:25 dilution, or mouse and rabbit IgG control, followed by incubation with Alexa Fluor 555‐conjugated goat anti‐mouse and Alexa Fluor 680‐conjugated donkey anti‐rabbit antibodies, both at 1:1,000 dilution. Co‐staining of FadA (red) and amyloid oligomers (green) were observed in the carcinoma, but not normal, tissues. The images were taken using a 60X objective. Scale bar equals 10 μm.
Amyloid‐FadA facilitates biofilm formation
In both periodontal disease and CRC, biofilms play important roles, allowing bacteria to secure colonization in the host, resist host immune defense, and acquire nutrients from other members of the microbial community (Tomkovich et al, 2019). We hypothesize that amyloid‐like FadA may serve as a scaffold for biofilm formation. Sarkosyl‐insoluble pellets were obtained from Fn 12230, US1 (ΔfadA), and lam as described above and were used to coat 96‐well plates, followed by inoculation of Fn12230 (experimental scheme outlined in Fig EV3A). Fn 12230 by itself did not form biofilm under the test conditions, possibly due to insufficient secreted FadA. Sarkosyl‐resistant pellets purified from wild‐type Fn readily facilitated biofilm growth, but the sarkosyl‐insoluble pellets obtained from US1 (ΔfadA) and lam were defective in facilitating biofilm formation (Fig 4A). These results indicate that secreted amyloid‐like FadA support biofilm growth in trans, independent of surface‐expressed FadA. Notably, the sarkosyl‐resistant pellet obtained from US1 (ΔfadA) supported partial biofilm formation, thus, within the total amyloid‐like assemblies produced by Fn, FadA itself plays a primary role, but additional components may also contribute to the scaffold. For example, genome sequence of Fn 12230 revealed two additional FadA paralogs, FadA2 and FadA3, with 24.5 and 25% identity with FadA, respectively (Table EV3, Fig EV3B). This is consistent with previous reports of detecting multiple copies of fadA genes on the Fusobacterium genomes and may explain some of the residual activities observed in US1 (ΔfadA) (Manson McGuire et al, 2014; Umana et al, 2019).
Figure EV3. Experimental illustration of preparation of detergent insoluble pellets for biofilm assay, alignment of FadA and its paralogs, and effect of Congo Red on Fn viability.

-
ASchematic illustration of preparation of detergent‐resistant bacterial pellets and its use as scaffold for biofilm growth.
-
BAlignment of FadA (1), FadA2 (2) and FadA3 (3). Identical aa residues are highlighted. cov: the coverage of sequence in the alignment; pid: percent identity of the sequence compared to reference (DOJBCGPA_00667). On the top of alignment: “.” is 10‐aa mark, “:” is a 50‐aa mark and “1” is a 100‐aa mark; the colors and letters of consensus sequences and background colors of protein sequences are defined in https://desmid.github.io/mview/manual/manual.html.
-
CEffects of Congo Red on Fn viability. Fn 12230 was incubated in PBS (top row), 10 mg/ml Congo Red (10 CR), or 50 mg/ml Congo Red (50 CR) for 48 h (left panel) or 72 h (right panel), followed by measurement of OD600 (OD) and viable counts (CFU).
Figure 4. Functional analysis of amyloid‐like FadA in vitro .

-
AFacilitation of Fn biofilm formation by amyloid‐like FadA. An aliquot of 10 μg each of the detergent‐resistant pellets prepared from Fn 12230 (wild type), US1 (ΔfadA) and lam, or PBS, was added to 96‐well plates and incubated at 37°C for 5 h. Fresh culture of Fn 12230 was diluted to OD600 of 0.2, and an aliquot of 200 μl was added to the wells, followed by incubation at 37°C for 48 or 72 h under anaerobic conditions. Following washes, the biofilms were incubated with 100 μl 0.1% crystal violet for 15 min. After washing, 100 μl 95% ethanol was added to each well and the optical density at OD550 was measured in a microplate reader. The results shown are the average of four experiments each performed in duplicate. The error bars indicate SD. A representative image following 72 h of incubation is shown in the top panel. **P < 0.01. ***P < 0.001 (t‐test).
-
BInhibition of biofilm formation by Congo Red. An aliquot of 10 μg detergent‐resistant pellets prepared from Fn 12230 (pellet) was added to 96‐well plates in the presence of 0, 10, or 50 µg/ml Congo Red (CR), followed by addition of Fn 12230 and incubated as described above. The results shown are the average of four experiments each performed in duplicate. The error bars indicate SD. A representative image following 72 h of incubation is shown in the top panel. **P < 0.01, ***P < 0.001 (t‐test).
-
CEffects of pH on Fn survival. Wild‐type Fn 12230 (Fn) or its fadA‐deletion mutant US1 (ΔfadA) grown to log or stationary phase were washed and incubated in PBS at pH7 or pH4 for 1 or 2 h. The live bacterial counts at time 0 were designated as 100%, and those after 1 or 2 h of incubation were expressed relative to time 0. The results shown are the average of 5 (for log) or 4 (for stationary) independent experiments, each performed in triplicate. The error bars indicate SD. *P < 0.05, **P < 0.01, ***P < 0.001 (one‐way ANOVA).
-
DEffects of Congo Red on Fn survival in acidic environment. Wild‐type Fn 12230 (Fn) or its fadA‐deletion mutant US1 (ΔfadA) grown to stationary phase were washed and incubated in PBS at pH4 in the presence of absence of 50 μg/ml Congo Red (CR) for 1 or 2 h. The live bacterial counts at time 0 were designated as 100%, and those after 1 or 2 h of incubation were expressed relative to time 0. The results shown are the average of 5 independent experiments, each performed in duplicate. The error bars indicate SD. *P < 0.05, **P < 0.01, ***P < 0.001 (one‐way ANOVA).
-
EAttachment of Fn 12230 grown to log or stationary phase to CRC cells DLD1. Fn 12230 (Fn) was either unwashed or washed twice with PBS or 1 mg/ml Congo Red (CR) or mixed with 20 μl mAb 7H7, before adding to the monolayers at multiplicity of infection (MOI) of 50:1. US1 (ΔfadA) was included as a negative control. The attachment level by untreated log phase Fn was designated as 100%. All other values were expressed relative to 100%. Data shown are mean values ± SEM. The experiment was performed in duplicate and repeated four times. *P < 0.05, **P < 0.01, ***P < 0.001 (t‐test).
-
FInhibition of Fn attachment by FadAc in the presence or absence of anti‐amyloid antibody OC. An aliquot of 50 μg purified recombinant FadAc was pre‐incubated with DLD1 for 45 min either alone, or mixed with 20 μl OC or rabbit IgG control, before Fn was added, and the attachment assay performed as above. The attachment value by Fn alone was designated as 100%, with all other values expressed relative to it. FadA‐mutant L14A was included as a negative control. Data shown are mean values ± SEM. The experiment was performed in duplicate and repeated three times. **P < 0.01 (one‐way ANOVA).
To confirm it was indeed the amyloid‐like FadA that promoted biofilm formation, we repeated the experiment in the presence of Congo Red (scheme detailed in Fig EV3A), which has been used to disrupt and inhibit the cytotoxicity associated with amyloid‐like polyglutamine aggregates (Sanchez et al, 2003). Congo Red inhibited biofilm formation in a dose‐dependent manner (Fig 4B), without affecting the viability of Fn (Fig EV3C).
Amyloid‐like FadA confers acid tolerance
Since Fn and fadA are detected in the fecal microbiome, it indicates transmission through the GI tract (Thomas et al, 2019; Wirbel et al, 2019). We speculate that secreted amyloid‐like FadA may serve as a protective “shield” enhancing Fn survival in the acidic gastric environment (see Fig 1F). Although the survival of wild‐type Fn at neutral pH (7.0) was indistinguishable between the two growth phases, increased survival in acidic pH (4.0) was observed in stationary phase compared to log phase (Fig 4C, compare the gray bars between left and right panels). Furthermore, survival of wild‐type Fn was dramatically better than US1 in stationary phase than in log phase at pH 4.0 (Fig 4C).
To determine whether and to what extent amyloid‐like FadA plays a role in the observed acid‐resistance of Fn, the viability of wild type and US1 (ΔfadA) in stationary phase at pH 4.0 was tested in the presence of Congo Red. Remarkably, Congo Red drastically reduced viability of wild‐type Fn to the similar levels as US1, without affecting the latter, indicating that the FadA‐mediated acid‐resistance relies solely on its amyloid‐like properties (Fig 4D).
Amyloid‐like FadA facilitates Fn binding to CRC cells
Fn binding to human CRC cells DLD1 increased by ˜ 2.5‐fold in stationary phase compared to log phase (Fig 4E). Furthermore, while washing the bacteria with PBS or Congo Red had little or no effect on binding in log phase, it reduced binding in stationary phase by ˜ 2.5 and 5‐fold, respectively (Fig 4E). These results indicate that the secreted amyloid‐like assemblies are critical for Fn to bind CRC cells. Furthermore, anti‐FadA mAb 7H7 only mildly inhibited Fn binding in log phase, but dramatically inhibited binding in stationary phase, supporting that the enhanced binding in stationary phase was mediated by FadA (Fig 4E). These results are consistent with previous observations that Fn binds CRC cells and promotes tumor progression through FadAc (Rubinstein et al, 2013; Rubinstein et al, 2019). When DLD1 cells were pre‐incubated with FadAc, Fn binding was effectively inhibited. However, when FadAc was pre‐incubated with anti‐amyloid antibody OC, its inhibitory activity was significantly reduced, while the rabbit IgG control had no such effect (Fig 4F). These results demonstrate that the amyloid properties of FadAc mediates Fn binding to CRC cells.
Amyloid‐like FadA facilitates Fn colonization and stimulation of CRC in vivo
To examine the role of amyloid‐like FadA in CRC progression in vivo, a mouse xenograft model was used. Consistent with the in vitro observations, Congo Red significantly inhibited Fn colonization in xenograft tumors (Fig 5A). To determine whether amyloid‐like FadA is a driver of CRC progression, wild‐type Fn 12230, US1 (ΔfadA), and lam were injected into xenograft tumors in nude mice. Fn 12230 significantly stimulated tumor growth while US1 (ΔfadA) and lam were defective (Fig 5B). The fadA‐complementing strain USF81 restored tumor growth to similar levels as Fn 12230 (Fig EV4). These results demonstrate that the secreted amyloid‐like FadA is required for promoting tumor progression. We have shown previously that FadAc, but not mFadA, stimulated xenograft tumor growth (Rubinstein et al, 2013). The effect of FadAc on xenograft CRC tumors was assessed in the presence or absence of the OC antibody. Compared to rabbit IgG control, the OC antibody significantly attenuated tumor growth stimulated by FadAc (Fig 5C). Similarly, the sarkosyl‐resistant pellets prepared from wild‐type Fn significantly stimulated tumor growth compared to the pellets of US1 (ΔfadA), but the enhanced stimulation was completely attenuated by the OC antibody (Fig 5D). These results elucidate the critical role of amyloid‐like FadA in promoting CRC progression.
Figure 5. Amyloid‐like FadA mediates colonization in CRC and promotes tumor growth in vivo .

-
AAnalysis of Fn colonization in mouse xenograft tumors by IHC. An inoculum of 5 × 106 HCT116 cells was injected subcutaneously and bilaterally into the nude mice. Once the tumors became visible (after 3–4 days), approximately 5 × 106 CFU of Fn 12230 washed in PBS or 1 mg/ml Congo Red (CR) were injected into the tumors. The tumors were extracted one week later, and the formalin‐fixed paraffin‐embedded (FFPE) tissues were stained with mAb 7H7. Note the brown stains in tumor injected with PBS‐washed Fn but not with CR‐washed Fn. Scale bar = 20 μM.
-
BEffects of secreted amyloid‐like FadA on tumor growth. HCT116 cells were injected into the nude mice as described above, followed by injection of 5 × 106 CFU of Fn 12230 (Fn, n = 7), lam (n = 3), or US1 (ΔfadA, n = 4). The tumor volumes were measured immediately before bacteria injection (Day 0, designated as 100%) and at 3 days after injection (Day 3). The horizontal lines represent averages of tumor volumes (left panel). The individual tumors are shown (right panel). *P < 0.05, **P < 0.01, ***P < 0.001 (t‐test).
-
CEffect of anti‐amyloid antibody OC on FadAc‐mediated tumor growth. HCT116 cells were injected into the nude mice (n = 6) as described above, followed by injection of 4 μl of 2 mg/ml FadAc mixed with 6 μl OC antibody or rabbit IgG control (rIgG) into tumors on the opposite sides. The tumor volumes were measured on Day 0 and Day 5 as described above. The horizontal lines represent the averages (left panel). The individual tumor pairs are shown (right panel). *P < 0.05, **P < 0.01 (t‐test).
-
DEffect of detergent‐resistant pellets prepared from Fn 12230 (Fn) and US1 (ΔfadA) on tumor growth. HCT116 cells were inoculated into the nude mice as described above. In one group of mice (n = 3), sarkosyl‐resistant pellets prepared from Fn 12230 and US1 (ΔfadA) were injected into the tumors on opposite sides. In another group (n = 5), sarkosyl‐resistant pellets prepared from Fn 12230 were mixed with OC or rabbit IgG control (rIgG) before injecting into the tumors on opposite sides. The tumor volumes were measured as described above. The vertical lines represent the standard deviations. The individual tumor pairs are shown (right panel). *P < 0.05, **P < 0.01, ***P < 0.001 [compared to US1 (ΔfadA), t‐test].
Figure EV4. The fadA‐complementing strain restores CRC growth stimulation.

HCT116 cells were inoculated subcutaneously and bilaterally into the nude mice (n = 4), followed by injection of 5 × 106 CFU of Fn 12230 and the fadA‐complementing clone USF81 on opposite sides as described in Fig 5B. The tumor volumes were measured on Day 0 and 3 as described. The horizontal lines represent averages of tumor volumes (top panel). The individual tumors are shown (bottom panel). **P < 0.01, ***P < 0.001 (t‐test).
Amyloid‐like FadA induces periodontal bone loss in mice
To assess the virulence potential of amyloid‐like FadA in periodontal disease, a mouse periodontitis model was established. Fn 12230, US1 (ΔfadA), and lam were inoculated continuously into the oral cavity of C56BL/6 mice for 10 weeks. Significant periodontal bone loss was detected in the molars of mice treated with Fn 12230, but not in those treated with US1 (ΔfadA) or lam (Figs 6A and B, and EV5A–C). These results demonstrate that secreted amyloid‐like FadA plays a key role in inducing periodontal bone loss.
Figure 6. Amyloid‐like FadA induces periodontal bone loss in mice.

-
AApproximately 1 × 109 CFU of Fn 12230, US1 (ΔfadA), and lam suspended in carboxymethylcellulose (CMC) were orally administered to C57BL/6 mice three times a week for 10 weeks. CMC alone was administered as a control. Maxillae from mice inoculated with CMC alone (n = 5), Fn 12230 (n = 5), US1 (ΔfadA; n = 5), and lam (n = 4) were harvested and fixed in 4% paraformaldehyde and stored in 70% ethanol, followed by microCT (μCT) scanning using a Scanco vivaCT 80 system at 55 kVp, 145 µA, and 250 ms integration time. Shown in the figure are reconstructed grayscale images. ImageJ was used to measure differences in bone height from the cementoenamel junction (CEJ) to the alveolar crest between palatal roots of first and second molars (see arrows).
-
BThe average bone loss of each group shown in (A), with the lines above each bar representing standard deviations. *P < 0.05. **P < 0.01 (t‐test).
Figure EV5. Fn 12230 induces periodontal bone loss in mice.

-
AImage showing first, second, and third molars in the upper jaw of a mouse.
-
BApproximately 1 × 109 CFU of Fn 12230 suspended in carboxymethylcellulose were orally administered to C57BL/6 mice four times a week for 10 weeks. CMC alone was administered as a control. Maxillae from mice inoculated with CMC alone (n = 3) and Fn 12230 (n = 3) were harvested and fixed in 4% paraformaldehyde and stored in 70% ethanol, followed by μCT scanning using a Scanco vivaCT 80 system at 55 kVp, 145 µA, and 250 ms integration time. Shown in the figure are reconstructed grayscale images. ImageJ was used to measure differences in bone height from the cementoenamel junction (CEJ) to the alveolar crest of first and second molars (see arrows). Buccal, the side facing cheek; palatal, the side facing palate.
-
CThe average bone loss from both buccal and palatal sides of CMC control and Fn12230 shown in (B) is shown by the colored bars, with the lines above each bar representing standard deviations. *P < 0.05. **P < 0.01 (t‐test).
Discussion
Fn is an abundant commensal bacterium dwelling in the oral cavity of both periodontal healthy and periodontal disease‐affected individuals. It is present in gingivitis, a reversible form of periodontal disease, but also in more severe forms of periodontitis where irreversible tissue damages occur. Once outside the oral cavity, Fn causes infections that are often life‐threatening. The motivation of this study was to determine how this bacterium functions both as a ubiquitous oral commensal and a rampant systemic pathogen. Here, we show that the virulence factor FadA undergoes a dramatic biochemical transformation into an amyloid‐like structure to enhance its virulence potential. The amyloid‐like FadA exhibits common amyloid properties such as binding to amyloid dyes Congo Red and Thioflavin‐T, resistance to ionic detergents, and formation of large heterogeneous filamentous aggregates. In addition, it reacts with antibodies raised against human amyloid β42, which shares no primary sequence homology with FadA. The emergent amyloid‐like properties of FadA facilitate several critical pathogenic attributes that ultimately allow Fn to survive under stressful conditions, form biofilms, bind host cells, colonize CRC tumors, stimulate CRC progression and induce periodontal bone loss. The enhanced virulence can be inhibited by the amyloid dyes or the anti‐amyloid antibodies, indicating the critical role of the amyloid properties in pathogenesis. Amyloid‐like FadA is specifically enriched in diseased periodontal sites and in colonic carcinomas, but not in the non‐diseased controls, supporting that amyloid‐like FadA may function as a molecular switch for the commensal‐to‐pathogen conversion. Alternatively, expressing of amyloid‐like FadA could also be an adaptive response to the changing environment, which then enhances the virulence potential of Fn. These two scenarios are not mutually exclusive. Expression of amyloid‐like FadA in stationary phase suggests that the trigger may be a stress signal.
Fn has long been recognized as an essential component of subgingival biofilm. The discovery of amyloid‐like FadA as a scaffold sheds mechanistic light on the critical role of Fn in dental plaque development. Furthermore, our data from the mouse periodontitis model indicate that Fn plays more than just a scaffolding role in periodontal disease; instead, it is actively involved in the disease process. Our results clearly demonstrate that amyloid‐like FadA, rather than FadA monomers, is the key driver of pathogenesis, not only in periodontal disease, but also in CRC progression. Recent studies have linked periodontal disease to CRC (Kim et al, 2019). The causal role of amyloid‐FadA in both periodontal disease and CRC provides a possible mechanistic link between these two diseases. Elevated amyloid‐like FadA levels in periodontal disease may facilitate systemic dissemination of Fn thus increasing the risk for developing systemic infections such as pregnancy complications and cancer.
The current study reveals that in the presence of amyloid‐FadA, Fn could survive the acidic environment and disseminate through the GI tract. While Fn can also disseminate via circulation, the saliva, which contains high titers of bacteria, is constantly being swallowed, providing a means for GI dissemination. Findings of significantly increased Fn and fadA in the fecal microbiome of CRC patients support the GI translocation (Thomas et al, 2019; Wirbel et al, 2019). Furthermore, oral inoculation of Fn stimulated colonic tumor formation in mice (Kostic et al, 2013; Rubinstein et al, 2019). Bacterial conglomerates, as they are in saliva, could be even more resistant to acidic environment than a singular species in planktonic broth. We have recently demonstrated that the Fusobacteria communities in stomach and colon share more similarities, with their community diversities significantly reduced compared to in saliva (Richardson et al, 2020). It is plausible that Fn strains expressing elevated amyloid‐FadA may have selective advantage during transmission, through either circulation or the GI tract. Identification of amyloid‐producing Fn may help identify individuals at risk for developing diseases implicated by Fn.
It should be pointed out that acid tolerance is not only beneficial to the bacteria during GI translocation, but also helpful for survival in the tumor microenvironment. Due to increased glycolysis in cancer, high levels of lactate are secreted, leading to acidic extracellular environment (Jiang, 2017). The enhanced colonization and growth stimulation of CRC by amyloid‐FadA may be due to a combination of enhanced biofilm formation, acid tolerance, and binding to the cancer cells.
Previous studies showed that pre‐FadA is required for FadA function (Xu et al, 2007; Temoin et al, 2012b). We now know that it is a key component of amyloid‐like FadA, required for the formation of mature fibrils. It is unclear if pre‐FadA alone, or a mixture of pre‐FadA and mFadA, is present in the secreted amyloid‐like FadA. We speculate that the mFadA filaments observed in the crystal structure could be intercalated with pre‐FadA. The highly hydrophobic signal peptides from two neighboring filaments may form anti‐parallel β‐sheets serving as “hooks” to crosslink these filaments, thus stacking multiple FadA filaments into a fibrous structure (Fig 7). This model is similar to the phenol‐soluble modulin α3 (PSMα3) peptide secreted by Staphalococcus aureus, which forms amyloid‐like fibrils with α‐helices stacked perpendicular to the fibril axis (Tayeb‐Fligelman et al, 2017). Such an amyloid “lattice” is consistent with the plaque‐like coating we observe on Fn. This model explains all the observations we have made with FadA so far. We have reported previously that FadAc, but not mFadA, is the active form, and that the size and function of FadAc is proportional to the amount of pre‐FadA in the complex (Xu et al, 2007; Temoin et al, 2012b). Our model elucidates the crucial role of pre‐FadA in the formation of mature fibrils. With increasing quantities of pre‐FadA, more singular FadA filaments crosslink with one another, thus forming increasingly large, heterogeneous and stable fibrous structures. Without pre‐FadA, each FadA oligomer filament can be easily dissociated. This could explain the instability of mFadA, which was unable to bind Thioflavin‐T in the presence of SDS, while FadAc and the mutants that retained pre‐FadA did.
Figure 7. A proposed model of the structure of amyloid‐like FadA.
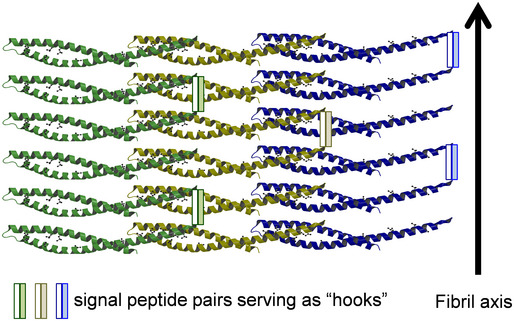
The FadA filaments were based on the crystal structure of mFadA (Nithianantham et al, 2009), with the addition of intercalating pre‐FadA. The signal peptide pairs from two neighboring filaments (one clear bar and one shaded bar) bind to each other through hydrophobic interactions, serving as “hooks” crosslinking the filaments so that the filaments are stacked perpendicular to the fibril axis into fibrous sheets.
The finding that amyloid‐like FadA is secreted via autotransporter Fap2 is unexpected and intriguing. Since the Fap2 mutants are defective in producing pre‐FadA, without affecting mFadA, Fap2 may be required for secreting pre‐FadA and/or amyloid‐like FadA. Fap2 mutants have been found to be defective for colonizing murine placentas and colorectal tumors, similar as the fadA‐deletion mutant (Coppenhagen‐Glazer et al, 2015; Abed et al, 2016). It is plausible that some effects exhibited by the Fap2 mutant may be due to lack of amyloid‐FadA. Since wild‐type Fn produces significantly larger polymers than E. coli expressing FadAc, additional bacterial components may be involved in the fibril formation and/or secretion. Fn encodes an unusually large number of Type V autotransporters (Umana et al, 2019). Given the size of the amyloid‐like FadA aggregates, it is possible that multiple autotransporters form multimeric complexes with pore sizes sufficiently large to allow the amyloid‐like oligomers to pass through. Alternatively, FadA may be secreted as monomers or small oligomers, which then assemble into large polymers after secretion. The structure of FadA fibrils and the interactions between FadA and Fap2‐like Type V autotransporters will be investigated in future studies.
Unveiling the significance of amyloid‐like FadA in Fn pathogenesis opens new doors for therapies. CRC implicated with Fn often has poor prognosis, due to the positive feedback interactions between FadA and the oncotarget Annexin A1, induction of chemo‐resistance and stimulation of metastasis (Mima et al, 2016; Bullman et al, 2017; Yu et al, 2017; Rubinstein et al, 2019; Casasanta et al, 2020). However, the use of broad‐spectrum antimicrobials to treat cancer is undesirable due to disturbance of the entire flora. A specific therapy against Fn virulence factors may be more effective. Our results suggest that targeting amyloid‐like FadA could substantially and specifically inhibit the pathogenic potential of Fn in multiple diseases. While Congo Red is very unlikely to be useful due to its carcinogenicity, the anti‐FadA antibody 7H7 and many other anti‐amyloid compounds, such as those inhibiting polymerization of the curli fibers produced by E. coli (Cegelski et al, 2009), present untapped potential as anti‐cancer agents. Some anti‐amyloid drugs that have failed as treatments for neurodegenerative disease because of the immense challenge of traversing the blood–brain barrier could have unexpected utility against Fn‐driven CRC and other debilitating diseases. Since FadA is uniquely encoded by Fusobacterium (Han et al, 2005), it is an ideal therapeutic target to specifically inhibit Fn‐mediated pathogenesis.
In summary, we present evidence that Fn regulates its virulence by controlling the production of amyloid‐like FadA. With increasing evidence of commensal microbes turning pathogenic in various human diseases, the discovery that Fn secretes an amyloid‐like adhesin to enhance its pathogenicity represents a novel paradigm. Furthermore, the discovery that an amyloid‐like bacterial protein promotes both periodontal disease and tumorigenesis not only sheds remarkable new lights on how oral health could impact extra‐oral infections, but also broadens the scope for therapeutic development for a variety of human diseases.
Materials and Methods
Bacterial strains and cell cultures
The bacterial strains used in this study are listed in Table EV4. Fn strains were grown as described (Han, 2006). Human CRC cells DLD1 were maintained as previously described (Rubinstein et al, 2013; Rubinstein et al, 2019). E. coli MC4100, Δcsg, and ΔcsgA were kindly provided by Dr. Matthew Chapman at University of Michigan.
Congo Red depletion assay
Fn 12230, US1 (ΔfadA), and lam were grown to stationary phase, followed by resuspension in PBS to OD600 of 0.5, 1.0, 1.5, and 2.0. Five microliters of 2 mg/ml Congo Red (Sigma‐Aldrich CAT# C6277) was added to 1 ml bacterial suspension to reach a final concentration of 10 μg/ml and incubated at room temperature for 10 min. The bacterial suspensions were centrifuged at 10,000 g for 5 min. An aliquot of 200 μl of supernatant was transferred to a 96‐well plate to measure OD500 in a microplate reader. Each experiment was performed in duplicate and repeated five times.
Semi‐denaturing detergent agarose gel electrophoresis (SDD‐AGE)
Fn 12230, Fn 23726, and their respective mutants were grown to 0.3‐0.4 OD600 (log phase) or > 0.8 OD600 (stationary phase). Recombinant E. coli BL21 expressing FadAc or mFadA were grown to 0.6 OD600, followed by induction with 1 mM IPTG for 3 h. E. coli MC4100, Δcurli, and ΔcsgA were grown for 48 h at 26°C on YESCA plates. The bacteria were then collected by centrifugation at 5,000 g for 5 min and lysed by incubation in the lysis buffer (10 mM Tris pH8.0, 0.1 M NaCl, 0.1 mM EDTA, 0.5% Triton X‐100, and 2 mg/ml lysozyme) at 4°C for 20 min with rotation. Sarkosyl was then added to a final concentration of 1% and incubated at 4°C for 20 min with rotation. The samples were centrifuged at 18,000 g at 4°C for 20 min. After measuring the protein concentrations by BCA, the pellets were stored at −80°C until use. An aliquot of 50 μg (for E. coli, Fn 23726 and its mutants) or 100 μg (for Fn 12230 and its mutants) of the pellets prepared above were incubated with 4×loading buffer [0.5xTAE (20 mM Tris pH 8.3, 20 mM acetic acid, 0.5 mM EDTA), 8% SDS, 20% glycerol, bromophenol blue] for 10 min before loading onto 1.7% agarose gel containing 0.5x TAE and 0.1% SDS. The electrophoresis was carried out overnight at 4°C with circulating 0.5 × TAE containing 0.1% SDS. Components on the gel were then immobilized onto nitrocellulose membrane by capillary transfer in TBS (50 mM Tris–HCl, 150 mM NaCl pH 7.4) for 24 h at room temperature, followed by Western blot analysis (see below).
Western blot analysis
One milliliter each of Fn 12230, US1, and lam in stationary phase was pelleted by centrifugation and loaded onto 12% SDS–PAGE. Following electrophoresis, the bacterial components were transferred to PVDF membranes (Bio‐Rad, Hercules, CA). The membrane was blocked with 5% skim milk in TBS containing 0.1% Tween‐20 (TBST) at room temperature for 1 h followed by incubation with anti‐FadA monoclonal antibody (mAb) 7H7 at 1:4,000 dilution in 0.5% skim milk in TBST at 4°C overnight. After washing three times with TBST, the membrane was incubated with HRP‐conjugated goat anti‐mouse IgG at 1:4,000 dilution in TBST at room temperature for 1 h. Following washes, the immune‐reactive components were detected with ECL Western Blotting Substrate (Thermo Fisher Scientific).
Time course study
Fn 12230 and US1 were sub‐cultured to OD600 of 0.1 and grown in Columbia Broth supplemented with 5 µg/ml hemin and 1µg/ml menadione at 37°C under anaerobic conditions. Ten OD600 units of bacteria were harvested by centrifugation at 12‐h intervals, followed by centrifugation at 5,000 g for 5 min. The bacteria were lysed with 500 μl lysis buffer containing 2 mg/ml lysozyme at 4°C for 20 min with rotation, followed by addition of sarkosyl to a final concentration of 1% and incubated at 4°C for 20 min with rotation. An aliquot of 30 μl homogeneous mixture was saved as “Total” sample. The remaining mixture was centrifuged at 100,000 g for 20 min at 4°C. An aliquot of 30 μl supernatant was saved as “Supernatant.” The pellet was resuspended in 470 μl lysis buffer, and an aliquot of 30 μl was taken as “Pellet” sample. The samples were then analyzed by Western blot analysis as described above. Ponceau staining of lysozyme was used as a loading control.
Slot blot assay
Ten micrograms each of purified recombinant protein was slot‐blotted onto nitrocellulose membrane (Bio‐Rad) and assayed for reactivity with polyclonal anti‐amyloid fibril antibodies OC (α‐amyloid fibril OC, Sigma‐Aldrich CAT# AB2286) at 1:5,000 dilution and goat anti‐rabbit IgG (GE Healthcare) at 1:10,000 dilution, or polyclonal anti‐amyloid oligomer antibody A11 (α‐oligomer A11 at 1:5,000 dilution, Thermo Fisher Scientific CAT# AHB0052) and goat anti‐rabbit IgG at 1:10,000 dilution, followed by detection with ECL Prime (GE Healthcare). Amyloid‐like recombinant Rim4 was used as a positive control (Berchowitz et al, 2015), and histone H1 was used as a negative control. Loading was assessed by Ponceau stain (Sigma CAT# P7170).
Thioflavin‐T binding assay
Thioflavin‐T assay was conducted as previously described (Tukel et al, 2009). A mixture of 50 μl of FadA proteins and 50 μl of 10 μM Thioflavin‐T was added to a black 96‐well plate and incubated for 10 min at room temperature. After incubation, the fluorescence intensity was determined using a SpectraMax M5 microplate reader at excitation wavelength of 440 nm and emission wavelength of 500 nm. For SDS stability test, the FadA proteins were pre‐incubated in 0.1% SDS for 30 min at room temperature, followed by Thioflavin‐T binding assay as described above.
Immunohistochemistry (IHC)
Bacteria and dental plaque samples were fixed in 4% paraformaldehyde, resuspended in PBS, and placed on electro‐charged slides. Endogenous peroxidase was quenched in 3% hydrogen peroxide for 30 min, followed by washing. Slides were incubated with 5% skim milk for 1 h at room temperature, followed by anti‐FadA mAb 7H7 raised against FadAc at 1:800 dilution or mouse IgG isotype control (R&D Systems, Minneapolis, MN) overnight at 4°C. After washing, slides were blocked in 2.5% horse serum for 1 h at room temperature and incubated with ImmPRESS HRP anti‐mouse IgG (Vector Lab) for 30 min at room temperature followed by washes, before being developed with the DAB Peroxidase Substrate Kit (Vector Lab), counter stained with hematoxylin, dried by sequential incubation in 70, 95, 100% ethanol and xylene, and covered in Permount mounting medium (Fisher chemical).
For formalin‐fixed paraffin‐embedded (FFPE) mouse xenograft tumors, the slides were incubated at 60°C for 1 h and de‐paraffinized by incubation in xylene for three times, followed by rehydration with sequential incubation in 100, 95, and 70% ethanol, ddH2O, and PBS. Endogenous peroxidase was quenched in 3% hydrogen peroxide for 15 min, followed by washing. Slides were incubated with 2.5% horse serum in PBS containing 0.5% BSA for 2 h at room temperature, followed by anti‐Fn polyclonal antibodies at 1:500 dilution or rabbit IgG isotype control (Invitrogen) overnight at 4°C. After washing, slides were incubated with ImmPRESS HRP anti‐rabbit IgG (Vector Lab) and developed as described above.
Double‐immunofluorescent staining
Bacterial cells were fixed, washed with PBS, and placed on slides followed by incubation with 3% hydrogen peroxide and 5% skim milk as described above. Slides were incubated with anti‐FadA mAb 7H7 antibody at 1:800 dilution and anti‐amyloid oligomer antibody A11 (Thermo Fisher Scientific CAT# AHB0052) at 1:500 dilution or mouse IgG (R&D Systems, Minneapolis, MN) and rabbit IgG isotype (Invitrogen) controls overnight at 4°C. After washing, slides were incubated with Alexa Fluor 680‐conjugated donkey anti‐rabbit (Invitrogen) and Alexa Fluor 555‐conjugated goat anti‐mouse (Invitrogen), both at 1:1,000 dilutions, washed, and covered in mounting medium containing DAPI.
For human colonic specimens, frozen tissue sections were fixed in 4% paraformaldehyde for 15 min, sequentially washed by 70% ethanol, ddH2O, and PBS, and permeabilized by 0.1% Triton X‐100 in PBS followed by blocking in 5% skim milk. Slides were incubated with α‐FadA mAb 7H7 antibody at 1:50 dilution and α‐amyloid antibody A11 at 1:25 dilution, or mouse and rabbit IgG controls, overnight at 4°C. Then, slides were washed, incubated with secondary antibodies as described above.
Scanning Electron Microscope (SEM)
Bacteria cultures were harvested by gentle centrifugation, washed in PBS, resuspended in 2% (v/v) glutaraldehyde in PBS and fixed for 1 h at 4°C. Bacteria were fixed again by 1% osmium tetroxide in PBS for 1 h at 4°C. Fixed bacteria were dehydrated with 25, 50, 75, and 100% ethanol and then dried with the Critical Point Dryer (CPD) to remove ethanol. Before imaging, samples were coated with Au/Pd to be conductive during electron microscopy. Zeiss Sigma VP SEM instrument was used at an operating voltage of 3 kV with an InLense detector to obtain more surface information of the bacteria exterior. Thermo Fisher/FEI Talos F200X FEI instrument operating at 200 kV was also utilized to observe both interior and exterior of bacteria at higher magnifications.
Bacterial genome sequencing and variation identification
Fn12230 and lam DNA were extracted using modified protocol of the Qiagen MagAttract PowerMicrobiome DNA/RNA Kit (Qiagen 27500‐4‐EP). To obtain the genome reference of Fn strain, paired‐end library was prepared based on a low‐volume Nextera sequencing protocol (Baym et al, 2015) and sequenced on Illumina HiSeq 4000 or HiSeq X platform to generate more than 3 million paired‐end reads for each sample. Raw reads were then processed by Cutadapt v2.1 with following parameters “‐‐minimum‐length 25:25 ‐u 15 ‐u ‐5 ‐U 15 ‐U ‐5 ‐q 15 ‐‐max‐n 0 ‐‐pair‐filter=any” to remove low‐quality bases and Nextera adapters. PacBio long‐read sequencing was performed for the reference strain Fn 12230 by SNPsaurus to improve the performance of de novo genome assembling. Illumina reads passing quality filtering and PacBio long reads were assembled using Unicycler with hybrid mode to generate the genome sequence of reference strain and the genome were then annotated using Prokka v1.13.3 with parameter “‐‐rnammer –rfam.” A complete genome of Fn 12230 (2.42 Mbp) and two circular plasmids (46.3 Kbp and 5.85 Kbp) was obtained. To identify the genomic variation of lam, the reads obtained with lam were aligned to the assembled reference genome using Bowtie2 v2.3.4 in paired‐end mode with default setting. Resulting reads alignments were then processed by SAMtools v1.9 and Picard to fix paired‐end mate and remove PCR duplicates. Remaining alignments in BAM format were processed using BCFtools v1.9 with following parameters “‐‐ploidy 1” and VCFtools v0.1.17 to call genomic variation (SNPs and Indels). Resulting variations were further subjected to manual inspection to remove any false positive due to low reads coverage and misalignment of homologous regions. The illumina reads for both strains, Pacbio reads for Fn12230, and de novo genome assembly of Fn12230 are deposited in NCBI database with the accession number SRA: PRJNA632750.
Verification of SNPs between Fn 12230 and lam
For each SNP in lam identified by genome sequencing, primers were designed (Table EV5) to amplify the SNP‐containing region from Fn 12230 and lam by PCR. The PCR amplicons were submitted for Sanger sequencing (Vendor) to verify the sequence variations.
Circular dichroism (CD) test
CD was used to evaluate the secondary structure of FadA and its mutants. One hundred microliter of 0.2 mg/ml purified FadA was analyzed by Chirascan V100 Spectrometer using a 1 mm path‐length fused quartz cell. The measurements are an average of three scans for each sample, captured at a scan rate of 1 s, bandwidth and step size of 1 nm, over a wavelength range of 190‐250 nm. CD spectra were recorded and analyzed with BestSel server (http://bestsel.elte.hu/).
Collection of dental plaque samples
This study was reviewed and approved by the Institutional Review Board of Columbia University Medical Center (IRB‐AAAR7153). Subjects were recruited from patients who presented to Columbia University College of Dental Medicine. Inclusion criteria included subjects of 18 years of age or older who were in good general health or controlled common systemic conditions. Subjects with chronic periodontitis were defined as those with presence of proximal attachment loss ≥ 3 mm in ≥ 2 non‐adjacent teeth. Chronic periodontitis participants must have both diseased (probing depth ≥ 7 mm, with clinical attachment loss) and healthy (probing depth ≤ 3 mm, without clinical attachment loss and bleeding on probing) sites in their oral cavity. Periodontally healthy subjects were defined as those with no site with probing depth > 3 mm, no site with attachment loss and bleeding on probing at ≤ 10% sites. Subjects who had been on antibiotics within 3 months of enrollment, were diagnosed with infectious diseases, such as HIV and hepatitis, and had history of periodontal treatment within 6 months, smoked more than 10 cigarettes per day, had uncontrolled or poorly controlled diabetes or other metabolic diseases, as well as women who were pregnant or nursing a child, were excluded. Written consent form was obtained from all the subjects before the plaque collection. Subgingival plaque samples were collected using sterilized curettes from the interproximal areas from a group of 40 patients: 20 diagnosed with chronic periodontitis and 20 periodontally healthy participants, matched for age, gender, and race. From each patient with chronic periodontitis, two subgingival plaque samples were collected, with one pooled from the diseased sites and another pooled from healthy sites.
Real‐time quantitative PCR
DNA was extracted from plaque samples, and the concentration was measured using NanoDrop ND 1000 spectrophotometer. Real‐time quantitative PCR (qPCR) was performed in duplicates, using StepOnePlus™ (Applied Biosystems). Each 20 μl reaction was prepared with Power SYBR® Green PCR Master Mix kit according to the manufacturer’s instructions (Applied Biosystems). Specific forward and reverse primers, listed in Table EV5, were used at concentration of 500 nM. PCR amplifications were performed as following: 95°C for 10 min followed by 40 cycles of 95°C for 30 s, 60°C for 30 s, and 72°C for 30 s, then 95°C for 15 s and 60°C for 1 min, ending with a 95°C for 15 s. To quantify gene copies, standard curves using plasmids carrying 16S rRNA gene or fadA gene were generated. Each experiment was performed in duplicate and repeated at least twice.
Biofilm assay
An aliquot of 50 μl 0.2 mg/ml detergent‐resistant pellet, or PBS, was added into each well in a 96‐well plate and incubated at 37°C for 5 h. A total of 200 μl Fn 12230 (OD600 0.2), or Columbia broth alone, was added to the wells in the presence of 0, 10, and 50 μg/ml Congo Red, followed by anaerobic incubation at 37°C for 48 or 72 h. The wells were then washed, and the biofilms were stained with 100 μl 0.1% crystal violet solution for 15 min. After washing, 100 μl 95% ethanol was added to each well and incubated for 15 min at room temperature, and the optical density at OD550 was measured in a microplate reader. Each experiment was performed in duplicate and repeated four times.
pH tolerance assay
Fn 12230 and US1 were grown to 0.4 OD600 (log phase) or > 0.8 OD600 (stationary phase), resuspended in pre‐reduced PBS at pH 4.0 or 7.0, and incubated at room temperature with or without 50 μg/ml Congo Red. At 0, 1, or 2 h, aliquots of the bacterial suspension were taken for serial dilutions and plating on blood agar plates. The plates were incubated anaerobically at 37°C to enumerate the viable cell counts. Each experiment was performed in duplicate or triplicate and repeated 3–5 times.
Cell culture attachment assay
The assay was performed as previously described (Han et al, 2000). Briefly, DLD1 cells were seeded in 24‐well plates at 1 × 105 cells per well and grown to 90% confluent. The bacteria were either used or washed twice with PBS or 1 mg/ml Congo Red, before adding to the monolayers at a multiplicity of infection (MOI) of 50:1 and incubated for 1 h at 37°C in 5% CO2. Following washes with PBS, the monolayers were lysed with water, and serial dilutions of the lysates were plated onto blood agar plates to enumerate the total cell‐associated bacteria. The level of attachment was expressed as the percentage of bacteria recovered following cell lysis relative to the total number of bacteria initially added. Each experiment was performed in duplicate and repeated four times. For the inhibitory binding assays, 20 μl mAb 7H7, or 50 μg FadA with or without 20 μl rabbit IgG (1 mg/ml) or OC were mixed with Fn before adding to the cells.
CRC xenograft model
Four‐ to five‐week‐old female NCR/NU mice were purchased from Taconic Biosciences (NY, USA). An inoculum of 5 × 106 HCT116 cells was injected subcutaneously and bilaterally into the mice. After 3–5 days, the following were injected into the tumors: 5 × 106 CFU bacterial cells washed twice with PBS or 1 mg/ml Congo Red, 4 μl (2 mg/ml) FadAc mixed with 6 μl α‐amyloid OC antibody or rabbit IgG control, or 10 μl (2 mg/ml) sarkosyl‐resistant pellets prepared from Fn or US1 (ΔfadA) alone, or 4 μl (5 mg/ml) sarkosyl‐resistant pellets mixed with 6 μl rabbit IgG or OC. The tumor length and width were measured using calipers, and tumor volumes were calculated using the following formula: Volume = (width)2 × length/2. After the tumors were taken out, they were fixed in 4% PFA for 24 h and embedded in paraffin for IHC as described above.
Mouse periodontitis model
This study was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) under Animal Care Protocol AC‐AAAT1453 (Y2 M01). C57BL/6 mice were premedicated with prophylactic antibiotic regimen 1 week prior to bacterial inoculation. Approximately 1 × 109 CFU each of Fn 12230, US1, and lam were resuspended in 2% carboxymethylcellulose (CMC). The oral cavity of 8‐week‐old C57BL/6 mice was prophylactically swabbed with 0.12% chlorhexidine gluconate in order to reduce endogenous bacterial flora prior to bacterial inoculation. Fn 12230, US1, and lam resuspended in CMC were administered via oral inoculation three‐four times a week for 10 weeks, with CMC alone being administered as a control. At the conclusion of the 10‐week period, inoculated C57BL/6 mice were sacrificed via CO2 and cervical dislocation, and were decapitated. Jaws were defleshed, maxillae were harvested, fixed in 4% paraformaldehyde (PFA) for 24 h, and stored in 70% ethanol for prior to μCT analysis.
MicroCT (μCT) analysis of mouse bone
Maxillae harvested from CMC alone, Fn 12230, US1, and lam inoculated C57BL/6 mice were scanned for μCT using a Scanco vivaCT 80 System (Brüttisellen, Switzerland) using machine settings of 55 kVp, 145 µA, and 250 ms integration time. Grayscale images were reconstructed with a voxel size of 10.4 µm, smoothed using a Gaussian filter to remove noise (sigma = 0.8, support = 1), and threshold was set at 40% of the maximum grayscale value to isolate mineralized tissue. CEJ to alveolar crest measurements were made manually on reconstructed images using ImageJ. Measurements for each mesiobuccal, distobuccal, buccal, and palatal root, or palatal root alone, among all mice were averaged, and the standard deviation was determined. The differences between groups were examined by two‐tailed t‐test, and P < 0.05 was considered statistically significant.
Statistical analysis
The differences between groups were examined by paired t‐test or one‐way ANOVA. P value < 0.05 was considered statistically significant.
Author contributions
QM performed the Fn Congo Red depletion, time course, SDD‐AGE, IHC, biofilm, CD spectrum and immunofluorescent studies, purified recombinant proteins, and inoculated mice for the mouse periodontitis study; QG conducted the E.coli Congo Red depletion, Thioflavin‐T binding, cell attachment and CRC xenograft studies, purified recombinant proteins, and prepared Fn samples for SEM; SM, SR, and XEG conducted the mouse periodontitis experiments; KT, YW, and PNP collected and analyzed periodontal bacterial plaque samples; YH and HW sequenced Fn 12230 and lam and analyzed the genomic data; YP performed pH tolerance studies and designed primers for PCR and Sanger sequencing; ZL designed primers for PCR and Sanger sequencing; AZ performed SEM; RL provided the Δfap2 mutant of Fn 23726; LEB performed slot blot and Western blot for the time course study and assisted with SDD‐AGE, and wrote the manuscript. YWH designed most of the experiments, supervised the entire study and wrote the manuscript.
Conflict of interest
YWH is a scientific advisor to Emanate Biomedical which is not involved in this work. HHW is a scientific advisor and equity holder of SNIPR Biome and Imvela who are not involved with this work. Otherwise, the authors declare no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Acknowledgements
The authors thank Matthew Chapman for the generous gift of E. coli curli strains and anti‐CsgA antibodies, Jia Ma at Columbia University Precision Molecular Characterization Core for assisting with circular dichroism analysis. This study was supported in part by NIH grants R01CA192111 and R01DE029532 to Y.W.H., R35 GM124633‐01 and Irma T. Hirschl Career Scientist Award to L.E.B, R01AI132403 and Burroughs Wellcome Fund (PATH1016691) to H.W., R01AR065564 to X.E.G., 1S10OD025102 to the Precision Molecular Characterization Core, and a fellowship from China Scholarship Council to Q.M (No.201806790025).
EMBO reports (2021) 22: e52891.
Contributor Information
Luke E Berchowitz, Email: leb2210@cumc.columbia.edu.
Yiping W Han, Email: ywh2102@cumc.columbia.edu.
Data availability
The illumina reads of Fn 12230 and the lam mutant, PacBio reads for Fn 12230, and de novo genome assembly of Fn12230 are deposited in NCBI BioProject database with the accession number PRJNA632750. https://www.ncbi.nlm.nih.gov/bioproject/PRJNA632750.
References
- Abed J, Emgård J, Zamir G, Faroja M, Almogy G, Grenov A, Sol A, Naor R, Pikarsky E, Atlan Ket al (2016) Fap2 mediates Fusobacterium nucleatum colorectal adenocarcinoma enrichment by binding to tumor‐expressed Gal‐GalNAc. Cell Host Microbe 20: 215–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baym M, Kryazhimskiy S, Lieberman TD, Chung H, Desai MM, Kishony R (2015) Inexpensive multiplexed library preparation for megabase‐sized genomes. PLoS One 10: e0128036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berchowitz LE, Kabachinski G, Walker MR, Carlile TM, Gilbert WV, Schwartz TU, Amon A (2015) Regulated Formation of an Amyloid‐like Translational Repressor Governs Gametogenesis. Cell 163: 406–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boke E, Ruer M, Wuhr M, Coughlin M, Lemaitre R, Gygi SP, Alberti S, Drechsel D, Hyman AA, Mitchison TJ (2016) Amyloid‐like Self‐Assembly of a Cellular Compartment. Cell 166: 637–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullman S, Pedamallu CS, Sicinska E, Clancy TE, Zhang X, Cai D, Neuberg D, Huang K, Guevara F, Nelson Tet al (2017) Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 358: 1443–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casasanta MA, Yoo CC, Udayasuryan B, Sanders BE, Umana A, Zhang Y, Peng H, Duncan AJ, Wang Y, Li Let al (2020) Fusobacterium nucleatum host‐cell binding and invasion induces IL‐8 and CXCL1 secretion that drives colorectal cancer cell migration. Sci Signal 13: eaba9157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellarin M, Warren Rl, Freeman Jd, Dreolini L, Krzywinski M, Strauss J, Barnes R, Watson P, Allen‐Vercoe E, Moore Raet al (2012) Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res 22: 299–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cegelski L, Pinkner JS, Hammer ND, Cusumano CK, Hung CS, Chorell E, Åberg V, Walker JN, Seed PC, Almqvist Fet al (2009) Small‐molecule inhibitors target Escherichia coli amyloid biogenesis and biofilm formation. Nat Chem Biol 5: 913–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppenhagen‐Glazer S, Sol A, Abed J, Naor R, Zhang X, Han YW, Bachrach G (2015) Fap2 of Fusobacterium nucleatum is a galactose‐inhibitable adhesin involved in coaggregation, cell adhesion, and preterm birth. Infect Immun 83: 1104–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans ML, Gichana E, Zhou Y, Chapman MR (2018) Bacterial Amyloids, In Amyloid proteins: methods and protocols, Sigurdsson EM, Calero M, Gasset M (eds), pp 267–288. New York, NY: Springer; [Google Scholar]
- Fardini Y, Wang X, Temoin S, Nithianantham S, Lee D, Shoham M, Han YW (2011) Fusobacterium nucleatum adhesin FadA binds vascular endothelial cadherin and alters endothelial integrity. Mol Microbiol 82: 1468–1480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YW (2006) Laboratory maintenance of fusobacteria. Curr Protoc Microbiol Chapter 13: Unit 13A.1 [DOI] [PubMed] [Google Scholar]
- Han YW, Fardini Y, Chen C, Iacampo KG, Peraino VA, Shamonki JM, Redline RW (2010) Term stillbirth caused by oral Fusobacterium nucleatum . Obstet Gynecol 115: 442–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YW, Ikegami A, Rajanna C, Kawsar HI, Zhou Y, Li M, Sojar HT, Genco RJ, Kuramitsu HK, Deng CX (2005) Identification and characterization of a novel adhesin unique to oral fusobacteria. J Bacteriol 187: 5330–5340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YW, Shen T, Chung P, Buhimschi IA, Buhimschi CS (2009) Uncultivated bacteria as etiologic agents of intra‐amniotic inflammation leading to preterm birth. J Clin Microbiol 47: 38–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YW, Shi W, Huang GT, Kinder Haake S, Park NH, Kuramitsu H, Genco RJ (2000) Interactions between periodontal bacteria and human oral epithelial cells: Fusobacterium nucleatum adheres to and invades epithelial cells. Infect Immun 68: 3140–3146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh YY, Tung SY, Pan HY, Yen CW, Xu HW, Lin YJ, Deng YF, Hsu WT, Wu CS, Li C (2018) Increased abundance of Clostridium and Fusobacterium in gastric microbiota of patients with gastric cancer in Taiwan. Sci Rep 8: 158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami A, Chung P, Han YW (2009) Complementation of the fadA mutation in Fusobacterium nucleatum demonstrates that the surface‐exposed adhesin promotes cellular invasion and placental colonization. Infect Immun 77: 3075–3079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang B (2017) Aerobic glycolysis and high level of lactate in cancer metabolism and microenvironment. Genes Dis 4: 25–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan CW, Lux R, Haake SK, Shi W (2009) The Fusobacterium nucleatum outer membrane protein RadD is an arginine‐inhibitable adhesin required for inter‐species adherence and the structured architecture of multispecies biofilm. Mol Microbiol 71: 35–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan CW, Ma X, Paranjpe A, Jewett A, Lux R, Kinder‐Haake S, Shi W (2010) Fusobacterium nucleatum outer membrane proteins Fap2 and RadD induce cell death in human lymphocytes. Infect Immun 78: 4773–4778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R, Head E, Sarsoza F, Saing T, Cotman CW, Necula M, Margol L, Wu J, Breydo L, Thompson JLet al (2007) Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener 2: 18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim GW, Kim YS, Lee SH, Park SG, Kim DH, Cho JY, Hahm KB, Hong SP, Yoo JH (2019) Periodontitis is associated with an increased risk for proximal colorectal neoplasms. Sci Rep 9: 7528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic A, Chun E, Robertson L, Glickman J, Gallini C, Michaud M, Clancy T, Chung D, Lochhead P, Hold Get al (2013) Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor‐immune microenvironment. Cell Host Microbe 14: 207–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, Ojesina AI, Jung J, Bass AJ, Tabernero Jet al (2012) Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res 22: 292–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Baba Y, Ishimoto T, Iwatsuki M, Hiyoshi Y, Miyamoto Y, Yoshida N, Wu R, Baba H (2019) Progress in characterizing the linkage between Fusobacterium nucleatum and gastrointestinal cancer. J Gastroenterol 54: 33–41 [DOI] [PubMed] [Google Scholar]
- Manson McGuire A, Cochrane K, Griggs AD, Haas BJ, Abeel T, Zeng Q, Nice JB, MacDonald H, Birren BW, Berger BWet al (2014) Evolution of invasion in a diverse set of Fusobacterium species. MBio 5: e01864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mima K, Nishihara R, Qian ZR, Cao Y, Sukawa Y, Nowak JA, Yang J, Dou R, Masugi Y, Song Met al (2016) Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut 65: 1973–1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsuhashi K, Nosho K, Sukawa Y, Matsunaga Y, Ito M, Kurihara H, Kanno S, Igarashi H, Naito T, Adachi Yet al (2015) Association of Fusobacterium species in pancreatic cancer tissues with molecular features and prognosis. Oncotarget 6: 7209–7220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nithianantham S, Xu M, Yamada M, Ikegami A, Shoham M, Han YW (2009) Crystal structure of FadA adhesin from Fusobacterium nucleatum reveals a novel oligomerization motif, the leucine chain. J Biol Chem 284: 3865–3872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz P, Bissada NF, Palomo L, Han YW, Al‐Zahrani MS, Panneerselvam A, Askari A (2009) Periodontal therapy reduces the severity of active rheumatoid arthritis in patients treated with or without tumor necrosis factor inhibitors. J Periodontol 80: 535–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson M, Ren J, Rubinstein MR, Taylor JA, Friedman RA, Shen B, Han YW (2020) Analysis of 16S rRNA genes reveals reduced Fusobacterial community diversity when translocating from saliva to GI sites. Gut Microbes 12: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein MR, Baik JE, Lagana SM, Han RP, Raab WJ, Sahoo D, Dalerba P, Wang TC, Han YW (2019) Fusobacterium nucleatum promotes colorectal cancer by inducing Wnt/beta‐catenin modulator Annexin A1. EMBO Rep 20: e47638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW (2013) Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E‐cadherin/β‐catenin signaling via its FadA adhesin. Cell Host Microbe 14: 195–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez I, Mahlke C, Yuan J (2003) Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature 421: 373–379 [DOI] [PubMed] [Google Scholar]
- Segata N, Haake SK, Mannon P, Lemon KP, Waldron L, Gevers D, Huttenhower C, Izard J (2012) Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples. Genome Biol 13: R42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondheimer N, Lindquist S (2000) Rnq1: an epigenetic modifier of protein function in yeast. Mol Cell 5: 163–172 [DOI] [PubMed] [Google Scholar]
- Strauss J, Kaplan GG, Beck PL, Rioux K, Panaccione R, Devinney R, Lynch T, Allen‐Vercoe E (2011) Invasive potential of gut mucosa‐derived fusobacterium nucleatum positively correlates with IBD status of the host. Inflamm Bowel Dis 17: 1971–1978 [DOI] [PubMed] [Google Scholar]
- Swidsinski A, Dorffel Y, Loening‐Baucke V, Theissig F, Ruckert Jc, Ismail M, Rau Wa, Gaschler D, Weizenegger M, Kuhn Set al (2011) Acute appendicitis is characterised by local invasion with Fusobacterium nucleatum/necrophorum. Gut 60: 34–40 [DOI] [PubMed] [Google Scholar]
- Tayeb‐Fligelman E, Tabachnikov O, Moshe A, Goldshmidt‐Tran O, Sawaya MR, Coquelle N, Colletier JP, Landau M (2017) The cytotoxic Staphylococcus aureus PSMalpha3 reveals a cross‐alpha amyloid‐like fibril. Science 355: 831–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temoin S, Chakaki A, Askari A, El‐Halaby A, Fitzgerald S, Marcus RE, Han YW, Bissada NF (2012a) Identification of oral bacterial DNA in synovial fluid of patients with arthritis with native and failed prosthetic joints. J Clin Rheumatol 18: 117–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temoin S, Wu KL, Wu V, Shoham M, Han YW (2012b) Signal peptide of FadA adhesin from Fusobacterium nucleatum plays a novel structural role by modulating the filament's length and width. FEBS Lett 586: 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas AM, Manghi P, Asnicar F, Pasolli E, Armanini F, Zolfo M, Beghini F, Manara S, Karcher N, Pozzi Cet al (2019) Metagenomic analysis of colorectal cancer datasets identifies cross‐cohort microbial diagnostic signatures and a link with choline degradation. Nat Med 25: 667–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomkovich S, Dejea CM, Winglee K, Drewes JL, Chung L, Housseau F, Pope JL, Gauthier J, Sun X, Mühlbauer Met al (2019) Human colon mucosal biofilms from healthy or colon cancer hosts are carcinogenic. J Clin Invest 129: 1699–1712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tukel C, Wilson RP, Nishimori JH, Pezeshki M, Chromy BA, Baumler AJ (2009) Responses to amyloids of microbial and host origin are mediated through toll‐like receptor 2. Cell Host Microbe 6: 45–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umana A, Sanders BE, Yoo CC, Casasanta MA, Udayasuryan B, Verbridge SS, Slade DJ (2019) Utilizing whole Fusobacterium genomes to identify, correct, and characterize potential virulence protein families. J Bacteriol 201: e00273‐19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Buhimschi CS, Temoin S, Bhandari V, Han YW, Buhimschi IA (2013) Comparative microbial analysis of paired amniotic fluid and cord blood from pregnancies complicated by preterm birth and early‐onset neonatal sepsis. PLoS One 8: e56131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirbel J, Pyl PT, Kartal E, Zych K, Kashani A, Milanese A, Fleck JS, Voigt AY, Palleja A, Ponnudurai Ret al (2019) Meta‐analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer. Nat Med 25: 679–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Yamada M, Li M, Liu H, Chen SG, Han YW (2007) FadA from Fusobacterium nucleatum utilizes both secreted and nonsecreted forms for functional oligomerization for attachment and invasion of host cells. J Biol Chem 282: 25000–25009 [DOI] [PubMed] [Google Scholar]
- Yu TC, Guo F, Yu Y, Sun T, Ma D, Han J, Qian Y, Kryczek I, Sun D, Nagarsheth Net al (2017) Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell 170: 548–563.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Data Availability Statement
The illumina reads of Fn 12230 and the lam mutant, PacBio reads for Fn 12230, and de novo genome assembly of Fn12230 are deposited in NCBI BioProject database with the accession number PRJNA632750. https://www.ncbi.nlm.nih.gov/bioproject/PRJNA632750.