

Abstract
Background
Tracing of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) transmission chains is still a major challenge for public health authorities, when incidental contacts are not recalled or are not perceived as potential risk contacts. Viral sequencing can address key questions about SARS-CoV-2 evolution and may support reconstruction of viral transmission networks by integration of molecular epidemiology into classical contact tracing.
Methods
In collaboration with local public health authorities, we set up an integrated system of genomic surveillance in an urban setting, combining a) viral surveillance sequencing, b) genetically based identification of infection clusters in the population, c) integration of public health authority contact tracing data, and d) a user-friendly dashboard application as a central data analysis platform.
Results
Application of the integrated system from August to December 2020 enabled a characterization of viral population structure, analysis of 4 outbreaks at a maximum care hospital, and genetically based identification of 5 putative population infection clusters, all of which were confirmed by contact tracing. The system contributed to the development of improved hospital infection control and prevention measures and enabled the identification of previously unrecognized transmission chains, involving a martial arts gym and establishing a link between the hospital to the local population.
Conclusions
Integrated systems of genomic surveillance could contribute to the monitoring and, potentially, improved management of SARS-CoV-2 transmission in the population.
Keywords: genomic epidemiology, infection chain, community transmission, rapid sequencing, Nanopore sequencing
Tracing of SARS-CoV-2 population transmission chains is still a major challenge. We present an integrated system of genomic surveillance and show it to be capable of detecting infection chains in a large city during ongoing community transmission.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a pandemic coronavirus first detected in late 2019 [1, 2], has infected >135 million individuals and led to >2.9 million associated deaths [3]. Until the wide availability of vaccines, non-pharmaceutical interventions to limit SARS-CoV-2 transmission will continue to play an important role in pandemic management. The specific source of SARS-CoV-2 infections during community transmission, however, often remains unknown even in public health systems that operate effective contact tracing regimes (eg, for around 40% of cases in the city of Düsseldorf; Düsseldorf Health Department internal data).
Genomic epidemiology [4, 5], that is, the application of modern genomic technologies to characterize viral transmission chains [6–10], can crucially contribute to the design and evaluation of viral containment strategies. Its possible applications include the targeted investigation of putative outbreaks, for example, in hospitals [11] and care homes, as well as untargeted “surveillance sequencing” to monitor transmission dynamics and viral evolution in the population at large [12–16]. In an integrated genomic epidemiology approach, the joint analysis of surveillance, outbreak, and contact tracing data can enable the improved analysis of infection chains in the population and healthcare settings [17].
In summer 2020, we established and tested a fully integrated SARS-CoV-2 genomic epidemiology system in Düsseldorf, the capital of North Rhine Westphalia, a city of about 600 000 inhabitants in Germany’s largest metropolitan area. Our approach combined untargeted longitudinal surveillance sequencing, implemented in collaboration with a large commercial diagnostic laboratory, analysis of putative SARS-CoV-2 outbreaks from the city’s largest hospital, the integration of local public health authorities, and the development of a user-friendly dashboard to facilitate data analysis and exchange by all participating stakeholders (Figure 1).
Figure 1.

Integrated genomic surveillance in the Düsseldorf area. Population surveillance sequencing enables the characterization of local SARS-CoV-2 population structure, facilitating the discrimination between clonal hospital outbreaks (here: putative outbreak 1) or simultaneously detected but unrelated SARS-CoV-2 hospital ward cases (here: putative outbreak 2). Viral population surveillance data can also enable the de novo identification of infection clusters in the population based on the genetic data. Added value of genomic surveillance is maximized when genetic data are integrated with complementary epidemiological data or approaches, such as contact tracing or hospital outbreak data. Utilization of viral genetic data by diverse stakeholders is facilitated by providing a user-friendly real-time web application (“dashboard”) for analysis and visualization of the generated viral genomes. Abbreviation: SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
METHODS
Surveillance Sample Collection
Surveillance sample collection was implemented in collaboration with the local diagnostic laboratory Zotz | Klimas, the largest commercial SARS-CoV-2 testing laboratory in Düsseldorf. A convenience sampling approach, arbitrarily targeting 20 – 30 samples per week with Ct value <32, was implemented; sample selection was typically carried out on a single day and no metadata were used to determine sampling choices. Selected samples were shipped to the Institute of Virology at the Heinrich Heine University for amplification.
Outbreak Sample Collection
Samples from outbreaks at Düsseldorf University Hospital were collected by local clinical staff and the employee health department and sent to the Institute of Virology at the University Hospital of Düsseldorf, which is responsible for diagnostic testing of patients and staff. All 4 putative SARS-CoV-2 outbreaks identified by the hospital’s hygiene staff between September and December 2020 are included in this article. Outbreak samples were sequenced locally on the Oxford Nanopore platform or externally in collaboration with SeqIT GmbH (Kaiserslautern, Germany); see Supplementary Table 2.
Sequencing and Assembly
Full sequencing and assembly protocols for Nanopore and Illumina are specified in Supplementary Text 3. Nanopore sequencing was based on the Artic protocol [18–20].
Quality Control and Isolate Assembly Inclusion Criteria
All consensus sequences with >3000 undefined (“N”) characters were classified as low quality and excluded from all further analyses, leading to the exclusion of 21 surveillance isolate assemblies (pre-filtering: 341 surveillance assemblies; post-filtering: 320 surveillance assemblies). For the remaining isolates, higher Ct values are associated with increased numbers of “N” characters (Supplementary Figure 5).
Clade Assignment and Variant Calling
Sample clades, variants and other summary statistics were computed with the NextClade tool of NextStrain [21]. Pangolin (https://github.com/cov-lineages/pangolin) was used to screen for the presence of B.1.1.7 and B.1.351.
SARS-CoV-2 Dashboard Application
The implementation of the SARS-CoV-2 dashboard web application is described in Supplementary Text 4 and visualized in Supplementary Figure 6.
Phylogenetic and Minimum Spanning Tree Analyses
Details of phylogenetic and minimum spanning tree analyses are described in Supplementary Text 3.
Identification of Putative Population Infection Clusters
Putative infection clusters in the surveillance sequencing data were identified by greedily clustering all isolate genomes with edit distance 0, using the dashboard distance matrix (see above), and filtering for clusters with ≥4 members. All candidate clusters were manually inspected.
Integration of Contact Tracing Data
We integrated contact tracing and case information data available at and collected by Düsseldorf Health Department (Gesundheitsamt Düsseldorf). All personally identifiable information remained at Düsseldorf Health Department.
RESULTS
Genomic Surveillance in the Düsseldorf Region
In collaboration with a local diagnostic lab and employing a convenience sampling approach, we obtained 320 high-quality SARS-CoV-2 viral isolate genomes from samples collected in Düsseldorf between August and December 2020 (median: 19 samples/week). The collected genomes represented 3.1% of 10 276 newly diagnosed polymerase chain reaction (PCR)-confirmed cases during the sampling period; the proportion of sequenced cases on a weekly basis varied between 0% and 20% (Supplementary Table 1), and complete or near-complete dropout due to challenges with sampling logistics during periods of high incidence was observed for 3 weeks in Fall 2020 (Supplementary Table 1). During the last 4 weeks of the sampling period, the proportion of sequenced cases stabilized between 2% and 3%. Sequencing data, sample metadata, and assembly quality are summarized in Supplementary Tables 2 and 3. By sample genome inclusion criteria (see Methods), all included isolate genomes were of high quality (<3000Ns). In total, 80/320 isolate genome consensus sequences contained at least one ambiguous character (average: 0.48 ambiguous characters / genome), indicating potential intra-patient strain variability.
The development of viral population structure was largely consistent with the developments in Europe at large; for example, although clade 20E was initially found at low frequencies, it accounted for nearly half of the sequenced genomes towards the end of the sampling period (Figure 2C, Figure 3A). One notable exception was the absence of clade 20I/501Y.V1 (equivalent to B.1.1.7/“Alpha” in Pangolin nomenclature; https://github.com/cov-lineages/pangolin), which had already reached significant frequencies in some European countries by the end of December 2020 (eg, 48% in the UK or 15% in the Netherlands); the viral variants B.1.351/“Beta,” first identified in South Africa, and P.1/“Gamma” were also not detected. A comparison between the locally generated data and 385 109 non-Düsseldorf samples from GISAID [23] showed that many of the detected isolate genomes were not yet represented in global databases (see Supplementary Table 4 and Supplementary Text 1 for details).
Figure 2.

Local development of SARS-CoV-2 from September to December 2020. A, Newly diagnosed (red line) and sequenced (blue bars; by sample collection week) cases of SARS-CoV-2 by calendar week of 2020 in Düsseldorf. Horizontal bars indicate sample collection times for 4 hospital outbreaks on different wards (A–D) of Düsseldorf University Hospital. B, Sequenced samples by sample origin. C, Clade composition of surveillance samples by sample collection week, using the NextStrain [21] color scheme. D, Substitutions per sequenced surveillance sample and sample collection line; each dot represents one viral genome, blue line: linear fit. Abbreviation: SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
Figure 3.

A, Phylogenetic tree of the 320 surveillance samples collected during this study; colours are assigned according to the NextStrain [21] clade system. B, Joint phylogenetic tree of 44 samples from 4 hospital outbreaks (Ward A–D) and 320 surveillance samples. For a description of the outbreaks, see main text. Putative population infection clusters are highlighted in yellow (1–5). Gaps in the corresponding shaded areas correspond to related samples not identified by the greedy clustering algorithm (see Methods). Tree visualization based on iTol [22]. C, Minimum spanning tree (calculated with the Python library networkx version 2.5; visualized with Cytoscape version 3.8.2 and Inkscape version 0.92) visualization of the 4 hospital outbreaks, including all identical or near-identical (distance = 0 or distance = 1) from GISAID and the surveillance sequencing cohort. Samples from GISAID are labelled with their country of origin (Lux = Luxemburg; Net = Netherlands; Swi = Switzerland; Eng = England). The large gray circle represents a cluster of identical and near-identical GISAID samples. Solid lines without number indicate distance = 1 and dashed lines indicate distance = 0 between samples. Abbreviation: SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
SARS-CoV-2 Dashboard
To enable the independent interrogation of viral sequencing data by all involved stakeholders, we developed a user-friendly web application (“Dashboard”; see Methods; https://covgen.hhu.de and https://covgen.hhu.de/paper for the version used here). The dashboard was continuously updated with sequenced population and outbreak isolate genomes and enabled the targeted development of hypotheses about the genetic structure of outbreaks or transmission chains by hospital hygiene staff or local health authorities. Inter-sample genetic relatedness was visualized using a minimum spanning tree (MST), and the interface supported various filtering and visualization options, for example, enabling the targeted display of outbreaks and genetically related samples from the local viral population (Supplementary Figure 2). During the sampling period, the dashboard was a key tool to assist the interrogation of the genetic structure of the sequenced viral genomes.
Hospital Outbreak Analysis
We used the developed integrated system to characterize the genetic structure of 4 SARS-CoV-2 hospital outbreaks at Düsseldorf University Hospital and to search for putative transmission chains connecting the local population to the hospital. For 3 of 4 characterized outbreaks (Wards A, B, and C), sequencing (Supplementary Tables 2 and 3) confirmed the clonal structure of the outbreaks (Figure 3B, Figure 3C) within 15 to 32 days (Supplementary Table 3, Supplementary Text 2), but no links between the outbreaks and the local viral population were identified. Infection control and prevention measures that were put into place included staff re-training, improved room ventilation, and upgrades to patient protective equipment; a detailed description of the outbreaks and improved hospital infection control and prevention measures is given in Supplementary Text 2.
For 1 outbreak (Ward D) that affected 16 patients and 13 healthcare workers in mid-December, sequencing confirmed the clonal nature of the outbreak and identified 2 potential links between the outbreak and the local population (Figure 3C); first, analyzed sequencing data were available within 26 days after the detection of the outbreak. For a detailed analysis of the outbreak samples in the context of the Düsseldorf surveillance data, we split the outbreak samples into 2 subgroups. Subgroup-01 represented the majority viral type of the outbreak, and all samples within Subgroup-01 were genetically identical; Subgroup-02 represented samples with a distance (see Supplementary Text 1) of 1 single-nucleotide polymorphism (SNP) to Subgroup-01. An analysis of the surveillance data showed that the viral type of Subgroup-01 was present in the local population as early as 1 October (sample Z164; distance to majority of outbreak samples = 0) and that the viral type of Subgroup-02 was detected again in the surveillance data, although the outbreak was ongoing (sample Z794, sampled on 16 December). Contact tracing data collected by Düsseldorf public health authorities showed that a family member of the individual that sample Z164 was taken from was treated at another ward of the clinical department of Düsseldorf University Hospital in October 2020, establishing a possible link between the surveillance samples and the hospital outbreak on ward “D”; a SARS-CoV-2 antibody test of this family member was weakly positive for immunoglobulin A (IgA). Contact tracing data also showed that a close relative of the individual who provided sample Z794 was treated at a coronavirus disease 2019 (COVID-19) ward of Düsseldorf University Hospital, to which SARS-CoV-2-positive cases from ward “D” had been transferred to. Interestingly, 5 highly related isolates, identical (distance = 0) to samples part of the ward “D” outbreak, were also identified in Switzerland (GISAID IDs 796188, 728868, 737730 768145, 737914; sampled between 9 November and 28 December). Near-identical samples (distance = 1) were also found in the Netherlands and the United Kingdom (Supplementary Table 4). Implemented infection control and prevention measures included comprehensive screening, a temporary stop of admissions to the ward, and re-education of staff in nonpharmaceutical interventions. Further details on the Ward D outbreak are presented in Supplementary Text 2.
Genetically Based Identification of Population Infection Clusters
To evaluate whether untargeted surveillance sequencing data could enable the genetically based identification of infection clusters and transmission chains in the population during community transmission, we applied a simple greedy clustering algorithm (Methods) to the viral sequence surveillance data, identifying groups of genetically identical samples. This analysis identified 5 putative infection clusters within the Düsseldorf samples (Figure 3B; Supplementary Table 5). Routine public health authority contact tracing data were subsequently integrated and showed that the identified clusters reflected epidemiologically relevant associations.
PopClust#1 consisted of 7 patient samples collected in late August; contact tracing data revealed that this cluster corresponded to a known transmission event during a school excursion in August 2020.
PopClust#2 consisted of 6 patient samples collected in mid-/late September; of these, 3 were linked via a care home, 2 were collected from members of the same family, and the remaining sample was linked to an otherwise unrelated primary school outbreak.
PopClust#4 and PopClust#5, consisting of 4 and 5 patient samples collected in November and December, respectively, contained samples that were linked via joint household membership, as well as samples without any obvious connections to the identified households.
Finally, investigation of PopClust#3, representing five samples collected in early October, enabled the discovery of a previously unrecognized population transmission chain. For samples Z132 and Z177, a reanalysis of the originally collected contact tracing data pointed to a connection between the cases involving another positively tested individual X (not part of the study): (Z132, X) were connected via active membership of the same martial arts gym, and (X, Z177) had a close personal relationship. Z132 and X had not identified each other as direct contacts; the identified putative link between Z132 and X was further supported by the timing of the infections and highlights the potential for fomite and/or aerosol transmission in martial arts contexts. Two additional samples of PopClust#3 were members of the same household, and for the remaining sample in PopClust#3, no obvious connection to the other samples was identified.
Sample ID and contact tracing information for the identified clusters are summarized in Supplementary Table 5.
Rapid Nanopore Sequencing Experiment
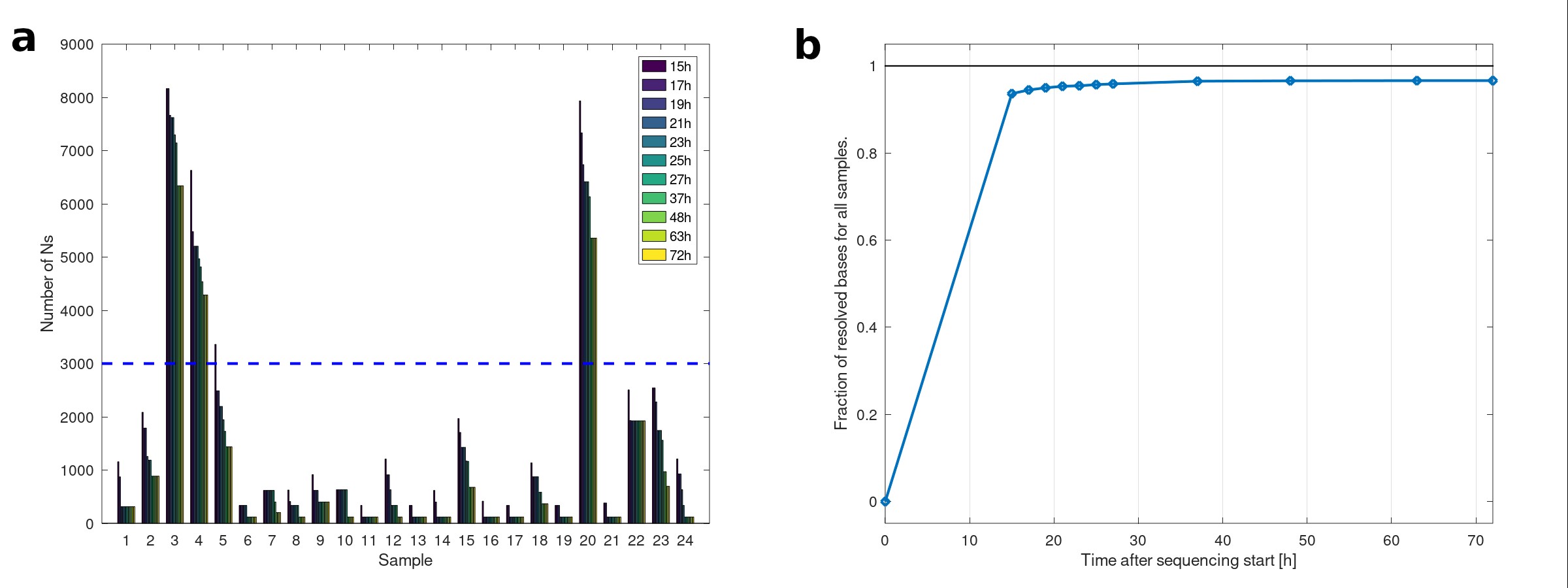
To investigate whether a rapid viral surveillance sequencing workflow could be implemented, we measured total turnaround time for 24 samples with different cycle threshold (Ct) values (range 17–31) from the surveillance cohort from sample receipt to bioinformatic analysis when using streamlined workflows and processes (Supplementary Text 3). After a total time of 28 hours (11 hours for sample and library preparation, 15 hours for sequencing, and ≤2 hours for bioinformatic analysis; Supplementary Figure 3), 19 of 24 genomes were resolved to high quality (<3000 N positions; Supplementary Figure 4A). Increasing the sequencing time by at least 2 hours increased the number of high-quality resolved genomes to 20; the number of resolved bases across all samples saturated after 37 hours of sequencing (Supplementary Figure 4B).
DISCUSSION
An improved understanding of SARS-CoV-2 transmission chains is key to effective viral containment. We have shown that an integrated local SARS-CoV-2 genomic epidemiology system implemented in the state capital city of Düsseldorf could enable the retrospective detection of SARS-CoV-2 infection chains through hospitals and the local population during ongoing community transmission. We could confirm the clonal nature of 4 outbreaks in a regional maximum care hospital and contribute to the design and implementation of refined infection control intervention measures, minimizing the risk of nosocomial SARS-CoV-2 transmission (see Supplementary Text 3). We also developed a simple algorithmic approach to identify putative infection clusters in the local population based on genetic data alone and found 5 such clusters in the generated surveillance data from August to December 2020. Integration with contact tracing data showed that the untargeted sequencing data captured epidemiologically relevant viral transmissions in settings of societal importance, such as care homes, schools, and recreational physical activities. Intriguingly, we found 2 potential links between the local population and a hospital outbreak and identified a previously unrecognized population transmission chain in a martial arts gym, confirming the potential utility of untargeted sequencing for identification of transmission chains in the population during ongoing community transmission. Key features of our approach include the joint analysis of population and outbreak sequencing data, the integration of genetic data with contact tracing data, and the availability of a user-friendly dashboard as a central data analysis platform for all participating stakeholders.
When applied at scale, fully integrated local genomic epidemiology systems could contribute to a more effective management of the SARS-CoV-2 pandemic on multiple levels. First, significant uncertainties remain with respect to the relative importance of viral transmission in various settings of societal importance, such as restaurants, public transport, childcare facilities, or schools. Routine large-scale untargeted surveillance sequencing could contribute to a more quantitative understanding of transmission risks in such settings and thus enable the design of improved infection prevention measures. Second, as shown here, genomic epidemiology can enable the identification of large infection clusters and superspreading events in the population, which play an important role in driving the pandemic [24]. Third, genetically characterizing a large proportion of local cases may enable locally adapted strategies for containing the spread of variants of concern [25]. Fourth, genomic epidemiology could increase accountability for the prevention and management of infection chains at the individual or organizational level, as sequencing can provide supporting evidence as well as evidence against individual transmissions or transmission contexts suggested by classical epidemiological approaches. A simple model calculation (see Supplementary Table 6 for an interactive Excel sheet) shows that a strategy of sequencing all positive cases would increase the total cost of the testing system by only 30–40%. Although not insignificant, these additional costs could potentially be offset by the economic benefits of improved pandemic management; as the current costs of lockdown-like measures in many countries are significant (eg, estimated at EUR 25–75 billion per week for Germany; see Florian et al [26]), even a small improvement could translate into overall cost-effectiveness.
Important remaining challenges include the logistics of achieving turnaround times compatible with public health authority decision making [27], as well as the downstream integration of viral sequencing data. As we and others have demonstrated, the Nanopore technology can enable turnaround times as low as 28 hours for SARS-CoV-2 [17, 28, 29]. Further improvements may be possible in the fields of sample logistics, real-time control of sequencing runs (eg, with RAMPART; https://artic.network/rampart), and streaming data analysis. The developed SARS-CoV-2 dashboard with automatic detection of infection clusters could be a first step toward a more routine integration of viral sequencing into the processes of local public health authorities; further work, however, is required, for example, for integration with classical contact tracing software. In addition, the simple greedy algorithm used here to identify candidate population infection clusters could be improved by population genetics modeling [30, 31], as well as by the incorporation of additional dimensions such as sampling time.
Limitations of this study include the utilized convenience sampling scheme, potentially associated with a biased selection of samples; the relatively low proportion of sequenced positive cases over the sampling period, potentially limiting generalizability; the retrospective nature of the study, limiting the potential actionability of the generated surveillance data from the perspective of public health; and the fact that intervention based on genetic data was limited to the investigated hospital outbreaks.
Based on the methods and results developed here, these limitations are currently addressed in a follow-up study started in Spring 2021. Representing a natural extension of the work presented here, the follow-up study will enable a further investigation of the full potential of “real-time” genomic surveillance for supporting public health decision making, based on an improved characterization of pathogen transmission chains in the population at large. Preliminary results (n = 4260) of the study show that case sequencing rates of over 50% can be achieved even at 7-day newly diagnosed case incidence rates >150 per 100 000 population; that “swab-to-sequence” times of <72 hours can be achieved in routine sequencing setups; and that real-time integration of these sequencing data can enable the routine detection of transmission chains in settings such as care homes, primary schools and families, consistent with the results presented here. Although a full analysis will be presented after the conclusion of the follow-up study in late 2021, a continuously updated view of local viral population structure and putative infection clusters is provided via the publicly available dashboard web application presented here (https://covgen.hhu.de).
Supplementary Data
Supplementary materials are available at Clinical Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
{kind=link}
{kind=link}
{kind=link}
Notes
Author Contributions. A. W., T. H., P. F., and L. E. contributed to the design of the study and contributed to the data collection, data analysis, data interpretation, literature review, and contributed to the writing of the manuscript. A. T. and J. N. contributed to the data collection, data analysis, data interpretation, figures, and contributed to the writing of the manuscript. M. D., D. S., A. D., T. W., T. T., D. K. M., L. H., K. H., N. L., S. H., M. A., M. P. D., A. T., and S. K.-B. contributed to the data collection, data analysis, and data interpretation. J. T., K. P., R. Z., and K. G. contributed to the design and supervision of the study and to the writing of the manuscript. A. T. D. contributed to the design and the supervision of the study, to data collection, data analysis, data interpretation, literature review, and wrote the first draft of the manuscript. All authors reviewed and approved the final manuscript.
Deutsche COVID-19 Omics Initiative (DeCOI)
Janine Altmüller, Angel Angelov, Anna C Aschenbrenner, Robert Bals, Alexander Bartholomäus, Anke Becker, Daniela Bezdan, Michael Bitzer, Helmut Blum, Ezio Bonifacio, Peer Bork, Nicolas Casadei, Thomas Clavel, Maria Colome-Tatche, Inti Alberto De La Rosa Velázquez, Andreas Diefenbach, Alexander Dilthey, Nicole Fischer, Konrad Förstner, Sören Franzenburg, Julia-Stefanie Frick, Gisela Gabernet, Julien Gagneur, Tina Ganzenmüller, Marie Gauder, Alexander Goesmann, Siri Göpel, Adam Grundhoff, Hajo Grundmann, Torsten Hain, André Heimbach, Michael Hummel, Thomas Iftner, Angelika Iftner, Stefan Janssen, Jörn Kalinowski, René Kallies, Birte Kehr, Andreas Keller, Oliver Keppler, Sarah Kim-Hellmuth, Christoph Klein, Michael Knop, Oliver Kohlbacher, Karl Köhrer, Jan Korbel, Peter G Kremsner, Denise Kühnert, Ingo Kurth, Markus Landthaler, Yang Li, Kerstin Ludwig, Oliwia Makarewicz, Manja Marz, Alice McHardy, Christian Mertes, Maximilian Münchhoff, Sven Nahnsen, Markus Nöthen, Francine Ntoumi, Peter Nürnberg, Uwe Ohler, Stephan Ossowski, Jörg Overmann, Silke Peter, Klaus Pfeffer, Anna R Poetsch, Ulrike Protzer, Alfred Pühler, Nikolaus Rajewsky, Markus Ralser, Olaf Rieß, Stephan Ripke, Ulisses Rocha, Philip Rosenstiel, Emmanuel Saliba, Leif Erik Sander, Birgit Sawitzki, Simone Scheithauer, Philipp Schiffer, Jonathan Schmid-Burgk, Wulf Schneider, Eva-Christina Schulte, Joachim Schultze, Alexander Sczyrba, Mariam L Sharaf, Yogesh Singh, Michael Sonnabend, Oliver Stegle, Jens Stoye, Fabian Theis, Janne Vehreschild, Thirumalaisamy P Velavan, Jörg Vogel, Max von Kleist, Andreas Walker, Jörn Walter, Dagmar Wieczorek, Sylke Winkler, John Ziebuhr.
Acknowledgments. The authors acknowledge Jeff Barrett for discussions and for introducing the notion of clonality / heterozygosity as a useful summary statistic of surveillance sequencing data. They would like to thank the hospital hygiene staff and the employee health department medical officers for their support.
The authors gratefully acknowledge the authors, the originating and submitting laboratories for their sequence and metadata shared through GISAID. All submitters of data may be contacted directly via GISAID. The Acknowledgments Table for GISAID is part of Supplementary Table 7. Computational support and infrastructure were provided by the “Centre for Information and Media Technology” (ZIM) at the University of Düsseldorf (Germany). Sequencing and the design of sequencing workflows were supported by the Biologisch-Medizinisches Forschungszentrum der Heinrich Heine University Düsseldorf (BMFZ). Viral genome sequences are available on GISAID. Accessions are listed in Supplementary Table 3. All source code for the dashboard is available under the MIT license and can be downloaded from https://github.com/DiltheyLab/SARS-CoV2-Dashboard-Releases. The version of the dashboard and the data for the analyses presented here are archived under https://covgen.hhu.de/paper. This study was approved by the ethics committee of the Medical Faculty of Heinrich Heine University Düsseldorf (#2020–839).
Financial support. Jürgen Manchot Foundation German Research Foundation award number 428994620 German Federal Ministry of Education and Research award numbers 031L0184B and B-FAST (grant number 01KX2021).
Potential conflicts of interests. The authors: No reported conflicts of interest.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest.
Contributor Information
German COVID-19 OMICS Initiative (DeCOI):
Janine Altmüller, Angel Angelov, Anna C Aschenbrenner, Robert Bals, Alexander Bartholomäus, Anke Becker, Daniela Bezdan, Michael Bitzer, Helmut Blum, Ezio Bonifacio, Peer Bork, Nicolas Casadei, Thomas Clavel, Maria Colome-Tatche, Inti Alberto De La Rosa Velázquez, Andreas Diefenbach, Alexander Dilthey, Nicole Fischer, Konrad Förstner, Sören Franzenburg, Julia-Stefanie Frick, Gisela Gabernet, Julien Gagneur, Tina Ganzenmüller, Marie Gauder, Alexander Goesmann, Siri Göpel, Adam Grundhoff, Hajo Grundmann, Torsten Hain, André Heimbach, Michael Hummel, Thomas Iftner, Angelika Iftner, Stefan Janssen, Jörn Kalinowski, René Kallies, Birte Kehr, Andreas Keller, Oliver Keppler, Sarah Kim-Hellmuth, Christoph Klein, Michael Knop, Oliver Kohlbacher, Karl Köhrer, Jan Korbel, Peter G Kremsner, Denise Kühnert, Ingo Kurth, Markus Landthaler, Yang Li, Kerstin Ludwig, Oliwia Makarewicz, Manja Marz, Alice McHardy, Christian Mertes, Maximilian Münchhoff, Sven Nahnsen, Markus Nöthen, Francine Ntoumi, Peter Nürnberg, Uwe Ohler, Stephan Ossowski, Jörg Overmann, Silke Peter, Klaus Pfeffer, Anna R Poetsch, Ulrike Protzer, Alfred Pühler, Nikolaus Rajewsky, Markus Ralser, Olaf Rieß, Stephan Ripke, Ulisses Rocha, Philip Rosenstiel, Emmanuel Saliba, Leif Erik Sander, Birgit Sawitzki, Simone Scheithauer, Philipp Schiffer, Jonathan Schmid-Burgk, Wulf Schneider, Eva-Christina Schulte, Joachim Schultze, Alexander Sczyrba, Mariam L Sharaf, Yogesh Singh, Michael Sonnabend, Oliver Stegle, Jens Stoye, Fabian Theis, Janne Vehreschild, Thirumalaisamy P Velavan, Jörg Vogel, Max von Kleist, Andreas Walker, Jörn Walter, Dagmar Wieczorek, Sylke Winkler, and John Ziebuhr
References
- 1. Wu F, Zhao S, Yu B, et al. . A new coronavirus associated with human respiratory disease in China. Nature 2020; 579:265–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhou P, Yang XL, Wang XG, et al. . A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020; 579:270–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. World Health Organization. WHO COVID-19 Weekly Epidemiological Update (11 April 2021). Available at: https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---13-april-2021. Accessed 26 January 2021.
- 4. Gardy JL, Loman NJ. Towards a genomics-informed, real-time, global pathogen surveillance system. Nat Rev Genet 2018; 19:9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grubaugh ND, Ladner JT, Lemey P, et al. . Tracking virus outbreaks in the twenty-first century. Nat Microbiol 2019; 4:10–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Quick J, Loman NJ, Duraffour S, et al. . Real-time, portable genome sequencing for Ebola surveillance. Nature 2016; 530:228–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Grubaugh ND, Ladner JT, Kraemer MUG, et al. . Genomic epidemiology reveals multiple introductions of Zika virus into the United States. Nature 2017; 546:401–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gonzalez-Reiche AS, Hernandez MM, Sullivan M, et al. . Introductions and early spread of SARS-CoV-2 in the New York city area. medRxiv 2020: 2020.04.08.20056929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lu J, du Plessis L, Liu Z, et al. . Genomic epidemiology of SARS-CoV-2 in Guangdong province, China. Cell 2020; 181:997–1003 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Popa A, Genger JW, Nicholson MD, et al. . Genomic epidemiology of superspreading events in Austria reveals mutational dynamics and transmission properties of SARS-CoV-2. Sci Transl Med 2020; 12:eabe2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. MacFadden DR, McGeer A, Athey T, et al. . Use of genome sequencing to define institutional influenza outbreaks, Toronto, Ontario, Canada, 2014–15. Emerg Infect Dis 2018; 24:492–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. COVID-19 Genomics UK (COG-UK) consortium. An integrated national scale SARS-CoV-2 genomic surveillance network. Lancet Microbe 2020; 1:e99–e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Geoghegan JL, Ren X, Storey M, et al. . Genomic epidemiology reveals transmission patterns and dynamics of SARS-CoV-2 in Aotearoa New Zealand. Nat Commun 2020; 11:6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pattabiraman C, Habib F, P K H, et al. . Genomic epidemiology reveals multiple introductions and spread of SARS-CoV-2 in the Indian state of Karnataka. PLoS One 2020; 15:e0243412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Oude Munnink BB, Nieuwenhuijse DF, Stein M, et al. ; Dutch-Covid-19 response team . Rapid SARS-CoV-2 whole-genome sequencing and analysis for informed public health decision-making in the Netherlands. Nat Med 2020; 26:1405–10. [DOI] [PubMed] [Google Scholar]
- 16. Gudbjartsson DF, Stefansson K. Early spread of SARS-CoV-2 in the Icelandic population. Reply. N Engl J Med 2020; 383:2184–5. [DOI] [PubMed] [Google Scholar]
- 17. Meredith LW, Hamilton WL, Warne B, et al. . Rapid implementation of SARS-CoV-2 sequencing to investigate cases of health-care associated COVID-19: a prospective genomic surveillance study. Lancet Infect Dis 2020; 20:1263–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Quick J. ARTIC amplicon sequencing protocol for MinION for nCoV-2019. Available at: https://dx.doi.org/10.17504/protocols.io.bbmuik6w. Accessed 20 June 2021. [Google Scholar]
- 19. Quick J, Grubaugh ND, Pullan ST, et al. . Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat Protoc 2017; 12:1261–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tyson JR, James P, Stoddart D, et al. . Improvements to the ARTIC multiplex PCR method for SARS-CoV-2 genome sequencing using nanopore. bioRxiv 2020. doi:10.1101/2020.09.04.283077 [Google Scholar]
- 21. Hadfield J, Megill C, Bell SM, et al. . Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 2018; 34:4121–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 2016; 44:W242–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shu Y, McCauley J. GISAID: Global initiative on sharing all influenza data: from vision to reality. Euro Surveill 2017; 22:30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Laxminarayan R, Wahl B, Dudala SR, et al. . Epidemiology and transmission dynamics of COVID-19 in two Indian states. Science 2020; 370:691–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rambaut A, Loman N, Pybus O, et al. . Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. Available at: https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563. Accessed 20 June 2021.
- 26. Florian D, Clemens F, Marcell G, et al. . Die volkswirtschaftlichen Kosten des corona-shutdown für Deutschland: eine szenarienrechnung. ifo Schnelldienst 2020; 73:29–35. [Google Scholar]
- 27. Köser CU, Holden MT, Ellington MJ, et al. . Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak. N Engl J Med 2012; 366:2267–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Greninger AL, Naccache SN, Federman S, et al. . Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis. Genome Med 2015; 7:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Quick J, Ashton P, Calus S, et al. . Rapid draft sequencing and real-time nanopore sequencing in a hospital outbreak of Salmonella. Genome Biol 2015; 16:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bouckaert R, Heled J, Kühnert D, et al. . BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol 2014; 10:e1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Drummond AJ, Rambaut A, Shapiro B, Pybus OG. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol 2005; 22:1185–92. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.