

Abstract
The genesis of the headache phase in migraine with aura is thought to be mediated by cortical spreading depression (CSD) and the subsequent activation and sensitization of primary afferent neurons that innervate the intracranial meninges and their related large vessels. Yet, the exact mechanisms underlying this peripheral meningeal nociceptive response remain poorly understood. We investigated the relative contribution of cortical astrocytes to CSD-evoked meningeal nociception using extracellular single-unit recording of meningeal afferent activity and 2-photon imaging of cortical astrocyte calcium activity, in combination with 2 pharmacological approaches to inhibit astrocytic function. We found that fluoroacetate and l-α-aminoadipate, which inhibit astrocytes through distinct mechanisms, suppressed CSD-evoked afferent mechanical sensitization, but did not affect afferent activation. Pharmacological inhibition of astrocytic function, which ameliorated meningeal afferents’ sensitization, reduced basal astrocyte calcium activity but had a minimal effect on the astrocytic calcium wave during CSD. We propose that calcium-independent signaling in cortical astrocytes plays an important role in driving the sensitization of meningeal afferents and the ensuing intracranial mechanical hypersensitivity in migraine with aura.
Keywords: Migraine, Cortical spreading depression, Meningeal afferents, Mechanosensitivity, Astrocytes, Two-photon calcium imaging
1. Introduction
Migraine is one of the most common neurological disorders and the leading cause of disability in people younger than 50 years.9,41 Migraine consists of episodic attacks characterized by moderate to severe throbbing headache, which is aggravated by physical activity and accompanied by several other symptoms, including nausea and light sensitivity. Although the origin of migraine remains unclear, a large body of work supports the notion that the severe headache and its exacerbation during physical activity are driven by the prolonged activation and sensitization of trigeminal nociceptive afferent neurons that innervate the intracranial meninges and their large vessels.27 The mechanisms underlying the genesis of these migraine-related meningeal nociceptive responses are thought to involve the local action of algesic mediators released near the afferent’s meningeal nerve endings.27 Yet, the endogenous conditions leading to the release of these factors remain unclear.
One key to better understanding the endogenous processes underlying the genesis of meningeal nociception in migraine is the phenomenon of cortical spreading depression (CSD). This abnormal cortical event involves a massive wave of neuronal depolarization, followed by a transient depression of cortical synaptic activity.34 Cortical spreading depression is considered the underlying cause of the aura symptoms, which precede the headache in some attacks, and a major event that produces prolonged activation and mechanical sensitization of meningeal nociceptive afferents52,54,55 and the ensuing headache pain of migraine.17
The massive neural activation during a CSD event is associated with a robust intracellular astrocyte Ca2+ wave.11,37 Activation of cortical astrocytes and the ensuing intracellular astrocyte Ca2+ elevations during CSD have been suggested to mediate the associated vasoconstriction of meningeal pial arteries.7 In addition to releasing vasoactive mediators that act on meningeal vessels, activated astrocytes can also release a host of proinflammatory algesic molecules47 that could potentially interact with meningeal nociceptive afferents and drive migraine headache.23 Here, we tested whether astrocyte-to-meninges signaling might contribute to the CSD-evoked activation and sensitization of meningeal afferents. Using in vivo single-unit recording in rats, we show that pharmacological inhibition of cortical astrocytic function can block the CSD-evoked meningeal afferent mechanical sensitization without affecting the associated increase in ongoing activity. Furthermore, data obtained using in vivo 2-photon calcium imaging suggest that Ca2+-independent signaling in cortical astrocytes drives mechanical sensitization of meningeal afferents after CSD.
2. Material and methods
2.1. Animals
All experiments were approved by the Institutional Animal Care and Use Committee of the Beth Israel Deaconess Medical Centre and followed the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines.25 Animals (Sprague-Dawley male rats, Taconic, Germantown, New York) were housed in pairs with food and water available ad libitum under a constant 12-hour light/dark cycle (lights on at 7:00 AM) at room temperature. All procedures and testing were conducted during the light phase of the cycle (9: 00 AM to 4:00 PM). Experimental animals were randomly assigned to different treatment groups. Based on our previous data, as well as power calculations (effect size d > 0.8, α < 0.05), the minimal sample size for the electrophysiological experiments was n = 15 (1 afferent/animal). No statistical methods were used to predetermine sample size for the imaging experiments, but our sample sizes are similar to those reported in previous publications, using similar techniques.40,49
2.2. Surgical preparation for electrophysiological experiments
Animals (220–250 g) were deeply anesthetized with urethane (1.5 g/kg, i.p.), as in all our previous recordings of meningeal afferents, and mounted on a stereotaxic frame (Kopf Instruments, Tujunga, CA). Core temperature was kept at 37.5 to 38°C using a homoeothermic control system. Animals were intubated and breathed spontaneously room air enriched with O2. Physiological parameters were collected throughout the experiments using PhysioSuite (Kent Scientific, Torrington, CT) and CapStar-100 (CWE). Data used in this report were obtained from animals exhibiting physiological levels of oxygen saturation (>95%), heart rate (350–450 beats/minute), and end-tidal CO2 (3.5%–4.5%). A saline-cooled dental drill was used to perform 3 separate craniotomies. One craniotomy was used to expose the left transverse sinus and the posterior part of the superior sagittal sinus, including the adjacent cranial dura, extending ~2 mm rostral to the transverse sinus. Another small craniotomy (1 × 1 mm) was centered 2 mm caudal and 2 mm lateral to bregma to allow insertion of the recording electrode into the trigeminal ganglion. An additional small burr hole (diameter, 0.5 mm) was drilled above the frontal cortex to induce CSD.57 The exposed dura was bathed with a modified synthetic interstitial fluid (SIF) containing 135 mM NaCl, 5 mM KCl, 1 mM MgCl2, 5 mM CaCl2, 10 mM glucose, and 10 mM HEPES, pH 7.2.
2.3. In vivo recording of meningeal afferent activity
Single-unit activity of meningeal afferents (1 afferent/rat) was recorded from their cell body in the ipsilateral (left) trigeminal ganglion. We used a contralateral approach, as previously described57 in which a platinum-coated tungsten microelectrode (impedance, 50–100 kΩ; FHC, Bowdoin, ME) is advanced through the right hemisphere into the left trigeminal ganglion using an angled (22.5°) trajectory. The insertion of the recording electrode using this approach does not produce CSD in the left cortical hemisphere.57 Meningeal afferents were identified by their constant response latency to electrical stimuli applied to the dura above the ipsilateral transverse sinus (0.5 ms pulse, 1–3 mA, 0.5 Hz). The response latency was used to calculate conduction velocity (CV), based on a conduction distance to the trigeminal ganglion of 12.5 mm.42 Neurons were classified as Aδ (1.5 ≤ CV ≤ 5 m/second) or C afferents (CV < 1.5 m/second). Neural activity was digitized and sampled at 10 kHz using power 1401/Spike2 interface (CED, Cambridge, United Kingdom). A real-time waveform discriminator (Spike2 software) was used to create and store a template for the action potential evoked by electrical stimulation, which was used to acquire and analyze afferent activity.
2.4. Assessment of changes in meningeal afferent mechanosensitivity
Mechanical receptive fields (RFs) of meningeal afferents were first identified by probing the exposed dura with a 2.0 g von Frey monofilament (Stoelting, Wood Dale, IL). The lowest mechanical threshold site was further determined using monofilaments, which exert lower forces (≥0.02 g). This site was then used to quantify the afferents’ mechanical responsiveness, using a servo force-controlled mechanical stimulator (Series 300B Dual Mode Servo System; Aurora Scientific, Aurora, ON). Mechanical stimuli were delivered using a flat-ended cylindrical plastic probe attached to the tip of the stimulator arm. One of 3 probe diameters (0.5, 0.8, or 1.1 mm) was selected for each neuron, depending on its sensitivity.57 Two ramp-and-hold mechanical stimuli were applied in each trial (rise time, 100 ms; stimulus width, 2 seconds; interstimulus interval, 120 seconds) and included an initial threshold stimulus (which typically evoked at baseline 1–4 Hz responses), followed by a suprathreshold stimulus (2–3× of the threshold pressure). Stimulus trials were delivered every 15 minutes throughout the experiment to minimize response desensitization.57 Ongoing activity was recorded continuously between the stimulation trials. Basal responses to mechanical stimuli were determined during at least 4 consecutive trials before the induction of CSD. Data were analyzed only from afferents that exhibited consistent responses (variation of <0.5 Hz for threshold responses and <1.5 Hz for suprathreshold responses) during baseline recordings.57 Post-CSD data were collected for at least 2 hours based on previous data.54–56
2.5. Induction and monitoring of cortical spreading depression
A single CSD episode was induced in the left frontal cortex by briefly inserting a glass micropipette (50 μm diameter) ~2 mm deep into the cortex for 2 seconds.57 Successful elicitation of CSD was ensured by simultaneously recording changes in cerebral blood flow with a laser Doppler flowmetry probe positioned within the craniotomy, just above the exposed dura, ~1 mm from the RF of the recorded unit. The laser Doppler signal was digitized (100 Hz) and recorded using the power 1401/Spike2. Laser Doppler recordings were obtained with the ambient lights turned off. Baseline LDF data were based on recordings conducted for at least 30 minutes before CSD. Induction of CSD was considered successful when the typical hemodynamic signature characterized by a large transient (~1–2 minutes) cortical hyperemia, followed by persistent (>1 hours) post-CSD oligemia was observed (Fig. 1A).13
Figure 1.
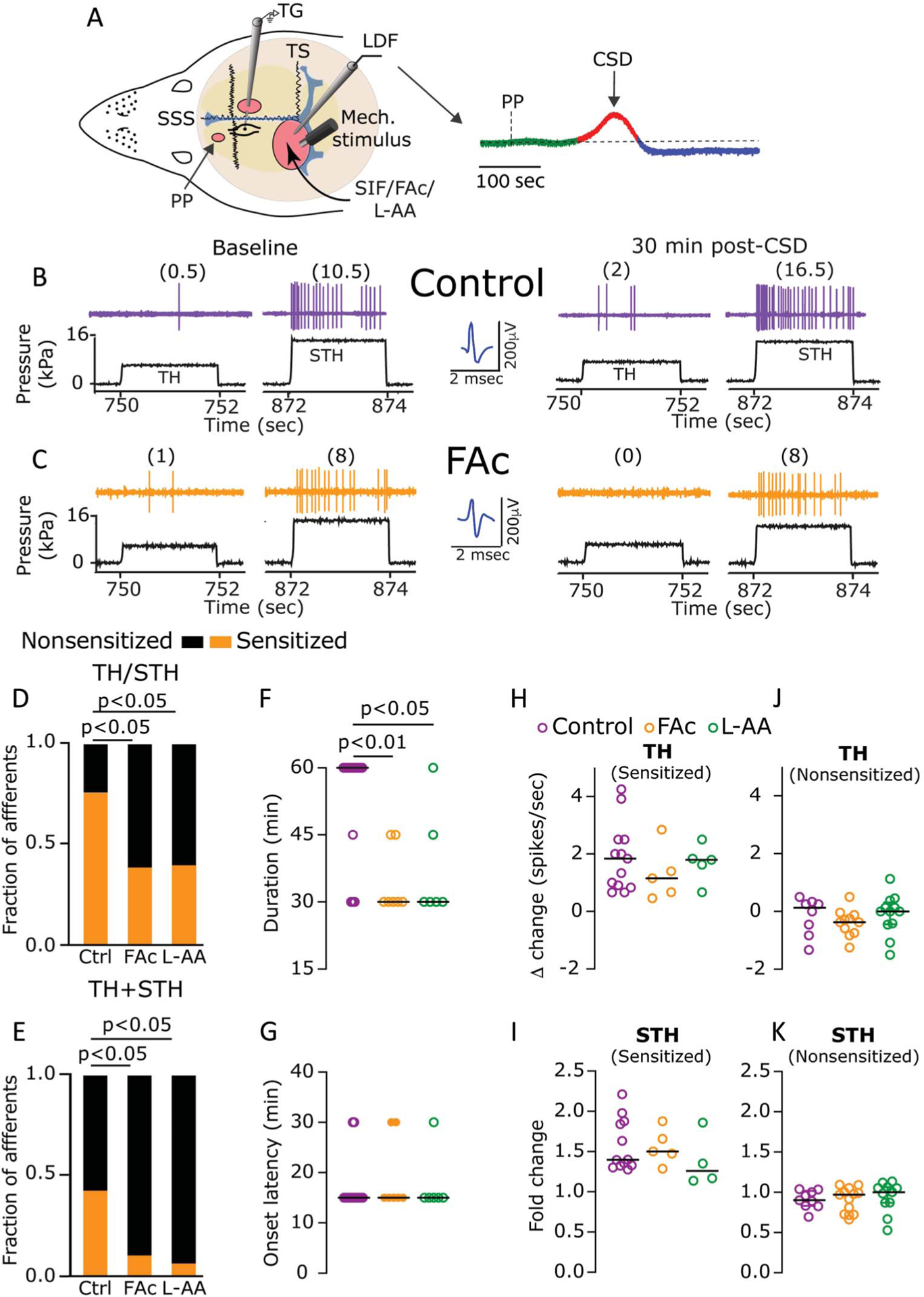
Astrocytes contribute to CSD-evoked mechanical sensitization of meningeal dural afferents. (A) Experimental setup: 3 skull openings (red ovals) were made. A small burr hole was drilled over the left frontal cortex to elicit a single CSD event using a pinprick (PP) stimulation. Afferent activity was recorded in the left trigeminal ganglion (TG) using a tungsten microelectrode inserted through a craniotomy made over the contralateral hemisphere. An ipsilateral craniotomy was made to expose a part of the left transverse sinus (TS) and superior sagittal sinus (SSS) and their vicinity to find and stimulate mechanosensitive meningeal afferents. Quantitative mechanical stimuli were delivered to the receptive field of afferents using a feedback-controlled mechanical stimulator. A laser Doppler flowmeter (LDF) probe was placed over the cortex near the receptive field of afferents to record changes in cerebral blood flow and validate CSD induction noninvasively. Induction of CSD was considered successful when the typical hemodynamic signature characterized by a large transient (~1–2 minutes) cortical cerebral hyperemia (red trace), followed by persistent (>1 hours) post-CSD oligemia (blue trace) was observed. (B and C) Examples of experimental trials depicting the responses of 2 afferents to threshold (TH) and suprathreshold (STH) mechanical stimuli (black traces) applied to their receptive field during baseline recording and then at 30 minutes after CSD elicitation in control (B) and FAc-treated animals (C). Responses in spikes/second are in parentheses. Note the sensitization in control but not after FAc treatment. When compared with controls (n = 21), the fraction of afferents that became sensitized at the TH or STH level (TH/STH, D), or at both levels (TH + STH, E) was lower in animals treated with FAc (n = 18), or L-AA (n = 15) (P < 0.05, χ2 test, treatment vs control). Inhibition of astrocytic function with FAc or L-AA also decreased the duration of the sensitization response (F); (P values indicate the Dunn post hoc test after a significant [P < 0.01] Kruskal–Wallis test between all treatments). The onset latency (G) or magnitude (H and I) of the sensitization response was not different between the 3 treatments (P > 0.05, Kruskal–Wallis). Inhibition of astrocytic function with FAc or L-AA did not affect TH or STH mechanical responses in nonsensitized afferents (J and K), (P > 0.05, Kruskal–Wallis). Data in (F–K) include all data points. Lines represent the median. CBF, cerebral blood flow; CSD, cortical spreading depression; FAc, fluoroacetate; L-AA, L-α-aminoadipate.
2.6. Adeno-associated virus injections
Three-week-old male rats were anesthetized with isoflurane (3.5% for induction, 1.5%–2% for maintenance) and placed in a stereotaxic apparatus. Body temperature was controlled by a heating pad. A lubricant eye ointment was applied to protect the cornea. An incision was made down the midline of the scalp to expose the skull. A 1-mm burr hole was drilled over the visual cortex, and a Hamilton syringe fitted with an injecting glass pipette was lowered 300 μm below the pia. To image astrocytic Ca2+ activity, we used the adeno-associated virus (AAV) 2/5-GfaABC1D-cyto-GCaMP6f viral vector. The viral prep was made using pZac2.1 gfaABC1D-cyto-GCaMP6f plasmid, a gift from Dr. Baljit Khakh (Addgene viral prep #52925-AAV5; http://n2t.net/addgene:52925;RRID:Addgene_52925).18 The human GFAP promoter (GfaABC1D) is highly selective for astrocytes.39 The AAV (5 μL, 7 × 1012 vg/mL) was injected into layer 2/3 of the visual cortex using a microinjection syringe pump (WPI, Ardmore, PA). The incision was then sutured, and rats were left to recover for 3 to 4 weeks.
2.7. In vivo 2-photon imaging of intracellular astrocyte Ca2+ activity
Rats previously injected with the AAV were anesthetized with urethane (1.5 g/kg, i.p.). Body temperature was maintained using a heating pad. A skin incision was made to expose the calvaria, and a small burr hole was drilled above the frontal cortex to induce CSD with a pinprick as above. To image GCaMP6f-labeled astrocyte Ca2+ activity, a 3-mm craniotomy was made, centered at the AAV injection site. At the end of the epidural pharmacological treatment (see below), the dura mater was removed, and low-melting agarose (2% dissolved in artificial cerebrospinal fluid) was placed on the exposed cortex. A cover glass was then fixed above the cortex together with a custom-made 2-pronged titanium headpost using C&B-Metabond (Parkell, Edgewood, NY) as previously described.15 A rubber imaging well was glued to the headpost to accommodate the water-immersion objective. Two-photon Ca2+ imaging of astrocytes in layers 2/3 of the visual cortex was performed in head-fixed rats using a resonant-scanning 2-photon microscope (Neurolabware: 15.5 frames/second; 796 × 512 pixels/frame). Single plane movies were made using a Nikon 16X, 0.8 NA objective, at a 4.8× digital zoom (~310 × 220 μm). Laser power at 920 nm (Mai Tai laser, Spectra Physics, Santa Clara, CA) measured below the objective was <40 mW. For capturing basal astrocyte intracellular Ca2+ activity, we obtained 5 minutes movies from 3 to 5 regions in each animal. To examine CSD-related astrocyte intracellular Ca2+ activity, we imaged one region for 5 minutes before CSD. The same region was then imaged again for 5 minutes to capture changes in Ca2+ activity during and after the CSD wave. Only one CSD event was triggered in each animal.
2.8. Drugs and treatment approaches
Molecular strategies that allow manipulation of astrocytic functions in a large area of the cortex in our rat model are currently unavailable. Hence, we used 2 pharmacological tools, fluoroacetate (FAc, MP Biomedicals, Solon, OH) and L-α-aminoadipate (L-AA, Sigma, St. Louis, MO), to test the role of astrocytes. FAc is preferentially taken up by astrocytes by a specific transporter, and disrupts their metabolism by inhibiting the tricarboxylic acid cycle enzyme aconitase.12 FAc has been widely used as an astrocyte-specific inhibitor.12,33 L-α-aminoadipate (L-AA, Sigma) is selectively incorporated into astrocytes by the cystine–glutamate antiporter. It induces astrocyte toxicity by affecting glutamate-dependent metabolism and protein synthesis, leading to loss of cellular integrity.1,3,33 L-α-aminoadipate has been previously used to selectively block astrocyte-mediated function in the adult rodent brain.1,5,21,24,30,51 The astrocyte inhibitors were applied topically to the dura mater to avoid damage to the meningeal afferents’ RF due to intracortical injection. FAc (100 mM) was diluted in SIF. L-α-aminoadipate (9 mM) was made in 1N HCl and further diluted in SIF with pH adjusted to 7.4. Drug concentrations applied to the dura were higher than those used previously for targeting astrocytes1,12 but were considered nontoxic to cortical neurons given an estimated dilution factor of 1 × 104 to 2.5 × 105 for such transmeningeal delivery approach.36,53 To further minimize potential off-target neurotoxic effects of FAc, we applied it for 15 minutes, beginning 60 minutes before the onset of CSD. L-α-aminoadipate, which requires prolonged treatment to affect astrocytes, was left on the dura for 4 hours before evoking CSD. Indirect neurotoxic effects of L-AA have been documented, but only beyond 24 hours after treatment.29
2.9. Quantification and statistical analysis
For analysis of afferent activity, N refers to the number of afferents. For comparisons of astrocytic intracellular Ca2+ activity, N refers to the number of Ca+2 events. All data were analyzed and plotted using Prism 8 software and are presented as the median and interquartile range (IQR). Group data in graphs show all data points and are plotted as scatter dot plots and the median. Statistical analyses are described in the results, figure legends, and below in this section. Differences in CSD-evoked afferent activation and sensitization propensities and rate of propagating/nonpropagating astrocytic Ca+2 events were analyzed using a 2-tailed χ2 test. All other data were analyzed using a nonparametric 2-tailed Mann–Whitney test or a Kruskal–Wallis test, followed by the Dunn post hoc test. P < 0.05 was considered significant.
2.9.1. Afferent data analyses
Offline analyses for meningeal afferent responses were conducted using template matching in Spike 2. Criteria used to consider meningeal afferent activation and sensitization responses were based on our previous studies.55–57 In brief, a prolonged increase in the ongoing activity of afferents was considered if the firing rate increased above the upper endpoint of the 95% CI calculated for the baseline mean for at least 10 minutes. During the CSD wave, acute afferent activation was considered when the discharge rate increased ≥30 seconds after the pinprick and lasted ≤120 seconds. The development of mechanical sensitization and its duration were based on the following criteria: threshold and/or suprathreshold responses increased to a level greater than the upper endpoint of the 95% CI calculated for the baseline mean, increased responsiveness began during the first 60 minutes post-CSD, and lasted for at least 30 minutes.
2.9.2. Astrocyte GCaMP imaging data processing and analyses
Videos of baseline and CSD-evoked astrocyte intracellular Ca2+ activity were denoised using a 3D Gaussian filter and then aligned using a discrete Fourier transform registration algorithm. The registered videos were then spatially downsampled by a factor of 4 and fed into the Ca2+ imaging analysis pipeline. Given the uniqueness of the large CSD-evoked astrocytic Ca2+ wave and the notion that the ROI-based analysis approach is limited to signals with fixed size and shape, we opted to use a recently developed non-ROI–based analysis approach that uses the Astrocyte Quantitative Analysis (AQuA) algorithm.49 The AQuA method captures regions of astrocytic Ca2+ signals (events) that can change size or location across time and allows assessment of event propagation. We used this method to perform a nonbiased population-level analysis of basal and CSD-evoked astrocytic intracellular Ca2+ spatiotemporal activity and their response to FAc treatment. Data reported include standard measurements used to describe astrocytic event physiology (amplitude and duration) and additional biologically relevant unique AQuA outputs related to event size and propagation.
3. Results
3.1. Astrocyte inhibitors curtail cortical spreading depression–evoked meningeal afferent mechanical sensitization
We first asked whether astrocytes contribute to the development of the CSD-evoked sensitization of meningeal afferents by studying the effects of FAc. We compared the mechanical responsiveness of 18 afferents (10 Aδ and 8 C) recorded from FAc-treated animals with 21 meningeal afferents (7 Aδ and 14 C afferents) recorded from vehicle-treated animals. The data obtained from the Aδ and C afferents were combined as they show similar sensitization characteristics after CSD.55 FAc pretreatment did not affect the afferent’s basal mechanosensitivity (Dunn post hoc test, P > 0.05 vs control for both TH and STH stimuli levels; Table 1), indicating no effect on the basal excitability of meningeal afferents. FAc-treated animals did not exhibit spontaneous CSD events, or more than a single event after the pinprick stimulus (18/18 cases, not shown), suggesting no direct or indirect neuronal effects of FAc that could increase vulnerability to spreading depression.4,45 FAc pretreatment, however, significantly inhibited the mechanical sensitization of meningeal afferents that developed after CSD (compare the examples in Figs. 1B and C). As Figure 1D depicts, when sensitization was defined as enhanced responsiveness at either the threshold (TH) or suprathreshold (STH) stimuli level, fewer sensitized afferents were detected in the FAc group when compared with the control group (7/18 vs 16/21; χ2 = 5.6; P < 0.05). A similar inhibition was detected when sensitization was defined as enhanced responsiveness at both the TH and STH stimuli levels (FAc: 2/18, Control: 9/21; χ2 = 4.8; P < 0.05, Fig. 1E). In addition to decreasing the propensity to develop sensitization, in afferents deemed sensitized, FAc treatment led to a decrease in the duration of the sensitized state when compared with the control group (30 [15.0] min vs 60 [15.0] min; Dunn post hoc test, P < 0.01, Fig. 1F). Astrocytic inhibition, however, had no effect on the latency for developing mechanical sensitization (15 [0.0] min vs 15 [15.0] minute, Dunn post hoc test, P > 0.05, Fig. 1G), or its magnitude, (TH, 1.2 (1.6) spikes/sec vs 1.8 [1.6] spikes/sec; STH, 1.5 [0.4]-fold vs 1.4 [0.5]-fold, Dunn post hoc test, P > 0.05 for both; Figs. 1H and I). Importantly, the inhibition of the CSD-evoked meningeal afferent sensitization was not due to a desensitization effect of repeated stimulation28 or delayed toxic effects of FAc, given that the post-CSD changes in the mechanical responses of nonsensitized afferents were similar in the control and FAc-treated animals (TH, −0.4 [1.6] spikes/sec vs 0.1 [1.1] spikes sec; STH 96.7 [30.6] % vs 90.0 [18.8] %, Dunn post hoc test, P > 0.05 for both, Figs. 1J and K).
Table 1.
Baseline mechanical response properties of meningeal dural afferents in animals treated with vehicle (control), FAc, or L-AA.
| Dural RFs | TH responses | STH responses | |
|---|---|---|---|
| Control (n = 21) | 2 (1) | 1.4 (1.3) | 8 (5.1) |
| FAc (n = 18) | 2 (1) | 1.2 (1.5) | 5 (5.8) |
| L-AA (n = 15) | 2 (1) | 1.6 (2.8) | 11 (9.1) |
Data are expressed as median (IQR). N represent the number of afferents tested (1 afferent/animal). The number of distinct dural receptive fields (RFs) was tested using a suprathreshold von Frey stimulus. Threshold and suprathreshold responses (in spikes/second) are based on activity recorded during a 2-s stimulation and represent the average of 4 trials conducted before CSD. Group differences were analyzed using the 2-tailed Kruskal–Wallis test and were not significant (P > 0.05) for the 3 parameters tested.
FAc, fluoroacetate; RFs, receptive fields; STH, suprathreshold; TH, threshold.
To further examine the role of astrocytes in CSD-evoked meningeal nociception, we employed another pharmacological approach to inhibit cortical astrocyte function using the astrocyte toxin, L-AA. Baseline mechanical responses of meningeal afferents after L-AA treatment were similar to those recorded in the control group (Table 1), indicating the lack of an off-target toxic effect on the afferents. Inhibition of cortical astrocyte-related function with L-AA did not affect the induction of CSD (15/15 cases, not shown), but significantly inhibited the development of the ensuing mechanical sensitization, similar to the effect observed after FAc treatment. When sensitization was defined as enhanced responsiveness at either TH or STH stimuli levels, only 6 of the 15 afferents were deemed sensitized (vs. 16/21 in controls, χ2 = 4.8; P < 0.05, Fig. 1D). Similarly, when sensitization was assessed as increased responsiveness at both the TH and STH stimuli levels, only 1 of the 16 were sensitized (compared with 9/21 in controls, χ2 = 6.9; P < 0.05, Fig. 1E). Similar to the effect observed in FAc-treated animals, the duration of the CSD-evoked afferent sensitization in L-AA-treated animals was also shorter when compared with controls (30.0 [18.8] min vs 60.0 [15.0] min, Dunn post hoc test, P < 0.05, Fig. 1F). In the few neurons that displayed sensitization after L-AA, their onset latency was similar to that observed in control (15.0(4) min, Kruskal–Wallis test, P > 0.05 L-AA vs control, Fig. 1G). Sensitization magnitudes were also similar at the TH level (1.8 [1.2] spikes/sec) and STH level (125.8 [58.9] %, Dunn post hoc test, P > 0.05 L-AA vs control, for both, Figs. 1H and I). Finally, L-AA treatment did not decrease mechanical responsiveness in nonsensitized afferents (TH, 0.0 [0.7] spikes/sec, STH 100.0 [26.6] %, Dunn post hoc test, P > 0.05 vs control for both, Figs. 1J and K), suggesting that the inhibitory effect on the sensitization was related to the blockade of CSD-evoked astrocytic function rather than a nonspecific delayed effect on the afferents.
3.2. Cortical spreading depression–evoked meningeal afferent activation is not affected by metabolic inhibition of astrocytes
Cortical spreading depression–evoked activation of meningeal afferents may involve distinct mechanisms from those mediating the accompanied mechanical sensitization response.55,57 Therefore, we asked next whether inhibition of astrocyte-related function, using FAc or L-AA, can also inhibit the CSD-evoked increases in the afferents’ ongoing activity. When compared with control animals treated with vehicle, FAc or L-AA did not affect baseline ongoing activity of Aδ or C afferents (Table 2). Inhibition of astrocyte function with FAc or L-AA did not affect the CSD-evoked acute activation of meningeal afferents. In FAc-treated animals we observed acute activation in 2 of the 10 Aδ and 2 of the 8 C afferents, ratios, not different from those observed in control animals (6/21 Aδ, χ2 = 0.26; P > 0.05; 11/32 C, χ2 = 0.26; P > 0.05; χ2 = 0.63; P > 0.05 for the combined population, Fig. 2C). Similarly, in L-AA-treated animals we observed acute activation in 2 of the 7 Aδ and 2 of the 8 C afferents, not different from that observed in the control group (χ2 = 0.0; P > 0.05 for Aδ; χ2 = 0.04; P > 0.05 for C afferents; χ2 = 0.45; P > 0.05 for the combined population, Fig. 2C).
Table 2.
Baseline ongoing activity in animals treated with vehicle, FAc, or L-AA.
| Aδ | C | Combined population | |
|---|---|---|---|
| Control | (n = 21); 0.05 (0.23) | (n = 32); 0.77 (1.4) | 0.16 (1.18) |
| FAc | (n = 10); 0.08 (0.50) | (n = 8); 0.88 (1.83) | 0.16 (1.23) |
| L-AA | (n = 7); 0.1 (0.70) | (n = 8); 0.85 (1.18) | 0.77 (0.98) |
Data are expressed as median (IQR). N represent the number of afferents tested (1 afferent/animal). Ongoing activity (in spikes/sec) is based on data collected for at least 45 minutes before CSD. Group differences were analyzed using 2-tailed Kruskal–Wallis test and were not significant (P > 0.05) for the population tested.
FAc, fluoroacetate; L-AA, L-α-aminoadipate.
Figure 2.
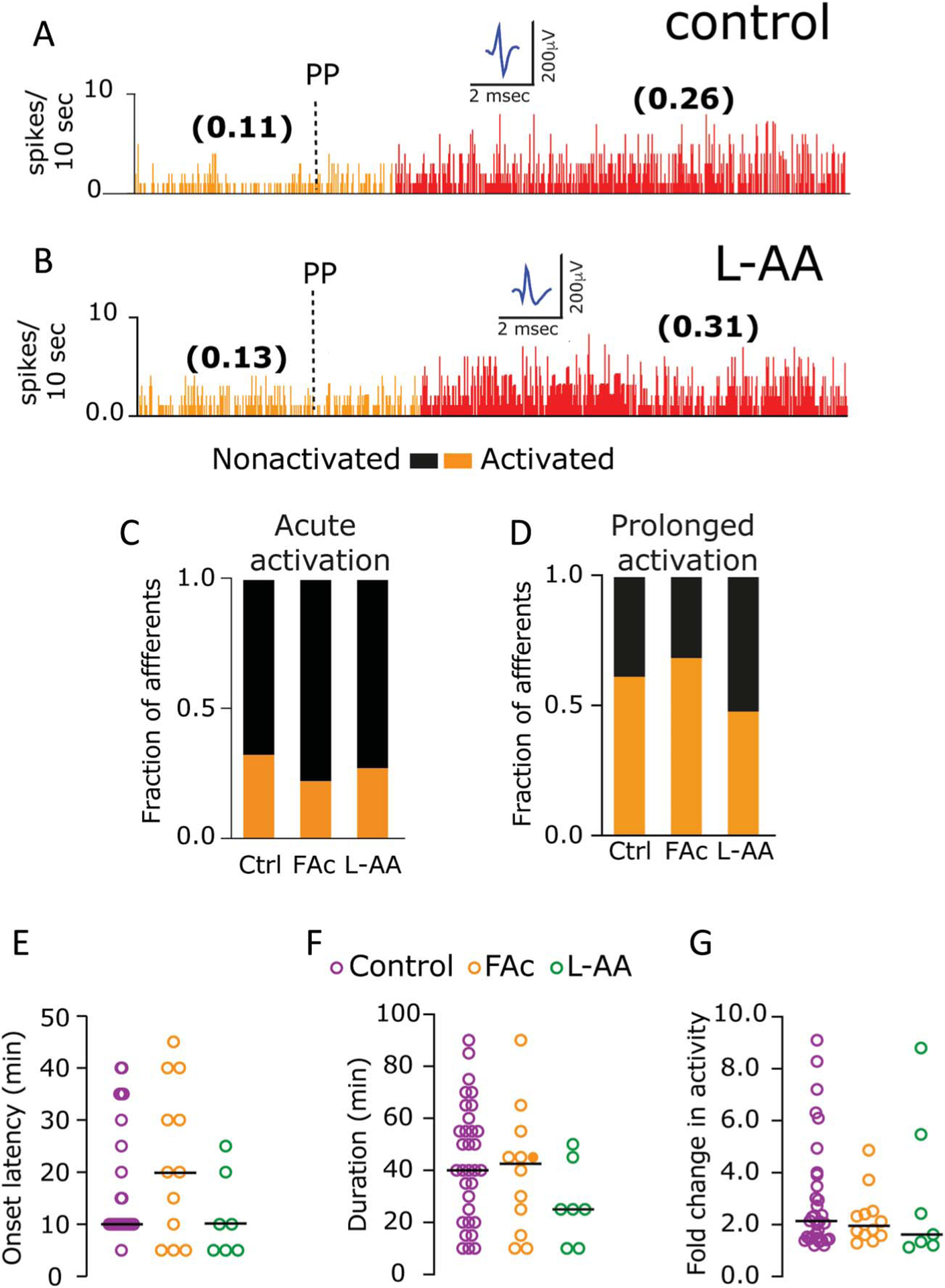
Cortical spreading depression-evoked meningeal afferent activation is not affected by pharmacological inhibition of astrocytes. (A and B) Examples of experimental trials depicting the CSD-evoked prolonged activation of meningeal afferents in control (A) and L-AA-treated animals (B). Ongoing activity (spikes/second) is in parentheses. Inhibition of astrocytic function with FAc or L-AA did not affect the propensity of the afferents to develop acute (C) or prolonged activity (D) after CSD (P > 0.05, χ2 test treatment vs control). None of the prolonged activation parameters were affected by the treatments, including onset latency (E), duration (F), and magnitude of activation (G) (P > 0.05 for all comparisons, Kruskal–Wallis test). Data in (E–G) include all data points. Lines represent the median. CSD, cortical spreading depression; FAc, fluoroacetate; L-AA, L-α-aminoadipate.
The induction of CSD-evoked prolonged increase in ongoing afferent activity was also not affected by the astrocytic inhibitors. In FAc-treated animals, we observed prolonged activation in 7 of the 10 Aδ and 5 of the 8 C afferents, not different from those observed in control animals (12/21 Aδ, χ2 = 0.47; P > 0.05; 20/32 C, χ2 = 0.01; P > 0.5; χ2 = 0.22; P > 0.05 for the combined population, Fig. 2D). In L-AA-treated animals, CSD-evoked prolonged activation was observed in 3 of the 7 Aδ and 4 of the 8 C afferents, not different from the control group (χ2 = 0.43; P > 0.05 for Aδ; χ2 = 0.42; P > 0.05 for C; χ2 = 0.90; P > 0.05 for the combined populations, Fig. 2D). Further analyses of the CSD-evoked prolonged activation of meningeal afferents revealed no significant differences in the response characteristics among the 3 treatment groups. These include similar onset latencies (control: 10.0 [13.8] min; FAc: 20.0 [32.5] min; L-AA: 10.0 [15.0] min; H = 4.7, Kruskal–Wallis, P > 0.05, Fig. 2E), durations (control: 40.0 [37.5] min; FAc: 42.5 [35.0] min; L-AA, 25.0 [35.2] min; H = 2.8, Kruskal–Wallis, P > 0.05, Fig. 2F), and response magnitudes (control: 2.2 [0.2]-fold; FAc: 1.8 [0.9]-fold; L-AA: 1.6 [3.2]-fold; H = 1.1, Kruskal–Wallis, P > 0.05, Fig. 2G).
3.3. Inhibition of cortical astrocyte metabolism decreases basal astrocytic intracellular Ca2+ signals but minimally affects cortical spreading depression–evoked astrocytic intracellular Ca2+ wave
A key mechanism by which astrocytes release their mediators and promote paracrine signaling is through an increase in intracellular Ca2+ activity.47 Therefore, we asked whether the large astrocyte Ca2+ wave during CSD could mediate meningeal afferents’ sensitization. FAc has been shown to inhibit astrocytic intracellular Ca2+ activity.38,46 Hence, to test the effect of FAc on CSD-evoked astrocytic intracellular Ca2+ activity, we expressed the genetically encoded Ca2+ indicator GCaMP6f6 in cortical astrocytes and used in vivo 2-photon microscopy to image Ca2+ activity of cortical astrocytes in anesthetized rats pretreated with FAc or vehicle control. Of note, because L-AA leads to astrocyte ablation,1,20,24 its effect could not be tested using this imaging approach.
We first examined whether the treatment approach and dose of FAc we used in the electrophysiological study affects basal astrocyte Ca2+ signal in vivo. We used a nonbiased, population-level analysis of astrocytic intracellular Ca2+ activity49 to compare 1109 events imaged in vehicle-treated rats (21 regions in 4 animals) with 692 events imaged in FAc-treated rats (22 imaging regions in 6 animals). FAc-treated animals displayed a significant reduction of basal astrocyte intracellular Ca2+ activity, reflected in all tested parameters. These include reduced number of events [FAc: 24.0(12.3) events/5 minutes, control: 44.0(71.1) events/5 minutes, Mann–Whitney P < 0.01, Fig. 3D], smaller events size [FAc: 21.1(18.1) √μm2, control: 31.4(27.1) √μm2, Mann–Whitney P < 0.0001, Fig. 3E], magnitude [FAc 0.2(1.5) dF/F, control: 0.3(1.2) dF/F, Mann–Whitney P < 0.0001, Fig. 3F], and shorter duration [FAc: 17.0(12.8) sec, control: 21.5(13.0) sec, Mann–Whitney P < 0.0001, Fig. 3G]. Analysis of the 500 largest events also revealed a significantly smaller number of propagating events (FAc: 280/500 propagating events, control: 360/500 propagating events, χ2 = 27.8; P < 0.0001, Fig. 3H).
Figure 3.
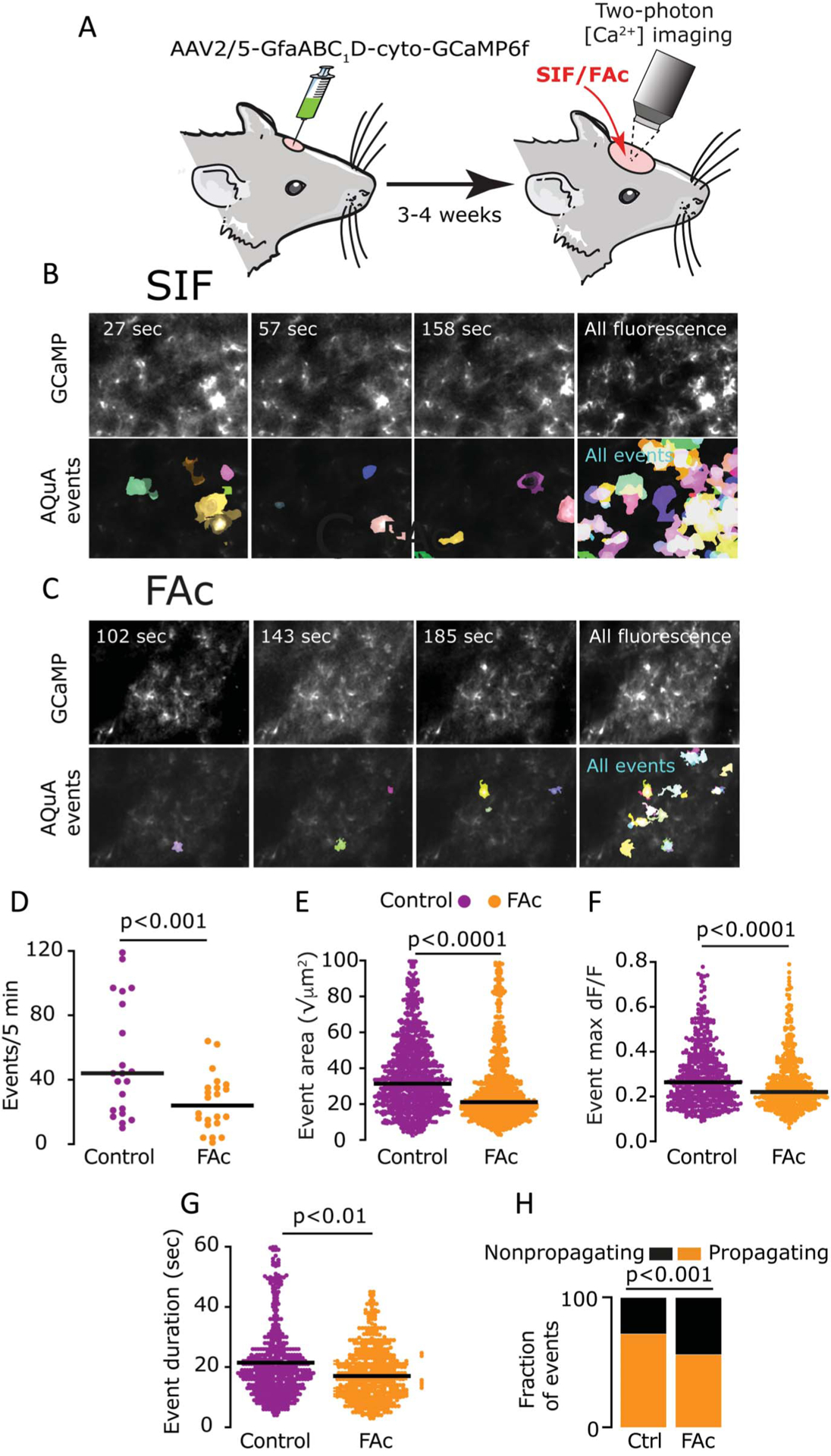
Acute FAc treatment curtails basal cortical astrocyte Ca2+ activity. (A) Experimental approach. A craniotomy was made to expose the visual cortex, and AAV2/5-gfaABC1D-cyto-GCaMP6f was injected to transduce cortical astrocytes for visualization of astrocytic Ca2+ signals. Two-photon imaging of basal astrocyte Ca2+ activity was conducted through a cranial window, 3 to 4 weeks later. (B and C) Representative frames (310 × 220 μm) from 5-min imaging experiments of astrocytic GCaMP in vehicle (B) and FAc-treated animals (C). AQuA-detected events (in color) are shown below the GCaMP fluorescence. The right columns display average GCaMP fluorescence (top) and all AQuA-detected events (bottom) from the entire video. (D–G) AQuA-based detection of basal cortical astrocytes’ Ca2+ dynamics showing the inhibitory effect of FAc (FAc, 692 events from 6 rats; vehicle, 1109 events from 4 rats), including a reduced number of events (D), decreased event area (E), maximum magnitude (dF/F) (F), and duration between 10% onset time to 10% offset time (G); P values are based on a Mann–Whitney test. (H) Data from the 500 largest events showing a reduction in the relative number of propagating events after FAc treatment (FAc, 280/500, control, 360/500, P < 0.001, χ2 test). FAc, fluoroacetate.
Unexpectedly, despite a robust inhibitory effect of basal intracellular Ca2+ activity, FAc treatment was largely ineffective in curtailing the CSD-evoked astrocytic Ca2+ wave. Overall, we observed a comparable ratio of propagating/nonpropagating Ca2+ events (FAc, 42/141, [6 animals], control, 50/153 [4 animals], χ2 = 0.5; P > 0.05, Fig. 4D), and similar event size (FAc: 8.5 [12.5] √μm2, control: 9.4 [11.2] √μm2, Mann–Whitney; P > 0.05, Fig. 4E) and magnitude (FAc 3.9 [2.1] dF/F, control: 3.6 [1.1] dF/F, Mann–Whitney; P > 0.05, Fig. 4F). There was, however, a modest reduction in event duration in response to FAc treatment (FAc: 11.0 [3.1] sec, control: 12.0 [5.2] sec, Mann–Whitney; P < 0.01, Fig. 4G).
Figure 4.
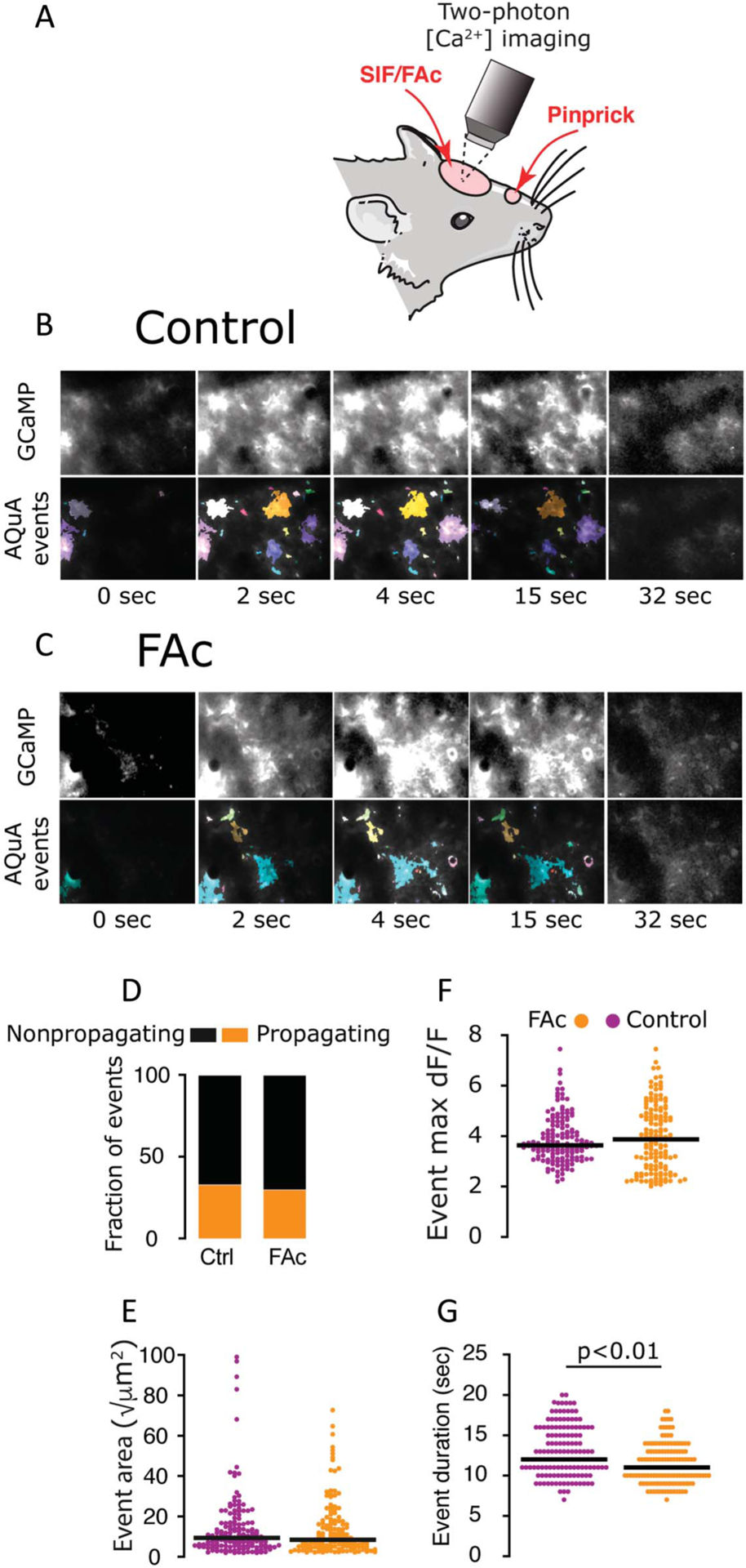
Effects of FAc treatment on CSD-evoked cortical astrocyte Ca2+ activity. (A) Experimental setup. A single CSD was triggered in the frontal cortex, and the ensuing astrocytic Ca2+ wave was imaged in the visual cortex, which was pretreated with vehicle (SIF) or FAc. (B and C) Representative frames capturing the astrocytic GCaMP fluorescence during the passage of the CSD wave in control (B, top) and FAc-treated animal (C, top). AQuA-detected events are shown in the bottom panels (image frames are 310 × 220 μm). (D) FAc treatment did not affect the ratio of propagating/nonpropagating Ca2+ events (FAc, 42/141, 6 animals, control, 50/153, 4 animals, P > 0.05, χ2 test. FAc treatment also had not effect on event size (E) or maximum dF/F (F); P > 0.05 vs control, Mann–Whitney. (G) FAc treatment was associated with a mild decrease in event duration (10% onset time to 10% offset time); P < 0.01 vs control, Mann–Whitney test. CSD, cortical spreading depression; FAc, fluoroacetate; SIF, synthetic interstitial fluid.
4. Discussion
Here, using in vivo single-unit recording in anesthetized rats coupled with 2 distinct pharmacological approaches to inhibit astrocytic function, we provide evidence for an astrocyte-dependent process that mediates CSD-evoked mechanical sensitization of meningeal afferents. Our data further support the idea of an astrocyte-independent activation of meningeal afferents after CSD and is congruent with the notion that the mechanisms underlying the activation and sensitization of meningeal afferents after CSD involve distinct mechanisms.57
Astrocytes release many of their signaling mediators in a Ca2+-dependent manner. However, there is also evidence for astrocytic signaling involving Ca2+-independent mediator release.47 Our finding that the same acute FAc treatment approach that blocked the CSD-evoked sensitization of meningeal afferents had a minimal effect on the astrocytic Ca2+ wave during CSD points to an astrocytic mechanism that does not depend on intracellular Ca2+ signaling in astrocytes. However, we cannot exclude the possibility that even the minor decrease in the duration of Ca2+ events observed after FAc treatment (<10%) could play a role. The negligible effect of FAc on the CSD-evoked astrocytic Ca2+ wave was surprising given previous studies documenting its ability to inhibit astrocyte Ca2+ elevations (this study14,38). The robustness of the CSD-evoked astrocytic Ca2+ elevations and its reliance on multiple intracellular processes, including IP3 receptor type 2 (IP3R2) and aquaporin-4 signaling10 may explain the poor responsiveness to FAc treatment. Which Ca2+-independent astrocytic signaling might play a role in mediating the sensitization of meningeal afferents after CSD? Recent work suggests that astrocytes can release arachidonic acid–derived lipid mediators, such as PGE2, in a Ca2+-independent manner.48 Our previous finding that blocking cyclooxygenase and ensuing synthesis of arachidonic acid–related mediators, including PGE2, can selectively inhibit CSD-evoked sensitization of meningeal afferents57 points to a possible contribution of astrocyte-derived prostanoids. The finding that FAc can block the release of PGE2 from astrocytes8 further supports this notion. Another astrocyte mediator that may contribute to the sensitization of meningeal afferents is ATP.22 Astrocytes can release ATP through a Ca2+-independent process involving membrane pores, such as the P2X7 purinergic channel, pannexin-1, and anion channels.26,31,32,50 Importantly, FAc treatment can impair astrocytic ATP production and activity-dependent release,16,44 potentially by blocking P2X7 activity.19 Whether ATP released by astrocytes arrives at the dural level and directly contributes to the sensitization of meningeal dural afferents remains unclear given that local action of ATP causes dural afferent activation,54 a CSD-evoked nociceptive response not blocked by astrocytes inhibitors.
Although FAc and L-AA are widely considered selective astrocyte inhibitors,33 in large part because of preferential uptake by astrocytes, the possibility of off-target neurotoxic effects remains a concern. Three lines of reasoning argue against off-target effects as a major confounding variable in this study. First, studies demonstrating neurotoxic effects were conducted using concentrations that are orders of magnitude greater than those expected to reach the cortex using our transmeningeal delivery approach.12,29 Although the concentrations of the inhibitors applied to the dura were higher and potentially toxic to meningeal afferents, we did not observe any nonspecific effects on the afferents’ responsiveness. Second, prolonged exposure to higher concentrations of FAc has been shown to increase vulnerability to spreading depression,3,43 but in our study, FAc treatment neither led to spontaneous CSD events nor to more than one event following the triggering stimulus. Third, because FAc and L-AA affect astrocytes through distinct mechanisms, the finding that both treatments had similar effects on the afferents’ responses to CSD cannot be easily explained as the result of a specific off-target neurotoxic effect.
The cortex-to-meninges signaling pathways by which astrocytes contribute to the sensitization of meningeal afferents after CSD remain to be defined. According to a key migraine hypothesis, CSD leads to release of proinflammatory mediators in the parenchyma that diffuse into the CSF-filled subarachnoid space and stimulate trigeminal sensory nerve endings that innervate this leptomeningeal compartment. The subsequent antidromic activation of trigeminal afferent collaterals in the dura mater gives rise to dural neurogenic inflammation2,23 that sustains the activation meningeal dural afferents and promotes their sensitization.27 Our data, however, does not support this hypothesis, given that inhibition of astrocytic function did not affect the prolonged activation of meningeal afferents. This finding further argues against a mechanism linking astrocyte activation and the development of neurogenic dural vasodilation after CSD.23 We propose 2 alternative hypotheses to explain the sensitization of meningeal dural afferents after CSD. The first scenario involves the direct nociceptive action of astrocytic mediators that traverse the arachnoid layer near the dural sinuses,35 which is densely innervated by mechanosensitive dural afferents.43 In a second scenario, astrocytes release gliotransmitters that affect other cells, including cortical neurons and nonneuronal cells in the parenchyma, subarachnoid space, and dura, which produce the final sensitizing mediators.
Astrocytes are thought to regulate numerous events related to CSD, including the vasoconstriction of cortical arterioles and recovery of cortical neural activity.37 Our findings support another critical aspect of astrocytic function in CSD, namely the development of meningeal afferents’ mechanical sensitization. This nociceptive change is believed to mediate the exacerbations of the migrainous headache pain during intracranial mechanical deformations such as those occurring during coughing, straining, bending over, or rapid head movement. Our findings may shed new light on the pathophysiological mechanisms underlying migraine headache and support further research on the development of novel therapeutic approaches.
Acknowledgments
The study was funded by NIH grants: NS086830, NS078263, NS101405 to DL. The authors have no conflict of interest to declare.
Footnotes
Conflict of interest statement
The authors have no conflicts of interest to declare.
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
References
- [1].Banasr M, Duman RS. Glial loss in the prefrontal cortex is sufficient to induce depressive-like behaviors. Biol Psychiatry 2008;64:863–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bolay H, Reuter U, Dunn AK, Huang Z, Boas DA, Moskowitz MA. Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat Med 2002;8:136–42. [DOI] [PubMed] [Google Scholar]
- [3].Brown DR, Kretzschmar HA. The glio-toxic mechanism of alpha-aminoadipic acid on cultured astrocytes. J Neurocytol 1998;27:109–18. [DOI] [PubMed] [Google Scholar]
- [4].Canals S, Larrosa B, Pintor J, Mena MA, Herreras O. Metabolic challenge to glia activates an adenosine-mediated safety mechanism that promotes neuronal survival by delaying the onset of spreading depression waves. J Cereb Blood Flow Metab 2008;28:1835–44. [DOI] [PubMed] [Google Scholar]
- [5].Chen G, Park CK, Xie RG, Berta T, Nedergaard M, Ji RR. Connexin-43 induces chemokine release from spinal cord astrocytes to maintain late-phase neuropathic pain in mice. Brain 2014;137:2193–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, Looger LL, Svoboda K, Kim DS. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 2013;499:295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chuquet J, Hollender L, Nimchinsky EA. High-resolution in vivo imaging of the neurovascular unit during spreading depression. J Neurosci 2007;27: 4036–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Clasadonte J, Poulain P, Hanchate NK, Corfas G, Ojeda SR, Prevot V. Prostaglandin E2 release from astrocytes triggers gonadotropin-releasing hormone (GnRH) neuron firing via EP2 receptor activation. Proc Natl Acad Sci U S A 2011;108:16104–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Collaborators GBDH. Global, regional, and national burden of migraine and tension-type headache, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 2018;17:954–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Enger R, Dukefoss DB, Tang W, Pettersen KH, Bjørnstad DM, Helm PJ, Jensen V, Sprengel R, Vervaeke K, Ottersen OP, Nagelhus EA. Deletion of aquaporin-4 curtails extracellular glutamate elevation in cortical spreading depression in awake mice. Cereb Cortex 2017;27:24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Enger R, Tang W, Vindedal GF, Jensen V, Johannes Helm P, Sprengel R, Looger LL, Nagelhus EA. Dynamics of ionic shifts in cortical spreading depression. Cereb Cortex 2015;25:4469–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fonnum F, Johnsen A, Hassel B. Use of fluorocitrate and fluoroacetate in the study of brain metabolism. Glia 1997;21:106–13. [PubMed] [Google Scholar]
- [13].Gariepy H, Zhao J, Levy D. Differential contribution of COX-1 and COX-2 derived prostanoids to cortical spreading depression-Evoked cerebral oligemia. J Cereb Blood Flow Metab 2017;37:1060–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ghosh A, Wyss MT, Weber B. Somatotopic astrocytic activity in the somatosensory cortex. Glia 2013;61:601–10. [DOI] [PubMed] [Google Scholar]
- [15].Goldey GJ, Roumis DK, Glickfeld LL, Kerlin AM, Reid RC, Bonin V, Schafer DP, Andermann ML. Removable cranial windows for long-term imaging in awake mice. Nat Protoc 2014;9:2515–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gordon GRJ, Baimoukhametova DV, Hewitt SA, Rajapaksha WRAKJS, Fisher TE, Bains JS. Norepinephrine triggers release of glial ATP to increase postsynaptic efficacy. Nat Neurosci 2005;8:1078–86. [DOI] [PubMed] [Google Scholar]
- [17].Harriott AM, Chung DY, Uner A, Bozdayi RO, Morais A, Takizawa T, Qin T, Ayata C. Optogenetic spreading depression elicits trigeminal pain and anxiety behavior. Ann Neurol 2020;89:99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Haustein MD, Kracun S, Lu XH, Shih T, Jackson-Weaver O, Tong X, Xu J, Yang XW, O’Dell TJ, Marvin JS, Ellisman MH, Bushong EA, Looger LL, Khakh BS. Conditions and constraints for astrocyte calcium signaling in the hippocampal mossy fiber pathway. Neuron 2014;82:413–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Heinrich A, Ando RD, Turi G, Rozsa B, Sperlagh B. K+ depolarization evokes ATP, adenosine and glutamate release from glia in rat hippocampus: a microelectrode biosensor study. Br J Pharmacol 2012; 167:1003–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Jakel S, Dimou L. Glial cells and their function in the adult brain: a journey through the history of their ablation. Front Cell Neurosci 2017;11:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Jiang BC, Cao DL, Zhang X, Zhang ZJ, He LN, Li CH, Zhang WW, Wu XB, Berta T, Ji RR, Gao YJ. CXCL13 drives spinal astrocyte activation and neuropathic pain via CXCR5. J Clin Invest 2016;126:745–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Joseph EK, Green PG, Levine JD. ATP release mechanisms of endothelial cell-mediated stimulus-dependent hyperalgesia. J Pain 2014;15:771–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Karatas H, Erdener SE, Gursoy-Ozdemir Y, Lule S, Eren-Kocak E, Sen ZD, Dalkara T. Spreading depression triggers headache by activating neuronal Panx1 channels. Science 2013;339:1092–5. [DOI] [PubMed] [Google Scholar]
- [24].Khurgel M, Koo AC, Ivy GO. Selective ablation of astrocytes by intracerebral injections of alpha-aminoadipate. Glia 1996;16:351–8. [DOI] [PubMed] [Google Scholar]
- [25].Kilkenny C, Browne WJ, Cuthi I, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. Vet Clin Pathol 2012;41:27–31. [DOI] [PubMed] [Google Scholar]
- [26].Kovacs G, Kornyei Z, Toth K, Baranyi M, Brunner J, Neubrandt M, Denes A, Sperlagh B. Modulation of P2X7 purinergic receptor activity by extracellular Zn(21) in cultured mouse hippocampal astroglia. Cell Calcium 2018;75:1–13. [DOI] [PubMed] [Google Scholar]
- [27].Levy D, Labastida-Ramirez A, MaassenVanDenBrink A. Current understanding of meningeal and cerebral vascular function underlying migraine headache. Cephalalgia 2019;39:1606–22. [DOI] [PubMed] [Google Scholar]
- [28].Levy D, Strassman AM. Mechanical response properties of A and C primary afferent neurons innervating the rat intracranial dura. J Neurophysiol 2002;88:3021–31. [DOI] [PubMed] [Google Scholar]
- [29].Lima A, Sardinha VM, Oliveira AF, Reis M, Mota C, Silva MA, Marques F, Cerqueira JJ, Pinto L, Sousa N, Oliveira JF. Astrocyte pathology in the prefrontal cortex impairs the cognitive function of rats. Mol Psychiatry 2014;19:834–41. [DOI] [PubMed] [Google Scholar]
- [30].Liu T, Han Q, Chen G, Huang Y, Zhao LX, Berta T, Gao YJ, Ji RR. Toll-like receptor 4 contributes to chronic itch, alloknesis, and spinal astrocyte activation in male mice. PAIN 2016;157:806–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Nikolic L, Nobili P, Shen W, Audinat E. Role of astrocyte purinergic signaling in epilepsy. Glia 2020;68:1677–91. [DOI] [PubMed] [Google Scholar]
- [32].Pan HC, Chou YC, Sun SH. P2X7 R-mediated Ca(2+) -independent d-serine release via pannexin-1 of the P2X7 R-pannexin-1 complex in astrocytes. Glia 2015;63:877–93. [DOI] [PubMed] [Google Scholar]
- [33].Pena-Ortega F, Rivera-Angulo AJ, Lorea-Hernandez JJ. Pharmacological tools to study the role of astrocytes in neural network functions. Adv Exp Med Biol 2016;949:47–66. [DOI] [PubMed] [Google Scholar]
- [34].Pietrobon D, Moskowitz MA. Pathophysiology of migraine. Annu Rev Physiol 2013;75:365–91. [DOI] [PubMed] [Google Scholar]
- [35].Ringstad G, Eide PK. Cerebrospinal fluid tracer efflux to parasagittal dura in humans. Nat Commun 2020;11:354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Roth TL, Nayak D, Atanasijevic T, Koretsky AP, Latour LL, McGavern DB. Transcranial amelioration of inflammation and cell death after brain injury. Nature 2014;505:223–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Seidel JL, Escartin C, Ayata C, Bonvento G, Shuttleworth CW. Multifaceted roles for astrocytes in spreading depolarization: a target for limiting spreading depolarization in acute brain injury? Glia 2016;64:5–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Shigetomi E, Bowser DN, Sofroniew MV, Khakh BS. Two forms of astrocyte calcium excitability have distinct effects on NMDA receptor-mediated slow inward currents in pyramidal neurons. J Neurosci 2008; 28:6659–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Shigetomi E, Bushong EA, Haustein MD, Tong X, Jackson-Weaver O, Kracun S, Xu J, Sofroniew MV, Ellisman MH, Khakh BS. Imaging calcium microdomains within entire astrocyte territories and endfeet with GCaMPs expressed using adeno-associated viruses. J Gen Physiol 2013;141:633–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Srinivasan R, Huang BS, Venugopal S, Johnston AD, Chai H, Zeng H, Golshani P, Khakh BS. Ca(2+) signaling in astrocytes from Ip3r2(−/−) mice in brain slices and during startle responses in vivo. Nat Neurosci 2015;18:708–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Steiner TJ, Stovner LJ, Vos T, Jensen R, Katsarava Z. Migraine is first cause of disability in under 50s: will health politicians now take notice? J Headache Pain 2018;19:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Strassman AM, Raymond SA, Burstein R. Sensitization of meningeal sensory neurons and the origin of headaches. Nature 1996;384:560–4. [DOI] [PubMed] [Google Scholar]
- [43].Strassman AM, Weissner W, Williams M, Ali S, Levy D. Axon diameters and intradural trajectories of the dural innervation in the rat. J Comp Neurol 2004;473:364–76. [DOI] [PubMed] [Google Scholar]
- [44].Swanson RA, Graham SH. Fluorocitrate and fluoroacetate effects on astrocyte metabolism in vitro. Brain Res 1994;664:94–100. [DOI] [PubMed] [Google Scholar]
- [45].Szerb JC. Glutamate release and spreading depression in the fascia dentata in response to microdialysis with high K1: role of glia. Brain Res 1991;542:259–65. [DOI] [PubMed] [Google Scholar]
- [46].Vance KM, Rogers RC, Hermann GE. PAR1-activated astrocytes in the nucleus of the solitary tract stimulate adjacent neurons via NMDA receptors. J Neurosci 2015;35:776–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Verkhratsky A, Nedergaard M. Physiology of astroglia. Physiol Rev 2018; 98:239–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wang F, Bradshaw HB, Pena S, Jablonska B, Xavier J, Gong S, Li B, Chandler-Militello D, Bekar LK, Smith NA. Calcium-independent astrocytic lipid release modulates neuronal activity through Kv channels. BiorXiv 2020. doi: 10.1101/2020.01.12.903393. [DOI] [Google Scholar]
- [49].Wang Y, DelRosso NV, Vaidyanathan TV, Cahill MK, Reitman ME, Pittolo S, Mi X, Yu G, Poskanzer KE. Accurate quantification of astrocyte and neurotransmitter fluorescence dynamics for single-cell and population-level physiology. Nat Neurosci 2019;22:1936–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Xiong Y, Sun S, Teng S, Jin M, Zhou Z. Ca(2+)-Dependent and Ca(2+)-independent ATP release in astrocytes. Front Mol Neurosci 2018;11:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Xu HL, Koenig HM, Ye S, Feinstein DL, Pelligrino DA. Influence of the glia limitans on pial arteriolar relaxation in the rat. Am J Physiol Heart Circ Physiol 2004;287:H331–339. [DOI] [PubMed] [Google Scholar]
- [52].Zhang X, Levy D, Noseda R, Kainz V, Jakubowski M, Burstein R. Activation of meningeal nociceptors by cortical spreading depression: implications for migraine with aura. J Neurosci 2010;30:8807–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Zhao J, Bree D, Harrington MG, Strassman AM, Levy D. Cranial dural permeability of inflammatory nociceptive mediators: potential implications for animal models of migraine. Cephalalgia 2017;37:1017–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zhao J, Levy D. Modulation of intracranial meningeal nociceptor activity by cortical spreading depression: a reassessment. J Neurophysiol 2015; 113:2778–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhao J, Levy D. Cortical spreading depression promotes persistent mechanical sensitization of intracranial meningeal afferents: implications for the intracranial mechanosensitivity of migraine. eNeuro 2016;3: ENEURO.0287–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zhao J, Levy D. The CGRP receptor antagonist BIBN4096 inhibits prolonged meningeal afferent activation evoked by brief local K(+) stimulation but not cortical spreading depression-induced afferent sensitization. Pain Rep 2018;3:e632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zhao J, Levy D. Dissociation between CSD-evoked metabolic perturbations and meningeal afferent activation and sensitization: implications for mechanisms of migraine headache onset. J Neurosci 2018;38:5053–66. [DOI] [PMC free article] [PubMed] [Google Scholar]