

Abstract
Background
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variants concerning for enhanced transmission, evasion of immune responses, or associated with severe disease have motivated the global increase in genomic surveillance. In the current study, large-scale whole-genome sequencing was performed between November 2020 and the end of March 2021 to provide a phylodynamic analysis of circulating variants over time. In addition, we compared the viral genomic features of March 2020 and March 2021.
Methods
A total of 1600 complete SARS-CoV-2 genomes were analyzed. Genomic analysis was associated with laboratory diagnostic volumes and positivity rates, in addition to an analysis of the association of selected variants of concern/variants of interest with disease severity and outcomes. Our real-time surveillance features a cohort of specimens from patients who tested positive for SARS-CoV-2 after completion of vaccination.
Results
Our data showed genomic diversity over time that was not limited to the spike sequence. A significant increase in the B.1.1.7 lineage (alpha variant) in March 2021 as well as a transient circulation of regional variants that carried both the concerning S: E484K and S: P681H substitutions were noted. Lineage B.1.243 was significantly associated with intensive care unit admission and mortality. Genomes recovered from fully vaccinated individuals represented the predominant lineages circulating at specimen collection time, and people with those infections recovered with no hospitalizations.
Conclusions
Our results emphasize the importance of genomic surveillance coupled with laboratory, clinical, and metadata analysis for a better understanding of the dynamics of viral spread and evolution.
Keywords: SARS-CoV-2, COVD-19, variant of concern, sequencing
Genomic surveillance of severe acute respiratory syndrome coronavirus 2 in the US National Capital Region showed transient circulation of regional variants, emergence and predominance of lineage B.1.1.7, genomic diversity that is not limited to the spike sequence, and an association of lineage B.1.243 with severe disease.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has caused a rapidly evolving pandemic and within a little over 1 year, >162 million cases were globally confirmed, with >3 million deaths worldwide (https://coronavirus.jhu.edu/map.html). Since its first introduction, genomic sequencing revealed global diversity and identified variations in different regions of SARS-CoV-2 genomes [1–5]. The aspartate-to-glycine change at position 614 (S: D614G) substitution in the spike (S) protein garnered much attention in mid-2020 and rapidly became globally predominant [2, 6, 7]. Most recently, variants with higher transmissibility are raising a major concern (variants of concern [VOCs]). Lineage B.1.1.7 (alpha variant), characterized by an unusual large number of changes in its genome [8] and first detected in the United Kingdom in September 2020, is globally circulating [9–12]. Along with multiple mutations in the spike protein, there are 3 specific changes of particular concern: the S: N501Y, shown to enhance the binding affinity to angiotensin-converting enzyme 2 (ACE2), the S: 69–70del that could potentially cause immune escape, and the S: P681H that is close to the furin cleavage site [8, 13, 14]. Other S: N501Y emerging VOCs include B.1.351 (beta) and P.1 (gamma), which both carry S: E484K and became predominant in their countries of origin [15, 16].
In addition, regional variants of interest (VOIs) and VOCs evolved in the United States and showed marked spread and concerning genomic changes. California variants were associated with a spike in the number of cases and were reported from other regions in the United States [17]. These variants are characterized by the S: L452R, which could affect neutralization by monoclonal antibodies [18, 19]. A large percentage of screened isolates in New York City belonged to VOI (B.1.526 [iota] and B.1.525 [eta]) [20]. The significance of the evolution of regional variants and VOCs has become an area of international focus.
We previously reported the genomic diversity of SARS-CoV-2 at its early introduction in March 2020 to the National Capital Region [3]. In this report, we provide an update on the diversification of SARS-CoV-2 after 1 year of the pandemic.
MATERIALS AND METHODS
Ethical Considerations and Data Availability
Research was conducted under protocol IRB00221396 with a waiver of consent. Whole genomes were deposited at GISAID (Global Initiative on Sharing All Influenza Data) (Supplementary Table 1). Whole-genome data were made available publicly, and raw genomic data requests may be directed to the correspondence author (H. H. M.)
Sample Selection
Remnant nasopharyngeal or lateral midturbinate nasal clinical specimens that tested positive for SARS-CoV-2 after standard of care diagnostic or screening assays were performed across the Johns Hopkins Medical System (representing a wide geographic area in the National Capital Region—Maryland, Washington, DC, and Virginia). Different molecular assays are used for SARS-CoV-2 detection, including the NeuMoDx (Qiagen) [21, 22], cobas (Roche) [21], Aptima (Hologic), Xpert Xpress SARS-CoV-2/Flu/RSV (Cepheid) [23], ePlex respiratory pathogen panel 2 (GenMark) [24], Accula, and RealStar SARS-CoV-2 assays (altona Diagnostics) [25]. Testing was performed in accordance with the manufacturer instructions and our in house validated protocols. Specimen selection was random except for cycle threshold (Ct), where values <20 were preferentially selected when available.
Genome Sequencing and Analysis
Automated nucleic acid extraction was performed using the chemagic 360 instrument (PerkinElmer), following the manufacturer’s protocol. Libraries were prepared using the ARTIC protocol, as described elsewhere [3] Nanopore reads were base-called with MinKNOW and demultiplexed with Guppy v3.5.2 barcoder software, requiring barcodes at both ends. Reads were size restricted, and alignment and variant calling were performed with the artic-ncov2019 medaka protocol. Thresholds were set to a minimum of 90% coverage and 100 mean depth. Mutations were visually confirmed with the Integrated Genomics viewer (version 2.8.10). Clades were determined using Nextclade beta version 0.12.0 (clades.nextstrain.org) [26], and lineages were determined with the Pangolin coronavirus disease 2019 (COVID-19) lineage assigner (COG-UK; cog-uk.io).
Clinical Data Analysis
Clinical data were retrieved from the electronic medical records manually. Severity index scores were assigned as follows: 0 indicated asymptomatic, 1, outpatient or admitted for another reason without oxygen requirement; 2, inpatient (or oxygen requirement for COVID-19); 3, intensive care unit (ICU) admission; and 4, death. Severity scores were determined at the time of clinical data analysis, which was >30 days after the date of sample collection in all patients.
Statistical Analysis
Fisher exact, χ 2, and Kruskal-Wallis tests were performed to show associations with Bonferroni correction, depending on the type and number of results evaluated. Post hoc analysis was performed using Conover analysis with Holmes adjustment or χ 2 analysis. Odds ratios were calculated with MedCalc’s odds ratio calculator, using a method described elsewhere [27].
RESULT
SARS-CoV-2 Molecular Testing at Johns Hopkins Laboratory
A total of 378 107 tests were performed as of 1 April 2021, with 23 947 positive results identified. The positivity rates showed 2 distinctive peaks, in April 2020 and December 2020 to January 2021, with maximum 15-day rolling averages of 20.1% and 10.0%, respectively (Figure 1A). The end of January witnessed a reduction in the positivity that plateaued at a 15-day rolling average of 3.1% (Figure 1A).
Figure 1.

Severe acute respiratory syndrome coronavirus 2 positivity and genotypes at Johns Hopkins Hospital. A, Percentage positivity among total molecular tests. B, Percentage clade distribution between November 2020 and March 2021. C, Stack plot of estimated number of cases per clade based on total number of cases per day and percentage sequenced within each clade. Data shown as 15-day rolling average.
SARS-CoV-2 Sequencing and Demographics
A total of 1600 complete or near-complete genomes were obtained from samples collected between 26 October 2020 and 31 March 2021, constituting 14.3% of positive results during this time frame. Initially, clade 20A predominated, until December 2020 when 20G became the dominant clade (Figure 1B). This was associated with a peak in December and January (Figure 1C). The decline in 20G in February and March coincided with decreased positivity rates, and as clade 20I/501Y.V1 started to increase in frequency, positivity rates increased, proportions of positive samples from black patients increased, and the mean age of patients dropped (Figure 2A and 2B). The mean age for black patients was significantly lower than that of white patients (43.6 vs 50.1 years, respectively; t test, P < .001) (Figure 2C and 2D). The mean Ct values were comparable in different race populations, with consistently lower Ct values in symptomatic versus asymptomatic patients (as determined by the ordering test codes that differentiate symptomatic from asymptomatic patients at the time of sample collection, Figure 2E and 2F), and 82%–85% of patients were symptomatic.
Figure 2.

Patient demographics in all patients with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) diagnosed at Johns Hopkins Hospital (A, C, E) and SARS-CoV-2–positive results characterized by whole-genome sequencing (B, D, F), from November 2020 to the end of March 2021. A, B, Patient race by percentage (white [blue line], black [orange line], Asian [green line], or other/unknown [dark red line]) and 15-day rolling mean age (purple line). C, D, Age of symptomatic and asymptomatic patients, by race. E, F, Cycle threshold (Ct) for positive results in symptomatic and asymptomatic patients, by race.
Genomic Diversity in the National Capital Region Between November 2020 and March 2021
A high degree of diversity was noted from November 2020 until the end of March 2021 (Figure 3 and Supplementary Table 2). The most common clades were 20G (491 samples), 20I (318 samples), 20C (302 samples), 20A (262 samples), and 20B (193 samples). Within each clade, lineage frequencies varied (Supplementary Table 2). Although the proportion of lineages changed drastically over this period (Figure 4A), the most common lineages were B.1.2 (396 samples), B.1.1.7 (318 samples), B.1.243 (126 samples), B.1.596 (95 samples), and B.1.526.1 (78 samples) (Figure 4B). The average patient age was 40.7 years in this data set, with a significant association between age and lineage (Kruskal-Wallis test, P = .002). Post hoc Conover analysis with Holmes adjustment on lineages with >50 samples showed significant differences between the dominant lineages, B.1.2 (mean patient age, 43.3 years) and B.1.1.7 (37.5 years) (Figure 4C). Lineages had comparable Ct value distributions (Figure 4D). Lineages B.1.2 and B.1.596 were associated with white race, with 54% (P = 4.4 × 10−11) and 62.5% (P = .03) of samples from white patients (Figure 4E). In contrast, B.1.1.7 was associated with being black (P = 1.3 × 10−7).
Figure 3.

Phylogenetic relatedness of severe acute respiratory syndrome coronavirus 2 genomes sequenced at Johns Hopkins between November 2020 and the end of March 2021. The tree was generated using Nextstrain [26].
Figure 4.

Genomic diversity over time and associated patient demographics. A, Percentage of predominant lineages over time. B, Total numbers of key lineages over the surveillance period. C, Patient ages for predominant and key lineages, colored by clade. D, Cycle threshold (Ct) values in predominant and key lineages, colored by clade. E, Patient race by percentage, in association with predominant and key lineages. *P < .05.
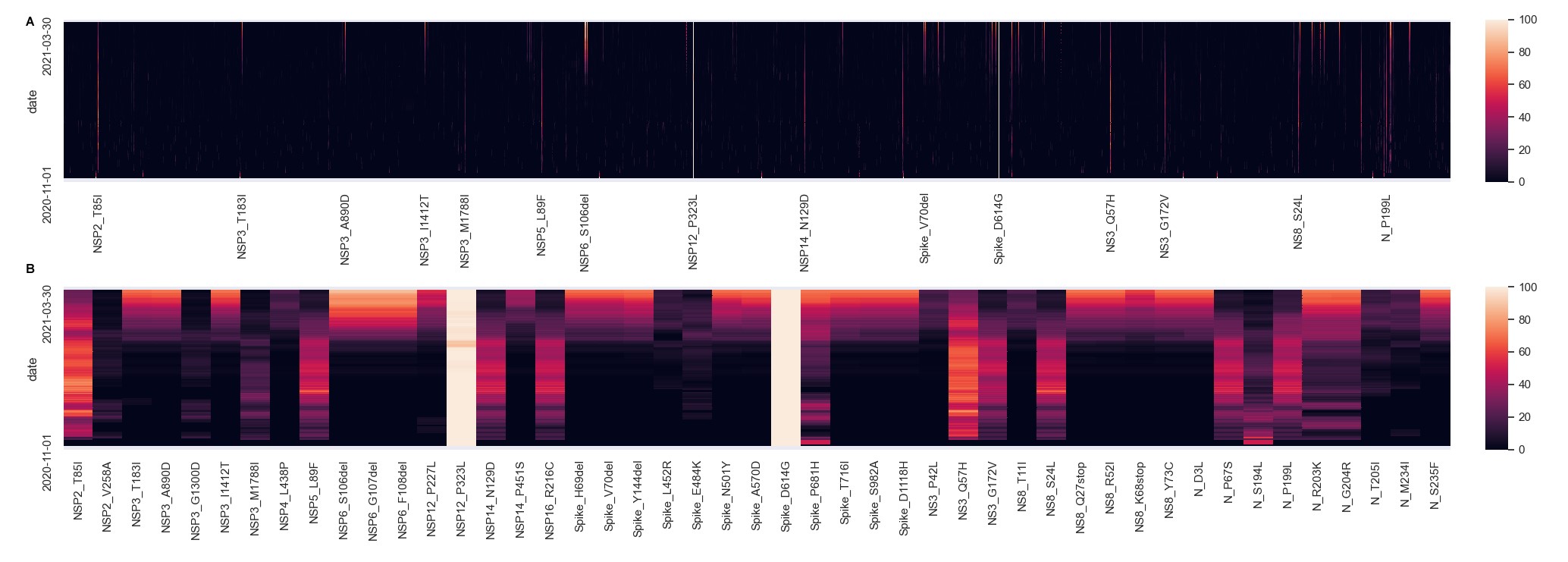
There were 1973 distinct individual amino acid substitutions, 78% of present in ≤5 samples. A heat map of the percentage of samples positive for each amino acid substitution over time (rolling 7-day average) highlights the rarity of most of these mutations (Supplementary Figure 1). Filtering for more common substitutions (Supplementary Figure 1B) showed a major increase in February in changes associated with B.1.1.7 (eg, S:N501Y, S:A570D, S:T716I, S:S982A, and S:D1118H) while substitutions associated with B.1.2 declined. Others, such as S:P681H, S:L452R, N:R203K, and S:E484K, were present in multiple lineages and showed an undulating pattern (Supplementary Table 2). The most common amino acid substitutions across lineages were S:D614G, NSP12:P323L, NSP2:T85I, and NS3:Q57H. In Supplementary Table 2, we list “shared” mutations for each lineage (defined as present in >90% of the samples of that lineage within our data set). The lineages with the highest number of shared amino acid changes were B.1.1.7 [24], B.1.1.318 [23], B.1.351 [20], and B.1.526.1 [20].
Genomic Changes in the National Capital Region Over 1 Year
Genomes from March 2021 were compared with those from March 2020 [3]. Of the 20 lineages circulating in this area in March 2020, only 3 (B.1, B.1.1, and B.1.1.207) were seen but rarely during March 2021 (Figure 5A). Twenty-eight lineages were present in March 2021 compared with 20 in March 2020. Diversity increased in NSP3, NS3 and the spike protein, but was similar in NSP12 and NSP14 in the 2 time frames (Figure 5B). Only 6 spike protein mutations were present in March 2020, compared with 105 in March 2021.
Figure 5.

Genomic changes in the National Capital Region over 1 year. A, Characterized lineages from March 2020 and March 2021. B, Number of unique amino acid substitutions and deletions viral encoded proteins from March 2020 and March 2021. C, Heat map of amino acid substitutions and deletions present in March 2021 and March 2020.
The average number of amino acid substitutions increased from 5.2 to 23.3, with 93 unique substitutions present in March 2020, and 733 in March 2021. Of the 93 substitutions that were present in 2020, 61 were no longer present during March 2021 (Figure 5C), and only 8 were present in >5% of samples during March 2021 (NSP2:T85I, NSP12:P323L, S:L5F, S:D614G, NS3aQ57H, NS8:S24L, N:R203K, and N:G204R).
VOCs, VOIs, and Regionally Circulating Variants
The first VOC detected in our cohort was B.1.1.7 in January 2021 which increased to approximately 70% of our new samples in the end of March (Figure 1B). While B.1.351 was also first noted in January, only 15 samples of this lineage were sequenced in this time frame. B.1.429 first appeared in December 2021 and peaked in January 2021 (24 samples total). P.1 and B.1.427 were each present in only 1 sample. The VOI B.1.526.1, present in 78 samples since first being detected in late January, has become more predominant over time. This lineage, similar to B.1.429, harbors the S: L452R. Other lineages with L452R included A.2.5, B.1.526.1, and B.1.1.487 (Supplementary Figure 2).
The most common lineage to carry S:E484K was a subset of B.1.1.207 (46 samples), which also carried the S:P681H mutation, followed by lineage R.1 (40 samples). The presence of 2 lineages with S:P681H and S:E484K (B.1.1.207 and B.1.1.318) within this region was initially concerning, as these changes were found in VOCs but almost never reported together (Supplementary Figure 2). Only a subset of B.1.1.207 carried S:E484K, and this lineage has dropped off in frequency. None of the patients with these lineages required hospitalization for COVID-19 symptoms (Supplementary Table 1 and Table 1; local Maryland variant). The other lineage, B.1.1.318 within our population with both S:E484K and S:P681H, was present in 6 samples between mid-February and mid-March. Variants carrying S: P681H but not the S: E484K belonged to diverse lineages (Supplementary Figure 2).
Table 1.
Demographic and Clinical Characteristics of Patients Infected With a Local Maryland Variant
| Demographic and Clinical Characteristics | Patients, No. (%)a (n = 44) |
|---|---|
| Age, median (range), y | 49 (1.2–84) |
| No. (%) of | |
| Sex | |
| Male | 18 (40.9) |
| Female | 26 (59.1) |
| Comorbid conditions (n = 34)b | |
| Total | 19/34 (55.9) |
| Obesity (BMI, >30c) | 7 (20.6) |
| Hypertension | 10 (29.4) |
| Cardiovascular disease | 4 (11.7) |
| Diabetes | 2 (5.9) |
| Asthma/allergic rhinitis | 4 (11.8) |
| Transplant recipient | 1 (2.9) |
| Admission status (basis for severity index) | |
| Outpatient | 40 (90.9) |
| Inpatient | 4 (9.1) |
| ICU | 1 (2.3) |
| Admission status by age | |
| ≤40 y | 17 (100) |
| >40–55 y | |
| Outpatient | 12 (92.3) |
| Inpatient | 1 (7.7) |
| >55 y | |
| Outpatient | 11 (78.6) |
| Inpatient | 3 (21.4) |
| ICU | 1 (7.1) |
| Positive after COVID-19 vaccine | 4 (9.1)d |
| Travel history | 0 (0) |
Abbreviations: BMI, body mass index; COVID-19, coronavirus disease 2019; ICU, intensive care unit.
aData represent no. (%) of patients unless otherwise specified.
bInformation on comorbid conditions was not available for 10 patients.
cBMI was calculated as weight in kilograms divided by height in meters squared.
dPositive results occurred 3 days after the first vaccine dose in 2 patients, 7 days after the first dose in 1, and 3 weeks after the first dose in 1.
Mutations, Lineages, and COVID-19 Outcomes
We performed detailed record reviews in 116 patients (≥30 days after the date of sample collection) with hospital admissions in our cohort associated with the characterized samples. Sixteen patients were asymptomatic, and 87 were admitted for COVID-19. Eleven were admitted to the ICU with no associated deaths, and another 8 died. As expected, infections associated with hospitalization were caused by the more prevalent lineages (Figure 6A). However, B.1.243 was associated with noticeably high levels of ICU admissions or death, given its prevalence (Fisher exact test with Bonferroni correction P = .04; accounting for the total number of samples from each lineage) (Figure 6A; for lineage prevalence, refer to Figure 4B and Supplementary Table 2). Amino acid changes in hospitalized patients compared with all patients with ≥10% prevalence and a 5% change between the 2 groups showed that 4 mutations (S: P681H, NSP6: 106–108del, N: S194L, and N: T205I) were present to a higher degree, but not significantly higher or didn’t reach statistical significance. Record reviews on all B.1.243-infected patients (122 patients) compared with all B.1.2-infected patients (as a control group; 395 patients) showed that the odds ratio for ICU admission or death associated with B.1.243 compared with B.1.2 infection was 4.9 (95% confidence interval, 1.4–17.9; P = .02) (Supplementary Figure 3). Notably, this analysis did not adjust for patients’ metadata or underlying conditions.
Figure 6.

Association of lineages with severe coronavirus disease 2019. A, Number of samples by lineage with hospital admission status separated by disease severity. Numbers above bars represent percentages of the total for each lineage. *P < .05. Abbreviation: ICU, intensive care unit. B, Heat map of percentages of samples with amino acid changes that showed at ≥5% of samples from hospitalized patients, compared with all samples.
Genomes From Fully Vaccinated Patients
Thirty-six positive samples were collected between January and April from patients who tested positive after completing 2 doses of the COVID-19 Pfizer or Moderna vaccine. Only 14 of these had complete or near-complete genomes, with most genomes belonging to the B.1.1.7 variant (Table 2). The Ct values from the clinical assays of positive samples after full vaccination ranged from 14.4 to 37.9 (23 samples with available Ct values), and low Ct values were correlated with higher depth and coverage of genome sequencing. More than half were in symptomatic patients, but all patients experienced only mild disease, which did not require hospitalization (Table 2).
Table 2.
Demographic and Clinical Characteristics of Patients With Positive Results for Coronavirus Disease 2019 After Full Vaccination
| Demographic and Clinical Characteristics | Patients, No. (n = 36)a |
|---|---|
| Age, median (range) | 47 (23 to >90) |
| Sex | |
| Male | 15 |
| Female | 21 |
| Symptomatic | |
| Yes | 19 |
| No | 17 |
| Comorbid conditions | |
| Obesity (BMI, >30b) | 11 |
| Hypertension | 11 |
| Cardiovascular disease | 7 |
| Diabetes | 2 |
| Asthma | 2 |
| Allergic rhinitis | 2 |
| History of cancer/ autoimmune disease/HIV/possible immunocompromise | 4 |
| Outpatient status (no admission) | 36 |
| Interval between 2nd vaccine dose and positive COVID-19 result, median (range), d | 31.5 (4–75) |
| Clinical assay Ct, median (range)c | 26.13 (14.35–37.88) |
| Data for 14 genomes with >90% coverage | |
| B.1.1.7 (collection in March 2021) | 9 |
| B.1.526 (collection in March 2021) | 2 |
| B.1.526.1 (collection in March 2021) | 1 |
| B.1.2 (collection in January 2021) | 1 |
| B.1.1 (collection in January 2021) | 1 |
Abbreviations: BMI, body mass index; COVID-19, coronavirus disease 2019; Ct, cycle threshold; HIV, human immunodeficiency virus.
aData represent no. of patients unless otherwise specified.
bBMI was calculated as weight in kilograms divided by height in meters squared.
cData available for only 23 samples.
DISCUSSION
Whole-genome sequencing for surveillance has been critical for monitoring the evolution of SARS-CoV-2 [3]. Our data showed an increase in the diversity of the spike protein and other genomic regions that include the NSP3 and NS3 over time. A shift from 20C clade in March 2020 to 20G by the end of the year and then to 20I/50Y.V1 in March 2021 was evident. The most common amino acid substitutions were extra-spike and included stable changes (eg, NS3:Q57H) and changes that increased (multiple NSP3 and NS8 changes). The spike amino acid changes that were most common after S: D614G included N501Y and P681H, with a notable increase in diversity compared with March 2020. A marked increase in B.1.1.7 started in February 2021. A regional variant was detected with increased prevalence in February 2021 that combined S: P681H and S: E484K. Notably, the circulation of variants carrying S: E484K has been temporary and the prevalence of this change remains sparse. On the other hand, a global increase in S: P681H is notable as a part of B.1.1.7.
Among the most frequently detected polymorphisms were the NSP6 deletions 106–108 that were present at a higher prevalence in hospitalized patients’ genomes. NSP6 was shown to have a role in autophagosome generation [28] and these 3 amino acids are in a loop predicted to be external to the autophagy vesicles [28]. NSP6 was also shown to antagonize type I interferon and hence has a role in the evasion of the innate immune responses [29]. NSP3 showed the most increased diversity in March 2021, compared with March 2020. This protein is the largest protein encoded by coronaviruses, has multiple domains and functions, and is essential for viral replication [30].
The evolution of VOCs was associated with a displacement of previous lineages. B.1.1.7 has become predominant by the end of March 2021. It is interesting that this variant was more successful than B.1.351, as both were circulating at the end of January 2021 [31]. Our data show that variants that carried S: E484K showed temporary circulation, in contrast to variants with S: P681H. The S: E484K variants B.1.351 and P1, however, were more successful in South Africa and South America, respectively. A newly emerging variant from India (21A, B.1.617.2; delta) is currently reported from multiple regions and is displacing B.1.1.7 (https://www.gisaid.org/hcov19-variants/). The genomic determinants of the success of certain variants in specific geographic locations or specific demographic groups remain an enigma, and factors that include previous natural infections, vaccination status, and vaccine efficacy for a geographic location likely affect these associations [32, 33]. Specifically, the association of lineage B.1.1.7 with younger age may be the result of preferential vaccination of older individuals before the emergence of this lineage in our geographic region.
The relationship between viral genomic polymorphism and the change in disease severity has been an area of debate. Earlier research proposed an association of the S:D614G with mortality [34]; however, this might be difficult to interpret owing to this substitution’s early global dominance [35]. The evolution of VOCs emphasizes a natural selection in favor of these variants. Early studies concluded that there was no association between the B.1.1.7 and enhanced severity [36], but other studies proposed a correlation with higher mortality [37] and hospitalization [38] rates. Signature mutations in the B.1.1.7, P1 and B.1.351 variants, including S: N501Y, could affect binding to ACE2 [39, 40]. Polymorphisms in ACE2 were shown to affect the COVID-19 outcome [41]; hence, variants that affect ACE2 binding might have an effect on disease severity [42].
Interestingly, we noticed a significant association of B.1.243 with ICU admission and mortality rate. This lineage shows amino acid substitutions NSP12:P323L, N:S194L,S:D614G, and S:P681H in >90% of specimens. It was also the only lineage with the occasional co-occurrence of N:T205I and N:S194L, which were both seen in higher percentages of sequences from known hospitalized patients. B.1.243 has substitution NSP3:G1300D, which was present in the genomes of all of the hospitalized patients, but in only about 80% of the total genomes of this lineage. Ongoing work by our group using cell culture and hamster models aims at examining the direct association of lineage B.1.243 with an increase in viral fitness or pathogenesis in well-controlled experiments. In addition, large-scale retrospective whole-genome sequencing between April 2020 and November 2020 is currently in progress to validate our observations.
With the nationwide increase in vaccination, the correlation between certain variants and breakthrough infections is an area of investigation. As of 26 April 2021, the Centers for Disease Control and Prevention reported 9245 positive cases after full vaccination, of a total of >95 million vaccinated individuals in the United States. Whether VOCs are more associated with infection after vaccination or escape of vaccine-induced immune responses will be challenging to investigate, owing to the relative infrequency of breakthrough cases and the increased prevalence of VOCs. A study from Israel proposed that vaccine breakthrough cases are more frequent with B.1.1.7 and B.1.351 [32]. Our data showed that among the 36 cases only 14 complete genomes were recovered, which was tightly correlated with the viral load in the respiratory specimens. The lineages detected represented the commonly circulating lineages in the time frame of specimen collection (Table 2).
In conclusion, it is essential to characterize SARS-CoV-2 evolving variants in real time. We have implemented a surveillance protocol that allows us to identify predominant and novel variants. The limitations of our study include the relatively small number of patients with severe disease, which limited the adjustment of lineage associations with disease outcome to patients’ metadata and underlying conditions and limited the restricted analysis to genomes of good quality, which might be tightly associated with viral loads at the time of sample collection.
Supplementary Data
Supplementary materials are available at Clinical Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
{kind=link}
{kind=link}
{kind=link}
Notes
Author contributions. Study design: H. H. M. Data collection: M. S. Data acquisition: N. G. Cell cultures: A. P. Data collection and analysis: C. P. M., C. H. L., A. A., and H. H. M. Data interpretation: C. P. M. and H. H. M. Figures: C. P. M. Manuscript writing: C. P. M. and H. H. M. Scientific revision: S. C. R. and A. P. Fund acquisition: S. C. R. and H. H. M.
Acknowledgments. This study was possible only with the unique efforts of the Johns Hopkins Clinical Microbiology Laboratory faculty and staff.
Disclaimer. The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Institute of Biomedical Imaging and Bioengineering; the National Heart, Lung, and Blood Institute; the National Institutes of Health, or the US Department of Health and Human Services.
Financial support. This work was supported by the intramural research program of the National Institutes of Health (NIH) and the Centers for Disease Control and Prevention.
S. C. R. is supported by the Maryland Department of Health (funding of H. H. M.’s work, under a contract they helped craft). H. H. M. is supported by the following: the HIV Prevention Trials Network, sponsored by the National Institute of Allergy and Infectious Diseases , the National Institute on Drug Abuse, and the National Institute of Mental Health, and the Office of AIDS Research, NIH (grant UM1 AI068613), the NIH RADx-Tech program (grant 3U54HL143541-02S2), the NIH RADx-UP initiative (grant R01 DA045556-04S1), the Johns Hopkins University President’s Fund Research Response, the Johns Hopkins Department of Pathology, the Maryland Department of Health, Johns Hopkins University, and NIH grants, paid as salary and indirectly to H. H. M.’s institution (grants UM1AI0686, UM1 AI068613-14, R01 DA045556-04S1, and 3U54HL143541-02S2).
Potential conflicts of interest. S. C. R. reports the following: support from the NIH (to Johns Hopkins University, including K08, R01, and U19 grants) and miDiagnostics (research collaboration governed by a contract between miDiagnostics and Johns Hopkins University for which S. C. R. is the principal investigator); travel support from the NIH Fogarty International Center (for teaching science writing at Makerere University); the following patents: USPTO 8168771 (Use of consensus sequence as vaccine antigen to enhance recognition of virulent viral variants), USPTO 9512183 (Synthetic hepatitis C genome and methods of making and use), USPTO 10788480 (Aggregation-assisted separation of plasma from whole blood; Application 20200222528 (Nucleoside-modified mRNA-lipid nanoparticle lineage vaccine for hepatitis C virus), and PCT 16/078 760 (Novel antiviral proteins and their uses in therapeutic methods); and participation in a therapeutics and prevention data and safety monitoring board for the National Institute of Allergy and Infectious Diseases, all outside the submitted work. H. H. M. reports the following: receiving research contract, reagents, and equipment from DiaSorin Molecular; participation in clinical trials with NIH/Centers for Disease Control and Prevention/Biomedical Advanced Research and Development Authority; serving as co–principal investigator for Johns Hopkins Center of Excellence in Influenza Research and Surveillance HHSN272201400007C; receiving an Fisher Center Discovery Program grant, to Johns Hopkins University, as principal investigator for enterovirus research; receiving research contract, reagents, and equipment from BioRad; receiving NIH grants for human papillomavirus research (grants 4UH3CA211396-03 and R01 CA243393); receiving consulting fees from Hologic, AlphaSight, and Guidepoint; and receiving honoraria/payments from Qiagen and GenMark for speaking, manuscript writing, or educational events, all outside the submitted work. All other authors report no potential conflicts. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1. Wang C, Liu Z, Chen Z, et al. The establishment of reference sequence for SARS-CoV-2 and variation analysis. J Med Virol 2020; 92:667–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Morais Júnior IJ, Polveiro RC, Souza GM, Bortolin DI, Sassaki FT, Lima ATM. The global population of SARS-CoV-2 is composed of six major subtypes. Sci Rep 2020; 10:18289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Thielen PM, Wohl S, Mehoke T, et al. Genomic diversity of SARS-CoV-2 during early introduction into the Baltimore-Washington metropolitan area. JCI Insight 2021; 6:e144350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brufsky A. Distinct viral clades of SARS-CoV-2: implications for modeling of viral spread. J Med Virol 2020; 92:1386–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Laamarti M, Alouane T, Kartti S, et al. Large scale genomic analysis of 3067 SARS-CoV-2 genomes reveals a clonal geo-distribution and a rich genetic variations of hotspots mutations. PLoS One 2020; 15:e0240345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Korber B, Fischer WM, Gnanakaran S, et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020; 182:812–827.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lokman SM, Rasheduzzaman M, Salauddin A, et al. Exploring the genomic and proteomic variations of SARS-CoV-2 spike glycoprotein: a computational biology approach. Infect Genet Evol 2020; 84:104389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ostrov DA. Structural consequences of variation in SARS-CoV-2 B.1.1.7. J Cell Immunol 2021; 3:103–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Moore JP, Offit PA. SARS-CoV-2 vaccines and the growing threat of viral variants. JAMA 2021; 325:821–2. [DOI] [PubMed] [Google Scholar]
- 10. Leung K, Shum MH, Leung GM, Lam TT, Wu JT. Early transmissibility assessment of the N501Y mutant strains of SARS-CoV-2 in the United Kingdom, October to November 2020. Euro Surveill 2021; 26:2002106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhao S, Lou J, Cao L, et al. Quantifying the transmission advantage associated with N501Y substitution of SARS-CoV-2 in the United Kingdom: an early data-driven analysis. J Travel Med 2021; 28:taab011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Challen R, Brooks-Pollock E, Read JM, Dyson L, Tsaneva-Atanasova K, Danon L. Risk of mortality in patients infected with SARS-CoV-2 variant of concern 202012/1: matched cohort study. BMJ 2021; 372:n579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huang Y, Yang C, Xu XF, Xu W, Liu SW. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol Sin 2020; 41:1141–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang Q, Qiu Y, Li JY, Zhou ZJ, Liao CH, Ge XY. A unique protease cleavage site predicted in the spike protein of the novel pneumonia coronavirus (2019-nCoV) potentially related to viral transmissibility. Virol Sin 2020; 35:337–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wibmer CK, Ayres F, Hermanus T, et al. SARS-CoV-2 501Y.V2 escapes neutralization by South African COVID-19 donor plasma. Nat Med 2021; 27:622–5. [DOI] [PubMed] [Google Scholar]
- 16. Tada T, Dcosta BM, Samanovic-Golden M, et al. Neutralization of viruses with European, South African, and United States SARS-CoV-2 variant spike proteins by convalescent sera and BNT162b2 mRNA vaccine-elicited antibodies. bioRxiv [Preprint]. February 7, 2021. Available from: https://www.biorxiv.org/content/10.1101/2021.02.05.430003v1. [Google Scholar]
- 17. Zhang W, Davis BD, Chen SS, Sincuir Martinez JM, Plummer JT, Vail E. Emergence of a novel SARS-CoV-2 variant in Southern California. JAMA 2021; 325:1324–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li Q, Wu J, Nie J, et al. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell 2020; 182:1284–1294.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Deng X, Garcia-Knight MA, Khalid MM, et al. Transmission, infectivity, and antibody neutralization of an emerging SARS-CoV-2 variant in California carrying a L452R spike protein mutation. medRxiv [Preprint]. March 9, 2021. Available from: https://www.medrxiv.org/content/10.1101/2021.03.07.21252647v1. [Google Scholar]
- 20. West AP, Barnes CO, Yang Z, Bjorkman PJ. SARS-CoV-2 lineage B.1.526 emerging in the New York region detected by software utility created to query the spike mutational landscape. bioRxiv [Preprint]. February 23, 2021. Available from: https://www.biorxiv.org/content/10.1101/2021.02.14.431043v2. [Google Scholar]
- 21. Mostafa HH, Hardick J, Morehead E, Miller JA, Gaydos CA, Manabe YC. Comparison of the analytical sensitivity of seven commonly used commercial SARS-CoV-2 automated molecular assays. J Clin Virol 2020; 130:104578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mostafa HH, Lamson DM, Uhteg K, et al. Multicenter evaluation of the NeuMoDx™ SARS-CoV-2 Test. J Clin Virol 2020; 130:104583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mostafa HH, Carroll KC, Hicken R, et al. Multi-center evaluation of the cepheid Xpert(R) Xpress SARS-CoV-2/Flu/RSV test. J Clin Microbiol 2020; 59:e02955-20. [Google Scholar]
- 24. Jarrett J, Uhteg K, Forman MS, et al. Clinical performance of the GenMark Dx ePlex respiratory pathogen panels for upper and lower respiratory tract infections. J Clin Virol 2021; 135:104737. [DOI] [PubMed] [Google Scholar]
- 25. Uhteg K, Jarrett J, Richards M, et al. Comparing the analytical performance of three SARS-CoV-2 molecular diagnostic assays. J Clin Virol 2020; 127:104384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hadfield J, Megill C, Bell SM, et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 2018; 34:4121–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sheskin DJ. Handbook of parametric and nonparametric statistical procedures. 3rd ed. Boca Raton, FL: CHC, 2003. [Google Scholar]
- 28. Benvenuto D, Angeletti S, Giovanetti M, et al. Evolutionary analysis of SARS-CoV-2: how mutation of non-structural protein 6 (NSP6) could affect viral autophagy. J Infect 2020; 81:e24–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xia H, Cao Z, Xie X, et al. Evasion of type I interferon by SARS-CoV-2. Cell Rep 2020; 33:108234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lei J, Kusov Y, Hilgenfeld R. Nsp3 of coronaviruses: structures and functions of a large multi-domain protein. Antiviral Res 2018; 149:58–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Feder KA, Pearlowitz M, Goode A, et al. Linked clusters of SARS-CoV-2 variant B.1.351—Maryland, January–February 2021. MMWR Morb Mortal Wkly Rep 2021; 70:627–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kustin T, Harel N, Finkel U, et al. Evidence for increased breakthrough rates of SARS-CoV-2 variants of concern in BNT162b2 mRNA vaccinated individuals. medRxiv [Preprint]. April 9, 2021. Available from: https://www.medrxiv.org/content/10.1101/2021.04.06.21254882v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hacisuleyman E, Hale C, Saito Y, et al. Vaccine breakthrough infections with SARS-CoV-2 variants. N Engl J Med 2021; 384:2212–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Toyoshima Y, Nemoto K, Matsumoto S, Nakamura Y, Kiyotani K. SARS-CoV-2 genomic variations associated with mortality rate of COVID-19. J Hum Genet 2020; 65:1075–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lauring AS, Hodcroft EB. Genetic variants of SARS-CoV-2—what do they mean? JAMA 2021; 325:529–31. [DOI] [PubMed] [Google Scholar]
- 36. Davies NG, Abbott S, Barnard RC, et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021; 372:eabg3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Davies NG, Jarvis CI, van Zandvoort K, et al. Increased mortality in community-tested cases of SARS-CoV-2 lineage B.1.1.7. Nature 2021; 593:270–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Frampton D, Rampling T, Cross A, et al. Genomic characteristics and clinical effect of the emergent SARS-CoV-2 B.1.1.7 lineage in London, UK: a whole-genome sequencing and hospital-based cohort study. Lancet Infect Dis 2021; doi: 10.1016/S1473-3099(21)00170-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ali F, Kasry A, Amin M. The new SARS-CoV-2 strain shows a stronger binding affinity to ACE2 due to N501Y mutant. Med Drug Discov 2021; 10:100086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Williams AH, Zhan CG. Fast prediction of binding affinities of the SARS-CoV-2 spike protein mutant N501Y (UK variant) with ACE2 and miniprotein drug candidates. J Phys Chem B 2021; 125:4330–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Devaux CA, Rolain JM, Raoult D. ACE2 receptor polymorphism: Susceptibility to SARS-CoV-2, hypertension, multi-organ failure, and COVID-19 disease outcome. J Microbiol Immunol Infect 2020; 53:425–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brest P, Refae S, Mograbi B, Hofman P, Milano G. Host polymorphisms may impact SARS-CoV-2 infectivity. Trends Genet 2020; 36:813–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.