

SUMMARY
Single-cell technologies are emerging as powerful tools for cancer research. These technologies characterize the molecular state of each cell within a tumor, enabling new exploration of tumor heterogeneity, microenvironment cell type composition, and cell state transitions that impact therapeutic response — particularly in the context of immunotherapy. Analyzing clinical samples has great promise for precision medicine but is technically challenging. Successfully identifying predictors of response requires well-coordinated, multi-disciplinary teams to ensure adequate sample processing for high-quality data generation, and computational analysis for data interpretation. Here, we review current approaches to sample processing and computational analysis regarding their application to translational cancer immunotherapy research.
Introduction
Single-cell analysis has become a widespread tool used in cancer research to characterize the cellular and molecular composition of tumors (Marx, 2021; Sandberg, 2014; Zhu et al., 2020). Technologies to profile single-cells are currently able to measure tumor heterogeneity across molecular levels, including DNA (Navin and Hicks, 2011), RNA (Tang et al., 2009), protein (Bandura et al., 2009), and epigenetics (Buenrostro et al., 2015). Whereas bulk technologies are limited to an averaged signal often representing the molecular states of the most abundant cell populations, single-cell approaches resolve the cellular composition of the tumor microenvironment (TME). This characterization holds particular promise for the field of tumor immunology, as comprehensive profiling can determine the cell types and pathways involved in anti-tumor responses and immune evasion. In addition, recent spatial transcriptomics and proteomics approaches preserve tissue architecture, enabling the analysis of cell-to-cell interactions and cellular neighborhoods reflective of the interactions in immune responses (Schürch et al., 2020). Samples derived from immunotherapy clinical trials can benefit from using single-cell-based technologies to capture the nuances of therapeutic immune cell responses in cancer. The development of immune checkpoint inhibitors (ICIs) enhanced cancer therapy by providing clinical benefits to a portion of previously incurable cancers; however, most patients do not respond to ICIs (Ribas and Wolchok, 2018). Understanding the complex immune cell composition and molecular pathways associated with cell state transitions during these therapies can potentially identify mechanistic predictors of response and elucidate new druggable targets to overcome immunotherapy resistance (Giladi and Amit, 2018).
Current single-cell technologies span a wide array of rapidly advancing methodologies, with the most common examples for tumor immunotherapy including single-cell RNA-sequencing (scRNA-seq) for transcriptional profiling (Tang et al., 2009), mass cytometry (CyTOF) for proteomics profiling (Bandura et al., 2009), and spatial molecular profiling (Giesen et al., 2014; Marx, 2021; Ståhl et al., 2016) (Figure 1). Each of these technologies provides a high-dimensional molecular profile for individual cells, which can be computationally sorted into distinct cell populations. These technologies profile more than the canonical cell type markers that are commonly measured in multi-parameter flow cytometry experiments, for example. The high-dimensional nature of these approaches can enable more refined annotation of cell types, inference of cellular state transitions, and association of molecular pathways. These characterizations require complementary computational techniques to determine the pathways that drive the behavior of each distinct cell type and infer the intra- and inter-cellular interactions associated with transitions in cell states. The inference of these pathways mirrors the current clinical research in tumor immunology, where precision medicine strategies are being developed to use combination therapeutics to rewire the tumor microenvironment to enable immunotherapy sensitization (Gohil et al., 2021).
Figure 1 -.
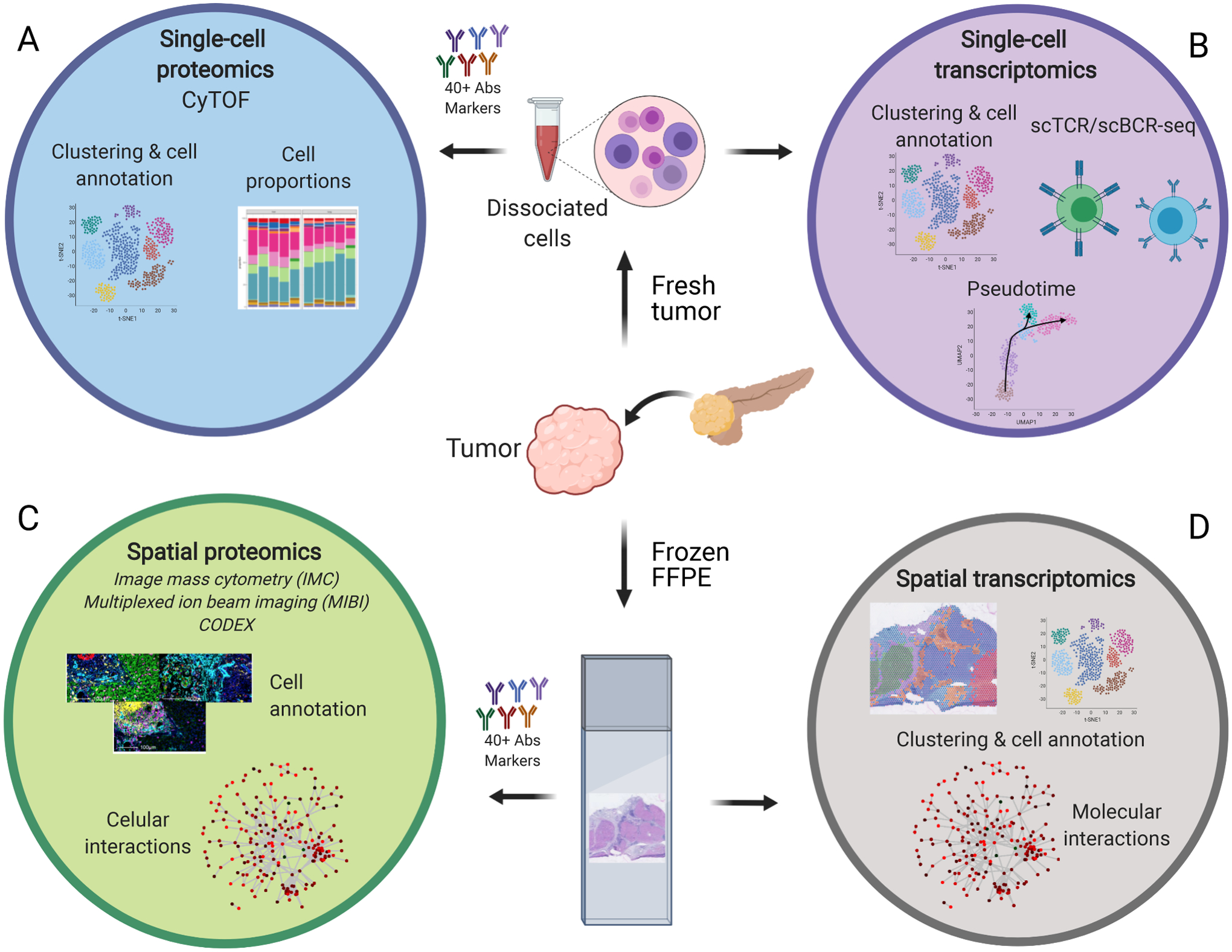
High dimensional transcriptomics and proteomics approaches for cancer profiling. Several high dimensional approaches are currently available to understand cancers cellular composition and inter-cellular interactions. A. Single-cell proteomics (CyTOF) provides cell composition and cell state information. B. Single-cell transcriptomics allows the same type of analysis, but its genome-wide coverage can also deliver cell trajectory predictions and T and B cell repertoires. In order to correlate cell composition and states to cellular interactions, spatial technologies are more informative than single-cell suspension analysis. C. With spatial proteomics and its single-cell resolution, it is possible to identify individual cell types and determine specific cell-to-cell interactions. D. Although it lacks single-cell resolution, spatial transcriptomics can predict cell interactions based on the molecular expression of receptors and ligands between different cell neighbors and discover driving oncogenic pathways among the different cell niches because it is not restricted to previously selected markers. The selection of which approach to apply will depend on what samples are available, how they are preserved, and what biological questions need answered.
While single-cell approaches hold promise for precision immunotherapy, the selection of profiling technology and computational analysis methods influence the features that can be characterized from tumor samples. In the case of clinical samples, single-cell study designs must balance cost, requirements for sample preservation, labor-intensity of the protocols, and information content. This review describes technology and analysis approaches for single-cell analysis in clinical cancer research, with a focus on tumor immunology. Specifically, we describe the steps involved in single-cell analysis from sample collection to computational analysis, including recent spatial transcriptomics and proteomics approaches. We cover the capabilities of different approaches and discuss additional factors that can bias the biological interpretation of single-cell data. Overall, this review aims to highlight the experimental and computational best practices, using benchmarked technologies and computational tools, in order to ensure that clinical data captures biologically relevant and reproducible findings.
Single-cell and spatial technologies for immune profiling
Single-cell and spatial approaches can be used to examine tumors in great detail, characterizing cell type composition and tumor heterogeneity by gene or protein expression (Berglund et al., 2018; Patel et al., 2014). These approaches have already been implemented to profile the tumor microenvironment of multiple cancer types, including leukemia, melanoma, breast, lung, and gastrointestinal, among others.
Here, we summarize benchmarked technologies currently employed for high-dimensional characterization of tumors in the context of immunotherapy research (Table 1).
Table 1 -.
High dimensional gene and protein expression technologies and application in human cancer studies.
| Technology | Reference | Cancer Type | Tissue | Cell types profiled | Therapy | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Tumor | Adj. normal | Lymph node | Blood | Immune | Tumor | Stromal | |||||
| Single-cell proteomics | CyTOF | Gadalla et al. 2019 | Ovarian, melanoma, breast | - | |||||||
| Subrahmanyam et al. 2018 | Melanoma | Anti-CTLA4, anti-PD-1 | |||||||||
| Krieg et al. 2018 | Melanoma | Anti-PD-1 | |||||||||
| Wu et al. 2020 | Pancreatic | GVAX, anti-CTLA4 | |||||||||
| Single-cell transcriptomics | scRNAseq | Nagaoka et al. 2020 | Gastric | Anti-PD-1, anti-IL-17 | |||||||
| Kieffer et al. 2020 | Breast | - | |||||||||
| Schelker at al. 2017 | Ovarian | - | |||||||||
| Peng et al. 2019 | Pancreatic | - | |||||||||
| Patel et al. 2014 | Glioblastoma | - | |||||||||
| Tirosh et al. 2016 | Melanoma | - | |||||||||
| Ma et al. 2019 | Hepatocellular, Cholangiocarcinoma | Anti-CTLA4, anti-PD-1 | |||||||||
| Bernard et al. 2019 | Pancreatic | - | |||||||||
| Schlesinger et al. 2020 | Pancreatic | - | |||||||||
| Sun et al. 2021 | Hepatocellular | - | |||||||||
| Davidson et al. 2020 | Melanoma | - | |||||||||
| Savas et al. 2018 | Breast | - | |||||||||
| Zheng et al. 2017 | Hepatocellular | - | |||||||||
| Guo et al. 2018 | NSCLC | - | |||||||||
| Sade-Feldman et al. 2018 | Melanoma | Anti-CTLA4, anti-PD-1 | |||||||||
| Spatial proteomics | MIBI | Angelo et al. 2014 | Breast | - | |||||||
| Keren et al. 2019 | Breast | - | |||||||||
| IMC | Giesen et al. 2014 | Breast | - | ||||||||
| Jackson et al. 2020 | Breast | - | |||||||||
| Xiang et al. 2020 | NSCLC | - | |||||||||
| Ho et al. 2020 | Hepatocellular carcinoma | Cabozantinib, anti-PD-1 | |||||||||
| CODEX | Schürch et al. 2020 | Colorectal | - | ||||||||
| MxIF | Yan et al. 2019 | Melanoma | - | ||||||||
| Spatial transcriptomics | Spatial transcriptomics | Moncada et al. 2020 | Pancreatic | - | |||||||
| Multi-omics | CITE-seq, scRNAseq | Cadot et al. 2020 | Leukemia (CLL) | Ibrutinib | |||||||
| CyTOF, scRNAseq | Gubin et al. 2018 * | Sarcoma | anti-CTLA4, anti-PD-1 | ||||||||
| scRNAseq, TCR, CyCIF | Yost et al. 2019 | Basal cell | Anti-PD-1 | ||||||||
| Azizi et al. 2018 | Breast cancer | - | |||||||||
| Jerby-Arnon et al. 2018 | Melanoma | Anti-PD-1 | |||||||||
| Wu et al. 2020 | NSCLC, endometrial, colorectal, renal | - | |||||||||
| scRNAseq, spatial transcriptomics, MIBI | Ji et al. 2020 | Squamous cell | - | ||||||||
| scRNAseq, IMC | Aoki et al. 2020 | Hodgkin lymphoma | - | ||||||||
Single-cell proteomics
Fluorescent-based flow cytometry is currently the gold-standard method for cell type identification. It remains the most commonly used single-cell method for cell type annotation and sorting in immunology (Gadalla et al., 2019). Although this is a reproducible approach, fluorescent flow cytometry is limited by the number of features that can be simultaneously analyzed (up to 30 markers) due to the inherent limitations related to channel spillover and equipment throughput. Thus, a high-parameter study often requires complex compensation strategies or splitting panels into subpanels with redundancy of key markers to obtain high-dimensional single-cell proteomic characterization. Sampling strategies designed to increase the dimensionality of fluorescence-based characterization ultimately require larger numbers of cells, limiting application for patient biopsies, which have a limited number of cells (Gadalla et al., 2019).
As an alternative to fluorescent-based flow cytometry, CyTOF detects metal intensities from antibodies conjugated with isotopically enriched heavy-metal reporter ions. This design enables CyTOF to profile up to 50 markers simultaneously (Bendall et al., 2011). Based on the mass range of the reporter ions used when conjugating the antibody panel, CyTOF methods can theoretically be developed to detect >100 markers in the same cell to enable high-dimensional molecular profiling. Another advantage of the reliance on heavy-metal conjugated antibodies over fluorescence-based technologies is the fact that they are rarely present in biological samples, eliminating analytical challenges resulting from false signals from intrinsic cellular background (Bandura et al., 2009; Bendall et al., 2011). As antibody-based technologies, both CyTOF and fluorescence-based cytometry can evaluate protein isoforms (e.g., CD45RO) and post-translational modifications (e.g., phosphorylation) (Bendall et al., 2011). CyTOF profiling relies on antibody panels that are ultimately limited by the number of isotopically enriched metals that can be reliably conjugated and is highly dependent on antibody quality. This reliance on pre-selected antibody panels restricts analysis to anticipated cell types, which limits the discovery of new cell types and molecular changes due to immunotherapy treatment (Hartmann and Bendall, 2020). Still, the metal reporters in CyTOF are robust to freezing and thawing, and to a variety of fixation protocols, making this technology versatile in application and storage needs when compared to other single-cell methods (Leipold et al., 2018; Sumatoh et al., 2017).
The multi-parameter profiling of CyTOF makes it a powerful technique to understand variations in immune cell composition before and after immunotherapy (Figure 1A). This technology has been used to model changes in the distribution of cell type abundances in preclinical models (Gubin et al., 2018) and peripheral blood mononuclear cells (PBMCs) of immunotherapy treated tumors (Krieg et al., 2018; Subrahmanyam et al., 2018; Wu et al., 2020a). Notably, application of a panel of 40 markers for analysis of PBMCs from melanoma patients before anti-CTLA-4 or anti-PD-1 therapies identified that PBMCs from anti-CTLA-4 responders were enriched for naive and effector T cells when compared to non-responders. Among anti-PD-1 responders, central memory and effector memory T cells were more frequent, suggesting that different cell type compositions are potential predictors of response to distinct immunotherapies (Subrahmanyam et al., 2018).
Single-cell transcriptomics
Single-cell sequencing approaches perform genome-wide profiling of individual cells. As a result, they are not limited by pre-determined markers and can be applied to globally characterize transcriptional profiles (scRNA-seq) (Tang et al., 2009), mutational burden (single-cell DNA-sequencing) (Navin et al., 2011), and chromatin states (single-cell ATAC-sequencing) (Buenrostro et al., 2015). Of these technologies, gene expression profiling with scRNA-seq is the most commonly used to identify cell types in the tumor microenvironment. In contrast to previous studies with bulk RNA-seq data, scRNA-seq profiling does not require experimental protocols to sort cells before sequencing (Avila Cobos et al., 2020). The comprehensive, whole-transcriptome profiling of cell types with scRNA-seq allows for inference of cell state transitions, differential gene expression, and functional oncogenic and immunologic pathway analysis (Lim et al., 2020; Trapnell, 2015) (Figure 1B). Such analyses can be performed from scRNA-seq data directly using computational approaches.
Different technologies have been developed for scRNA-seq and the choice of which platform to apply depends on the biological questions that need to be addressed. SMART-seq allows for single-cell analysis of full-transcripts of hundreds of cells that are FACS sorted into microtiter plates for library preparations (Ramsköld et al., 2012). Massively Parallel RNA Single-Cell Sequencing (MARS-seq) also requires cell sorting and sequencing is restricted to the 3’ end of the transcript. MARS-seq introduced transcript tagging with cell-specific barcodes and a unique molecular identifier (UMI) that allows sequencing counts to be assigned to the respective gene (Islam et al., 2014). Fluidigm C1 became an attractive option as its microfluidic platform automated cell capture and increased the number of cells profiled from a few hundred cells to nearly a thousand cells (Xin et al., 2016). This microfluidic platform allows full transcript sequencing or 3’ sequencing. The development of droplet-based methods such as inDrop (Klein et al., 2015), Drop-seq (Macosko et al., 2015) and the widely used 10X Genomics platform (Zheng et al., 2017b) increased the scalability of single-cell profiling. Droplet-based approaches allow thousands of single-cells to be sequenced from an individual sample. In these methods, cells are captured and encapsulated in gel emulsion beads, inside which barcoding and UMI tagging occur. A limitation of these platforms is that sequencing will only capture the 3’ or 5’ ends of the transcripts. Still, the barcoding strategies of UMI-based approaches enable the adaptation to single-cell multi-omics profiling across numerous molecular scales (Lee et al., 2020). Concurrent profiling of protein and RNA with CITE-seq (Stoeckius et al., 2017) as well as T and B cell receptor (TCR/BCR) sequencing and RNA (Goldstein et al., 2019; Tu et al., 2019) are particularly applicable to tumor immunology.
All scRNA-seq technologies can be used to characterize the cellular composition of tumors. Both the study cohort and underlying biological question should determine which platform to select for analysis. Methods covering full-transcripts (SMART-seq and Fluidigm C1) are ideal for identifying rare gene variants and splicing isoforms at a trade-off of profiling a relatively small number of cells. Therefore, these full-transcript technologies are ideally suited for high-resolution characterization of rare cell populations. UMI-based methods have higher cellular resolution, but lower molecular resolution and are subject to signal dropout that can result in failed detection of genes. Still, the high-dimensional cellular profiling makes these UMI-based technologies more suitable than full-transcript counterparts to annotate the diverse cell types in the tumor microenvironment and measure gene expression changes between treatment conditions (See et al., 2018).
Numerous scRNA-seq studies have examined tumor heterogeneity and identified new cell types or functional subtypes that are a result of tumor progression (Azizi et al., 2018; Bernard et al., 2019; Davidson et al., 2020; Guo et al., 2018; Li et al., 2017; Ma et al., 2019; Patel et al., 2014; Peng et al., 2019; Puram et al., 2017; Tirosh et al., 2016; Zhao et al., 2020). Since tumor heterogeneity is crucial to understand tumor evolution and anti-tumor immune responses, scRNA-seq has been extensively applied to tumor-infiltrating leukocytes (TILs) in order to identify the immunosuppressive and effector cell types that populate different tumors and to associate cell types with specific transcriptional signatures to understand immune modulation (Azizi et al., 2018; Guo et al., 2018; Peng et al., 2019; Savas et al., 2018; Tirosh et al., 2016; Yost et al., 2019; Zheng et al., 2017a). ScRNA-seq analysis has also been used to uncover the mechanisms driving resistance to immunotherapy (Gubin et al., 2018; Jerby-Arnon et al., 2018; Sade-Feldman et al., 2018). For example, Gubin et al. performed both CyTOF and scRNA-seq profiling of tumors from a preclinical sarcoma model to assess the cellular composition and functional changes induced by different ICIs (anti-PD-1, anti-CTLA4, and the combination) (Gubin et al., 2018). The combined use of scRNA-seq and CyTOF allowed cross-validation of cell type abundances associated with ICI response in different data modalities. The measurement of additional molecular parameters through scRNA-seq enabled de novo discovery of cell state transitions conserved between mouse and human tumors in data reanalysis by Davis-Marcisak et al., which included a subset of activated NK cells associated with anti-CTLA4 response (Davis-Marcisak et al., 2020).
Spatial analysis platforms
Single-cell approaches like CyTOF and scRNA-seq that are widely applied to characterize tumors rely on the profiling of dissociated tumor specimens, which results in the loss of the spatial organization of cells within a sample. New technologies that maintain the spatial organization of cells are essential to infer cell-to-cell interactions within the TME. Thus, in recent years spatial proteomics and transcriptomics analysis are emerging as powerful tools to characterize the spatial distribution of cell types within a tumor. These technologies allow for direct measurement of spatial co-localization of cells, which is often associated with inter-cellular interactions. The emerging high-molecular coverage of these technologies enables further inference of the cellular and molecular pathways as well as cell-state transitions associated with interactions between cells (Ji et al., 2020).
Chromogenic immunohistochemistry (IHC) has been the gold-standard approach for clinical spatial proteomics profiling (Ramos-Vara and Miller, 2014). However, it has limited multiplexing capacity (4 markers), which represents a challenge for research into the comprehensive cellular composition of the TME. The development of fluorescent IHC increased the number of proteins that could be interrogated at the same time (8 markers), but similar to fluorescent-based flow cytometry the overlap between wavelengths limits the isolation of large numbers of proteins (Gorris et al., 2018; Viratham Pulsawatdi et al., 2020). Sequential IHC techniques were developed to profile up to 12 proteins simultaneously and then can be stripped to allow for restaining, which increases the molecular resolution (Tsujikawa et al., 2017), but the number of markers is still limited by the quality of the tissue after multiple cycles of antibody stripping which ultimately limits the resolution of these technologies. Recent advances have led to the development of protein multiplex technologies that allow the mapping of roughly 50 markers in the same section. Image mass cytometry (IMC) (Giesen et al., 2014), multiplexed ion beam imaging (MIBI) (Angelo et al., 2014), and cyclic imaging detection (CODEX, CyCIF, and MxIF) (Gerdes et al., 2013; Goltsev et al., 2018; Lin et al., 2015) are approaches that can measure protein levels of up to 50 markers at the same time and provide the spatial distribution of the signal as well as information on which cells are in contact with each other (cell neighbors) (Figure 1C).
High-dimensional spatial proteomics technologies have been applied to characterize cellular interactions in melanoma (Yan et al., 2019), breast (Jackson et al., 2020; Keren et al., 2018), colorectal (Schürch et al., 2020), cutaneous squamous cell carcinoma (Ji et al., 2020), Hodgkin lymphoma (Aoki et al., 2020), liver (Ho et al., 2020), and lung tumors (Xiang et al., 2020). In the context of immunotherapy treatment, Ho et al. leveraged IMC to identify cellular neighborhoods containing B cells, helper T cells, and CD68+CD163− myeloid cells suggestive of an immune response in an immunotherapy responsive liver tumor (Ho et al., 2020). In the case of immunotherapy treated melanoma, Jerby-Arnon et al. (Jerby-Arnon et al., 2018) used t-CyCIF to demonstrate that tumor cells can express markers that decrease T cell infiltration, creating immune cold cell neighborhoods that are detectable prior to immunotherapy initiation.
Similar to the comparison between CyTOF and scRNA-seq, spatial transcriptional (ST) profiling provides higher molecular resolution than spatial proteomics technologies and are reviewed in detail by Maniatis et al. 2021 (Maniatis et al., 2021). Approaches such as Slide-seq and the 10X Genomics Visium platform enable whole-transcriptome characterization within spots on a slide that provide near single-cell resolution in fresh frozen samples (Figure 1D). These technologies use specially designed slides spotted with DNA-barcoded beads (Slide-seq) (Rodriques et al., 2019) or oligo-dT/UMI tags (10X Genomics) (Islam et al., 2014) that will capture the tissue RNA on the slide. The barcoded spots are around 50 to 100um in size, allowing 2 to 10 cells to be captured in each spot, and the sequencing counts will refer to the population of cells mapped to the slide spots. Computational deconvolution methods to estimate the molecular profile of single-cells from each spot are currently an active area of research. Even though the technology lacks single-cell resolution, it is still possible to identify cellular neighborhoods and the cell types frequently interacting within such niches directly from the expression profiles of the spots (Moncada et al., 2020) (Figure 1D).
The development of high-dimensional RNA in-situ hybridization technologies led to single-cell resolution ST analysis with near genome-wide capabilities. Although these in-situ approaches do not involve transcript sequencing, their ability to detect thousands of transcripts in tissues allows their classification as ST platforms (Marx, 2021). Lubeck et al. (Lubeck et al., 2014) developed SeqFISH that uses sequential hybridization and fluorescent signal detection for single-cell in situ RNA measurement of a few hundred pre-selected genes. The improved SeqFISH+ (Eng et al., 2019) allows for profiling up to 10,000 genes nearing the resolution of the whole transcriptome but still requires prior selection of the genes. Another in situ technique that provides accurate spatial single-cell resolution with genome-wide coverage is MERFISH (Xia et al., 2019). MERFISH requires multiple steps of hybridization and imaging, resulting in extensive experimental labor depending on the number of genes to be profiled (Xia et al., 2019). Emerging multi-omics technologies, such as DBit-seq allow for concurrent proteomics and transcriptomics spatial molecular profiling, merging the strengths of both spatial transcriptomics and spatial proteomics for cellular characterization (Liu et al., 2020).
Tumor sample processing for single-cell profiling in clinical research
Although single-cell profiling has spread rapidly in tumor immunology, the intensive sample processing required limits application to clinical specimens. Notably, the majority of non-spatial single-cell technologies, such as scRNA-seq and CyTOF, require viably dissociated cells for profiling (Lafzi et al., 2018) (Figure 1A and B). The most commonly used methods for sample dissociation apply enzymatic-based digestion and heated incubation. The sample storage prior to dissociation, type of enzyme, and time of incubation all impact the single-cell profiling and must be optimized carefully for each tumor type (Lafzi et al., 2018). Digested samples must consist of single-cells upon microscopy examination and accurate characterization of the molecular states can only be achieved for live cells, with viability greater than 70%. Nonetheless, dead cells can be filtered as part of preprocessing after analysis, allowing for lower cellular viability in the case of assays with high-throughput cellular characterization such as CyTOF. The requirement of viable cells for dissociation poses a further barrier for the analysis of samples that are most typically preserved non-viably, such as biopsies. In a clinical environment, maintaining cell viability requires rapid sample acquisition from the surgical or clinical team, pathological assessment, transportation to the lab, tumor dissociation, sample resuspension, and sequencing library preparation. Thus, a highly coordinated routine is required to obtain, process, and preserve samples rapidly enough to maintain cellular viability — a challenging process for staff limited groups and for multi-site clinical trials. An additional challenge posed by the need for this immediate profiling is that all single-cell technologies are subject to technical artifacts that arise from processing samples at different times, in distinct profiling batches, or by different technicians. In clinical research, biospecimens necessarily arise at the time of treatment making it impossible to control for technical artifacts in experimental design in cohort studies or time-course profiling during treatment. These batch effects can be overcome by optimizing preservation protocols so that samples can be processed simultaneously or including a control sample in each batch that can be used to correct for technical artifacts computationally. Alternative strategies such as flash freezing for nuclei isolation and single-nuclei RNA sequencing (snRNA-seq) have been shown to compare to scRNA-seq and are emerging as alternatives for single-cell analysis of cryopreserved samples that can overcome some of these limitations (Denisenko et al., 2020; Slyper et al., 2020).
Spatial molecular profiling relies on slide-based technologies that retain the cellular architecture, without requiring tumor dissociation. Requirements for sample preservation and preparation in spatial proteomic assays depend on the technology. Spatial proteomics can be performed for both frozen and formalin-fixed paraffin-embedded (FFPE) samples (Figure 1C). Most current spatial transcriptomics approaches rely on frozen samples, with approaches to use FFPE samples under development (Gracia Villacampa et al., 2020) (Figure 1D). The ability to profile FFPE preserved samples enables clinical research on samples processed for long term storage.
Computational pipelines for single-cell and spatial analysis
The high-dimensional nature of single-cell data makes computational pipelines a critical component for obtaining cellular and molecular interpretation. Analysis methods are advancing in step with new technologies, providing a wide range of pipelines to choose from. These diverse analysis methods fall under several main classifications that together enable biological interpretation (Figure 2). First, the single-cell data from all platforms must be preprocessed from raw outputs into estimates of the molecular expression for each cell while removing poor quality cells. Subsequently, the data are clustered and visualized with marker genes for annotation of cell types in distinct clusters. Next, differential expression analysis can estimate changes in molecular markers among and within cell types between treatment groups. For single-cell transcriptomics, the increased number of molecular markers allows for in-depth analysis of cell state transitions and intra-cellular gene regulatory networks. Single-cell network inference algorithms also include inter-cellular interactions, relying on indirect inference based on ligand-receptor pairs (Browaeys et al., 2020; Cherry et al., 2020; Efremova et al., 2020). Finally, spatial molecular analysis algorithms utilize additional spatial statistics and neighborhood analysis for cellular co-localization that can provide more direct evidence of inter-cellular interactions (Dries et al., 2019; Luecken and Theis, 2019; Van Gassen et al., 2015).
Figure 2 -.
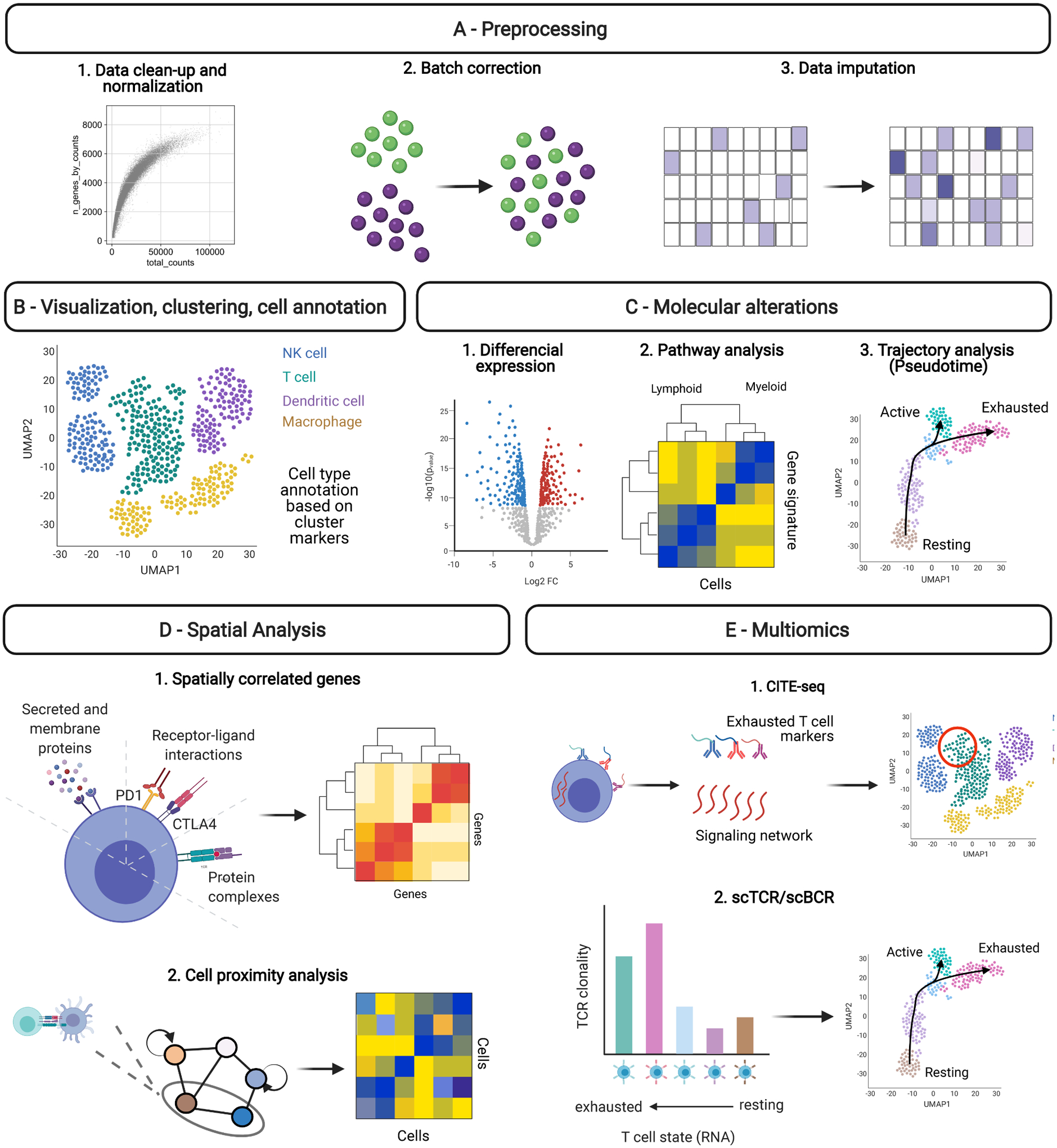
Computational workflow and methods for single-cell and spatial analysis. Several open source benchmarked computational tools are available for high dimensional datasets analysis. Independent of the tools of choice, analytical steps are required in order to obtain reproducible results and identify markers to predict response and targets for new therapeutics. A. Single-cell and spatial data analysis will start with raw data preprocessing for (1) data clean-up to remove poor quality cells and normalization to correct for low or high numbers of reads associated with experimental artifacts; (2) batch correction to remove unwanted variation among samples due to experimental discrepancies; (3) and data imputation to correct for the real data dropouts (zeros in the data). B. Subsequently, dimensionality reduction will allow data visualization and cell type annotation using clusterization tools that assign annotations based on specific markers expressed by each cluster. From there, the data is ready for downstream analysis depending on the methodology applied and biological questions. C. Molecular alterations can be identified using (1) differential expression analysis. In the case of transcriptomics data, it is also possible (2) to perform pathway analysis to identify drivers of cancer progression and responses to therapies and (3) to predict cell fate trajectories to understand tumor and TME modulation across time. D. From proteomics and transcriptomics data, it is possible to take a snapshot of the (1) molecular (e.g.: protein markers expression, cytokine genes expression, receptor-ligand expression), and (2) cellular interactions (e.g.: cell proximity analysis) that potentially drive the different features associated with cancer progression and response to therapies. E. Finally, multi-omics approaches allowing (1) protein and gene expression analysis from the same samples (CITE-seq) or (2) T and B cell repertoire analysis in combination with transcriptional profile add an additional layer of information that increases accuracy for cell types annotation and investigation of their role in cancer evolution and therapeutic responses.
While commercial software for single-cell analysis exists, the majority of analysis approaches are implemented in free, open-source software. To ensure broad adoption, this software is often built upon bioinformatics ecosystems such as the R/Bioconductor project that provide community standards and peer-review (Amezquita et al., 2020) or community curated pipelines in R (ie., Seurat, Monocle, and Giotto) (Butler et al., 2018; Cao et al., 2019; Dries et al., 2019; Hao et al., 2020; McInnes et al., 2018; Qiu et al., 2017; Satija et al., 2015; Stuart et al., 2019; Trapnell et al., 2014) and Python (ie., Scanpy) (Wolf et al., 2018). Implementing these analysis pipelines can require extensive compute resources and computer programming experience. To make these pipelines more generally accessible, platforms such as Galaxy (Tekman et al., 2020) and GenePattern Notebook (Reich et al., 2017) provide these methods in interactive, user-friendly interfaces for single-cell analysis with direct access to cloud computing. Further improvements in creating user-friendly databases, such as the developing CellxGene platform (Megill et al., 2021), remain an active area of development for the single-cell community.
Pre-processing and batch correction
The first step of single-cell and spatial data analysis is pre-processing the raw data output from each technology into measurements of protein or transcript abundances for each cell or spatial spot in the respective sample (Figure 2A). All downstream analyses rely on these data summaries, making preprocessing critical to the accuracy of the resulting findings. The pre-processing approaches depend on machine-specific data outputs and biases, requiring techniques that are tailored to each technology
Single-cell proteomics technologies typically output FCS files, following the standards of lower throughput flow cytometry experiments. Whereas these FCS files are the final output of CyTOF, in spatial proteomics (ie., IMC, CODEX) the data are obtained as images. Subsequently, a segmentation step is used to determine cellular boundaries prior to protein quantification and exported into the FCS file formats. The downstream analyses of FCS files require additional primary analysis steps to obtain protein abundances for each cell: bead-based normalization to standardize the intensities for each signal, de-barcoding to isolate the cells for each experiment in a single batch if multiplexed, and in some cases, compensation to account for spill-over of signal between channels (Nowicka et al., 2017). Altogether, this pipeline provides an estimate of normalized antibody intensities for each cell that can be carried forward to subsequent analysis of cell types.
Most single-cell transcriptomics technologies are sequencing-based and provide FASTQ files containing short reads. The preprocessing of FASTQ files involves alignment to the human transcriptome and quantification of reads for each transcript or UMI, depending on the technology. Some software also performs quality control while preprocessing the raw data (Gao et al., 2020). These alignment and preprocessing steps return a matrix containing barcodes specific to each cell captured and the counts for detected transcripts. Whereas single-cell proteomics relies on control beads to normalize the data, single-cell transcriptomics leverages the higher-dimensional nature of the data to derive a distribution for data normalization to correct for the overall differences in read depth for each cell (Hafemeister and Satija, 2019). In normalizing single-cell data, it is important to note that a value of zero read counts means either that a gene is not expressed in a cell or that it is randomly not detected among the sequencing short reads. Imputation methods were developed to estimate missing expression values and well suited to gene-level visualization. However, these methods can introduce false positives into the data, potentially introducing statistical biases if they are used for downstream analysis (Hou et al., 2020). Another step in pre-processing scRNA-seq is ensuring that barcodes refer to a unique cell and not to more than one cell that was captured in the same droplet (doublet) or to empty droplets (no cell). Those barcodes must be detected and filtered prior to analysis (Luecken and Theis, 2019). Likewise, dead cells quantified through quantifying the fraction of mitochondrial transcript counts relative to the total transcript counts must also be filtered for accurate analysis (Ilicic et al., 2016).
Spatial technologies based on imaging require an initial cell segmentation step to isolate the cell boundaries in which protein or RNA abundances are estimated. Segmentation tools are under rapid development for both spatial proteomics and spatial transcriptomics, building upon frameworks developed for microscopy (Caicedo et al., 2017). After segmentation, many of the same pre-processing and analysis methods for single-cell technologies can then be applied directly to estimates of molecular abundances. Sequencing-based, spot-level spatial transcriptomics technologies rely on alignment and quantification of the short reads associated with the barcode for each spot. These transcriptional profiles yield a semi-bulk estimate for all the cells captured in an individual spot. Spot-based deconvolution processes are required to estimate the transcriptional profile at single-cell resolution (Elosua-Bayes et al., 2021).
Normalization procedures are being developed to correct for discrepancies in molecular abundances or signal variation to ensure that cells obtained from a single processing batch (CyTOF) or library (scRNA-seq) in single-cell assays are comparable. Nonetheless, different experimental covariates can introduce unwanted bias, or batch effects, into the estimated molecular profiles from each data modality. Batch effects can arise due to differences in incubation periods during dissociation, handling personnel, reagent lots, or timing of sample processing. Batch effects are pervasive in high-throughput datasets and have been long recognized in previous generations of bulk technologies (Leek et al., 2010). Technical noise is amplified in the case of single-cell technologies, requiring even greater attention to study design and batch correction methods to remove these technical artifacts (Hicks et al., 2018). The choice of normalization and batch correction methods can have a more substantial impact on the experimental results than the choice of downstream method for differential expression, making it a critical step in single-cell analysis pipelines (Bullard et al., 2010). Experimental designs that ensure each batch shares cells from the same biological condition allow remaining batch effects to be corrected computationally. Several batch correction tools have been designed to remove technical artifacts in the low-dimensional embeddings used to visualize single-cell data. Others correct the data itself by either leveraging the correlation structures between genes to preserve only biological variation or explicitly incorporating the batch as a covariate in the model. Batch-aware analysis pipelines should utilize batch correction techniques to standardize the data visualization and then model batch as a covariate for downstream differential expression analyses (Tran et al., 2020).
Visualization of data through low dimensional embeddings
Single-cell and spatial data pre-processing, filtering, and normalization methods yield high dimensional matrices representing an abundance of molecular species (proteins or transcripts as rows) by cells (columns). In the case of spatial datasets, the tissue position is added as an additional layer to these complex matrices and is used for visualization purposes. The high dimensionality of the matrices limits the direct application of standard data visualization techniques that often rely on 2- or 3-dimensional plots. Typically, the number of markers measured with these technologies is higher than the number of distinct biological processes (e.g., cell types, cell state transitions, etc) captured. These features introduce correlations between the molecular species measured, which can be captured through a smaller number of features than the total number of markers in the data, enabling the use of dimensionality reduction techniques for visualization and analysis (Cleary et al., 2017; Stein-O’Brien et al., 2019; Wagner et al., 2016).
The most commonly used dimension reduction techniques for single-cell data, and also for spatial datasets, are t-distributed stochastic neighbor embedding (tSNE) and uniform manifold approximation and embedding (UMAP) (Figure 2B). Dimensional reduction techniques are the first step in analysis to enable visual inspection and data interpretation. Briefly, these methods transform high-dimensional data into a lower dimension embedding for visualization. In the resulting plot, each point represents a single-cell that is plotted using a computational method that ensures the distance between cells along the coordinate axes corresponds to the distance they would have from one another if computed for the entire molecular profile. In computing these distances, UMAP balances the global structure of more distant points, whereas the balance between preserving the distance of nearby points and more distant clusters is a tunable parameter in t-SNE (Becht et al., 2018; van der Maaten and Hinton, 2008).
Both t-SNE and UMAP are well suited to visualizing clusters for distinct cell types within the data. Other manifold learning approaches, such as PHATE, have been designed with additional constraints to ensure that the embeddings not only model clusters but also preserve continuous transitions between cell states (Moon et al., 2019). Overall, these embeddings provide a visualization tool to explore the variation and structure of the data but require further analysis methods to infer biological insights from the representations. Most translational analyses select a single embedding, typically UMAP, that best distinguishes cell types and cell states in the tumor microenvironment. This embedding is used to anchor the visualization of results from subsequent analysis, coloring cells based on cell type annotations or expression values for genes or proteins that significantly change due to treatment. In spatial single-cell analysis, after applying the same dimensionality techniques, these low-dimensional visualizations are often paralleled by visualization of the selected features directly on the tissue image.
Annotation of cell types in the tumor microenvironment
Accurate cell type identification in scRNA-seq provides the first step to inferring changes in cell proportions between samples and from perturbations such as therapies. Gating strategies used to identify cell types in flow cytometry can also be applied to the proteins or genes in single-cell assays. However, gating approaches fail to realize the potential of the high-throughput profiling to comprehensively identify cell types present in the data, characterize cellular heterogeneity, and discover new cell types. Mirroring the mathematical assumptions of t-SNE and UMAP, cells of the same cell type can be expected to have similar gene expression profiles (Becht et al., 2018; van der Maaten and Hinton, 2008). Thus, clustering algorithms are effective tools for cell type identification (Figure 2B). The large scale of single-cell data can require specialized implementations of clustering algorithms to ensure that these algorithms can run quickly, without requiring extensive computing resources. Many clustering algorithms employed for single-cell analysis leverage tools from social network analysis to identify groups of cells with similar molecular profiles and to mitigate noise from rare cells in the analysis (Blondel et al., 2008; Van Gassen et al., 2015; Xu and Su, 2015). Genes or proteins that are uniquely expressed in each cluster serve as marker genes that can be used to annotate the cell types associated with those clusters. These cell type definitions and labels will depend on the number of clusters used for analysis. Determining this optimal number of clusters remains an open question. Indeed, the hierarchical nature of cell types (e.g., subclassification of lymphoid cells into B cells, T cells, and NK cells, and subsequent subclassification of CD8+ and CD4+ T cells) suggests that different dimensions will capture different granularity of cell type delineation, reflected in emerging methods for ensemble-based clustering (Mohammadi et al., 2020; Way et al., 2020). Therefore, standard practice for cell type assignment currently relies on an iterative process of clustering cells at multiple dimensions and assessing the expression of marker genes for known immunological and stromal populations in the resident tissue type. To avoid the manual nature of this approach, several methods have emerged to leverage reference cell databases to infer identities of individual cells or clusters (Huang et al., 2020). By using curated signatures, cell type annotation becomes robust and reproducible across studies. However, these signature-based methods will not identify cell types that were not previously included in the signatures and the non-annotated cluster of cells will have to be manually verified and annotated.
As reference atlases of cell types emerge for tumors through projects such as the Human Tumor Atlas Network (Rozenblatt-Rosen et al., 2020), the first waves of annotation will rely heavily on prior biological knowledge for classifying cell types. However, as new relationships between cell types are discovered new tools will be necessary to help characterize novel biology and approaches to distinguish stable cell types from cell state transitions (e.g., between activated and exhausted T cells) remains an open area of research for single-cell analysis (Trapnell, 2015). The common lineages of tumor cells with their normal counterpart can make them difficult to identify through marker genes or clustering analysis alone. In order to distinguish cancer cells from normal cells in scRNA-seq, copy number variation (CNV) analysis is a robust approach that detects large chromosomal variations (gains and losses of large DNA segments) by examining the gene expression distribution along chromosomes. These methods use RNA expression levels to infer DNA copy number at a given genomic region, which can separate cells with extensive CNV alterations, such as cancer cells, from diploid cells (Fan et al., 2018; Gao et al., 2021; Patel et al., 2014; Tickle et al., 2019). In order to perform CNV inference, it is important to use methods designed to scale with the size of data being used. Early approaches for CNV inference (Fan et al., 2018; Patel et al., 2014) were developed using first-generation scRNA-seq technologies (Fluidigm C1, SMART-seq) (Ramsköld et al., 2012; Xin et al., 2016), which have lower cell throughput than the more recent high-throughput technologies (inDrop, Drop-seq, 10X Genomics platform) (Klein et al., 2015; Macosko et al., 2015; Zheng et al., 2017b). The development of computational tools with improved speed and accuracy for large-scale datasets with sparse molecular coverage remains a critical area of research, with new approaches, such as CopyKAT, starting to emerge that are compatible with widely used high-throughput platforms (Gao et al., 2021).
Analysis of cell-type dependent molecular changes
After cell type identification is performed, functional changes from perturbations such as treatment can be determined through differential expression analysis comparing treatment conditions within each cell type (Figure 2C). Briefly, these analysis methods compare the distribution of expression values for each protein or gene between treatment groups for the subset of cells annotated as a given cell type. The optimal statistical test for this differential expression analysis remains an open question, although approaches based upon negative binomial tests are emerging as providing the best model of the distribution of the molecular abundances of both scRNA-seq (Hafemeister and Satija, 2019; Risso et al., 2018) and CyTOF data (Crowell et al., 2020; Nowicka et al., 2017). Standard pathway analysis tools can then be applied to determine the molecular pathways that were altered based upon the results of these differential expression analyses (Irizarry et al., 2009; Subramanian et al., 2005).
In patients with the same cancer type, tumor heterogeneity can contribute to dramatic differences in therapeutic outcomes. Thus, characterization of cellular heterogeneity within the tumor microenvironment is necessary to gain a deeper understanding of tumor progression and treatment. Metrics to assess differences in heterogeneity between sample groups detect molecular variability across overall transcriptional profiles (Azizi et al., 2018) or at the pathway level (Davis-Marcisak et al., 2019; Fan et al., 2016) within individual cell types. Immune cell populations within tumors can also be highly heterogeneous, making it valuable to use these methods to determine the heterogeneity among these cells as well.
Whereas differential expression analysis can infer molecular changes from cells of a pre-specified cell type, changes in molecular pathways and state transitions may occur for multiple cell types simultaneously resulting in incomplete identification through these analysis approaches. In contrast, non-negative matrix factorization (NMF) approaches seek potentially overlapping, but low-dimensional patterns that contribute additively to the sources of variation in the data. As a result, they capture patterns that may co-occur, better modeling hierarchies in cellular lineages. Each of the patterns learned from matrix factorization analysis can represent a distinct biological process, which can be interpreted biologically through the gene weights of the corresponding features (Stein-O’Brien et al., 2018, 2019; Zhu et al., 2017). For example, NMF approach was used to identify NK cell activation in anti-CTLA4 response in our re-analysis of the scRNA-seq data from Gubin et al. (Davis-Marcisak et al., 2020; Gubin et al., 2018). The gene signatures from these NMF approaches are often robust across multiple datasets, allowing for transfer learning approaches to identify the gene signatures associated with these inferred cell states in new datasets (Stein-O’Brien et al., 2019). This approach has been leveraged for cross-species analysis relating pre-clinical and clinical models (Davis-Marcisak et al., 2020), and indeed transfer learning is at the core of many supervised signature-based cellular annotations leveraging single-cell atlases. New non-linear approaches can also learn molecular changes from perturbations independent of cell type annotations (Burkhardt et al., 2021).
Trajectory inference and pseudotemporal ordering for cell state transitions
The heterogeneous nature of single-cell data allows us to observe not only the diverse cell types in the sample but also a range of molecular states within each cell type. While scRNA-seq data represents a single snapshot of the overall sample, it can still contain individual cells that correspond to a broad range of molecular states (Ji and Ji, 2016). Trajectory inference methods computationally order the individual cells along a biological process according to their molecular states (Figure 2B) (Qiu et al., 2017; Setty et al., 2016; Shin et al., 2015). Many trajectory inference methods also assign a “pseudotime” value to each cell that represents its relative position along the trajectory. This process allows us to observe gene expression dynamics and identify cell states on a continuum along biological processes more directly than the inferences of cell state transitions from NMF methods. Numerous trajectory inference methods have been developed in recent years, and they differ on the basis of their underlying algorithms, required prior information, and the expected topology (e.g., cyclic, linear, bifurcating) of the output trajectories. Although some recent methods (Qiu et al., 2017) also infer the topology of the trajectory, most methods order cells along an assumed topology (Setty et al., 2016; Shin et al., 2015). Thus, the accuracy of the inferred trajectories is dependent on the choice of appropriate analysis method for the dataset and its associated biological process (Saelens et al., 2019). Since cancer datasets contain a heterogeneous mix of cell types, trajectory inference methods cannot be directly applied to the data. Instead, the common approach is to isolate certain cell types (Savas et al., 2018) and perform trajectory inference only with respect to these cell types.
The determination of cellular state in these analyses relies on successful trajectory inference. The key challenge for successful trajectory inference is its dependence on the embedding from techniques such as UMAP. As a result, they only follow cell state transitions in cells if the shape of that embedding matches the topology of the trajectory. Other dimension reduction approaches that explicitly model cell state transitions could be better suited for this inference. For example, RNA velocity (La Manno et al., 2018) uses the spliced and unspliced mRNAs to calculate a high-dimensional vector representing the time-derivative of the gene expression state of each cell in the dataset. RNA velocity has recently been generalized to model gene-specific kinetics (Bergen et al., 2020) and cellular transport mechanisms for spatial transcriptomics data (Xia et al., 2019). Estimates from RNA velocity can be used for more detailed visualization of the kinetic state of each cell using directional arrows in the low-dimensional embeddings. The length and direction of these arrows correspond to the high dimensional RNA velocity vector of the cell. scMomentum (Soto et al., 2020) incorporates RNA velocity estimates computed by scVelo for predicting cell-type-specific directed gene regulatory networks. For every cell-type-specific network, an energy landscape is generated, where a cell’s energy represents its differentiation potential. Extending the concept of RNA velocity, the first and second-order kinetics of protein translation in single-cell multi-omics datasets can be estimated using protein velocity and acceleration (Gorin et al., 2020). Whereas the unspliced mRNA level of a cell is said to represent its future spliced mRNA levels, the current protein expression in a cell can represent the past spliced mRNA levels. The combination of protein and RNA velocity can be visualized as a curve calculated from the three points corresponding to past, present, and future values of the spliced mRNA which represent the kinetics of the cell state. Overall, the trajectory inference or velocity analyses are relevant for identifying cell state dynamics and predicting cell fates from the analyses of a single “snapshot” in time, with potential to estimate the evolution of tumor and immune cells during cancer immunotherapy.
Inferring intra- and intercellular interaction networks from single-cell and spatial technologies
Gene regulatory network (GRN) inference is a key step to understand the interactions between genes within and between cells, allowing for inference of the biological processes underlying molecular regulation (Figure 2E). Numerous GRN inference methods have been developed in single-cell data with the goal of learning the structure of gene networks from data directly. Many approaches have been adapted from techniques that were originally developed for bulk transcriptomics analysis, which quantify network structure based upon the correlation between pairs of genes (Langfelder and Horvath, 2008) or use machine learning methods to determine which genes can modify expression the profile of one another (Huynh-Thu and Sanguinetti, 2015; Huynh-Thu et al., 2010). Newer methods have extended these approaches to specifically model the heterogeneity of single-cell data (Chan et al., 2017), including explicit extensions for time-course data (Papili Gao et al., 2018). Notably, the temporal ordering of cells by trajectory inference methods enables further inference of GRNs that can use the relative timing of gene expression changes to infer which gene controls the expression of another based on which is expressed first (Deshpande et al., 2019; Matsumoto et al., 2017; Specht and Li, 2017).
Whereas data-driven methods for GRN analysis infer intra-cellular signaling networks, regulatory processes may also occur between cells as through paracrine signaling or direct cell-to-cell interactions. Consider the case of interactions between dendritic cells (DCs) and T cells as an example of interacting cell types during immune response. DCs are antigen presenting cells that stimulate the clonal expansion and cytotoxic function of T cells (Wculek et al., 2020). To estimate these interactions from scRNA-seq data, a number of approaches attempt to infer intercellular interactions by identifying co-expressed ligand-receptor pairs (Cherry et al., 2020; Efremova et al., 2020; Kumar et al., 2018) between cell types. The incomplete ability of transcriptional data to model receptor activation and the noisy nature of single-cell data pose limitations to the inference of inter-cellular signaling networks from single-cell data alone. Spatial molecular technologies provide a promising source of information to enhance these estimates by modeling inter-cellular interactions more directly through observations of cellular co-localization. To that end, recently developed methods (Li et al., 2020; Yuan and Bar-Joseph, 2020) use spatial transcriptomic data to identify spatially informed GRNs and intercellular signaling genes. Other interpretations of networks include spatially proximal or interacting cell types to recognize pairs of cells that have a higher likelihood of colocalization and spatially informed identification of co-expressed ligand-receptor pairs (Dries et al., 2019). Thus, it is possible to infer the interactions between DC and T cells from single-cell data through ligand receptor network methods, while the direct visualization of cellular colocalization from spatial datasets can confirm such interactions.
Single-cell multi-omics
One of the advantages of UMI-based scRNA-seq is the ability to attach additional oligonucleotides barcodes to cells to allow for concurrent measures of multiple molecular modalities in the same cell with single-cell multi-omics technologies (Zhu et al., 2020). A notable multi-omics technology for studying the TME is Cellular Indexing of Transcriptomes and Epitopes by Sequencing (CITE-seq) (Stoeckius et al., 2017) (Figure 2D). CITE-seq simultaneously obtains antibody-based proteomics and transcriptional profiling, combining the benefits of a priori identification of cell types using proteomics with the unsupervised analysis of scRNA-seq. This technology has been applied to monitor the temporal changes in PBMC composition during chronic lymphocytic leukemia (CLL) therapy with the targeted agent ibrutinib, demonstrating clonal heterogeneity among leukemic cells and therapeutic perturbations in cancer and immune cells (Cadot et al., 2020).
The behavior of certain immune cells can also be traced from multi-omics by using genetic identifiers. T cells and B cells undergo germline DNA recombination that results in a broad repertoire of T and B cell receptors (TCRs, BCRs). Multi-omics technologies enable simultaneous transcriptional profiling of T cells and B cells and their respective receptors (Figure 2D). TCR sequences can be acquired directly from platforms such as 10x Genomics TCR/BCR and paired transcriptome sequencing, or they can be inferred from raw sequencing reads of scRNA-seq data by computational algorithms such as TraCeR (Stubbington et al., 2016), BraCeR (Lindeman et al., 2018), and VDJPuzzle (Eltahla et al., 2016; Rizzetto et al., 2018). The availability of combined scRNA-seq and scTCR-seq data in these approaches enables the association of cellular states with clonal expansion. In these analyses, the specific TCR and BCR profiling generates information about antigen-specific anti-tumor responses (Azizi et al., 2018; Sade-Feldman et al., 2018; Yost et al., 2019).
Paired scRNA and TCR-seq is gaining traction and being applied to a variety of cancers. A recent pan-cancer study of ICI-treated patients used this combined analysis to identify the clonal expansion of effector T cells in patients that respond to anti-PDL1 therapy (Wu et al., 2020b). In addition, expanded clonotypes were detectable across tumor, normal adjacent tissue, and peripheral blood, suggesting a potential minimally invasive biomarker of immunotherapy response (Wu et al., 2020b). Clonotype information can also complement trajectory inference analysis of intratumoral T cells and B cells to track the dynamic relationships of these lymphocytes as they mount anti-tumor responses and respond to therapy. For example, a recent study using paired TCR- and RNA-seq to profile CD8+ CAR-T cells from the blood of patients undergoing CD19 CAR-T immunotherapy found distinct clonal transcriptional dynamics and expansion after adoptive transfer (Sheih et al., 2020). In basal cell carcinoma, combined scRNA-seq of CD8+ T cells treated with anti-PD-1 found an increased presence of activated and exhausted populations, as well as a hybrid population expressing activation and exhaustion markers (Yost et al., 2019), an expected effect of anti-PD-1 therapy (Sade-Feldman et al., 2018; Wei et al., 2017). TCR analysis indicated that the largest clones presented exhaustion gene signatures. Also using TCR clonality, the authors were able to track those clones in pre- and post-treatment samples and observed that anti-PD-1 therapy did not convert exhausted T cells to a non-exhausted state. There was no expansion of the exhausted T cell clones but new clonotypes, absent in the pre-treatment samples, were detected suggesting that anti-PD-1 therapy attracts new T cells to the tumor with the potential to identify a new panel of antigens (Yost et al., 2019). These findings provide an immense contribution to understanding responses to ICIs and indicate that these therapeutic agents enhance the ability of tumors to attract additional T cells as opposed to reactivating exhausted T cells already present in the tumor (Yost et al., 2019).
While the ability to sequence both TCR chains provides an advantage over bulk single-chain methods, determining the antigen specificity of captured T cells remains a critical area of research. Advances into new multi-omics technologies are also actively being developed, with emerging methods for various combinations of proteomics, transcriptomics, spatial, and immune receptor profiling. These advances include technologies for resolve the multi-scale pathways in the tumor microenvironment, including intracellular phospho-proteomic states (Gerlach et al., 2019), intranuclear sequencing of transcription factors (Chung et al., 2021), chromatin (Cao et al., 2018), CRISPR-based screens (Hill et al., 2018), barcoding for lineage tracing of single-cells (Al’Khafaji et al., 2018; Kong et al., 2020), and concurrent spatial profiling of RNA and protein (Liu et al., 2020).
Future perspectives
Single-cell and spatial molecular profiling technologies and complementary computational analysis pipelines are rapidly advancing as tools for cancer research. The inferences from these technologies rely on the study design, sample processing, and analysis pipelines selected for profiling. Due to the rapid advance of these technologies, many of the computational pipelines that enable interpretation of this data are still being developed. As single-cell data evolve as translational tools, computational methodologies will play a role in driving new discoveries. While powerful, these high-throughput technologies primarily serve as profiling tools to generate new hypotheses about the tumor microenvironment and therapeutic modalities. Therefore, complementary mechanistic bench studies remain an important complement to translate single-cell research into actionable therapeutic targets.
In translational immunotherapy research, the ultimate test of mechanism is that a therapeutic intervention yields the hypothesized immune modulation on the tumor microenvironment within a patients’ tumor. While single-cell technologies can be applied to measure these effects, full mechanistic characterization requires time-course profiling that would involve serial sample collection from the same patient which are unethical and unfeasible to perform. Although monitoring the immune cell repertoire from a patient’s peripheral blood is more feasible for time-course studies, single-cell studies comparing the immune cell composition of human tumors and peripheral blood have identified intrinsic differences (Azizi et al., 2018). Therefore, future studies are needed to provide a more comprehensive comparison between the tumor immune landscape and that of the periphery to enable the use of single-cell technologies as therapeutic biomarkers. The heterogeneity between patient tumors and inability to test more than one treatment regimen in a patient further challenges mechanistic single-cell studies in translational research. Single-cell atlas studies pooling clinical trials studies and perturbation studies from preclinical models can provide important references to support such human profiling studies. Single-cell profiling of pre-clinical models treated with immunotherapies can point to the cell types and pathways relevant to therapeutic response, while cross-species analysis of human samples treated with the same therapies can reveal which therapeutic responses are conserved. Emerging computational tools to identify shared responses in mice and humans from single-cell datasets can further support model selection for preclinical analysis to inform the design of human clinical trials (Davis-Marcisak et al., 2020; Peng et al., 2021) (Figure 3).
Figure 3 -.
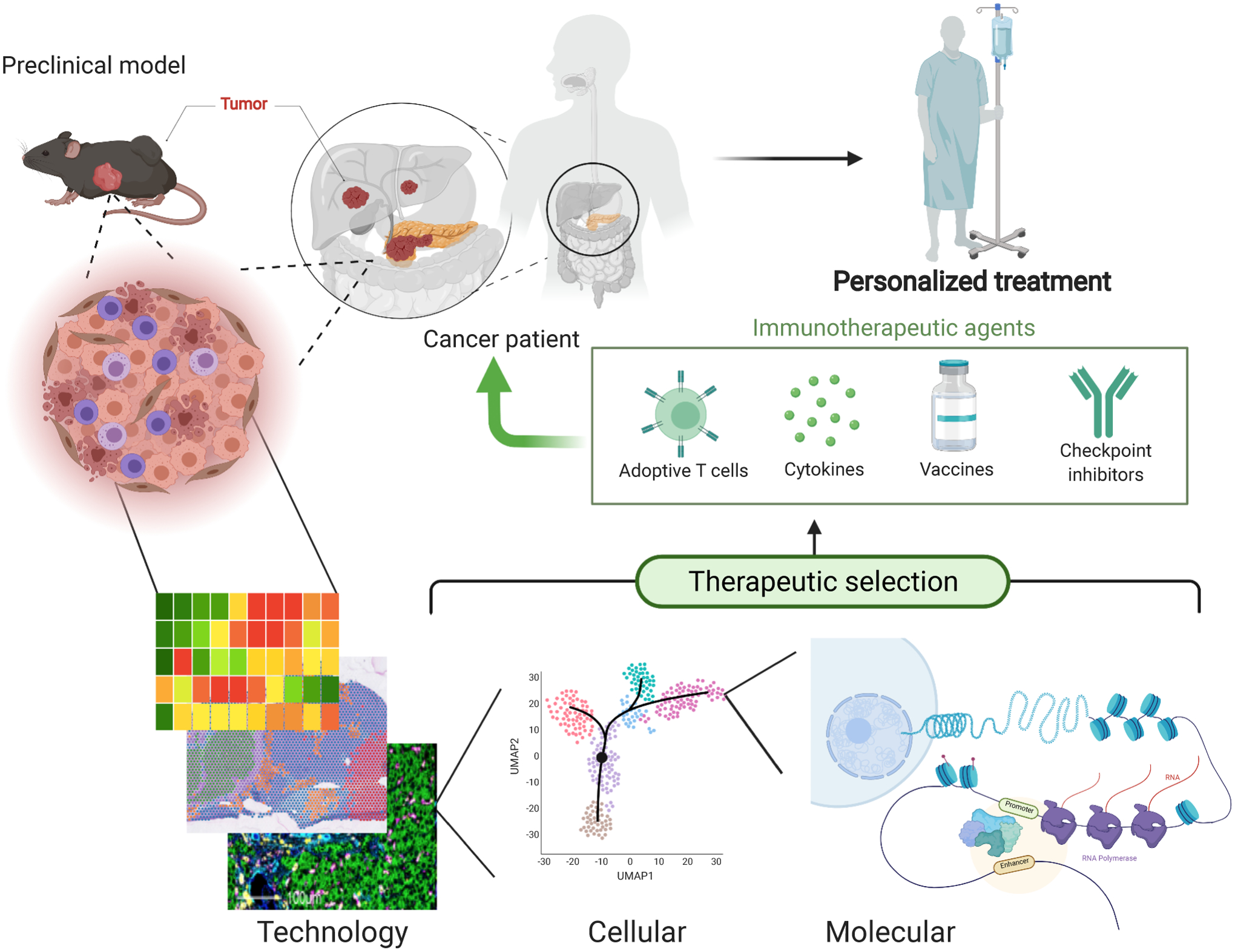
Mouse to human studies using high dimensional analysis will drive the next generation of precision cancer immunotherapies. Single-cell and spatial technologies have the power to drive discoveries based on the cell types that are commonly affected by immunotherapies in preclinical and human tumors. Following identification of the commonalities between models, studies can focus on identifying molecular and cellular markers of response using multi-omics approaches. The combination of different layers of data will drive patient selection for the most adequate therapy, better clinical trial designs, and development of new immunotherapies.
Single-cell experiments must be carefully designed to achieve the desired depth of immune characterization and to avoid confounding technical biases with phenotypic covariates. Technology selection should align with the underlying hypothesis of the study. For example, single-cell and spatial transcriptomics is well suited to drive genome-wide discovery across unbiased cell populations, and single-cell proteomics is better suited for studies that aim to profile known molecular targets and cell types. Pathologists can play an important role in selecting regions that capture the appropriate biological region (e.g., tumor dense regions) and account for tumor-heterogeneity. Statistical evaluation of sample size is also an important consideration of study design. Single-cell datasets from large cohorts are important for biomarker discovery and applications of machine learning to predict patient outcomes, particularly to avoid overfitting these models with the large number of molecular features they measure. However, the significant costs of these technologies impose a practical limitation to designing powered cohorts in single-cell atlas studies. Thus, a balance of low-dimensional profiling with proteomics technologies on large cohorts and leveraging selected samples for higher-dimensional, mechanistic studies is a critical step during experimental design. Computational algorithms for cross-platform data integration can provide an important complement to balance molecular depth with sample size in these mixed designs. Close collaboration between experimental, computational, and statistical scientists can support optimal study design and also prioritize new computational approaches for data analysis tailored to the translational research goals of each study. A multi-disciplinary approach will help the cancer research community to overcome these challenges and use single-cell and spatial platforms to make new discoveries for cancer immunotherapy.
Table 2 -.
Single-cell and spatial technologies sample requirements and data analysis opportunities.
| TECHNOLOGY | Single-cell proteomics | Single-cell transcriptomics | Spatial proteomics | Spatial transcriptomics | ||
|---|---|---|---|---|---|---|
| scRNA-seq | snRNA-seq | Slide-seq, 10× Genomics | SeqFISH, SeqFISH+, MERFISH | |||
| Sample | Dissociated cells | Viable dissociated cells | Viable nuclei (fresh or frozen tumor) | Frozen, FFPE | Frozen | Frozen, FFPE |
| Tissue type | Tumor and normal tissue, blood | Tumor and normal tissue | ||||
| Genome coverage | Pre-selected markers (~40 markers) |
Genome-wide | Pre-selected markers (~40 markers) |
Genome-wide | Pre-selected markers (up to 10,000 genes) |
|
| DATA ANALYSIS | ||||||
| Cell composition | Yes | |||||
| Cell states | Yes | |||||
| Differential expression | Yes | |||||
| Pathway analysis | No | Yes | No | Yes | ||
| Cell fate trajectories | No | Yes | No | |||
| Molecular interaction | No | Yes | No | Yes | ||
| Cellular interactions | No | Yes | ||||
| scTCR/scBCR | No | Yes | No | |||
Acknowledgements
We thank Janelle Montagne, Ben K. Johnson, Jackie Zimmerman and Jacob Mitchell for feedback on the manuscript. Figures were created with BioRender.com.
Funding
This work was supported by Lustgarten Foundation: Pancreatic Cancer Research Grant (to EMJ), Sol Goldman Pancreatic Cancer Research Center Grant (to LTK), Emerson Collective Cancer Research Fund (to EMJ), Allegheny Health Network (AHN) Grant (to EJF), U01CA212007 (to EJF), U01CA253403 (to EJF), the JHU Discovery Award (to EJF), P30CA006973, F31CA250135-01A1 (to EFDM), Kavli NDI Postdoctoral Fellowship (to GSO), and JHU Provost Postdoctoral Fellowship (to GSO).
Declaration of Interests
WH is a co-inventor of patents with potential for receiving royalties from Rodeo Therapeutics/Amgen, is a consultant for Exelixis, and receives research funding from Sanofi. EMJ is a paid consultant for Adaptive Biotech, CSTONE, Achilles, DragonFly, and Genocea; receives funding from Lustgarten Foundation and Bristol Myer Squibb; is the Chief Medical Advisor for Lustgarten and SAB advisor to the Parker Institute for Cancer Immunotherapy (PICI) and for the C3 Cancer Institute. EJF is a member of the Scientific Advisory Board of Vioscera Therapeutics / ResistanceBio. All other authors have nothing to disclose.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Al’Khafaji AM, Deatherage D, and Brock A (2018). Control of Lineage-Specific Gene Expression by Functionalized gRNA Barcodes. ACS Synth. Biol 7, 2468–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amezquita RA, Lun ATL, Becht E, Carey VJ, Carpp LN, Geistlinger L, Marini F, Rue-Albrecht K, Risso D, Soneson C, et al. (2020). Orchestrating single-cell analysis with Bioconductor. Nat. Methods 17, 137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelo M, Bendall SC, Finck R, Hale MB, Hitzman C, Borowsky AD, Levenson RM, Lowe JB, Liu SD, Zhao S, et al. (2014). Multiplexed ion beam imaging of human breast tumors. Nat. Med 20, 436–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki T, Chong LC, Takata K, Milne K, Hav M, Colombo A, Chavez EA, Nissen M, Wang X, Miyata-Takata T, et al. (2020). Single-Cell Transcriptome Analysis Reveals Disease-Defining T-cell Subsets in the Tumor Microenvironment of Classic Hodgkin Lymphoma. Cancer Discov 10, 406–421. [DOI] [PubMed] [Google Scholar]
- Avila Cobos F, Alquicira-Hernandez J, Powell JE, Mestdagh P, and De Preter K (2020). Benchmarking of cell type deconvolution pipelines for transcriptomics data. Nat. Commun 11, 5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, Nainys J, Wu K, Kiseliovas V, Setty M, et al. (2018). Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 174, 1293–1308.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandura DR, Baranov VI, Ornatsky OI, Antonov A, Kinach R, Lou X, Pavlov S, Vorobiev S, Dick JE, and Tanner SD (2009). Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal. Chem 81, 6813–6822. [DOI] [PubMed] [Google Scholar]
- Becht E, McInnes L, Healy J, Dutertre C-A, Kwok IWH, Ng LG, Ginhoux F, and Newell EW (2018). Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol 37, 38–44. [DOI] [PubMed] [Google Scholar]
- Bendall SC, Simonds EF, Qiu P, Amir ED, Krutzik PO, Finck R, Bruggner RV, Melamed R, Trejo A, Ornatsky OI, et al. (2011). Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 332, 687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergen V, Lange M, Peidli S, Wolf FA, and Theis FJ (2020). Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol 38, 1408–1414. [DOI] [PubMed] [Google Scholar]
- Berglund E, Maaskola J, Schultz N, Friedrich S, Marklund M, Bergenstrahle J, Tarish F, Tanoglidi A, Vickovic S, Larsson L, et al. (2018). Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat. Commun 9, 2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard V, Semaan A, Huang J, San Lucas FA, Mulu FC, Stephens BM, Guerrero PA, Huang Y, Zhao J, Kamyabi N, et al. (2019). Single-Cell Transcriptomics of Pancreatic Cancer Precursors Demonstrates Epithelial and Microenvironmental Heterogeneity as an Early Event in Neoplastic Progression. Clin. Cancer Res 25, 2194–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondel VD, Guillaume J-L, Lambiotte R, and Lefebvre E (2008). Fast unfolding of communities in large networks. J. Stat. Mech 2008, P10008. [Google Scholar]
- Browaeys R, Saelens W, and Saeys Y (2020). NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods 17, 159–162. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, and Greenleaf WJ (2015). Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullard JH, Purdom E, Hansen KD, and Dudoit S (2010). Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics 11, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt DB, Stanley JS, Tong A, Perdigoto AL, Gigante SA, Herold KC, Wolf G, Giraldez AJ, van Dijk D, and Krishnaswamy S (2021). Quantifying the effect of experimental perturbations at single-cell resolution. Nat. Biotechnol 39, 619–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadot S, Valle C, Tosolini M, Pont F, Largeaud L, Laurent C, Fournie JJ, Ysebaert L, and Quillet-Mary A (2020). Longitudinal CITE-Seq profiling of chronic lymphocytic leukemia during ibrutinib treatment: evolution of leukemic and immune cells at relapse. Biomark. Res 8, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caicedo JC, Cooper S, Heigwer F, Warchal S, Qiu P, Molnar C, Vasilevich AS, Barry JD, Bansal HS, Kraus O, et al. (2017). Data-analysis strategies for image-based cell profiling. Nat. Methods 14, 849–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Cusanovich DA, Ramani V, Aghamirzaie D, Pliner HA, Hill AJ, Daza RM, McFaline-Figueroa JL, Packer JS, Christiansen L, et al. (2018). Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361, 1380–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, Zhang F, Mundlos S, Christiansen L, Steemers FJ, et al. (2019). The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan TE, Stumpf MPH, and Babtie AC (2017). Gene Regulatory Network Inference from Single-Cell Data Using Multivariate Information Measures. Cell Syst 5, 251–267.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry C, Cahan P, Garmire LX, and Elisseeff JH (2020). Domino: reconstructing intercellular signaling dynamics with transcription factor activation in model biomaterial environments. BioRxiv [Google Scholar]
- Chung H, Parkhurst C, Magee EM, Phillips D, Habibi E, Chen F, Yeung B, Waldman JA, Artis D, and Regev A (2021). Simultaneous single cell measurements of intranuclear proteins and gene expression. BioRxiv [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary B, Cong L, Cheung A, Lander ES, and Regev A (2017). Efficient generation of transcriptomic profiles by random composite measurements. Cell 171, 1424–1436.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowell HL, Chevrier S, Jacobs A, Sivapatham S, Tumor Profiler Consortium, Bodenmiller B, and Robinson MD (2020). An R-based reproducible and user-friendly preprocessing pipeline for CyTOF data. F1000Res. 9, 1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson S, Efremova M, Riedel A, Mahata B, Pramanik J, Huuhtanen J, Kar G, Vento-Tormo R, Hagai T, Chen X, et al. (2020). Single-Cell RNA Sequencing Reveals a Dynamic Stromal Niche That Supports Tumor Growth. Cell Rep 31, 107628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis-Marcisak EF, Sherman TD, Orugunta P, Stein-O’Brien GL, Puram SV, Roussos Torres ET, Hopkins AC, Jaffee EM, Favorov AV, Afsari B, et al. (2019). Differential Variation Analysis Enables Detection of Tumor Heterogeneity Using Single-Cell RNA-Sequencing Data. Cancer Res 79, 5102–5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis-Marcisak EF, Fitzgerald AA, Kessler MD, Danilova L, Jaffee EM, Zaidi N, Weiner LM, and Fertig EJ (2020). A novel mechanism of natural killer cell response to anti-CTLA-4 therapy identified by integrative analysis of mouse and human tumors. BioRxiv. [Google Scholar]
- Denisenko E, Guo BB, Jones M, Hou R, de Kock L, Lassmann T, Poppe D, Clement O, Simmons RK, Lister R, et al. (2020). Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus RNA-seq workflows. Genome Biol 21, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande A, Chu L-F, Stewart R, and Gitter A (2019). Network Inference with Granger Causality Ensembles on Single-Cell Transcriptomic Data. BioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dries R, Zhu Q, Eng C-HL, Sarkar A, Bao F, George RE, Pierson N, Cai L, and Yuan G-C (2019). Giotto, a pipeline for integrative analysis and visualization of single-cell spatial transcriptomic data. BioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efremova M, Vento-Tormo M, Teichmann SA, and Vento-Tormo R (2020). CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc 15, 1484–1506. [DOI] [PubMed] [Google Scholar]
- Elosua-Bayes M, Nieto P, Mereu E, Gut I, and Heyn H (2021). SPOTlight: seeded NMF regression to deconvolute spatial transcriptomics spots with single-cell transcriptomes. Nucleic Acids Res [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltahla AA, Rizzetto S, Pirozyan MR, Betz-Stablein BD, Venturi V, Kedzierska K, Lloyd AR, Bull RA, and Luciani F (2016). Linking the T cell receptor to the single cell transcriptome in antigen-specific human T cells. Immunol. Cell Biol 94, 604–611. [DOI] [PubMed] [Google Scholar]
- Eng C-HL, Lawson M, Zhu Q, Dries R, Koulena N, Takei Y, Yun J, Cronin C, Karp C, Yuan G-C, et al. (2019). Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 568, 235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Salathia N, Liu R, Kaeser GE, Yung YC, Herman JL, Kaper F, Fan J-B, Zhang K, Chun J, et al. (2016). Characterizing transcriptional heterogeneity through pathway and gene set overdispersion analysis. Nat. Methods 13, 241–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Lee H-O, Lee S, Ryu D-E, Lee S, Xue C, Kim SJ, Kim K, Barkas N, Park PJ, et al. (2018). Linking transcriptional and genetic tumor heterogeneity through allele analysis of single-cell RNA-seq data. Genome Res 28, 1217–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadalla R, Noamani B, MacLeod BL, Dickson RJ, Guo M, Xu W, Lukhele S, Elsaesser HJ, Razak ARA, Hirano N, et al. (2019). Validation of cytof against flow cytometry for immunological studies and monitoring of human cancer clinical trials. Front. Oncol 9, 415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Ling M, Tang X, Wang S, Xiao X, Qiao Y, Yang W, and Yu R (2020). Comparison of high-throughput single-cell RNA sequencing data processing pipelines. Brief. Bioinformatics. [DOI] [PubMed] [Google Scholar]
- Gao R, Bai S, Henderson YC, Lin Y, Schalck A, Yan Y, Kumar T, Hu M, Sei E, Davis A, et al. (2021). Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes. Nat. Biotechnol 39, 599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes MJ, Sevinsky CJ, Sood A, Adak S, Bello MO, Bordwell A, Can A, Corwin A, Dinn S, Filkins RJ, et al. (2013). Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proc Natl Acad Sci USA 110, 11982–11987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach JP, van Buggenum JAG, Tanis SEJ, Hogeweg M, Heuts BMH, Muraro MJ, Elze L, Rivello F, Rakszewska A, van Oudenaarden A, et al. (2019). Combined quantification of intracellular (phospho-)proteins and transcriptomics from fixed single cells. Sci. Rep 9, 1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giesen C, Wang HAO, Schapiro D, Zivanovic N, Jacobs A, Hattendorf B, Schuffler PJ, Grolimund D, Buhmann JM, Brandt S, et al. (2014). Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 11, 417–422. [DOI] [PubMed] [Google Scholar]
- Giladi A, and Amit I (2018). Single-Cell Genomics: A Stepping Stone for Future Immunology Discoveries. Cell 172, 14–21. [DOI] [PubMed] [Google Scholar]
- Gohil SH, Iorgulescu JB, Braun DA, Keskin DB, and Livak KJ (2021). Applying high-dimensional single-cell technologies to the analysis of cancer immunotherapy. Nat. Rev. Clin. Oncol 18, 244–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein LD, Chen Y-JJ, Wu J, Chaudhuri S, Hsiao Y-C, Schneider K, Hoi KH, Lin Z, Guerrero S, Jaiswal BS, et al. (2019). Massively parallel single-cell B-cell receptor sequencing enables rapid discovery of diverse antigen-reactive antibodies. Commun. Biol 2, 304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goltsev Y, Samusik N, Kennedy-Darling J, Bhate S, Hale M, Vazquez G, Black S, and Nolan GP (2018). Deep Profiling of Mouse Splenic Architecture with CODEX Multiplexed Imaging. Cell 174, 968–981.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorin G, Svensson V, and Pachter L (2020). Protein velocity and acceleration from single-cell multiomics experiments. Genome Biol 21, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorris MAJ, Halilovic A, Rabold K, van Duffelen A, Wickramasinghe IN, Verweij D, Wortel IMN, Textor JC, de Vries IJM, and Figdor CG (2018). Eight-Color Multiplex Immunohistochemistry for Simultaneous Detection of Multiple Immune Checkpoint Molecules within the Tumor Microenvironment. J. Immunol 200, 347–354. [DOI] [PubMed] [Google Scholar]
- Gracia Villacampa E, Larsson L, Kvastad L, Andersson A, Carlson J, and Lundeberg J (2020). Genome-wide Spatial Expression Profiling in FFPE Tissues. BioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubin MM, Esaulova E, Ward JP, Malkova ON, Runci D, Wong P, Noguchi T, Arthur CD, Meng W, Alspach E, et al. (2018). High-Dimensional Analysis Delineates Myeloid and Lymphoid Compartment Remodeling during Successful Immune-Checkpoint Cancer Therapy. Cell 175, 1014–1030.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, Kang B, Liu Z, Jin L, Xing R, et al. (2018). Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med 24, 978–985. [DOI] [PubMed] [Google Scholar]
- Hafemeister C, and Satija R (2019). Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 20, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zagar M, et al. (2020). Integrated analysis of multimodal single-cell data. BioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann FJ, and Bendall SC (2020). Immune monitoring using mass cytometry and related high-dimensional imaging approaches. Nat. Rev. Rheumatol 16, 87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks SC, Townes FW, Teng M, and Irizarry RA (2018). Missing data and technical variability in single-cell RNA-sequencing experiments. Biostatistics 19, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill AJ, McFaline-Figueroa JL, Starita LM, Gasperini MJ, Matreyek KA, Packer J, Jackson D, Shendure J, and Trapnell C (2018). On the design of CRISPR-based single-cell molecular screens. Nat. Methods 15, 271–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou W, Ji Z, Ji H, and Hicks SC (2020). A systematic evaluation of single-cell RNA-sequencing imputation methods. Genome Biol 21, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho WJ, Sharma G, Zhu Q, Stein-O’Brien G, Durham J, Anders R, Popovic A, Mo G, Kamel I, Weiss M, et al. (2020). Integrated immunological analysis of a successful conversion of locally advanced hepatocellular carcinoma to resectability with neoadjuvant therapy. J. Immunother. Cancer 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q, Liu Y, Du Y, and Garmire LX (2020). Evaluation of Cell Type Annotation R Packages on Single-cell RNA-seq Data. Genomics Proteomics Bioinformatics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh-Thu VA, and Sanguinetti G (2015). Combining tree-based and dynamical systems for the inference of gene regulatory networks. Bioinformatics 31, 1614–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh-Thu VA, Irrthum A, Wehenkel L, and Geurts P (2010). Inferring regulatory networks from expression data using tree-based methods. PLoS ONE 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilicic T, Kim JK, Kolodziejczyk AA, Bagger FO, McCarthy DJ, Marioni JC, and Teichmann SA (2016). Classification of low quality cells from single-cell RNA-seq data. Genome Biol 17, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Wang C, Zhou Y, and Speed TP (2009). Gene set enrichment analysis made simple. Stat. Methods Med. Res 18, 565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam S, Zeisel A, Joost S, La Manno G, Zajac P, Kasper M, Lonnerberg P, and Linnarsson S (2014). Quantitative single-cell RNA-seq with unique molecular identifiers. Nat. Methods 11, 163–166. [DOI] [PubMed] [Google Scholar]
- Jackson HW, Fischer JR, Zanotelli VRT, Ali HR, Mechera R, Soysal SD, Moch H, Muenst S, Varga Z, Weber WP, et al. (2020). The single-cell pathology landscape of breast cancer. Nature 578, 615–620. [DOI] [PubMed] [Google Scholar]
- Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su M-J, Melms JC, Leeson R, Kanodia A, Mei S, Lin J-R, et al. (2018). A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell 175, 984–997.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z, and Ji H (2016). TSCAN: Pseudo-time reconstruction and evaluation in single-cell RNA-seq analysis. Nucleic Acids Res 44, e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji AL, Rubin AJ, Thrane K, Jiang S, Reynolds DL, Meyers RM, Guo MG, George BM, Mollbrink A, Bergenstrahle J, et al. (2020). Multimodal analysis of composition and spatial architecture in human squamous cell carcinoma. Cell 182, 497–514.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren L, Bosse M, Marquez D, Angoshtari R, Jain S, Varma S, Yang S-R, Kurian A, Van Valen D, West R, et al. (2018). A Structured Tumor-Immune Microenvironment in Triple Negative Breast Cancer Revealed by Multiplexed Ion Beam Imaging. Cell 174, 1373–1387.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA, and Kirschner MW (2015). Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong W, Biddy BA, Kamimoto K, Amrute JM, Butka EG, and Morris SA (2020). CellTagging: combinatorial indexing to simultaneously map lineage and identity at single-cell resolution. Nat. Protoc 15, 750–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg C, Nowicka M, Guglietta S, Schindler S, Hartmann FJ, Weber LM, Dummer R, Robinson MD, Levesque MP, and Becher B (2018). High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat. Med 24, 144–153. [DOI] [PubMed] [Google Scholar]
- Kumar MP, Du J, Lagoudas G, Jiao Y, Sawyer A, Drummond DC, Lauffenburger DA, and Raue A (2018). Analysis of Single-Cell RNA-Seq Identifies Cell-Cell Communication Associated with Tumor Characteristics. Cell Rep 25, 1458–1468.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafzi A, Moutinho C, Picelli S, and Heyn H (2018). Tutorial: guidelines for the experimental design of single-cell RNA sequencing studies. Nat. Protoc 13, 2742–2757. [DOI] [PubMed] [Google Scholar]
- Langfelder P, and Horvath S (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lonnerberg P, Furlan A, et al. (2018). RNA velocity of single cells. Nature 560, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek JT, Scharpf RB, Bravo HC, Simcha D, Langmead B, Johnson WE, Geman D, Baggerly K, and Irizarry RA (2010). Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet 11, 733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Hyeon DY, and Hwang D (2020). Single-cell multiomics: technologies and data analysis methods. Exp. Mol. Med 52, 1428–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leipold MD, Obermoser G, Fenwick C, Kleinstuber K, Rashidi N, McNevin JP, Nau AN, Wagar LE, Rozot V, Davis MM, et al. (2018). Comparison of CyTOF assays across sites: Results of a six-center pilot study. J. Immunol. Methods 453, 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim B, Lin Y, and Navin N (2020). Advancing Cancer Research and Medicine with Single-Cell Genomics. Cancer Cell 37, 456–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindeman I, Emerton G, Mamanova L, Snir O, Polanski K, Qiao S-W, Sollid LM, Teichmann SA, and Stubbington MJT (2018). BraCeR: B-cell-receptor reconstruction and clonality inference from single-cell RNA-seq. Nat. Methods 15, 563–565. [DOI] [PubMed] [Google Scholar]
- Lin J-R, Fallahi-Sichani M, and Sorger PK (2015). Highly multiplexed imaging of single cells using a high-throughput cyclic immunofluorescence method. Nat. Commun 6, 8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Yang M, Deng Y, Su G, Enninful A, Guo CC, Tebaldi T, Zhang D, Kim D, Bai Z, et al. (2020). High-Spatial-Resolution Multi-Omics Sequencing via Deterministic Barcoding in Tissue. Cell 183, 1665–1681.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Courtois ET, Sengupta D, Tan Y, Chen KH, Goh JJL, Kong SL, Chua C, Hon LK, Tan WS, et al. (2017). Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet 49, 708–718. [DOI] [PubMed] [Google Scholar]
- Li Y, Ma A, Mathe EA, Li L, Liu B, and Ma Q (2020). Elucidation of Biological Networks across Complex Diseases Using Single-Cell Omics. Trends Genet 36, 951–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M, and Cai L (2014). Single-cell in situ RNA profiling by sequential hybridization. Nat. Methods 11, 360–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luecken MD, and Theis FJ (2019). Current best practices in single-cell RNA-seq analysis: a tutorial. Mol. Syst. Biol 15, e8746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Maaten L, and Hinton G (2008). Visualizing Data using t-SNE. Journal of Machine Learning Research. [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. (2015). Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis S, Petrescu J, and Phatnani H (2021). Spatially resolved transcriptomics and its applications in cancer. Curr. Opin. Genet. Dev 66, 70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx V (2021). Method of the Year: spatially resolved transcriptomics. Nat. Methods 18, 9–14. [DOI] [PubMed] [Google Scholar]
- Matsumoto H, Kiryu H, Furusawa C, Ko MSH, Ko SBH, Gouda N, Hayashi T, and Nikaido I (2017). SCODE: an efficient regulatory network inference algorithm from single-cell RNA-Seq during differentiation. Bioinformatics 33, 2314–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Hernandez MO, Zhao Y, Mehta M, Tran B, Kelly M, Rae Z, Hernandez JM, Davis JL, Martin SP, et al. (2019). Tumor cell biodiversity drives microenvironmental reprogramming in liver cancer. Cancer Cell 36, 418–430.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes L, Healy J, and Melville J (2018). UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. ArXiv. [Google Scholar]
- Megill C, Martin B, Weaver C, Bell S, Prins L, Badajoz S, McCandless B, Pisco AO, Kinsella M, Griffin F, et al. (2021). cellxgene: a performant, scalable exploration platform for high dimensional sparse matrices. BioRxiv. [Google Scholar]
- Mohammadi S, Davila-Velderrain J, and Kellis M (2020). A multiresolution framework to characterize single-cell state landscapes. Nat. Commun 11, 5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncada R, Barkley D, Wagner F, Chiodin M, Devlin JC, Baron M, Hajdu CH, Simeone DM, and Yanai I (2020). Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat. Biotechnol 38, 333–342. [DOI] [PubMed] [Google Scholar]
- Moon KR, van Dijk D, Wang Z, Gigante S, Burkhardt DB, Chen WS, Yim K, Elzen A van den, Hirn MJ, Coifman RR, et al. (2019). Visualizing structure and transitions in high-dimensional biological data. Nat. Biotechnol 37, 1482–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navin N, and Hicks J (2011). Future medical applications of single-cell sequencing in cancer. Genome Med 3, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, et al. (2011). Tumour evolution inferred by single-cell sequencing. Nature 472, 90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowicka M, Krieg C, Crowell HL, Weber LM, Hartmann FJ, Guglietta S, Becher B, Levesque MP, and Robinson MD (2017). CyTOF workflow: differential discovery in high-throughput high-dimensional cytometry datasets. [version 3; peer review: 2 approved]. F1000Res. 6, 748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papili Gao N, Ud-Dean SMM, Gandrillon O, and Gunawan R (2018). SINCERITIES: inferring gene regulatory networks from time-stamped single cell transcriptional expression profiles. Bioinformatics 34, 258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. (2014). Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng D, Gleyzer R, Tai W-H, Kumar P, Bian Q, Isaacs B, da Rocha EL, Cai S, DiNapoli K, Huang FW, et al. (2021). Evaluating the transcriptional fidelity of cancer models. Genome Med 13, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Sun B-F, Chen C-Y, Zhou J-Y, Chen Y-S, Chen H, Liu L, Huang D, Jiang J, Cui G-S, et al. (2019). Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res 29, 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, Rodman C, Luo CL, Mroz EA, Emerick KS, et al. (2017). Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 171, 1611–1624.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, and Trapnell C (2017). Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Vara JA, and Miller MA (2014). When tissue antigens and antibodies get along: revisiting the technical aspects of immunohistochemistry--the red, brown, and blue technique. Vet. Pathol 51, 42–87. [DOI] [PubMed] [Google Scholar]
- Ramsköld D, Luo S, Wang Y-C, Li R, Deng Q, Faridani OR, Daniels GA, Khrebtukova I, Loring JF, Laurent LC, et al. (2012). Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol 30, 777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich M, Tabor T, Liefeld T, Thorvaldsdottir H, Hill B, Tamayo P, and Mesirov JP (2017). The genepattern notebook environment. Cell Syst 5, 149–151.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, and Wolchok JD (2018). Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risso D, Perraudeau F, Gribkova S, Dudoit S, and Vert J-P (2018). A general and flexible method for signal extraction from single-cell RNA-seq data. Nat. Commun 9, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzetto S, Koppstein DNP, Samir J, Singh M, Reed JH, Cai CH, Lloyd AR, Eltahla AA, Goodnow CC, and Luciani F (2018). B-cell receptor reconstruction from single-cell RNA-seq with VDJPuzzle. Bioinformatics 34, 2846–2847. [DOI] [PubMed] [Google Scholar]
- Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, Vanderburg CR, Welch J, Chen LM, Chen F, and Macosko EZ (2019). Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenblatt-Rosen O, Regev A, Oberdoerffer P, Nawy T, Hupalowska A, Rood JE, Ashenberg O, Cerami E, Coffey RJ, Demir E, et al. (2020). The Human Tumor Atlas Network: Charting Tumor Transitions across Space and Time at Single-Cell Resolution. Cell 181, 236–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, Lieb DJ, Chen JH, Frederick DT, Barzily-Rokni M, et al. (2018). Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 175, 998–1013.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saelens W, Cannoodt R, Todorov H, and Saeys Y (2019). A comparison of single-cell trajectory inference methods. Nat. Biotechnol 37, 547–554. [DOI] [PubMed] [Google Scholar]
- Sandberg R (2014). Entering the era of single-cell transcriptomics in biology and medicine. Nat. Methods 11, 22–24. [DOI] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savas P, Virassamy B, Ye C, Salim A, Mintoff CP, Caramia F, Salgado R, Byrne DJ, Teo ZL, Dushyanthen S, et al. (2018). Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat. Med 24, 986–993. [DOI] [PubMed] [Google Scholar]
- Schürch CM, Bhate SS, Barlow GL, Phillips DJ, Noti L, Zlobec I, Chu P, Black S, Demeter J, McIlwain DR, et al. (2020). Coordinated cellular neighborhoods orchestrate antitumoral immunity at the colorectal cancer invasive front. Cell 182, 1341–1359.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See P, Lum J, Chen J, and Ginhoux F (2018). A Single-Cell Sequencing Guide for Immunologists. Front. Immunol 9, 2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setty M, Tadmor MD, Reich-Zeliger S, Angel O, Salame TM, Kathail P, Choi K, Bendall S, Friedman N, and Pe’er D (2016). Wishbone identifies bifurcating developmental trajectories from single-cell data. Nat. Biotechnol 34, 637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheih A, Voillet V, Hanafi L-A, DeBerg HA, Yajima M, Hawkins R, Gersuk V, Riddell SR, Maloney DG, Wohlfahrt ME, et al. (2020). Clonal kinetics and single-cell transcriptional profiling of CAR-T cells in patients undergoing CD19 CAR-T immunotherapy. Nat. Commun 11, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J, Berg DA, Zhu Y, Shin JY, Song J, Bonaguidi MA, Enikolopov G, Nauen DW, Christian KM, Ming G, et al. (2015). Single-Cell RNA-Seq with Waterfall Reveals Molecular Cascades underlying Adult Neurogenesis. Cell Stem Cell 17, 360–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slyper M, Porter CBM, Ashenberg O, Waldman J, Drokhlyansky E, Wakiro I, Smillie C, Smith-Rosario G, Wu J, Dionne D, et al. (2020). A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat. Med 26, 792–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto LM, Bernal-Tamayo JP, Lehmann R, Balsamy S, Martinez-de-Morentin X, Vilas-Zornoza A, San-Martin P, Prosper F, Gomez-Cabrero D, Kiani N, et al. (2020). scMomentum: Inference of Cell-Type-Specific Regulatory Networks and Energy Landscapes. BioRxiv. [Google Scholar]
- Specht AT, and Li J (2017). LEAP: constructing gene co-expression networks for single-cell RNA-sequencing data using pseudotime ordering. Bioinformatics 33, 764–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl PL, Salmen F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, Giacomello S, Asp M, Westholm JO, Huss M, et al. (2016). Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82. [DOI] [PubMed] [Google Scholar]
- Stein-O’Brien GL, Arora R, Culhane AC, Favorov AV, Garmire LX, Greene CS, Goff LA, Li Y, Ngom A, Ochs MF, et al. (2018). Enter the Matrix: Factorization Uncovers Knowledge from Omics. Trends Genet 34, 790–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein-O’Brien GL, Clark BS, Sherman T, Zibetti C, Hu Q, Sealfon R, Liu S, Qian J, Colantuoni C, Blackshaw S, et al. (2019). Decomposing Cell Identity for Transfer Learning across Cellular Measurements, Platforms, Tissues, and Species. Cell Syst 8, 395–411.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R, and Smibert P (2017). Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbington MJT, Lonnberg T, Proserpio V, Clare S, Speak AO, Dougan G, and Teichmann SA (2016). T cell fate and clonality inference from single-cell transcriptomes. Nat. Methods 13, 329–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subrahmanyam PB, Dong Z, Gusenleitner D, Giobbie-Hurder A, Severgnini M, Zhou J, Manos M, Eastman LM, Maecker HT, and Hodi FS (2018). Distinct predictive biomarker candidates for response to anti-CTLA-4 and anti-PD-1 immunotherapy in melanoma patients. J. Immunother. Cancer 6, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumatoh HR, Teng KWW, Cheng Y, and Newell EW (2017). Optimization of mass cytometry sample cryopreservation after staining. Cytometry A 91, 48–61. [DOI] [PubMed] [Google Scholar]
- Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, Wang X, Bodeau J, Tuch BB, Siddiqui A, et al. (2009). mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 6, 377–382. [DOI] [PubMed] [Google Scholar]
- Tekman M, Batut B, Ostrovsky A, Antoniewski C, Clements D, Ramirez F, Etherington GJ, Hotz H-R, Scholtalbers J, Manning JR, et al. (2020). A single-cell RNA-seq Training and Analysis Suite using the Galaxy Framework. BioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tickle T, Tirosh I, Georgescu C, Brown M, and Haas B (2019). inferCNV of the Trinity CTAT Project. Https://Github.Com/Broadinstitute/InferCNV.
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran HTN, Ang KS, Chevrier M, Zhang X, Lee NYS, Goh M, and Chen J (2020). A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol 21, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C (2015). Defining cell types and states with single-cell genomics. Genome Res 25, 1491–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol 32, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujikawa T, Kumar S, Borkar RN, Azimi V, Thibault G, Chang YH, Balter A, Kawashima R, Choe G, Sauer D, et al. (2017). Quantitative Multiplex Immunohistochemistry Reveals Myeloid-Inflamed Tumor-Immune Complexity Associated with Poor Prognosis. Cell Rep 19, 203–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu AA, Gierahn TM, Monian B, Morgan DM, Mehta NK, Ruiter B, Shreffler WG, Shalek AK, and Love JC (2019). TCR sequencing paired with massively parallel 3’ RNA-seq reveals clonotypic T cell signatures. Nat. Immunol 20, 1692–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gassen S, Callebaut B, Van Helden MJ, Lambrecht BN, Demeester P, Dhaene T, and Saeys Y (2015). FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A 87, 636–645. [DOI] [PubMed] [Google Scholar]
- Viratham Pulsawatdi A, Craig SG, Bingham V, McCombe K, Humphries MP, Senevirathne S, Richman SD, Quirke P, Campo L, Domingo E, et al. (2020). A robust multiplex immunofluorescence and digital pathology workflow for the characterisation of the tumour immune microenvironment. Mol. Oncol 14, 2384–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner A, Regev A, and Yosef N (2016). Revealing the vectors of cellular identity with single-cell genomics. Nat. Biotechnol 34, 1145–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Way GP, Zietz M, Rubinetti V, Himmelstein DS, and Greene CS (2020). Compressing gene expression data using multiple latent space dimensionalities learns complementary biological representations. Genome Biol 21, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, and Sancho D (2020). Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol 20, 7–24. [DOI] [PubMed] [Google Scholar]
- Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang N-AAS, Andrews MC, Sharma P, Wang J, Wargo JA, Pe’er D, et al. (2017). Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 170, 1120–1133.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf FA, Angerer P, and Theis FJ (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biol 19, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu AA, Bever KM, Ho WJ, Fertig EJ, Niu N, Zheng L, Parkinson RM, Durham JN, Onners BL, Ferguson A, et al. (2020a). A Phase 2 Study of Allogeneic GM-CSF Transfected Pancreatic Tumor Vaccine (GVAX) with Ipilimumab as Maintenance Treatment for Metastatic Pancreatic Cancer. Clin. Cancer Res [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TD, Madireddi S, de Almeida PE, Banchereau R, Chen Y-JJ, Chitre AS, Chiang EY, Iftikhar H, O’Gorman WE, Au-Yeung A, et al. (2020b). Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature 579, 274–278. [DOI] [PubMed] [Google Scholar]
- Xiang H, Ramil CP, Hai J, Zhang C, Wang H, Watkins AA, Afshar R, Georgiev P, Sze MA, Song XS, et al. (2020). Cancer-Associated Fibroblasts Promote Immunosuppression by Inducing ROS-Generating Monocytic MDSCs in Lung Squamous Cell Carcinoma. Cancer Immunol. Res 8, 436–450. [DOI] [PubMed] [Google Scholar]
- Xia C, Fan J, Emanuel G, Hao J, and Zhuang X (2019). Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression. Proc Natl Acad Sci USA 116, 19490–19499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin Y, Kim J, Ni M, Wei Y, Okamoto H, Lee J, Adler C, Cavino K, Murphy AJ, Yancopoulos GD, et al. (2016). Use of the Fluidigm C1 platform for RNA sequencing of single mouse pancreatic islet cells. Proc Natl Acad Sci USA 113, 3293–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, and Su Z (2015). Identification of cell types from single-cell transcriptomes using a novel clustering method. Bioinformatics 31, 1974–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Leontovich AA, Gerdes MJ, Desai K, Dong J, Sood A, Santamaria-Pang A, Mansfield AS, Chadwick C, Zhang R, et al. (2019). Understanding heterogeneous tumor microenvironment in metastatic melanoma. PLoS ONE 14, e0216485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, McNamara KL, Granja JM, Sarin KY, Brown RA, et al. (2019). Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med 25, 1251–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, and Bar-Joseph Z (2020). GCNG: graph convolutional networks for inferring gene interaction from spatial transcriptomics data. Genome Biol 21, 300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Zhang S, Liu Y, He X, Qu M, Xu G, Wang H, Huang M, Pan J, Liu Z, et al. (2020). Single-cell RNA sequencing reveals the heterogeneity of liver-resident immune cells in human. Cell Discov 6, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C, Zheng L, Yoo J-K, Guo H, Zhang Y, Guo X, Kang B, Hu R, Huang JY, Zhang Q, et al. (2017a). Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 169, 1342–1356.e16. [DOI] [PubMed] [Google Scholar]
- Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et al. (2017b). Massively parallel digital transcriptional profiling of single cells. Nat. Commun 8, 14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Preissl S, and Ren B (2020). Single-cell multimodal omics: the power of many. Nat. Methods 17, 11–14. [DOI] [PubMed] [Google Scholar]
- Zhu X, Ching T, Pan X, Weissman SM, and Garmire L (2017). Detecting heterogeneity in single-cell RNA-Seq data by non-negative matrix factorization. PeerJ 5, e2888. [DOI] [PMC free article] [PubMed] [Google Scholar]