

Abstract
Defining how interactions between tumor subpopulations contribute to invasion is essential for understanding how tumors metastasize. Here, we find that the heterogeneous expression of the transcription factor ΔNp63 confers distinct proliferative and invasive EMT states in subpopulations that establish a leader-follower relationship to collectively invade. A ΔNp63-high EMT program coupled the ability to proliferate with an interleukin 1α (IL-1α) and miR-205-dependent suppression of cellular protrusions that are required to initiate collective invasion. An alternative ΔNp63-low EMT program conferred cells with the ability to initiate and lead collective invasion. However, this ΔNp63-low EMT state triggered a collateral loss of fitness. Importantly, rare growth-suppressed ΔNp63-low EMT cells influenced tumor progression by leading the invasion of proliferative ΔNp63-high EMT cells in heterogeneous primary tumors. Thus, heterogeneous activation of distinct EMT programs promotes a mode of collective invasion that overcomes cell intrinsic phenotypic deficiencies to induce the dissemination of proliferative tumor cells.
INTRODUCTION
Tumors are composed of phenotypically diverse communities of neoplastic cells (1). This heterogeneity influences tumor evolution and responses to extrinsic stressors, such as chemotherapy (2). It is well established that clonal expansion occurs when a heritable trait provides a fitness advantage, with repeated rounds of clonal expansion resulting in the accumulation of tumor promoting traits (3). However, this Darwinian framework alone does not explain the full impact of phenotypic variability (4). Functional interactions between subpopulations are also an essential feature of tumor development (5–8). Notably, relationships between distinct subpopulations promote local invasion (9,10) and metastasis (11,12). While there has been progress in defining the modes of interaction between subpopulations that can induce collaborative forms of invasion, the signaling networks responsible for initiating these invasive interactions and how these relationships shape tumor evolution remain poorly defined. We therefore sought to define the regulatory programs that promote alliances between tumor subpopulations.
We focused our investigation on a relationship between subpopulations that induces the collective invasion of cohesive groups of tumor cells. Collective invasion is the predominant form of dissemination observed in primary tumors (13), and tumor explants (14), and can be detected by intravital imaging (15). Notably, highly invasive subpopulations we term “trailblazer” cells create microtracks in the extracellular matrix (ECM) that promote the invasion of intrinsically less invasive “opportunist” siblings (9,16). This relationship permits a subpopulation of trailblazer cells to promote a transition from a benign to an invasive malignant stage of growth in orthotopic tumors (9), consistent with the simultaneous invasion of distinct subpopulations observed in breast cancer patients (17,18). Trailblazer and opportunist populations can both express EMT inducing transcription factors (EMT-TFs) and the microfilament protein vimentin (9). Thus, these trailblazer and opportunist phenotypes can be distinct EMT states that are dictated by regulatory programs that exist outside the established EMT signaling framework (19). The components of trailblazer EMT programs that promote collective invasion, ECM reorganization and metastasis have begun to be defined (9,20). By comparison, factors that specify the opportunist EMT state and the contribution of opportunist EMT cells during tumor progression are poorly understood. Defining these unique opportunist EMT program factors is necessary to understand how interactions between trailblazer and opportunist cells are established and contribute to tumor progression.
Here, we find that heterogeneous expression of the transcription factor ΔNp63 confers a relationship between proliferative opportunist cells and growth-deficient trailblazer cells that promotes local dispersion. Thus, trailblazer-opportunist interactions can confer properties to the bulk population that are not found in either cell type.
METHODS
Cell Culture.
Hs578T and HCC1806 cells were a gift from John Minna (UTSW). SUM159 and SUM229 cells were a gift from Angelique Whitehurst (UTSW). MCFDCIS cells were purchased from Asterand. Cells were cultured as described (9,21) and verified by Powerplex genotyping. Cells were tested for mycoplasma (Lonza, LT07–703) the creation of freezedown stocks, with the latest test being 2/14/18. Cells from these frozen stocks were routinely used within 25 passages. ΔNp63α (Addgene, 26979), (22), was cloned into a PLX403 (Addgene, 41395), (23), using the Gateway cloning system according to manufacturers’ protocol (Invitrogen). pTRIPZ non-targeting control (RHS4743) and ΔNp63 targeting shRNA (24246) are from Dharmacon. Virus was produced and cells were infected to generate stable cell lines as described (24).
3D culture experiments.
Growth factor reduced Matrigel (Corning, 10–12 mg/ml stock concentration, #354230) and bovine (Corning, #354231) or rat tail (Corning, #354236) collagen I were used for organotypic culture experiments. Vertical invasion assays and experiments in 3D culture were performed and quantified as described previously using a Matrigel/Collagen I matrix (3–5 mg/ml Matrigel and 1.8–2.1 mg/ml Collagen I) (9,20). A 120 μm span on the z-axis is shown for the vertical invasion assays. For fluorescent bead experiments, cells were resuspended in 50 ul of a Matrigel/Collagen I mix containing 0.03% Fluoro-Max Dyed Blue Aqueous Fluorescent Particles (Thermo Scientific, B0100), and then plated on 30 μl of a basement layer of Matrigel/Collagen I. For spheroid cluster experiments, 96-well Nunclon Sphera low adhesion plates (Thermo Scientific, 174925) were used to form clusters. 2000–4000 cells per well were plated and incubated at 37˚C for 24–72 h. Clusters were then resuspended in 50 μl of Matrigel/Collagen I mix and plated on 30 μl of a basement layer of Matrigel/Collagen I and allowed to invade for 24 h.
Immunoblot analysis, IF and IHC.
Experiments and analysis were performed as described (9) using antibodies detailed in Table S6. Anti-ΔNp63 antibodies were used except as indicated in Fig. S1D where an anti-p63α was used in parallel with an anti-ΔNp63 antibody.
siRNA experiments.
Cells were transfected with 50 nM of siRNA using RNAiMax transfection reagent (Invitrogen) for 48–72 h. The siRNAs were from Dharmacon and Sigma. Cells in all conditions designated as “Control” were transfected with a pool of siRNAs that does not target human genes. The p63 shRNA and siRNAs target all p63 isoforms. The details of the sequences and catalog numbers for each siRNA and shRNA are located in Table S7.
Flow cytometry.
Analyses were conducted following standard flow cytometry procedures as described (9) using 5 x 105 cells. Samples were resuspended in Hoechst 33342 [1ug/ml] diluted in 2% Fetal Bovine Serum (FBS)/Phosphate Buffered Saline (PBS). At least 50,000 events were collected on an LSRII flow cytometer (Becton Dickinson), and analyzed using Flow Jo software (Tree Star Technologies).
Quantitative real-time PCR.
Experiments were performed as described (9) using primer sequences listed in Table S8.
Colony formation assay.
Cells were plated at density of 300 cells (MCDCIS) or 1000 cells (SUM229) per well in a 6-well plate. Colonies grew 7 days (MCFDCIS) or 10 days (SUM229), and were fixed with formalin and stained with Giemsa (Sigma Aldrich).
ELISA assay.
Cells were lysed in RIPA buffer and the resulting lysates were analyzed to determine IL-1α expression in cells following the manufacturer’s protocol (Biolegend, 445804).
Identification of ΔNp63 regulated genes.
mRNA expression was determined using Human HT-12 v4 Expression BeadChips (Illumina Inc.). Data was processed with a model-based background correction approach (25), quantile-quantile normalization and log2 transformation. The raw data for ΔNp63 induced expression of genes in SUM159T cells is available at the GEO (GSE128190). The raw data for MCFDCIS and HCC1806 cells transfected with p63 siRNA pools was reported in (21) and is available at the GEO (GSE58643, GSE62569). ΔNp63 binding peaks (GSE72009) were defined in (26) using a p63α antibody (Santa Cruz, H-129) in cells that exclusively express high levels of ΔNp63α. Heatmaps showing the relative expression of genes analyzed with Illumina BeadChips were generated with GenePattern software using the HeatMapImage module. Median values of replicate probe sets for the same genes were used to summarize expression values for each gene. The predicted miR205 seed sequences were identified using TargetScan 7.1. (http://www.targetscan.org/vert_71/ ).
Mice.
The C3-Tag [FVB-Tg(C3–1-TAg)cJeg/JegJ] mice (27) were a gift from Jeffrey E. Green. Age-matched female NSG [NOD.Cg-Prkdcscid IL2rgtm1Wjl/Szj] mice were obtained from the Jackson Laboratory (Bar Harbor, ME) and bred and maintained under specific pathogen-free conditions in a barrier facility at the University of Texas Southwestern Medical Center (Dallas, TX). Mice were housed and bred in accordance with a protocol approved by the Institutional Animal Use and Care Committee at Georgetown University and the University of Texas Southwestern Medical Center in compliance with the NIH Guide for the Care and Use of Laboratory animals. Female mice were used for all analyses.
Xenograft experiments.
Female NSG mice (6–8-weeks old) were injected in the #4 fat pad. In Fig. 4C, 50,0000 MCFDCISO or MCFDCIST cells were injected. In Fig. 4F 100,000 MCFDCISO or 500,000 578T cells were injected. In Fig. 5 100,000 MCFDCISO-H2B:mCherry were injected alone or with 500,000 578T-H2B:GFP cells. In Fig. S5, 50,000 MCFDCISO-H2B:mCherry were injected alone or with 500,000 DCIST-H2B:GFP cells. Mice were sacrificed and primary tumors were excised for analysis 28 days after injection.
Figure 4. The loss of ΔNp63 expression that occurs during induction of the ΔNp63-low EMT trailblazer state can cause a collateral loss of cell fitness.
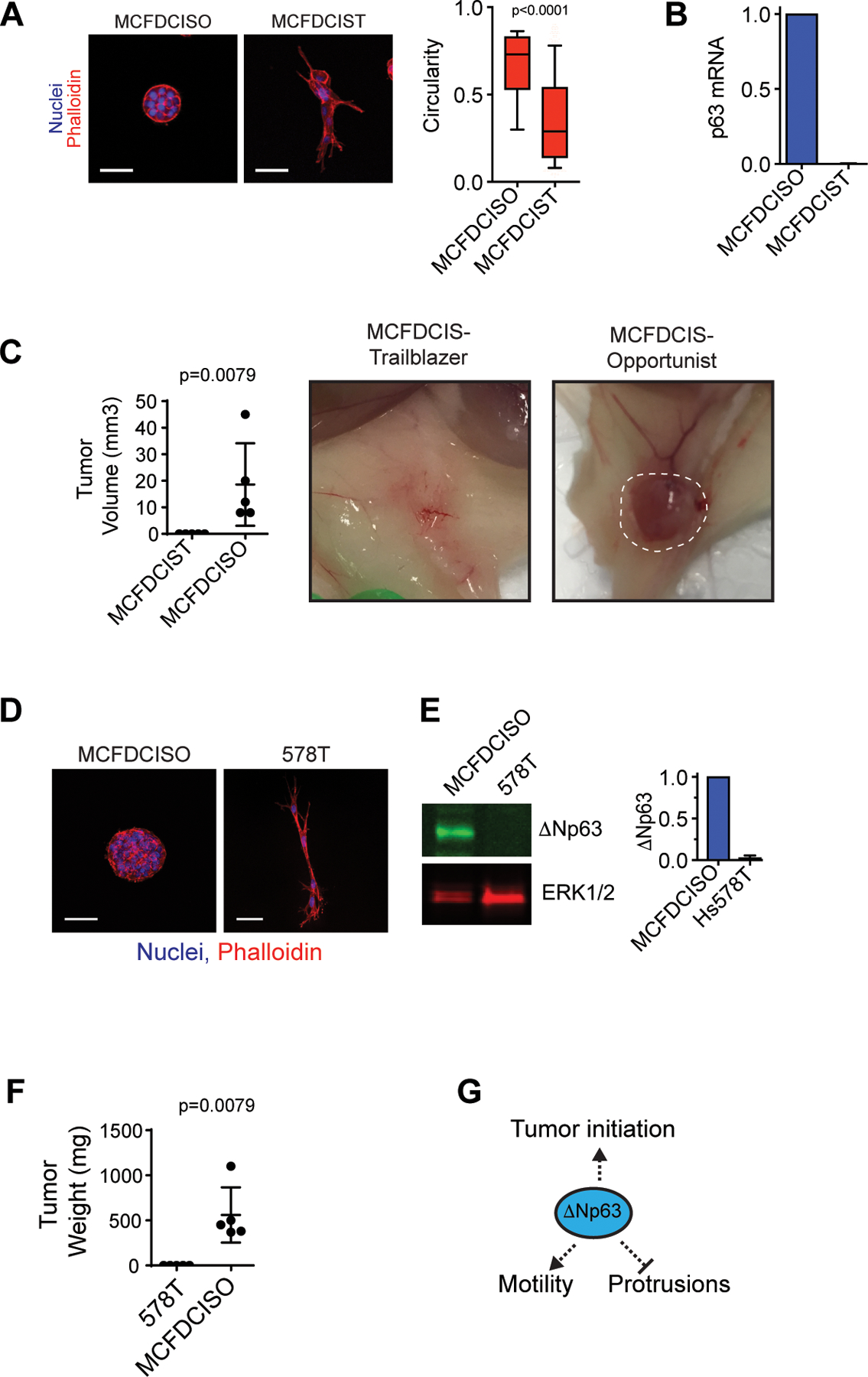
(A) MCFDCISO and MCFDCIST cells plated on ECM for 48 h. Box and whisker plots (10–90 percentile). MCFDCISO (n=1177 spheroids), MCFDCIST (n=841 spheroids). (B) ΔNp63 mRNA in MCFDCISO cells and MCFDCIST cells determined by qPCR (mean±range, n=2). (C) Tumors formed by MCFDCIST and MCFDCISO cells (mean ± SD, n=5 mice per condition). The dashed line shows the boundary of the MCFDCISO tumor. (D) MCFDCISO and 578T spheroids (mean±SD, n=3). (E) ΔNp63 expression in MCFDCISO and 578T cells (mean±SD, n=3). (F) Tumors formed by 578T and MCFDCISO cells (mean±SD, n=5 mice per condition). (G) Model showing conversion to the trailblazer state results in ΔNp63 suppression and reduced tumor initiating activity. Scale bars, 50 μm. P-values determined by Mann-Whitney U test.
Figure 5. Proliferative ΔNp63-high EMT opportunist cells collectively invade in response to the presence of slow-cycling ΔNp63-low EMT trailblazer cells in primary tumors.
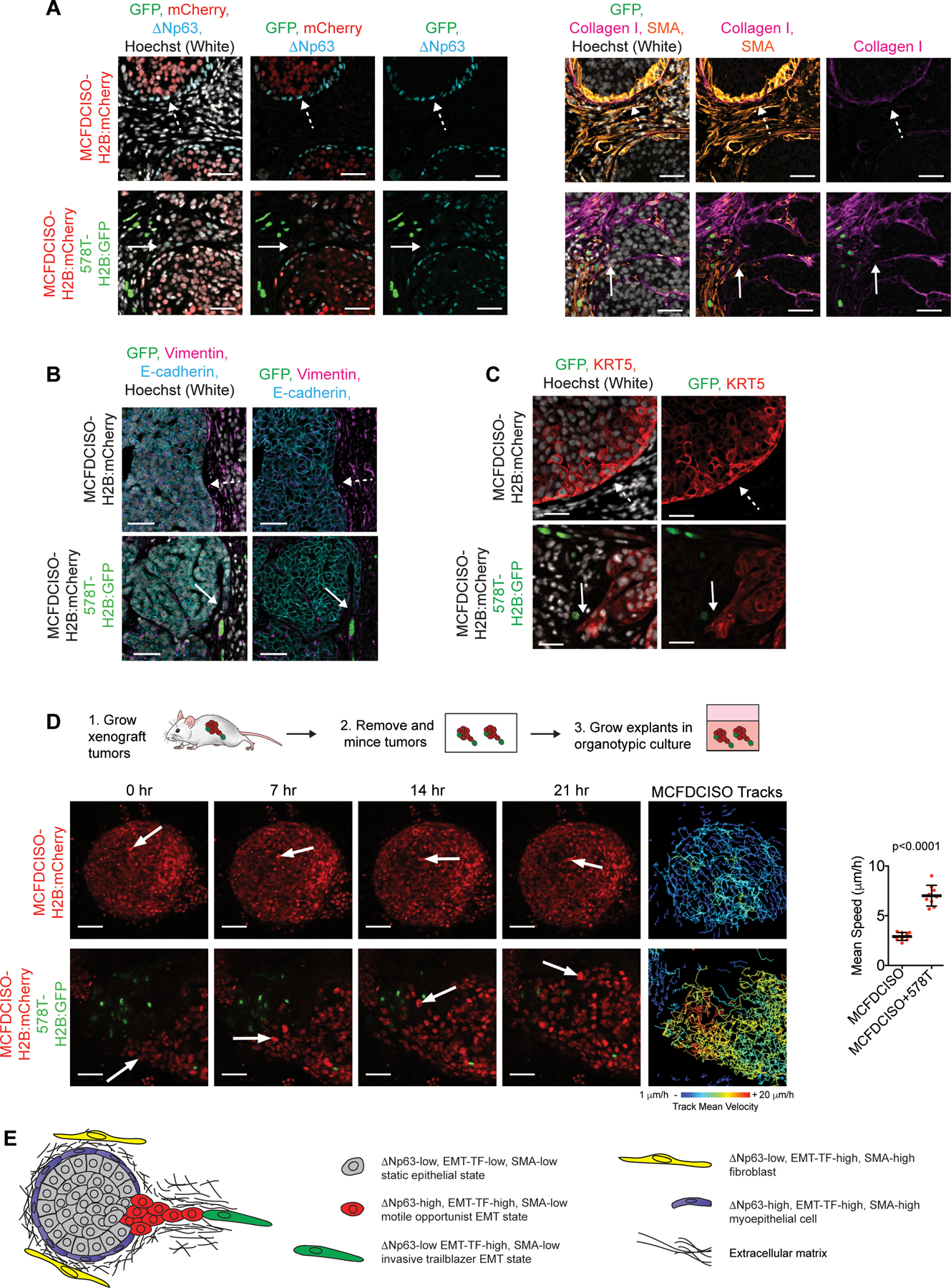
(A) Immunostaining of primary tumors composed of MCFDCISO cells alone or a mixture of MCFDCISO and 578T cells (1:5 ratio). Sequential sections were used for the GFP/mCherry/ΔNp63 and GFP/Collagen I/SMA immunostaining to facilitate direct comparison of the antibody combinations. Solid arrows, opportunistic collective invasion. Dashed arrows, noninvasive tumor-ECM boundary (n=5 mice per condition). (B and C) Immunostaining of the MCFDCISO and MCFDCISO/578T tumors. Solid arrows, opportunistic collective invasion. Dashed arrows, noninvasive tumor-ECM boundary. (D) Time-lapse imaging of MCFDCISO and MCFDCISO/578T tumor organoids. Arrows, representative cell movement. Tracking of MCFDCISO cell movement is shown. Graph shows mean speed of MCFDCISO cells (mean±SD cell speed for n=9 spheroids for each condition). P-value, Mann-Whitney U test. (E) Model showing how the ΔNp63-low EMT program confers rare trailblazer cells with the ability to promote the collective invasion of ΔNp63-high EMT state opportunist cells in primary tumors. Scale bars, 50 µm.
Tumor explants.
The largest tumors from female C3-Tag mice were minced and tumors were digested for up to 120 min at 37°C in a mixture of 1mg/ml Collagenase, 2U/µl DNase, 5% FBS in DMEM/F12. Digested tumors were pelleted at 80 x g for 1 min and the supernatant was discarded. Tumor organoids were then rinsed up to 5 times in 10 ml 5% FBS in DMEM/F12. Organoids were plated in a mixture of 50 ul of 2.4 mg/ml Rat tail collagen (354236, Corning) and 3 mg/ml growth factor reduced Matrigel onto a base layer of 30 µl of Collagen I/Matrigel. The organoids in ECM were overlaid with DMEM/F12 supplemented with 1% FBS, 1X ITS (Sigma, I3146), non-essential amino acids (Sigma, M7145) and 10 ng/ml FGF2 (Peprotech, 100–18C). Organoids were allowed to invade for 24 h, fixed and imaged (9). For analysis of the MCFDCIS model, at day 28 tumors were excised and minced into approximately 1 mm3 pieces using a scalpel. The minced tumors were then resuspended in 50 µl ECM (2.1 mg/ml Collagen I + 3 mg/ml Matrigel), and plated onto 30 µl of solidified ECM in 8-well chamber slides and overlaid with MEGM (Lonza, CC-3150) + 5% FBS and imaged.
Gene expression analysis in patient tumors.
The cBioPortal web resource (http://www.cbioportal.org) (28,29) was used to generate heatmap representations of gene expression from the METABRIC dataset (30) and classification was based on ER, HER2 and PAM50 intrinsic subtype.
Patient survival analysis.
Analysis of breast cancer patient survival times was performed using the KM-plotter meta-analysis database (31). Patients were stratified into “high” and “low” groups based on the upper quartile of IL-1α expression using probe 210118. Estrogen receptor (ER) and progesterone receptor (PR) status was judged by mRNA expression. Survival differences were compared by log-rank test.
Statistical Methods.
Survival differences were compared using the Mantel-Cox log-rank test (Graphpad, Prism). Data with a normal distribution determined by Shapiro-Wilk test were analyzed by two tailed Student’s t-test (Graphpad Prism). Data that did not pass a normality test were analyzed by Mann-Whitney U test.
RESULTS
Expression of the transcription factor ΔNp63 distinguishes opportunist and trailblazer EMT programs.
While genes that are induced as part of the invasive trailblazer EMT program have been identified (9,20), traits that confer the opportunist EMT state are poorly understood. To address this question, we defined which genes are more highly expressed in opportunist cells relative to sibling trailblazer cells in the SUM159 cell line model of triple negative breast cancer (TNBC). The SUM159 trailblazer (SUM159T) and opportunist (SUM159O) cells are epigenetically distinct subpopulations that we previously derived (9) based on their invasive phenotype when grown in 3D culture (Fig. 1A and Table S1). The transcription factor p63 was identified among a set of 30 genes with a ≥ 5-fold higher expression in opportunist cells relative to trailblazer cells with a false discovery rate (FDR) of <0.01 (Table S2 and Fig. S1A). p63 mRNA is detected in TNBC, estrogen-receptor positive and HER2 positive tumors and all PAM50 subtypes (Fig. S1B), indicating that p63 can influence invasive traits across the clinically relevant breast cancer subtypes, consistent with previous observations (21,26,32). There are two p63 isoforms, TAp63 and ΔNp63, which are distinguished by unique N-terminal domains that arise from the usage of different promoters (33). TAp63 and ΔNp63 have distinct expression patterns (33) and regulate different gene expression programs (34), with TAp63 functioning as a tumor suppressor and ΔNp63 acting as tumor promoter (35). Indeed, ΔNp63 was expressed at a higher level in opportunist cells relative to trailblazer siblings (Fig. 1B, S1C–G and Table S1). ΔNp63 is expressed in subpopulations of breast tumor cells and contributes to mammary tumor initiating activity and metastasis (32,36,37). Thus, understanding how ΔNp63 influences the invasive properties of breast tumor cells is important for understanding the mechanism of dissemination of a critical tumor subpopulation.
Figure 1. Expression of the transcription factor ΔNp63 distinguishes opportunist and trailblazer EMT programs.
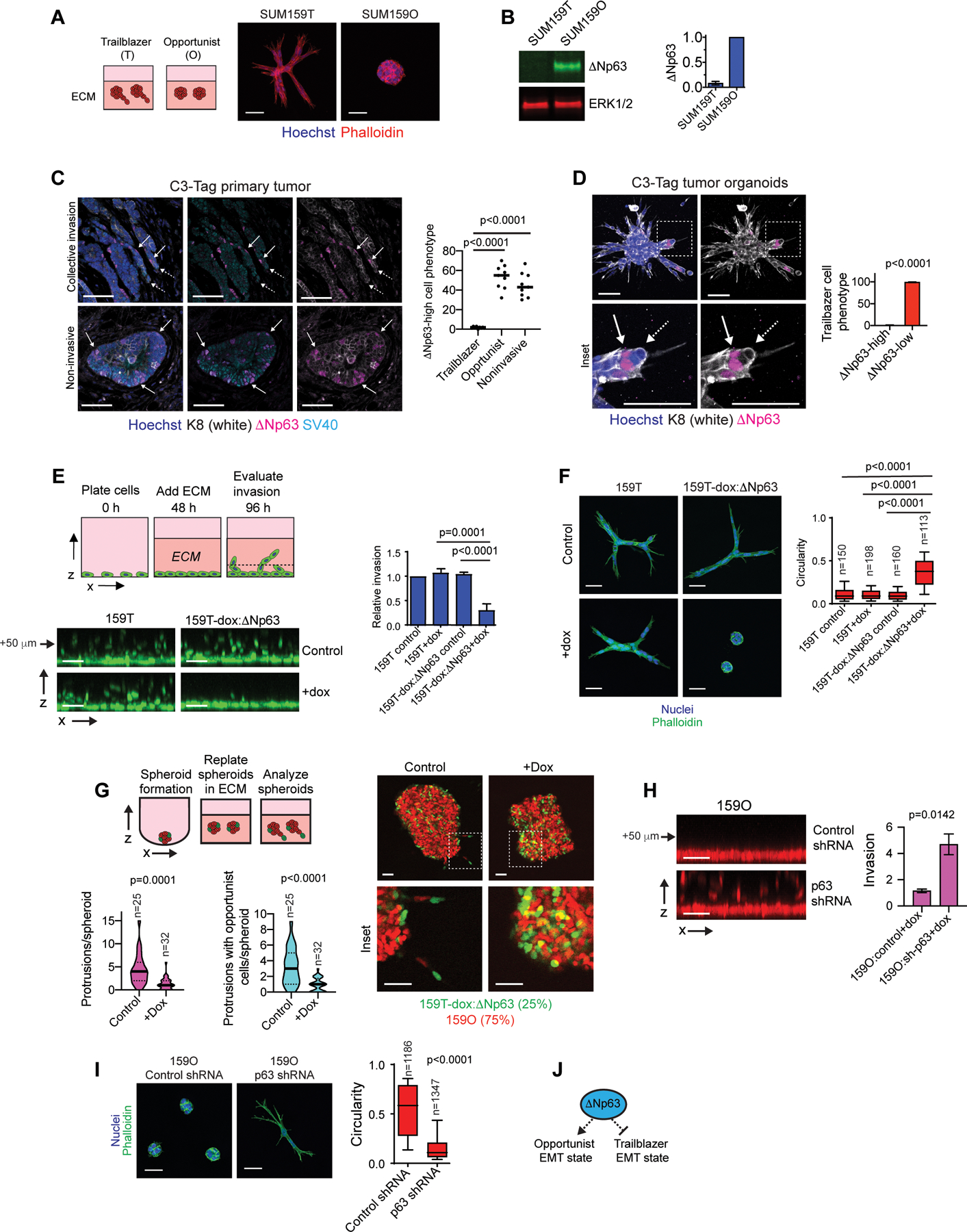
(A) Representative spheroids at day 4 (n=3). (B) ΔNp63 expression in SUM159 trailblazer (SUM159T) and opportunist (SUM159O) populations (n=3). (C) ΔNp63 expression in C3-Tag tumors. Graph shows quantification of the ΔNp63-high cell phenotype from at least 2 10x fields of view at the tumor-stroma interface (mean±SD, n=9 mice). Solid arrow= ΔNp63-high, dashed arrow=ΔNp63-low. (D) Opportunistic invasion of ΔNp63-high cells in C3Tag tumor organoids. Graph shows quantification of ΔNp63 expression in trailblazer cells as a percentage of at least 528 total organoids per mouse (mean±SD, n=4 mice). (E) Doxycycline (dox) induced ΔNp63 expression suppresses vertical invasion (mean±SD, n=4). Arrow indicates distance of monolayer. (F) Dox induced ΔNp63 expression suppresses spheroid invasion over 72 h in ECM. Box and whisker plots (10–90 percentile) show the quantification of spheroid morphology (n=spheroids shown on graph). (G) Dox induced ΔNp63 expression in SUM159T cells (green) reduces the number of invasive LCPs and the opportunistic invasion of SUM159O cells (red). Violin plots show quantification of LCPs and the frequency of opportunistic SUM159O invasion. (H) Dox induced p63 shRNA promotes SUM159O invasion (mean±SEM, n=12). Arrow indicates distance of monolayer. (I) Dox induced p63 shRNA promotes SUM159O spheroid invasion over 48 h in ECM. Box and whisker plots (10–90 percentile) show quantification of spheroid circularity (n=spheroids shown on graph). (J) ΔNp63 prevents induction of a trailblazer state by restricting the ability of cells to form LCPs while promoting cell motility. P-values, two-tailed Student’s t-test (C-E and H) or Mann Whitney U test (F, G and I). Scale bars, 50 μm.
To define the relationship between ΔNp63 expression and intrinsic invasiveness in spontaneous tumors, we evaluated ΔNp63 expression in the C3(1)/SV40 Large T antigen (C3-Tag) genetically engineered mouse (GEM) model of basal-type TNBC (27,38). Subpopulations of cells expressed ΔNp63 (Fig. 1C), as has been observed in primary breast tumors (32,39). Notably, ΔNp63-low cells led the invasion of ΔNp63-high cells in regions displaying pathological features of strand-like collective invasion at the tumor-stroma interface (Fig. 1C). Moreover, ΔNp63-low trailblazer cells led the collective invasion of ΔNp63-high cells in C3-Tag organoids, which invaded under conditions that support the normal noninvasive architecture of mammary epithelial organoids (Fig. 1D and S1H–I). By comparison, ΔNp63 expressing cells at the tumor-stromal interface and on the surface of C3-Tag organoids, as well as normal mammary epithelial organoids did not form long cellular protrusions (LCPs) or invade, consistent with ΔNp63 restricting the initiation of collective invasion (Fig. 1C and S1H and J). These results indicate that ΔNp63 expression distinguishes trailblazer and opportunist cells during collective invasion in C3-Tag tumors.
Induction of exogenous ΔNp63 restricted the ability of SUM159T cells to vertically invade into ECM from cell monolayers, extend LCPs into the ECM, and promote the cooperative invasion of SUM159O siblings (Fig. 1E–G and S1K–M). Similar results were observed when ΔNp63 was exogenously expressed in 578T cells, which lack detectable endogenous ΔNp63 expression (Fig. S1K, N and O). Thus, reduced ΔNp63 expression is necessary to promote a trailblazer state. The expression of a p63 shRNA or transfection with p63 siRNAs was sufficient to induce SUM159O collective invasion and the formation of LCPs (Fig. 1H–I and S1D, P–R). However, reduced ΔNp63 expression alone is not sufficient to promote a trailblazer phenotype (21,40). Estrogen receptor positive (ER-pos) breast cancer cells that lack the expression of ΔNp63 and EMT regulatory factors are not invasive (Fig. S1S–T and Table S1) and incapable of opportunistic invasion (24). Similarly, cells dependent on ΔNp63 to induce EMT-TF expression revert to a static epithelial state when ΔNp63 is experimentally depleted, rather than convert to a trailblazer EMT state (21,26). Thus, our findings indicate that the induction of the trailblazer EMT state requires the expression of EMT-TFs in conjunction with reduced ΔNp63 expression (Fig. S1U). Together, our results show that a ΔNp63-high EMT state confers an opportunist phenotype while an alternative ΔNp63-low EMT state is necessary to induce a trailblazer state (Fig. 1J).
The ΔNp63-high EMT state specifically restricts autonomous invasive ability while promoting cell migration.
ΔNp63 induction in SUM159T cells suppressed the formation of LCPs, which provide traction and exert tensile forces that reorganize the ECM into parallel fibrils that promote collective invasion (Fig. 2A, S2A–B, and Videos S1–4). However, ΔNp63 expressing SUM159T cells actively migrated within spheroids and the monolayers of our vertical invasion model (Fig. 2A–B, S2C–D and Videos S1–2). Consistent with these results, p63 shRNA expression in SUM159O cells promoted LCP formation and collective invasion while also reducing the rate of migration in spheroids and in the cell monolayers (Fig. 2C–D, S2E–G and Videos S5–6). The rapid migration of ΔNp63 expressing SUM159T and SUM159O cells is consistent with previous results from us and others showing that ΔNp63 can promote cell migration (21,40). Together, these results indicate that ΔNp63 promotes cell migration while restricting the ability of cells to form LCPs and exert tensile forces on the surrounding ECM (Fig. 2E). Thus, ΔNp63 dictates the mode of invasion rather than restricting all types of invasive behavior.
Figure 2. The ΔNp63-high EMT state specifically restricts autonomous invasive ability while permitting cell movement in spheroids.
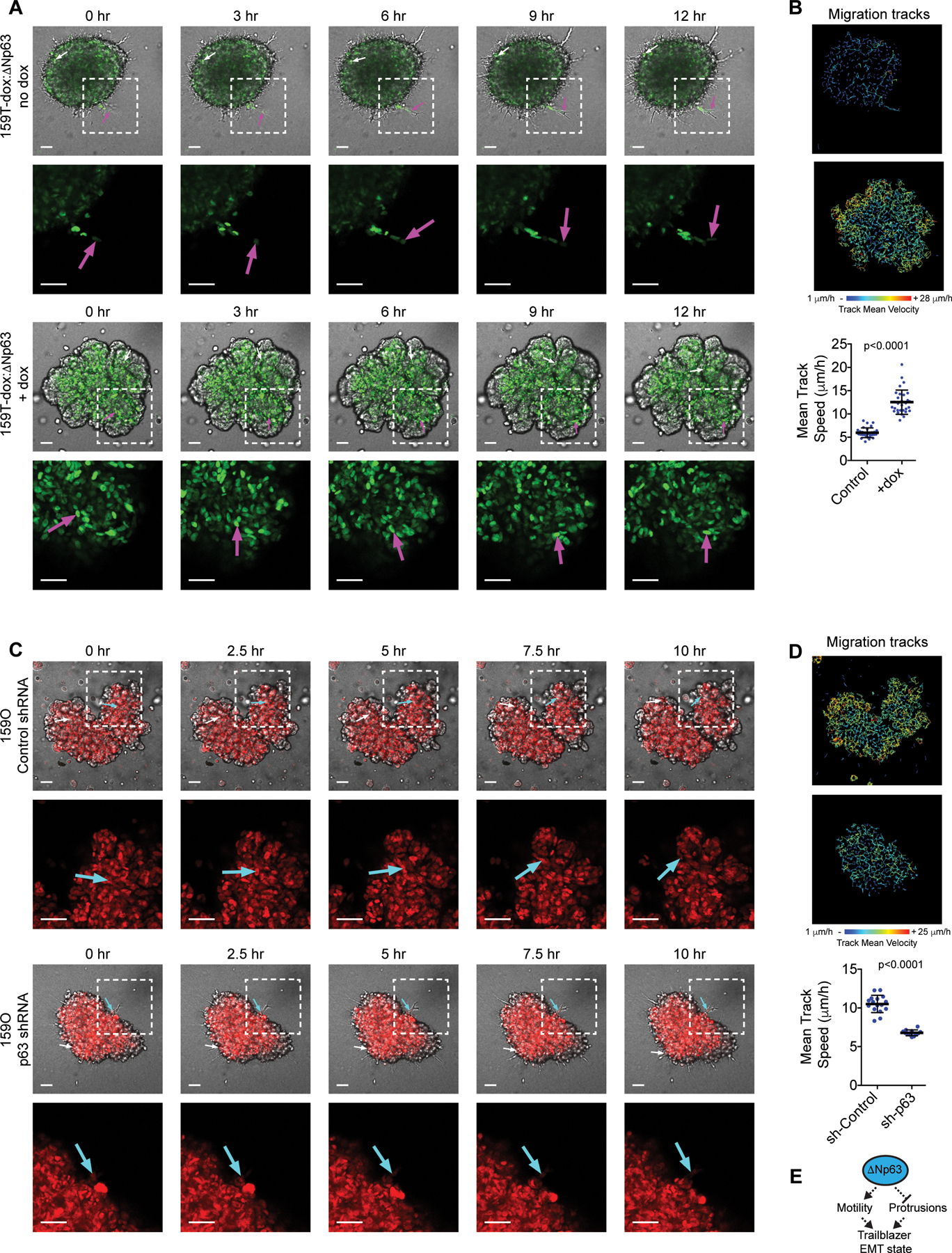
(A-B) Time-lapse confocal imaging and tracking of cell movement showing the influence of dox induced ΔNp63 expression on SUM159T invasion and motility (see Fig. S1L for methodology). Arrows indicate movement of representative cells. Graph shows cell speed (mean±SD, n=30 spheroids). (C-D) Time-lapse confocal imaging and tracking of cell movement showing the influence of dox induced p63 shRNA expression on SUM159O invasion and motility. Arrows indicate movement of representative cells. Graph shows cell speed (mean±SD cell, n=16 spheroids (Control) and 12 spheroids (p63 shRNA)). (E) Model showing that conversion to a trailblazer state requires ΔNp63 suppression to permit LCP formation and ECM reorganization. P-values, Mann-Whitney U test. Scale bars, 50 μm.
IL-1α and miR-205 are ΔNp63 target genes that restrict trailblazer invasion.
To identify ΔNp63 regulated genes that confer the opportunist phenotype we performed genome-wide mRNA expression analysis on SUM159T cells in the presence or absence of exogenous ΔNp63. Thirty-three genes were induced and 15 genes that were suppressed at least 2-fold (Fig. 3A, S3A and Tables S3–4). Genes previously identified as being required for trailblazer invasion (9,20) were not included in the ΔNp63 suppressed gene set (Fig. S3A) and the expression of proteases that promote ECM degradation were unchanged in expression (Table S5). After determining which differentially expressed genes had associated ΔNp63 binding sites (26) and were influenced by ΔNp63 expression in at least 2 opportunist populations, we prioritized the innate immunity cytokine IL-1α (41) for investigation. ChIP-seq experiments (21) showed a ΔNp63 binding peak upstream of the TSS for IL-1α and the ΔNp63-dependent regulation of IL-1α mRNA and protein was confirmed by qPCR and ELISA analysis of cell lysates (Fig. 3B–D and S3B). ΔNp63 is also required for IL-1α expression in MCFDCIS and HCC1806 breast cancer cells (Fig. S3C and Table S1), as well as keratinocytes (42), further supporting the robustness of ΔNp63 dependent regulation of IL-1α.
Figure 3. IL-1α and miR-205 are ΔNp63 targets that restrict invasion.
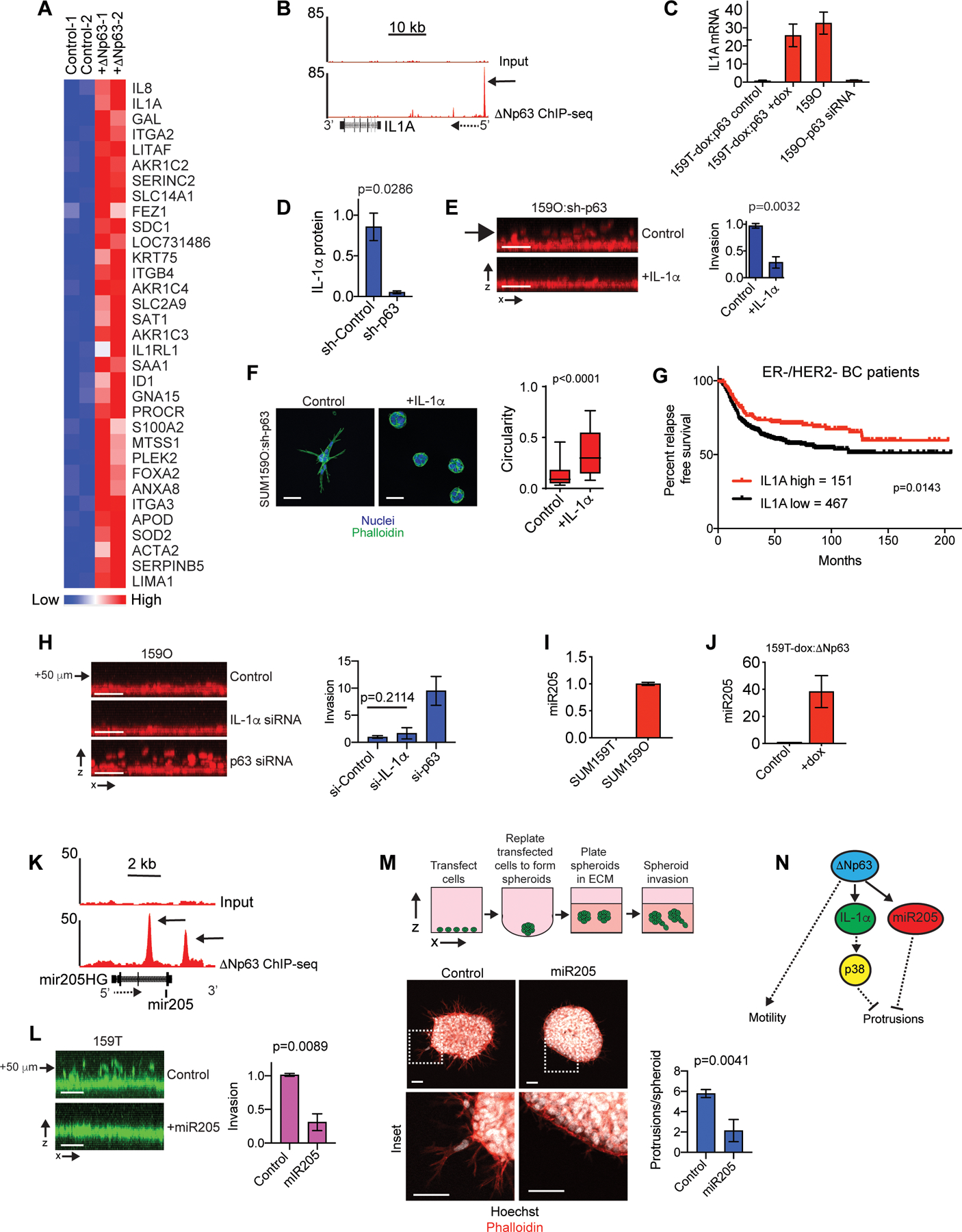
(A) Heatmap showing genes with a ≥2-fold increase in expression following 5 days of dox induced ΔNp63 expression. Biological replicates are shown. (B) ΔNp63 ChIP-seq and input DNA signals in the genomic regions surrounding the IL-1α gene. Solid arrow indicates the ΔNp63 binding site (C) qPCR results showing how dox induced ΔNp63 expression and p63 siRNA transfection influences IL-1α expression (mean±range, n=2). (D) ELISA results showing p63 shRNA reduces IL-1α in cell lysates. (E) IL-1α [10 ng/ml] suppresses SUM159O-shp63 invasion (mean ±SD, n=3). (F) Images showing how IL-1α influences the phenotype of ΔNp63 depleted SUM159O-shp63 cells plated onto a layer of ECM for 72 h. Scale bars, 50 μm. Box and whisker plot (10–90 percentile). Control (n=1569 spheroids) and + IL-1α (n=1461 spheroids). (G) Kaplan-Meier curves showing relapse-free survival of ER-/HER2- breast cancer patients classified as “IL-1α-high” (red) and “IL-1α-low” (black) based on IL-1α mRNA expression using KM Plotter. (H) x-z projections and quantification showing how IL-1α depletion influences invasion (mean±SD, n=5). Arrow indicates distance from monolayer. (I) qPCR showing miR205 expression (mean±range, n=2). (J) qPCR showing that ΔNp63 promotes miR205 expression, (mean±range, n=2). (K) ΔNp63 ChIP-seq and input DNA signals in the genomic regions surrounding miR205 and the miR205 host gene (miR205HG). Solid arrow indicates the ΔNp63 binding site and dashed arrow indicates gene orientation. (L) miR205 mimic transfection suppresses vertical invasion (mean±SD, n=3). Arrow indicates distance from monolayer. (M) miR205 mimic transfection suppresses SUM159T cells LCP formation (mean±SEM, n=19 spheroids (Control) and 14 spheroids (miR205)). (N) The ΔNp63 targets IL-1α and miR205 restrict the formation of LCPs that are necessary to initiate collective invasion. P-values, two-tailed Student’s t-test (E, H, L), Mann-Whitney U test (D, M), and log-rank (Mantel-Cox) test (I). Scale bars, 50 μm.
IL-1α is anchored to the plasma membrane where it mediates signaling through the autocrine activation of IL1R1 receptors or localizes to the nucleus where it interacts with histone acetyltransferases to regulate gene expression (41). Recombinant IL-1α treatment for 48 h suppressed SUM159O:p63 shRNA cell invasion and LCP formation (Fig. 3E–F), indicating that canonical activation of IL1R1 suppressed invasion. Recombinant IL-1α also suppressed 578T invasion and protrusion formation (Fig. S3D–E). Consistent with this suppression of invasion, patients in the top quartile of IL-1α expression had better odds of relapse free survival (Fig. 3G and S3F). IL-1α increased the activity of p38 MAP kinases (Fig. S3G) and treatment with 2 distinct inhibitors of p38 partially prevented the suppression of invasion by IL-1α (Fig. S3D, E and H). However, IL-1α depletion and p38 inhibition failed to enhance SUM159O invasion (Fig. 3H and S3I–K). IL-1α depletion was also unable to restore the invasion of 578T cells expressing ΔNp63 (Fig. S3L). Thus, our results indicate that IL-1α mediated p38 activation suppresses the ability of tumor cells to initiate collective invasion into the ECM. However, additional factors regulated by ΔNp63 contributed to the restriction of invasion as well.
We shifted our focus to potential functions of ΔNp63 regulated microRNAs (miRNAs). Based on previous results showing that ΔNp63-induced miR205–5p (miR205) promotes migration (21), we tested whether miR205 expression influenced the mode of collective invasion. Consistent with miR205 contributing to the ΔNp63-dependent suppression of trailblazer cell invasion, miR205 was expressed at a higher level in SUM159O cells, induced by exogenous ΔNp63 in SUM159T cells, ΔNp63 binding sites were found proximal to miR205 and a miR205 mimic suppressed SUM159T invasion (Fig. 3I–M). Three ΔNp63 suppressed genes, THBS1, FSTL1 and COL1A1 had predicted miR205 seed sequences in their 3’ UTRs (Fig. S3A and M), indicating that miR205 induction was potentially responsible for their suppression by ΔNp63. Notably, these genes encode ECM proteins that can enhance invasion (14,43,44). Thus, in addition to preventing ECM reorganization, ΔNp63 may also suppress invasion by influencing the composition of the ECM. Together, these results suggest that ΔNp63 confers an opportunist phenotype by regulating two targets, IL-1α, which activates p38, and miR205, which modulates gene expression (Fig. 3N). Thus, there is redundancy in the ΔNp63 regulated pathways that suppress the trailblazer phenotype.
The loss of ΔNp63 expression that occurs during induction of the ΔNp63-low EMT trailblazer state can cause a collateral loss of cell fitness.
The ability of ΔNp63 to promote tumor initiating activity and regulate breast cancer cell proliferation (32,36), prompted us to investigate the potential collateral effects of reduced ΔNp63 expression in trailblazer cells. We had previously found that ΔNp63 was required for colony formation activity of MCFDCIS cells (21). Given the established consequence of reduced ΔNp63 in these cells, we focused on the ability of MCFDCIS trailblazer populations to form tumors. A trailblazer subpopulation of MCFDCIS cells (MCFDCIST) derived from a clone that overexpresses Slug, and lacks ΔNp63 expression was evaluated (Fig. 4A–B and Table S1). MCFDCIST cells formed fewer tumors than MCFDCISO cells and the tumors that did form were substantially smaller in size (Fig. 4C). 578T cells, which are another ΔNp63 deficient trailblazer cell line, were also diminished in tumor forming ability relative to MCFDCISO cells (Fig. 4D–F).
To extend on these results we investigated the properties of sibling SUM229 trailblazer and opportunist populations in vitro. SUM229T cells and p63 depleted SUM229O cells were enriched at the G0/G1 checkpoint when compared to control SUM229O cells, indicating that they were in a slow-cycling state (Fig. S4A–C). Entry into a slow-cycling or quiescent state is a property of normal stem-like populations and is a proposed feature of tumor initiating cells (45). However, SUM229T cells and ΔNp63 depleted SUM229O cells had reduced colony formation activity relative to control SUM229O cells (Fig. S4D–E). Treatment with the p38 inhibitor BIRB796 also reduced SUM229O colony formation, suggesting that ΔNp63 regulated signaling pathways that suppress invasion are required for cell growth (Fig. S4F). These findings suggest that the reduction in ΔNp63 expression that is essential for cells to convert to a trailblazer state can have the subordinate outcome of inactivating necessary pro-growth signaling pathways in these populations (Fig. 4G). It should be noted that trailblazer cells can acquire tumor initiating capability, as SUM159T cells are capable of forming tumors under similar conditions (9). Nevertheless, our results show that in multiple contexts, the trailblazer state has reduced growth capacity. Thus, our observations also reveal that the ΔNp63-high EMT and ΔNp63-low EMT states can have distinct growth and tumor initiating activities in addition to unique invasive properties (Fig. 4G).
Proliferative ΔNp63-high EMT opportunist cells collectively invade in response to the presence of slow-cycling ΔNp63-low EMT trailblazer cells in primary tumors.
Given the weak tumor initiating activity of 578T and MCFDCIST cells, we asked whether these populations could influence opportunist subpopulation invasion during tumor development. To do this we evaluated orthotopic xenograft tumors that formed when 578T or MCFDCIST cells were co-injected with MCFDCISO cells. When injected alone, MCFDCISO cells formed non-invasive DCIS-like lesions encircled by myoepithelial cells expressing ΔNp63 and smooth muscle actin (SMA) (Fig. 5A and S5A–D) as has been previously reported (46,47). The luminal ΔNp63-low cells do not express Slug and thus have not entered a ΔNp63-low EMT state that supports the trailblazer phenotype, but rather have entered a luminal epithelial state (21). The 578T and MCFDCIST populations were injected at a 5-fold excess to the MCFDCISO cells yet remained a small proportion of the combined population (Fig. 5A and S5B–D) that would not amount to a detectable tumor, as was also observed when they were injected alone (Fig. 4C, F). Nonetheless, rare 578T and MCFDCIST cells were sufficient to induce ΔNp63-high/SMA-low MCFDCISO invasion proximal to trailblazer cells through tracks of collagen I, consistent with a cooperative mode of collective invasion (Fig. 5A and S5B–D). The opportunistically invasive ΔNp63-high MCFDCISO cells were in a hybrid EMT state, as indicated by simultaneous expression of the mesenchymal marker vimentin, and the epithelial markers, E-cadherin and KRT5 (Fig. 5B–C and S5E–F). The 578T and MCFDCIST cells also expressed vimentin; however, they lacked detectable E-cadherin and KRT5, indicating that ΔNp63-low EMT trailblazer populations sustained a more mesenchymal state than ΔNp63-high EMT opportunist cells, as is detected in vitro here (Fig. 5B–C and S5E–F) and previously (9). Fibroblasts can promote opportunistic invasion (24) and SMA and vimentin expressing fibroblasts were present in tumors formed by the MCFDCISO cells injected alone (Fig. 5A–B). However, invasion was not detected proximal to fibroblasts at this time point.
Interestingly, analysis of ΔNp63 expression in the tumors indicated that trailblazer cells can promote the conversion of static epithelial tumor cells into a motile ΔNp63-high opportunist state. This is indicated by the observation that ΔNp63-high cells lack SMA, suggesting that SMA-negative ΔNp63-low cells convert to a ΔNp63-high state, as is detected in vitro (Fig. 5A and S5B–D). The induction of ΔNp63, rather than a conversion of SMA-positive ΔNp63 cells into SMA-negative ΔNp63 cells, is supported by the relative increase in the number of ΔNp63 expressing cells rather than a loss of the SMA-positive myoepithelial population (Fig. 5A and S5B–D). ΔNp63 can be induced by paracrine signaling and the creation of cell contacts with the ECM (47). The trailblazer cells are positioned in the tumor to initiate either of these ΔNp63 induction mechanisms directly or through the recruitment of non-tumor cells. Indeed, time-lapse imaging of tumor explants revealed that the presence of 578T cells increased the motility of MCFDCISO cells (Fig. 5D and Video S7). This observation further indicates that trailblazer cells induce a motile ΔNp63-high EMT state in tumors. Interestingly, the MCFDCISO cells from co-injected tumors also moved at a greater rate than the proximal trailblazer cells yet did not invade into the exogenous ECM lacking trailblazer cells in our explant experiments (Fig. 5D and Video S7). This finding reinforces our earlier results demonstrating that migration rate is not directly correlated with the ability of cells to initiate invasion of siblings into the ECM. Importantly, the rapid movement of the ΔNp63-high EMT MCFDCISO cells suggests that they have the potential to metastasize once access to the vasculature is created by trailblazer siblings. Together these results reveal that subpopulations in two distinct EMT states can functionally interact to induce the invasion of highly proliferative cells into the ECM (Fig. 5E).
DISCUSSION
We have found that a highly invasive growth-deficient ΔNp63-low EMT subpopulation induces the opportunistic collective invasion of a less invasive, yet highly proliferative, ΔNp63-high EMT subpopulation. EMT program heterogeneity, including ΔNp63-high and ΔNp63-low EMT states, have been detected in a range of tumor types (48,49). Our results reveal that this EMT state heterogeneity has the potential to promote subpopulation interactions that confer properties not induced by any single EMT program. Thus, our findings suggest a new function for EMT state heterogeneity in primary tumors.
ΔNp63 specifies the opportunist EMT state by inducing Slug and Axl, which promote cell migration (21,26), while simultaneously restricting the formation of LCPs that initiate collective invasion through the parallel activation of IL-1α or miR205. These results suggest a new signaling framework for defining how EMT programs promote invasion, with the nature of the program potentially having both activating and suppressive components that can be coordinated by a single transcription factor. Notably, ΔNp63 may also activate this opportunist EMT program in lung, squamous, bladder and prostate cancer cells, all of which contain ΔNp63-high populations (50). Thus, this mechanism for specifying an opportunist state may be a conserved feature across tumor types. TAp63 can be expressed in cells that lack significant ΔNp63 expression (35). However, TAp63 is unlikely to induce a similar type of opportunist EMT state in these cells given that TAp63 and ΔNp63 regulate distinct gene expression programs (34). A less understood feature of p63 regulation is the alternative splicing that takes place at the C-terminus that produces α, β and γ spliceforms (35). The ΔNp63α spliceform promotes an opportunist phenotype in our models. Whether the ΔNp63β and ΔNp63γ spliceforms have a similar activity requires further investigation. The anchoring of ΔNp63-induced IL-1α to the plasma membrane (51) limits potential IL-1α mediated suppression to directly adjacent cells. IL-1α plasma membrane attachment may also promote sustained p38 activation, which is necessary to alter gene expression in response to IL1R stimulation (52). The ability of miR205 to suppress invasion also provides redundancy in ΔNp63-high cells if intrinsic IL-1α signaling is insufficient to reach the threshold necessary to confer an opportunist phenotype.
Induction of the ΔNp63-low trailblazer EMT state can be a spontaneous epigenetic event (9), and may also be induced by microenvironmental cues or genetic abnormalities that suppress ΔNp63 expression (53,54). Morphogenesis programs can confer leading invasive cells with unique molecular and functional properties (55). However, the ΔNp63-low EMT state does not appear to be part of the adult mammary gland morphogenesis program, which is driven by a non-protrusive form of branching morphogenesis (56). Indeed, the expression of ΔNp63 on the periphery of mammary epithelial organoids that we observed suggests that ΔNp63 may normally suppress protrusion formation to promote the retention of epithelial architecture in response to acute pathogenic insults. The enhanced ability to reorganize the ECM (9) makes a trailblazer EMT different from regulatory programs that induce a leader cell phenotype in other contexts, such as during wound closure in vitro (21), or when ECMs have been remodeled by extrinsic factors, including the recruitment of fibroblasts (24). Trailblazer cells and fibroblasts require Cdc42 to reorganize the ECM in a manner that induces opportunistic invasion, indicating overlap in mechanism of action for trailblazer cells and fibroblasts (9,24). However, rare trailblazer cells were able to promote invasion, whereas the abundant fibroblasts around MCFDCIS tumors did not induce invasion at the same time point. This does not exclude the possibility that fibroblasts promote invasion, either in collaboration with trailblazer cells or at a point later in tumor progression. However, our findings do suggest that rather than undergoing a transdifferentiation to acquire general traits of fibroblasts, ΔNp63-low trailblazer cells have unique features that promote ΔNp63-high collective invasion, which requires further investigation.
Reduced proliferation is a feature of invasive tumor cells in multiple contexts (57,58), including the ability to induce collaborative modes of invasion in vitro (16). Our results provide a new mechanism to explain how proliferation is coupled to invasive phenotype and reveal for the first time that rare growth restricted cells can promote the collective invasion of proliferative siblings in a primary tumor model. Critically, the induction of invasion by rare trailblazer cells provides proliferative ΔNp63-high EMT cells access to vessels that can ferry them to distant tissues for colonization. E-cadherin may confer ΔNp63-high cells with an enhanced ability to metastasize relative to the ΔNp63-low cells (59). Indeed, cells expressing ΔNp63 are capable of initiating metastatic growth (39) and have reduced sensitivity to chemotherapy (60). Thus, the interaction between trailblazer and opportunist cells reveals a mechanism that allows cells to bypass the need to acquire additional cell intrinsic perturbations that confer invasive ability. Importantly, our results support a shift towards identifying subpopulation interactions to improve prognostic accuracy. High IL-1α expression correlated with improved outcome and trailblazer EMT gene expression correlates with worse outcome (9), suggesting that defining the nature of opportunist and trailblazer EMT states has potential utility. Further analysis in additional data sets and the development of testing at single cell resolution is required to evaluate this possibility.
In summary, our results uncover a new way in which EMT program heterogeneity promotes tumor development by revealing a functional mechanism that has the potential to eliminate evolutionary bottlenecks through collaborative interactions.
Supplementary Material
SIGNIFICANCE.
These findings reveal how an interaction between cells in different EMT states confers properties that are not induced by either EMT program alone.
ACKNOWLEDGEMENTS.
Work was supported by NIH R01CA218670, Georgetown Women and Wine (G. Pearson), NIH T32CA009686 (S. Camacho), NIH R01CA205632 and R21CA226542 (A. Riegel), NIH R01CA192381 and U54CA210181 Project 2 (R. Brekken) and NIH P30CA051008 for shared resources (G. Pearson and A. Riegel).
Footnotes
The authors declare no potential conflicts of interest.
REFERENCES
- 1.Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer 2012;12:323–34. [DOI] [PubMed] [Google Scholar]
- 2.McGranahan N, Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 2015;27:15–26. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100:57–70. [DOI] [PubMed] [Google Scholar]
- 4.Marusyk A, Polyak K. Tumor heterogeneity: causes and consequences. Biochim Biophys Acta 2010;1805:105–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Almendro V, Marusyk A, Polyak K. Cellular heterogeneity and molecular evolution in cancer. Annu Rev Pathol 2013;8:277–302. [DOI] [PubMed] [Google Scholar]
- 6.Cleary AS, Leonard TL, Gestl SA, Gunther EJ. Tumour cell heterogeneity maintained by cooperating subclones in Wnt-driven mammary cancers. Nature 2014;508:113–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marusyk A, Tabassum DP, Altrock PM, Almendro V, Michor F, Polyak K. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature 2014;514:54–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang M, Tsimelzon A, Chang CH, Fan C, Wolff A, Perou CM, et al. Intratumoral heterogeneity in a Trp53-null mouse model of human breast cancer. Cancer discovery 2015;5:520–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Westcott JM, Prechtl AM, Maine EA, Dang TT, Esparza MA, Sun H, et al. An epigenetically distinct breast cancer cell subpopulation promotes collective invasion. J Clin Invest 2015;125:1927–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neelakantan D, Zhou H, Oliphant MUJ, Zhang X, Simon LM, Henke DM, et al. EMT cells increase breast cancer metastasis via paracrine GLI activation in neighbouring tumour cells. Nature communications 2017;8:15773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calbo J, van Montfort E, Proost N, van Drunen E, Beverloo HB, Meuwissen R, et al. A functional role for tumor cell heterogeneity in a mouse model of small cell lung cancer. Cancer Cell 2011;19:244–56. [DOI] [PubMed] [Google Scholar]
- 12.Celia-Terrassa T, Meca-Cortes O, Mateo F, de Paz AM, Rubio N, Arnal-Estape A, et al. Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J Clin Invest 2012;122:1849–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bronsert P, Enderle-Ammour K, Bader M, Timme S, Kuehs M, Csanadi A, et al. Cancer cell invasion and EMT marker expression: a three-dimensional study of the human cancer-host interface. J Pathol 2014;234:410–22. [DOI] [PubMed] [Google Scholar]
- 14.Nguyen-Ngoc KV, Cheung KJ, Brenot A, Shamir ER, Gray RS, Hines WC, et al. ECM microenvironment regulates collective migration and local dissemination in normal and malignant mammary epithelium. Proc Natl Acad Sci U S A 2012;109:E2595–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ilina O, Campanello L, Gritsenko PG, Vullings M, Wang C, Bult P, et al. Intravital microscopy of collective invasion plasticity in breast cancer. Dis Model Mech 2018;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Konen J, Summerbell E, Dwivedi B, Galior K, Hou Y, Rusnak L, et al. Image-guided genomics of phenotypically heterogeneous populations reveals vascular signalling during symbiotic collective cancer invasion. Nature communications 2017;8:15078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park SY, Gonen M, Kim HJ, Michor F, Polyak K. Cellular and genetic diversity in the progression of in situ human breast carcinomas to an invasive phenotype. J Clin Invest 2010;120:636–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Casasent AK, Schalck A, Gao R, Sei E, Long A, Pangburn W, et al. Multiclonal Invasion in Breast Tumors Identified by Topographic Single Cell Sequencing. Cell 2018;172:205–17e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pearson GW. Control of Invasion by Epithelial-to-Mesenchymal Transition Programs during Metastasis. Journal of clinical medicine 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maine EA, Westcott JM, Prechtl AM, Dang TT, Whitehurst AW, Pearson GW. The cancer-testis antigens SPANX-A/C/D and CTAG2 promote breast cancer invasion. Oncotarget 2016;7:14708–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dang TT, Esparza MA, Maine EA, Westcott JM, Pearson GW. DeltaNp63alpha Promotes Breast Cancer Cell Motility through the Selective Activation of Components of the Epithelial-to-Mesenchymal Transition Program. Cancer Res 2015;75:3925–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chatterjee A, Upadhyay S, Chang X, Nagpal JK, Trink B, Sidransky D. U-box-type ubiquitin E4 ligase, UFD2a attenuates cisplatin mediated degradation of DeltaNp63alpha. Cell Cycle 2008;7:1231–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang X, Boehm JS, Yang X, Salehi-Ashtiani K, Hao T, Shen Y, et al. A public genome-scale lentiviral expression library of human ORFs. Nat Methods 2011;8:659–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dang TT, Prechtl AM, Pearson GW. Breast cancer subtype-specific interactions with the microenvironment dictate mechanisms of invasion. Cancer Res 2011;71:6857–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie Y, Wang X, Story M. Statistical methods of background correction for Illumina BeadArray data. Bioinformatics 2009;25:751–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dang TT, Westcott JM, Maine EA, Kanchwala M, Xing C, Pearson GW. DeltaNp63alpha induces the expression of FAT2 and Slug to promote tumor invasion. Oncotarget 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Green JE, Shibata MA, Yoshidome K, Liu ML, Jorcyk C, Anver MR, et al. The C3(1)/SV40 T-antigen transgenic mouse model of mammary cancer: ductal epithelial cell targeting with multistage progression to carcinoma. Oncogene 2000;19:1020–7. [DOI] [PubMed] [Google Scholar]
- 28.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery 2012;2:401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nature communications 2016;7:11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast cancer research and treatment 2010;123:725–31. [DOI] [PubMed] [Google Scholar]
- 32.Chakrabarti R, Wei Y, Hwang J, Hang X, Andres Blanco M, Choudhury A, et al. DeltaNp63 promotes stem cell activity in mammary gland development and basal-like breast cancer by enhancing Fzd7 expression and Wnt signalling. Nat Cell Biol 2014;16:1004–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crum CP, McKeon FD. p63 in epithelial survival, germ cell surveillance, and neoplasia. Annu Rev Pathol 2010;5:349–71. [DOI] [PubMed] [Google Scholar]
- 34.Abbas HA, Bui NHB, Rajapakshe K, Wong J, Gunaratne P, Tsai KY, et al. Distinct TP63 Isoform-Driven Transcriptional Signatures Predict Tumor Progression and Clinical Outcomes. Cancer Res 2018;78:451–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Su X, Chakravarti D, Flores ER. p63 steps into the limelight: crucial roles in the suppression of tumorigenesis and metastasis. Nat Rev Cancer 2013;13:136–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Memmi EM, Sanarico AG, Giacobbe A, Peschiaroli A, Frezza V, Cicalese A, et al. p63 Sustains self-renewal of mammary cancer stem cells through regulation of Sonic Hedgehog signaling. Proc Natl Acad Sci U S A 2015;112:3499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gatti V, Bongiorno-Borbone L, Fierro C, Annicchiarico-Petruzzelli M, Melino G, Peschiaroli A. p63 at the Crossroads between Stemness and Metastasis in Breast Cancer. International journal of molecular sciences 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herschkowitz JI, Simin K, Weigman VJ, Mikaelian I, Usary J, Hu Z, et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol 2007;8:R76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Di Franco S, Turdo A, Benfante A, Colorito ML, Gaggianesi M, Apuzzo T, et al. DeltaNp63 drives metastasis in breast cancer cells via PI3K/CD44v6 axis. Oncotarget 2016;7:54157–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheung KJ, Gabrielson E, Werb Z, Ewald AJ. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell 2013;155:1639–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Di Paolo NC, Shayakhmetov DM. Interleukin 1alpha and the inflammatory process. Nat Immunol 2016;17:906–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barton CE, Johnson KN, Mays DM, Boehnke K, Shyr Y, Boukamp P, et al. Novel p63 target genes involved in paracrine signaling and keratinocyte differentiation. Cell death & disease 2010;1:e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang T, Sun L, Yuan X, Qiu H. Thrombospondin-1 is a multifaceted player in tumor progression. Oncotarget 2017;8:84546–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gu C, Wang X, Long T, Wang X, Zhong Y, Ma Y, et al. FSTL1 interacts with VIM and promotes colorectal cancer metastasis via activating the focal adhesion signalling pathway. Cell death & disease 2018;9:654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A 2009;106:13820–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miller FR, Santner SJ, Tait L, Dawson PJ. MCF10DCIS.com xenograft model of human comedo ductal carcinoma in situ. J Natl Cancer Inst 2000;92:1185–6. [DOI] [PubMed] [Google Scholar]
- 47.Hu M, Yao J, Carroll DK, Weremowicz S, Chen H, Carrasco D, et al. Regulation of in situ to invasive breast carcinoma transition. Cancer Cell 2008;13:394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aiello NM, Maddipati R, Norgard RJ, Balli D, Li J, Yuan S, et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev Cell 2018;45:681–95e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S, et al. Identification of the tumour transition states occurring during EMT. Nature 2018;556:463–68. [DOI] [PubMed] [Google Scholar]
- 50.Flores ER. The roles of p63 in cancer. Cell Cycle 2007;6:300–4. [DOI] [PubMed] [Google Scholar]
- 51.Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity 2013;39:1003–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tomida T, Takekawa M, Saito H. Oscillation of p38 activity controls efficient pro-inflammatory gene expression. Nature communications 2015;6:8350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yoh KE, Regunath K, Guzman A, Lee SM, Pfister NT, Akanni O, et al. Repression of p63 and induction of EMT by mutant Ras in mammary epithelial cells. Proc Natl Acad Sci U S A 2016;113:E6107–E16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hu L, Liang S, Chen H, Lv T, Wu J, Chen D, et al. DeltaNp63alpha is a common inhibitory target in oncogenic PI3K/Ras/Her2-induced cell motility and tumor metastasis. Proc Natl Acad Sci U S A 2017;114:E3964–E73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rorth P Collective cell migration. Annu Rev Cell Dev Biol 2009;25:407–29. [DOI] [PubMed] [Google Scholar]
- 56.Ewald AJ, Brenot A, Duong M, Chan BS, Werb Z. Collective epithelial migration and cell rearrangements drive mammary branching morphogenesis. Dev Cell 2008;14:570–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Giampieri S, Manning C, Hooper S, Jones L, Hill CS, Sahai E. Localized and reversible TGFbeta signalling switches breast cancer cells from cohesive to single cell motility. Nat Cell Biol 2009;11:1287–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 2012;22:725–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Padmanaban V, Krol I, Suhail Y, Szczerba BM, Aceto N, Bader JS, et al. E-cadherin is required for metastasis in multiple models of breast cancer. Nature 2019;573:439–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leong CO, Vidnovic N, DeYoung MP, Sgroi D, Ellisen LW. The p63/p73 network mediates chemosensitivity to cisplatin in a biologically defined subset of primary breast cancers. J Clin Invest 2007;117:1370–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.