

Abstract
Regulator of G-protein signaling 10 (RGS10) is a member of the superfamily of RGS proteins that canonically act as GTPase activating proteins (GAPs). RGS proteins accelerate GTP hydrolysis on the G-protein α subunits and result in termination of signaling pathways downstream of G protein-coupled receptors. Beyond its GAP function, RGS10 has emerged as an anti-inflammatory protein by inhibiting LPS-mediated NF-κB activation and expression of inflammatory cytokines, in particular TNF-α. Although RGS10 is abundantly expressed in resting macrophages, previous studies have shown that RGS10 expression is suppressed in macrophages following Toll-like receptor 4 (TLR4) activation by LPS. However, the molecular mechanism by which LPS induces Rgs10 silencing has not been clearly defined. The goal of the current study was to determine whether LPS silences Rgs10 expression through an NF-κB-mediated proinflammatory mechanism in pulmonary macrophages, a unique type of innate immune cells. We demonstrate that Rgs10 transcript and RGS10 protein levels are suppressed upon LPS treatment in the murine MH-S alveolar macrophage cell line. We show that pharmacological inhibition of PI3K/ NF-κB/p300 (NF-κB co-activator)/TNF-α signaling cascade and the activities of HDAC (1–3) enzymes block LPS-induced silencing of Rgs10 in MH-S cells as well as microglial BV2 cells and BMDMs. Further, loss of RGS10 generated by using CRISPR/Cas9 amplifies NF-κB phosphorylation and inflammatory gene expression following LPS treatment in MH-S cells. Together, our findings strongly provide critical insight into the molecular mechanism underlying RGS10 suppression by LPS in pulmonary macrophages.
Keywords: Regulator of G-protein Signaling (RGS)10, Macrophages, Alveolar macrophage, Microglia, Lipopolysaccharide (LPS), Toll-like receptor (TLR)-4, cytokines
1. Introduction
Tissue-resident macrophages (TRMs) emerge from embryonic precursors that exert vital and specific functions in tissue homeostasis, inflammation, and regeneration [1, 2]. Among TRMs, alveolar macrophages (AMs) are the specialized cells that reside in the pulmonary alveoli and the dominant innate immune cells, as they represent 90–95% of the cellular numbers under normal conditions, making them the natural sentinels of the respiratory system [3, 4]. Like other TRMs, AMs are involved in maintaining lung homeostasis by phagocytosing apoptotic cells and cells debris resulting from lung infection or epithelial injury while also maintaining a dominant immunosuppressive phenotype [5, 6]. In particular, AMs have a central role in clearing the alveolar environment from an excessive production of lipid-rich molecules (surfactants) that are produced by type II alveolar epithelial cells and functionally prevent alveolar collapse during exhalation [7]. Despite these physiological functions, dysregulation of AM activation or their impaired clearance function are associated with initiation and progression of several respiratory pathologies, such as acute lung injury (ALI), pulmonary alveolar proteinosis [8], and chronic obstructive pulmonary disease [9].
Regulator of G-protein signaling (RGS) proteins are a large family of proteins containing RGS domain that binds and deactivates heterotrimeric G-protein subunits [10, 11]. Canonically, RGS proteins terminate signaling pathways downstream of G protein-coupled receptors (GPCRs) by acting as GTPase activating proteins (GAPs) on active form, GTP-bound Gα-subunits, through enhancing their intrinsic GTPase activity for GTP hydrolysis and returning G-proteins to their inactive form, GDP-bound Gα-subunits [12]. Due to enormous implications of GPCRs and G-protein signaling in diverse systems, RGS proteins have emerged to play a wide range of roles in regulation of physiological processes and pathologies [13].
Among RGS proteins, RGS10, a member of D/R12 subfamily of RGS proteins, is a small RGS protein that lacks structural domains and functional motifs outside of RGS domain. RGS10 has been shown to be selective to interact with and inactivate Gαi family of G-proteins via its classical GAP [14–16], and has a high enrichment in the brain [17, 18] and peripheral macrophages [19]. Beyond its GAP function, RGS10 has an anti-inflammatory role, as loss of RGS10 in microglia and macrophages amplifies NF-κB transcriptional activity and the generation of, pro-inflammatory mediators, such as TNF-α, interleukins, and COX-2-mediated prostaglandin E2 (PGE2) upon Toll-like receptor 4 (TLR4) activation [20–22]. Further, following macrophage activation, RGS10 acts as a key regulator of macrophage polarization by suppressing classical M1 activation and promoting alternative M2 activation, a phenotype also similar to that of AMs [19].
Induction of the classically activated macrophages (pro-inflammatory M1 phenotype) is triggered by pathogen-associated molecular patterns (i.e. bacterial lipopolysaccharide (LPS)) recognized by pattern recognition receptors (i.e. TLR4). This, in turn, initiates activation of certain inflammatory events involving kinase activity of PI3K, NF-κB and MAPK signaling pathways, ultimately leading to enhance inflammatory genes expression, including TNF-α. More importantly, previous studies have implicated the direct action of TLR4 stimulation by LPS in regulating RGS10 expression. First, Lee et al. showed early [19, 20] that RGS10 protein is naturally expressed at high levels in resting microglia and macrophages, but its expression is suppressed following LPS treatment in microglia and macrophages. Alqinyah et al. [23] subsequently demonstrated that the inhibition in the level of RGS10 protein in response to LPS in microglia is a result of LPS-induced Rgs10 transcript silencing. Although these studies highlight the effect of activated microglia and macrophages on RGS10 expression, the mechanistic basis for this effect have not defined yet.
In this study, we aimed to determine inflammatory responses that are involved in LPS-mediated Rgs10 suppression in macrophages. First, we observe a reduction in the level of RGS10 expression in the MH-S alveolar macrophage cell line upon LPS stimulation, similar to what have been reported in activated microglia and bone marrow derived macrophages (BMDMs) [19, 20, 23]. Furthermore, we show for the first time that the pharmacological inhibition of PI3K activity, NF-κB-dependent TNF-α secretion, and histone deacetylase (HDAC) class I activity reverse suppression of Rgs10 expression upon LPS stimulation in MH-S cells, as well as BV2 microglial cells and BMDMs, suggesting that these inflammatory mediators are required for the suppressive effect of LPS on RGS10 expression. Finally, consistent with its anti-inflammatory role in microglia and BMDMs, we demonstrate that loss of RGS10 in MH-S cells significantly enhances LPS-induced upregulation of NF-κB phosphorylation and inflammatory genes expression. The findings presented here provide novel insights into the molecular basis and the pharmacological approaches that regulate Rgs10 expression in activated macrophages.
2. Materials and Methods
2.1. Cells
MH-S mouse alveolar macrophage cell line, which is used as a model to study alveolar macrophage functions [24], was purchased from the American Type Culture Collection (ATCC® CRL-2019™). The mouse microglial BV2 cell line, which is extensively used to study microglia functions [25], was derived from primary microglial cell cultures infected with v-raf/v-myc oncogene-carrying retrovirus (J2) [26]. L-929 mouse fibroblast cell line was generously provided by Dr. Biao He lab at University of Georgia (Athens, GA). MH-S, BV2, and L-929 cells were grown in Dulbecco’s modified Eagle’s medium (Millipore Sigma. D6429) supplemented with 10% low-endotoxin fetal bovine serum (Thermo Fisher Scientific, cat#: 10082147) and an antibiotics combination of (1% penicillin/streptomycin) (Thermo Fisher Scientific, cat#: 15140122) and incubated at 37 °C in a humidified atmosphere 5% CO2.
2.2. Reagents
Lipopolysaccharide (LPS) (from E. coli O111:B4 strain, cat#: L2630) was obtained from Millipore Sigma. Recombinant mouse TNF-alpha (aa 80–235) protein (410-MT) was purchased from R&D systems. Puromycin dihydrochloride (cat#: 4089) was purchased from Tocris Bioscience. LY294002 (cat#: 70920), BAY 11–7082 (cat#: 10010266), A-485 (cat#: 24119), R-7050 (cat#: 16870), PD 98059 (cat#: 10006726), SB 239063 (cat#: 19142), SP 600125 (cat#: 10010466), Trichostatin A (TSA) (cat#: 89730), JSH-23 (cat#: 15036), wortmannin (cat#: 10010591), and 5-Azacytidine (5-Aza) (cat#: 11164) were obtained from Cayman Chemical.
2.3. Isolation and culture of BMDMs
Bone marrow was isolated from tibiae and femurs of mice by flushing with 1X ice-cold PBS supplemented with 2% low-endotoxin fetal bovine serum (FBS) (Thermo Fisher Scientific, cat#: 10082147) using a 10-ml syringe and 25-gauge needle followed by centrifugation at 450 × g for 10 min at 4 °C. The cells were resuspended in 1X ice-cold PBS supplemented with 2% low-endotoxin FBS, passed through a 70 μm cell strainer to remove solid fragments and then centrifuged again at 450 × g for 10 min at 4 °C. The cell pellet was then resuspended in ammonium-chloride-potassium (ACK) lysing buffer (Thermo Fisher Scientific, cat#: A1049201) for 30 sec to lyse red blood cells. Cells were then washed in 1X ice-cold PBS (20 ml) and centrifuged again at 450 g for 10 min at 4 °C. The cell pellet was disaggregated in ice-cold complete DMEM (containing 10% low-endotoxin FBS and 1% penicillin/streptomycin) for cell counting. Differentiated macrophages were obtained by culturing 6 × 106 bone marrow cells on sterile petri dishes in 10 ml of complete DMDM supplemented with 20% of conditioned L-929 cell media as a source of M-CSF (conditioned media collected from confluent L-929 cells grown for 12 days) for three days at 37 °C and 5% CO2. On day 3, 5 ml of complete DMEM and supplemented with 20% of conditioned L-929 cell media were added, and the cells were grown for four additional days. On day 7, the medium of cells was removed, and the adherent cells were washed with 1X PBS and then harvested by incubating in 10 ml of cell dissociation buffer (CORNING, cat#: 25–056-CI) for 15 min at 37 °C. Plates were washed with 1X PBS to collect dislodged cells, and cells were centrifuged at 250 g for 5 min. Cell pellet was resuspended in complete DEME medium for cell counting.
2.4. Generation of CRISPR/Cas9 control and RGS10 deficient MH-S cells
Scrambled control vector (K010) and 20-bp guide RNA sequence (49 TGTTTTGCAGATATCCATGA) targeting mouse RGS10 in one-lentivector (cat#: K4107305) were purchased from ABM Inc. (Richmond, BC, Canada). 10ng of vectors were transformed into Proclone competent DH5 alpha cells and the transformants were incubated in 2 ml of LB with 100 μg/ml of carbenicillin overnight at 37 °C. The following day, glycerol stocks were prepared by resuspended 500 μl of bacterial culture in 500 μl of 50% glycerol and stored in 1.5 ml freezing tube at −80 °C. Furthermore, one ml of bacterial culture was transformed into 250 ml of LB with 100 μg/ml of carbenicillin and allowed to grow for 15 hours at 37 °C. The plasmids were extracted from the bacterial culture using the GeneJET plasmid maxiprep kit according to the manufacturer’s instructions. The plasmid concentration and purity were quantified using a Nano Drop spectrophotometer. To produce lentiviral particles, 293T cells plated in two 10 cm2 dishes transfected with 10μg of expression sgRNA CRISPER/Cas9 vectors and 10 μg of ABM Inc.’s third generation (cat#: LVO53) packaging mix in presence of Lentifectin™ transfection reagent (cat#: G074) to facilitate plasmid uptake by cells following company’s lentiviral packaging protocol. 24 hours post-infection, the medium was removed and replaced with fresh complete growth medium for another 24 hours. Viral medium was collected 48 hours post-infection followed by centrifugation at 3,000 RPM for 15 minutes at 4°C. The viral supernatant was separated from cell debris by using a low-protein binding 0.45 μm sterile filter and then subsequently concentrated by centrifugation at 25,000 RPM for two hours at 4°C (~120,000g; SW28 rotor, Beckman, Brea, C). The viral pellets were resuspended with PBS and stored at −80 °C until cells transfection. Different volumes of lentiviral particles were transfected into MH-S cells in the presence of 0.8 μg/ml of polybrene. 24 hours post-infection, the medium was removed and replaced with fresh complete culture medium. 48 hours following transfection, cells were split and selected for stable expression using 4 μg/ml puromycin selection media as determined by a kill curve assay. Genome-editing was checked using qRT-PCR and western blot analysis.
2.5. Small interfering RNA transfection
The small interfering RNA (siRNA) targeting mouse p300 (cat#: sc-29432) and non-targeting control siRNA (cat#: sc-37007) were purchased from Santa Cruz Biotechnology. The siRNA transfection was performed using Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific, cat#: 13–778-030) according to the manufacturer’s recommended protocol. The mouse p300 siRNA used herein is the same as described and validated in [27–30]. The final concentration of siRNA in an antibiotic-free culture medium was 60 nM. Following transfection, MH-S cells were cultured for an additional 48 hours in an antibiotic-free culture medium before cells harvesting. The efficiency of knockdown was assessed by measuring p300 mRNA using quantitative RT-PCR.
2.6. Quantitative Real-Time Polymerase Chain Reaction
Total RNA was isolated from cells using TRIzol reagent (Invitrogen, cat#: 15596018), and cDNA was synthesized from 2μg of total RNA using the High-capacity Reverse Transcriptase cDNA kit (Applied Biosystem, cat#: 4368814). Each cDNA sample was diluted 10-fold, and a 5μl was used in a 14μl PCR reaction (SYBR™ Green PCR Master Mix.) (Thermo Fisher Scientific, cat#: 4309155) containing primers at concentration of 5μM each. All the reactions were run in triplicates. The mRNA expression levels were normalized to the housekeeping β-actin gene and were calculated using the 2−ΔΔCT method. Mouse TNF-α, IL-1β, IL-6, INOS, RGS10 and β-actin primers were obtained from Millipore Sigma. The primer sequences used for gene amplification are listed as follows: TNF-α forward, 5′-CCTGTAGCCCACGTCGTAG-3’, TNF-α reverse, 5′-GGGAGTAGACAAGGTACAACCC-3’, IL-1β forward, 5′- GAAATGCCACCTTTTGACAGTG-3’, IL-1β reverse, 5′-TGGATGCTCTCATCATCAGGACAG-3’, IL-6 forward, 5’-CTGCAAGAGACTTCCATCCAG −3’, IL-6 reverse, 5’- AGTGGTATAGACAGGTCTGTTGG −3’, INOS forward, 5′-TGACGGCAAACATGACTTCAG-3’, INOS reverse, 5′-GCCATCGGGCATCTGGTA-3’, RGS10 forward, 5’-CCCGGAGAATCTTCTGGAAGACC-3’, RGS10 reverse, 5’-CTGCTTCCTGTCCTCCGTTTTC-3’, p300 forward, 5’-AGGCAGAGTAGGACAGTGAA-3’, p300 reverse, 5’-CTCAGTCTGGGTCACTCAAT-3’, β-actin forward, 5′-GGCTGTATTCCCCTCCATCG-3’, β-actin reverse, 5′-CCAGTTGGTAACAATGCCATGT-3’.
2.7. Western Blot
Cells were washed twice with 1X cold-PBS and then were lysed with RIPA lysis buffer containing (50mM Tris HCl pH 6.8, 150mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% NP-40, and 1X proteases/phosphatase inhibitors cocktail (Cell Signaling technology, cat#: 5872S). Cell lysates were incubated for 30 minutes on ice followed by centrifugation at 20,000 g for 15 min at 4°C. Proteins concentration was measured using Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, cat#: 23225). The cell lysates were normalized and mixed with an equal volume of 2X SDS-PAGE sample buffer containing (0.5 M Tris HCl pH 6.8, 10% SDS, 20% glycerol, 200 mM β-mercaptoethanol, and 1% bromophenol blue). The lysates were boiled for 10 mins at 95 °C, and 20 μl of protein sample was separated on 12% SDS-PAGE gels followed by transfer to nitrocellulose membranes (Biorad, cat#: 1620115) using standard protocol. Blotted membranes were blocked with 5% nonfat dry milk, shaking at room temperature for one hour, then incubated overnight with the following primary antibodies: goat anti-RGS10 (diluted 1:1,000, Santa Cruz Biotechnology, cat#: sc-6206), rabbit anti-phospho-NF-κB p65 (diluted 1:1000, Cell signaling technology, cat#: 93H1 #3033), rabbit-anti-NF-κB p65 (diluted 1:1000, Cell Signaling technology, cat#: D14E12 #8242), and mouse anti-β-actin (diluted 1:3000, Santa Cruz Biotechnology, cat#: sc-47778). Following primary antibodies incubation, blotted membranes were washed with 1X-TBST buffer for three times (each time for 10min) and incubated at room temperature for one hour with the following suitable secondary-HRP conjugated antibodies (diluted 1:5,000) mouse anti-goat IgG-HRP (Santa Cruz Biotechnology, cat#: sc-2354), goat anti-mouse IgG HRP (Bethyl Laboratories, cat#: A90–116P), and goat anti-rabbit IgG-HRP (Millipore Sigma, cat#$: 12–348). After washing with 1X-TBS-T buffer three times, SuperSignal™ West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific, cat#: 34580) was applied to detect immunoreactivity of HRP. Image lab Software from Bio-Rad was used to quantify western blot bands that were normalized to the endogenous control β -actin.
2.8. TNF-α Enzyme-Linked Immunosorbent Assay (ELISA)
Culture supernatants collected from MH-S cells following LPS treatment with or without inhibitors were centrifuged at 250 g for 10 min at 4°C and assayed for TNF-α levels using mouse TNF-α DuoSet ELISA (R&D systems, cat#: DY410) according to the manufacturer’s instructions. Briefly, high binding ELISA plate (CORNING, cat#: 9018) was coated with the mouse anti-TNF-α capture antibody overnight and blocked with 1X BSA for 3 hours at room temperature. Mouse TNF-α standards or samples of medium were applied to the plate for 2 hours, and then incubated with detection antibody for 2 hours followed by streptavidin HRP for 20 min at room temperature. Between steps, plate was washed with 0.05% Tween® 20 in PBS. The plate was visualized with TMB substrate (Thermo Fisher Scientific, cat#: 34021), stopped with 2N H2SO4, and analyzed on a microplate photometer at 450 nm (EPOCH2, BioTek Instruments, Inc).
2.9. Immunofluorescence
MH-S cells were grown overnight on 4-well glass Millicell EZ Slide (Millipore corporation, cat#: PEZGS0416) and then treated with LPS (100 ng/ml) for 60 minutes with or without a one-hour pretreatment with LY-294002 (15 μM). Following the treatment, MH-S cells were fixed with 4% paraformaldehyde for 10 min and washed with PBS. Cells were subsequently permeabilized with 0.1% Triton X100 while blocking in PBS with 5% BSA and 10% normal horse serum for 1 h. Primary antibody staining to detect rabbit-anti-NF-κB p65 was performed at a 1:250 dilution in PBS with 1% BSA, 1% normal horse serum, and 0.3% Triton X100 overnight at 4 °C. Slides were washed in PBS, and secondary antibody staining was performed at a 1:500 dilution in PBS with 1% BSA, 1% normal horse serum for 1 h with horse anti-rabbit IgG antibody (H+L), DyLight® 488 (Vector Laboratories, cat#: DI-1088–1.5). Slides were washed with PBS and then vectashield™ anti-fade mounting medium with DAPI (Vector Laboratories, cat#: H-1200–10) was applied to the cell’s prior coverslip addition. All digital images were acquired at the University of Georgia College of Veterinary Medicine Cytometry Core on a Nikon A1R confocal microscope (Nikon Eclipse Ti-E inverted microscope) and examined with NIS Element software (Nikon, Version 6.4).
2.10. Statistical Analysis
All quantified data compiled from three independent repeats, unless otherwise noted, were analyzed for statistical difference between groups using student’s t-test or one-way ANOVA followed by Tukey post hoc analysis. Prism software was used to carry out statistical analyses. Data are expressed as mean ± S.E.M. where *, p<0.05; **, p<0.01; and ***, p<0.001 indicate the levels of significance.
3. Results
3.1. RGS10 expression is silenced in response to LPS stimulation in MH-S cells
RGS10 protein is expressed at high levels in resting macrophages and microglia, the brain’s resident macrophages. Strikingly, RGS10 expression is silenced in microglia and BMDMs following LPS stimulation [19, 20]. To test whether RGS10 expression is suppressed in response to LPS in the pulmonary macrophage MH-S cells, we treated MH-S cells with vehicle or LPS (10 ng/ml) to analyze RGS10 gene and protein expression levels by quantitative RT-PCR and western blot analysis, respectively. The transcript level of Rgs10 was silenced 6 hours or 24 hours post LPS stimulation (Figure 1A). Consistent with the diminished Rgs10 transcript level in activated MH-S cells, LPS treatment also triggered RGS10 protein suppression, in which the maximal effect of LPS-induced RGS10 protein suppression was observed at 48 hours (Figure 1B and Figure S1). Densitometry confirmed a significant down-regulation of RGS10 protein by LPS (Figure 1C). Therefore, polarization of alveolar macrophage to pro-inflammatory M1 phenotype upon LPS treatment negatively regulates RGS10 expression.
Fig. 1. RGS10 expression is silenced in response to LPS stimulation in MH-S cells.

(A) MH-S cells were treated with vehicle (serum-free media) or LPS (10 ng/ml) for 6 and 24 hours. RNA was isolated from the cells using TRIzol reagent, and cDNA was synthesized from the extracted RNA. RGS10 transcript level was measured using quantitative RT-PCR and normalized to an endogenous housekeeping gene β-actin. The relative expression was calculated by using the 2−ΔΔCt method. (B) MH-S cells were treated with vehicle (serum-free media) or LPS (10 ng/ml) for 24 and 48 hours. Cells were lysed and subjected to SDS-PAGE followed by immunoblotting using specific antibodies against RGS10 and β-actin. Densitometry of RGS10 band was normalized to β-actin. Fold differences of qRT-PCR and immunoblot densitometry data were calculated after normalizing to vehicle conditions and compiled from three independent experimental repeats. Data were analyzed for statistical differences using an analysis of variance (ANOVA) followed by Tukey post hoc test between groups. Data are presented as mean ± SEM where *, p< 0.05; **, p< 0.01; ***, p< 0.001.
3.2. PI3K mediates RGS10 silencing in response to TLR-4 activation
A significant body of evidence has linked the activation of PI3K to TLR4 signaling [31]. To determine whether PI3K activation contributes to RGS10 suppression by LPS, we measured Rgs10 transcript and RGS10 protein expression following LPS treatment with or without pharmacological inhibition of PI3K kinase activity. MH-S cells were treated with vehicle or the PI3K inhibitor LY294002 (15 μM) for one hour prior to LPS (10 ng/ml) stimulation for 6 hours and 48 hours, for experiments measuring transcript and protein levels, respectively. LY294002 treatment significantly increased the basal level of Rgs10 mRNA and restored both Rgs10 transcript expression (6 hours) (Figure 2A) and RGS10 protein levels (48 hours) upon LPS treatment (Figure 2B). Because LPS has been shown previously to suppress Rgs10 transcript in microglia and BMDMs, we investigated whether inhibition of PI3K kinase activity blocks LPS-induced Rgs10 silencing in BV2 microglial cells and BMDMs, as well. Addition of the PI3K kinase inhibitor LY294002 stabilized the transcript level of Rgs10 upon LPS stimulation in both BV2 (Figure 2C)and BMDMs (Figure 2D). To validate the observed effects of LY294002, we treated MH-S cells with LPS in the presence or absence of another PI3K inhibitor, wortmannin (10 μM), and observed a similar effect, in which wortmannin significantly blocked LPS-stimulated Rgs10 suppression (Figure 2E). Overall, these data indicate that activation of PI3K activity is required for LPS-dependent Rgs10 silencing.
Fig. 2. PI3K mediates RGS10 silencing in response to TLR4 activation.

(A) MH-S cells were plated in 12-well plate and allowed to adhere overnight before treatment with vehicle (serum-free media) or LPS (10 ng/ml) for 6 hours with or without a one hour-pretreatment with 15 μM LY294002 (LY). RNA was isolated from the cells using TRIzol reagent, and cDNA was synthesized from the extracted RNA. RGS10 transcript level was measured using qRT-PCR and normalized to an endogenous housekeeping gene β-actin. The relative expression was calculated by using the 2−ΔΔCt method. (B) MH-S cells were plated in six-well plate and allowed to adhere overnight before treatment with vehicle (serum-free media) or LPS (10 ng/ml) for 48 hours with or without a one hour-pretreatment with 15 μM LY294002 (LY). Cells were lysed and subjected to SDS-PAGE followed by immunoblotting using specific antibodies against RGS10 and β-actin. Densitometry of RGS10 band was normalized to β-actin. (C) BV2 cells were treated with vehicle (serum-free media) or LPS (10 ng/ml) for 6 hours with or without a one hour-pretreatment with 15 μM LY294002 (LY). (D) Murine BMDMs were treated with vehicle (serum-free media) or LPS (1 ng/ml) for 3 hours with or without LY294002 (15 μM). (E) MH-S cells were treated with vehicle (serum-free media) or LPS (10 ng/ml) for 6 hours with or without a one hour-pretreatment with 10 μM wortmannin (Wort.). RGS10 transcript level in (C, D, and E) was measured using qRT-PCR and normalized to an endogenous housekeeping gene β-actin. The relative expression was calculated by using the 2−ΔΔCt method. Fold differences of qRT-PCR and IB densitometry data were calculated after normalizing to vehicle conditions and pooled from three independent experiments. Data were analyzed for statistical differences using an analysis of variance (ANOVA) followed by Tukey post hoc test between groups. Data are presented as mean ± SEM where *, p< 0.05; **, p< 0.01; ***, p< 0.001.
3.3. Blocking NF-κB activation restores RGS10 suppression following LPS stimulation
NF-κB is a central transcription factor that is activated upon LPS exposure and subsequently enhances the expression of inflammatory mediators, contributing to the activation and function of macrophages. Activation of PI3K has been shown to be a crucial modulator of NF-κB activation and nuclear translocation [32, 33]. To confirm whether PI3K regulates NF-κB(p65) nuclear translocation following LPS stimulation, MH-S were pretreated with LY294002 for one hour followed by LPS treatment to determine nuclear localization of NF-κB(p65) by immunofluorescence. As expected, LPS evoked the nuclear translocation of the p65 subunit of NF-κB, while LY294002 pretreatment inhibited LPS-induced NF-κB(p65) nuclear translocation (Figure 3A). To address whether the activation of NF-κB plays a role in mediating inhibitory effects of LPS on RGS10 expression, we used the NF-κB inhibitor, BAY 11–7082. We first validated the inhibitor’s effect on NF-κB by measuring phosphorylation of the p65-NF-κB subunit (a cytoplasmic event of NF-κB signal activation) following BAY 11–7082 incubation and subsequent 20 minute-long LPS stimulation. The data indicate that BAY 11–7082-inhibited LPS-enhanced phosphorylation of p65-NF-κB in MH-S cells (Figure 3B).
Fig. 3. Blocking NF-κB activation restores RGS10 suppression following LPS stimulation.
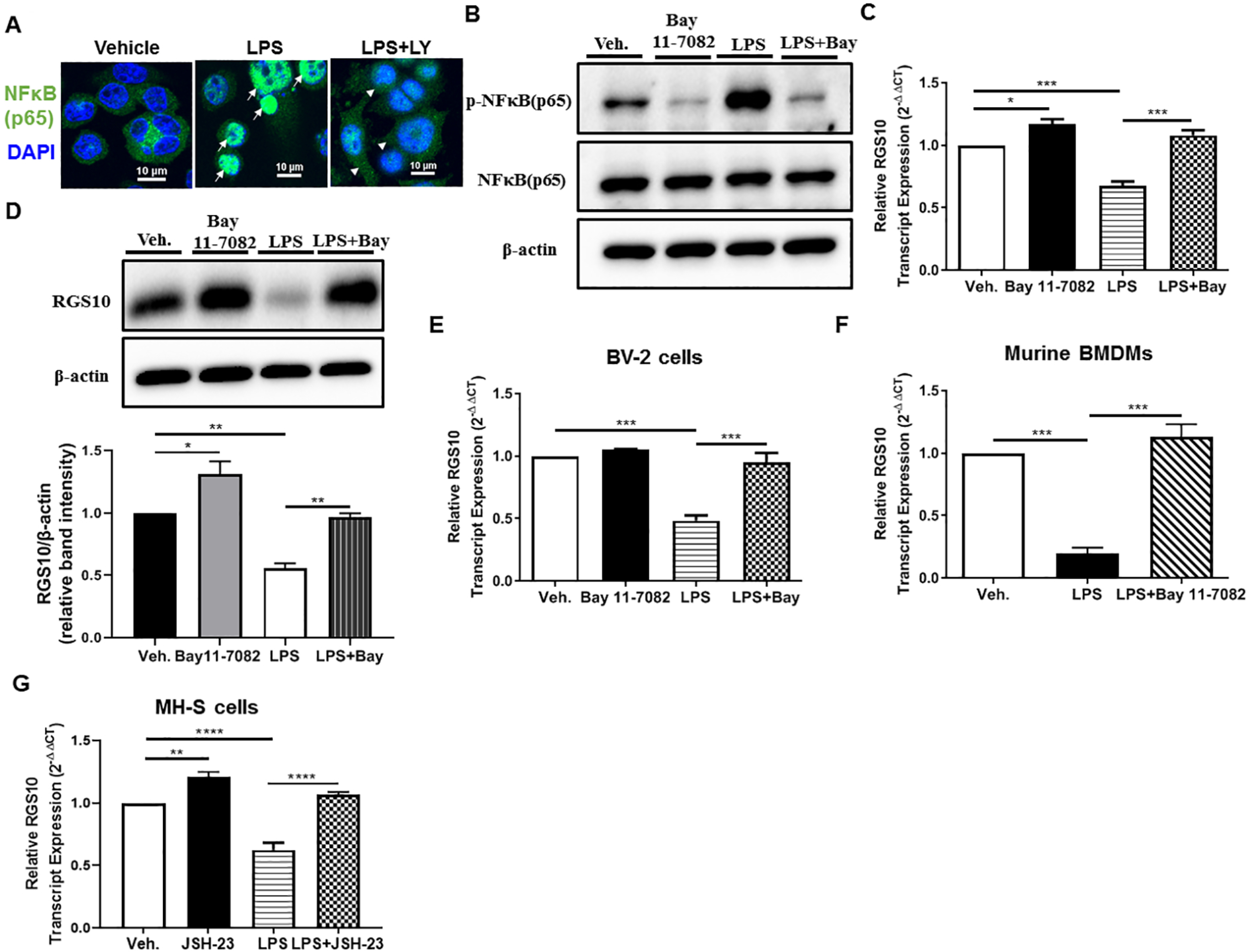
(A) LY294002 inhibits LPS-induced NF-κB (p65) nuclear localization. Immunofluorescence staining for NF-κB (p65) in MH-S cells were treated with LPS (100 ng/ml) for 60 minutes in the presence of absence of LY294002 (15 μM). IF images presented in (A) are representative two independent repeats. (B) Protein expression levels of p-NF-κB (p65) (phospho-NF-κB p65 (Ser536)), NF-κB (p65), and β-actin following treatment of LPS (10 ng/ml) for 20 minutes with or without Bay 11–7082 in MH-S cells. Blot presented in (B) is representative of two independent experiments. (C) MH-S cells were plated in 12-well plate and allowed to adhere overnight before treatment with vehicle (serum-free media) or LPS (10 ng/ml) for 6 hours with or without a one hour-pretreatment with 20 μM Bay 11–7082 (Bay). RNA was isolated from the cells using TRIzol reagent, and cDNA was synthesized from the extracted RNA. RGS10 transcript level was measured using qRT-PCR and normalized to an endogenous housekeeping gene β-actin. The relative expression was calculated by using the 2−ΔΔCt method. (D) MH-S cells were plated in six-well plate and allowed to adhere overnight before treatment with vehicle (serum-free media) or LPS (10 ng/ml) for 48 hours with or without a one hour-pretreatment with 20 μM Bay 11–7082 (Bay). Cells were lysed and subjected to SDS-PAGE followed by immunoblotting using specific antibodies against RGS10 and β-actin. Densitometry of RGS10 band was normalized to β-actin. (E) BV2 cells were treated with vehicle (serum-free media) or LPS (10 ng/ml) for 6 hours with or without a one hour-pretreatment with 20 μM Bay 11–7082 (Bay). (F) Murine BMDMs were treated with vehicle (serum-free media) or LPS (1 ng/ml) for 3 hours with or without Bay 11–7082 (20 μM). (G) MH-S cells were treated with vehicle (serum-free media) or LPS (10 ng/ml) for 6 hours with or without a one hour-pretreatment with 7 μM JSH-23. RGS10 transcript level in (E, F, and G) was measured using qRT-PCR and normalized to an endogenous housekeeping gene β-actin. The relative expression was calculated by using the 2−ΔΔCt method. Fold differences of qRT-PCR and IB densitometry data were calculated after normalizing to vehicle conditions. All the representative data in (C-G) are pooled from three independent experiments. Data were analyzed for statistical differences using an analysis of variance (ANOVA) followed by Tukey post hoc test between groups. Data are presented as mean ± SEM where *, p< 0.05; **, p< 0.01; ***, p< 0.001.
To assess whether NF-κB activation is needed for the LPS-induced suppression of RGS10, we next pretreated MH-S cells with the NF-κB inhibitor BAY 11–7082 (20 μM) for one hour followed by LPS (10 ng/ml) stimulation to measure mRNA (6 hours) or protein (48 hours) levels of RGS10. Our results showed that NF-κB inhibition significantly enhanced the basal expression of RGS10 and abolished LPS-induced RGS10 downregulation (Figure 3C and 3D). Consistent with its suppressive activity against LPS-mediated Rgs10 silencing in MH-S cells, a similar effect was observed in microglia (Figure 3E) and BMDMs (Figure 3F), as BAY 11–7082 significantly impaired the LPS-induced decrease of Rgs10 expression in both cell types. To further confirm the involvement of NF-κB activation in LPS-induced RGS10 silencing, we examined the effect of a second and well-established NF-κB inhibitor, JSH-23 (7 μM). As expected, and demonstrated in (Figure 3G), we observed a similar effect of Bay 11–7082, in which JSH-23 completely blunted Rgs10 suppression in response to LPS in MH-S cells. In addition to NF-κB signaling, LPS activates downstream MAPK signaling pathways, including extracellular signal-regulated kinase (ERK) pathway, the c-Jun N-terminal kinase (JNK) pathway, and the p38 pathway [34]. Due to the importance of these pathways in regulating inflammatory responses including NF-κB activity, we examined whether inhibitors specifically targeting each of these pathways would restore decreased RGS10 expression in LPS-stimulated MH-S cells. To test this, we stimulated MH-S cells with LPS in the presence or absence of the following inhibitors (10 μM): PD 98059 (MEK1/2 inhibitor-blocking ERK activity), SB 23906 (p38 inhibitor), and SP 600125 (JNK inhibitor). At the same concentration and time point of LPS treatment described above, the inhibition of the signaling pathways of ERK (Figure S2A), p38 (Figure S2B), and JNK (Figure S2C) had no impact on the basal or LPS-suppressed RGS10 expression. Altogether, these findings suggest that LPS activation of TLR4 induces RGS10 suppression in a NF-κB-dependent manner that is independent of MAPK pathways.
3.4. The inhibition of p300 abrogates LPS-triggered RGS10 suppression
The p300 and its paralog CREB-binding protein (CBP) co-activators contribute to enhancement of genes transcription through their intrinsic histone acetyltransferase (HAT) activity. They catalyze acetylation of histone proteins around the promoter of genes, resulting in chromatin relaxation for transcription factors (TFs) access and acetylation of nonhistone proteins, such as TFs for their activation [35]. Because of the observed involvement of NF-κB activation in LPS-induced RGS10 suppression, we next questioned whether the p300 co-activator required for full NF-kB-dependent transcriptional activity [36–38], promotes LPS-triggered RGS10 suppression. We cultured pulmonary macrophages MH-S cells with or without A-485, a highly selective inhibitor of p-300 co-activator that has been validated to target the p-300 HAT domain [39] in multiple cells including macrophages, and treated them with LPS (10 ng/ml) for 6 hours or 48 hours. Stimulation of MH-S cells with LPS resulted in a significant decrease in the mRNA and protein expression levels of RGS10, as expected. However, 1-hour pretreatment with 10 μM A-485 abrogated the downregulation of RGS10 in response to LPS on both the mRNA (Figure 4A) and the protein levels (Figure 4B). Similarly, A-485 treatment impaired LPS-stimulated Rgs10 silencing in both BV2 (Figure 4C) and BMDM cells (Figure 4D). To expand these results, and to further prove that the suppressive effect of A-485 on LPS-induced RGS10 silencing was due to targeted inhibition of p300 function, we assessed the effect of siRNA mediated knockdown of p300 on LPS-triggered TNF-α amplification and RGS10 suppression. Transient transfection of siRNA targeted at p300 in MH-S cells consistently resulted in 60% reduction of p300 mRNA level (Figure 4E). As previously reported [29, 39], p300 knockdown significantly inhibited LPS-induced upregulation of TNF-α mRNA (Figure 4F). More importantly, siRNA-mediated p300 knockdown strongly blocked the inhibitory effect of LPS on RGS10 expression (Figure 4G). Taken together, in line with facilitating the transcription of NF-κB-dependent inflammatory genes, our data demonstrate the essential function of the NF-κB co-activator p-300 in mediating LPS-sensitive RGS10 expression.
Fig. 4. The inhibition of p300 abrogates LPS-triggered RGS10 suppression.

(A) MH-S cells were plated in 12-well plate and allowed to adhere overnight before treatment with vehicle (serum-free media) or LPS (10 ng/ml) for 6 hours with or without a one hour-pretreatment with 10 μM A-485. RNA was isolated from the cells using TRIzol reagent, and cDNA was synthesized from the extracted RNA. RGS10 transcript level was measured using qRT-PCR and normalized to an endogenous housekeeping gene β-actin. The relative expression was calculated by using the 2−ΔΔCt method. (B) MH-S cells were plated in six-well plate and allowed to adhere overnight before treatment with vehicle (serum-free media) or LPS (10 ng/ml) for 48 hours with or without a one hour-pretreatment with 10 μM A-485. Cells were lysed and subjected to SDS-PAGE followed by immunoblotting using specific antibodies against RGS10 and β-actin. Densitometry of RGS10 band was normalized to β-actin. (C) BV2 cells were treated with vehicle (serum-free media) or LPS (10 ng/ml) for 6 hours with or without a one hour-pretreatment with 10 μM A-485. (D) Murine BMDMs were treated with vehicle (serum-free media) or LPS (1 ng/ml) for 3 hours with or without A-485 (10 μM). RGS10 transcript level in (C and D) was measured using qRT-PCR and normalized to an endogenous housekeeping gene β-actin. The relative expression was calculated by using the 2−ΔΔCt method. (E-G) MH-S cells were plated in 12-well plates and simultaneously transiently transfected with control or p300-targeting siRNA constructs. 48 hours post-transfection, cells were treated with vehicle (serum-free media) or LPS (10 ng/ml) for 6 hours. The relative expression of p300 transcript (E), TNF-α transcript (F), and RGS10 transcript (G) were measured using qRT-PCR and normalized to an endogenous housekeeping gene β-actin. The relative expression was calculated by using the 2−ΔΔCt method. Fold differences of qRT-PCR and IB densitometry data were calculated after normalizing to vehicle conditions and pooled from three independent experiments. Data were analyzed for statistical differences using an analysis of variance (ANOVA) followed by Tukey post hoc test between groups. Data are presented as mean ± SEM where *, p< 0.05; **, p< 0.01; ***, p< 0.001.
3.5. LPS-mediated RGS10 suppression is TNF-α-dependent
LPS is known to lead to the production of pro-inflammatory cytokines in pulmonary macrophages, such as TNF-α that is also known to act via NF-κB activation. To explore whether LPS suppresses RGS10 expression -at least in part- through a TNF-α-mediated autocrine loop, we next quantified TNF-α protein secretion in MH-S cells in response to LPS in the presence of Bay 11–7082 (NF-κB inhibitor) or A-485 (p300 inhibitor) by using ELISA. The results indicate that inhibition of either NF-κB activation or its co-activator p300 blocked LPS-stimulated TNF-α release (Figure 5A). To explore whether TNF-α signaling affects LPS-stimulated Rgs10 suppression, we blocked the tumor necrosis factor receptor (TNFR) by preincubating MH-S cells with R-7050 (TNFR inhibitor) followed by LPS priming. Pretreatment of MH-S cells with R-7050 upregulated endogenous RGS10 levels and fully abrogated RGS10 suppression by LPS (Figure 5B and Figure 5C). In addition to MH-S cells, we also confirmed these results in BV2 cells (Figure 5D) and BMDMs (Figure 5E), in which the TNFR inhibitor also blocked LPS-induced suppression of Rgs10. To further confirm that LPS downregulates RGS10 in MH-S cells through a mechanism involving TNF-α and TNFR, we treated MH-S cells with TNF-α (100 ng/ml) and examined its effect on the transcript and protein levels of RGS10. TNF-α treatment reduced Rgs10 transcript (Figure 5F) and RGS10 protein (Figure 5G) expression levels. Given the ability of NF-κB to regulate RGS10 expression upon TLR4 activation and the fact that TNF-α is also an inducer of NF-κB signaling, we evaluated TNF-α-triggered RGS10 reduction following pharmacological inhibition of NF-κB by Bay 11–7082. Similar to the regulatory effect of NF-κB on LPS-induced RGS10 suppression, inhibition of NF-κB activation blunted RGS10 suppression by TNF-α in MH-S cells (Figure 5H), suggesting the involvement of NF-κB activation in both LPS-and TNF-α-induced RGS10 suppression. Thus, our results collectively suggest that LPS induces the down-regulation of RGS10 expression through a mechanism that requires TNF-α.
Fig. 5. LPS-mediated RGS10 suppression is TNF-α-dependent.

(A) MH-S cells were cultured overnight and then incubated with vehicle (serum-free media) or LPS (10 ng/ml) for 24 hours with or without Bay 11–7082 (20 μM) or A-485 (10 μM). Culture medium was collected for detecting TNF-α level using ELISA. The data is representative of two independent experiments. (B) MH-S cells were plated in 12-well plate and allowed to adhere overnight before treatment with vehicle (serum-free media) or LPS (10 ng/ml) for 6 hours with or without a one hour-pretreatment with 10 μM R-7050. RNA was isolated from the cells using TRIzol reagent, and cDNA was synthesized from the extracted RNA. RGS10 transcript level was measured using qRT-PCR and normalized to an endogenous housekeeping gene β-actin. The relative expression was calculated by using the 2−ΔΔCt method. (C) MH-S cells were plated in six-well plate and allowed to adhere overnight before treatment with vehicle (serum-free media) or LPS (10 ng/ml) for 48 hours with or without a one hour-pretreatment with 10 μM R-7050. Cells were lysed and subjected to SDS-PAGE followed by immunoblotting using specific antibodies against RGS10 and β-actin. Densitometry of RGS10 band was normalized to β-actin. (D) BV2 cells were treated with vehicle (serum-free media) or LPS (10 ng/ml) for 6 hours with or without a one hour-pretreatment with 10 μM R-7050. (E) Murine BMDMs were treated with vehicle (serum-free media) or LPS (1 ng/ml) for 3 hours with or without R-7050 (10 μM). (F) MH-S cells were treated with vehicle or TNF-α (100 ng/ml) for 24 hours. RGS10 transcript level in (D-F) was measured using qRT-PCR and normalized to an endogenous housekeeping gene β-actin. The relative expression was calculated by using the 2−ΔΔCt method. (G) MH-S cells were treated with vehicle or TNF-α (100 ng/ml) for 48 hours. Cells were lysed and subjected to SDS-PAGE followed by immunoblotting using specific antibodies against RGS10 and β-actin. Densitometry of RGS10 band was normalized to β-actin. Fold differences of qRT-PCR and IB densitometry data were calculated after normalizing to vehicle conditions and pooled from three independent experiments. Data were analyzed for statistical differences using unpaired t-test or an analysis of variance (ANOVA) followed by Tukey post hoc test between groups. Data are presented as mean ± SEM where *, p< 0.05; **, p< 0.01; ***, p< 0.001.
3.6. HDAC activity is required for Rgs10 expression silencing by LPS
Our results, along with previous studies [19, 20] have demonstrated that LPS-induced reduction of RGS10 protein expression is a result of Rgs10 transcript suppression, suggesting epigenetic mechanisms behind Rgs10 regulation. Given that histone deacetylation mediated by HDACs is primarily implicated in epigenetic transcriptional repression, we investigated whether HDAC inhibitors reverse Rgs10 silencing by LPS. First, to gain a global insight into the role of HDAC enzymes in promoting LPS-mediated RGS10 suppression, we treated MH-S cells with vehicle or LPS (10 ng/ml) following pretreatment with the pan-HDAC inhibitor (trichostatin, TSA, 100 nM) for one hour. We found that the inhibition of the global HDAC activity by TSA increased the basal level of RGS10 and completely blocked the suppressive effect of LPS on Rgs10 transcript (Figure 6A) and RGS10 protein (Figure 6B) expressions. The activity or expression of HDACs 1–3 has been shown to be enhanced by LPS stimulation in macrophage cells, where they have an essential role in facilitating LPS-activated transcription of inflammatory gene expression including TNF-α [40–42]. Since TSA is a non-selective and general HDAC inhibitor, we next examined the effect of a selective inhibitor targeting class I HDACs (HDACs 1–3) on the suppression of RGS10 by LPS. We co-treated MH-S cells with LPS and the selective HDACs (1–3) inhibitor apicidin [43]. We found that apicidin treatment fully inhibited RGS10 suppression by LPS in MH-S cells (Figure 6C and 6D). Consistent with these MH-S cells data, we also observed a similar effect in BV2 (Figure 6E) and BMDM (Figure 6F) cells, where apicidin significantly blocked LPS-induced Rgs10 silencing. In addition to histone deacetylation, DNA methylation is another epigenetic mechanism that is mediated by DNA methyltransferases (DNMTs) and negatively regulate genes transcription. To determine the involvement of DNMTs in the regulation of RGS10 expression in LPS-activated MH-S cells, we pretreated the cells with the DNMTs inhibitor 5-Azacytidine (5-Aza) followed by LPS stimulation. 5-Aza pretreatment does not have a significant effect on LPS-mediated suppression of RGS10 expression (Figure S3). These findings suggest that Rgs10 silencing following TLR4 activation is mediated by class I HDACs (1–3), not DNMTs.
Fig. 6. HDAC activity is required for Rgs10 suppression by LPS.

(A and C) MH-S cells were plated in 12-well plate and allowed to adhere overnight before treatment with vehicle (serum-free media) or LPS (10 ng/ml) for 6 hours with or without a one hour-pretreatment with 100nM TSA (A) or 500nM apicidin (C). RNA was isolated from the cells using TRIzol reagent, and cDNA was synthesized from the extracted RNA. RGS10 transcript level was measured using qRT-PCR and normalized to an endogenous housekeeping gene β-actin. The relative expression was calculated by using the 2−ΔΔCt method. (B and D) MH-S cells were plated in six-well plate and allowed to adhere overnight before treatment with vehicle (serum-free media) or LPS (10 ng/ml) for 48 hours with or without a one hour-pretreatment with 100nM TSA (B) or 500nM apicidin (D). Cells were lysed and subjected to SDS-PAGE followed by immunoblotting using specific antibodies against RGS10 and β-actin. Densitometry of RGS10 band was normalized to β-actin. (E) BV2 cells were treated with vehicle (serum-free media) or LPS (10 ng/ml) for 6 hours with or without a one hour-pretreatment with 500nM apicidin. (F) Murine BMDMs were treated with vehicle (serum-free media) or LPS (1 ng/ml) for 3 hours with or without 500nM apicidin. RGS10 transcript level in (E and F) was measured using qRT-PCR and normalized to an endogenous housekeeping gene β-actin. The relative expression was calculated by using the 2−ΔΔCt method. Fold differences of qRT-PCR and IB densitometry data were calculated after normalizing to vehicle conditions and pooled from three independent experiments. Data were analyzed for statistical differences using an analysis of variance (ANOVA) followed by Tukey post hoc test between groups. Data are presented as mean ± SEM where *, p< 0.05; **, p< 0.01; ***, p< 0.001.
3.7. Loss of RGS10 amplifies phosphorylation of p65-NF-κB and upregulates pro-inflammatory gene expression in pulmonary macrophages
Beyond its canonical function against Gαi-subunits, RGS10 acts as an anti-inflammatory regulator in microglia and BMDMs, where RGS10 particularly inhibits LPS-stimulated expression of various inflammatory genes, such as TNF-α and interleukins [19, 20]. In order to confirm the anti-inflammatory action of RGS10 in MH-S AM cells, we generated MH-S stable control and RGS10-deficient (RGS10 KO) cells using the CRISPR-Cas9 gene editing system and examined inflammatory responses in LPS-stimulated control and RGS10 KO MH-S cells. First, we confirmed that RGS10 protein expression was entirely absent in MH-S KO cells (Figure 7A). We next treated control and RGS10 KO MH-S cells with LPS (10 ng/ml) for 20 minutes to determine the extent of phosphorylation of the p65-NF-κB subunit. Immunoblot analysis showed that loss of RGS10 enhanced LPS-induced NF-κB(p65) phosphorylation (Figure 7A). RGS10 protein levels did not change in the parental MH-S cells during the short time incubation of LPS treatment (Figure 7A). We subsequently measured transcript levels of pro-inflammatory genes following 24 hours of LPS stimulation and found that a significant upregulation of TNF-α (Figure 7B), IL-1β (Figure 7C), IL-6 (Figure 7D), and INOS (Figure 7E) in RGS10 KO AM MH-S cells compared to control cells expressing RGS10. Our results strongly suggest that RGS10 suppresses pulmonary macrophages activation.
Fig. 7. Loss of RGS10 amplifies phosphorylation of p65-NF-κB and upregulation of pro-inflammatory gene expression in pulmonary macrophages.

(A) MH-S cells were stably infected with control or RGS10 targeted CRISPR/Cas9 lentivirus as described in materials and methods. Control and RGS10 knockout (KO) MH-S cells were treated with vehicle (serum-free media) or LPS (10 ng/ml) for 20 minutes. Cells were lysed and subjected to SDS-PAGE followed by immunoblotting using specific antibodies against RGS10, p-NF-κB (p65) (phospho-NF-κB p65 (Ser536)), NF-κB (p65), and β-actin. Densitometry of p-NF-κB (p65) intensity was normalized to NF-κB (p65). (B-E) Control and RGS10 knockout (KO) MH-S cells were treated with vehicle (serum-free media) or LPS (10 ng/ml) for 24 hours. RNA was isolated from the cells using TRIzol reagent, and cDNA was synthesized from the extracted RNA. Relative expression of TNF-α transcript (B), IL-1β transcript (C), IL-6 transcript (D), and INOS transcript (E) were analyzed using qRT-PCR and normalized to an endogenous housekeeping gene β-actin. The relative expression was calculated by using the 2−ΔΔCt method. Fold differences of qRT-PCR and IB densitometry data were calculated after normalizing to vehicle conditions and pooled from three independent experiments. Data were analyzed for statistical differences using an analysis of variance (ANOVA) followed by Tukey post hoc test between groups. Data are presented as mean ± SEM where *, p< 0.05; **, p< 0.01; ***, p< 0.001.
4. Discussion
RGS10 is a small protein that is implicated in multiple disease states [44] and controls physiology of diverse cells, including macrophages [19, 45], neurons [46], osteoclasts [47–49], T-lymphocytes [50], cancer cells [51, 52], platelets [53–55], and cardiomyocytes [56, 57]. Among these cells, the highest endogenous expression of RGS10 is found in immune cells, with abundant expression in macrophages [19, 20]. Macrophage cells in different organs, such as microglia and alveolar macrophage maintain normal organ homeostasis and become activated in response to injury, infection, and environmental toxins. Under acute activation conditions, they play an important role in neutralizing those assaults and engulfing dead cells and cells debris. However, sustained activation of macrophages leads to an overproduction of inflammatory mediators, such as TNF-α and interleukins, resulting in persistent inflammatory responses, inflammatory diseases, and organs damage.
Macrophages can be activated following TLR4 stimulation by microbial molecules, such as the bacterial LPS that triggers multiple inflammatory signaling pathways, leading to the transcription of inflammatory genes. RGS10 is highly enriched in macrophage cells under resting conditions, but the expression of RGS10 is silenced following stimulation with LPS [19, 20]. Although TLR-4 activation by LPS results in the suppression of RGS10 expression in macrophages, the inflammatory responses that are required for LPS-induced silencing of RGS10 are unknown.
In this study, we confirmed and further expanded the abundant expression of RGS10 in macrophages, as we detected its high expression in another unique type of TRMs, alveolar macrophages. While the expression of RGS10 is high in the resting state of MH-S cells, its expression is down-regulated following MH-S cells polarization to classically activated macrophages characterized by a pro-inflammatory M1 phenotype. Consistent with the suppressive effect of LPS on RGS10 expression in microglia and BMDMs, we also showed that LPS induced suppression of Rgs10 transcript and RGS10 protein expression levels in pulmonary macrophages.
The interaction of TLR4 with LPS results in the activation of multiple intracellular inflammatory signaling pathways, which ultimately induce inflammatory genes expression, including TNF-α. In this study, we identified the signaling steps that are essential for LPS-stimulated RGS10 suppression. In particular, we demonstrated that LPS facilitates RGS10 silencing through setting off a cascade of events: activation of PI3K, subsequent activation and initiation of NF-κB nuclear translocation, and enhancement of HDACs enzyme activity. In the nucleus, NF-κB acts as transcription factor that drives the expression of TNF-α and several other pro-inflammatory genes via an interaction with its co-activator, p-300. TNF-α, in turn, binds its TNFR receptor and in a positive feedback loop amplifies NF-κB signaling and enhances HDAC (1–3) activities, subsequently mediating histones deacetylation at Rgs10 promoter and resulting in transcriptional silencing of Rgs10 expression. Collectively, our data substantiate that pharmacological inhibition of the activities of these inflammatory responses enhance the basal expression of RGS10 and/or blocks LPS-mediated silencing of RGS10 (Figure 8).
Fig. 8. A proposed model of TLR-4 activation-induced RGS10 suppression in microglia and macrophages.

RGS10 canonically interacts with Gαi proteins and acts as GTPase-activating protein (GAP) to diminish the activation of Gαi proteins by G-protein Coupled Receptors (GPCRs) (1). Beyond its GAP activity, RGS10 is highly enriched in macrophages and plays an anti-inflammatory role by inhibiting the pro-inflammatory M1 phenotype activation by suppressing LPS-stimulated NF-κB activity, pro-inflammatory genes expression and promoting the anti-inflammatory M2 phenotype activation (2). Activation of macrophages is triggered by LPS-stimulated TLR-4 (3). Upon LPS stimulation, PI3K is activated (4), which in turn promotes NF-κB activation and its subsequent nuclear translocation (5). In the nucleus, NF-κB binds to its cognate κB DNA site and interacts with its co-activator p-300 (6) to initiate a plethora of pro-inflammatory genes expression including TNF-α. TNF-α binds its receptor (7) in an autocrine manner that further amplifies NF-κB signals in a positive feedback loop (5) and upregulates HDACs (1–3) activity (8). HDACs (1–3) mediates histones deacetylation at the Rgs10 promoter and results in transcriptional silencing of Rgs10 expression (9). Suppression of Rgs10 expression will indirectly amplifies G protein-coupled receptor signaling that is regulated by RGS10 (1) and will lead to the loss of its anti-inflammatory activity (2), resulting in an amplification of pro-inflammatory M1 activation of macrophages in a feed-forward mechanism that contribute to dysregulation of inflammatory signaling (NF-κB-dependent pro-inflammatory mediators) and inhibition of anti-inflammatory M2 activation of macrophages. Our data substantiate that pharmacological inhibition of inflammatory signal responses in (4), (5), (6), (7), and (8) enhance the basal expression of RGS10 and/or blocks LPS-mediated silencing of RGS10. The effects of pharmacological agents LY294002, wortmannin, Bay 11–7082, JSH-23, A-485, R-7050, and apicidin are shown.
RGS10 regulates microglia and macrophages activation by acting as a critical regulator of inflammatory signaling. More specifically, following LPS stimulation, RGS10 strongly suppresses the production of pro-inflammatory mediators, in particular TNF-α and interleukins and inhibits the activity of NF-κB, a critical transcription factor and signaling link between the activation of TLR-4 by LPS and inflammatory mediator’s expression [19–21, 58]. Our results expand the anti-inflammatory function of RGS10 to alveolar macrophages, where a complete loss of RGS10 expression results in a robust upregulation of LPS-induced phosphorylation of p65-NF-κB and inflammatory genes expression, such as TNF-α. Our data in combination with previous studies have showed that RGS10 expression is suppressed following macrophages activation, which will lead to the loss of ant-inflammatory effect of RGS10 and thereby amplifies inflammatory signal responses that contribute to macrophages activation in a vicious cycle. Thus, understanding the mechanism underlying the silencing of RGS10 expression in activated macrophages is critical to identify therapeutic targets that can prevent RGS10 loss and thereby diminish multiple inflammatory signaling pathways. Our study is the first to set the basis for identifying inflammatory targets that facilitate RGS10 suppression in response to LPS-activated macrophages and provide valuable insight for guiding future efforts to screen and develop small molecule regulators of RGS10 expression that could serves as anti-inflammatory therapeutics.
Numerous studies have demonstrated the localization of RGS10 to both the cytoplasm and the nucleus of the cells, with no significant plasma membrane localization [59, 60]. In microglia, RGS10 is evenly expressed in both the cytoplasmic and the nuclear compartments under the resting condition. However, in response to LPS stimulation, much of RGS10 translocates from the cytoplasm to the nucleus [20]. More importantly, an early study has reported that nuclear localization of RGS10 is driven by the cyclic AMP-dependent protein kinase A (PKA)-mediated phosphorylation of RGS10 on serine 168 [61]. In dopaminergic neurons, overexpression of RGS10-S168A (RGS10SA, resistant to phosphorylation by PKA) or blocking PKA-triggered RGS10 phosphorylation and the subsequent nuclear translocation limits the prosurvival role of RGS10 in TNF-α-induced neurotoxicity [46]. In terms of NF-κB signaling regulation, PKA is known to phosphorylate NF-κB(p65) and modulates its transcriptional activity via potentiation the interaction of the phosphorylated NF-κB(p65) and the CBP/p300 co-activators [62, 63]. Therefore, it is important to determine if PKA regulates LPS-triggered RGS10 nuclear localization and whether PKA is involved in LPS-induced RGS10 suppression in macrophages.
DNA methylation and histone modifications are epigenetic marks involved in the regulation of gene transcription [64, 65]. In particular, DNMTs-mediated DNA hypermethylation and loss of histone acetylation via HDAC enzymes represent critical mechanisms that alter chromatin structure, TF-DNA interactions and generally result in gene suppression. Given their role in the transcriptional repression, we tested the effect of DNA methylation and histone deacetylation inhibition on LPS-induced Rgs10 silencing. Blocking HDACs activity by the pan-HDACs inhibitor (TSA) stabilizes the expression of RGS10 upon LPS exposure, whereas DNMTs inhibitor does not have a significant effect on LPS-induced RGS10 suppression. Stimulation of several cell types including microglia and macrophages with LPS has shown to induce expression and activity of HDAC enzymes [66], mainly three class I HDACs (1–3), HDAC1, HDAC2, and HDAC3 [41, 42]. We showed that the selective HDAC (1–3) enzymes inhibitor (Apicidin) blocks the ability of LPS to suppress RGS10 transcription, demonstrating that the activity of HDACs 1–3 is required for LPS silencing of RGS10. Class I HDACs (1–3) are highly homologous nuclear proteins that form homo- and heterodimers between each other and constitute the catalytic activity of multiple protein repressor complexes [67–69]. Ongoing studies including isoform-selective inhibitor(s) and chromatin immunoprecipitation assay are examining which class I HDAC (1–3) isoform(s) are involved in LPS-induced suppression of RGS10. Growing evidence strongly implicates class I HDACs (1–3) in the regulation of differentiation and activation of macrophages, as they support pro-inflammatory M1 responses and inhibit M2-phenotype polarization [70, 71]. Due to the reported anti-inflammatory HDAC inhibitors in activated macrophages and the fact that loss of RGS10 enhances inflammatory mediators’ expression, it will be interesting to test whether the anti-inflammatory action of HDAC inhibitors is due to reversing RGS10 silencing in macrophages.
In addition to macrophages, suppression of RGS10 expression can occur in multiple cells, including microglia [20, 21], neurons [46], cardiomyocytes [56], and ovarian cancer cells [51]. Loss of RGS10 expression in these cells contributes to microglia overactivation and subsequent neuroinflammation-mediated neurodegeneration [20, 21, 46, 72], cardiac hypertrophy [56], and ovarian cancer chemoresistance [73, 74], respectively. Critically, inflammatory signaling, in particular NF-κB activity and pro-inflammatory cytokines are implicated in neurodegenerative diseases (such as Parkinson’s disease) [75], heart failure [76, 77], and cancer progression and chemoresistance [78]. Our findings in elucidating inflammatory responses facilitating RGS10 suppression in microglia and macrophages may have broad effects to include neurons, cardiomyocytes, and ovarian cancer cells, where loss of RGS10 expression is observed, its physiological role is protective, and the inflammatory signaling has a major influence. Therefore, RGS10 is a valuable and novel drug target, as restoring or stabilizing RGS10 expression could be an effective therapeutic strategy in the treatment of these pathologies associated with low level of RGS10 expression and high levels of inflammatory mediators.
This study has limitations, as all the data have generated in mouse cells not human cells. Further, due to the lower yield of murine primary pulmonary macrophages, the focus of this study was solely done on in vitro alveolar macrophage MH-S cells. However, we confirmed the main findings in primary mouse BMDMs and microglia, suggesting that the regulation of RGS10 expression by LPS is not macrophage type-specific.
5. Conclusion
This study showed the suppression of RGS10 expression in response to LPS activation in alveolar macrophages and identified the inflammatory responses that are required for the inhibitory effect of LPS on RGS10 expression. Pharmacological inhibition of PI3K activity, NF-κB-dependent TNF-α expression and HDACs (1–3) activities stabilized RGS10 expression following LPS stimulation in AMs, as well as microglia and BMDMs. MH-S cells lacking functional RGS10 are hypersensitive to LPS stimulation demonstrated by amplification of phospho-NF-κB(p65) and upregulation of pro-inflammatory gene expression. Collectively, these findings provide important information about the molecular mechanism underlying RGS10 suppression in activated macrophages and will help in the development of future small molecule RGS10 stabilizers in order to exploit its strong anti-inflammatory activity.
Supplementary Material
Fig. S1. Validation of the RGS10 antibody.
MH-S cells were treated with vehicle (serum-free media) or LPS (10 ng/ml) for 24 and 48 hours. Cells were lysed and subjected to SDS-PAGE followed by immunoblotting using specific antibodies against RGS10 and β-actin. The pictures represent the full-length gels with the indicated molecular weight markers (BioRad, cat#: 1610374) for Figure 1B. The picture was taken using a BioRad Molecular Imager ChemiDoc™ XRS+ System with Image Lab™ Software.
Fig. S2. Blocking MAPK signaling pathways does not affect LPS-induced RGS10 suppression.
(A-C) MH-S cells were plated in six-well plate and allowed to adhere overnight before treatment with vehicle (serum-free media) or LPS (10 ng/ml) for 48 hours with or without a one hour-pretreatment with 10 μM of ((A) PD98059 (PD), (B) SB23906 (SB), and (C) SP600125 (SP)). Cells were lysed and subjected to SDS-PAGE followed by immunoblotting using specific antibodies against RGS10 and β-actin. One blot of two similar independent experiments is shown in each panel.
Fig. S3. The effect of 5-Aza, DNMTs inhibitor, on LPS-stimulated silencing of RGS10.
MH-S cells were plated in six-well plate and allowed to adhere overnight before treatment with vehicle (serum-free media) or LPS (10 ng/ml) for 48 hours with or without a one hour-pretreatment with 10 μM 5-Aza-2’-deoxyytidine (5-Aza). Cells were lysed and subjected to SDS-PAGE followed by immunoblotting using specific antibodies against RGS10 and β-actin. One blot of two similar independent experiments is shown.
Highlights:
RGS10 expression is suppressed in response to LPS-stimulated pulmonary macrophages.
Pharmacological inhibition of inflammatory responses involving PI3K and NF-κB-dependent TNF-α stabilizes RGS10 expression following TLR4 activation by LPS.
Blocking histone deacetylase activity restores RGS10 expression upon LPS stimulation.
Loss of RGS10 amplifies LPS-induced NF-κB activation and pro-inflammatory gene expression in pulmonary macrophages.
Acknowledgements:
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Funding:
This work was supported by the University of Georgia Research Foundation startup grant to B.R. and the National Institutes of Health (to B.R., R01AI146857-01A1).
Abbreviations:
- 5-Aza
5-aza-2’-deoxycytidine
- CNS
central nervous system
- COX-2
cyclooxygenase-2
- ERK
extracellular signal-regulated kinase
- GAP
GTPase-activating protein
- GPCR
G protein coupled receptor
- HDAC
histone deacetylase
- IL
interleukin
- INOS
inducible nitric oxide synthase
- LPS
lipopolysaccharide
- MAPK
mitogen-activated protein kinase
- NF-κB
nuclear factor-kappa B
- PGE2
prostaglandin E2
- RGS
regulator of G-protein signalling
- RT-PCR
reverse transcription-polymerase chain reaction
- TF
transcription factor
- TLR
toll-like receptor
- TNF-α
tumor necrosis factor-alpha
- TSA
trichostatin A
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Ginhoux F and Guilliams M, Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity, 2016. 44(3): p. 439–449. [DOI] [PubMed] [Google Scholar]
- [2].Mosser DM, Hamidzadeh K, and Goncalves R, Macrophages and the maintenance of homeostasis. Cell Mol Immunol, 2021. 18(3): p. 579–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hussell T and Bell TJ, Alveolar macrophages: plasticity in a tissue-specific context. Nat Rev Immunol, 2014. 14(2): p. 81–93. [DOI] [PubMed] [Google Scholar]
- [4].Kopf M, Schneider C, and Nobs SP, The development and function of lung-resident macrophages and dendritic cells. Nat Immunol, 2015. 16(1): p. 36–44. [DOI] [PubMed] [Google Scholar]
- [5].Westphalen K, et al. , Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature, 2014. 506(7489): p. 503–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Rubins JB, Alveolar macrophages: wielding the double-edged sword of inflammation. Am J Respir Crit Care Med, 2003. 167(2): p. 103–4. [DOI] [PubMed] [Google Scholar]
- [7].Whitsett JA, Wert SE, and Weaver TE, Diseases of pulmonary surfactant homeostasis. Annu Rev Pathol, 2015. 10: p. 371–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Trapnell BC, Whitsett JA, and Nakata K, Pulmonary alveolar proteinosis. N Engl J Med, 2003. 349(26): p. 2527–39. [DOI] [PubMed] [Google Scholar]
- [9].Vlahos R and Bozinovski S, Role of alveolar macrophages in chronic obstructive pulmonary disease. Front Immunol, 2014. 5: p. 435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Watson N, et al. , RGS family members: GTPase-activating proteins for heterotrimeric G-protein alpha-subunits. Nature, 1996. 383(6596): p. 172–5. [DOI] [PubMed] [Google Scholar]
- [11].Hollinger S and Hepler JR, Cellular regulation of RGS proteins: modulators and integrators of G protein signaling. Pharmacol Rev, 2002. 54(3): p. 527–59. [DOI] [PubMed] [Google Scholar]
- [12].Gilman AG, G proteins: transducers of receptor-generated signals. Annu Rev Biochem, 1987. 56: p. 615–49. [DOI] [PubMed] [Google Scholar]
- [13].Squires KE, et al. , Genetic Analysis of Rare Human Variants of Regulators of G Protein Signaling Proteins and Their Role in Human Physiology and Disease. Pharmacol Rev, 2018. 70(3): p. 446–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hunt TW, et al. , RGS10 is a selective activator of G alpha i GTPase activity. Nature, 1996. 383(6596): p. 175–7. [DOI] [PubMed] [Google Scholar]
- [15].Masuho I, et al. , A Global Map of G Protein Signaling Regulation by RGS Proteins. Cell, 2020. 183(2): p. 503–521 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Popov S, et al. , The regulators of G protein signaling (RGS) domains of RGS4, RGS10, and GAIP retain GTPase activating protein activity in vitro. Proc Natl Acad Sci U S A, 1997. 94(14): p. 7216–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Waugh JL, et al. , Regional, cellular, and subcellular localization of RGS10 in rodent brain. J Comp Neurol, 2005. 481(3): p. 299–313. [DOI] [PubMed] [Google Scholar]
- [18].Butovsky O, et al. , Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci, 2014. 17(1): p. 131–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lee JK, et al. , Critical role of regulator G-protein signaling 10 (RGS10) in modulating macrophage M1/M2 activation. PLoS One, 2013. 8(11): p. e81785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lee JK, et al. , Regulator of G-protein signaling 10 promotes dopaminergic neuron survival via regulation of the microglial inflammatory response. J Neurosci, 2008. 28(34): p. 8517–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lee JK, et al. , Regulator of G-protein signaling-10 negatively regulates NF-kappaB in microglia and neuroprotects dopaminergic neurons in hemiparkinsonian rats. J Neurosci, 2011. 31(33): p. 11879–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Alqinyah M, et al. , RGS10 Regulates the Expression of Cyclooxygenase-2 and Tumor Necrosis Factor Alpha through a G Protein-Independent Mechanism. Mol Pharmacol, 2018. 94(4): p. 1103–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Alqinyah M, et al. , Regulator of G Protein Signaling 10 (Rgs10) Expression Is Transcriptionally Silenced in Activated Microglia by Histone Deacetylase Activity. Mol Pharmacol, 2017. 91(3): p. 197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mbawuike IN and Herscowitz HB, MH-S, a murine alveolar macrophage cell line: morphological, cytochemical, and functional characteristics. J Leukoc Biol, 1989. 46(2): p. 119–27. [DOI] [PubMed] [Google Scholar]
- [25].Henn A, et al. , The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. ALTEX, 2009. 26(2): p. 83–94. [DOI] [PubMed] [Google Scholar]
- [26].Blasi E, et al. , Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J Neuroimmunol, 1990. 27(2–3): p. 229–37. [DOI] [PubMed] [Google Scholar]
- [27].Kadiyala CS, et al. , Acetylation of retinal histones in diabetes increases inflammatory proteins: effects of minocycline and manipulation of histone acetyltransferase (HAT) and histone deacetylase (HDAC). J Biol Chem, 2012. 287(31): p. 25869–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jang SM, et al. , KAT5-mediated SOX4 acetylation orchestrates chromatin remodeling during myoblast differentiation. Cell Death Dis, 2015. 6: p. e1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lee KH, et al. , Cigarette smoke extract-induced downregulation of p300 is responsible for the impaired inflammatory cytokine response of macrophages. Cell Signal, 2021. 85: p. 110050. [DOI] [PubMed] [Google Scholar]
- [30].Lee Y, et al. , Coactivation of the CLOCK-BMAL1 complex by CBP mediates resetting of the circadian clock. J Cell Sci, 2010. 123(Pt 20): p. 3547–57. [DOI] [PubMed] [Google Scholar]
- [31].Cianciulli A, et al. , Microglia Mediated Neuroinflammation: Focus on PI3K Modulation. Biomolecules, 2020. 10(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Dilshara MG, et al. , Downregulation of NO and PGE2 in LPS-stimulated BV2 microglial cells by trans-isoferulic acid via suppression of PI3K/Akt-dependent NF-kappaB and activation of Nrf2-mediated HO-1. Int Immunopharmacol, 2014. 18(1): p. 203–11. [DOI] [PubMed] [Google Scholar]
- [33].Xu S, et al. , TLR4 promotes microglial pyroptosis via lncRNA-F630028O10Rik by activating PI3K/AKT pathway after spinal cord injury. Cell Death Dis, 2020. 11(8): p. 693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kyriakis JM and Avruch J, Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev, 2012. 92(2): p. 689–737. [DOI] [PubMed] [Google Scholar]
- [35].Dancy BM and Cole PA, Protein lysine acetylation by p300/CBP. Chem Rev, 2015. 115(6): p. 2419–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Vanden Berghe W, et al. , The nuclear factor-kappaB engages CBP/p300 and histone acetyltransferase activity for transcriptional activation of the interleukin-6 gene promoter. J Biol Chem, 1999. 274(45): p. 32091–8. [DOI] [PubMed] [Google Scholar]
- [37].Hoberg JE, et al. , IkappaB kinase alpha-mediated derepression of SMRT potentiates acetylation of RelA/p65 by p300. Mol Cell Biol, 2006. 26(2): p. 457–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Chen LF, et al. , NF-kappaB RelA phosphorylation regulates RelA acetylation. Mol Cell Biol, 2005. 25(18): p. 7966–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Peng J, et al. , p300/CBP inhibitor A-485 alleviates acute liver injury by regulating macrophage activation and polarization. Theranostics, 2019. 9(26): p. 8344–8361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Durham BS, Grigg R, and Wood IC, Inhibition of histone deacetylase 1 or 2 reduces induced cytokine expression in microglia through a protein synthesis independent mechanism. J Neurochem, 2017. 143(2): p. 214–224. [DOI] [PubMed] [Google Scholar]
- [41].Wu C, et al. , Histone deacetylase 2 is essential for LPS-induced inflammatory responses in macrophages. Immunol Cell Biol, 2019. 97(1): p. 72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chen X, et al. , Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc Natl Acad Sci U S A, 2012. 109(42): p. E2865–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bradner JE, et al. , Chemical phylogenetics of histone deacetylases. Nat Chem Biol, 2010. 6(3): p. 238–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Almutairi F, Lee JK, and Rada B, Regulator of G protein signaling 10: Structure, expression and functions in cellular physiology and diseases. Cell Signal, 2020. 75: p. 109765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Vural A, et al. , Galphai2 Signaling Regulates Inflammasome Priming and Cytokine Production by Biasing Macrophage Phenotype Determination. J Immunol, 2019. 202(5): p. 1510–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lee JK, et al. , RGS10 exerts a neuroprotective role through the PKA/c-AMP response-element (CREB) pathway in dopaminergic neuron-like cells. J Neurochem, 2012. 122(2): p. 333–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Yang S, et al. , Specificity of RGS10A as a key component in the RANKL signaling mechanism for osteoclast differentiation. J Cell Sci, 2007. 120(Pt 19): p. 3362–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yang S and Li YP, RGS10-null mutation impairs osteoclast differentiation resulting from the loss of [Ca2+]i oscillation regulation. Genes Dev, 2007. 21(14): p. 1803–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yang S, et al. , Inhibition of Rgs10 Expression Prevents Immune Cell Infiltration in Bacteria-induced Inflammatory Lesions and Osteoclast-mediated Bone Destruction. Bone Res, 2013. 1(3): p. 267–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Garcia-Bernal D, et al. , RGS10 restricts upregulation by chemokines of T cell adhesion mediated by alpha4beta1 and alphaLbeta2 integrins. J Immunol, 2011. 187(3): p. 1264–72. [DOI] [PubMed] [Google Scholar]
- [51].Hooks SB, et al. , Regulators of G-Protein signaling RGS10 and RGS17 regulate chemoresistance in ovarian cancer cells. Mol Cancer, 2010. 9: p. 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Altman MK, et al. , Suppression of the GTPase-activating protein RGS10 increases Rheb-GTP and mTOR signaling in ovarian cancer cells. Cancer Lett, 2015. 369(1): p. 175–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ma P, et al. , RGS10 shapes the hemostatic response to injury through its differential effects on intracellular signaling by platelet agonists. Blood Adv, 2018. 2(16): p. 2145–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Hensch NR, et al. , RGS10 Negatively Regulates Platelet Activation and Thrombogenesis. PLoS One, 2016. 11(11): p. e0165984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].DeHelian DJ, et al. , RGS10 and RGS18 differentially limit platelet activation, promote platelet production, and prolong platelet survival. Blood, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Miao R, et al. , Regulator of G-Protein Signaling 10 Negatively Regulates Cardiac Remodeling by Blocking Mitogen-Activated Protein Kinase-Extracellular Signal-Regulated Protein Kinase 1/2 Signaling. Hypertension, 2016. 67(1): p. 86–98. [DOI] [PubMed] [Google Scholar]
- [57].Bender K, et al. , A role for RGS10 in beta-adrenergic modulation of G-protein-activated K+ (GIRK) channel current in rat atrial myocytes. J Physiol, 2008. 586(8): p. 2049–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ren J, et al. , Inhibition of regulator of G protein signaling 10, aggravates rheumatoid arthritis progression by promoting NF-kappaB signaling pathway. Mol Immunol, 2021. 134: p. 236–246. [DOI] [PubMed] [Google Scholar]
- [59].Chatterjee TK and Fisher RA, Cytoplasmic, nuclear, and golgi localization of RGS proteins. Evidence for N-terminal and RGS domain sequences as intracellular targeting motifs. J Biol Chem, 2000. 275(31): p. 24013–21. [DOI] [PubMed] [Google Scholar]
- [60].Haller C, et al. , Structure, chromosomal localization and expression of the mouse regulator of G-protein signaling10 gene (mRGS10). Gene, 2002. 297(1–2): p. 39–49. [DOI] [PubMed] [Google Scholar]
- [61].Burgon PG, et al. , Phosphorylation and nuclear translocation of a regulator of G protein signaling (RGS10). J Biol Chem, 2001. 276(35): p. 32828–34. [DOI] [PubMed] [Google Scholar]
- [62].Zhong H, et al. , The transcriptional activity of NF-kappaB is regulated by the IkappaB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell, 1997. 89(3): p. 413–24. [DOI] [PubMed] [Google Scholar]
- [63].Zhong H, Voll RE, and Ghosh S, Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell, 1998. 1(5): p. 661–71. [DOI] [PubMed] [Google Scholar]
- [64].Venkatesh S and Workman JL, Histone exchange, chromatin structure and the regulation of transcription. Nat Rev Mol Cell Biol, 2015. 16(3): p. 178–89. [DOI] [PubMed] [Google Scholar]
- [65].Smith ZD and Meissner A, DNA methylation: roles in mammalian development. Nat Rev Genet, 2013. 14(3): p. 204–20. [DOI] [PubMed] [Google Scholar]
- [66].Zhu H, et al. , Histone deacetylase-3 activation promotes tumor necrosis factor-alpha (TNF-alpha) expression in cardiomyocytes during lipopolysaccharide stimulation. J Biol Chem, 2010. 285(13): p. 9429–9436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Delcuve GP, Khan DH, and Davie JR, Roles of histone deacetylases in epigenetic regulation: emerging paradigms from studies with inhibitors. Clin Epigenetics, 2012. 4(1): p. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Khan DH, et al. , Protein kinase CK2 regulates the dimerization of histone deacetylase 1 (HDAC1) and HDAC2 during mitosis. J Biol Chem, 2013. 288(23): p. 16518–16528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Thomas EA, Involvement of HDAC1 and HDAC3 in the Pathology of Polyglutamine Disorders: Therapeutic Implications for Selective HDAC1/HDAC3 Inhibitors. Pharmaceuticals (Basel), 2014. 7(6): p. 634–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Chen S, et al. , Epigenetic regulation of macrophages: from homeostasis maintenance to host defense. Cell Mol Immunol, 2020. 17(1): p. 36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Datta M, et al. , Histone Deacetylases 1 and 2 Regulate Microglia Function during Development, Homeostasis, and Neurodegeneration in a Context-Dependent Manner. Immunity, 2018. 48(3): p. 514–529 e6. [DOI] [PubMed] [Google Scholar]
- [72].Lee JK, et al. , RGS10 deficiency ameliorates the severity of disease in experimental autoimmune encephalomyelitis. J Neuroinflammation, 2016. 13: p. 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Ali MW, et al. , Transcriptional suppression, DNA methylation, and histone deacetylation of the regulator of G-protein signaling 10 (RGS10) gene in ovarian cancer cells. PLoS One, 2013. 8(3): p. e60185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Hooks SB and Murph MM, Cellular deficiency in the RGS10 protein facilitates chemoresistant ovarian cancer. Future Med Chem, 2015. 7(12): p. 1483–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Chitnis T and Weiner HL, CNS inflammation and neurodegeneration. J Clin Invest, 2017. 127(10): p. 3577–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Gordon JW, Shaw JA, and Kirshenbaum LA, Multiple facets of NF-kappaB in the heart: to be or not to NF-kappaB. Circ Res, 2011. 108(9): p. 1122–32. [DOI] [PubMed] [Google Scholar]
- [77].Gaspar-Pereira S, et al. , The NF-kappaB subunit c-Rel stimulates cardiac hypertrophy and fibrosis. Am J Pathol, 2012. 180(3): p. 929–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Harrington BS and Annunziata CM, NF-kappaB Signaling in Ovarian Cancer. Cancers (Basel), 2019. 11(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Validation of the RGS10 antibody.
MH-S cells were treated with vehicle (serum-free media) or LPS (10 ng/ml) for 24 and 48 hours. Cells were lysed and subjected to SDS-PAGE followed by immunoblotting using specific antibodies against RGS10 and β-actin. The pictures represent the full-length gels with the indicated molecular weight markers (BioRad, cat#: 1610374) for Figure 1B. The picture was taken using a BioRad Molecular Imager ChemiDoc™ XRS+ System with Image Lab™ Software.
Fig. S2. Blocking MAPK signaling pathways does not affect LPS-induced RGS10 suppression.
(A-C) MH-S cells were plated in six-well plate and allowed to adhere overnight before treatment with vehicle (serum-free media) or LPS (10 ng/ml) for 48 hours with or without a one hour-pretreatment with 10 μM of ((A) PD98059 (PD), (B) SB23906 (SB), and (C) SP600125 (SP)). Cells were lysed and subjected to SDS-PAGE followed by immunoblotting using specific antibodies against RGS10 and β-actin. One blot of two similar independent experiments is shown in each panel.
Fig. S3. The effect of 5-Aza, DNMTs inhibitor, on LPS-stimulated silencing of RGS10.
MH-S cells were plated in six-well plate and allowed to adhere overnight before treatment with vehicle (serum-free media) or LPS (10 ng/ml) for 48 hours with or without a one hour-pretreatment with 10 μM 5-Aza-2’-deoxyytidine (5-Aza). Cells were lysed and subjected to SDS-PAGE followed by immunoblotting using specific antibodies against RGS10 and β-actin. One blot of two similar independent experiments is shown.