

Abstract
The rapid spread of SARS-CoV-2 infection around the globe has caused a massive health and socioeconomic crisis. Identification of phosphorylation sites is an important step for understanding the molecular mechanisms of SARS-CoV-2 infection and the changes within the host cells pathways. In this study, we present DeepIPs, a first specific deep-learning architecture to identify phosphorylation sites in host cells infected with SARS-CoV-2. DeepIPs consists of the most popular word embedding method and convolutional neural network-long short-term memory network architecture to make the final prediction. The independent test demonstrates that DeepIPs improves the prediction performance compared with other existing tools for general phosphorylation sites prediction. Based on the proposed model, a web-server called DeepIPs was established and is freely accessible at http://lin-group.cn/server/DeepIPs. The source code of DeepIPs is freely available at the repository https://github.com/linDing-group/DeepIPs.
Keywords: SARS-CoV-2, phosphorylation, word embedding, CNN, LSTM
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a highly transmissible and pathogenic coronavirus that emerged in late 2019 and has caused a pandemic of acute respiratory disease, named ‘coronavirus disease 2019’ (COVID-19), which presents a massive health and socioeconomic crisis [1, 2]. To devise therapeutic strategies to conquer SARS-CoV-2 infection and the associated COVID-19 pathology, it is urgent to develop new drugs and repurpose existing ones to dampen the disease course and reduce the burden of medical institutions [3]. As of 2 October 2020, there were about 405 therapeutic drugs in development for COVID-19 but mostly remain computational without tests in infection models [4]. Comprehensive understanding of the molecular mechanisms of SARS-CoV-2 infection and the changes within the host cell pathways is essential to rationally repurpose drugs [5].
Proteomics approaches are powerful tools to elucidate mechanisms of pathogenesis by quantifying changes in protein abundance and phosphorylation [6]. For instance, Stukalov et al. [7] characterized interactome, proteome and signaling process in a systems wide manner to study the relationship of SARS-CoV-2 and host cells. Bouhaddou et al. [8] presented a quantitative mass spectrometry-based phosphoproteomics survey of SARS-CoV-2 infection in Vero E6 cells to reveal dramatic rewiring of phosphorylation on host and viral proteins. Klann et al. [5] used a SARS-CoV-2 infection system in Caco-2 human cells to study signaling changes by phosphoproteomics. Hekman et al. [9] performed a quantitative phosphoproteomics survey of SARS-CoV-2 infection in iAT2 cells to exploit the mechanisms driving infection and pathology. The high-throughput Mass Spectrometry techniques used in the above studies can annotate phosphorylation sites accurately, therefore accumulating a large number of phosphorylation examples. However, traditional experimental methods are labor-intensive and time-consuming especially applied in verifying huge amounts of candidate phosphorylation sites. Alternately, as a complementary technique to traditional experimental strategies, the computational approach is a better choice.
To date, a considerable number of predictors for identifying phosphorylation sites have been proposed. Most of them show a common strategy that can be summarized as two steps: (i) to encode original sequence based on artificially designed feature extraction method and (ii) to choose an optimized machine learning algorithm for classification and prediction. For example, PhosPred-RF used information theory feature, overlapping property feature, 20-bit features, 21-bit features and Skip-n-gram features, trained by random forest-based algorithm for phosphorylation sites prediction [10]. Quokka applied a variety of sequence scoring functions combined with an optimized logistic regression algorithm for the prediction of phosphorylation sites [11]. GPS 5.0 utilized two novel methods named position weight determination and scoring matrix optimization followed by logistic regression algorithm to identify phosphorylation sites [12]. Although features involved in these methods achieved good performance phosphorylation sites predictions, there is limitation of ‘feature engineering’, which requires artificially design that may result in biased features [13].
One promising and attractive solution for such a challenge is the deep-learning-based approach. Compared with the cumbersome ‘feature engineering’ of conventional machine-learning techniques, deep-learning shows a distinctive advantage. It can automatically generate complex patterns and capture the high-level abstraction adaptively from the training data. Based on these, several deep-learning-based models have been proposed for phosphorylation sites identification. For example, MusiteDeep took raw sequence data as input and used convolutional neural networks (CNNs) with a novel two-dimensional attention mechanism for predicting phosphorylation sites [13]. CapsNet introduced a capsule network with multi-layer CNN for protein post-translational modification site identification and presented some outstanding properties of capsules in characterizing biologically meaningful features [14]. DeepPSP designed a global–local information-based deep neural network for the prediction of phosphorylation sites [15]. These approaches using only raw sequence have shown superior to the previous traditional machine learning methods. However, there is no specific deep-learning architecture to identify phosphorylation sites in host cells infected with SARS-CoV-2.
Here, we present a novel CNN-long short-term memory network (LSTM) architecture, DeepIPs, to accurately predict phosphorylation sites in host cells infected with SARS-CoV-2 (Figure 1). Different from aforementioned deep-learning methods, DeepIPs uses word embedding approaches in natural language processing to obtain protein sequence representation, which avoids the limitation of ‘feature engineering’ and effectively improves the performance of the model. To evaluate the performance of DeepIPs, we built different independent datasets to assess the model. The evaluation results reveal that the robust representations generated by word embedding and CNN-LSTM architecture have a strong discriminant power in recognizing general phosphorylation sites. We believe that the proposed architecture can also address other bioinformatics problems better than previous methods. In addition, our study provides an early example use-case of popular word embedding methods in biological sequence analysis and may shed light on other biological prediction problems.
Figure 1 .
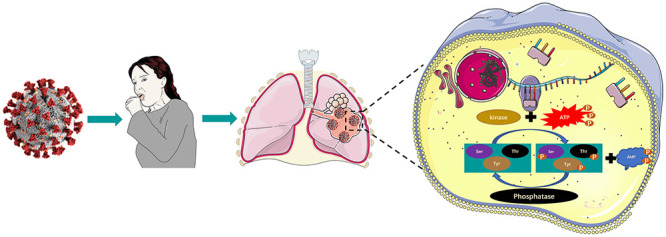
Schematic diagram of the change process of phosphorylation modification levels in vivo after host cells are infected with SARS-CoV-2.
Materials and methods
Benchmark dataset construction
In this study, the experimentally verified phosphorylation sites of human A549 cells infected with SARS-CoV-2 were collected from literature [7]. The dataset included 14 119 phosphorylation sites. To reduce the sequence redundancy of phosphorylation proteins and avoid model overfitting, the CD-HIT program [16] was used with the sequence identity threshold of 30%. To facilitate comparison with other existing methods on phosphorylation site prediction, the processed sequences were truncated into 33-residue-long sequence segments with S/T or Y located at the center. A segment was defined as a positive sample if its central S/T or Y was phosphorylation; otherwise, it was defined as a negative sample. As a result, a great number of negative samples were obtained. To balance the positive and negative data, we randomly selected a subset of non-redundant negative samples to match the number of positive samples [17–19]. After doing all of these, 5387 positive samples and 5387 negative samples of S/T sites, 102 positive samples and 102 negative samples of Y sites were obtained. Meanwhile, a common used performance evaluation strategy in deep-learning frameworks for sequence analysis was adopted in this study, which separates the dataset into strictly non-overlapping training set and independent testing set randomly in a ratio of 8:2 [20]. The detailed description of data is listed in Table 1.
Table 1.
Phosphorylation data collected in this study. The benchmark datasets of S/T sites and Y sites were divided into training set and independent testing set randomly in a ratio of 8:2, respectively
| Data type | Residue type | Positive samples | Negative samples |
|---|---|---|---|
| Training | S/T | 4308 | 4308 |
| Y | 81 | 81 | |
| Testing | S/T | 1079 | 1079 |
| Y | 21 | 21 |
The representation of proteins with word embedding vectors
Word embedding is a set of techniques in natural language processing in which words from a vocabulary are represented as vectors using a large corpus of text as the input. Our previous study has demonstrated that word embedding method which convert each amino acid (aa) into a fixed-length vector of a defined size along with reduced feature dimensions can produce satisfactory prediction performance [21]. Thus, in this study, two strategies were implemented to encode protein sequences: one is a supervised embedding layer (SEL); another is an unsupervised embedding layer based on pre-trained word embedding methods such as Word2Vec [22], GloVe [23] and fastText [24, 25]. Details were described as follows.
Supervised embedding layer
The essence of embedding layer in Keras [26] is a fully connected neural network, which turn positive
integers (indexes) into dense vectors of fixed size. For a given protein
sequence, a fixed-length digital vector was generated by replacing the amino
acids with their corresponding encoders. If the length is less than
‘max_length’, we used function
‘pad_sequence’ to amplify the length of
protein sequence to 200 aa. By doing this, a protein sequence is converted
to a sparse vector with many zeros. However, this ordinary encoding scheme
cannot reflect the relationship between protein residues and their
sequential and spatial neighbors. Thus, we used embedding layer to map amino
acids to dense vectors by simulating protein sequences as documents and
amino acids as words [27]. The
semantic similarity between two arbitrary amino acids learned from
large-scale sequences allows us to use the continuous metric notions of
similarity to assess the semantic quality of individual amino acids.
Embedding an amino acid can be done by multiplying the one-hot vector from
the left with a weight matrix 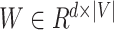 , where
, where
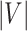 is the number of
unique amino acids and
is the number of
unique amino acids and 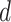 is the
embedding size. Supporting that
is the
embedding size. Supporting that 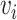 is the
one-hot vector of an amino acid
is the
one-hot vector of an amino acid 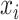 in a
given protein sequence
in a
given protein sequence 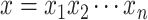 , the
embedding of
, the
embedding of 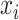 can be represented
as follows:
can be represented
as follows:
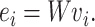 |
(1) |
The weight matrix is randomly initialized and updated in a back-propagation
fashion. After the embedding layer, an input sequence can be presented by a
dense matrix 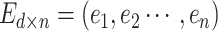 .
.
Word2Vec
Word2Vec is a machine learning model based on feed-forward neural network
that can be used to generate vector representations of words in a text and
has been widely used in bioinformatics problems [28–31]. The basic idea for
training such a model is to assign similar vector representations to words
in similar contexts according to word proximity collected from a large
corpus of documents. Here, we utilized Word2Vec to train a distributed
representation and embedding for protein sequences. We considered
subsequences of fixed-length 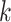 as amino
acid ‘word’ (also referred to as
as amino
acid ‘word’ (also referred to as 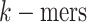 ). The
collection of all possible
). The
collection of all possible 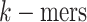 was defined
as the vocabulary (size of vocabulary =
was defined
as the vocabulary (size of vocabulary = 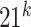 ). We then used a
). We then used a
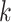 sized sliding window to
scan protein sequence as well as its flanking region with step size 1. After
protein sequences and their flanking regions were built, we adopted CBOW
model which has the advantage over the skip-gram model of uniformly
organizing the information distributed in the dataset to pre-train the
embedding layer. The CBOW model aims to predict the current word using a few
surrounding context words. There are three layers in the model: the input
layer, hidden layer and output layer.
sized sliding window to
scan protein sequence as well as its flanking region with step size 1. After
protein sequences and their flanking regions were built, we adopted CBOW
model which has the advantage over the skip-gram model of uniformly
organizing the information distributed in the dataset to pre-train the
embedding layer. The CBOW model aims to predict the current word using a few
surrounding context words. There are three layers in the model: the input
layer, hidden layer and output layer. 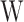 and
and
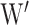 are the shared input weight matrix and output weight matrix, respectively.
The input layer of the model is a word vector. Since the CBOW model produces
the target word through
are the shared input weight matrix and output weight matrix, respectively.
The input layer of the model is a word vector. Since the CBOW model produces
the target word through 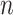 predictions
before and after the target word as shown in Eq. (2), the target function of
the model can be easily obtained as follows:
predictions
before and after the target word as shown in Eq. (2), the target function of
the model can be easily obtained as follows:
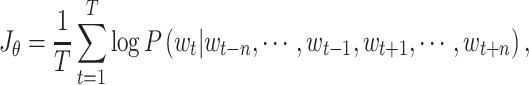 |
(2) |
where
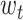 is target word and
is target word and
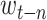 , …,
, …,
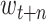 represent the
context words. Because the hidden layer does not involve any non-linear
transformation, it can be regarded as a softmax layer, so that
represent the
context words. Because the hidden layer does not involve any non-linear
transformation, it can be regarded as a softmax layer, so that
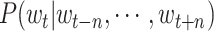 can be defined by
can be defined by
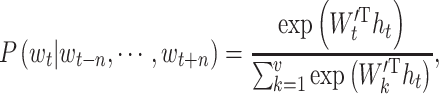 |
(3) |
where
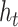 is the value of the
input word vector mapped to the hidden layer vector. The input vector is
first subjected to matrix operation with the matrix
is the value of the
input word vector mapped to the hidden layer vector. The input vector is
first subjected to matrix operation with the matrix
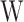 , and the
average value of all input vectors after matrix operation is obtained to
obtain
, and the
average value of all input vectors after matrix operation is obtained to
obtain 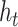 ;
;
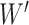 is the
hidden layer to the output weight matrix between layers.
is the
hidden layer to the output weight matrix between layers.
After training the CBOW model, the optimized parameters were transferred as the initial weights of the embedding layer, and fine tuning is done with the subsequent layers together under the supervision of the label of fragments. In our work, the Word2Vec was implemented with genism 3.8.0.
GloVe
GloVe is an unsupervised learning algorithm to produce vector representations of words. Learning is performed in global word-word co-occurrence statistics counted from a corpus [23]. The GloVe model learns items on the non-zero entries of a global word-word co-occurrence matrix, which shows how frequently words co-occur in the given corpus in a table. In general, the number of matrix entries that are non-zero is much smaller than the total number of words in the corpus. Thus, the loss function based on the weighted least-squares regression model converges faster
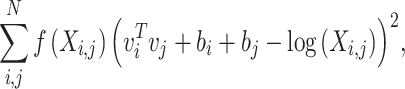 |
(4) |
where
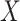 is a word co-occurrence
matrix,
is a word co-occurrence
matrix, 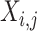 is the
frequency of word
is the
frequency of word 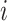 co-occurring with word
co-occurring with word
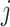 and
and
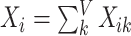 is
the total number of occurrences of word
is
the total number of occurrences of word 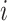 in the corpus. The
probability of word
in the corpus. The
probability of word 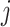 that occurs in the
context of word
that occurs in the
context of word 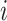 is
is
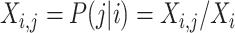 .
.
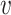 is word embedding,
is word embedding,
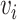 and
and
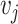 are the word
vectors of word
are the word
vectors of word 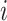 and word
and word
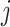 ,
,
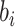 and
and
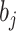 are constant terms,
are constant terms,
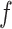 is the weight function
and
is the weight function
and 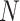 is the size of the
vocabulary. In our experiment, we set the vector size to 100 and the window
size to 15.
is the size of the
vocabulary. In our experiment, we set the vector size to 100 and the window
size to 15.
fastText
fastText is a library created by the Facebook Research Team that allows us to
create an unsupervised learning algorithm for obtaining vector
representations for words [32]. The
model utilizes low-rank matrix to reduce the computation burden while
sharing parameters among features and classes. This is especially useful in
the case of large output space, where rare classes may have only a few
training examples [25]. fastText
uses architecture similar to the CBOW model, which minimizes the softmax
loss 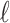 over
over
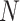 documents
documents
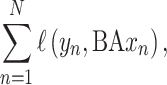 |
(5) |
where
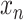 is a bag of one-hot
vectors and
is a bag of one-hot
vectors and 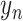 is the label of the
is the label of the
 document.
Unlike Word2Vec and GloVe, which are based on word-level representation,
fastText uses a smaller unit of character level to obtain word
representation. In this study, we implemented a bag of 1 g to capture
some partial information about the local word order.
document.
Unlike Word2Vec and GloVe, which are based on word-level representation,
fastText uses a smaller unit of character level to obtain word
representation. In this study, we implemented a bag of 1 g to capture
some partial information about the local word order.
Architecture design
Here, we presented a hybrid deep-learning architecture consisted of CNNs followed by a LSTM layer, where CNNs were used to extract high-level motif features and LSTM was used to learn long-range dependencies (Figure 2). The details of the architecture are as follows:
Figure 2 .
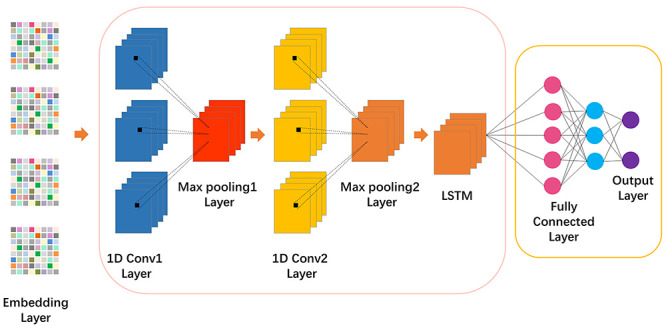
Visualization of the detailed architecture of DeepIPs. The input of DeepIPs is four different word embedding methods. The protein sequences are encoded as vectors that are fed into CNN-LSTM block. The convolution block was used for initial feature extraction and LSTM block was used to further capture the features from convolutional layer. Finally, the output of CNN-LSTM is fed into an additional fully connected layer and a Softmax layer to produce the final output.
(i) Convolutional layer: The convolutional layer is a major building block of CNN, which contains a set of learnable filters where each filter is convolved with the input of the layer to encode the local knowledge of the small receptive field. This process helps conserve the dimensional relationship between numeric values in the vectors [33]. Thus, a 1D convolutional layer was used to construct a convolution kernel and then derive features encoded in the embedding layer [34].
(ii) Rectified Linear Unit (ReLU): An additional non-linear operation was presented after every convolution operation. It aims to introduce the property of non-linearity into the model and produce a more desirable output. The output function of ReLU is as follows:
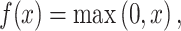 |
(6) |
where
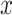 is the number of inputs in
a neural network.
is the number of inputs in
a neural network.
(iii) Pooling layer: Max pooling is a sample-based discretization process. It was used to down-sample the hidden-layer output matrix, reducing its dimensionality and allowing for assumptions to be made about features contained in the sub-regions binned. In this step, we set Max pooling stride equal to 2.
(iv) Dropout layer: A technique which probabilistically dropping out nodes in the network for reducing overfitting and improving the generalization of deep neural networks. In this step, we set the dropout size equal to 0.5.
(v) LSTM layer: An LSTM layer consists of a set of recurrently connected blocks, which contain one or more recurrently connected memory cells and three multiplicative units—the input, output and forget gates. The output of each LSTM cell encodes the observed short- and long-term dependence on that cell’s input. The outputs of the LSTM layers are fused using concatenation to obtain the final feature vector. In this step, we set the output size equal to 70.
(vi) Dense layer: A neural network layer that is connected deeply, which means that each neuron in the dense layer receives input from all neurons of its previous layer. Our task is to train a binary classification model to distinguish phosphorylation sites and non-phosphorylation sites. Therefore, in this step, we set the number of nodes equal to 2.
In our work, the CNN-LSTM architecture was implemented with Keras library 2.2.2 [26], TensorFlow 1.2.1 and sklearn 0.22.1. Detailed parameter information can be obtained from https://github.com/linDing-group/DeepIPs.
Performance evaluation
To assess the performance of phosphorylation site prediction, several commonly
used evaluation metrics were employed in this study, including sensitivity
(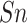 ), specificity
(
), specificity
(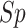 ), overall accuracy
(
), overall accuracy
(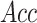 ) and Matthew’s
correlation coefficient (
) and Matthew’s
correlation coefficient (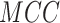 ) [35–39]. The detailed
definitions are
) [35–39]. The detailed
definitions are
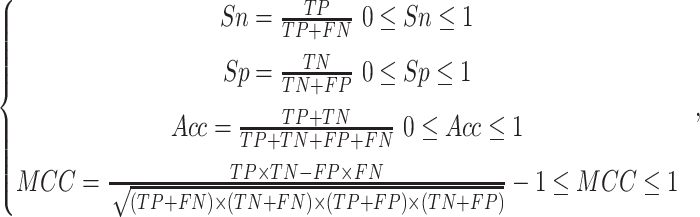 |
(7) |
where
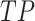 ,
, 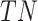 ,
, 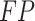 and
and 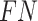 represent the number of true
positive samples, true negative samples, false positive samples and false
negative samples, respectively. Furthermore, we also used receiver operating
characteristic (ROC) curve as well as the area under ROC curve
(AUC) to assess the overall performance [40–42], the closer the
AUC value to 1, which demonstrates that the overall
performance is better.
represent the number of true
positive samples, true negative samples, false positive samples and false
negative samples, respectively. Furthermore, we also used receiver operating
characteristic (ROC) curve as well as the area under ROC curve
(AUC) to assess the overall performance [40–42], the closer the
AUC value to 1, which demonstrates that the overall
performance is better.
Results
Performance evaluation of different word embedding methods
We evaluated and compared the prediction performance of four different word embedding methods used by CNN-LSTM architecture with 5-fold cross-validation based on S/T and Y phosphorylation sites datasets. The confusion matrices were shown in Figure 3 and Table 2. From Figure 3 and Table 2, the following points were observed.
Figure 3 .
ROC curves of SEL, Word2Vec, GloVe and fastText with 5-fold cross-validation on S/T sites and Y sites, respectively.
Table 2.
Confusion matrices of SEL, Word2Vec, GloVe and fastText with 5-fold cross-validation
| Residue type | Algorithm |
 (%) (%) |
 (%) (%) |
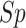 (%) (%) |
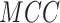
|
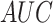
|
|---|---|---|---|---|---|---|
| S/T | SEL | 80.45 | 79.70 | 81.19 | 0.6102 | 0.8871 |
| Word2Vec | 76.04 | 70.53 | 81.49 | 0.5264 | 0.8397 | |
| GloVe | 79.54 | 73.50 | 85.52 | 0.5954 | 0.8849 | |
| fastText | 78.26 | 70.33 | 86.11 | 0.5740 | 0.8723 | |
| Y | SEL | 73.44 | 75.00 | 72.55 | 0.4988 | 0.8199 |
| Word2Vec | 74.35 | 73.33 | 76.18 | 0.5160 | 0.8057 | |
| GloVe | 75.22 | 74.85 | 76.18 | 0.5183 | 0.8414 | |
| fastText | 59.17 | 35.00 | 80.00 | 0.1680 | 0.6537 |
For S/T sites, the SEL, Word2Vec, GloVe and fastText, all could produce
satisfactory performance, indicating that word embedding methods have ability to
capture information hidden in the protein sequence by mapping the truncated
phosphorylation and non-phosphorylation peptides from high-dimensional space to
low-dimensional space. In particular, the supervised learning-based method SEL
outperformed the other three unsupervised learning-based methods in terms of
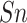 ,
, 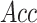 ,
, 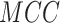 ,
, 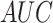 , except for
, except for
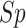 . This result suggested
that SEL method with more information utilization and robust embedding
architecture may serve as efficient approach for S/T phosphorylation site
prediction. Additionally, compared with fastText and GloVe methods, Word2Vec
performs worse in all evaluation metrics. The reason is that the sequential
information among the textual units is discarded inside the CBOW model. Even if
the word vector sampled by the sliding window contains certain sequential
information, the embedding constraint on words is very small, and it is not
enough to capture sufficient sequence order features. In contrast, fastText
introduces a strongly constrained n-gram to extract sequential
features, which helps it improve 4.62, 2.22, 4.76 and 3.26% compare with
Word2Vec in
. This result suggested
that SEL method with more information utilization and robust embedding
architecture may serve as efficient approach for S/T phosphorylation site
prediction. Additionally, compared with fastText and GloVe methods, Word2Vec
performs worse in all evaluation metrics. The reason is that the sequential
information among the textual units is discarded inside the CBOW model. Even if
the word vector sampled by the sliding window contains certain sequential
information, the embedding constraint on words is very small, and it is not
enough to capture sufficient sequence order features. In contrast, fastText
introduces a strongly constrained n-gram to extract sequential
features, which helps it improve 4.62, 2.22, 4.76 and 3.26% compare with
Word2Vec in 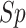 ,
, 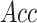 ,
, 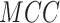 and
and 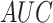 , respectively. Furthermore,
it is clear that GloVe exhibits an advantage over Word2Vec and fastText in
identifying S/T phosphorylation sites. The reason is that GloVe can mine better
word representations from global corpus, which is different from Word2Vec and
fastText based on local corpus. These results indicated that sequential features
provide significant contribution to improve the predictive ability of the model.
Meanwhile, co-occurrence-based GloVe is superior to the distributed
assumption-based Word2Vec and fastText in S/T phosphorylation site prediction
problem.
, respectively. Furthermore,
it is clear that GloVe exhibits an advantage over Word2Vec and fastText in
identifying S/T phosphorylation sites. The reason is that GloVe can mine better
word representations from global corpus, which is different from Word2Vec and
fastText based on local corpus. These results indicated that sequential features
provide significant contribution to improve the predictive ability of the model.
Meanwhile, co-occurrence-based GloVe is superior to the distributed
assumption-based Word2Vec and fastText in S/T phosphorylation site prediction
problem.
For Y sites, we found that GloVe achieves the best performance
(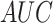 = 0.8414),
while SEL produces the second best result (
= 0.8414),
while SEL produces the second best result (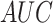 = 0.8199).
This result suggested that both SEL and GloVe have the ability to learn specific
embedding function that maps all peptides containing in training data into a
joint low-dimensional embedding space. Interestingly, we noticed that this
result is similar to the S/T sites prediction task, where SEL and GloVe perform
better than other word embedding methods, which intuitively shows that these two
approaches are robust and effective in learning from small and large training
data. As we all know, the pre-training step involved in unsupervised
learning-based embedding serves as a prior for the parameter space, which is
useful for generalization to small training data. However, we observed the
opposite result that fastText exhibits worst performance as the number of
training data decreases. This indicated that fastText is not practically
suitable for Y phosphorylation sites prediction. We speculated that the reasons
for this situation include two aspects: different Y-containing sequences have
similar sequential information, fastText’s operation of averaging the
word vectors of each sequence leads to a large amount of information loss;
fastText’s linear network structure is not capable of complex learning
task such as Y phosphorylation sites prediction. In addition, Word2Vec performs
considerably better on such small training data, indicating that Word2Vec can
achieve stable performance when being applied to both small and large datasets.
Taken together, in this study, we established the final models for S/T sites and
Y sites based on SEL and GloVe, respectively.
= 0.8199).
This result suggested that both SEL and GloVe have the ability to learn specific
embedding function that maps all peptides containing in training data into a
joint low-dimensional embedding space. Interestingly, we noticed that this
result is similar to the S/T sites prediction task, where SEL and GloVe perform
better than other word embedding methods, which intuitively shows that these two
approaches are robust and effective in learning from small and large training
data. As we all know, the pre-training step involved in unsupervised
learning-based embedding serves as a prior for the parameter space, which is
useful for generalization to small training data. However, we observed the
opposite result that fastText exhibits worst performance as the number of
training data decreases. This indicated that fastText is not practically
suitable for Y phosphorylation sites prediction. We speculated that the reasons
for this situation include two aspects: different Y-containing sequences have
similar sequential information, fastText’s operation of averaging the
word vectors of each sequence leads to a large amount of information loss;
fastText’s linear network structure is not capable of complex learning
task such as Y phosphorylation sites prediction. In addition, Word2Vec performs
considerably better on such small training data, indicating that Word2Vec can
achieve stable performance when being applied to both small and large datasets.
Taken together, in this study, we established the final models for S/T sites and
Y sites based on SEL and GloVe, respectively.
Evaluation of DeepIPs for phosphorylation site prediction
In this section, we first compared DeepIPs with different deep-learning network
architectures including CNN [43] and
LSTM [44] on the training data as
described in Benchmark dataset construction section. The
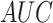 values of these methods
on residues S/T and Y were shown in Figure
4. All detailed evaluation indicators were listed in Table 3. In general, DeepIPs obtained higher
values of these methods
on residues S/T and Y were shown in Figure
4. All detailed evaluation indicators were listed in Table 3. In general, DeepIPs obtained higher
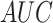 value than other
deep-learning architectures, showing that DeepIPs has better overall
performance. For instance, on S/T sites, the
value than other
deep-learning architectures, showing that DeepIPs has better overall
performance. For instance, on S/T sites, the 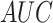 value of our architecture is
0.8871, which is 0.98 and 2.16% higher than CNN and LSTM, respectively.
In addition to
value of our architecture is
0.8871, which is 0.98 and 2.16% higher than CNN and LSTM, respectively.
In addition to 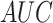 values, it is obvious
that DeepIPs consistently achieved higher performance in terms of
values, it is obvious
that DeepIPs consistently achieved higher performance in terms of
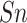 ,
, 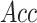 ,
, 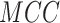 than other deep-learning
architectures. For Y sites,
than other deep-learning
architectures. For Y sites, 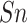 ,
,
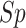 ,
, 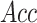 ,
, 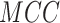 and
and 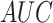 value of DeepIPs at the
high-stringency level are 74.85, 76.18, 75.22, 0.5183 and 0.8414, respectively.
These metrics are higher than CNN and LSTM on all the measurements, which
demonstrates the efficient architecture of the constructed model. Furthermore,
we also found that the performance of LSTM is not as good as other deep-learning
approaches on S/T and Y sites, indicating that LSTM may not be an ideal
architecture for phosphorylation site prediction.
value of DeepIPs at the
high-stringency level are 74.85, 76.18, 75.22, 0.5183 and 0.8414, respectively.
These metrics are higher than CNN and LSTM on all the measurements, which
demonstrates the efficient architecture of the constructed model. Furthermore,
we also found that the performance of LSTM is not as good as other deep-learning
approaches on S/T and Y sites, indicating that LSTM may not be an ideal
architecture for phosphorylation site prediction.
Figure 4 .
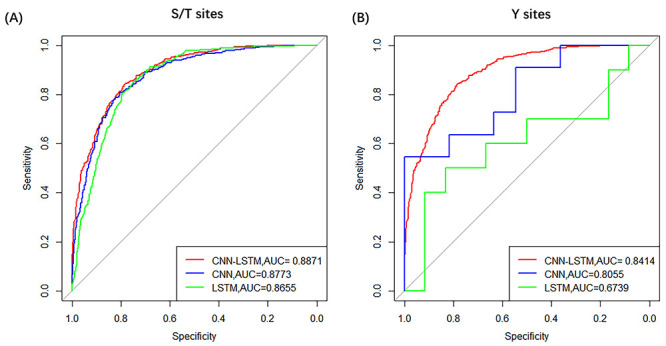
ROC curves of different deep-learning network architectures with 5-fold cross-validation on S/T sites and Y sites, respectively.
Table 3.
Evaluation indicators of different deep-learning network architectures with 5-fold cross-validation, including CNN- LSTM, CNN and LSTM, respectively
| Residue type | Algorithm |
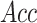 (%) (%) |
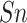 (%) (%) |
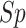 (%) (%) |
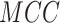
|
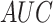
|
|---|---|---|---|---|---|---|
| S/T | CNN-LSTM | 80.45 | 79.70 | 81.19 | 0.6102 | 0.8871 |
| CNN | 80.05 | 76.53 | 83.54 | 0.6035 | 0.8773 | |
| LSTM | 79.22 | 73.50 | 84.89 | 0.5903 | 0.8655 | |
| Y | CNN-LSTM | 75.22 | 74.85 | 76.18 | 0.5183 | 0.8414 |
| CNN | 76.05 | 76.36 | 76.18 | 0.5371 | 0.8055 | |
| LSTM | 66.48 | 68.33 | 65.09 | 0.3630 | 0.6739 |
To further assess the performance of DeepIPs, we should compare DeepIPs with
several existing phosphorylation site prediction tools using independent test
data. However, most of models used different training data and did not provide
standalone tools or web-server, thereby making it difficult to provide a direct
comparison. To solve it, we only chose three representative deep-learning-based
tools that are DeepPSP [15],
MusiteDeep2020 [45] and MusiteDeep2017
[13]. The code or webserver of
these predictors has been provided and available online. For fair comparison, we
rebuilt the models of these three tools, and the corresponding performances were
obtained. The 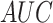 values on independent
data were plotted in Figure 5. All detailed
evaluation metrics were shown in Table
4. We noticed that DeepIPs is superior to other three predictors. For S/T
sites, the
values on independent
data were plotted in Figure 5. All detailed
evaluation metrics were shown in Table
4. We noticed that DeepIPs is superior to other three predictors. For S/T
sites, the 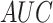 value of DeepIPs is
0.8937, which is 1.75, 0.7 and 1.39% higher than DeepPSP, MusiteDeep2020
and MusiteDeep2017, respectively. For Y sites, AUC values of
DeepPSP, MusiteDeep2020 and MusiteDeep2017 are 0.8209, 0.8730 and 0.8141,
respectively, while DeepIPs can produce a higher value of 0.9252. These results
indicated that DeepIPs has excellent prediction ability when comparing with
existing tools.
value of DeepIPs is
0.8937, which is 1.75, 0.7 and 1.39% higher than DeepPSP, MusiteDeep2020
and MusiteDeep2017, respectively. For Y sites, AUC values of
DeepPSP, MusiteDeep2020 and MusiteDeep2017 are 0.8209, 0.8730 and 0.8141,
respectively, while DeepIPs can produce a higher value of 0.9252. These results
indicated that DeepIPs has excellent prediction ability when comparing with
existing tools.
Figure 5 .
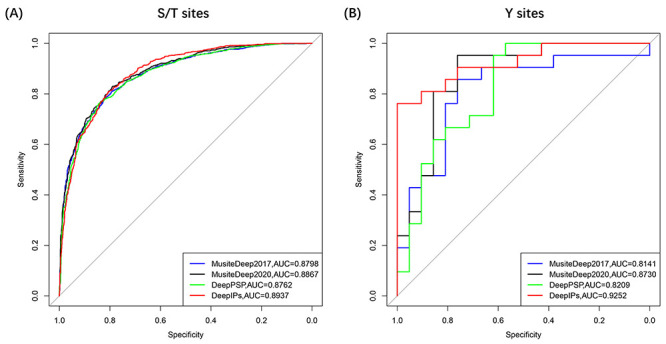
ROC curves of existing tools for phosphorylation site prediction.
Table 4.
Performance of existing tools for phosphorylation site prediction with 5-fold cross-validation
| Residue type | Method |
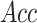 (%) (%) |
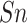 (%) (%) |
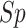 (%) (%) |
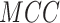
|
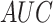
|
|---|---|---|---|---|---|---|
| S/T | DeepIPs | 80.63 | 79.61 | 83.50 | 0.6316 | 0.8937 |
| DeepPSP | 80.21 | 76.65 | 83.78 | 0.6058 | 0.8762 | |
| MusiteDeep2020 | 80.95 | 82.95 | 78.96 | 0.6196 | 0.8867 | |
| MusiteDeep2017 | 80.17 | 78.87 | 81.46 | 0.6035 | 0.8798 | |
| Y | DeepIPs | 83.33 | 90.48 | 80.95 | 0.7175 | 0.9252 |
| DeepPSP | 76.19 | 95.24 | 57.14 | 0.5665 | 0.8209 | |
| MusiteDeep2020 | 85.71 | 95.24 | 76.19 | 0.7276 | 0.8730 | |
| MusiteDeep2017 | 80.95 | 85.71 | 76.19 | 0.6219 | 0.8141 |
Additionally, we noticed that DeepIPs has only a slight improvement in model performance compared with the MusiteDeep2020 on S/T sites. The possible reason is that the independent dataset could be included in the training procedure of these tools, so that their performance is similar. To make an equal and objective evaluation of the performance, we collected the experimentally verified S/T phosphorylation sites of Vero E6, Caco-2 and iAT2 cell lines infected with SARS-CoV-2 from literatures [5, 8, 9]. Next, we performed very rigorous procedure to eliminate the overlap entries between this dataset and the training data of DeepIPs and MusiteDeep2020. Thus, an unseen independent dataset was constructed. Subsequently, we inputted the non-redundant dataset into DeepIPs and MusiteDeep2020 for examining their performance. The results showed that DeepIPs can correctly identify 58.66% (210/358) modification sites which is better than the results generated from MusiteDeep2020 43.58% (156/358). Apart from this, we also integrated the unseen independent dataset into training dataset and built the corresponding model. The result showed that the model can correctly differentiate 79.05 (283/358) modification sites contained in unseen independent dataset. Overall, these results further demonstrate the stability and generalization ability of our proposed method.
Discover potential therapeutic targets
Previous study has shown that some kinase inhibitors, such as Gilteritinib (a designated FLT3/AXL inhibitor, Ipatasertib (AKT inhibitor)), can be used as potential drugs for the treatment of COVID-19 by hindering the replication of SARS-CoV-2 and interfering with its required host pathway [7]. Therefore, by integrating different database resources, narrowing the scope of antiviral compounds and discovering host kinases that act as therapeutic targets will lay the foundation for the development of new therapeutic strategies. Inspired by this idea, we utilized the gene names and protein accession numbers in the benchmark dataset used in this work as indexes to search the corresponding kinases in the databases of PhosphoSitePlus [46] and phosphor.ELM [47], and categorized the kinase families. The detailed results were shown in Supplementary Data. We found that most of the phosphorylation process is mediated by cyclin-dependent kinases, indicating that viral proteins accelerate the host cell cycle through interaction with host kinases. In addition, PKC, CK2, PKA and Src are also involved in the phosphorylation reaction. Thus, the development of specific inhibitors of these kinases may be a promising approach to treat SARS-CoV-2 infection.
Conclusion and discussion
Phosphorylation is of significance in biological process, which relates to the occurrence of SARS-CoV-2 infection. Due to the limitations of experimental verifying sites that cost time and money, it is very urgent to develop effective computational methods for phosphorylation identification in SARS-CoV-2 infection. Hence, in this study, we propose DeepIPs, which consists of the most popular word embedding methods and CNN-LSTM architecture, to predict phosphorylation sites. The independent test demonstrates that DeepIPs has a better performance than existing phosphorylation sites predictors. Furthermore, a freely accessible web-server called DeepIPs was established.
The major contributions of our study can be summarized as follows. Firstly, we systematically compared the pros and cons of four word embedding methods in predicting S/T or Y phosphorylation sites. Due to the better transferability of word embedding, our analysis can promote its application research in other bioinformatics classification problems. Secondly, we compared the CNN-LSTM architecture used in this work with other deep-learning algorithms, such as CNN and LSTM. The results show that CNN-LSTM can comprehensively capture short- and long-range correlation information which once again proves that the architecture has capacity in identifying phosphorylation sites. The last and the most important is that the model we built has special value in predicting phosphorylation sites in host cells infected with SARS-CoV-2.
In addition, the following aspects can be further improved in the future. Firstly, the word embedding methods, such as SEL, Word2Vec, fastText and GloVe used in this study, are all based on fixed representations of word vectors, which cannot represent the different meanings of word in different contexts. The dynamic word representation methods, such as ELMo, GPT and BERT, can extract contextual semantic information based on the words in the context, thus have stronger word representation capabilities. Secondly, the CNN-LSTM architecture we designed cannot explain meaningful biological process well due to ‘black box’ property. Therefore, we will use some interpretable deep-learning algorithms in future works, such as generating adversarial network.
Key Points
We provided a systematic comparison of the most popular word embedding methods in identifying phosphorylation sites in host cells infected with SARS-CoV-2.
We proposed DeepIPs, a new CNN-LSTM architecture for phosphorylation sites prediction.
The independent test demonstrates the excellent performance of DeepIPs.
We established a freely accessible web-server called DeepIPs.
Supplementary Material
Acknowledgements
We would like to thank the anonymous reviewers for valuable suggestions.
Hao Lv is a PhD candidate of the Center for Informational Biology at the University of Electronic Science and Technology of China. His research interests include bioinformatics.
Fu-Ying Dao is a PhD candidate of the Center for Informational Biology at the University of Electronic Science and Technology of China. Her research interests include bioinformatics.
Hasan Zulfiqar is a PhD candidate of the Center for Informational Biology at the University of Electronic Science and Technology of China. His research interests include bioinformatics.
Hao Lin is a Professor of the Center for Informational Biology at the University of Electronic Science and Technology of China. His research is in the areas of bioinformatics and system biology.
Contributor Information
Hao Lv, Center for Informational Biology at the University of Electronic Science and Technology of China, Chengdu 610054, China.
Fu-Ying Dao, Center for Informational Biology at the University of Electronic Science and Technology of China, Chengdu 610054, China.
Hasan Zulfiqar, Center for Informational Biology at the University of Electronic Science and Technology of China, Chengdu 610054, China.
Hao Lin, Center for Informational Biology at the University of Electronic Science and Technology of China, Chengdu 610054, China.
Data availability
We provide the Python source code of DeepIPs model training, which is freely available at https://github.com/linDing-group/DeepIPs.
Authors’ contributions
Conceptualization, H. Lin.; Investigation, H. Lv., F.-Y.D.; Coding, H. Lv.; Writing—Original Draft, H. Lv., F.-Y.D. H. Z; Writing—Review & Editing, H. Lin.; Funding Acquisition, H. Lin.
Funding
National Natural Science Foundation of China (61772119); Sichuan Provincial Science Fund for Distinguished Young Scholars (2020JDJQ0012).
References
- 1.Barnes CO, Jette CA, Abernathy ME, et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature 2020;588:682–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hu B, Guo H, Zhou P, et al. Characteristics of SARS-CoV-2 and COVID-19. Nat Rev Microbiol 2021;19:141–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gordon DE, Jang GM, Bouhaddou M, et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020;583:459–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith M, Smith JC. Repurposing therapeutics for COVID-19: supercomputer-based docking to the SARS-CoV-2 viral spike protein and viral spike protein-human ACE2 interface. ChemRxiv 2020. doi: 10.26434/chemrxiv.11871402.v4. [DOI] [Google Scholar]
- 5.Klann K, Bojkova D, Tascher G, et al. Growth factor receptor signaling inhibition prevents SARS-CoV-2 replication. Mol Cell 2020;80:164–174 e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bojkova D, Klann K, Koch B, et al. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature 2020;583:469–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stukalov A, Girault V, Grass V, et al. Multi-level proteomics reveals host-perturbation strategies of SARS-CoV-2 and SARS-CoV. Nature, 2021;594:246–52. doi: 10.1101/2020.06.17.156455. [DOI] [PubMed] [Google Scholar]
- 8.Bouhaddou M, Memon D, Meyer B, et al. The global phosphorylation landscape of SARS-CoV-2 infection. Cell 2020;182:685–712 e619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hekman RM, Hume AJ, Goel RK, et al. Actionable cytopathogenic host responses of human alveolar type 2 cells to SARS-CoV-2. Mol Cell 2020;80:1104–1122 e1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wei L, Xing P, Tang J, et al. PhosPred-RF: a novel sequence-based predictor for phosphorylation sites using sequential information only. IEEE Trans Nanobioscience 2017;16:240–7. [DOI] [PubMed] [Google Scholar]
- 11.Li F, Li C, Marquez-Lago TT, et al. Quokka: a comprehensive tool for rapid and accurate prediction of kinase family-specific phosphorylation sites in the human proteome. Bioinformatics 2018;34:4223–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang C, Xu H, Lin S, et al. GPS 5.0: An update on the prediction of kinase-specific phosphorylation sites in proteins. Genomics Proteomics Bioinformatics 2020;18:72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang D, Zeng S, Xu C, et al. MusiteDeep: a deep-learning framework for general and kinase-specific phosphorylation site prediction. Bioinformatics 2017;33:3909–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang D, Liang Y, Xu D. Capsule network for protein post-translational modification site prediction. Bioinformatics 2019;35:2386–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo L, Wang Y, Xu X, et al. DeepPSP: a global-local information-based deep neural network for the prediction of protein phosphorylation sites. J Proteome Res 2021;20:346–56. [DOI] [PubMed] [Google Scholar]
- 16.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006;22:1658–9. [DOI] [PubMed] [Google Scholar]
- 17.Basith S, Manavalan B, Hwan Shin T, et al. Machine intelligence in peptide therapeutics: a next-generation tool for rapid disease screening. Med Res Rev 2020;40:1276–314. [DOI] [PubMed] [Google Scholar]
- 18.Wei L, He W, Malik A, et al. Computational prediction and interpretation of cell-specific replication origin sites from multiple eukaryotes by exploiting stacking framework. Brief Bioinform 2020. doi: 10.1093/bib/bbaa275. [DOI] [PubMed] [Google Scholar]
- 19.Mei S, Li F, Xiang D, et al. Anthem: a user customised tool for fast and accurate prediction of binding between peptides and HLA class I molecules. Brief Bioinform 2021. doi: 10.1093/bib/bbaa415. [DOI] [PubMed] [Google Scholar]
- 20.Luo F, Wang M, Liu Y, et al. DeepPhos: prediction of protein phosphorylation sites with deep learning. Bioinformatics 2019;35:2766–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lv H, Dao FY, Guan ZX, et al. Deep-Kcr: accurate detection of lysine crotonylation sites using deep learning method. Brief Bioinform 2020. doi: 10.1093/bib/bbaa255. [DOI] [PubMed] [Google Scholar]
- 22.Mikolov T, Chen K, Corrado G, et al. Efficient estimation of word representations in vector space. arXiv. 2013. doi: 1301.3781v3.
- 23.Pennington J, Socher R, Manning CD. Glove: global vectors for word representation. In: Proceedings of the 2014 Conference on Empirical Methods in Natural Language Processing (EMNLP). Doha, Qatar: Association for Computational Linguistics, 2014, 1532–43.
- 24.Bojanowski P, Grave E, Joulin A, et al. Enriching word vectors with subword information. Trans Assoc Comput Linguist. 2017;5:135–46. [Google Scholar]
- 25.Joulin A, Grave E, Bojanowski P, et al. Fasttext. zip: Compressing text classification models. arXiv. 2016. doi: 1612.03651v1.
- 26.Chollet FJASCL . Keras: the python deep learning library, 2018, ascl: 1806.1022.
- 27.Li H, Gong XJ, Yu H, et al. Deep neural network based predictions of protein interactions using primary sequences. Molecules 2018;23:1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang H, Wang Z, Li Z, et al. Incorporating deep learning with word embedding to identify plant ubiquitylation sites. Front Cell Dev Biol 2020;8:572195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu Y, Song J, Wilson C, et al. PhosContext2vec: a distributed representation of residue-level sequence contexts and its application to general and kinase-specific phosphorylation site prediction. Sci Rep 2018;8:8240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang R, Wang Y, Yang Y, et al. Predicting CTCF-mediated chromatin loops using CTCF-MP. Bioinformatics 2018;34:i133–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dao FY, Lv H, Zhang D, et al. DeepYY1: a deep learning approach to identify YY1-mediated chromatin loops. Brief Bioinform 2020. doi: 10.1093/bib/bbaa356. [DOI] [PubMed] [Google Scholar]
- 32.Joulin A, Grave E, Bojanowski P, et al. Bag of tricks for efficient text classification. arXiv. 2016. doi: 1607.01759v3.
- 33.Abbasi K, Razzaghi P, Poso A, et al. DeepCDA: deep cross-domain compound-protein affinity prediction through LSTM and convolutional neural networks. Bioinformatics 2020;36:4633–42. [DOI] [PubMed] [Google Scholar]
- 34.Le NQK, Yapp EKY, Nagasundaram N, et al. Classifying promoters by interpreting the hidden information of DNA sequences via deep learning and combination of continuous FastText N-Grams. Front Bioeng Biotechnol 2019;7:305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Charoenkwan P, Chiangjong W, Lee VS, et al. Improved prediction and characterization of anticancer activities of peptides using a novel flexible scoring card method. Sci Rep 2021;11:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Charoenkwan P, Kanthawong S, Nantasenamat C, et al. iDPPIV-SCM: a sequence-based predictor for identifying and analyzing dipeptidyl peptidase IV (DPP-IV) inhibitory peptides using a scoring card method. J Proteome Res 2020;19:4125–36. [DOI] [PubMed] [Google Scholar]
- 37.Charoenkwan P, Chiangjong W, Nantasenamat C, et al. StackIL6: a stacking ensemble model for improving the prediction of IL-6 inducing peptides. Brief Bioinform 2021. doi: 10.1093/bib/bbab172. [DOI] [PubMed] [Google Scholar]
- 38.Charoenkwan P, Yana J, Nantasenamat C, et al. iUmami-SCM: a novel sequence-based predictor for prediction and analysis of umami peptides using a scoring card method with propensity scores of dipeptides. J Chem Inf Model 2020;60:6666–78. [DOI] [PubMed] [Google Scholar]
- 39.Li F, Chen J, Ge Z, et al. Computational prediction and interpretation of both general and specific types of promoters in Escherichia coli by exploiting a stacked ensemble-learning framework. Brief Bioinform 2021;22:2126–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Charoenkwan P, Nantasenamat C, Hasan MM, et al. BERT4Bitter: a bidirectional encoder representations from transformers (BERT)-based model for improving the prediction of bitter peptides. Bioinformatics 2021. doi: 10.1093/bioinformatics/btab133. [DOI] [PubMed] [Google Scholar]
- 41.Hasan MM, Basith S, Khatun MS, et al. Meta-i6mA: an interspecies predictor for identifying DNA N6-methyladenine sites of plant genomes by exploiting informative features in an integrative machine-learning framework. Brief Bioinform 2020. doi: 10.1093/bib/bbaa202. [DOI] [PubMed] [Google Scholar]
- 42.Zhu Y, Li F, Xiang D, et al. Computational identification of eukaryotic promoters based on cascaded deep capsule neural networks. Brief Bioinform 2020. doi: 10.1093/bib/bbaa299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.LeCun Y, Bengio Y, Hinton G. Deep learning. Nature 2015;521:436–44. [DOI] [PubMed] [Google Scholar]
- 44.Hochreiter S. Schmidhuber J. LSTM can solve hard long time lag problems. Advances in Neural Information Processing Systems 1996;9:473–9. [Google Scholar]
- 45.Wang D, Liu D, Yuchi J, et al. MusiteDeep: a deep-learning based webserver for protein post-translational modification site prediction and visualization. Nucleic Acids Res 2020;48:W140–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hornbeck PV, Kornhauser JM, Latham V, et al. 15 years of PhosphoSitePlus(R): integrating post-translationally modified sites, disease variants and isoforms. Nucleic Acids Res 2019;47:D433–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Diella F, Gould CM, Chica C, et al. Phospho.ELM: a database of phosphorylation sites--update 2008. Nucleic Acids Res 2008;36:D240–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We provide the Python source code of DeepIPs model training, which is freely available at https://github.com/linDing-group/DeepIPs.