

Abstract
Background
Remdesivir is an antiviral medicine with properties to inhibit viral replication of SARS‐CoV‐2. Positive results from early studies attracted media attention and led to emergency use authorisation of remdesivir in COVID‐19. A thorough understanding of the current evidence regarding the effects of remdesivir as a treatment for SARS‐CoV‐2 infection based on randomised controlled trials (RCTs) is required.
Objectives
To assess the effects of remdesivir compared to placebo or standard care alone on clinical outcomes in hospitalised patients with SARS‐CoV‐2 infection, and to maintain the currency of the evidence using a living systematic review approach.
Search methods
We searched the Cochrane COVID‐19 Study Register (which comprises the Cochrane Central Register of Controlled Trials (CENTRAL), PubMed, Embase, ClinicalTrials.gov, WHO International Clinical Trials Registry Platform, and medRxiv) as well as Web of Science (Science Citation Index Expanded and Emerging Sources Citation Index) and WHO COVID‐19 Global literature on coronavirus disease to identify completed and ongoing studies without language restrictions. We conducted the searches on 16 April 2021.
Selection criteria
We followed standard Cochrane methodology.
We included RCTs evaluating remdesivir for the treatment of SARS‐CoV‐2 infection in hospitalised adults compared to placebo or standard care alone irrespective of disease severity, gender, ethnicity, or setting.
We excluded studies that evaluated remdesivir for the treatment of other coronavirus diseases.
Data collection and analysis
We followed standard Cochrane methodology.
To assess risk of bias in included studies, we used the Cochrane RoB 2 tool for RCTs. We rated the certainty of evidence using the GRADE approach for outcomes that were reported according to our prioritised categories: all‐cause mortality at up to day 28, duration to liberation from invasive mechanical ventilation, duration to liberation from supplemental oxygen, new need for mechanical ventilation (high‐flow oxygen, non‐invasive, or invasive mechanical ventilation), new need for invasive mechanical ventilation, new need for non‐invasive mechanical ventilation or high‐flow oxygen, new need for oxygen by mask or nasal prongs, quality of life, serious adverse events, and adverse events (any grade).
Main results
We included five RCTs with 7452 participants diagnosed with SARS‐CoV‐2 infection and a mean age of 59 years, of whom 3886 participants were randomised to receive remdesivir. Most participants required low‐flow oxygen (n=4409) or mechanical ventilation (n=1025) at baseline. Studies were mainly conducted in high‐ and upper‐middle‐income countries. We identified two ongoing studies, one was suspended due to a lack of COVID‐19 patients to recruit.
Risk of bias assessments were considered to be some concerns or high risk for clinical status and safety outcomes because participants who had died did not contribute information to these outcomes. Without adjustment, this leads to an uncertain amount of missing values and the potential for bias due to missing data.
Effects of remdesivir in hospitalised individuals
Remdesivir probably makes little or no difference to all‐cause mortality at up to day 28 (risk ratio (RR) 0.93, 95% confidence interval (CI) 0.81 to 1.06; risk difference (RD) 8 fewer per 1000, 95% CI 21 fewer to 7 more; 4 studies, 7142 participants; moderate‐certainty evidence). There was limited evidence for a beneficial effect of remdesivir on mortality in a subset of 435 participants who received low flow oxygen at baseline in one study (RR 0.32, 95% CI 0.15 to 0.66). We could not confirm this finding due to restricted availability of relevant subgroup data from other studies.
Remdesivir may have little or no effect on the duration to liberation from invasive mechanical ventilation (2 studies, 1298 participants, data not pooled, low‐certainty evidence). We are uncertain whether remdesivir increases or decreases the chance of clinical improvement in terms of duration to liberation from supplemental oxygen at up to day 28 (3 studies, 1691 participants, data not pooled, very low‐certainty evidence).
We are very uncertain whether remdesivir decreases or increases the risk of clinical worsening in terms of new need for mechanical ventilation at up to day 28 (high‐flow oxygen or non‐invasive ventilation or invasive mechanical ventilation) (RR 0.78, 95% CI 0.48 to 1.24; RD 29 fewer per 1000, 95% CI 68 fewer to 32 more; 3 studies, 6696 participants; very low‐certainty evidence); new need for non‐invasive mechanical ventilation or high‐flow oxygen (RR 0.70, 95% CI 0.51 to 0.98; RD 72 fewer per 1000, 95% CI 118 fewer to 5 fewer; 1 study, 573 participants; very low‐certainty evidence); and new need for oxygen by mask or nasal prongs (RR 0.81, 95% CI 0.54 to 1.22; RD 84 fewer per 1000, 95% CI 204 fewer to 98 more; 1 study, 138 participants; very low‐certainty evidence). Remdesivir may decrease the risk of clinical worsening in terms of new need for invasive mechanical ventilation (67 fewer participants amongst 1000 participants; RR 0.56, 95% CI 0.41 to 0.77; 2 studies, 1159 participants; low‐certainty evidence).
None of the included studies reported quality of life.
Remdesivir probably decreases the serious adverse events rate at up to 28 days (RR 0.75, 95% CI 0.63 to 0.90; RD 63 fewer per 1000, 95% CI 94 fewer to 25 fewer; 3 studies, 1674 participants; moderate‐certainty evidence). We are very uncertain whether remdesivir increases or decreases adverse events rate (any grade) (RR 1.05, 95% CI 0.86 to 1.27; RD 29 more per 1000, 95% CI 82 fewer to 158 more; 3 studies, 1674 participants; very low‐certainty evidence).
Authors' conclusions
Based on the currently available evidence remdesivir probably has little or no effect on all‐cause mortality at up to 28 days in hospitalised adults with SARS‐CoV‐2 infection. We are uncertain about the effects of remdesivir on clinical improvement and worsening. There were insufficient data available to examine the effect of remdesivir on mortality across subgroups defined by respiratory support at baseline.
Future studies should provide additional data on efficacy and safety of remdesivir for defined core outcomes in COVID‐19 research, especially for different population subgroups. This could allow us to draw more reliable conclusions on the potential benefits and harms of remdesivir in future updates of this review. Due to the living approach of this work, we will update the review periodically.
Plain language summary
Remdesivir to treat people with COVID‐19
Is remdesivir (an antiviral medicine) an effective treatment for COVID‐19?
Key messages
• For adults hospitalised with COVID‐19, remdesivir probably has little or no effect on deaths from any cause up to 28 days after treatment compared with placebo (sham treatment) or usual care.
• We are uncertain whether remdesivir improves or worsens patients’ condition, based on whether they needed more or less help with breathing.
• Researchers should agree on key outcomes to be used in COVID‐19 research, and future studies should investigate these areas. This would allow future updates of this review to draw more certain conclusions about the use of remdesivir to treat COVID‐19.
What is remdesivir?
Remdesivir is a medicine that fights viruses. It has been shown to prevent the virus that causes COVID‐19 (SARS‐CoV‐2) from reproducing. Medical regulators have approved remdesivir for emergency use to treat people with COVID‐19.
What did we want to find out?
We wanted to know if remdesivir is an effective treatment for people in hospital with COVID‐19 and if it causes unwanted effects compared to placebo or usual care.
People with COVID‐19 are given different kinds of breathing support, depending on how severe their breathing difficulties are. We used the types of breathing support people received as a measure of the success of remdesivir in treating COVID‐19. Types of breathing support included:
• for severe breathing difficulties: invasive mechanical ventilation, when a breathing tube is put into patients’ lungs, and a machine (ventilator) breathes for them. Patients are given medicine to make them sedated whilst they are on a ventilator.
• for moderate to severe breathing difficulties: non‐invasive mechanical ventilation through a mask over the nose and/or mouth, or a helmet. Air or oxygen is pushed through the mask. Patients are generally awake for this treatment.
• for moderate breathing difficulties: oxygen via a mask or prongs that sit in the nostrils. Patients can still breathe room air.
We were interested in the following outcomes:
• deaths from any cause in the 28 days after treatment;
• whether patients got better after treatment, measured by how long they spent on mechanical ventilation or oxygen;
• whether patients’ condition worsened so that they needed oxygen or mechanical ventilation;
• quality of life;
• any unwanted effects; and
• serious unwanted effects.
What did we do?
We searched for studies that investigated remdesivir to treat adults with COVID‐19 compared to placebo or standard care. Patients were hospitalised with COVID‐19 and could be of any gender or ethnicity.
We compared and summarised the results of the studies and rated our confidence in the evidence, based on factors such as study methods and sizes.
What did we find?
We found 5 studies with 7452 people hospitalised with COVID‐19. Of these, 3886 people were given remdesivir. The average age of patients was 59 years. Studies took place around the world, mainly in high‐ and upper‐middle‐income countries.
Main results
The included studies compared remdesivir to placebo or usual care in people hospitalised with COVID‐19 for up to 28 days.
Deaths from any cause
• Remdesivir probably makes little or no difference to deaths from any cause (4 studies, 7142 people). In 1000 people, 8 fewer die with remdesivir compared to placebo or standard care.
Did patients get better with remdesivir?
• Remdesivir may have little or no effect on the length of time patients spent on invasive mechanical ventilation (2 studies, 1298 people).
• We do not know whether remdesivir increases or decreases time on supplemental oxygen (3 studies, 1691 people).
Did patients get worse with remdesivir?
• We do not know whether patients are more or less likely to need any mechanical ventilation (invasive or non‐invasive) with remdesivir (3 studies, 6696 people).
• Patients may be less likely to need invasive mechanical ventilation (2 studies, 1159 people).
• We do not know whether patients are more or less likely to need non‐invasive mechanical ventilation (1 study, 573 people).
• We do not know whether patients are more or less likely to need oxygen by mask or nasal prongs (1 study, 138 people).
Quality of life
• None of the included studies reported quality of life.
Unwanted effects
• We do not know whether remdesivir leads to more or fewer unwanted effects of any level (3 studies, 1674 people).
• Patients are probably less likely to experience serious unwanted effects with remdesivir than with placebo or standard care (3 studies, 1674 people). In 1000 people, 63 fewer would experience a serious unwanted effect compared to placebo or standard care.
What are the limitations of the evidence?
We are moderately confident in the evidence for deaths from any cause and serious unwanted effects; however, our confidence in the other evidence is limited because studies used different methods to measure and record their results, and we did not find many studies for some of our outcomes of interest.
How up‐to‐date is this evidence?
The evidence is current to 16 April 2021.
Summary of findings
Summary of findings 1. Remdesivir compared to placebo or standard care alone for hospitalised adults with confirmed SARS‐CoV‐2 infection.
| Remdesivir compared to placebo or standard care alone for hospitalised adults with confirmed SARS‐CoV‐2 infection | ||||||
| Patient or population: hospitalised adults with confirmed SARS‐CoV‐2 infection Settings: in‐hospital Intervention: remdesivir (10 days) Comparator: placebo or standard care alone | ||||||
| Outcomes | Anticipated absolute effects | Relative effect 95% CI | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | ||||||
| Placebo or standard care alone | Risk difference with remdesivir | |||||
| All‐cause mortality at up to day 28 | 108 per 1000i | 8 fewer per 1000 (21 fewer to 7 more) | RR 0.93 (0.81 to 1.06) | 7142 (4 RCTs) | ⊕ ⊕ ⊕ ⊖ MODERATE Due to serious imprecision1 | Remdesivir probably makes little or no difference to all‐cause mortality. |
| Improvement of clinical status: duration to liberation from invasive mechanical ventilation at up to day 28 | 2 studies reported this outcome as median, which could not be included in meta‐analysis. 1 study reported a median of 17 days (IQR 9 to 28) in the remdesivir group and 20 days (IQR 8 to 28) in the control group (rate difference −3.0, 95% CI −9.3 to 3.3). The other study reported a median of 7 days (IQR 4 to 16) in the remdesivir group and 15.5 days (IQR 6 to 21) in the control group (rate difference –4.0, 95% CI –14 to 2). | 1298 (2 RCTs) | ⊕ ⊕ ⊖ ⊖ LOW Due to serious risk of bias and serious imprecision2,3 | Remdesivir may have little or no effect on improvement of clinical status: duration to liberation from invasive mechanical ventilation. | ||
| Improvement of clinical status: duration to liberation from supplemental oxygen at up to day 28 | 3 studies reported this outcome as median, which could not be included in meta‐analysis. 1 study reported a median of 13 days (IQR 5 to 28) in the remdesivir group and 21.0 days (IQR 8 to 28) in the control group (rate difference −8.0, 95% CI −11.8 to ‐4.2). 1 study reported a median of 19 days (IQR 11 to 30) in the remdesivir and 21 days (IQR 14 to 30.5) in the control group (rate difference −2, 95% CI −6 to 1). The third study reported time to room air regardless of the initial respiratory support: 4 days (IQR 2 to 6) in the remdesivir group and 6 days (IQR 4 to 14) in the control group (HR 1.93, 95% CI 1.11 to 3.36). | 1691 (3 RCTs) | ⊕ ⊖ ⊖ ⊖
VERY LOW Due to serious risk of bias, serious imprecision, and other considerations2,4,5 |
We are uncertain as to whether remdesivir increases or decreases the chance of clinical improvement: duration to liberation from supplemental oxygen . | ||
| Clinical worsening: new need for mechanical ventilation at day 28 (defined as high‐flow oxygen, non‐invasive, or invasive mechanical ventilation) | 131 per 1000 | 29 fewer per 1000 (68 fewer to 32 more) |
RR 0.78 (0.48 to 1.24) | 6696 (3 RCTs) | ⊕ ⊖ ⊖ ⊖
VERY LOW Due to serious risk of bias, serious imprecision, and serious inconsistency1,4,6 |
We are very uncertain as to whether remdesivir decreases or increases the risk of clinical worsening: new need for mechanical ventilation. |
| Clinical worsening: new need for invasive mechanical ventilation at up to day 28 | 152 per 1000 | 67 fewer per 1000 (90 fewer to 35 fewer) |
RR 0.56 (0.41 to 0.77) | 1159 (2 RCTs) | ⊕ ⊕ ⊖ ⊖ LOW Due to serious risk of bias and other considerations4,5 | Remdesivir may decrease the risk of clinical worsening: new need for invasive mechanical ventilation. |
| Clinical worsening: new need for non‐invasive mechanical ventilation or high‐flow oxygen at up to day 28 | 241 per 1000 | 72 fewer per 1000 (118 fewer to 5 fewer) |
RR 0.70 (0.51 to 0.98) | 573 (1 RCT) | ⊕ ⊖ ⊖ ⊖
VERY LOW Due to serious risk of bias and very serious imprecision3,7 |
We are very uncertain as to whether remdesivir decreases or increases the risk of clinical worsening: new need for non‐invasive mechanical ventilation or high‐flow oxygen. |
| Clinical worsening: new need for oxygen by mask or nasal prongs at up to day 28 | 444 per 1000 | 84 fewer per 1000 (204 fewer to 98 more) |
RR 0.81 (0.54 to 1.22) | 138 (1 RCT) | ⊕ ⊖ ⊖ ⊖
VERY LOW Due to serious risk of bias and very serious imprecision3,8 |
We are very uncertain as to whether remdesivir decreases or increases the risk of clinical worsening: new need for oxygen by mask or nasal prongs. |
| Quality of life | NA | NA | NA | NA | NA | None of the included studies reported quality of life, therefore we do not know whether remdesivir has any impact on this outcome. |
| Serious adverse events at up to day 28 | 253 per 1000 | 63 fewer per 1000 (94 fewer to 25 fewer) |
RR 0.75 (0.63 to 0.90) | 1674 (3 RCTs) | ⊕ ⊕ ⊕ ⊖ MODERATE Due to serious risk of bias3 | Remdesivir probably decreases the risk of serious adverse events. |
| Adverse events (any grade) at up to day 28 | 587 per 1000 | 29 more per 1000 (82 fewer to 158 more) |
RR 1.05 (0.86 to 1.27) | 1674 (3 RCTs) | ⊕ ⊖ ⊖ ⊖
VERY LOW Due to serious risk of bias, serious inconsistency, and serious imprecision1,3,9 |
We are very uncertain as to whether remdesivir increases or decreases adverse events (any grade). |
| CI: confidence interval; HR: hazard ratio; IQR: interquartile range; NA: not applicable; RCT: randomised controlled trial; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of effect. Moderate certainty: We are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: Our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: We have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
i. All‐cause mortality at hospital discharge: RR 0.98, 95% CI 0.84 to 1.14; 1 study, 5451 participants; I² not applicable. All‐cause mortality (time‐to‐event): HR 0.93, 95% CI 0.80 to 1.07; 2 studies, 6513 participants; I² = 57%.
1Downgraded one level due to serious imprecision because of wide confidence intervals in the studies and the 95% confidence interval includes both benefits and harms. 2Downgraded one level due to serious imprecision because the 95% confidence interval includes both benefits and harms. 3Downgraded one level due to serious risk of bias because of competing risk of death. 4Downgraded one level due to serious risk of bias because of inadequate blinding of participants, personnel, and outcome assessors and possible deviation in time point of measuring in one study, and competing risk of death. 5Downgraded one level due to other considerations, as studies reported outcomes differently because of missing standards. 6Downgraded one level due to serious inconsistency because of statistical heterogeneity (I2 = 85%). 7Downgraded two levels due to serious imprecision because of few participants and data from only one study. 8Downgraded two levels due to very serious imprecision because of wide confidence intervals and data from only one study. 9Downgraded one level due to serious inconsistency because of statistical heterogeneity (I2 = 77%).
Background
This work is part of a series of Cochrane Reviews investigating treatments and therapies for coronavirus disease 2019 (COVID‐19). Reviews of this series share information in the background section and methodology based on the first published reviews about monoclonal antibodies, Kreuzberger 2021, and convalescent plasma (Chai 2020), and are part of the German research project “CEOsys” (COVID‐19 Evidence‐Ecosystem; CEOsys 2021).
Description of the condition
COVID‐19 is a rapidly spreading infectious disease caused by the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2). On 11 March 2020, the World Health Organization (WHO) declared the current COVID‐19 outbreak as a pandemic (WHO 2020a). COVID‐19 is unprecedented to previous coronavirus outbreaks, such as severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS, Table 2), with 813 and 858 deaths, respectively (WHO 2003; WHO 2019). In particular with respect to public health, socio‐economic conditions, and severity of the disease, it has surpassed the aforementioned outbreaks. Despite intensive international efforts to contain its spread, as of July 2021, the cumulative number of cases reported globally is almost 200 million, and the number of deaths is more than 4 million (WHO 2020b; WHO 2021a; WHO 2021b).
1. Glossary.
| Phrase/Word | Meaning/Description |
| Acute respiratory distress syndrome (ARDS) | ARDS is characterised by a massive response of the respiratory system to a wide variety of external and internal noxious stimuli. There is a disturbance of oxygen uptake and an acute onset. ARDS is the common end route of a wide variety of diseases leading to a severe systemic inflammatory response. The condition should be distinguished from disturbances of respiration caused by cardiac diseases. |
| Adverse event | An adverse event in the context of clinical trials is an unwanted medical occurrence in patients receiving a pharmacological or non‐pharmacological treatment, or both. An adverse event may not necessarily be considered to be related to the treatment. |
| Antimicrobials | Drugs used to treat diseases caused by micro‐organisms (bacteria, fungi, viruses, parasites). |
| Antiviral (medicine) | An agent that is directed against viruses |
| Bias | (Unconscious) distortion and misinterpretation of research results, especially those obtained experimentally. The most important sources for bias are as follows.
|
| Controlled non‐randomised study | A study in which the effects of a pharmacological or non‐pharmacological measure, or both, are compared between different groups of participants. The term 'controlled' means that the measure under investigation (intervention, verum) is compared with another measure (placebo or another intervention). The group of participants receiving the intervention under study is known as the intervention group. The group of participants who do not receive the intervention is known as the control group. A controlled non‐randomised study is easier to conduct than a randomised controlled trial, but has much less power (see bias). |
| Convalescent plasma | Blood plasma from patients who have had a disease (e.g. COVID‐19). Transfer of convalescent plasma to naive patients (patients who do not have antibodies themselves) leads to an increase in the immune defence of the receiving patient because convalescent plasma contains antibodies. |
| Corticosteroids | Hormones that are mainly produced in the adrenal cortex. Corticosteroids influence many biological processes in the organism, and are in particular closely linked to the immune system. Important naturally occurring representatives are cortisone and cortisol. Examples of synthetically produced corticosteroids are dexamethasone and budesonide. |
| Dichotomous | Dichotomy describes a system that can have exactly two mutually exclusive states. Example: either one has a certain disease (state A), or one does not have this disease (state B). The co‐occurrence of state A and state B is impossible. |
| Ebola | Ebola is a viral disease that is often severe. The Ebola virus belongs to the Filoviridae (from Latin 'filum' = filamentous). There are at least six different species of the virus. Ebola virus was previously called haemorrhagic fever because it is accompanied by high fever and severe internal and external bleeding. |
| Heterogeneous | Heterogeneity can be translated as 'non‐uniformity'. It is the opposite of homogeneity. In the context of meta‐analyses, heterogeneity is a measure of the comparability of clinical trials. For example, studies that examine different populations (e.g. children versus adults) have limited comparability and can lead to misleading conclusions when the data from such studies are pooled in a meta‐analysis. |
| Hydroxychloroquine | A drug related to chloroquine, which is used mainly for the treatment of rheumatoid arthritis, lupus erythematosus, and the prevention of malaria |
| Immunocompromised status | Immunocompromised are people who have a congenital or acquired disorder of the immune response. Examples of acquired disorders include infection with HIV. Long‐term treatment with certain drugs (e.g. corticosteroids) can also lead to disorders/weakening of the immune response. |
| Interventions | The term 'intervention' in the context of clinical trials refers to the measure whose effect (superiority, inferiority, non‐inferiority) on a specific condition is to be assessed in comparison to other measures. An intervention need not always consist of the administration of a specific drug (so‐called non‐pharmacological interventions). |
| Mechanical ventilation | Mechanical ventilation is the term used to describe a procedure in which oxygen is supplied to the patient with the aid of ventilators or other devices. This measure is very restrictive and not without risk, and is therefore used only if the patient can no longer take in enough oxygen through his or her natural breathing (spontaneous respiration). In this review, the following procedures are subsumed under the term 'mechanical ventilation'.
|
| Middle East respiratory syndrome (MERS) | MERS is a respiratory disease caused by a coronavirus (MERS‐CoV). Most cases of the disease are asymptomatic. Diarrhoea is a common accompanying symptom. In severe cases, pneumonia develops. |
| Monoclonal antibody (MAB) | Antibodies in general are produced by the organism (specifically the immune system) when it is exposed to an antigen (for example, pathogenic microorganisms and viruses). By reacting with specific parts of the antigen, the antibody can render it harmless. So‐called monoclonal antibodies are produced by infecting mice with an antigen, for example. The immune system (especially the B cells) of the infected mouse then produces antibodies that are specifically active against the antigen. These cells accumulate in the spleen of the infected mouse. These cells are then isolated from the animal's spleen in a complicated process and multiplied in vitro (i.e. in the test tube). The resulting monoclonal antibodies are all derived from genetically identical cells and are directed against a specific antigen. Monoclonal antibodies are administered in medicine when the patient does not produce any antibodies or produces too few of his or her own. In addition, these specific antibodies also enable the identification of antigens in the detection of various diseases. |
| Nasal prongs | Nasal prongs, or nasal cannula, is a device used to deliver low‐flow oxygen to the nose through a small plastic tube. |
| Observational study | Data collection in a specific population under a specific research question. The essential characteristic of an observational study is that no intervention/experiment is carried out. |
| Placebo | A placebo is a dummy drug that does not contain a pharmacologically active substance. |
| Randomised controlled trial | A randomised controlled trial is the best way to obtain conclusions regarding the efficacy and effectiveness of a pharmacological or non‐pharmacological intervention, or both. The term 'controlled' means that the measure under investigation (intervention, verum) is compared with another measure (placebo or another intervention). The term 'randomised' means that the participants in the study are randomly assigned to one of two or more prespecified treatment groups. The group of participants receiving the intervention under study is known as the intervention group. The group of participants who do not receive the intervention is known as the control group. |
| Severe acute respiratory syndrome (SARS) | A disease caused by SARS‐CoV, which, similar to COVID‐19, results in fever and muscle pain in combination with other flu‐like signs. In severe cases, atypical pneumonia may occur. |
| Systematic review | Scientific process of critical judgement of the data available with regard to a specific question. A 'systematic' approach is taken. This includes:
A systematic review can include a meta‐analysis, but this is not required. The aim of a systematic review is to answer the defined research question, or, if this is not possible, to identify gaps in the scientific coverage of the research question. |
SARS‐CoV‐2 is a positive‐sense, double‐stranded ribonucleic acid (RNA) virus that belongs to the Coronaviridae family (Chen 2020; Kumar 2020). There is widespread consensus that SARS‐CoV‐2 is closely related to a beta coronavirus detected in bat faeces. However, the host of origin and the intermediate host remain unclear (Lundstrom 2020; Malaiyan 2020; WHO 2021c). SARS‐CoV‐2 binds to angiotensin‐converting enzyme 2 receptors, which are expressed in lung, heart, kidney, intestine, as well as endothelium, by means of its spike glycoprotein (Yan 2020). Viral variants mainly present mutational changes in the spike glycoprotein (WHO 2021d). Infection with SARS‐CoV‐2 results in an immune response involving CD4+ and CD8+ T cells, which may lead to acute respiratory distress syndrome (ARDS) and multiple organ dysfunction syndrome through cytokine storm syndrome as a result of the release of pro‐inflammatory cytokines and chemokines (Li 2020b).
The median incubation time is estimated at between five and six days, and 97.5% of symptomatic cases develop symptoms within 11.5 days of exposure (Lauer 2020). Signs and symptoms can include sore throat, cough, fever, headache, fatigue, and myalgia or arthralgia. The presence of anosmia and ageusia, with an overall low sensitivity (lower than 50%), has a specificity greater than 90% and may be useful as a red flag for COVID‐19 (Struyf 2021). Other symptoms include shortness of breath, chills, nausea or vomiting, diarrhoea, nasal congestion, haemoptysis, and conjunctival congestion (WHO 2020c).
A large proportion of infected individuals remain asymptomatic throughout the course of the disease, depending on the time of the investigation, the cohort investigated, and the dominant circulating virus variants (Chen 2020a; Pan 2020; Wu 2020; Funk 2021). The reported frequency of asymptomatic courses also varies greatly and ranges between 6% and 96% (Oran 2020; Funk 2021). In a meta‐analysis, Buitrago‐Garcia and colleagues estimated the proportion of persistently asymptomatic infected individuals at 20%, with a prediction interval of 3% to 67% (Buitrago‐Garcia 2020). Despite the absence of clinical signs, asymptomatic individuals show typical findings on chest computed tomography (CT) in up to 50% of cases (Hu 2020; Meng 2020).
A smaller proportion of infected individuals are affected by severe (approximately 11% to 20%) or critical (approximately 1% to 5%) disease with hospitalisation and intensive care unit (ICU) admittance due to respiratory failure, septic shock, or multiple organ dysfunction syndrome (Wu 2020; Funk 2021). In a case series from 12 New York hospitals, 14% of patients hospitalised due to COVID‐19 were treated in ICU (Richardson 2020). Evaluations of patients during the first COVID‐19 wave in Germany show an estimate of 14% to 37% of this proportion (Schilling 2020; Tolksdorf 2020). In an observational study of 10,021 hospitalised adult patients in Germany with a confirmed COVID‐19 diagnosis, 17% received mechanical ventilation (non‐invasive and invasive). In this study, 27% of ventilated patients required dialysis due to acute renal failure. Mortality in patients not receiving mechanical ventilation was 16%, and up to 53% in ventilated patients. Mortality in patients receiving mechanical ventilation (non‐invasive and invasive) and dialysis was 73% (Karagiannidis 2020). In a systematic review and meta‐analysis of international studies, the proportion of patients who died was estimated at 34% amongst those treated in ICU, and 83% amongst those receiving invasive mechanical ventilation (Potere 2020).
The infection fatality ratio varies widely between countries and reporting periods (from 0.01% to more than 25%). However, these numbers may be misleading as they tend to overestimate the infection fatality ratio due to varying testing frequency, lag in reporting dates, and variations in case definitions, especially in the beginning of the pandemic, as clinicians were mainly focused on severe cases (Wu 2020; WHO 2020b).
Risk for severe disease, hospitalisation, and mortality is higher for individuals aged 65 years or older, males, smokers, and individuals with certain underlying medical conditions, such as cancer, chronic kidney disease, chronic obstructive pulmonary disease (COPD), heart conditions, immunocompromised state, obesity, sickle cell disease, or type 2 diabetes mellitus (Huang 2020; Karagiannidis 2020; Liang 2020; Petrilli 2020; WHO 2020c; Williamson 2020a).
Vaccination has been shown to be highly effective at reducing severe illness and death from COVID‐19. As of July 2021, more than 2.95 billion doses of COVID‐19 vaccines have been administered at the global level (https://covid19.who.int/). However, the majority of vaccines have been administered in a few high‐income countries. The majority of the world's population still remains susceptible to SARS‐CoV‐2 infection and at risk of developing COVID‐19. Moreover, the duration and degree of protection against the disease, but also against infection and transmission, is still not well‐defined, and vaccine hesitancy poses direct and indirect threats to health (Grubaugh 2020). Besides unequal access to vaccines, there is evidence indicating a significant impact of certain circulating variants of SARS‐CoV‐2 on immunity that is likely to have an impact on the epidemiological situation. (Grubaugh 2020; Schwarz 2021).
In light of the extent of the pandemic, including evolving virus variants and a scarcity of effective treatments as well as issues related to the global availability of vaccines, the role of effective therapies is of utmost interest for combating COVID‐19.
Description of the intervention
Remdesivir (GS‐5734) is an antiviral agent derived from a small‐molecule library and designed to target the replication of pathogenic RNA viruses (Siegel 2017). It evinced a broad‐spectrum in vitro efficacy against various emerging viruses, such as Filoviridae (e.g. Ebolavirus and Marburgvirus), Pneumoviridae (respiratory syncytial virus), and Coronaviridae (MERS‐CoV, SARS‐CoV) (Sheahan 2017; Choy 2020).
In the large and complex 2014 to 2016 outbreak in West Africa, remdesivir showed promising in vivo activity against the Ebola virus (Warren 2016; Tchesnokov 2019). In rhesus monkeys infected with Ebola virus, the novel nucleoside analog led to reduced plasma viral RNA and beneficial impact on clinical progression (Warren 2016). However, when transferred to patients, the remdesivir arm of a randomised controlled trial (RCT) was stopped early due to significant inferiority to monoclonal antibody treatment on mortality (Mulangu 2019).
Sheahan and colleagues outlined the possible relevance of remdesivir in the prevention and treatment of existing and emerging coronaviruses in a mouse model in 2017 (Sheahan 2017). Studies on murine models of SARS infection as well as MERS infection models in rhesus monkeys showed a significant reduction in virus replication, improvement of clinical symptoms, and reduction of lung tissue damage rate (de Wit 2020; Sheahan 2020). In terms of these effects, remdesivir was superior to other antiviral substances such as ribavirin or lopinavir/ritonavir (Sheahan 2020). Two years later, the SARS‐CoV‐2 pandemic led to rapid investigation of remdesivir as a potential virostatic drug. In vitro testing supported its efficacy against different clinical isolates, partially at low‐micromolar concentration (Choy 2020; Ogando 2020; Wang 2020). Early administration in SARS‐CoV‐2‐inoculated macaques reduced respiratory symptoms and lung damage compared to vehicle‐treated controls (Williamson 2020b). However, pharmacokinetic data in humans, precisely COVID‐19 patients, is rare, and the impact of impaired organ function on drug availability in infected cells is yet not well understood.
During the course of the COVID‐19 pandemic, the antiviral agent was initially administered to hospitalised patients with COVID‐19 in a compassionate‐use attempt. The Adaptive COVID‐19 Treatment Trial (ACTT‐1) was one of the first multicentre RCTs to report a shortened time to recovery in hospitalised COVID‐19 patients compared to standard care (Beigel 2020). Shortly after its publication, the US Food and Drug Administration released an Emergency Use Authorisation on 1 May 2020 (EUA 2021). Based on the recommendation of the European Medicines Agency, the European Union Commission followed in July 2020 with the authorisation of remdesivir as the first treatment option in patients at least 12 years of age with COVID‐19 pneumonia and the need for supplementary oxygen (EUA 2020). Later that year, the Committee for Medicinal Products for Human Use narrowed the indication to patients with low‐ or high‐flow oxygen or other non‐invasive ventilation (EMA 2020). The recommended dosing regimen is 200 mg intravenously (loading dose), followed by 100 mg over five to 10 days. According to the manufacturer, Gilead Science, phase one clinical trials revealed good tolerability and safety of intravenous administration of remdesivir in healthy individuals (EUA 2021). Reported side effects included phlebitis, constipation, headache, ecchymosis, nausea, and pain in the extremities, as well as transient increase in transaminases, prothrombin time, and blood glucose in laboratory findings (Malin 2020).
Meanwhile, further RCTs relativised the initial euphoria. Amongst them were the interim results of the WHO Solidarity trial, which could not find a benefit for time to clinical improvement, need for mechanical ventilation, or mortality (WHO Solidarity Trial Consortium 2021). Based on a meta‐analysis of four RCTs, the WHO updated its COVID‐19 treatment guidelines in January 2021 and recommended against the use of remdesivir in hospitalised patients (WHO 2021).
How the intervention might work
Remdesivir (GS‐5734) is a mono phosphoramidate nucleoside prodrug which inhibits the synthesis of viral RNA. By competing with its natural analog adenosine triphosphate, it blocks the RNA‐polymerase and leads to delayed chain termination, hence inhibiting the virus replication (Siegel 2017). The addition of the monophosphate prodrug improves the intracellular uptake, where phosphorylation turns it into its active metabolite (McGuigan 2006; Lo 2017).
In the early stage of a SARS‐CoV‐2‐associated pneumonia, the reduction of the viral load is postulated to prevent a systemic inflammatory reaction and, in particular, alveolar damage. The clinical presentation of COVID‐19 in the late pulmonary phase as well as in the hyper inflammatory phase are dominated by immunological processes, so that antiviral therapy strategies are no longer likely to be effective (Gautret 2020).
In summary, the broad‐spectrum nucleoside analog remdesivir could be beneficial in the early stages of SARS‐CoV‐2‐infection by inhibiting virus replication. This hypothesis is supported by promising in vitro and animal experiments (Choy 2020; Wang 2020; Williamson 2020b).
Why it is important to do this review
There is a clear and urgent need for more evidence‐based information to guide clinical decision‐making for COVID‐19 patients. Current treatment consists of supportive care with oxygen supply in moderately severe cases, and non‐invasive ventilation or invasive mechanical ventilation and extracorporeal membrane oxygenation (ECMO) in severe cases (CDC 2020; WHO 2020b). To date, few drugs have been shown to be of clear benefit in the treatment of COVID‐19, such as corticosteroids. Few drugs are approved for the treatment of COVID‐19, and international guidelines are constantly updated.
The application of remdesivir in COVID‐19 patients aims to reduce symptom severity as well as disease progression through inhibited virus replication. Whilst early clinical trials seemed to reproduce positive effects on clinical improvement, leading to widespread authorisation of emergency use, the currently available data are conflicting and uncertain. In part, expectations raised by in vitro findings were not met. Extensive work in the field of systematic reviews for interventions for COVID‐19 has already been undertaken, including on remdesivir. Assessment of the available data is not trivial due to inconsistent endpoint definitions, making it difficult to compare conducted trials. One review saw a reduction in mortality (Bansal 2021), whilst other reviews saw no or very small effects on mortality (Piscoya 2020; Siemieniuk 2020; Bhimrai 2021; Vegivinti 2021; Wilt 2021). A similarly inconsistent picture emerges for clinical improvement and incidence of (serious) adverse events (Elsawah 2020; Yokoyama 2020; Al‐Abdouh 2021; Kaka 2021; Wilt 2021).
These differences in the assessment of evidence can be attributed in part to the fact that some reviews also included non‐randomised studies (e.g. Bansal 2021). Furthermore, data regarding important subgroups are not readily available.
This systematic review will fill current gaps by identifying, describing, evaluating, and synthesising all evidence for remdesivir on clinical outcomes in COVID‐19. There is a need for a thorough understanding and an extensive review of the current body of evidence regarding the use of remdesivir for the treatment of COVID‐19. The primary goal of this review is to provide practising clinicians, healthcare providers, and interested laypersons with reliable and evidence‐based information that will lead to improvement in the treatment of COVID‐19.
Objectives
To assess the effects of remdesivir compared to placebo or standard care alone on clinical outcomes in hospitalised patients with SARS‐CoV‐2 infection, and to maintain the currency of the evidence using a living systematic review approach.
Methods
Criteria for considering studies for this review
Types of studies
The main description of methods is based on a template from the Cochrane Haematology working group in line with the series of Cochrane Reviews investigating treatments and therapies for COVID‐19. We made specific adaptations related to the research question where necessary. The protocol for this review was registered with PROSPERO on 26 February 2021 (CRD42021238065).
To assess the effects of remdesivir for treatment in hospitalised individuals with SARS‐CoV‐2 infection, we included RCTs, as this study design, if performed appropriately, provides the best evidence for experimental therapies in highly controlled therapeutic settings. We used the methods recommended in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2021a). We had planned to also accept non‐standard RCT designs, such as cluster‐randomised trials (methods as recommended in Chapter 23 of the Cochrane Handbook for Systematic Reviews of Interventions) and cross‐over trials (Higgins 2021b). We would only have considered results from the first period for cross‐over trials, because COVID‐19 is not a chronic condition, and its exact course and long‐term effects have yet to be defined.
We excluded controlled non‐randomised studies of intervention and observational studies. We also excluded animal studies, pharmacokinetic studies, and in vitro studies.
We included the following formats, if sufficient information was available on study design, characteristics of participants, interventions, and outcomes.
Full‐text publications
Preprint articles
Abstract publications
Results published in trials registries
Personal communication with investigators
We included preprints and conference abstracts to have a complete overview of the ongoing research activity, especially for tracking newly emerging studies about remdesivir in COVID‐19. We did not apply any limitation with respect to length of follow‐up.
Types of participants
We included adults with a confirmed diagnosis of COVID‐19 (as described in the study) and did not exclude any studies based on gender, ethnicity, disease severity, or setting.
We excluded studies that evaluated remdesivir for the treatment of other coronavirus diseases such as SARS or MERS, or other viral diseases, such as Ebola. We planned that if studies enrolled populations with or who were exposed to mixed viral diseases, we would only include these if the trial authors provided subgroup data for SARS‐CoV‐2 infection.
Types of interventions
We included the following interventions:
Remdesivir for the treatment of SARS‐CoV‐2 infection.
We included the following comparisons:
Placebo or standard care alone.
Types of outcome measures
We evaluated core outcomes in accordance with the Core Outcome Measures in Effectiveness Trials (COMET) Initiative for COVID‐19 patients (COMET 2020; WHO 2020d), and additional important outcomes that have been prioritised by consumer representatives and the German guideline panel for inpatient therapy of people with SARS‐CoV‐2 infection. Outcomes critical to this review are in bold.
All‐cause mortality at up to day 28, day 60, time‐to‐event, and at hospital discharge.
-
Clinical status, assessed by need for respiratory support with standardised scales (e.g. WHO Clinical Progression Scale (WHO 2020d), WHO Ordinal Scale for Clinical Improvement (WHO 2020d) at up to day 28, day 60, and up to longest follow‐up), including:
-
improvement of clinical status:
liberation from invasive mechanical ventilation in surviving participants;
ventilator‐free days;
duration to liberation from invasive mechanical ventilation;
liberation from supplemental oxygen in surviving participants;
duration to liberation from supplemental oxygen.
-
worsening of clinical status:
new need for mechanical ventilation (defined as high‐flow oxygen, non‐invasive, or invasive mechanical ventilation);
new need for invasive mechanical ventilation;
new need for non‐invasive mechanical ventilation or high‐flow oxygen;
new need for oxygen by mask or nasal prongs.
-
Need for dialysis at up to day 28.
Quality of life, including fatigue and neurological status, assessed with standardised scales (e.g. WHOQOL‐100) at up to seven days, up to 30 days, and longest follow‐up available.
Need for admission to ICU.
Duration of ICU length of stay, or time to discharge from ICU.
Duration of hospitalisation, or time to discharge from hospital.
Viral clearance, assessed with reverse transcription polymerase chain reaction (RT‐PCR) test for SARS‐CoV‐2 at baseline, up to 3, 7, and 15 days.
Serious adverse events, defined as number of participants with event.
Adverse events (any grade, grade 1 to 2, grade 3 to 4), defined as number of participants with event.
Timing of outcome measurement
In the case of time‐to‐event analysis (e.g. for time to discharge from hospital and time to mortality), we included the outcome measure based on the longest follow‐up time. We also collected information on outcomes from all other time points reported in the publications.
We included adverse events occurring during active treatment and as well as long‐term adverse events. If sufficient data were available, we grouped the measurement time points of eligible outcomes, for example adverse events and serious adverse events, into those measured directly after treatment (up to 7 days after treatment), medium‐term outcomes (up to 15 days after treatment), and longer‐term outcomes (more than 30 days after treatment).
We combined three different types of advanced respiratory support (high‐flow oxygen, non‐invasive mechanical ventilation, and invasive mechanical ventilation) into one outcome measure, using the term 'mechanical ventilation' for clinical as well as patient‐oriented reasons (see Differences between protocol and review).
Search methods for identification of studies
Electronic searches
Our Information Specialist (MIM) conducted systematic searches in the following sources from the inception of each database to 16 April 2021 (date of last search for all databases), placing no restrictions on the language of publication.
-
Cochrane COVID‐19 Study Register (CCSR) (https://covid-19.cochrane.org/) comprising:
Cochrane Central Register of Controlled Trials (CENTRAL), monthly updates;
PubMed, daily updates;
Embase.com, weekly updates;
ClinicalTrials.gov (www.clinicaltrials.gov), daily updates;
World Health Organization International Clinical Trials Registry Platform (WHO ICTRP) (www.who.int/trialsearch), weekly updates;
medRxiv (www.medrxiv.org), weekly updates.
-
Web of Science Clarivate:
Science Citation Index Expanded;
Emerging Sources Citation Index.
WHO COVID‐19 Global literature on coronavirus disease (https://search.bvsalud.org/global-literature-on-novel-coronavirus-2019-ncov/).
For detailed search strategies, see Appendix 1.
Searching other resources
We identified other potentially eligible studies or ancillary publications by searching the reference lists of included studies, systematic reviews, and meta‐analyses. In addition, we contacted investigators of the included studies to obtain additional information on the retrieved studies.
We searched for grey literature, which we defined as searching study registries such as ClinicalTrials.gov and the WHO ICTRP, contained in the CCSR, as well as searching preprint servers and grey literature indexes contained in CCSR and WHO COVID‐19 Global literature on coronavirus disease. Once we established our set of included studies, we searched for preprints via Europe PubMed Central, to check if any preprints for included studies had been published since our database search.
Data collection and analysis
Selection of studies
Four review authors (KA, FG, KD, VT) independently screened the results of the search strategies for eligibility by reading the titles and abstracts using Covidence software (Covidence 2021). We coded the abstracts as either 'include' or 'exclude'. In the case of disagreement, or if it was unclear whether the abstract should be retrieved, we obtained the full‐text publication for further discussion. Several review authors (KA, FG, KD, VT) assessed the full‐text articles of the selected studies. If two review authors were unable to reach a consensus, they consulted a third review author to reach a final decision.
As recommended in the PRISMA statement (Moher 2009), we documented the study selection process in a flow chart showing the total numbers of retrieved references and the numbers of included and excluded studies. We listed all studies excluded after full‐text assessment and the reasons for their exclusion in the Excluded studies section.
Data extraction and management
We conducted data extraction according to the guidelines proposed by Cochrane (Li 2020a). Several review authors (KA, FG, KD, VT, AM) extracted data independently and in duplicate, using a customised data extraction form developed in Microsoft Excel (Microsoft 2018). Any disagreements were resolved by discussion or by consulting a third review author if necessary.
Two out of several review authors (KA, FG, KD, AM, VT, VP) independently assessed the included studies for methodological quality and risk of bias. If the review authors were unable to reach a consensus, a third review author was consulted.
We extracted the following information, where reported.
General information: author, title, source, publication date, country, language, duplicate publications.
Study characteristics: trial design, setting, and dates, source of participants, inclusion/exclusion criteria, comparability of groups, treatment cross‐overs, compliance with assigned treatment, length of follow‐up.
Participant characteristics: age, gender, ethnicity, number of participants recruited/allocated/evaluated, additional diagnoses, severity of disease, previous treatments, concurrent treatments, comorbidities (e.g. diabetes, respiratory disease, hypertension, immunosuppression, obesity, heart failure).
Interventions: dosage, frequency, timing, duration and route of administration, setting, duration of follow‐up.
Control interventions (placebo or standard care alone): dosage, frequency, timing, duration and route of administration, setting, duration of follow‐up.
Outcomes: as specified in Types of outcome measures section.
Risk of bias assessment: randomisation process, deviations from the intended interventions, missing outcome data, measurement of the outcome, selection of the reported result.
Assessment of risk of bias in included studies
We used the RoB 2 tool (beta version 7) to analyse the risk of bias of the included studies (Sterne 2019). Of interest in this review was the effect of the assignment to the intervention (the intention‐to‐treat effect), thus we performed all assessments with RoB 2 on this effect. The outcomes that we assessed are those specified for inclusion as described in the Methods section.
Two out of several review authors (KA, FG, KD, AM, VT, VP) independently assessed the risk of bias for each outcome using the RoB 2 Excel tool to manage and record assessments. In case of discrepancies amongst judgements and inability to reach consensus, a third review author was consulted reach a final decision. We assessed the following types of bias as outlined in Chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2021c).
Bias arising from the randomisation process
Bias due to deviations from the intended interventions
Bias due to missing outcome data
Bias in measurement of the outcome
Bias in selection of the reported result
For cluster‐RCTs, we had planned to add a domain to assess bias arising from the timing of identification and recruitment of participants in relation to timing of randomisation, as recommended in the archived RoB 2 guidance for cluster‐randomised trials and in Chapter 23 of the Cochrane Handbook for Systematic Reviews of Interventions (Eldridge 2016; Higgins 2021b).
To address these types of bias, we used the signalling questions recommended in RoB 2 and made a judgement according to the following options.
'Yes': if there is firm evidence that the question is fulfilled in the study (i.e. the study is at low or high risk of bias given the direction of the question).
'Probably yes': a judgement has been made that the question is fulfilled in the study (i.e. the study is at low or high risk of bias given the direction of the question).
'No': if there is firm evidence that the question is unfulfilled in the study (i.e. the study is at low or high risk of bias given the direction of the question).
'Probably no': a judgement has been made that the question is unfulfilled in the study (i.e. the study is at low or high risk of bias given the direction of the question).
'No information': if the study report does not provide sufficient information to permit a judgement.
We used the algorithms proposed by RoB 2 to assign each domain one of the following levels of bias.
Low risk of bias
Some concerns
High risk of bias
We subsequently derived an overall risk of bias rating for each prespecified outcome in each study in accordance with the following suggestions.
'Low risk of bias': we judge the trial to be at low risk of bias for all domains for the result.
'Some concerns': we judge the trial to raise some concerns in at least one domain for the result, but not to be at high risk of bias for any domain.
'High risk of bias': we judge the trial to be at high risk of bias in at least one domain for the result, or we judge the trial to have some concerns for multiple domains in a way that substantially lowers our confidence in the results.
We used the RoB 2 Excel tool to implement RoB 2 (beta version 7, available from riskofbias.info), and stored and presented our detailed RoB 2 assessments in the analyses section and as supplementary online material.
For domain three of the tool ('bias due to missing outcome data'), we considered death as a competing risk factor, especially for dichotomous clinical progression outcomes. We judged improvement to be at high risk of bias due to missing data because it is likely that death during follow‐up impeded liberation from respiratory support, and hence missing data on improvement depends on its true value.
Measures of treatment effect
For continuous outcomes, we recorded the mean, standard deviation, and total number of participants in both the treatment and control groups. Where continuous outcomes used the same scale, we performed analyses using the mean difference (MD) with 95% confidence intervals (CIs). For continuous outcomes measured with different scales, we performed analyses using the standardised mean difference (SMD). In our interpretation of SMDs, we re‐expressed SMDs in the original units of a particular scale with the most clinical relevance and impact (e.g. clinical symptoms with the WHO Clinical Progression Scale) (WHO 2020d).
For dichotomous outcomes, we recorded the number of events and the total number of participants in both the treatment and control groups. We reported the pooled risk ratio (RR) with its associated 95% CI, and risk difference (RD) with its associated 95% CI (Deeks 2020).
If sufficient information was available, we extracted and reported hazard ratios (HRs) for time‐to‐event outcomes (e.g. time to hospital discharge). If HRs were not available, we made every effort to estimate the HR as accurately as possible from available data using the methods proposed by Parmar and Tierney (Parmar 1998; Tierney 2007). If a sufficient number of studies provided HRs, we used HRs rather than RRs or MDs in a meta‐analysis, as they provide more information.
Unit of analysis issues
The aim of this review was to summarise trials that analyse data at the level of the individual. We would also have accepted cluster‐randomised trials for inclusion had any been identified. We collated multiple reports of a given study so that each study, rather than each report, was the unit of analysis.
Studies with multiple treatment groups
As recommended in Chapter 6 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2021d), for studies with multiple treatment groups of the same intervention (i.e. dose, route of administration), we planned to evaluate if study arms were sufficiently homogeneous to be combined. We planned that if study arms could not be pooled, we would compare each arm with the common comparator separately. For pair‐wise meta‐analysis, we planned to split the ‘shared’ group into two or more groups with a smaller sample size, and include two or more (reasonably independent) comparisons. For this purpose, both the number of events and the total number of participants would have been divided for dichotomous outcomes, and the total number of participants would have been divided with unchanged means and standard deviations for continuous outcomes.
One study included in the review had multiple treatment arms of the same intervention (5‐day course of remdesivir versus 10‐day course of remdesivir) (Spinner 2020). Given the small number of participants in this study, we did not perform meta‐analysis, but have reported the results for each treatment arm narratively in our subgroup analysis (see Effects of interventions, Duration of remdesivir application).
Dealing with missing data
In Chapter 10 of the Cochrane Handbook for Systematic Reviews of Interventions, a number of potential sources for missing data are suggested, which we took into account: at study level, at outcome level, and at summary data level (Deeks 2020). At all levels, it is important to differentiate between data 'missing at random', which may often be unbiased, and 'not missing at random', which may bias the study and in turn the review results.
In the case of missing data, we requested this information from the principal investigators; details are provided in the Included studies section. Beigel 2020 and Spinner 2020 provided additional data on all‐cause mortality at up to day 28 for subgroups of respiratory support, and Spinner 2020 provided data on clinical course. If after this data were still missing, we had to make explicit assumptions of any methods the included studies used.
Assessment of heterogeneity
We assessed heterogeneity of treatment effects between trials using a Chi² test with a significance level of P < 0.1. We used the I² statistic, Higgins 2003, and visual examination of the forest plot, to assess possible heterogeneity (I² > 30% to signify moderate heterogeneity, I² > 75% to signify considerable heterogeneity) (Deeks 2020). We planned that if the I2 was above 80%, we would explore possible causes of heterogeneity through sensitivity analyses. If we could not find a reason for heterogeneity, we would not perform a meta‐analysis, but instead would comment on the results from all studies and present these in tables.
Assessment of reporting biases
As mentioned above, we searched the trials registries to identify completed trials that have not been published elsewhere, to minimise publication bias or determine publication bias. We intended to explore potential publication bias by generating a funnel plot and statistically testing this by conducting a linear regression test for meta‐analyses involving at least 10 trials (Sterne 2019). We would consider P < 0.1 as significant for this test.
Data synthesis
If the clinical and methodological characteristics of individual studies were sufficiently homogeneous, we pooled the data in meta‐analysis. We performed analyses according to the recommendations in the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2020). We planned to treat placebo and no treatment as the same intervention, as well as standard care at different institutions and time points.
We used the Review Manager Web (RevMan Web) software for analyses (RevMan Web 2021). One review author entered the data into the software, and a second review author checked the data for accuracy. We used the random‐effects model for all analyses, as we anticipated that true effects would be related but not the same for included studies. We planned that if meta‐analysis was not possible, we would comment on the results narratively with the results from all studies, and present these in tables. If meta‐analysis was possible, we would assess the effects of potential biases in sensitivity analyses (see Sensitivity analysis). For binary outcomes, we based the estimation of the between‐study variance using the Mantel‐Haenszel method. We used the inverse‐variance method for continuous outcomes, outcomes that included data from cluster‐RCTs, or outcomes where HRs were available. We explored heterogeneity for the outcome clinical worsening: new need for mechanical ventilation (I²= 85%)
Subgroup analysis and investigation of heterogeneity
We conducted subgroup analyses for all‐cause mortality at up to day 28 exclusively. In the case of sufficient data, we performed subgroup analyses of the following characteristics for remdesivir versus placebo or standard care alone.
Age of participants (divided into applicable age groups, e.g. 18 to 65 years, 65 to 79 years, 80 years and older).
Pre‐existing conditions (e.g. diabetes, respiratory disease, hypertension, immunosuppression, obesity, cardiac injury).
Timing of first dose administration with illness onset.
-
Severity of condition:
no oxygen versus low‐flow oxygen versus mechanical ventilation (including high‐flow oxygen, non‐invasive ventilation, invasive mechanical ventilation, and extracorporeal membrane oxygenation).
-
Duration of remdesivir application:
5‐day course of remdesivir versus 10‐day course of remdesivir.
We used the tests for interaction to test for differences between subgroup results.
Sensitivity analysis
We performed sensitivity analysis of the following study characteristics for our prioritised outcomes, as described in the Types of outcome measures section.
Risk of bias assessment components (studies with a low risk of bias or some concerns versus studies with a high risk of bias).
Comparison of preprints versus peer‐reviewed articles.
Comparison of premature termination of studies with completed studies.
Comparison of adolescent and adult participants versus adult participants.
Summary of findings and assessment of the certainty of the evidence
We created Table 1 and evaluated the certainty of the evidence using the GRADE approach for interventions evaluated in RCTs.
Summary of findings
We used MAGICapp software to create summary of findings tables (MAGICapp). For time‐to‐event outcomes, we calculated absolute effects at specific time points, as recommended in the GRADE guidance 27 (Skoetz 2020).
Chapter 14 of the updated Cochrane Handbook for Systematic Reviews of Interventions specifies that the “most critical and/or important health outcomes, both desirable and undesirable, limited to seven or fewer outcomes” should be included in the summary of findings table(s) (Schünemann 2021). We included outcomes prioritised according to the Core Outcome Set for intervention studies, COMET 2020, and patient relevance; these are listed below.
All‐cause mortality: all‐cause mortality at hospital discharge most favourable; if not reported, we will include all‐cause mortality day 60, followed by day 28, or time‐to‐event estimate in the summary of findings table.
-
Improvement of clinical status, assessed with liberation from supplemental oxygen support or invasive mechanical ventilation, in accordance with WHO Clinical Progression Scale (WHO 2020d), at longest follow‐up available.
For all hospitalised individuals with oxygen support (WHO ≥ 5 at baseline on the WHO Clinical Progression Scale) (WHO 2020d): liberation from supplemental oxygen in surviving participants most favourable; if not reported, we will include duration to liberation from supplemental oxygen in the summary of findings table.
For the subgroup of severely ill individuals (WHO ≥ 7 at baseline on the WHO Clinical Progression Scale) (WHO 2020d): liberation from invasive mechanical ventilation in surviving participants most favourable; if not reported, we will include ventilator‐free days, followed by duration to liberation from invasive mechanical ventilation, in the summary of findings table.
-
Worsening of clinical status, assessed with new need for respiratory support, in accordance with the WHO Clinical Progression Scale (WHO 2020d), at longest follow‐up available.
New need for mechanical ventilation (non‐invasive ventilation or high‐flow oxygen or invasive ventilation).
New need for invasive mechanical ventilation.
New need for non‐invasive mechanical ventilation or high‐flow oxygen.
New need for oxygen by mask or nasal prongs.
Quality of life, including fatigue and functional independence, assessed with standardised scales (e.g. WHOQOL‐100) at longest follow‐up available.
Adverse events (any grade).
Serious adverse events.
Assessment of the certainty of the evidence
We used the GRADE approach to assess the certainty of the evidence for the outcomes listed above.
The GRADE approach uses five domains (risk of bias, consistency of effect, imprecision, indirectness, and publication bias) to assess the certainty of the body of evidence for each prioritised outcome.
We downgraded the certainty of the evidence for:
serious (−1) or very serious (−2) risk of bias;
serious (−1) or very serious (−2) inconsistency;
serious (−1) or very serious (−2) uncertainty about directness;
serious (−1) or very serious (−2) imprecise or sparse data;
serious (−1) or very serious (−2) probability of reporting bias.
The GRADE system uses the following criteria for assigning grades of evidence.
High: we are very confident that the true effect lies close to that of the estimate of the effect.
Moderate: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different.
Low: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect.
Very low: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect.
We followed the current GRADE guidance for these assessments in its entirety as recommended in Chapter 14 of the Cochrane Handbook for Systematic Reviews of Interventions (Schünemann 2021).
We used the overall risk of bias judgement, derived from the RoB 2 Excel tool, to inform our decision on downgrading the certainty of the evidence for risk of bias. We phrased the findings and certainty of the evidence as suggested in the informative statement guidance (Santesso 2020).
Methods for future updates
Living systematic review considerations
Our Information Specialist (MIM) will provide us with new search records each week, which two review authors will screen, extract, evaluate, and integrate following the guidance for Cochrane living systematic reviews (Cochrane LSR). We will manually check platform trials that were previously identified and listed as 'studies awaiting classification' for additional treatment arms. We will wait until the accumulating evidence changes our conclusions of the implications of research and practice before republishing the review. We will consider one or more of the following components to inform this decision.
Findings that change the estimated effect of one or more prioritised outcomes.
Findings that change the credibility (e.g. GRADE rating) of the estimated effect of one or more prioritised outcomes.
New settings, populations, interventions, comparisons, or outcomes studied.
In case of emerging policy relevance because of global controversies around the intervention, we will consider republishing an updated review even though our conclusions remain unchanged. We will review the review scope and methods approximately monthly, or more frequently if appropriate, in light of potential changes in COVID‐19 research (e.g. when additional comparisons, interventions, subgroups or outcomes, or new review methods become available).
Results
Description of studies
See Characteristics of included studies, Characteristics of excluded studies, and Characteristics of ongoing studies tables.
Results of the search
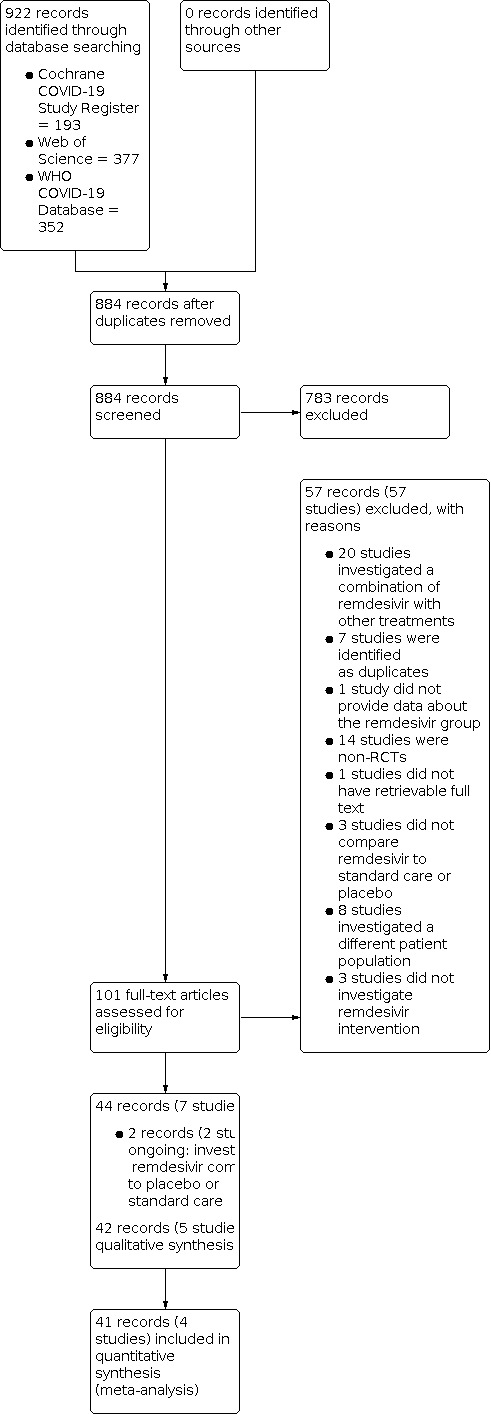
We performed the database searches for RCTs in April 2021 and identified 922 records. After removing duplicates, we screened 884 records based on title and abstract, of which 783 studies did not meet the prespecified inclusion criteria and were excluded. We screened the full texts, or if these were not available, the trial register entries, of the remaining 101 references. Reasons for exclusion of the studies excluded at full‐text stage are listed in Characteristics of excluded studies. We identified two ongoing records (two studies) (Characteristics of ongoing studies; Table 3). Overall, we included 42 records (five studies) in our narrative analysis and 41 records (four studies) in our meta‐analyses. We searched ClinicalTrials.gov and the WHO ICTRP for additional and ongoing trials that met our inclusion criteria. Details of our search strategy are provided in Appendix 1. We recorded the selection process in sufficient detail to complete a PRISMA flow diagram (Figure 1).
2. Characteristics of ongoing studies.
| Study ID | Comparison | Expected completion date |
| NCT04252664 | Remdesivir compared to placebo | Recruiting completed, no publication available yet |
| NCT04596839 | Remdesivir compared to standard care | Recruiting |
1.
Included studies
We included five RCTs with 7452 participants diagnosed with SARS‐CoV‐2 infection in the review (Beigel 2020; Spinner 2020; Wang 2020; Mahajan 2021; WHO Solidarity Trial Consortium 2021). The included participants (mean age 59.37 years, 63.31% male) were diagnosed with SARS‐CoV‐2 infection and were randomly assigned to receive either remdesivir or standard care alone. The majority of included studies were conducted in high‐ and upper‐middle‐income countries; the only reported lower‐middle‐income countries were Honduras, India, and the Philippines. A detailed overview of the characteristics of included studies is provided in Characteristics of included studies and Table 4.
3. Overview of included studies.
| Beigel 2020a | Spinner 2020 | Wang 2020 | WHO Solidarity Trial Consortium 2021 | Mahajan 2021 | |
| (By date of publication) | |||||
| Setting |
|
|
|
|
|
| Design |
|
|
|
|
|
| Study protocol | Reported | Reported | Reported | Reported | Not reported |
| Statistical analysis plan | Reported | Reported | Reported | Reported | Not reported |
|
Intervention (remdesivir) (duration of application (days)) |
10 | 5 or 10 | 10 | 10 | 5 |
| Control | SoC | Placebo + SoC | Placebo + SoC | SoC | SoC |
| Allocated participants (n) | 1062 | 596 | 236 | 5475 | 82 |
|
Number of participants per trial arm (allocated/evaluated) |
Intervention: 541/541 Placebo + SoC: 521/521 |
5‐day intervention: 199/191 10‐day intervention: 197/193 SoC: 200/200 |
Intervention: 158/158 Placebo + SoC: 78/78 |
Intervention: 2750/2743 SoC: 2725/2708 |
Intervention: 41/34 SoC: 41/36 |
| Clinical characteristics at baseline (all participants were hospitalised; ordered according to WHO Progression Scale, see Table 5) (n/N (%)) | |||||
|
No need for oxygen or medical care (not part of WHO 2020d) |
NA | 5‐day intervention: 0/191 (83.8) 10‐day intervention: 6/193 (3.2) SoC: 2/200 (1.0) |
NA | NA | NA |
| WHO 3 | NA | NA | NA | NA | NA |
| WHO 4 | Intervention: 75/541 (13.9) Placebo: 63/521 (12.1) |
5‐day intervention: 160/191 (83.8) 10‐day intervention: 163/193 (84.5) SoC: 160/200 (80.0) |
Intervention: 0/158 (0) Placebo + SoC: 3/78 (3.8) |
Intervention: 661/2743 (24.1) SoC: 664/2708 (24.5) |
NA |
| WHO 5 | Intervention: 232/541 (42.9) Placebo: 203/521 (39.0) |
5‐day intervention: 29/191 (15.2) 10‐day intervention: 23/193 SoC: 36/200 (18.0) |
Intervention: 129/158 (81.6) Placebo + SoC: 65/78 (83.3) |
Intervention: 1828/2743 (66.4) SoC: 1811/2708 (66.9) |
Intervention: 27/34 (79.4) SoC: 26/36 (72.2) |
| WHO 6 | Intervention: 95/541 (17.6) Placebo: 98/521 (18.8) |
5‐day intervention: 2/191 (1.0) 10‐day intervention: 1/193 (0.5) SoC: 2/200 (1.0) |
Intervention: 28/158 (17.2) Placebo + SoC: 9/78 (11.5) |
NA | Intervention: 7/34 (20.6) SoC: 10/36 (27.8) |
| WHO 7 | Intervention: 131/541 (24.2) Placebo: 154/521 (29.6) |
NA | Intervention: 0/158 (0) Placebo + SoC: 1/78 (1.3) |
Intervention: 254/2743 (9.3) SoC: 233/2708 (8.6) |
NA |
| WHO 8 | |||||
| WHO 9 | |||||
| WHO 10 | NA | NA | Intervention: 1/158 (0.6) Placebo + SoC: 0/78 (0) |
NA | NA |
| Demographics | |||||
| Age (years) |
Mean (SD) Intervention: 58.6 (14.6) Placebo: 59.2 (15.4) |
Median (IQR) 5‐day intervention: 58 (48 to 66) 10‐day intervention: 56 (45 to 66) SoC: 57 (45 to 66) |
Median (IQR) Intervention: 66 (57 to 73) Placebo: 64 (53 to 70) |
n/Total < 50 Intervention: 961/2743 SoC: 952/2708 50 to 69 Intervention: 1282/2743 SoC: 1287/2708 ≧ 70 Intervention: 500/2743 SoC: 268/2708 |
Mean (SD) Intervention: 58.09 (12.1) SoC: 57.41 (14.1) |
| Gender (male (n(%))) | Intervention: 352/541 (65.1) Placebo: 332/521 (63.7) |
5‐day intervention: 114/191 (59.7) 10‐day intervention: 118/193 (61.1) SoC: 125/200 (62.5) |
Intervention: 89/158 (56.3) Placebo: 51/78 (65.4) |
Intervention: 1706/2743 (62.2) SoC: 1725/2708 (63.7) |
Intervention: 21/34 (61.8) SoC: 27/36 (75.0) |
| Comorbidities at baseline (n (%)) | |||||
| Diabetes | Intervention: 164 (30.8) Placebo: 158 (30.4) |
5‐day intervention: 71 (37) 10‐day intervention: 85 (44) SoC: 76/200 (38) |
Intervention: 40 (25) Placebo: 16 (21) |
Intervention: 707 (26) SoC: 666 (25) |
Intervention: 21 (62) SoC: 21 (58) |
| Hypertension | Intervention: 269 (50.6) Placebo: 264 (50.9) |
5‐day intervention: 82 (43) 10‐day intervention: 85 (44) SoC: 81 (41) |
Intervention: 73 (46) Placebo: 30 (38) |
Not reported | Intervention: 15 (44) SoC: 17 (47) |
| CAD | Not reported | 5‐day intervention: 111 (58) 10‐day intervention: 111 (58) SoC: 107 (54) |
Intervention: 15 (9) Placebo: 2 (3) |
Not reported | Intervention: 4 (12) SoC: 5 (14) |
| COPD | Not reported | Not reported | Not reported | Intervention: 151 (6) SoC: 145 (5) |
Not reported |
| Asthma | Not reported | 5‐day intervention: 22 (12) 10‐day intervention: 31 (16) SoC: 28 (14) |
Not reported | Intervention: 139 (5) SoC: 139 (5) |
Intervention: 1 (3) SoC: 0 (0) |
| Obesity | Intervention: 242 (46) Placebo: 234 (45) |
BMI (median (IQR)) 5‐day intervention: 27 (24 to 30) 10‐day intervention: 28 (25 to 32) SoC: 27 (24 to 31) |
Not reported | Not reported | Not reported |
| CLD | Not reported | Not reported | Not reported | Intervention: 36 (1) SoC: 41 (2) |
Not reported |
| CKD | Not reported | Not reported | Not reported | Not reported | Intervention: 2 (6) SoC: 1 (3) |
| Other | Not reported | Not reported | Not reported |
Unspecified heart disease Intervention: 571 (21) SoC: 567 (21) |
Hyperlipidaemia Intervention: 4 (12) SoC: 3 (8) Hypothyroidism Intervention: 4 (12) SoC: 3 (8) |
| Concomitant medications (n(%)) | |||||
| Corticosteroids | Intervention: 115 (21.6) Placebo: 126 (24.4) |
5‐day intervention: 33 (17) 10‐day intervention: 29 (15) SoC: 38 (19) |
Intervention: 60 (38) Placebo: 31 (40) |
Intervention: 1310 (48) SoC: 1288 (48) |
Not reported |
| HCQ/CQ | Intervention: 184 (35) Placebo: 189 (37) |
5‐day intervention: 16 (8) 5‐day intervention: 22 (11) SoC: 89 (45) |
Not reported | Not reported | Not reported |
| Lopinavir‐ritonavir | Not reported | 5‐day intervention: 10 (5) 10‐day intervention: 11 (6) SoC: 43 (22) |
Intervention: 27 (17) Placebo: 31 (40) |
Not reported | Not reported |
| MAB (interleukin 6) | Intervention: 23 (4.3) Placebo: 26 (5.0) |
5‐day intervention: 1 (1) 10‐day intervention: 1 (1) SoC: 10 (5) |
Not reported | Intervention: 133 (5) SoC: 143 (5) |
Not reported |
| Azithromycin | Not reported | 5‐day intervention: 35 (18) 10‐day intervention: 41 (21) SoC: 62 (31) |
Not reported | Not reported | Not reported |
| Other |
Antibiotics Intervention: 420 (79) Placebo: 443 (86) Vasopressors Intervention: 147 (28) Placebo: 195 (38) Other anti‐inflammatory medications Intervention: 42 (8) Placebo: 37 (7) Other biologic therapies Intervention: 21 (4) Placebo: 13 (3) Other putative SARS‐CoV‐2 medications Intervention: 8 (2) Placebo: 14 (3) Other antiviral medications Intervention: 10 (2) Placebo: 8 (2) |
Not reported |
Antibiotics Intervention: 121 (77) Placebo: 63 (81) Interferon alfa‐2b Intervention: 29 (18) Placebo: 15 (19) |
Convalescent plasma Intervention: 52 (2) SoC: 58 (2) Interferon Intervention: 3 (0.1) SoC: 25 (1) Antiviral other than RDV Intervention: 65 (2) SoC: 152 (6) |
Not reported |
aMissing data at baseline (n/N): intervention: 8/541, placebo: 3/521.
BMI = body mass index
CAD = coronary artery disease
CKD = chronic kidney disease
CLD = chronic liver disease
COPD = chronic obstructive pulmonary disease
HCQ/CQ = hydroxychlorquine/chloroquine
IQR = interquartile range
MAB = monoclonal antibodies
NA = not available/not applicable
RDV = remdesivir
SD = standard deviation
SoC = standard of care
WHO = World Health Organization
Study design and control
All included RCTs used a parallel‐group design. Three studies had an open‐label design with comparison of remdesivir to standard care alone (Spinner 2020; Mahajan 2021; WHO Solidarity Trial Consortium 2021), whereas two studies were double‐blinded and placebo‐controlled (Beigel 2020; Wang 2020). In one study (Beigel 2020), participants in the control arm received a lyophilised placebo identical in physical appearance to the active lyophilised formulation and containing the same inactive ingredients; alternatively, a normal saline of equal volume was given if there were limitations on matching placebo supplies. In the other study (Wang 2020), participants in the control group received a placebo, which was provided by Gilead Sciences. Three studies compared remdesivir to standard care alone (Spinner 2020; Mahajan 2021; WHO Solidarity Trial Consortium 2021). Notably, two studies did not provide details on standard care (Wang 2020; Mahajan 2021). Three studies performed non‐specified standard care according to local guidelines (Beigel 2020; Spinner 2020; WHO Solidarity Trial Consortium 2021).
Intervention
A total of 3886 participants in the included RCTs were randomised to receive remdesivir. The majority of included studies applied a 10‐day course of remdesivir (Beigel 2020; Spinner 2020; Wang 2020; WHO Solidarity Trial Consortium 2021). Spinner 2020 (additionally) and Mahajan 2021 (solely) reported outcomes also for a five‐day treatment course. Participants in the WHO Solidarity trial were randomly assigned to receive either remdesivir (n = 2750), hydroxychloroquine (n = 954), lopinavir (n = 1411), or interferon beta‐1a (n = 2063) and were compared with a control group for each arm (WHO Solidarity Trial Consortium 2021). Because of an overlap of each control group with other groups, participants allocated for experimental treatments other than remdesivir and associated control groups were not included in the total calculation of participants in our review.
The treatment regimen in the interventional arms of all included studies consisted of standard care plus 200 mg remdesivir intravenously as a loading dose on day 1, followed by 100 mg daily. In three studies (Beigel 2020; Wang 2020; WHO Solidarity Trial Consortium 2021), the maintenance dose plus standard care was administered on days 2 through 10 or until hospital discharge or death. In one study (Spinner 2020), participants received maintenance dose plus standard care either for four or nine consecutive days or until hospital discharge or death; and in the remaining study (Mahajan 2021), the maintenance dose of 100 mg remdesivir plus standard care was administered daily on days 2 through 5.
Setting
Three studies were multicentre studies performed in several countries (Beigel 2020 in 73 sites in Denmark, Germany, Greece, Japan, Korea, Mexico, Singapore, Spain, the UK, and the USA; Spinner 2020 in 105 hospitals in Asia, Europe, and the USA; WHO Solidarity Trial Consortium 2021 in 405 hospitals in Argentina, Brazil, Canada, Germany, Honduras, India, Indonesia, Iran, Ireland, Israel, Italy, Kenya, Lebanon, Malaysia, Norway, Peru, the Philippines, Qatar, Saudi Arabia, South Africa, Spain, Switzerland, and Thailand) (Beigel 2020; Spinner 2020; WHO Solidarity Trial Consortium 2021). Wang 2020 performed a multicentre study in one country, China. Mahajan 2021 performed a single‐centre study in India.
Participants
All studies included hospitalised adults with SARS‐CoV‐2 infections (Beigel 2020; Spinner 2020; Wang 2020; Mahajan 2021; WHO Solidarity Trial Consortium 2021). Notably, Spinner 2020 included one participant younger than 18 years; this corresponds to 0.178% of all recruited participants in this RCT. Reported comorbidities across all studies included diabetes, hypertension and other cardiovascular diseases, chronic lung disease or liver disease, and obesity. Full details on comorbidities are provided in Table 4.
Diagnosis of SARS‐CoV‐2 infection
In four studies (Beigel 2020; Spinner 2020; Wang 2020; Mahajan 2021), SARS‐CoV‐2 infection diagnosis was confirmed by PCR and clinical or radiological signs of pneumonia. In two studies (Spinner 2020; WHO Solidarity Trial Consortium 2021), participants were included with “definite” SARS‐CoV‐2 infection without further specification, or rather with clinical signs of an acute respiratory infection. The included studies did not provide details on how many participants' SARS‐CoV‐2 infection was confirmed by PCR testing.
Severity of illness
No two studies reported similar subgroups of participants in terms of severity of illness. Three studies included participants with moderate to severe illness, although according to different definitions (Beigel 2020; Mahajan 2021; WHO Solidarity Trial Consortium 2021, Table 5). One study included participants with moderate illness (Spinner 2020); and one study included participants with severe illness (Wang 2020). In Beigel 2020, participants were considered to have severe disease if they required mechanical ventilation; if the oxygen saturation as measured by pulse oximetry (SpO2) was 94% or lower whilst they were breathing ambient air; or if they had tachypnoea (respiratory rate ≥ 24 breaths per minute). The majority of participants in this study met the aforementioned criteria and needed supplemental oxygen (intervention 42.9%, control 39.0%), non‐invasive ventilation or high‐flow oxygen (intervention 17.6%, control 18.8%), or invasive mechanical ventilation or extracorporeal membrane oxygenation (ECMO) (intervention 24.2%, control 29.6%). Only 13.9% of participants in the intervention arm and 12.1% of participants in the control arm were hospitalised without requiring supplemental oxygen. In Mahajan 2021, participants in both groups were classified as “highest disease severity”, but were excluded from the study if receiving mechanical ventilation or if having multi‐organ failure. The majority (79.4%) of participants in the intervention group versus 72.2% of participants in the control group received low‐flow supplemental oxygen, and 20.6% versus 27.8% received non‐invasive ventilation or high‐flow oxygen, respectively. In Spinner 2020, the majority (84%) of participants in the intervention group versus 80% of participants in the control group did not require supplemental oxygen. Although measured oxygen saturation at screening was above 94% whilst breathing room air, 13% of participants in the intervention group and 19% in the control group used supplemental oxygen because of deteriorating clinical status or for breathing comfort. In Wang 2020, severe SARS‐CoV‐2 infections was defined by oxygen saturation of 94% or lower on room air or a ratio of arterial oxygen partial pressure to fractional inspired oxygen of 300 mmHg or less. The majority of participants in this study needed oxygen supplementation (intervention 82%, control 83%), whilst non‐invasive ventilation or high‐flow oxygen was necessary in 18% and 12% of participants, respectively. Invasive mechanical ventilation or ECMO was only required in 1% of the control group and none in the intervention group. In WHO Solidarity Trial Consortium 2021, disease severity was not protocol‐defined, but baseline respiratory support was divided in “no supplemental oxygen”, “supplemental oxygen”, and “mechanical ventilation”. The majority of participants in the intervention group (66.6%) and the control group (66.9%) received supplemental oxygen at entry, whilst no supplemental oxygen was needed in 24.1% and 24.5% of participants in each group. The minority of participants received mechanical ventilation at entry: 9.3% and 8.6%, respectively.
4. Conversion between ordinal clinical progression scales used by included studies and WHO Clinical Progression Scale.
|
|
WHO 2020d | Description | Beigel 2020 | Spinner 2020 | Wang 2020 | WHO Solidarity Trial Consortium 2021 | Mahajan 2021 | |
| Day 1 to day 12 | Day 12 to day 24 | |||||||
|
Ambulatory: mild disease |
1 |
Asymptomatic Viral RNA detected |
1 | 7 | 1 | NA | NA | 1 |
| 2 |
Symptomatic Independent |
NA | NA | |||||
| 3 |
Symptomatic Assistance needed |
2 | NA | NA | ||||
|
Hospitalised: moderate disease |
NA | No oxygen or medical care | 3 | 6 | NA | No supplemental oxygen |
NA | NA |
| 4 | No oxygen therapy | 4 | 5 | 2 | 3 | 2 | ||
| 5 | Oxygen by mask or nasal prongs | 5 | 4 | 3 | Supplemental oxygen |
1 | 3 | |
|
Hospitalised: severe disease |
6 | Oxygen by NIV or high‐flow | 6 | 3 | 4 | NA | 2 | 4 |
| 7 |
Intubation and IMV pO2/FiO2 ≥ 150 or SpO2/FiO2 ≥ 200 |
7 | 2 | 5 | IMV | 4 | 5 | |
| 8 |
IMV and pO2/FiO2 < 150 (SpO2/FiO2 < 200) OR vasopressors |
|||||||
| 9 |
IMV and pO2/FiO2 < 150 and vasopressors, dialysis, or ECMO |
|||||||
| 10 | Dead | 8 | 1 | 6 | NA | NA | NA | |
ECMO = extracorporeal membrane oxygenation
FiO2 = fraction of inspired oxygen
IMV = invasive mechanical ventilation
NA = not applicable
NIV = non‐invasive mechanical ventilation
pO2 = partial pressure of oxygen
RNA = ribonucleic acid
SpO2 = oxygen saturation
Concomitant medications
Concomitant use of COVID‐19 medication was restricted to heparin and corticosteroids in one study (Mahajan 2021). Wang 2020 reported concomitant use of lopinavir–ritonavir, interferon, and corticosteroids. Two studies provided no details on concomitant therapy (Beigel 2020; WHO Solidarity Trial Consortium 2021). Additional therapy with traditional herbs including sho‐saiko‐to (or Xiao‐Shai‐Hu‐Tang) or investigational agents with putative antiviral activity against COVID‐19 was prohibited by protocol for participants receiving remdesivir in one study (Spinner 2020). However, concomitant use of lopinavir‐ritonavir, hydroxychloroquine/chloroquine, interferon, steroids, tocilizumab, and azithromycin was reported for all participants, predominantly in the control arm.
Outcomes
Primary outcomes differed significantly between included studies, with no two studies having chosen the same primary endpoint (Beigel 2020: time to recovery; Mahajan 2021: improvement in clinical outcomes; WHO Solidarity Trial Consortium 2021: all‐cause mortality; Spinner 2020: clinical status on day 11; Wang 2020: time to clinical improvement at day 28). A detailed narrative summary of all reported outcome measures for each of the included studies is provided in Table 6.
5. Narrative summary of outcomes of included studies.
| Beigel 2020 | |
| Study outcomes (time point, definition) | Reported outcome data (R: remdesivir group, C: control group) |
All‐cause mortality
|
1.Mortality at up to day 15, n/N (%); HR (95% CI)
2. Mortality at up to day 29: included in analysis (Analysis 1.1). 3. Mortality, time‐to‐event: included in analysis (Analysis 1.3). |
Clinical status with mean change in ordinal scale compared to baseline
8‐category ordinal scale:
|
Clinical status at day 15, n/N (%)
Mean ordinal scale (SD) at day 15; mean change from baseline scale (SD) at day 15: R: 3.7 (2.5); −1.9 (2.1) versus C: 4.3 (2.6); −1.4 (2.3)
Mean ordinal scale (SD) at day 29; mean change from baseline scale (SD) at day 29: R: 2.8 (2.5); −2.79 (2.3) versus C: 3.4 (2.7); −2.3 (2.6) |
Clinical improvement
|
|
|
Clinical worsening Incidence of new oxygen use, of non‐invasive ventilation or high‐flow oxygen, and of invasive ventilation or ECMO |
Initiation of respiratory support: Included in analysis (Analysis 1.5; Analysis 1.6; Analysis 1.7). |
Duration of hospitalisation up to day 29.
|
Initial length of hospital stay, median days (IQR)
Patients rehospitalised, per cent (95% CI); difference (95% CI)
|
Safety outcomes
|
Safety outcomes: included in analysis (Analysis 1.9; Analysis 1.10; Analysis 1.11). |
| Mahajan 2021 | |
| Study outcomes (time point, definition) | Reported outcome data |
All‐cause mortality
|
Death from day 12 to day 24, n/N (%)
|
|
Clinical status from day 12 to 24 6‐point ordinal scale
|
Clinical status from day 12 to 24, n/N (%)
|
| Duration of hospitalisation |
Admission days, mean (SD)
|
Safety outcomes
|
|
| Spinner 2020 | |
| Study outcomes (time point, definition) | Reported outcome data |
All‐cause mortality
|
1. Mortality at up to day 11, n/N (%)
2. Mortality at up to day 28, n/N (%): included in analysis (Analysis 1.1) 3. Kaplan‐Meier estimates of all‐cause mortality at day 28, % (95% CI)
|
|
Clinical status at day 11 with difference in clinical status distribution and days 14, 28 7‐point ordinal scale:
|
Clinical status at day 11, n/N (%)
Clinical status at day 14, n/N (%)
Clinical status at day 28, n/N (%)
|
Clinical improvement
|
|
| Duration of hospitalisation | Hospital discharges over time: reported in figures. |
Safety outcomes
|
Safety outcomes: included in analysis (Analysis 1.9; Analysis 1.10; Analysis 1.11). |
| Wang 2020 | |
| Study outcomes (time point, definition) | Reported outcome data |
All‐cause mortality
|
All‐cause mortality
|
|
Clinical status at day 7, 14, 28 with odds ratio (OR) 6‐category scale:
|
Clinical status at day 7, n/N (%)
Clinical status at day 14, n/N (%)
Clinical status at day 28, n/N (%)
|
Clinical improvement
|
Clinical Improvement
|
Clinical worsening
|
|
Duration of respiratory support
|
|
Duration of hospitalisation
|
|
Viral clearance
|
|
Safety outcomes
|
|
| WHO Solidarity Trial Consortium 2021 | |
| Study outcomes (time point, definition) | Reported outcome data |
All‐cause mortality
|
|
Clinical worsening
|
Initiation of mechanical ventilation: included in analysis (Analysis 1.4). |
Duration of hospitalisation
|
Time to discharge reported in figures. |
Abbreviations
ALT = alanine transaminase
AST = aspartate transaminase
C = control
CI = confidence interval
ECMO = extracorporeal membrane oxygenation
HR = hazard ratio
IMV = invasive mechanical ventilation
IQR = interquartile range
NIV = non‐invasive mechanical ventilation
OR = odds ratio
PCR = polymerase chain reaction
R = remdesivir
RNA = ribonucleic acid
SD = standard deviation
Ongoing studies
An overview of the characteristics of ongoing studies is provided in Characteristics of ongoing studies and Table 3. We included two records of ongoing studies comparing the effects of remdesivir with placebo or standard care alone (NCT04252664; NCT04596839). NCT04596839 was expected to be completed in 2021 and planned to evaluate 60 participants. NCT04252664 was discontinued: “The epidemic of COVID‐19 has been controlled well at present, no eligible patients can be recruited”. They had planned to evaluate 308 participants.
Excluded studies
We excluded 57 references (57 studies) that did not match our inclusion criteria (for details, see Characteristics of excluded studies):
Seven references were identified as duplicates;
For one reference the full‐text was not retrievable;
14 studies were non‐RCTs;
20 studies investigated a combination of remdesivir with other treatments;
Three studies did not compare remdesivir to standard care or placebo;
Eight studies investigated a different patient population;
One study did not provide data on the remdesivir group;
Three studies did not investigate remdesivir intervention.
Risk of bias in included studies
We assessed risk of bias for the results within the five included RCTs, Beigel 2020; Spinner 2020; Wang 2020; Mahajan 2021; WHO Solidarity Trial Consortium 2021, using the RoB 2 tool, as recommended in Chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions (Sterne 2019; Higgins 2021c). Outlined below are outcomes that were reported according to our review protocol. The completed RoB 2 tool with responses to all assessed signalling questions is available online at https://doi.org/10.5281/zenodo.5101320.
Remdesivir compared to placebo or standard care alone
All‐cause mortality
Four studies reported this outcome (see Table 9). Overall, we rated the risk of bias for mortality to be low for three studies (Beigel 2020; Spinner 2020; WHO Solidarity Trial Consortium 2021), and of some concerns for one study (Wang 2020). We assessed this outcome on a study level at up to day 28 and as time‐to‐event or at hospital discharge, if provided (see Table 10; Table 11), as well as for our subgroup analyses (see Table 20; Table 21; Table 22). For one study (Wang 2020), there were some concerns arising from baseline differences in gender distribution, respiratory status, comorbidities, and time from symptom onset, suggesting a possible problem with block wise and stratified randomisation process. We did not identify any concerns that could have biased the reported outcome in three studies (Beigel 2020; Spinner 2020; WHO Solidarity Trial Consortium 2021), and therefore judged the risk of bias to be low.
Risk of bias for analysis 1.1 All‐cause mortality at up to day 28.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Beigel 2020 | Low risk of bias | Randomisation was stratified by study site and disease severity in a 1:1 ratio and the allocation sequence was concealed. There were no baseline imbalances that would suggest a problem with randomisation. | Low risk of bias | Participants and care takers were adequately blinded. The analysis was appropriate (intention‐to‐treat population). | Low risk of bias | Outcome was available for all participants. | Low risk of bias | Measuring of the outcome was appropriate. The outcome was assessed using standardised methods and assessors were unaware of the treatment assignments. | Low risk of bias | The data was analysed in accordance with a pre‐specified protocol. | Low risk of bias | Overall judged low risk of bias. Information on randomisation process is not provided, but blinding was appropriate and outcome measurement was according to a pre‐specified protocol. |
| Spinner 2020 | Low risk of bias | Block‐randomisation was not stratified and web‐based. Sites did not have access to the randomisation list. There were no baseline imbalances that would suggest a problem with randomisation. | Low risk of bias | Participants and care takers were aware of the assigned intervention. There was a neglectable number of participants with deviations from the intended intervention and the analysis was appropriate (intention‐to‐treat population). | Low risk of bias | Data was available for nearly all participants (<5% missing) randomised. | Low risk of bias | Outcome assessors were aware of the treatment assignments. Knowledge of intervention received could not have affected the outcome measurement. | Low risk of bias | The trial protocol provided details of time points and analysis. Mortality was analysed in accordance with a prespecified analysis plan. | Low risk of bias | Overall judged low risk of bias. Details about randomisation, blinding of outcome assessors and a protocol were provided. It was an open‐label study, but this could not have affected the outcome measurement. |
| Wang 2020 | Some concerns | Randomisation was carried out 2:1 and in stratified blocks. Concealed envelopes were prepared. There were baseline differences in gender distribution, respiratory status at baseline, co‐morbidities and time from symptom onset suggesting possible randomisation problems. | Low risk of bias | The study was carried out double blinded. Analysis was done appropriately. | Low risk of bias | Data was available for nearly all participants (<5% missing) randomised. | Low risk of bias | Outcome assessors were unaware of the treatment assignments and could not have been affected by knowledge of intervention. | Low risk of bias | The trial protocol provided details of time points and analysis. However, the statistical analysis plan could not be found. | Some concerns | Overall judged some concerns for risk of bias. Details about randomisation, blinding of outcome assessors and a protocol were provided. There were baseline differences between intervention and control group suggesting a problem with block wise randomisation process. A statistical analysis plan could not be found. |
| WHO Solidarity Trial Consortium 2021 | Low risk of bias | Patients were randomised by a study website, which probably concealed the allocation sequence. There were no baseline imbalances that would suggest a problem with randomisation | Low risk of bias | Participants and care takers were aware of the assigned intervention, but there were no deviations from intended interventions and the analysis was appropriate (intention‐to‐treat population). | Low risk of bias | Data was available for nearly all participants (<5% missing) randomised. | Low risk of bias | Outcome assessors were aware of the intervention received. Knowledge of intervention received could not have affected the outcome measurement. | Low risk of bias | The data was analysed in accordance with a pre‐specified protocol. Multiple analyses were reported according to a pre‐defined protocol. | Low risk of bias | Overall judged low risk of bias. Details about randomisation and missing data were provided. It was an open‐label study, but this could not have affected the outcome measurement. Outcome measurement and analyses were carried out according to a pre‐defined study protocol. |
Risk of bias for analysis 1.2 All‐cause mortality at hospital discharge.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| WHO Solidarity Trial Consortium 2021 | Low risk of bias | Patients were randomised by a study website, which probably concealed the allocation sequence. There were no baseline imbalances that would suggest a problem with randomisation | Low risk of bias | Participants and care takers were aware of the assigned intervention, but there were no deviations from intended interventions and the analysis was appropriate (intention‐to‐treat population). | Low risk of bias | Data was available for nearly all participants (<5% missing) randomised. | Low risk of bias | Outcome assessors were aware of the intervention received. Knowledge of intervention received could not have affected the outcome measurement. | Low risk of bias | The data was analysed in accordance with a pre‐specified protocol. Multiple analyses were reported according to a pre‐defined protocol. | Low risk of bias | Overall judged low risk of bias. Details about randomisation and missing data were provided. It was an open‐label study, but this could not have affected the outcome measurement. Outcome measurement and analyses were carried out according to a pre‐defined study protocol. |
Risk of bias for analysis 1.3 All‐cause mortality (time‐to‐event).
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Beigel 2020 | Low risk of bias | Randomisation was stratified by study site and disease severity in a 1:1 ratio and the allocation sequence was concealed. There were no baseline imbalances that would suggest a problem with randomisation. | Low risk of bias | Participants and care takers were adequately blinded. The analysis was appropriate (intention‐to‐treat population). | Low risk of bias | Outcome available for all participants. | Low risk of bias | Measuring of the outcome was appropriate. The outcome was assessed using standardised methods and assessors were unaware of the treatment assignments. | Low risk of bias | The data was analysed in accordance with a pre‐specified protocol. | Low risk of bias | Overall judged low risk of bias. Information on randomisation process is not provided, but blinding was appropriate and outcome measurement was according to a pre‐specified protocol. |
| WHO Solidarity Trial Consortium 2021 | Low risk of bias | Patients were randomised by a study website, which probably concealed the allocation sequence. There were no baseline imbalances that would suggest a problem with randomisation | Low risk of bias | Participants and care takers were aware of the assigned intervention, but there were no deviations from intended interventions and the analysis was appropriate (intention‐to‐treat population). | Low risk of bias | Data was available for nearly all participants (<5% missing) randomised. | Low risk of bias | Outcome assessors were aware of the intervention received. Knowledge of intervention received could not have affected the outcome measurement. | Low risk of bias | The data was analysed in accordance with a pre‐specified protocol. Multiple analyses were reported according to a pre‐defined protocol. | Low risk of bias | Overall judged low risk of bias. Details about randomisation and missing data were provided. It was an open‐label study, but this could not have affected the outcome measurement. Outcome measurement and analyses were carried out according to a pre‐defined study protocol. |
Risk of bias for analysis 2.1 All‐cause mortality at up to day 28.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 2.1.1 Age <50 years | ||||||||||||
| WHO Solidarity Trial Consortium 2021 | Low risk of bias | Patients were randomised by a study website, which probably concealed the allocation sequence. There were no baseline imbalances that would suggest a problem with randomisation | Low risk of bias | Participants and care takers were aware of the assigned intervention, but there were no deviations from intended interventions and the analysis was appropriate (intention‐to‐treat population). | Low risk of bias | Data was available for nearly all participants (<5% missing) randomised. | Low risk of bias | Outcome assessors were aware of the intervention received. Knowledge of intervention received could not have affected the outcome measurement. | Low risk of bias | The data was analysed in accordance with a pre‐specified protocol. Multiple analyses were reported according to a pre‐defined protocol. | Low risk of bias | Overall judged low. Details about randomisation and missing data were provided. It was an open‐label study, but this could not have affected the outcome measurement. Outcome measurement and analyses were carried out according to a pre‐defined study protocol. |
| Subgroup 2.1.2 Age 50 to 69 years | ||||||||||||
| WHO Solidarity Trial Consortium 2021 | Low risk of bias | Patients were randomised by a study website, which probably concealed the allocation sequence. There were no baseline imbalances that would suggest a problem with randomisation | Low risk of bias | Participants and care takers were aware of the assigned intervention, but there were no deviations from intended interventions and the analysis was appropriate (intention‐to‐treat population). | Low risk of bias | Data was available for nearly all participants (<5% missing) randomised. | Low risk of bias | Outcome assessors were aware of the intervention received. Knowledge of intervention received could not have affected the outcome measurement. | Low risk of bias | The data was analysed in accordance with a pre‐specified protocol. Multiple analyses were reported according to a pre‐defined protocol. | Low risk of bias | Overall judged low. Details about randomisation and missing data were provided. It was an open‐label study, but this could not have affected the outcome measurement. Outcome measurement and analyses were carried out according to a pre‐defined study protocol. |
| Subgroup 2.1.3 Age >69 years | ||||||||||||
| WHO Solidarity Trial Consortium 2021 | Low risk of bias | Patients were randomised by a study website, which probably concealed the allocation sequence. There were no baseline imbalances that would suggest a problem with randomisation | Low risk of bias | Participants and care takers were aware of the assigned intervention, but there were no deviations from intended interventions and the analysis was appropriate (intention‐to‐treat population). | Low risk of bias | Data was available for nearly all participants (<5% missing) randomised. | Low risk of bias | Outcome assessors were aware of the intervention received. Knowledge of intervention received could not have affected the outcome measurement. | Low risk of bias | The data was analysed in accordance with a pre‐specified protocol. Multiple analyses were reported according to a pre‐defined protocol. | Low risk of bias | Overall judged low. Details about randomisation and missing data were provided. It was an open‐label study, but this could not have affected the outcome measurement. Outcome measurement and analyses were carried out according to a pre‐defined study protocol. |
Risk of bias for analysis 3.1 All‐cause mortality at up to day 28.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 3.1.1 ≤ 10 days of symptom onset | ||||||||||||
| Wang 2020 | Some concerns | Randomisation was carried out 2:1 and in stratified blocks. Concealed envelopes were prepared. There were baseline differences in gender distribution, respiratory status at baseline, co‐morbidities and time from symptom onset suggesting possible randomisation problems. | Low risk of bias | The study was carried out double blinded. Analysis was done appropriately. | Low risk of bias | Data for this outcome was available for nearly all participants randomised. | Low risk of bias | Outcome assessors were unaware of the treatment assignments and could not have been affected by knowledge of intervention. | Low risk of bias | The trial protocol provided details of time points and analysis. However, the statistical analysis plan could not be found. | Some concerns | Details about randomisation, blinding of outcome assessors and a protocol were provided. There were baseline differences between intervention and control group. |
| Subgroup 3.1.2 > 10 days of symptom onset | ||||||||||||
| Wang 2020 | Some concerns | Randomisation was carried out 2:1 and in stratified blocks. Concealed envelopes were prepared. There were baseline differences in gender distribution, respiratory status at baseline, co‐morbidities and time from symptom onset suggesting possible randomisation problems. | Low risk of bias | The study was carried out double blinded. Analysis was done appropriately. | Low risk of bias | Data for this outcome was available for nearly all participants randomised. | Low risk of bias | Outcome assessors were unaware of the treatment assignments and could not have been affected by knowledge of intervention. | Low risk of bias | The trial protocol provided details of time points and analysis. However, the statistical analysis plan could not be found. | Some concerns | Details about randomisation, blinding of outcome assessors and a protocol were provided. There were baseline differences between intervention and control group. |
Risk of bias for analysis 4.1 All‐cause mortality at up to day 28.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 4.1.1 No oxygen at baseline | ||||||||||||
| Beigel 2020 | Low risk of bias | Randomisation was stratified by study site and disease severity in a 1:1 ratio and the allocation sequence was concealed. There were no baseline imbalances that would suggest a problem with randomisation. | Low risk of bias | Participants and care takers were adequately blinded. The analysis was appropriate (intention‐to‐treat population). | Low risk of bias | Outcome was available for all participants. | Low risk of bias | Measuring of the outcome was appropriate. The outcome was assessed using standardised methods and assessors were unaware of the treatment assignments. | Low risk of bias | The data was analysed in accordance with a pre‐specified protocol. | Low risk of bias | Overall judged low risk of bias. Information on randomisation process is not provided, but blinding was appropriate and outcome measurement was according to a pre‐specified protocol. |
| Spinner 2020 | Low risk of bias | Block‐randomisation was not stratified and web‐based. Sites did not have access to the randomisation list. There were no baseline imbalances that would suggest a problem with randomisation. | Low risk of bias | Participants and care takers were aware of the assigned intervention. There was a neglectable number of participants with deviations from the intended intervention and the analysis was appropriate (intention‐to‐treat population). | Low risk of bias | Data was available for nearly all participants (<5% missing) randomised. | Low risk of bias | Outcome assessors were aware of the treatment assignments. Knowledge of intervention received could not have affected the outcome measurement. | Low risk of bias | The trial protocol provided details of time points and analysis. Mortality was analysed in accordance with a prespecified analysis plan. | Low risk of bias | Overall judged low risk of bias. Details about randomisation, blinding of outcome assessors and a protocol were provided. It was an open‐label study, but this could not have affected the outcome measurement. |
| WHO Solidarity Trial Consortium 2021 | Low risk of bias | Patients were randomised by a study website, which probably concealed the allocation sequence. There were no baseline imbalances that would suggest a problem with randomisation | Low risk of bias | Participants and care takers were aware of the assigned intervention, but there were no deviations from intended interventions and the analysis was appropriate (intention‐to‐treat population). | Low risk of bias | Data was available for nearly all participants (<5% missing) randomised. | Low risk of bias | Outcome assessors were aware of the intervention received. Knowledge of intervention received could not have affected the outcome measurement. | Low risk of bias | The data was analysed in accordance with a pre‐specified protocol. Multiple analyses were reported according to a pre‐defined protocol. | Low risk of bias | Overall judged low risk of bias. Details about randomisation and missing data were provided. It was an open‐label study, but this could not have affected the outcome measurement. Outcome measurement and analyses were carried out according to a pre‐defined study protocol. |
| Subgroup 4.1.2 Low‐flow oxygen at baseline | ||||||||||||
| Beigel 2020 | Low risk of bias | Randomisation was stratified by study site and disease severity in a 1:1 ratio and the allocation sequence was concealed. There were no baseline imbalances that would suggest a problem with randomisation. | Low risk of bias | Participants and care takers were adequately blinded. The analysis was appropriate (intention‐to‐treat population). | Low risk of bias | Outcome was available for all participants. | Low risk of bias | Measuring of the outcome was appropriate. The outcome was assessed using standardised methods and assessors were unaware of the treatment assignments. | Low risk of bias | The data was analysed in accordance with a pre‐specified protocol. | Low risk of bias | Overall judged low risk of bias. Information on randomisation process is not provided, but blinding was appropriate and outcome measurement was according to a pre‐specified protocol. |
| Subgroup 4.1.3 Mechanical ventilation at baseline | ||||||||||||
| Beigel 2020 | Low risk of bias | Randomisation was stratified by study site and disease severity in a 1:1 ratio and the allocation sequence was concealed. There were no baseline imbalances that would suggest a problem with randomisation. | Low risk of bias | Participants and care takers were adequately blinded. The analysis was appropriate (intention‐to‐treat population). | Low risk of bias | Outcome was available for all participants. | Low risk of bias | Measuring of the outcome was appropriate. The outcome was assessed using standardised methods and assessors were unaware of the treatment assignments. | Low risk of bias | The data was analysed in accordance with a pre‐specified protocol. | Low risk of bias | Overall judged low risk of bias. Information on randomisation process is not provided, but blinding was appropriate and outcome measurement was according to a pre‐specified protocol. |
| WHO Solidarity Trial Consortium 2021 | Low risk of bias | Patients were randomised by a study website, which probably concealed the allocation sequence. There were no baseline imbalances that would suggest a problem with randomisation | Low risk of bias | Participants and care takers were aware of the assigned intervention, but there were no deviations from intended interventions and the analysis was appropriate (intention‐to‐treat population). | Low risk of bias | Data was available for nearly all participants (<5% missing) randomised. | Low risk of bias | Outcome assessors were aware of the intervention received. Knowledge of intervention received could not have affected the outcome measurement. | Low risk of bias | The data was analysed in accordance with a pre‐specified protocol. Multiple analyses were reported according to a pre‐defined protocol. | Low risk of bias | Overall judged low risk of bias. Details about randomisation and missing data were provided. It was an open‐label study, but this could not have affected the outcome measurement. Outcome measurement and analyses were carried out according to a pre‐defined study protocol. |
Clinical status
Three studies reported this outcome (see Table 12; Table 13; Table 14; Table 15). We assessed this outcome on a study level by the need for respiratory support in accordance with a standardised scale (WHO 2020d), and included both clinical improvement and worsening in our assessment. Liberation of supplemental oxygen or invasive mechanical ventilation was reported in one study at day 15 (Beigel 2020), and therefore not included in our analyses. Duration to liberation from supplemental oxygen or invasive mechanical ventilation was reported by two and three studies, respectively (Beigel 2020; Spinner 2020; Wang 2020), but data could not be pooled and are therefore described narratively. Two studies provided data on clinical worsening (Beigel 2020; WHO Solidarity Trial Consortium 2021). Although detailed information on randomisation process was not provided in Beigel 2020, blinding was appropriate, and outcome measurement and analyses were according to a prespecified protocol. However, due to competing risk of death, we judged some concerns for risk of bias due to missing data (RoB 2, domain 3) for dichotomous worsening outcomes, and high risk of bias for dichotomous improvement outcomes. We assessed improvement as at high risk of bias due to missing data because it is likely that death during follow‐up impeded liberation from respiratory support, and hence the missing data on improvement depends on its true value. Spinner 2020 provided additional data on need for invasive mechanical ventilation after author enquiry. Given a relevant deviation of assessment time point, an open‐label study design, and a competing risk of death, we judged the outcome measurement as at high risk of bias.
Risk of bias for analysis 1.4 Worsening of clinical status: new need for mechanical ventilation.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 1.4.1 WHO 6 to 9 at day 28 (± 1 day), if ≤5 at baseline | ||||||||||||
| Beigel 2020 | Low risk of bias | Randomisation was stratified by study site and disease severity in a 1:1 ratio and the allocation sequence was concealed. There were no baseline imbalances that would suggest a problem with randomisation. | Low risk of bias | Participants and care takers were adequately blinded. The analysis was appropriate (intention‐to‐treat population). | Some concerns | There is an uncertain amount of missing data due to competing risk of death. It is uncertain, whether the result is biased or not. | Low risk of bias | Measuring of the outcome was appropriate. The outcome was assessed using standardised methods and assessors were unaware of the treatment assignments. | Low risk of bias | The data was analysed in accordance with a pre‐specified protocol. | Some concerns | Overall judged some concerns for risk of bias. Information on randomisation process is not provided, but blinding was appropriate and outcome measurement was according to a pre‐specified protocol. Impact of missing data due to competing risk of death is uncertain, but bias in outcome is judged unlikely. |
| Spinner 2020 | Low risk of bias | Block‐randomisation was not stratified and web‐based. Sites did not have access to the randomisation list. There were no baseline imbalances that would suggest a problem with randomisation. | Low risk of bias | Participants and care takers were aware of the assigned intervention. There was a neglectable number of participants with deviations from the intended intervention and the analysis was appropriate (intention‐to‐treat population). | Some concerns | There is an uncertain amount of missing data due to competing risk of death. It is uncertain, whether the result is biased or not. | High risk of bias | Outcome was assessed with a possible deviation of five days. Because assessors were aware of the treatment assignments, knowledge of intervention received could have affected the outcome measurement. | Low risk of bias | The trial protocol provided details of time points of measurement. Clinical status was reported in accordance with a prespecified analysis plan. | High risk of bias | Overall judged high risk of bias. Details about randomisation, blinding of outcome assessors and a protocol were provided. Impact of missing data due to competing risk of death is uncertain, but bias in outcome is judged unlikely. It was an open‐label study and therefore possible deviation in time point of measuring could have had an effect on the reported results. |
| WHO Solidarity Trial Consortium 2021 | Low risk of bias | Patients were randomised by a study website, which probably concealed the allocation sequence. There were no baseline imbalances that would suggest a problem with randomisation | Low risk of bias | Participants and care takers were aware of the assigned intervention, but there were no deviations from intended interventions and the analysis was appropriate (intention‐to‐treat population). | Some concerns | There is an uncertain amount of missing data due to competing risk of death. It is uncertain, whether the result is biased or not. | Low risk of bias | Outcome assessors were aware of the intervention received. Knowledge of intervention is unlikely to have affected the outcome measurement. | Some concerns | Outcome was reported as proposed by trial protocol, but time point and analysis was not pre‐specified. | Some concerns | Overall judged some concern. Details about randomisation and missing data were provided. It was an open‐label study, but this could not have affected the outcome measurement. Outcome was reported according to study protocol but time point and analysis method was not pre‐specified. |
Risk of bias for analysis 1.5 Worsening of clinical status: new need for invasive mechanical ventilation.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 1.5.1 WHO 7 to 9 at day 28 (± 1 day), if ≤6 at baseline | ||||||||||||
| Beigel 2020 | Low risk of bias | Randomisation was stratified by study site and disease severity in a 1:1 ratio and the allocation sequence was concealed. There were no baseline imbalances that would suggest a problem with randomisation. | Low risk of bias | Participants and care takers were adequately blinded. The analysis was appropriate (intention‐to‐treat population). | Low risk of bias | The proportion of missing values is less than 5% for both groups. | Low risk of bias | Measuring of the outcome was appropriate. The outcome was assessed using standardised methods and assessors were unaware of the treatment assignments. | Low risk of bias | The data was analysed in accordance with a pre‐specified protocol. | Low risk of bias | Overall judged low risk of bias. Information on randomisation process is not provided, but blinding was appropriate and outcome measurement was according to a pre‐specified protocol. |
| Spinner 2020 | Low risk of bias | Block‐randomisation was not stratified and web‐based. Sites did not have access to the randomisation list. There were no baseline imbalances that would suggest a problem with randomisation. | Low risk of bias | Participants and care takers were aware of the assigned intervention. There was a neglectable number of participants with deviations from the intended intervention and the analysis was appropriate (intention‐to‐treat population). | Low risk of bias | Data was available for nearly all participants (<5% missing) randomised to the interventions. | High risk of bias | Outcome was assessed with a possible deviation of five days. Because assessors were aware of the treatment assignments, knowledge of intervention received could have affected the outcome measurement. | Low risk of bias | The trial protocol provided details of time points of measurement. Clinical status was reported in accordance with a prespecified analysis plan. | High risk of bias | Overall judged high risk of bias. Details about randomisation, blinding of outcome assessors and a protocol were provided. It was an open‐label study and therefore possible deviation in time point of measuring could have influenced the reported results. |
Risk of bias for analysis 1.6 Worsening of clinical status: new need for non‐invasive mechanical ventilation or high‐flow oxygen.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 1.6.1 WHO= 6 at day 29, if ≤5 at baseline | ||||||||||||
| Beigel 2020 | Low risk of bias | Randomisation was stratified by study site and disease severity in a 1:1 ratio and the allocation sequence was concealed. There were no baseline imbalances that would suggest a problem with randomisation. | Low risk of bias | Participants and care takers were adequately blinded. The analysis was appropriate (intention‐to‐treat population). | Some concerns | There is an uncertain amount of missing data due to competing risk of death. It is uncertain, whether the result is biased or not. | Low risk of bias | Measuring of the outcome was appropriate. The outcome was assessed using standardised methods and assessors were unaware of the treatment assignments. | Low risk of bias | The data was analysed in accordance with a pre‐specified protocol. | Some concerns | Overall judged some concerns for risk of bias. Information on randomisation process is not provided, but blinding was appropriate and outcome measurement was according to a pre‐specified protocol. Impact of missing data due to competing risk of death is uncertain, but bias in outcome is judged unlikely. |
Risk of bias for analysis 1.7 Worsening of clinical status: new need for oxygen by mask or nasal prongs.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 1.7.1 WHO= 5 at day 29, if ≤ 4 at baseline | ||||||||||||
| Beigel 2020 | Low risk of bias | Randomisation was stratified by study site and disease severity in a 1:1 ratio and the allocation sequence was concealed. There were no baseline imbalances that would suggest a problem with randomisation. | Low risk of bias | Participants and care takers were adequately blinded. The analysis was appropriate (intention‐to‐treat population). | Some concerns | There is an uncertain amount of missing data due to competing risk of death. It is uncertain, whether the result is biased or not. | Low risk of bias | Measuring of the outcome was appropriate. The outcome was assessed using standardised methods and assessors were unaware of the treatment assignments. | Low risk of bias | The data was analysed in accordance with a pre‐specified protocol. | Some concerns | Overall judged some concerns for risk of bias. Information on randomisation process is not provided, but blinding was appropriate and outcome measurement was according to a pre‐specified protocol. Impact of missing data due to competing risk of death is uncertain, but bias in outcome is judged unlikely. |
Duration of hospitalisation
Three studies reported this outcome (see RoB 2.0 Tool assessment). We assessed this outcome on a study level for length of hospital stay in days. Since Beigel 2020 and Wang 2020 reported data as median, and WHO Solidarity Trial Consortium 2021 and Spinner 2020 only provided figures for this outcome, meta‐analysis could not be conducted. Together with the reporting of Mahajan 2021, we have presented data on this outcome narratively (see Table 6). Risk of bias has still been assessed where possible and was judged to be low for Beigel 2020. Some concerns arose with baseline differences in Wang 2020. Given an inexplicit randomisation process and concealment, a deviation from intended intervention, a relevant amount of missing data, and an inappropriate outcome measurement and analysis, we judged Mahajan 2021 as at high risk of bias.
Viral clearance
One study reported this outcome (see Table 16). We assessed this outcome on a study level with RT‐PCR test for SARS‐CoV‐2 at baseline and up to three, seven, 15, and 28 days. Only Wang 2020 provided data for this outcome and was judged as at high risk of bias due to baseline differences, a relevant amount of missing outcome data, and selective reporting.
Risk of bias for analysis 1.8 Viral clearance.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 1.8.1 Viral clearance at baseline | ||||||||||||
| Wang 2020 | Some concerns | Randomisation was carried out 2:1 and in stratified blocks. Concealed envelopes were prepared. There were baseline differences in gender distribution, respiratory status at baseline, co‐morbidities and time from symptom onset suggesting possible randomisation problems. | Low risk of bias | The study was carried out double blinded and the outcome was assessed by an analysis in “viral positive population” (at baseline). Not all viral positive participants at enrolment were included in the analysis since some of them were already negative at baseline. | High risk of bias | Data for this outcome was not available for all participants randomised. Missing participant data (17%) were balanced between groups but could have influenced the outcome. | Low risk of bias | Measurement was carried out as pre‐defined by protocol. Assessors and data analysts were blinded and could not have been affected by knowledge of intervention. | High risk of bias | The trial protocol provided details of time points and analysis. The provided analysis differed from the intended protocol. Only selected measurement was reported. | High risk of bias | Overall judged high risk of bias. Details about randomisation, blinding of outcome assessors and a protocol were provided. There were baseline differences between intervention and control group. The amount of missing data was relatively large and could have had an influence on the outcome. Measurement of the outcome was carried out according to pre‐defined protocol, but only selected analysis was reported. |
| Subgroup 1.8.2 Viral clearance at day 3 | ||||||||||||
| Wang 2020 | Some concerns | Randomisation was carried out 2:1 and in stratified blocks. Concealed envelopes were prepared. There were baseline differences in gender distribution, respiratory status at baseline, co‐morbidities and time from symptom onset suggesting possible randomisation problems. | Low risk of bias | The study was carried out double blinded and the outcome was assessed by an analysis in “viral positive population” (at baseline). Not all viral positive participants at enrolment were included in the analysis since some of them were already negative at baseline. | High risk of bias | Data for this outcome was not available for all participants randomised. Missing participant data (17%) were balanced between groups but could have influenced the outcome. | Low risk of bias | Measurement was carried out as pre‐defined by protocol. Assessors and data analysts were blinded and could not have been affected by knowledge of intervention. | High risk of bias | The trial protocol provided details of time points and analysis. The provided analysis differed from the intended protocol. Only selected measurement was reported. | High risk of bias | Overall judged high risk of bias. Details about randomisation, blinding of outcome assessors and a protocol were provided. There were baseline differences between intervention and control group. The amount of missing data was relatively large and could have had an influence on the outcome. Measurement of the outcome was carried out according to pre‐defined protocol, but only selected analysis was reported. |
| Subgroup 1.8.3 Viral clearance at day 7 | ||||||||||||
| Wang 2020 | Some concerns | Randomisation was carried out 2:1 and in stratified blocks. Concealed envelopes were prepared. There were baseline differences in gender distribution, respiratory status at baseline, co‐morbidities and time from symptom onset suggesting possible randomisation problems. | Low risk of bias | The study was carried out double blinded and the outcome was assessed by an analysis in “viral positive population” (at baseline). Not all viral positive participants at enrolment were included in the analysis since some of them were already negative at baseline. | High risk of bias | Data for this outcome was not available for all participants randomised. Missing participant data (17%) were balanced between groups but could have influenced the outcome. | Low risk of bias | Measurement was carried out as pre‐defined by protocol. Assessors and data analysts were blinded and could not have been affected by knowledge of intervention. | High risk of bias | The trial protocol provided details of time points and analysis. The provided analysis differed from the intended protocol. Only selected measurement was reported. | High risk of bias | Overall judged high risk of bias. Details about randomisation, blinding of outcome assessors and a protocol were provided. There were baseline differences between intervention and control group. The amount of missing data was relatively large and could have had an influence on the outcome. Measurement of the outcome was carried out according to pre‐defined protocol, but only selected analysis was reported. |
| Subgroup 1.8.4 Viral clearance at day 14 | ||||||||||||
| Wang 2020 | Some concerns | Randomisation was carried out 2:1 and in stratified blocks. Concealed envelopes were prepared. There were baseline differences in gender distribution, respiratory status at baseline, co‐morbidities and time from symptom onset suggesting possible randomisation problems. | Low risk of bias | The study was carried out double blinded and the outcome was assessed by an analysis in “viral positive population” (at baseline). Not all viral positive participants at enrolment were included in the analysis since some of them were already negative at baseline. | High risk of bias | Data for this outcome was not available for all participants randomised. Missing participant data (17%) were balanced between groups but could have influenced the outcome. | Low risk of bias | Measurement was carried out as pre‐defined by protocol. Assessors and data analysts were blinded and could not have been affected by knowledge of intervention. | High risk of bias | The trial protocol provided details of time points and analysis. The provided analysis differed from the intended protocol. Only selected measurement was reported. | High risk of bias | Overall judged high risk of bias. Details about randomisation, blinding of outcome assessors and a protocol were provided. There were baseline differences between intervention and control group. The amount of missing data was relatively large and could have had an influence on the outcome. Measurement of the outcome was carried out according to pre‐defined protocol, but only selected analysis was reported. |
We could not conduct risk of bias assessment for quality of life, need for dialysis, need for admission to ICU, duration of ICU length of stay, or time to discharge from ICU as none of these outcomes were reported.
Serious adverse events
Three studies reported this outcome (see Table 17) and were judged with some concerns (Beigel 2020; Spinner 2020; Wang 2020). This judgement in Beigel 2020 was based on inappropriate analyses and selection of participants, which did not comply with the appropriate safety population. We assessed Wang 2020 as some concerns due to baseline differences between the intervention and control group. For Spinner 2020, there was a low risk arising from the awareness of the assigned intervention (open‐label), which is unlikely to have affected the outcome measurement. However, we judged missing outcome data as some concerns of bias in all studies due to competing risk of death without evidence, that missing outcome data does not depend on its true value.
Risk of bias for analysis 1.9 Serious adverse events.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Beigel 2020 | Low risk of bias | Randomisation was stratified by study site and disease severity in a 1:1 ratio and the allocation sequence was concealed. There were no baseline imbalances that would suggest a problem with randomisation. | Some concerns | Participants and care takers were adequately blinded. The analysis was inappropriate (as‐treated population). | Some concerns | Selection of included participants is not plausible and there is an uncertain amount of missing data due to competing risk of death. It is uncertain, whether the result is biased or not. | Low risk of bias | Measuring of the outcome was appropriate. The outcome was assessed using standardised methods and assessors were unaware of the treatment assignments. | Low risk of bias | The data was analysed in accordance with a pre‐specified protocol. | Some concerns | Overall judged some concerns for risk of bias due to inappropriate analyses and non‐plausible participant selection. Information on randomisation process is not provided, but blinding was appropriate and outcome measurement was according to a pre‐specified protocol. |
| Spinner 2020 | Low risk of bias | Block‐randomisation was not stratified and web‐based. Sites did not have access to the randomisation list. There were no baseline imbalances that would suggest a problem with randomisation. | Low risk of bias | Participants and care takers were aware of the assigned intervention. There was a neglectable number of participants with deviations from the intended intervention and the analysis was appropriate (intention‐to‐treat population). | Some concerns | There is an uncertain amount of missing data due to competing risk of death. It is uncertain, whether the result is biased or not. | Low risk of bias | Outcome assessors were aware of the treatment assignments. Serious adverse events are unlikely to be influenced by diagnostic bias due to their severity. | Low risk of bias | Data was reported according to a study protocol providing details of time points, analyses and methods for categorisation and registration of adverse events. | Some concerns | Overall judged some concerns for isk of bias. Details about randomisation, blinding of outcome assessors and a protocol were provided. Impact of missing data due to competing risk of death is uncertain. It was an open‐label study, but it was unlikely to affect the outcome measurement. |
| Wang 2020 | Some concerns | Randomisation was carried out 2:1 and in stratified blocks. Concealed envelopes were prepared. There were baseline differences in gender distribution, respiratory status at baseline, co‐morbidities and time from symptom onset suggesting possible randomisation problems. | Low risk of bias | The study was carried out double‐blinded and the outcome was assessed by safety set. | Some concerns | There is an uncertain amount of missing data due to competing risk of death. | Low risk of bias | Outcome assessors were unaware of the treatment assignments and could not have been affected by knowledge of intervention. | Low risk of bias | The trial protocol provided details of time points and analysis. The statistical analysis plan could not be found. | Some concerns | Overall judged some concerns for risk of bias. Details about randomisation, blinding of outcome assessors and a protocol were provided. There were baseline differences between intervention and control group. There is an uncertain amount of missing data due to competing risk of death. |
Adverse events (any grade)
Three studies reported these outcomes (see Table 18; Table 19). We identified some concerns for risk of bias in Beigel 2020 and Wang 2020 due to inappropriate analysis (as‐treated population) and differences in baseline characteristics, respectively. Some concerns also arose from the open‐label study design in Spinner 2020, particularly in the reporting of lower‐grade adverse events in participants who were aware of the intervention. Additionally, adverse events grade three and higher were not compared between groups as stated by the statistical analysis plan. We judged missing outcome data as some concerns in all studies due to competing risk of death without evidence, that missing outcome data does not depend on its true value.
Risk of bias for analysis 1.10 Adverse events, any grade.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Beigel 2020 | Low risk of bias | Randomisation was stratified by study site and disease severity in a 1:1 ratio and the allocation sequence was concealed. There were no baseline imbalances that would suggest a problem with randomisation. | Some concerns | Participants and care takers were adequately blinded. The analysis was inappropriate (as‐treated population). | Some concerns | There is an uncertain amount of missing data due to competing risk of death. It is uncertain, whether the result is biased or not. | Low risk of bias | Measuring of the outcome was appropriate. The outcome was assessed using standardised methods and assessors were unaware of the treatment assignments. | Low risk of bias | The data was analysed in accordance with a pre‐specified protocol. | Some concerns | Overall judged some concerns due to inappropriate analyses and non‐plausible participant selection. There is an uncertain amount of missing data due to competing risk of death. Information on randomisation process is not provided, but blinding was appropriate and outcome measurement was according to a pre‐specified protocol. |
| Spinner 2020 | Low risk of bias | Block‐randomisation was not stratified and web‐based. Sites did not have access to the randomisation list. There were no baseline imbalances that would suggest a problem with randomisation. | Low risk of bias | Participants and care takers were aware of the assigned intervention. There was a neglectable number of participants with deviations from the intended intervention and the analysis was appropriate (intention‐to‐treat population). | Some concerns | There is an uncertain amount of missing data due to competing risk of death. It is uncertain, whether the result is biased or not. | Some concerns | Outcome assessors were aware of the treatment assignments. Knowledge of intervention received could have affected outcome measurement. | Low risk of bias | Data was reported according to a study protocol providing details of time points, analyses and methods for categorisation and registration of adverse events. | Some concerns | Overall judged some concerns for risk of bias. Details about randomisation, blinding of outcome assessors and a protocol were provided. It was an open‐label study, and knowledge of intervention received could have affected outcome measurement to some degree. There is an uncertain amount of missing data due to competing risk of death. |
| Wang 2020 | Some concerns | Randomisation was carried out 2:1 and in stratified blocks. Concealed envelopes were prepared. There were baseline differences in gender distribution, respiratory status at baseline, co‐morbidities and time from symptom onset suggesting possible randomisation problems. | Low risk of bias | The study was carried out double‐blinded and the outcome was assessed by safety set. | Some concerns | There is an uncertain amount of missing data due to competing risk of death. It is uncertain, whether the result is biased or not. | Low risk of bias | Outcome assessors were unaware of the treatment assignments and could not have been affected by knowledge of intervention. | Low risk of bias | The trial protocol provided details of time points and analysis. The statistical analysis plan could not be found. | Some concerns | Overall judged some concerns for risk of bias. Details about randomisation, blinding of outcome assessors and a protocol were provided. There were baseline differences between intervention and control group suggesting a problem with block wise randomisation process. There is an uncertain amount of missing data due to competing risk of death. |
Risk of bias for analysis 1.11 Adverse events, grade 3 to 4.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Beigel 2020 | Low risk of bias | Randomisation was stratified by study site and disease severity in a 1:1 ratio and the allocation sequence was concealed. There were no baseline imbalances that would suggest a problem with randomisation. | Some concerns | Participants and care takers were adequately blinded. The analysis was inappropriate (as‐treated population). | Some concerns | There is an uncertain amount of missing data due to competing risk of death. It is uncertain, whether the result is biased or not. | Low risk of bias | Measuring of the outcome was appropriate. The outcome was assessed using standardised methods and assessors were unaware of the treatment assignments. | Low risk of bias | The data was analysed in accordance with a pre‐specified protocol. | Some concerns | Overall judged some concerns due to inappropriate analyses and non‐plausible participant selection. Information on randomisation process is not provided, but blinding was appropriate and outcome measurement was according to a pre‐specified protocol. There is an uncertain amount of missing data due to competing risk of death. |
| Spinner 2020 | Low risk of bias | Block‐randomisation was not stratified and web‐based. Sites did not have access to the randomisation list. There were no baseline imbalances that would suggest a problem with randomisation. | Low risk of bias | Participants and care takers were aware of the assigned intervention. There was a neglectable number of participants with deviations from the intended intervention and the analysis was appropriate (intention‐to‐treat population). | Some concerns | There is an uncertain amount of missing data due to competing risk of death. It is uncertain, whether the result is biased or not. | Low risk of bias | Outcome assessors were aware of the treatment assignments. Adverse events grade 3‐4 are unlikely to be influenced by diagnostic bias due to their severity. | Some concerns | A study protocol provided details of time points, analyses and methods for categorisation and registration of adverse events. Adverse events grade 3 and higher were not compared between groups as stated by the statistical analysis plan. | Some concerns | Overall some concerns for risk of bias. Details about randomisation, blinding of outcome assessors and a protocol were provided. There is an uncertain amount of missing data due to competing risk of death. It was an open‐label study, but it was unlikely to affect the outcome measurement. Outcome was not reported as stated in a pre‐defined statistical analysis plan. |
| Wang 2020 | Some concerns | Randomisation was carried out 2:1 and in stratified blocks. Concealed envelopes were prepared. There were baseline differences in gender distribution, respiratory status at baseline, co‐morbidities and time from symptom onset suggesting possible randomisation problems. | Low risk of bias | The study was carried out double‐blinded and the outcome was assessed by safety set. | Some concerns | There is an uncertain amount of missing data due to competing risk of death. It is uncertain, whether the result is biased or not. | Low risk of bias | Outcome assessors were unaware of the treatment assignments and could not have been affected by knowledge of intervention. | Low risk of bias | The trial protocol provided details of time points and analysis. The statistical analysis plan could not be found. | Some concerns | Overall judged some concerns for risk of bias. Details about randomisation, blinding of outcome assessors and a protocol were provided. There were baseline differences between intervention and control group. There is an uncertain amount of missing data due to competing risk of death. |
Effects of interventions
See: Table 1
See: Table 1 Remdesivir compared to placebo or standard care alone for adult in‐hospital participants with confirmed SARS‐CoV‐2 infection.
Remdesivir compared to placebo or standard care alone
We have presented the summary of findings and the certainty of the evidence for adult in‐hospital participants with confirmed SARS‐CoV‐2 infection, comparing a 10‐day course of remdesivir to placebo or standard care alone.
All‐cause mortality
We assessed all‐cause mortality at up to day 28, as time‐to‐event and at hospital discharge. We did not find data for all‐cause mortality beyond day 28.
All‐cause mortality at up to day 28
Four studies reported this outcome for 7142 participants (see Analysis 1.1). Considering the reported event rates across studies, we found that remdesivir probably makes little or no difference to all‐cause mortality at up to day 28 compared to placebo or standard care (risk ratio (RR) 0.93, 95% confidence interval (CI) 0.81 to 1.06; risk difference (RD) 8 fewer per 1000, 95% CI 21 fewer to 7 more; 4 studies, 7142 participants; I² = 0%; moderate‐certainty evidence). Our main reasons for downgrading were serious imprecision because of wide confidence intervals in the studies, and the 95% confidence interval includes both benefits and harms.
1.1. Analysis.

Comparison 1: Remdesivir versus placebo or standard care alone, Outcome 1: All‐cause mortality at up to day 28
All‐cause mortality at hospital discharge
One study reported this outcome for 5451 participants (see Analysis 1.2). The outcome occurred in 301 of 2743 cases in the remdesivir group and 303 of 2708 cases in the control group. Treatment with remdesivir resulted in no difference in all‐cause mortality at hospital discharge (RR 0.98, 95% CI 0.84 to 1.14; 1 study, 5451 participants; I² not applicable).
1.2. Analysis.

Comparison 1: Remdesivir versus placebo or standard care alone, Outcome 2: All‐cause mortality at hospital discharge
All‐cause mortality, time to event
Two studies reported this outcome for 6513 participants (see Analysis 1.3). Treatment with remdesivir resulted in no difference in mortality when measured over time (hazard ratio (HR) 0.93, 95% CI 0.80 to 1.07; 2 studies, 6513 participants; I² = 57%). One study reported median number of days for 236 participants and was described narratively (Wang 2020). The median (interquartile (IQR)) number of days from randomisation to death for 158 participants in the remdesivir group and 78 participants in the control group was 9.5 days (IQR 6.0 to 18.5) and 11.0 days (IQR 7.0 to 18.0), respectively. A Kaplan‐Meier curve was not provided, and a hazard ratio could not be estimated.
1.3. Analysis.

Comparison 1: Remdesivir versus placebo or standard care alone, Outcome 3: All‐cause mortality (time‐to‐event)
Improvement of clinical status
For clinical improvement, the included studies reported various parameters with differing scales (see Table 6).
Liberation from invasive mechanical ventilation in surviving participants
Reporting of clinical status was not provided according to our outcome definition. Only one study reported this outcome (Beigel 2020), at day 15 for 1062 participants; for details, see Table 6.
Ventilator‐free days
We did not find any data for this outcome.
Duration to liberation from invasive mechanical ventilation
Two studies reported this outcome in median days, which did not allow meta‐analysis, and is therefore presented narratively. Beigel 2020 reported a median of 17 days (IQR 9 to 28) to liberation from invasive mechanical ventilation or extracorporeal membrane oxygenation (ECMO) after receiving remdesivir, compared to 20 days (IQR 8 to 28) in the control group (rate difference −3.0, 95% CI −9.3 to 3.3; 1062 participants). In Wang 2020, participants stayed on invasive mechanical ventilation or ECMO for a median of 7 days (IQR 4 to 16) in the remdesivir group versus 15.5 days (IQR 6 to 21) in the control group (rate difference −4.0, 95% CI −14.0 to 2.0; 236 participants). Remdesivir may have little or no effect on clinical improvement defined as duration to liberation from invasive mechanical ventilation at up to day 28 (2 studies, 1298 participants; low‐certainty evidence). Our main reasons for downgrading were serious risk of bias because of competing risk of death, and serious imprecision because the 95% confidence interval includes both benefits and harms.
Liberation from supplemental oxygen in surviving participants
Reporting of clinical status was not provided according to our outcome definition. Only one study reported this outcome (Beigel 2020), at day 15 for 1062 participants; for details, see Table 6.
Duration to liberation from supplemental oxygen up to day 28
Three studies reported this outcome in median days, which did not allow for meta‐analysis, and is therefore presented narratively. Beigel 2020 reported a median of 13 days (IQR 5 to 28) until liberation from supplemental oxygen after receiving remdesivir, compared to a median of 21 days (IQR 8 to 28) in the control group (rate difference −8.0, 95% CI −11.8 to −4.2; 1062 participants). In Wang 2020, participants stayed on supplemental oxygen for a median of 19 days (IQR 11 to 30) in the remdesivir group, compared to 21 days (IQR 14 to 30.5) in the control group (rate difference −2, 95% CI −6.0 to 1.0; 236 participants). Contrary to the aforementioned studies, Spinner 2020 provided time to room air regardless of the initial respiratory support. Participants in the 10‐day remdesivir arm needed a median of 4 days (IQR 2 to 6) of supplemental oxygen compared to a median of 6 days (IQR 4 to 14) in the control group (HR 1.93, 95% CI 1.11 to 3.36; 393 participants). We are uncertain whether remdesivir increases or decreases the chance of clinical improvement defined as duration to liberation from supplemental oxygen at up to day 28 (3 studies, 1691 participants; very low‐certainty evidence). Our main reasons for downgrading were serious risk of bias because of inadequate blinding of participants, personnel, and outcome assessors, and possible deviation in time point of measuring in one study, and because of competing risk of death. Further reasons for downgrading were serious imprecision because the 95% confidence interval includes both benefits and harms, and studies reported outcomes differently because of missing standards.
Worsening of clinical status
New need for mechanical ventilation up to day 28 (defined as high‐flow oxygen or non‐invasive ventilation or invasive mechanical ventilation)
Three studies reported new need for mechanical ventilation for 6696 participants, if not received at baseline (see Analysis 1.4). Considering the reported event rates across studies, the evidence is very uncertain regarding the effects of remdesivir on the risk of clinical worsening: new need for mechanical ventilation within 28 days when compared to placebo or standard care (RR 0.78, 95% CI 0.48 to 1.24; RD 29 fewer per 1000, 95% CI 68 fewer to 32 more; 3 studies, 6696 participants; I² = 85%; very low‐certainty evidence). Our main reasons for downgrading were serious imprecision because of wide confidence interval in the studies; serious inconsistency because of statistical heterogeneity; and serious risk of bias. Reasons for risk of bias were inadequate blinding of participants, personnel, and outcome assessors, as well as possible deviation in time point of measuring in one study and competing risk of death.
1.4. Analysis.

Comparison 1: Remdesivir versus placebo or standard care alone, Outcome 4: Worsening of clinical status: new need for mechanical ventilation
New need for invasive mechanical ventilation up to day 28
Two of the three aforementioned studies reported new need for invasive mechanical ventilation for 1159 participants, if not received at baseline (see Analysis 1.5). Considering the reported event rates across studies, we found that remdesivir may decrease the risk of clinical worsening: new need for invasive mechanical ventilation within 28 days when compared to placebo or standard care (RR 0.56, 95% CI 0.41 to 0.77; RD 67 fewer per 1000, 95% CI 90 fewer to 35 fewer; 2 studies, 1159 participants; I² = 0%; low‐certainty evidence). Our main reasons for downgrading were serious risk of bias because of inadequate blinding of participants, personnel, and outcome assessors, as well as possible deviation in time point of measuring in one study, and competing risk of death. Furthermore, studies reported outcomes differently because of missing standards.
1.5. Analysis.

Comparison 1: Remdesivir versus placebo or standard care alone, Outcome 5: Worsening of clinical status: new need for invasive mechanical ventilation
New need for non‐invasive mechanical ventilation or high‐flow oxygen up to day 28
One study reported new need for non‐invasive mechanical ventilation for 573 participants, if not received at baseline (see Analysis 1.6). Considering the reported event rates across studies, the evidence is very uncertain regarding the effects of remdesivir on the risk of clinical worsening: new need for non‐invasive mechanical ventilation or high‐flow oxygen within 28 days when compared to placebo or standard care (RR 0.70, 95% CI 0.51 to 0.98; RD 72 fewer per 1000, 95% CI 118 fewer to 5 fewer; 1 study, 573 participants; I² not applicable; very low‐certainty evidence). Our main reasons for downgrading were serious risk of bias because of competing risk of death, and serious imprecision due to few participants and data from only one study.
1.6. Analysis.

Comparison 1: Remdesivir versus placebo or standard care alone, Outcome 6: Worsening of clinical status: new need for non‐invasive mechanical ventilation or high‐flow oxygen
New need for oxygen by mask or nasal prongs (low‐flow oxygen) up to day 28
One study reported new need for oxygen by mask or nasal prongs for 138 participants, if not received at baseline (see Analysis 1.7). Considering the reported event rates across studies, the evidence is very uncertain regarding the effects of remdesivir on the risk of clinical worsening: new need for oxygen by mask or nasal prongs within 28 days when compared to placebo or standard care (RR 0.81, 95% CI 0.54 to 1.22; RD 84 fewer per 1000, 95% CI 204 fewer to 98 more; 1 study, 138 participants; I² not applicable; very low‐certainty evidence). Our main reasons for downgrading were serious imprecision because of wide confidence intervals, and data coming from only one study.
1.7. Analysis.

Comparison 1: Remdesivir versus placebo or standard care alone, Outcome 7: Worsening of clinical status: new need for oxygen by mask or nasal prongs
Need for dialysis at up to day 28
We did not find any data for this outcome.
Quality of life
We did not find any data for this outcome.
Need for admission to ICU
We did not find any data for this outcome.
Duration of ICU length of stay or time to discharge from ICU
We did not find any data for this outcome.
Duration of hospitalisation or time to discharge from hospital
Five studies reported this outcome (see Table 6). Beigel 2020 reported median (IQR) days for 1062 participants. Participants receiving remdesivir treatment were hospitalised for a median of 12 days (IQR 6 to 28) compared to a median of 17 days (18 to 28) in the control group (rate difference −5, 95% CI −7.7 to −2.3). Wang 2020 reported the outcome for 236 participants, with a median duration of hospitalisation of 25 days (IQR 16 to 38) in the remdesivir group versus a median of 24 days (18 to 36) in the control group (rate difference 0, 95% CI −4.0 to 4.0). Additionally, Wang 2020 provided a median time to hospital discharge of 21 days in both groups (remdesivir 21.0 (IQR 12 to 31), control 21.0 (IQR 13.5 to 28.5); rate difference 0, 95% CI −3.0 to 3.0). Mahajan 2021 reported mean (standard deviation) of a five‐day remdesivir course (remdesivir 11.55 (4.3) versus control 12.38 (5.2)).
Viral clearance
One study reported this outcome for 236 participants who tested positive at enrolment (see Analysis 1.8). Wang 2020 provided data on viral clearance (undetectable viral RNA from naso‐/oropharyngeal swab) at baseline and at several time points, of which day 3, 7, and 15 were included as predefined in this review. In Wang 2020, viral clearance at day 14 was reported, which we used in our analysis. The study also visualised viral RNA load over time from baseline by quantitative PCR on the upper respiratory tract and lower respiratory tract and by duration of illness (≤ 10 days versus > 10 days). Undetectable viral RNA in upper respiratory tract was reported for 131 participants in the remdesivir group and 65 participants in the placebo group; 40 data sets were missing. Nasopharyngeal PCR for remdesivir versus placebo was negative in 24 versus 13 (RR 0.92, 95% CI 0.50 to 1.68) at baseline; 37 versus 19 (RR 0.97, 95% CI 0.61 to 1.54) at day 3; 66 versus 32 (RR 1.02, 95% CI 0.76 to 1.38) at day 7; and 93 versus 49 (RR 0.94, 95% CI 0.79 to 1.12) at day 14.
1.8. Analysis.

Comparison 1: Remdesivir versus placebo or standard care alone, Outcome 8: Viral clearance
Overall, viral clearance in the positive population of the remdesivir and placebo arms increased over time, with 71% and 75.4% undetectable RNA at day 14, respectively. There was no significant difference between groups at any time point.
Serious adverse events
Three studies reported this outcome for 1674 participants (see Analysis 1.9). Considering the reported event rates across studies, we found that remdesivir probably decreases the risk of serious adverse events within 28 days when compared to placebo or standard care (RR 0.75, 95% CI 0.63 to 0.90; RD 63 fewer per 1000, 95% CI 94 fewer to 25 fewer; 3 studies, 1674 participants; I² = 0%; moderate‐certainty evidence). We downgraded the certainty of evidence due to serious risk of bias because of competing risk of death.
1.9. Analysis.

Comparison 1: Remdesivir versus placebo or standard care alone, Outcome 9: Serious adverse events
Adverse events
Adverse events, any grade
Three studies reported this outcome for 1674 participants (see Analysis 1.10). Considering the reported event rates across studies, the evidence is very uncertain regarding the effects of remdesivir on adverse events (any grade) within 28 days when compared to placebo or standard care (RR 1.05, 95% CI 0.86 to 1.27; RD 29 more per 1000, 95% CI 82 fewer to 158 more; 3 studies, 1674 participants; I² = 77%; very low‐certainty evidence). Our main reasons for downgrading were serious imprecision because of wide confidence intervals in the studies. We also downgraded for serious risk of bias because of competing risk of death, and serious inconsistency due to statistical heterogeneity.
1.10. Analysis.

Comparison 1: Remdesivir versus placebo or standard care alone, Outcome 10: Adverse events, any grade
Adverse events, grade 3 to 4
Three studies reported this outcome for 1674 participants (see Analysis 1.11). Considering the reported event rates across studies, we estimated that remdesivir results in 42 fewer participants sustaining at least one adverse event grade 3 to 4 compared to placebo or standard care alone amongst 1000 participants. Treatment with remdesivir probably results in little or no difference on the occurrence of adverse events grade 3 to 4 within 28 days when compared to placebo or standard care (RR 0.90, 95% CI 0.80 to 1.00; 3 studies, 1674 participants; I² = 0%; moderate‐certainty evidence). Our main reasons for downgrading were serious risk of bias because of inadequate blinding of participants, personnel, and outcome assessors; one study stopped earlier than scheduled, and one study used an inappropriate patient population.
1.11. Analysis.

Comparison 1: Remdesivir versus placebo or standard care alone, Outcome 11: Adverse events, grade 3 to 4
Subgroup analyses
We conducted subgroup analyses for prioritised effectiveness outcomes to explore heterogeneity between predefined subgroups. Since data were only available for mortality at up to day 28, analyses were performed exclusively for this outcome.
Age of participants
One study reported all‐cause mortality at up to day 28 divided by age groups (< 50 years, 50 to 69 years, > 69 years) for 5451 participants (see Analysis 2.1). There were no subgroup differences (Chi²= 0.10, df = 2, P = 0.95, I² not applicable).
2.1. Analysis.

Comparison 2: Subgroup analysis (age of participants): remdesivir versus placebo or standard care alone, Outcome 1: All‐cause mortality at up to day 28
Pre‐existing conditions
Protocol‐specified comorbidities included diabetes, respiratory disease, hypertension, immunosuppression, obesity, and cardiac injury. One study reported all‐cause mortality at up to day 28 subdivided by pre‐existing conditions of interest (WHO Solidarity Trial Consortium 2021). They compared the effect of remdesivir in one specific subgroup (e.g. with asthma) to a control without that condition (e.g. without asthma). However, since there is a partial overlap of comorbidities between subgroups, control groups might therefore involve participants with other pre‐existing conditions of interest. As a result, meta‐analysis not be conducted, and all‐cause mortality at up to day 28 is reported narratively (see Table 6).
Timing of first dose administration with illness onset
One study reported all‐cause mortality at up to day 28 divided by timing of first dose administration with illness onset for 233 participants (see Analysis 3.1). The evidence suggests a benefit for early initiation of treatment with remdesivir (≤ 10 days after symptom onset) compared to placebo or standard care alone (RR 0.76, 95% CI 0.29 to 1.95), whilst in the delayed initiation of treatment (> 10 days after symptom onset), the evidence suggests a potential inferiority of remdesivir (RR 1.48, 95% CI 0.45 to 4.88). However, there were no relevant subgroup differences (Chi²= 0.74, df = 1, P = 0.39, I² not applicable).
3.1. Analysis.

Comparison 3: Subgroup analysis (timing of first dose administration with illness onset): remdesivir versus placebo or standard care alone, Outcome 1: All‐cause mortality at up to day 28
Severity of condition
Three studies reported all‐cause mortality by day 28 subdivided by respiratory support at baseline for 3194 participants (see Analysis 4.1). The evidence suggests a benefit for remdesivir compared to placebo or standard care alone only in the subgroup with low‐flow oxygen at baseline (RR 0.32, 95% CI 0.15 to 0.66; 1 study, 435 participants; I² not applicable). The test for subgroup differences suggests relevant subgroup differences and reveals high heterogeneity: Chi² = 8.32, df = 2, P = 0.02, I² = 75.7%.
4.1. Analysis.

Comparison 4: Subgroup analysis (severity of condition, no oxygen versus low‐flow oxygen versus mechanical ventilation (including high‐flow oxygen, non‐invasive ventilation, invasive mechanical ventilation, and ECMO): remdesivir versus placebo or standard care alone, Outcome 1: All‐cause mortality at up to day 28
Duration of remdesivir application
We compared a 5‐day course of remdesivir to a 10‐day course for this subgroup. One study reported all‐cause mortality at up to day 28 subdivided by duration of remdesivir application for 584 participants (see Analysis 5.1). Two of 191 participants receiving remdesivir for 5 days versus to 3 of 193 participants receiving remdesivir for 10 days versus 4 of 200 participants receiving control died by day 28. There were no subgroup differences (Chi²= 0.09, df = 1, P = 0.09, I² not applicable).
5.1. Analysis.

Comparison 5: Subgroup analysis (duration of remdesivir application): remdesivir versus placebo or standard care alone, Outcome 1: All‐cause mortality at up to day 28
Sensitivity analysis
Risk of bias assessment components (studies with a low risk of bias or some concerns versus studies with a high risk of bias)
We performed sensitivity analysis for the outcome clinical worsening (new need for mechanical ventilation) to explore heterogeneity (I² = 85% and high risk of bias in Spinner 2020). The result in effect did not differ after exclusion of Spinner 2020.
Comparison of preprints versus peer‐reviewed articles
We did not include any preprints.
Comparison of premature termination of studies with completed studies
One study was stopped early because recruitment of participants was no longer possible due to infection incidences (Wang 2020). We performed sensitivity analysis for comparison of premature termination of studies with completed studies for the outcome all‐cause mortality at up to day 28. The result in effect did not differ after exclusion of Wang 2020.
Comparison of adolescent and adult participants versus adult participants
One study included one participant (n = 1/562) younger than 18 years (Spinner 2020), which corresponds to 0.178% of all recruited participants in this RCT. We performed sensitivity analysis for the comparison of adolescent and adult participants versus adult participants alone in included studies and the outcome all‐cause mortality at up to day 28. The result in effect did not differ after exclusion of Spinner 2020.
Discussion
Summary of main results
The aim of this living systematic review was to assess the effects of remdesivir compared to placebo or standard care alone on clinical outcomes in hospitalised adults with SARS‐CoV‐2 infection. This is the first version of this systematic review. We included five RCTs with 7452 participants diagnosed with SARS‐CoV‐2 infection, of whom 3886 were randomised to receive remdesivir (Beigel 2020; Spinner 2020; Wang 2020; Mahajan 2021; WHO Solidarity Trial Consortium 2021). We identified two ongoing studies, one of which was suspended (recruitment was not possible due to infection incidences).
Remdesivir versus placebo or standard care alone
Remdesivir probably makes little or no difference to all‐cause mortality at up to day 28 (RR 0.93, 95% CI 0.81 to 1.06; RD 8 fewer per 1000, 95% CI 21 fewer to 7 more; 4 studies, 7142 participants; moderate‐certainty evidence).
Data on improvement in clinical status defined as liberation from respiratory support were not provided according to our outcome definition. Duration to liberation was reported in median days, which did not allow meta‐analysis. Based on the available data, remdesivir may have little or no effect on the duration to liberation from invasive mechanical ventilation (Beigel 2020: 17 days versus 20 days, 1062 participants; Wang 2020: 7 days versus 15.5 days, 236 participants; low‐certainty evidence). We are uncertain whether remdesivir increases or decreases the chance of clinical improvement: duration to liberation from supplemental oxygen at up to day 28 (very low‐certainty evidence).
We are very uncertain whether remdesivir increases or decreases the risk of clinical worsening at up to day 28 defined by new need for mechanical ventilation (high‐flow oxygen or non‐invasive mechanical ventilation or invasive mechanical ventilation) (very low‐certainty evidence); new need for non‐invasive mechanical ventilation or high‐flow oxygen (very low‐certainty evidence); and new need for oxygen by mask or nasal prongs (very low‐certainty evidence). We found low‐certainty evidence for a decreased risk of clinical worsening in terms of new need for invasive mechanical ventilation, compared to placebo or standard care alone.
We identified subgroup differences for all‐cause mortality at up to day 28 in the subgroup analysis for severity of condition, although with high heterogeneity (Chi² = 8.32, df = 2, P = 0.02, I² = 75.7%). The evidence suggests a benefit for remdesivir compared to placebo or standard care alone only in the subgroup with low‐flow oxygen at baseline (RR 0.32, 95% CI 0.15 to 0.66; 1 study, 435 participants). However, these findings were based on data from one study only that reported the outcome equivalent to our predefined parameter (Beigel 2020). Data for this subgroup and outcome were not provided by any other matching study, hence uncertainty remains.
None of the included studies reported on quality of life, therefore we do not know whether remdesivir has any impact on this outcome.
We included results from three RCTs (1674 participants) to assess the adverse effects profile of remdesivir compared to placebo or standard care alone in people hospitalised with COVID‐19. Remdesivir probably decreases serious adverse events (moderate‐certainty evidence). We are very uncertain whether remdesivir increases or decreases adverse events (any grade) when compared to placebo or standard care alone (very low‐certainty evidence).
Overall completeness and applicability of evidence
We identified five RCTs, mainly from high‐ and upper‐middle‐income countries, investigating the therapeutic effects of remdesivir in hospitalised adults with SARS‐CoV‐2 infection. Of 7452 total participants, 3886 were randomised to receive remdesivir. The diagnosis of SARS‐CoV‐2 infection was confirmed by polymerase chain reaction (PCR) and, in some studies, radiological sign of COVID‐19 pneumonia. The proportion of PCR‐negative participants at baseline was reported in only one study (Wang 2020). The majority of participants received other experimental COVID‐19 treatment options, such as corticosteroids, antimicrobials, hydroxychloroquine, convalescent plasma, or combinations of these treatments.
All the included studies involved hospitalised, moderately or severely ill people with COVID‐19, or both, and compared the effect of remdesivir (3860 evaluated participants) to placebo (599 evaluated participants) or standard care alone (2944 evaluated participants). To assess the effects of remdesivir, we included data from four RCTs (7403 evaluated participants). The analysis of safety outcomes (serious adverse events, adverse events) was affected by a relevant lack of data. Since the largest study did not report safety data (WHO Solidarity Trial Consortium 2021), we could only include data for 1674 participants from three RCTs in our analysis.
Different scales of disease severity and progression were used amongst studies, and to the present day the terms 'moderate' and 'severe' are inconsistently used to define severity of disease in different guidelines and consensus statements of national and international organisations (e.g. WHO 2020d versus WHO 2021). For hospitalised patients, the need of respiratory support essentially determines their course within the hospital (e.g. ICU admission) and, from the individual patient's perspective, has a strong impact on acute health‐related quality of life, functional independence and autonomy. We therefore analysed respiratory support at baseline and during the observation period as a surrogate for COVID‐19 disease severity: no oxygen, low‐flow oxygen, and mechanical ventilation (including non‐invasive mechanical ventilation, high‐flow oxygen, and invasive mechanical ventilation). At baseline, 1957 participants did not need any additional oxygen; 4409 participants received low‐flow oxygen support; and 1025 participants were treated with mechanical ventilation. Only the low‐flow oxygen at baseline group showed a decreased mortality if treated with remdesivir. However, these findings were based on data from one study only (Beigel 2020). In contrast, a major subgroup comparison of participants receiving any type of oxygen (low‐ and high‐flow) in the WHO Solidarity Trial Consortium 2021 study did not show any significant difference in 28‐day mortality between remdesivir and standard care alone (n = 3639 participants; RR 0.85, 95% CI 0.66 to 1.09). Since the authors of WHO Solidarity Trial Consortium 2021 could not provide detailed data on mortality for the different levels of oxygen support, we were unable to include the reported group of low‐ and high‐flow oxygen in our subgroup analysis. Hence, the certainty of the evidence for the effects of remdesivir in COVID‐19 participants receiving low‐flow oxygen at baseline remains low for methodological reasons.
We detected no differences for mortality at up to day 28 in further participant subgroups relevant for daily clinical routine, namely age, timing of first remdesivir dose, and duration of remdesivir application.
Although we contacted all study authors, especially with regard to detailed description of the extent of respiratory support (e.g. low‐ versus high‐flow oxygen, non‐invasive versus invasive mechanical ventilation), there remained differences in reporting severity of illness and incomplete data sets, resulting in a relevant obstacle to the planned subgroup analysis. Hence, due to incompleteness of the data, uncertainty remains regarding a possible benefit of remdesivir treatment for COVID‐19 patients receiving low‐flow oxygen support only.
Certainty of the evidence
We included data from four RCTs to assess the effects of remdesivir for individuals with moderate and severe SARS‐CoV‐2 infection when compared to placebo or standard care alone. We evaluated the certainty of the evidence using the GRADE approach, with any downgrading substantiated (see Table 1). The evidence for effect outcomes was of moderate to very low certainty, and the evidence for safety outcomes was of moderate to very low certainty.
All‐cause mortality
We downgraded one level to moderate certainty for serious imprecision due to wide 95% confidence interval that included both benefits and harms.
Improvement of clinical status: duration to liberation from invasive mechanical ventilation
We downgraded to low certainty of evidence for serious risk of bias because of competing risk of death, and for serious imprecision because the 95% confidence interval includes both benefits and harms.
Improvement of clinical status: duration to liberation from supplemental oxygen
We downgraded to very low certainty of evidence due to serious risk of bias because of inadequate blinding of participants, personnel, and outcome assessors, and possible deviation in time point of measuring in one study, and because of competing risk of death. We also downgraded due to serious imprecision because the 95% confidence interval includes both benefits and harms, and studies reported outcomes differently because of missing standards.
Clinical worsening: new need for mechanical ventilation (high‐flow oxygen, non‐invasive mechanical ventilation, and invasive mechanical ventilation)
We downgraded to very low certainty because of serious imprecision due to wide confidence interval, serious inconsistency because of statistical heterogeneity, and serious risk of bias. Reasons for risk of bias were inadequate blinding of participants, personnel, and outcome assessors, as well as possible deviation in time point of measuring in one study, and competing risk of death.
Clinical worsening: new need for invasive mechanical ventilation
We downgraded to low certainty because of serious risk of bias due to inadequate blinding of participants, personnel, and outcome assessors, as well as possible deviation in time point of measuring in one study, and competing risk of death. Furthermore, studies reported outcomes differently because of missing standards.
Clinical worsening: new need for non‐invasive mechanical ventilation or high‐flow oxygen
We downgraded to very low certainty because of serious risk of bias due to competing risk of death, and serious imprecision due to wide confidence interval, and data coming from only one study.
Clinical worsening: new need for oxygen by mask or nasal prongs (low‐flow oxygen)
We downgraded to very low certainty because of risk of bias due to competing risk of death, serious imprecision due to wide confidence interval, and data coming from only one study.
Quality of life
None of the included studies reported quality of life, therefore we do not know whether remdesivir has any impact on this outcome.
Serious adverse events
We downgraded to moderate certainty for risk of bias through competing risk of death.
Adverse events (any grade)
We downgraded to very low certainty because of serious imprecision due to wide confidence interval. We also downgraded due to serious risk of bias because of competing risk of death, and serious inconsistency due to statistical heterogeneity (I² = 77%).
Publication of the identified ongoing RCTs will necessitate an update of this review. The conclusions of the updated review could differ from those of the present review and may allow for a better judgment regarding the effects of remdesivir administration for the treatment of COVID‐19.
Potential biases in the review process
Experienced medical information specialists of the CEOsys consortium developed an all‐encompassing search strategy to identify available evidence to answer our research question. We aimed at identifying all completed, but also ongoing studies, for inclusion in this review. The sensitive search included relevant electronic databases as well as clinical trial registries. As a supplementary search, we screened reference lists of included studies. We included preprints in addition to peer‐reviewed full‐text articles. We are aware of the potentially lower quality of preprint publications, and that results could change once the peer‐reviewed journal publications are available. Where data were missing, we contacted study authors; for details, see Characteristics of included studies. An overview of included studies is provided in Table 4. We are confident that we identified all relevant studies, and will monitor ongoing studies as well as full publication of preprints closely after the publication of this review.
Differences to review protocol
A prespecified protocol is available at an international prospective register of systematic reviews (CRD42021238065). As a major difference to the protocol, we plan a living approach for this review. Considering the current incompleteness of subgroup data regarding the effect of remdesivir for particular patient populations, we hope to reduce the uncertainty of evidence by regular updates. In contrast to our predefined inclusion criteria (adult participants), we did not exclude the study of Spinner 2020, which involved adolescent participants between 12 and 18 years. After an inquiry, we learned that only one participant (0.178%) was under the age of 18, which we presumed to have a non‐relevant impact on our results. Our main outcomes were extended for data inclusion at longest follow‐up for clinical status, which did not, however, exceed 28 days. Furthermore, ventilator‐free days were found to be of clinical relevance after completion of the protocol and were added to the prioritised outcomes. For clinical worsening, new need for non‐invasive and new need for invasive ventilation was extended by new need for low‐flow oxygen to address clinical worsening relevant for the transition from ambulatory to in‐hospital care. Also, after careful consideration, we decided to additionally report new need for mechanical ventilation (high‐flow oxygen or non‐invasive mechanical ventilation or invasive mechanical ventilation) for clinical (indication for organ dysfunction and need of intensive care) as well as patient‐oriented (loss of independence and quality of life) reasons. Moreover, the combination is in accordance with the WHO definition of “severe” COVID‐19 (WHO 2020d; WHO 2020e). Besides adverse events grade 3 to 4, we additionally evaluated the effect of remdesivir on adverse events of any grade to cover safety analysis regarding low‐grade adverse events. We added hazard ratio to measures of effects where this information was available. Categorising into age subgroups was not possible as predefined for subgroup analysis, and we concretised disease severity according to WHO progression scale (WHO 2020d). Any change of methodology was done before analysis. We identified no other potential sources of bias in our review process.
Agreements and disagreements with other studies or reviews
The results we found do not decisively differ from those of other systematic reviews, Elsawah 2020; Piscoya 2020; Roshanshad 2020; Al‐Abdouh 2021; Lai 2021; Vegivinti 2021; Welte 2021, or living guidelines (Siemieniuk 2020; Kaka 2021). Except for one review (Vegivinti 2021), which used the Scottish Intercollegiate Guidelines Network (SIGN) checklists for controlled clinical trials for the risk of bias and determination of the levels of evidence, meta‐analyses and systematic reviews were conducted based on Cochrane guidelines and using the Cochrane risk of bias tool. Roshanshad 2020 used also the Newcastle‐Ottawa scale for the assessment of the quality of non‐randomised studies in meta‐analyses. The levels of evidence were performed analogous to our review, mostly according to the GRADE approach. In contrast to our review, the cited reviews did not exclusively include RCTs with a placebo or standard care control arm, but also case studies, Elsawah 2020; Piscoya 2020; Roshanshad 2020, and simulated studies (Bansal 2021). The selection of the analysed RCTs was not always identical to that of our review. None of the other reviews cited the Mahajan 2021 study, due to its publication in March 2021. Our review excluded the publication Goldman 2020, which compared clinically used dosing schemes of remdesivir, but had no placebo or standard of care arm. The synthetic interpretation of the results of the aforementioned reviews and guidelines is difficult due to different methodological approaches, the type of subgroup formation, and the partial inclusion of non‐RCTs. We found major differences in the published reviews of Kaka 2021 and Bansal 2021. Kaka 2021 found a marginally increased mortality in participants already treated with mechanical ventilation or ECMO, or both, at baseline (studies analysed: Beigel 2020; Wang 2020; WHO Solidarity Trial Consortium 2021), and a benefit for remdesivir in terms of clinical improvement (studies analysed: Beigel 2020; Spinner 2020; Wang 2020). Contrary to our findings, Bansal 2021 concluded from their meta‐analysis that remdesivir significantly reduces mortality. Since the authors only included Beigel 2020, Wang 2020, and a simulated two‐arm study that is only available as preprint (Hsu 2020), their conclusions are not substantiated by the entire available evidence.
Authors' conclusions
Implications for practice.
We found moderate‐certainty evidence that remdesivir probably has little or no effect on all‐cause mortality at up to 28 days in hospitalised individuals with moderate and severe COVID‐19. We were unable to perform meta‐analysis of clinical improvement parameters, but considering the data provided, remdesivir may have little or no effect on the duration to liberation from invasive mechanical ventilation. We are uncertain whether remdesivir increases or decreases the chance of clinical improvement in terms of duration to liberation from supplemental oxygen at up to day 28 given the very low certainty of the evidence. We found low‐certainty evidence that remdesivir may decrease the risk of new need for invasive mechanical ventilation. However, we are very uncertain whether remdesivir affects the overall risk for clinical worsening. Remdesivir probably decreases the rate of serious adverse events; however, due to inconsistent reporting of safety data, the evidence regarding the effect of remdesivir is very uncertain when pooling any grade of adverse events. Due to incompleteness of subgroup data, we are uncertain whether there is a possible benefit of remdesivir for the treatment of COVID‐19 patients receiving low‐flow oxygen therapy only.
Implications for research.
In this first version of a systematic review on remdesivir in hospitalised individuals with SARS‐CoV‐2 infection, we included data from five randomised controlled trials. Each study reported different primary outcomes. Furthermore, different scales of disease severity were applied to characterise subgroups, and safety data reporting was incomplete. These aspects lower the certainty of the evidence and make it difficult to draw valid conclusions for important clinical questions during an ongoing pandemic. In particular, differences in potential benefits or harms of remdesivir for the treatment of COVID‐19 depending on disease severity could not be analysed sufficiently.
Additional data on efficacy and safety of remdesivir for different population subgroups (e.g. depending on age, severity of disease, or kidney function), timing of application of remdesivir in the course of the infection, and the establishment of core outcomes for COVID‐19 research, may allow us to reduce uncertainty in potentially beneficial or harmful effects of remdesivir in future updates of this review. In accordance with the living approach of this review, we will continually update our search and include eligible trials.
History
Review first published: Issue 8, 2021
Notes
Parts of the review's Methods section are adopted from templates of Cochrane Haematology and a similar protocol published by Piechotta 2020, and the corresponding review (Chai 2020).
Risk of bias
Risk of bias for analysis 5.1 All‐cause mortality at up to day 28.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 5.1.1 5‐day remdesivir | ||||||||||||
| Spinner 2020 | Low risk of bias | Block‐randomisation was not stratified and web‐based. Sites did not have access to the randomisation list. There were no baseline imbalances that would suggest a problem with randomisation. | Low risk of bias | Participants and care takers were aware of the assigned intervention. There was a neglectable number of participants with deviations from the intended intervention and the analysis was appropriate (intention‐to‐treat population). | Low risk of bias | Data was available for nearly all participants (<5% missing) randomised. | Low risk of bias | Outcome assessors were aware of the treatment assignments. Knowledge of intervention received could not have affected the outcome measurement. | Low risk of bias | The trial protocol provided details of time points and analysis. Mortality was analysed in accordance with a prespecified analysis plan. | Low risk of bias | Overall judged low. Details about randomisation, blinding of outcome assessors and a protocol were provided. It was an open‐label study, but this could not have affected the outcome measurement. |
| Subgroup 5.1.2 10‐day remdesivir | ||||||||||||
| Spinner 2020 | Low risk of bias | Block‐randomisation was not stratified and web‐based. Sites did not have access to the randomisation list. There were no baseline imbalances that would suggest a problem with randomisation. | Low risk of bias | Participants and care takers were aware of the assigned intervention. There was a neglectable number of participants with deviations from the intended intervention and the analysis was appropriate (intention‐to‐treat population). | Low risk of bias | Data was available for nearly all participants (<5% missing) randomised. | Low risk of bias | Outcome assessors were aware of the treatment assignments. Knowledge of intervention received could not have affected the outcome measurement. | Low risk of bias | The trial protocol provided details of time points and analysis. Mortality was analysed in accordance with a prespecified analysis plan. | Low risk of bias | Overall judged low. Details about randomisation, blinding of outcome assessors and a protocol were provided. It was an open‐label study, but this could not have affected the outcome measurement. |
Acknowledgements
This work is part of a series of reviews investigating treatments and therapies for COVID‐19 as part of the project CEOsys. Text passages in the Background section (e.g. Description of the intervention and Why it is important to do this review) are shared between reviews of this series. We thank the authors of the first published reviews of this series for providing and sharing this information. Moreover, we thank the Cochrane Haematology working group for use of the template for the description of methods.
This review was published in collaboration with the Cochrane Editorial and Methods Department.
We particularly thank Helen Wakeford and Lara Kahale of Cochrane's Central Editorial Service for their support. They managed the editorial process and contributed to the improvement of the review.
We thank Sarah Hodgkinson (Associate Editor for the Children and Family Network; Circulation and Breathing Network, Cochrane) for her valuable advice and comments regarding the presentation of the results and their discussion, which significantly improved the quality of the review.
We thank Robin Featherstone (Information Specialist, Cochrane) for commenting on the search strategy.
We thank Tess Moore (Methods Support Unit, Cochrane) for her comments regarding the risk of bias assessments and their presentation.
We thank Denise Mitchel (Senior Copy Editor, Cochrane Central Executive team) for editing the plain language summary so that it is understandable for the layperson.
We thank Lisa Winer (Cochrane's Freelance Medical Copy Editor) and Toby Lasserson (Deputy Editor in Chief, Cochrane Central Executive team) for their valuable advice and support.
We thank Jakob J Malin (Department I of Internal Medicine, Division of Infectious Diseases, University of Cologne) for his comments, and especially his clinically important advice regarding the subgroup analyses.
We thank Hayley B Gershenkorn (MD FCCM ATSF, Professor, University of Miami Miller School of Medicine) for her critical comments and her advice for improving the review.
We would like to thank consumer reviewer Hariklia Nguyen in particular for her comments, which have led to a better comprehensibility and readability of the review.
We thank all authors who provided additional information on their studies.
The research was part of a project supported by the German Federal Ministry of Education and Research (NaFoUniMedCovid19, funding number: 01KX2021; part of the project CEOsys). The contents of this document reflect only the authors' views, and the German Ministry is not responsible for any use that may be made of the information it contains.
FG, FF, and VT are thankful for the support of their colleagues with the Department of Anesthesia and Intensive Care at the University of Leipzig Medical Center whilst working on the manuscript of this review during the COVID‐19 pandemic. We thank Dr Sven Laudi at the Department of Anesthesia and Intensive Care at Leipzig University Medical Services for his biostatistical advice.
Appendices
Appendix 1. Search strategies
Cochrane COVID‐19 Study Register
Search string: remdesivir* OR GS5734 OR "GS 5734"
Study characteristics: 1) "Intervention assignment": "Randomised" OR "Unclear" 2) "Study type": "Interventional" AND "Study design": "Parallel/Crossover" OR "Unclear" OR "Other"
= 193 references
Web of Science Core Collection (Advanced search)
#1 TI=(remdesivir* OR GS5734 OR "GS 5734") OR AB=(remdesivir* OR GS5734 OR "GS 5734")
#2 TI=(COVID OR COVID19 OR "SARS‐CoV‐2" OR "SARS‐CoV2" OR SARSCoV2 OR "SARSCoV‐2" OR "SARS coronavirus 2" OR "2019 nCoV" OR "2019nCoV" OR "2019‐novel CoV" OR "nCov 2019" OR "nCov 19" OR "severe acute respiratory syndrome coronavirus 2" OR "novel coronavirus disease" OR "novel corona virus disease" OR "corona virus disease 2019" OR "coronavirus disease 2019" OR "novel coronavirus pneumonia" OR "novel corona virus pneumonia" OR "severe acute respiratory syndrome coronavirus 2") OR AB=(COVID OR COVID19 OR "SARS‐CoV‐2" OR "SARS‐CoV2" OR SARSCoV2 OR "SARSCoV‐2" OR "SARS coronavirus 2" OR "2019 nCoV" OR "2019nCoV" OR "2019‐novel CoV" OR "nCov 2019" OR "nCov 19" OR "severe acute respiratory syndrome coronavirus 2" OR "novel coronavirus disease" OR "novel corona virus disease" OR "corona virus disease 2019" OR "coronavirus disease 2019" OR "novel coronavirus pneumonia" OR "novel corona virus pneumonia" OR "severe acute respiratory syndrome coronavirus 2")
#3 #1 AND #2
#4 TI=(random* OR placebo OR trial OR groups OR "phase 3" or "phase3" or p3 or "pIII") OR AB=(random* OR placebo OR trial OR groups OR "phase 3" or "phase3" or p3 or "pIII")
#5 #3 AND #4 Indexes=SCI‐EXPANDED, ESCI Timespan=1945‐2021
= 377 references
WHO COVID‐19 Global literature on coronavirus disease
(remdesivir* OR GS5734 OR "GS 5734") AND (random* OR placebo OR trial OR groups OR "phase 3" or "phase3" or p3 or "pIII")
without MEDLINE and PubMed = 352 references
Data and analyses
Comparison 1. Remdesivir versus placebo or standard care alone.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 All‐cause mortality at up to day 28 | 4 | 7142 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.81, 1.06] |
| 1.2 All‐cause mortality at hospital discharge | 1 | 5451 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.84, 1.14] |
| 1.3 All‐cause mortality (time‐to‐event) | 2 | 6513 | Hazard Ratio (IV, Fixed, 95% CI) | 0.93 [0.80, 1.07] |
| 1.4 Worsening of clinical status: new need for mechanical ventilation | 3 | 6696 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.48, 1.24] |
| 1.4.1 WHO 6 to 9 at day 28 (± 1 day), if ≤5 at baseline | 3 | 6696 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.48, 1.24] |
| 1.5 Worsening of clinical status: new need for invasive mechanical ventilation | 2 | 1159 | Risk Ratio (M‐H, Random, 95% CI) | 0.56 [0.41, 0.77] |
| 1.5.1 WHO 7 to 9 at day 28 (± 1 day), if ≤6 at baseline | 2 | 1159 | Risk Ratio (M‐H, Random, 95% CI) | 0.56 [0.41, 0.77] |
| 1.6 Worsening of clinical status: new need for non‐invasive mechanical ventilation or high‐flow oxygen | 1 | 573 | Risk Ratio (M‐H, Random, 95% CI) | 0.70 [0.51, 0.98] |
| 1.6.1 WHO= 6 at day 29, if ≤5 at baseline | 1 | 573 | Risk Ratio (M‐H, Random, 95% CI) | 0.70 [0.51, 0.98] |
| 1.7 Worsening of clinical status: new need for oxygen by mask or nasal prongs | 1 | 138 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.54, 1.22] |
| 1.7.1 WHO= 5 at day 29, if ≤ 4 at baseline | 1 | 138 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.54, 1.22] |
| 1.8 Viral clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.8.1 Viral clearance at baseline | 1 | 196 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.50, 1.68] |
| 1.8.2 Viral clearance at day 3 | 1 | 196 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.61, 1.54] |
| 1.8.3 Viral clearance at day 7 | 1 | 196 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.76, 1.38] |
| 1.8.4 Viral clearance at day 14 | 1 | 196 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.79, 1.12] |
| 1.9 Serious adverse events | 3 | 1674 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.63, 0.90] |
| 1.10 Adverse events, any grade | 3 | 1674 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.86, 1.27] |
| 1.11 Adverse events, grade 3 to 4 | 3 | 1674 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.80, 1.00] |
Comparison 2. Subgroup analysis (age of participants): remdesivir versus placebo or standard care alone.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 All‐cause mortality at up to day 28 | 1 | 5451 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.84, 1.13] |
| 2.1.1 Age <50 years | 1 | 1913 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.72, 1.45] |
| 2.1.2 Age 50 to 69 years | 1 | 2569 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.78, 1.18] |
| 2.1.3 Age >69 years | 1 | 969 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.74, 1.28] |
Comparison 3. Subgroup analysis (timing of first dose administration with illness onset): remdesivir versus placebo or standard care alone.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 All‐cause mortality at up to day 28 | 1 | 233 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.47, 2.05] |
| 3.1.1 ≤ 10 days of symptom onset | 1 | 118 | Risk Ratio (M‐H, Random, 95% CI) | 0.76 [0.29, 1.95] |
| 3.1.2 > 10 days of symptom onset | 1 | 115 | Risk Ratio (M‐H, Random, 95% CI) | 1.48 [0.45, 4.88] |
Comparison 4. Subgroup analysis (severity of condition, no oxygen versus low‐flow oxygen versus mechanical ventilation (including high‐flow oxygen, non‐invasive ventilation, invasive mechanical ventilation, and ECMO): remdesivir versus placebo or standard care alone.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 4.1 All‐cause mortality at up to day 28 | 3 | 3194 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.52, 1.34] |
| 4.1.1 No oxygen at baseline | 3 | 1794 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.48, 1.89] |
| 4.1.2 Low‐flow oxygen at baseline | 1 | 435 | Risk Ratio (M‐H, Random, 95% CI) | 0.32 [0.15, 0.66] |
| 4.1.3 Mechanical ventilation at baseline | 2 | 965 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.71, 1.58] |
Comparison 5. Subgroup analysis (duration of remdesivir application): remdesivir versus placebo or standard care alone.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 5.1 All‐cause mortality at up to day 28 | 1 | 584 | Risk Ratio (M‐H, Random, 95% CI) | 0.65 [0.18, 2.41] |
| 5.1.1 5‐day remdesivir | 1 | 291 | Risk Ratio (M‐H, Random, 95% CI) | 0.52 [0.07, 3.66] |
| 5.1.2 10‐day remdesivir | 1 | 293 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.13, 4.58] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Beigel 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
Baseline characteristics
Inclusion criteria:
Has laboratory‐confirmed SARS‐CoV‐2 infection as determined by PCR or other commercial or public health assay in any specimen, as documented by either or the following:
Illness of any duration, and at least 1 of the following:
Exclusion criteria:
Previous treatments: lopinavir/ritonavir (Kaletra) |
|
| Interventions |
|
|
| Outcomes |
Primary study outcome: time to recovery: the day of recovery was defined as the first day on which the participant satisfies 1 of the following 3 categories from the ordinal scale:
Review outcomes Inpatient setting:
|
|
| Identification | ||
| Notes |
|
|
Mahajan 2021.
| Study characteristics | ||
| Methods |
|
|
| Participants |
Baseline characteristics
Inclusion criteria
Exclusion criteria
Previous treatments: NR |
|
| Interventions |
|
|
| Outcomes |
Primary study outcome: clinical status from day 12 to 24 on 6 ‐point ordinal scale, mortality from day 12 to day 24, adverse events, admission days, changes in oxygen‐support requirements, administration of high‐flow oxygen, non‐invasive positive pressure ventilation and invasive mechanical ventilation, hospital discharge, need for hospitalisation (if a participant was discharged before or on day 10, it was recorded as not hospitalised) Review outcomes
|
|
| Identification | ||
| Notes |
|
|
Spinner 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
Baseline characteristics
Inclusion criteria:
Exclusion criteria
Previous treatments: not reported |
|
| Interventions |
|
|
| Outcomes |
Primary study outcome: clinical status assessed by a 7‐point ordinal scale on day 11.
Review outcomes
|
|
| Identification | ||
| Notes |
|
|
Wang 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
Baseline characteristics
Inclusion criteria
Exclusion criteria:
Previous treatments (received before and after enrolment): injection of interferon alfa‐2b; lopinavir–ritonavir; vasopressors; renal replacement therapy; antibiotics; corticosteroids |
|
| Interventions |
|
|
| Outcomes |
Primary study outcome: time to clinical improvement at up to day 28. Clinical improvement was defined as a 2‐point reduction in participant's admission status on a 6‐point ordinal scale, or live discharge from the hospital, whichever came first. The scale is as follows: 6. death; 5. hospital admission for ECMO or mechanical ventilation; 4. hospital admission for non‐invasive ventilation or high‐flow oxygen therapy; 3. hospital admission for oxygen therapy (but not requiring high‐flow or non‐invasive ventilation); 2. hospital admission but not requiring oxygen therapy; 1. discharged or having reached discharge criteria (defined as clinical recovery, i.e. normalisation of pyrexia, respiratory rate < 24 breaths per minute, saturation of peripheral oxygen > 94% on room air, and relief of cough, all maintained for at least 72 hours). Review outcomes Inpatient setting:
|
|
| Identification | ||
| Notes |
|
|
WHO Solidarity Trial Consortium 2021.
| Study characteristics | ||
| Methods |
|
|
| Participants |
Baseline characteristics
Inclusion criteria:
Exclusion criteria:
Previous treatments: NR |
|
| Interventions |
|
|
| Outcomes |
Primary study outcome: all‐cause mortality, subdivided by the severity of disease at the time of randomisation, measured using patient records throughout the study Review outcomes
|
|
| Identification | ||
| Notes |
|
|
Abbreviations:
ALT = alanine transaminase
AST = aspartate transaminase
CAD = coronary artery disease
CKD = chronic kidney disease
CT = computed tomography
ECMO = extracorporeal membrane oxygenation
eGFR = estimated glomerular filtration rate
HR = hazard ratio
ICU = intensive care unit
IQR = interquartile range
IWRS = interactive web response system
N = total number of participants
n = number of participants
NA = not applicable
NR = not reported
NEWS = National Early Warning Score
NIAID = National Institute of Allergy and Infectious Diseases
OR = odd ratio
PaO2/FiO2 = ratio of arterial oxygen partial pressure to fractional inspired oxygen
PCR = polymerase chain reaction
RCT = randomised controlled trial
RT‐PCR = reverse transcription polymerase chain reaction
SaO2 = arterial oxygen saturation
SD = standard deviation
SoC = standard of care
SpO2 = peripheral oxygen saturation
ULN = upper limit of normal
WHO = World Health Organization
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Ader 2020 | Duplicate |
| Ader 2021 | No data about the remdesivir intervention |
| Alpern 2020 | Not a randomised controlled trial |
| Antinori 2020 | Not a randomised controlled trial |
| Banerjee 2020 | Not a randomised controlled trial |
| CTRI/2020/12/029613 | No intervention with remdesivir |
| CTRI/2020/12/029615 | Combination of remdesivir with other treatments |
| Deresinski 2020 | Not a randomised controlled trial |
| Elliott 2020 | Not a randomised controlled trial |
| Elliott 2021 | Combination of remdesivir with other treatments |
| EUCTR2020‐000841‐15‐ES | Intervention with remdesivir not compared to standard care or placebo |
| EUCTR2020‐000936‐23 | Duplicate |
| Euctr2020‐003510‐12‐dk | Wrong patient population |
| EUCTR2020‐004928‐42‐HU | Wrong patient population |
| Goldberg 2021 | Not a randomised controlled trial |
| Goldman 2020 | Combination of remdesivir with other treatments |
| Goldman 2020a | Intervention with remdesivir not compared to standard care or placebo |
| ISRCTN15874265 | Combination of remdesivir with other treatments |
| ISRCTN85762140 | Wrong patient population |
| Jang 2021 | Combination of remdesivir with other treatments |
| Kalil 2021 | Combination of remdesivir with other treatments |
| Lapadula 2020 | Not a randomised controlled trial |
| LBCTR2020043495 | Duplicate |
| Medical Brief | Full‐text not retrievable |
| NCT04252664a | Duplicate |
| NCT04256395 | Not a randomised controlled trial |
| NCT04280705 | Duplicate |
| NCT04292899 | Combination of remdesivir with other treatments |
| NCT04302766 | Wrong patient population |
| NCT04321928 | No intervention with remdesivir |
| NCT04323761 | Wrong patient population |
| NCT04353596 | No intervetion with remdesivir |
| NCT04401579 | Combination of remdesivir with other treatments |
| NCT04409262 | Combination of remdesivir with other treatments |
| NCT04410354 | Combination of remdesivir with other treatments |
| NCT04480333 | Wrong patient population |
| NCT04488081 | Combination of remdesivir with other treatments |
| NCT04492475 | Combination of remdesivir with other treatments |
| NCT04501952 | Wrong patient population |
| NCT04501978 | Combination of remdesivir with other treatments |
| NCT04539262 | Wrong patient population |
| NCT04583956 | Combination of remdesivir with other treatments |
| NCT04583969 | Combination of remdesivir with other treatments |
| NCT04640168 | Combination of remdesivir with other treatments |
| NCT04647695 | Combination of remdesivir with other treatments |
| NCT04678739 | Combination of remdesivir with other treatments |
| NCT04693026 | Combination of remdesivir with other treatments |
| NCT04713176 | Combination of remdesivir with other treatments |
| NCT04728880 | Not a randomised controlled trial |
| Olender 2020 | Intervention with remdesivir not compared to standard care or placebo |
| Pan 2020 | Duplicate |
| Pan 2021 | Duplicate |
| Saito 2020 | Not a randomised controlled trial |
| Shih 2020 | Not a randomised controlled trial |
| Soto 2020 | Not a randomised controlled trial |
| Sun 2020 | Not a randomised contolled trial |
| Tran 2020 | Not a randomised controlled trial |
Characteristics of ongoing studies [ordered by study ID]
NCT04252664.
| Study name | A trial of remdesivir in adults with mild and moderate COVID‐19 |
| Methods | Trial design: RCT
Sample size: NR
Setting: inpatient Language: Chinese Number of centres: 1 (Jin Yin‐tan hospital Wuhan, Hubei, China, 100013) Type of intervention: drug |
| Participants |
Inclusion criteria:
Exclusion criteria:
|
| Interventions | Details of intervention:
Treatment details of control group (e.g. dose, route of administration):
Concomitant therapy: NR |
| Outcomes |
Primary study outcome: Time to clinical recovery (TTCR) [time frame: up to 28 days] TTCR is defined as the time (in hours) from initiation of study treatment (active or placebo) until normalisation of fever, respiratory rate, and oxygen saturation, and alleviation of cough, sustained for at least 72 hours, or live hospital discharge, whichever comes first. Normalisation and alleviation criteria:
Secondary outcome measures:
Review outcomes: Inpatient setting:
|
| Starting date | 12 February 2020 |
| Contact information | Bin Cao, China‐Japan Friendship Hospital |
| Notes |
|
NCT04596839.
| Study name | Antiviral activity and safety of remdesivir in Bangladeshi patients with severe coronavirus disease (COVID‐19) |
| Methods | Trial design: RCT
Sample size: NR
Setting: inpatient Language: Bengali Number of centres: 1 (Combined Military Hospital Dhaka, Bangladesh, 1206) Type of intervention: drug |
| Participants |
Inclusion criteria:
Exclusion criteria:
|
| Interventions |
|
| Outcomes | Primary study outcome:
Secondary study outcomes:
Review outcomes Inpatient setting:
|
| Starting date | 4 September 2020 |
| Contact information | Dr Md. Alimur Reza, MBBS, MPH +8801711438139 NCT04596839,%20BEX‐06001,%20Antiviral%20Activity%20and%20Safety%20of%20Remdesivir%20in%20Bangladeshi%20Patients%20With%20Severe%20Coronavirus%20Disease%20(COVID‐19)" type="EXTERNAL">rea@bpl.net |
| Notes |
|
Abbreviations
ALT = alanine transaminase
AST = aspartate transaminase
ECMO = extracorporeal membrane oxygenation
eGFR = estimated glomerular filtration rate
ICU = intensive care unit
NIAID = National Institute of Allergy and Infectious Diseases
NR = not reported
PaO2/FiO2 = ratio of arterial oxygen partial pressure to fractional inspired oxygen
PCR = polymerase chain reaction
RCT = randomised controlled trial
RDV = remdesivir
RT‐PCR = reverse transcription polymerase chain reaction
SaO2 = arterial oxygen saturation
SAE = serious adverse events
SoC = standard of care
WHO = World Health Organization
Differences between protocol and review
Types of outcome measures
We specified outcomes regarding effectiveness and safety of remdesivir for individuals with COVID‐19 and either moderate to severe or mild to asymptomatic disease after a guideline consortium (CEOsys) that took place after protocol registration. This approach was implemented in all reviews of CEOsys. We created outcome categories and added/specified the following outcomes for hospitalised participants with moderate or severe COVID‐19, as follows.
All‐cause mortality at day 28, day 60, time‐to‐event, and at hospital discharge.
-
Clinical status, assessed by need for respiratory support with standardised scales (e.g. WHO Clinical Progression Scale (WHO 2020d), WHO Ordinal Scale for Clinical Improvement (WHO 2020d)) at day 28, day 60, and up to longest follow‐up, including:
-
improvement of clinical status:
weaning or liberation from invasive mechanical ventilation in surviving participants;
ventilator‐free days;
duration to liberation from invasive mechanical ventilation;
liberation from supplemental oxygen in surviving participants;
duration to liberation from supplemental oxygen.
-
worsening of clinical status:
new need for mechanical ventilation;
new need for invasive mechanical ventilation;
new need for non‐invasive mechanical ventilation or high‐flow oxygen;
new need for oxygen by mask or nasal prongs.
-
Need for dialysis at up to 28 days.
Quality of life, including fatigue and neurological status, assessed with standardised scales (e.g. WHOQOL‐100) at up to 7 days, up to 30 days, and the longest follow‐up available.
Need for admission to intensive care unit (ICU).
Duration of ICU length of stay, or time to discharge from ICU.
Duration of hospitalisation, or time to discharge from hospital.
Viral clearance, assessed with reverse transcription polymerase chain reaction (RT‐PCR) test for SARS‐CoV‐2 at baseline and up to 3, 7, and 15 days.
Serious adverse events, defined as number of participants with event.
Adverse events (any grade, grade 1 to 2, grade 3 to 4), defined as number of participants with event.
We combined three different types of advanced respiratory support (high‐flow oxygen, non‐invasive mechanical ventilation, and invasive mechanical ventilation) as one outcome measure with the term 'mechanical ventilation' for the following reasons.
Their application in clinical routine usually gives indirect evidence about a clinically relevant worsening of organ functions in an individual patient.
Their application is accompanied by a need for higher level of monitoring and care (e.g. admission to ICU).
For the individual patient, the application of each of these advanced respiratory support devices means a relevant loss of independence and quality of life, compared to application of low‐flow oxygen therapy or hospitalisation without any respiratory support.
Assessment of heterogeneity
We clarified our approach to exploring heterogeneity. We intended to conduct subgroups by type of respiratory support at baseline irrespective of the amount of statistical varaiation observed between the studies. We used sensitivity analysis rather than subgroup analysis to explore heterogeneity if the I square was over 80%.
Types of subgroup analyses
We expanded subgroup analysis, and additionally plan to conduct separate analysis if more data become available in the next updates of this review, for the following.
Age of participants (divided into applicable age groups, e.g. 18 to 65 years, 65 to 79 years, 80 years and older).
Pre‐existing conditions (e.g. diabetes, respiratory disease, hypertension, immunosuppression, obesity, cardiac injury).
Timing of first dose administration with illness onset.
-
Severity of condition:
no oxygen versus low‐flow oxygen versus mechanical ventilation (including high‐flow oxygen, non‐invasive ventilation, invasive mechanical ventilation, and extracorporeal membrane oxygenation).
-
Duration of remdesivir application:
5‐day course of remdesivir versus 10‐day course of remdesivir.
Although we contacted all study authors, especially in terms of detailed description of the extent of respiratory support (e.g. low‐ versus high‐flow oxygen, non‐invasive versus invasive mechanical ventilation), there remained differences in the reporting of severity of illness and incomplete data sets, which resulted in a relevant obstacle to the planned subgroup analysis.
Living systematic review considerations
Our Information Specialist (MIM) will provide us with new search records weekly, which two review authors will screen, extract, evaluate, and integrate following the guidance for Cochrane living systematic reviews (Cochrane LSR). We will manually check platform trials that were previously identified and listed as 'studies awaiting classification' for additional treatment arms. We will wait until the accumulating evidence changes our conclusions of the implications of research and practice before republishing the review. We will consider one or more of the following components to inform this decision.
Findings that change the estimated effect of one or more prioritised outcomes.
Findings that change the credibility (e.g. GRADE rating) of the estimated effect of one or more prioritised outcomes.
New settings, populations, interventions, comparisons, or outcomes studied.
In case of emerging policy relevance because of global controversies around the intervention, we will consider republishing an updated review even though our conclusions remain unchanged. We will review the review scope and methods approximately monthly, or more frequently if appropriate, in light of potential changes in COVID‐19 research (e.g. when additional comparisons, interventions, subgroups or outcomes, or new review methods become available).
Contributions of authors
KA: methodological expertise, study selection, data extraction and assessment, conception and writing of the manuscript.
FG: clinical expertise, study selection, data extraction and assessment, conception and writing of the manuscript.
KD: methodological expertise, study selection, data extraction and assessment.
AM: clinical expertise, data extraction and assessment, writing of the manuscript.
VT: clinical expertise, study selection, data extraction and assessment, writing of the manuscript.
VP: methodological expertise and advice, data extraction and assessment, conception and writing of the manuscript.
MIM: Information Specialist, development of the search strategy, writing of the manuscript.
MS: clinical expertise and advice, writing and proofreading of the manuscript.
CB: methodological expertise and advice, data extraction and assessment, conception, writing and proofreading of the manuscript.
FF: clinical expertise and advice, data extraction and assessment, conception, writing and proofreading of the manuscript.
Sources of support
Internal sources
-
University Hospital of Cologne, Germany
Cochrane Cancer, Department of Internal Medicine
-
University Hospital RWTH Aachen, Germany
Department of Intensive Care Medicine
-
Charité – Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt‐Universität zu Berlin, Germany
Department of Infectious Diseases and Respiratory Medicine
-
University Hospital Leipzig, Germany
Department of Anesthesiology and Intensive Care Medicine
External sources
-
Federal Ministry of Education and Research, Germany
This review is part of the CEOsys project funded by the Network of University Medicine (Nationales Forschungsnetzwerk der Universitätsmedizin (NUM)) by the Federal Ministry of Education and Research of Germany (Bundesministerium für Bildung und Forschung (BMBF)), grant number 01KX2021.
Declarations of interest
KA: is member of the CEOsys project funded by the Network of University Medicine (Nationales Forschungsnetzwerk der Universitätsmedizin (NUM)) by the Federal Ministry of Education and Research of Germany (Bundesministerium für Bildung und Forschung (BMBF)), grant number 01KX2021, paid to the institution.
FG: works as an Intensive Care Medicine Consultant and is member of the CEOsys project funded by the Network of University Medicine (Nationales Forschungsnetzwerk der Universitätsmedizin (NUM)) by the Federal Ministry of Education and Research of Germany (Bundesministerium für Bildung und Forschung (BMBF)), grant number 01KX2021, paid to the institution.
KD: is member of the CEOsys project funded by the Network of University Medicine (Nationales Forschungsnetzwerk der Universitätsmedizin (NUM)) by the Federal Ministry of Education and Research of Germany (Bundesministerium für Bildung und Forschung (BMBF)), grant number 01KX2021, paid to the institution.
AM: none known.
VT: works as an Intensive Care Medicine Consultant and is member of the CEOsys project (no direct funding).
VP: is member of the CEOsys project funded by the Network of University Medicine (Nationales Forschungsnetzwerk der Universitätsmedizin (NUM)) by the Federal Ministry of Education and Research of Germany (Bundesministerium für Bildung und Forschung (BMBF)), grant number 01KX2021, paid to the institution.
MIM: is member of the CEOsys project funded by the Network of University Medicine (Nationales Forschungsnetzwerk der Universitätsmedizin (NUM)) by the Federal Ministry of Education and Research of Germany (Bundesministerium für Bildung und Forschung (BMBF)), grant number 01KX2021, paid to the institution.
MS: has no known conflicts of interest to declare.
CB: none known.
FF: works as an Intensive Care Medicine Consultant and is member of the CEOsys project (no direct funding).
contributed equally as first authors
contributed equally as first authors
contributed equally as last authors
contributed equally as last authors
Edited (no change to conclusions)
References
References to studies included in this review
Beigel 2020 {published data only}2020‐001052‐18ISRCTN13035264jRCT2031190264
- Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC, et al. Remdesivir for the treatment of COVID-19 - final report. New England Journal of Medicine 2020;383:1813-26. [DOI: 10.1056/NEJMoa2007764] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beigel JH, Tomashek KM, Dodd LE. Remdesivir for the treatment of COVID-19 - preliminary report. New England Journal of Medicine 10 July 2020. [DOI: 10.1056/NEJMc2022236] [PMID: ] [DOI] [PubMed]
- Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC, et al. Remdesivir for the treatment of COVID-19 - preliminary report. New England Journal of Medicine 2020;383:1813-26. [CLINICALTRIALS.GOV: NCT04280705] [DOI: 10.1056/NEJMoa2007764] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein M. Avoiding the termination of ACTT. European Heart Journal 2020. [DOI: 10.1093/eurheartj/ehaa546] [DOI] [PubMed]
- EUCTR2020-001052-18-DE. A multicenter, adaptive, randomised blinded controlled trial of the safety and efficacy of investigational therapeutics for the treatment of COVID-19 in hospitalised adults - version for European Union/United Kingdom sites. www.clinicaltrialsregister.eu/ctr-search/search?query=EUCTR2020-001052-18-DE (first received 27 March 2020). [EUCTR: EUCTR2020-001052-18-DE]
- Galiuto L, Patrono C. Conflicting results on the efficacy of remdesivir in hospitalised COVID-19 patients: comment on the adaptive COVID-19 treatment trial. European Heart Journal 2020. [DOI: 10.1093/eurheartj/ehaa934] [DOI] [PMC free article] [PubMed]
- ISRCTN13035264. Adaptive COVID-19 treatment trial in the EU & UK. www.isrctn.com/ISRCTN13035264 (first received 24 July 2020). [ISRCTN: ISRCTN13035264]
- jRCT2031190264. A multicenter, adaptive, randomised blinded controlled trial of the safety and efficacy of investigational therapeutics for the treatment of COVID-19 in hospitalised adults. https://jrct.niph.go.jp/latest-detail/jRCT2031190264 (first received 24 March 2020).
- Ko F. Remdesivir reduces time to recovery in adults hospitalised with COVID-19: a meaningful step in therapeutic discovery. Journal of Clinical Outcomes Management 2020. [DOI: 10.1056/NEJMoa2007764] [DOI]
- NCT04280705. Adaptive COVID-19 treatment trial (ACTT). clinicaltrials.gov/ct2/show/NCT04280705 (first received 21 February 2020).
- Olalla J. Remdesivir for the treatment of COVID-19 - preliminary report. New England Journal of Medicine 10 July 2020. [DOI: 10.1056/NEJMc2022236] [DOI] [PubMed]
- Paladugu S, Donato AA. Remdesivir improved time to recovery in adults hospitalised with COVID-19 and lower respiratory tract involvement. Annals of Internal Medicine 2020;173(2):JC4-5. [DOI: 10.7326/ACPJ202007210-005] [PMID: ] [DOI] [PubMed]
Mahajan 2021 {published data only}
- Mahajan L, Singh AP, Gifty. Clinical outcomes of using remdesivir in patients with moderate to severe COVID-19: a prospective randomised study. Indian Journal of Anaesthesia 2021;65:41-6. [DOI: 10.4103/ija.IJA_149_21] [DOI] [PMC free article] [PubMed] [Google Scholar]
Spinner 2020 {published data only}EUCTR2020‐000842‐32ISRCTN85762140
- EUCTR2020-00084-32-ES. This study will test a drug named remdesivir (GS-5734TM) to evaluate the safety and effectiveness of the drug in treating patients with moderate coronavirus disease 2019 (COVID-19) compared to standard of care treatment. https://www.clinicaltrialsregister.eu/ctr-search/trial/2020-000842-32/ES (first received 11 March 2020).
- NCT04292730. Study to evaluate the safety and antiviral activity of remdesivir (GS-5734™) in participants with moderate coronavirus disease (COVID-19) compared to standard of care treatment. clinicaltrials.gov/ct2/show/results/NCT04292730 (first received 3 March 2020).
- Spinner CD, Gottlieb RL, Criner GJ, Arribas Lopez JR, Cattelan AM, Soriano Viladomiu A, et al. Effect of remdesivir vs standard care on clinical status at 11 days in patients with moderate COVID-19: a randomised clinical trial. JAMA 2020;324(11):1048-57. [DOI: 10.1001/jama.2020.16349] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wang 2020 {published data only}
- NCT04257656. A trial of remdesivir in adults with severe COVID-19. clinicaltrials.gov/ct2/show/NCT04257656 (first received 6 February 2020).
- Wang Y, Zhang D, Du G, Du R, Zhao J, Jin Y, et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. The Lancet 2020;395(10236):1569-78. [CLINICALTRIALS.GOV: NCT04257656] [DOI: 10.1016/s0140-6736(20)31022-9] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang D, Du G. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020;395(10238):1569, poster. [DOI] [PMC free article] [PubMed]
- Wang Y, Zhou F, Zhang D, Zhao J, Du R, Hu Y, et al. Evaluation of the efficacy and safety of intravenous remdesivir in adult patients with severe COVID-19: study protocol for a phase 3 randomised, double-blind, placebo-controlled, multicentre trial. Trials 2020;21(1):422. [CLINICALTRIALS.GOV: NCT04257656] [DOI: 10.1186/s13063-020-04352-9] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
WHO Solidarity Trial Consortium 2021 {published data only}ISRCTN83971151
- Ader F. Protocol for the discovery trial: multicentre, adaptive, randomised trial of the safety and efficacy of treatments for COVID-19 in hospitalised adults. BMJ Open 2020;10:e041437. [CLINICALTRIALS.GOV: NCT04315948] [DOI: 10.1136/bmjopen-2020-041437] [EUCTR: EUCTR2020-000936-23] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barratt-Due A, Olsen IC, Henriksen KN, Kåsine T, Lund-Johansen F, Hoel H, et al. Evaluation of remdesivir and hydroxychloroquine on viral clearance in COVID-19 patients: results from the NOR-solidarity randomised trial (preprint). SSRN 2021. [CLINICALTRIALS.GOV: NCT04321616] [DOI: 10.2139/ssrn.3774182] [DOI] [Google Scholar]
- CTRI/2020/04/024773. A clinical trial to study the effects of additional treatments for patients hospitalised and receiving treatment due to COVID -19. www.ctri.nic.in/Clinicaltrials/pmaindet2.php?trialid=42897 (first received 21 April 2020). [CTRI: CTRI/2020/04/024773]
- Deresinski S. Treatment of COVID-19 patients with remdesivir, hydroxychloroquine, lopinavir, or interferon beta-1a. Infectious Disease Alert 2021;40(4). [Google Scholar]
- EUCTR2020-000936-23-FR. Multi-centre, adaptive, randomised trial of the safety and efficacy of treatments of COVID-19 in hospitalised adults - discovery. www.clinicaltrialsregister.eu/ctr-search/trial/2020-000936-23/FR (first received 9 March 2020). [DOI] [PMC free article] [PubMed]
- EUCTR2020-000936-23-PT. Safety and efficacy of treatments of COVID-19 in hospitalised adults (discovery). www.clinicaltrialsregister.eu/ctr-search/trial/2020-000936-23/PT (first received 7 March 2020).
- EUCTR2020-000982-18-NO. A clinical study to evaluate the efficacy of different anti-viral drugs in SARS-CoV-2 infected patients (COVID-19). https://www.clinicaltrialsregister.eu/ctr-search/trial/2020-000982-18/NO (first received 24 March 2020).
- EUCTR2020-001366-11-IT. An international randomised trial of additional treatments for COVID-19 in hospitalised patients who are all receiving the local standard of care. www.clinicaltrialsregister.eu/ctr-search/trial/2020-001366-11/IT (first received 20 April 2020).
- EUCTR2020-001366-11-LT. An international randomised trial of additional treatments for COVID-19 in hospitalised patients who are all receiving the local standard of care. www.clinicaltrialsregister.eu/ctr-search/trial/2020-001366-11/LT (first received 7 April 2020).
- EUCTR2020-001549-38-DE. An international randomised trial of additional treatments for COVID-19 in hospitalised patients who are all receiving the local standard of care - WHO-solidarity-Germany. www.clinicaltrialsregister.eu/ctr-search/search?query=EUCTR2020-001549-38-DE (first received 6 April 2020).
- Harrington DP, Baden LR, Hogan JW. A large, simple trial leading to complex questions. New England Journal of Medicine 2021;384:576-7. [DOI: 10.1056/NEJMe2034294] [DOI] [PMC free article] [PubMed]
- ISRCTN83971151. Public health emergency solidarity trial of treatments for COVID-19 infection in hospitalised patients. www.isrctn.com/ISRCTN83971151 (first received 25 March 2020). [DOI: ]
- Kupferschmidt K. WHO's treatment megatrial is at a standstill. Science 2021;371(6533):972-3. [DOI: 10.1126/science.371.6533.972] [DOI] [PubMed] [Google Scholar]
- LBCTR2020043495. An international randomised trial of additional treatments for COVID-19 in hospitalised patients who are all receiving the local standard of care. https://lbctr.moph.gov.lb/Trials/Details/4522 (first received 22 April 2020).
- NCT04315948. Trial of treatments for COVID-19 in hospitalised adults (discovery). clinicaltrials.gov/ct2/show/NCT04315948 (first received 20 March 2020).
- NCT04321616. The efficacy of different anti-viral drugs in (severe acute respiratory syndrome-corona virus-2) SARS-CoV-2. clinicaltrials.gov/ct2/show/NCT04321616 (first received 25 March 2020).
- NCT04330690. Treatments for COVID-19: Canadian arm of the solidarity trial. clinicaltrials.gov/ct2/show/NCT04330690 (first received 1 April 2020).
- NCT04575064. An international randomised trial of additional treatments for COVID-19 in hospitalised patients who are all receiving the local standard of care - WHO-solidarity-Germany. clinicaltrials.gov/ct2/show/NCT04575064 (first received 5 October 2020).
- NCT04647669. WHO COVID-19 solidarity trial for COVID-19 treatments. clinicaltrials.gov/ct2/show/NCT04647669 (first received 1 December 2020).
- Pan H, Peto R, Karim QA, Alejandria M, Henao-Restrepo AM, García CH, et al. Repurposed antiviral drugs for COVID-19 – interim WHO solidarity trial results. medRxiv 2020:2020.10.15.20209817. [CLINICALTRIALS.GOV: NCT04315948] [DOI: 10.1101/2020.10.15.20209817] [ISRCTN: ISRCTN83971151] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Eynde JJ. COVID-19: an update about the discovery clinical trial. Pharmaceuticals 2020;13(5):98-107. [DOI: 10.3390/ph13050098] [DOI] [PMC free article] [PubMed]
- WHO Solidarity Trial Consortium. Repurposed antiviral drugs for COVID-19 - interim WHO solidarity trial results. New England Journal of Medicine 2021;384(8):497-511. [CLINICALTRIALS.GOV: NCT04315948] [DOI: 10.1056/NEJMoa2023184] [EUCTR: EUCTR2020-001366-11] [ISRCTN: ISRCTN83971151] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Ader 2020 {published data only}
- Ader F, Discovery French trial management team. Protocol for the DisCoVeRy trial: multicentre, adaptive, randomised trial of the safety and efficacy of treatments for COVID-19 in hospitalised adults. BMJ Open 2020;10(9):e041437. [DOI: 10.1136/bmjopen-2020-041437] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ader 2021 {published data only}
- Ader F, Peiffer-Smadja N, Poissy J, Bouscambert-Duchamp M, Belhadi D, Diallo A, et al. Antiviral drugs in hospitalised patients with COVID-19 - the DisCoVeRy trial. Medrxiv 2021. [CLINICALTRIALS.GOV: NCT04315948] [DOI: 10.1101/2021.01.08.20248149] [DOI] [Google Scholar]
Alpern 2020 {published data only}
- Alpern JD, Gertner E. Off‐label therapies for COVID‐19 - are we all in this together? Clinical Pharmacology & Therapeutics;108(2):182-4. [DOI] [PMC free article] [PubMed]
Antinori 2020 {published data only}
- Antinori S, Cossu MV, Ridolfo AL, Rech R, Bonazzetti C, Pagani G, et al. Compassionate remdesivir treatment of severe COVID-19 pneumonia in intensive care unit (ICU) and non-ICU patients: clinical outcome and differences in post-treatment hospitalisation status. Pharmacological Research 2020;158. [DOI] [PMC free article] [PubMed] [Google Scholar]
Banerjee 2020 {published data only}
- Banerjee I, Mohabeer P, Shukla A, Kashyap A, Robinson J. COVID-19: recent advances in epidemiology, virology, etiopathogenesis, clinical trials and vaccine development. Journal of Biomedical Sciences 2020;7(1):18-27. [Google Scholar]
CTRI/2020/12/029613 {published data only}
- CTRI/2020/12/029613. Drug to prevent lung injury secondary to COVID 19. www.ctri.nic.in/Clinicaltrials/pmaindet2.php?trialid=50026 (first received 7 December 2020).
CTRI/2020/12/029615 {published data only}
- CTRI/2020/12/029615. Remdesivir plus tocilizumab efficacy trial in moderate to severe COVID-19 patients. www.ctri.nic.in/Clinicaltrials/pmaindet2.php?trialid=49731 (first received 7 December 2020).
Deresinski 2020 {published data only}
- Deresinski S. Remdesivir and COVID-19. Infectious Disease Alert 2020;39(10). [Google Scholar]
Elliott 2020 {published data only}
- Elliott W, Chan J. Remdesivir injection. Internal Medicine Alert 2020;42(11). [Google Scholar]
Elliott 2021 {published data only}
- Elliot W, Chan J. Remdesivir injection and baricitinib tablets. Internal Medicine Alert 2021.
EUCTR2020‐000841‐15‐ES {published data only}
- EUCTR2020-000841-15-ES. A phase 3 randomised study to evaluate the safety and antiviral activity of remdesivir (GS-5734™) in participants with severe COVID-19 [Estudio de fase 3 aleatorizado para evaluar la seguridad y la actividad antiviral de remdesivir (GS-5734™) en participantes con infección grave por el COVID-19.]. www.clinicaltrialsregister.eu/ctr-search/trial/2020-000841-15/ES (first received 11 March 2020).
EUCTR2020‐000936‐23 {published data only}
- EUCTR2020-000936-23. Multi-centre, adaptive, randomised trial of the safety and efficacy of treatments of COVID-19 in hospitalised adults - DisCoVeRy. www.clinicaltrialsregister.eu/ctr-search/search?query=eudract_number:2020-000936-23 (first received 9 March 2020).
Euctr2020‐003510‐12‐dk {published data only}
- EUCTR2020-003510-12-DK. A study to test a drug named remdesivir to evaluate the efficacy and safety of the drug in treating patients with COVID-19 in an outpatient setting (non-hospitalised). www.clinicaltrialsregister.eu/ctr-search/search?query=EUCTR2020-003510-12-DK (first received 31 August 2020).
EUCTR2020‐004928‐42‐HU {published data only}
- EUCTR2020-004928-42-HU. Clinical trial of remdesivir in COVID-19 patients. (first received 12 October 2020).
Goldberg 2021 {published data only}
- Goldberg E, Ben Zvi H, Sheena L, Sofer S, Krause I, Sklan EH, et al. A real-life setting evaluation of the effect of remdesivir on viral load in COVID-19 patients admitted to a large tertiary centre in Israel. Clinical Microbiology and Infection 2021;27:917.e1-e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Goldman 2020 {published data only}
- Goldman JD, Lye DCB, Hui DS, Marks KM, Bruno R, Montejano R, et al. Remdesivir for 5 or 10 days in patients with severe COVID-19. New England Journal of Medicine 2020;383:1827-37. [CLINICALTRIALS.GOV: NCT04292899] [DOI: 10.1056/NEJMoa2015301] [DOI] [PMC free article] [PubMed] [Google Scholar]
- ISRCTN15874265. Study to assess the safety and effectiveness of remdesivir in people with severe COVID-19. www.isrctn.com/ISRCTN15874265 (first received 29 October 2020).
- NCT04292899. Study to evaluate the safety and antiviral activity of remdesivir (GS-5734™) in participants with severe coronavirus disease (COVID-19). clinicaltrials.gov/ct2/show/NCT04292899 (first received 3 March 2020).
Goldman 2020a {published data only}
- Goldman JD, Lye DCB, Hui DS‐C, Marks K, Bruno R, Montejano R, et al. Impact of baseline alanine aminotransferase levels on the safety and efficacy of remdesivir in severe COVID-19 patients. Hepatology 2020;72:279A. [Google Scholar]
ISRCTN15874265 {published data only}
- ISRCTN15874265. Study to assess the safety and effectiveness of remdesivir in people with severe COVID-19. www.isrctn.com/ISRCTN15874265 (first received 29 October 2020).
ISRCTN85762140 {published data only}
- ISRCTN85762140. Study to assess the safety and effectiveness of remdesivir in people with moderate COVID-19. www.isrctn.com/ISRCTN85762140 (first received 2 November 2020).
Jang 2021 {published data only}
- Jang Y, Shin JS, Lee MK, Jung E, An T, Kim U, et al. Comparison of antiviral activity of gemcitabine with 2′-gluoro-2′-deoxycytidine and combination therapy with remdesivir against SARS-CoV-2. International Journal of Molecular Sciences 2021;22(4):1581-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kalil 2021 {published data only}
- Elliott W, Chan J. Remdesivir injection and baricitinib tablets. Internal Medicine Alert 2021;43(1). [Google Scholar]
- Kalil AC, Patterson TF, Mehta AK, Tomashek KM, Wolfe CR, Ghazaryan V, ACTT-2 study group members. Baricitinib plus remdesivir for hospitalized adults with COVID-19. New England Journal of Medicine 2020;384(9):795-807. [CLINICALTRIALS.GOV: NCT04401579] [DOI: 10.1056/NEJMoa2031994] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT04401579. Adaptive COVID-19 treatment trial 2 (ACTT-2). clinicaltrials.gov/ct2/show/NCT04401579 (first received 26 May 2020).
Lapadula 2020 {published data only}
- Lapadula G, Bernasconi DP, Bellani G, Soria A, Rona R, Bombino M, et al. Remdesivir use in patients requiring mechanical ventilation due to COVID-19. Open Forum Infectious Diseases 2020;7(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
LBCTR2020043495 {published data only}
- LBCTR2020043495. An international randomised trial of additional treatments for COVID-19 in hospitalised patients who are all receiving the local standard of care. https://lbctr.moph.gov.lb/Trials/Details/4522 (first received 22 April 2020). [LBCTR: 2020043495]
Medical Brief {published data only}
- Medical Brief. Remdesivir for COVID-19 improves time to recovery – peer-reviewed adaptive COVID-19 teatment trial. www.medicalbrief.co.za/archives/remdesivir-for-covid-19-improves-time-to-recovery-peer-reviewed-adaptive-covid-19-treatment-trial/ 2020;304.
NCT04252664a {published data only}
- NCT04252664. A trial of remdesivir in adults with mild and moderate COVID-19. clinicaltrials.gov/ct2/show/NCT04252664 (first received 5 February 2020).
NCT04256395 {published data only}
- NCT04256395. Efficacy of a self-test and self-alert mobile applet in detecting susceptible infection of COVID-19 (COVID-19). clinicaltrials.gov/ct2/show/NCT04256395 (first received 5 February 2020).
NCT04280705 {published data only}
- NCT04280705. Adaptive COVID-19 treatment trial (ACTT). clinicaltrials.gov/ct2/show/NCT04280705 (first received 21 February 2020).
NCT04292899 {published data only}
- NCT04292899. Study to evaluate the safety and antiviral activity of remdesivir (GS-5734™) in participants with severe coronavirus disease (COVID-19). clinicaltrials.gov/ct2/show/NCT04292899 (first received 3 March 2020).
NCT04302766 {published data only}
- NCT04302766. Expanded access remdesivir (RDV; GS-5734™). clinicaltrials.gov/ct2/show/NCT04302766 (first received 10 March 2020).
NCT04321928 {published data only}
- NCT04321928. Personalised health education against the health damage of novel coronavirus (COVID-19) outbreak in Hungary (PROACTIVE-19). clinicaltrials.gov/ct2/show/NCT04321928 (first received 25 March 2020).
NCT04323761 {published data only}
- NCT04323761. Expanded access treatment protocol: temdesivir (RDV; GS-5734) for the treatment of SARS-CoV2 (CoV) infection (COVID-19). clinicaltrials.gov/ct2/show/NCT04323761 (first received 27 March 2020).
NCT04353596 {published data only}
- NCT04353596. Stopping ACE-inhibitors in COVID-19 (ACEI-COVID). clinicaltrials.gov/ct2/show/record/NCT04353596?view=record (first received 20 April 2020).
NCT04401579 {published data only}
- NCT04401579. Adaptive COVID-19 treatment trial 2 (ACTT-2). clinicaltrials.gov/ct2/show/NCT04401579 (first received 26 May 2020).
NCT04409262 {published data only}
- NCT04409262. A study to evaluate the efficacy and safety of remdesivir plus tocilizumab compared with remdesivir plus placebo in hospitalised participants with severe COVID-19 Pneumonia (REMDACTA). clinicaltrials.gov/ct2/show/NCT04409262 (first received 1 June 2020).
NCT04410354 {published data only}
- NCT04410354. Study of Merimepodib in combination with remdesivir in adult patients with advanced COVID-19. clinicaltrials.gov/ct2/show/NCT04410354 (first received 1 June 2020).
NCT04480333 {published data only}
- NCT04480333. Safety, tolerability and pharmacokinetics of inhaled nanoparticle formulation of remdesivir (GS-5734) and NA-831. clinicaltrials.gov/ct2/show/NCT04480333 (first received 21 July 2020).
NCT04488081 {published data only}
- NCT04488081. I-SPY COVID-19 trial: an adaptive platform trial for critically ill patients (I-SPY_COVID). clinicaltrials.gov/ct2/show/NCT04488081 (first received 27 July 2020).
NCT04492475 {published data only}
- NCT04492475. Adaptive COVID-19 treatment trial 3 (ACTT-3). clinicaltrials.gov/ct2/show/NCT04492475 (first received 30 July 2020).
NCT04501952 {published data only}
- NCT04501952. Study to evaluate the efficacy and safety of remdesivir (GS-5734™) treatment of coronavirus disease 2019 (COVID-19) in an outpatient setting. clinicaltrials.gov/ct2/show/NCT04501952 (first received 6 August 2020).
NCT04501978 {published data only}
- NCT04501978. ACTIV-3: therapeutics for inpatients with COVID-19 (TICO). clinicaltrials.gov/ct2/show/NCT04501978 (first received 6 August 2020).
NCT04539262 {published data only}
- NCT04539262. Study in participants with early stage coronavirus disease 2019 (COVID-19) to evaluate the safety, efficacy, and pharmacokinetics of remdesivir administered by inhalation. clinicaltrials.gov/ct2/show/NCT04539262 (first received 4 September 2020).
NCT04583956 {published data only}
- NCT04583956. ACTIV-5 / big effect trial (BET-A) for the treatment of COVID-19. clinicaltrials.gov/ct2/show/NCT04583956 (first received 12 October 2020).
NCT04583969 {published data only}
- NCT04583969. ACTIV-5 / big effect trial (BET-B) for the treatment of COVID-19. clinicaltrials.gov/ct2/show/NCT04583969 (first received 12 October 2020).
NCT04640168 {published data only}
- NCT04640168. Adaptive COVID-19 treatment trial 4 (ACTT-4). clinicaltrials.gov/ct2/show/NCT04640168 (first received 23 November 2020).
NCT04647695 {published data only}
- NCT04647695. IFN-beta 1b and remdesivir for COVID19. clinicaltrials.gov/ct2/show/NCT04647695 (first received 1 December 2020).
NCT04678739 {published data only}
- NCT04678739. Efficacy and safety of remdesivir and tociluzumab for the management of severe COVID-19: a randomised controlled trial. clinicaltrials.gov/ct2/show/NCT04678739 (first received 22 December 2020).
NCT04693026 {published data only}
- NCT04693026. Efficacy of remdisivir and Baricitinib for the treatment of severe COVID 19 patients. clinicaltrials.gov/ct2/show/NCT04693026 (first received 5 January 2021).
NCT04713176 {published data only}
- NCT04713176. Efficacy and safety of DWJ1248 with remdesivir in severe COVID-19 patients. clinicaltrials.gov/ct2/show/NCT04713176 (first received 19 January 2021).
NCT04728880 {published data only}
- NCT04728880. Remdesivir in adults with COVID-19: Mansoura university hospital experience. clinicaltrials.gov/ct2/show/NCT04728880 (first received 28 January 2021).
Olender 2020 {published data only}
- NCT04292899. Study to evaluate the safety and antiviral activity of remdesivir (GS-5734™) in participants with severe coronavirus disease (COVID-19). clinicaltrials.gov/ct2/show/NCT04292899 (first received 3 March 2020).
- Olender SA, Perez KK, Go AS, Balani B, Price-Haywood EG, Shah NS, et al. Remdesivir for severe coronavirus disease 2019 (COVID- 19) versus a cohort receiving standard of care. Clinical Infectious Diseases 2020;ciaa1041. [CLINICALTRIALS.GOV: NCT04292899] [DOI: 10.1093/cid/ciaa1041] [EUPAS: EUPAS34303] [DOI] [PMC free article] [PubMed] [Google Scholar]
Pan 2020 {published data only}
- Pan H, Peto R, Karim QA, Alejandria M, Henao-Restrepo AM, García CH, WHO Solidarity trial consortium. Repurposed antiviral drugs for COVID-19 –interim WHO SOLIDARITY trial results. medRxiv 2020;384:497-511. [DOI: 10.1101/2020.10.15.20209817] [ISRCTN83971151] [NCT04315948] [DOI] [PMC free article] [PubMed] [Google Scholar]
Pan 2021 {published data only}
- Pan H, Peto R, Henao-Restrepo AM, Preziosi MP, Sathiyamoorthy V, Karim QA, WHO Solidarity Trial Consortium. Repurposed antiviral drugs for COVID-19 - Interim WHO solidarity trial results. New England Journal of Medicine 2021;384:497-511. [DOI: 10.1056/NEJMoa2023184] [EUCTR2020-001366-11] [ISRCTN83971151] [NCT04315948] [DOI] [PMC free article] [PubMed] [Google Scholar]
Saito 2020 {published data only}
- Saito S, Hayakawa K, Mikami A, Izumi S, Funazaki H, Ashida S, et al. Investigator initiated clinical trial of remdesivir for the treatment of COVID-19 in Japan. Global Health & Medicine 2021;3:62-6. [DOI: 10.35772/ghm.2020.01106] [DOI] [PMC free article] [PubMed] [Google Scholar]
Shih 2020 {published data only}
- Shih WCJ, Yao C, Xie T. Data monitoring for the Chinese clinical trials of remdesivir in treating patients with COVID-19 during the pandemic crisis. Therapeutic Innovation & Regulatory Science 2020;54(5):1236-55. [DOI: 10.1007/s43441-020-00159-7] [DOI] [PMC free article] [PubMed] [Google Scholar]
Soto 2020 {published data only}
- Soto A, Quiñones-Laveriano DM, Garcia PJ, Gotuzzo E, Henao-Restrepo AM. Rapid responses to the COVID-19 pandemic through science and global collaboration: the solidarity clinical trial [Respuestas rápidas a la pandemia de COVID-19 a través de la ciencia y la colaboración global: el ensayo clínico solidaridad]. Revista Peruana de Medicina Experimental y Salud Pública 2020;37:356-60. [DOI: 10.17843/rpmesp.2020.372.5546] [DOI] [PubMed] [Google Scholar]
Sun 2020 {published data only}
- Sun D. Remdesivir for treatment of COVID-19: combination of pulmonary and IV administration may offer additional benefit. AAPS Journal 2020;22:1-6. [DOI: 10.1208/s12248-020-00459-8] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tran 2020 {published data only}
- Tran B. Early data on remdesivir for severe COVID-19: a promising start? Internal Medicine Alert 2020;42(13). [Google Scholar]
References to ongoing studies
NCT04252664 {published data only}
- A trial of remdesivir in adults with mild and moderate COVID-19. https://clinicaltrials.gov/ct2/show/NCT04252664.
NCT04596839 {published data only}
- Antiviral activity and safety of remdesivir in Bangladeshi patients with severe coronavirus disease (COVID-19). https://clinicaltrials.gov/ct2/show/NCT04596839.
Additional references
Al‐Abdouh 2021
- Al-Abdouh A, Bizanti A, Barbarawi M, Jabri A, Kumar A, Fashanu OE, et al. Remdesivir for the treatment of COVID-19: a systematic review and meta-analysis of randomised controlled trials. Contemporary Clinical Trials 2021;101:106272. [DOI: 10.1016/j.cct.2021.106272] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bansal 2021
- Bansal V, Mahapure KS, Bhurwal A, Gupta I, Hassanain S, Makadia J, et al. Mortality benefit of remdesivir in COVID-19: a systematic review and meta-analysis. Frontiers in Medicine 2021;7:606429. [DOI: 10.3389/fmed.2020.606429] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bhimrai 2021
- Bhimraj A, Morgan RL, Shumaker AH, Lavergne V, Baden L, Cheng VC, et al. IDSA guidelines on the treatment and management of patients with COVID-19. www.idsociety.org/practice-guideline/covid-19-guideline-treatment-and-management (accessed 20 April 2021).
Buitrago‐Garcia 2020
- Buitrago-Garcia D, Egli-Gany D, Counotte MJ, Hossmann S, Imeri H, Ipekci Aziz M, et al. Occurrence and transmission potential of asymptomatic and presymptomatic SARS-CoV-2 infections: a living systematic review and meta-analysis. PLoS Medicine 2020;17(9):e1003346. [DOI: 10.1371/journal.pmed.1003346] [DOI] [PMC free article] [PubMed] [Google Scholar]
CDC 2020
- Centers for Disease Control and Prevention. COVIDView - a weekly surveillance summary of U.S. COVID-19 activity (Key updates for week 43). www.cdc.gov/coronavirus/2019-ncov/covid-data/covidview/index.html (accessed 12 January 2020).
CEOsys 2021
- German COVID-19 evidence-ecosystem. www.covid-evidenz.de (accessed prior to 22 July 2021).
Chai 2020
- Chai KL, Valk SJ, Piechotta V, Kimber C, Monsef I, Doree C, et al. Convalescent plasma or hyperimmune immunoglobulin for people with COVID‐19: a living systematic review. Cochrane Database of Systematic Reviews 2020, Issue 7. Art. No: CD013600. [DOI: 10.1002/14651858.CD013600.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Chen 2020
- Chen Y, Liu Q, Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. Journal of Medical Virology 2020;92(4):418-23. [DOI: 10.1002/jmv.25681] [DOI] [PMC free article] [PubMed] [Google Scholar]
Chen 2020a
- Chen T, Wu D, Chen H, Yan D, Chen G. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: retrospective study. BMJ 2020;368:m1091. [DOI: 10.1136/bmj.m1091] [DOI] [PMC free article] [PubMed] [Google Scholar]
Choy 2020
- Choy KT, Wong AYL, Kaewpreedee P, Sia SF, Chen D, Hui KPY, et al. Remdesivir, lopinavir, emetine, and homoharringtonine inhibit SARS-CoV-2 replication in vitro. Antiviral Research 2020;178:104786. [DOI: 10.1016/j.antiviral.2020.104786] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cochrane LSR
- Guidance for the production and publication of Cochrane living systematic reviews: Cochrane Reviews in living mode. Available from community.cochrane.org/review-production/production-resources/living-systematic-reviews (accessed 12 July 2021).
COMET 2020
- Core outcome set developers’ response to COVID-19. www.comet-initiative.org/Studies/Details/1538 (accessed 2 November 2020).
Covidence 2021 [Computer program]
- Veritas Health Innovation Covidence. Version accessed 15 January 2021. Melbourne, Australia: Veritas Health Innovation. Available at covidence.org.
CRD42021238065
- CRD42021238065. Remdesivir for the treatment of hospitalized adults COVID-19 patients (part of German Ecosystem CEO-sys). www.crd.york.ac.uk/prospero/display_record.php?ID=CRD42021238065 (first received 26 February 2021).
Deeks 2020
- Deeks JJ, Higgins JP, Altman DG, editor(s). Chapter 10: Analysing data and undertaking meta-analyses. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions version 6.2 (updated February 2021). Available from training.cochrane.org/handbook. Cochrane, 2020. [Google Scholar]
de Wit 2020
- Wit E, Feldmann F, Cronin J, Jordan R, Okumura A, Thomas T, et al. Prophylactic and therapeutic remdesivir (GS5734) treatment in the rhesus macaque model of MERS-CoV infection. Proceedings of the National Academy of Sciences 2020;117(12):6771-6. [DOI: 10.1073/pnas.1922083117] [DOI] [PMC free article] [PubMed] [Google Scholar]
Eldridge 2016
- Eldridge S, Campbell M, Campbell M, Dahota A, Giraudeau B, Higgins JT, et al. Revised Cochrane risk of bias tool for randomized trials (RoB 2.0). Additional considerations for cluster-randomized trials. www.riskofbias.info/welcome/rob-2-0-tool/rob-2-for-cluster-randomized-trials (accessed 1 March 2021).
Elsawah 2020
- Elsawah KE, Elsoraky MA, Abdallah MS, ElShafie AH. Efficacy and safety of remdesivir in hospitalised COVID-19 patients: systematic review and meta-analysis including network meta-analysis. Reviews in Medical Virology 2021;31(4):e2187. [DOI: 10.1002/rmv.2187] [DOI] [PubMed] [Google Scholar]
EMA 2020
- European Medicines Agency. Summary on compassionate use remdesivir Gilead. www.ema.europa.eu/en/documents/other/summary-compassionate-use-remdesivir-gilead_en.pdf (accessed prior to 22 July 2021).
EUA 2020
- European Commission authorises first treatment against COVID-19. eeas.europa.eu/headquarters/headquarters-homepage/82135 (accessed 10 March 2021).
EUA 2021
- Fact sheet for health care providers Emergency Use Authorisazation (EUA) of remdesivir (GS-5734™). www.askgileadmedical.com/downloads/pdfs/EUA%20Fact%20Sheet%20for%20HCPs.pdf (accsessed 10 March 2021).
Funk 2021
- Funk T, Pharris A, Spiteri G, Bundle N, Melidou A, Carr M, et al. Characteristics of SARS-CoV-2 variants of concern B.1.1.7, B.1.351 or P.1: data from seven EU/EEA countries, weeks 38/2020 to 10/2021. Eurosurveillance 2021;26(16). [DOI: 10.2807/1560-7917.ES.2021.26.16.2100348] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gautret 2020
- Gautret P, Million M, Jarrot PA, Jau LC, Colson P, Fenollar F, et al. Natural history of COVID-19 and therapeutic options. Expert Review of Clinical Immunology 2020;16(12):1159-84. [DOI: 10.1080/1744666X.2021.1847640] [DOI] [PubMed] [Google Scholar]
Grubaugh 2020
- Grubaugh ND, Hanage WP, Rasmussen AL. Making sense of mutation: what D614G means for the COVID-19 pandemic remains unclear. Cell 2020;182(4):794-5. [DOI: 10.1016/j.cell.2020.06.040] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in metaanalyses. BMJ 2003;327:557-60. [DOI: 10.1136/bmj.327.7414.557] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2021a
- Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from training.cochrane.org/handbook.
Higgins 2021b
- Higgins JP, Eldridge S, Li T. Chapter 23: Including variants on randomized trials. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from training.cochrane.org/handbook.
Higgins 2021c
- Higgins JP, Savović J, Page MJ, Elbers RG, Sterne JA. Chapter 8: Assessing risk of bias in a randomized trial. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from training.cochrane.org/handbook.
Higgins 2021d
- Higgins JP, Tianging L, Deeks JJ, editor(s). Chapter 6: Choosing effect measures and computing estimates of effect. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from training.cochrane.org/handbook.
Hsu 2020
- Hsu CY, Lai CC, Yen AMF, Chen SLS, Chen HH. Efficacy of remdesivir in COVID-19 patients with a simulated two-arm controlled study (preprint). medRxiv 2021. [DOI: 10.1101/2020.05.02.20088559] [DOI]
Hu 2020
- Hu Z, Song C, Xu C, Jin G, Chen Y, Xu X, et al. Clinical characteristics of 24 asymptomatic infections with COVID-19 screened among close contacts in Nanjing, China. Science China Life Sciences 2020;63. [DOI: 10.1007/s11427-020-1661-4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Huang 2020
- Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020;395(10223):497-506. [DOI: 10.1016/S0140-6736(20)30183-5] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kaka 2021
- Kaka AS, MacDonnald R, Greer N, Vela K, Duan-Porter W, Obley A, et al. Major update: remdesivir for adults with COVID-19: a living systematic review and meta-analysis for the American College of Physicians Practice Points. Annals of Internal Medicine 2021;2021. [CORRECTION: www.acpjournals.org/doi/10.7326/L21-0130] [DOI: 10.7326/M20-8148] [DOI] [PMC free article] [PubMed] [Google Scholar]
Karagiannidis 2020
- Karagiannidis C, Mostert C, Hentschker C, Voshaar T, Malzahn J, Schillinger G, et al. Case characteristics, resource use, and outcomes of 10 021 patients with COVID-19 admitted to 920 German hospitals: an observational study. Lancet Respiratory Medicine 2020;8:853-62. [DOI: 10.1016/ S2213-2600(20)30316-7] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kreuzberger 2021
- Kreuzberger N, Hirsch C, Chai KL, Piechotta V, Valk SJ, Estcourt LJ, et al. SARS‐CoV‐2‐neutralising monoclonal antibodies for treatment of COVID‐19. Cochrane Database of Systematic Reviews 2021, Issue 1. Art. No: CD013825. [DOI: 10.1002/14651858.CD013825] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kumar 2020
- Kumar S, Maurya VK, Prasad AK, Bhat MLB. Structural, glycosylation and antigenic variation between 2019 novel coronavirus (2019-nCoV) and SARS coronavirus (SARS-CoV). VirusDisease 2020;31(1):13-21. [DOI: 10.1007/s13337-020-00571-5] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lai 2021
- Lai CC, Chen CH, Wang CY, Chen KH, Wang YH, Hsueh PR. Clinical efficacy and safety of remdesivir in patients with COVID-19: a systematic review and network meta-analysis of randomised controlled trials. Journal of Antimicrobial Chemotherapy 2021. [DOI: 10.1093/jac/dkab093] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lauer 2020
- Lauer SA, Grantz KH, Bi Q, Jones FK, Zheng Q, Meredith HR, et al. The incubation period of coronavirus disease 2019 (COVID-19) from publicly reported confirmed cases: estimation and application. Annals of Internal Medicine 2020;172(9):577–82. [DOI: 10.7326/M20-0504] [DOI] [PMC free article] [PubMed] [Google Scholar]
Li 2020a
- Li T, Higgins JP, Deeks JJ. Cochrane Handbook for Systematic Reviews of Interventions Version 6.2. (updated February 2021). Cochrane, 2020. Available from training.cochrane.org/handbook. [Google Scholar]
Li 2020b
- Li X, Geng M, Peng Y, Meng L, Lu S. Molecular immune pathogenesis and diagnosis of COVID-19. Journal of Pharmaceutical Analysis 2020;10(2):102-8. [DOI: 10.1016/j.jpha.2020.03.001] [DOI] [PMC free article] [PubMed] [Google Scholar]
Liang 2020
- Liang W, Guan W, Chen R, Wang W, Li J, Xu K, et al. Cancer patients in SARS-CoV-2 infection: a nationwide analysis in China. Lancet Oncology 2020;21(3):335-7. [DOI: 10.1016/S1470-2045(20)30096-6] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lo 2017
- Lo MK, Jordan R, Arvey A, Sudhamsu J, Shrivastava-Ranjan P, Hotard AL, et al. GS-5734 and its parent nucleoside analog inhibit Filo-, Pneumo-, and Paramyxoviruses. Scientific Reports 2017;7(1):1-7. [DOI: 10.1038/srep43395.] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lundstrom 2020
- Lundstrom K, Seyran M, Pizzol D, Adadi P, Mohamed Abd El-Aziz T, Hassan SS, et al. Viewpoint: origin of SARS-CoV-2. Viruses 2020;12(11):1203. [DOI: 10.3390/v12111203] [DOI] [PMC free article] [PubMed] [Google Scholar]
MAGICapp [Computer program]
- Norwegian MAGIC Evidence Ecosystem Foundation (powered by UserVoice Inc.) MAGICapp. Version accessed 25 February 2021. Brønnøysund (NOR): Norwegian MAGIC Evidence Ecosystem Foundation (powered by UserVoice Inc.). Available at magicapp.org.
Malaiyan 2020
- Malaiyan J, Arumugam S, Mohan K. An update on the origin of SARS-CoV-2: despite closest identity, bat (RaTG13) and pangolin derived coronaviruses varied in the critical binding site and O-linked glycan residues. Journal of Medical Virology 2020;93(1):499-505. [DOI: 10.1002/jmv.26261] [DOI] [PMC free article] [PubMed] [Google Scholar]
Malin 2020
- Malin JJ, Suárez I, Priesner V, Fätkenheuer G, Rybnike J. Remdesivir against COVID-19 and other viral diseases. Clinical Microbiology Reviews 2020;34(1):e00162-20. [DOI: 10.1128/CMR.00162-20] [DOI] [PMC free article] [PubMed] [Google Scholar]
McGuigan 2006
- McGuigan C, Hassan-Abdallah A, Srinivasan S, Wang Y, Siddiqui A, Daluge SM, et al. Application of phosphoramidate ProTide technology significantly improves antiviral potency of carbocyclic adenosine derivatives. Journal of Medicinal Chemistry 2006;49(24):7215-26. [DOI: 10.1021/jm060776w.] [DOI] [PubMed] [Google Scholar]
Meng 2020
- Meng H, Xiong R, He R, Lin W, Hao B, Zhang L, et al. CT imaging and clinical course of asymptomatic cases with COVID-19 pneumonia at admission in Wuhan, China. Journal of Infection 2020;81(1):e33-9. [DOI: 10.1016/j.jinf.2020.04.004] [DOI] [PMC free article] [PubMed] [Google Scholar]
Microsoft 2018 [Computer program]
- Mircosoft Excel. Microsoft Corporation, 2018. Available at office.microsoft.com/excel.
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG, The PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLOS Medicine 2009;6(7):e1000097. [DOI: 10.1371/journal.pmed.1000097] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mulangu 2019
- Mulangu S, Dodd LE, Davey RT, Mbaya OT, Proschan M, Mukadi D, et al. A randomised, controlled trial of Ebola virus disease therapeutics. New England Journal of Medicine 2019;381(24):2293-303. [DOI: 10.1056/NEJMoa1910993] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ogando 2020
- Ogando NS, Dalebout TJ, Zevenhoven-Dobbe JC, Limpens RWAL, van der Meer Y, Caly L, et al. SARS-coronavirus-2 replication in Vero E6 cells: replication kinetics, rapid adaptation and cytopathology. Journal of General Virology 2020;101(9):925-40. [DOI: 10.1099/jgv.0.001453] [DOI] [PMC free article] [PubMed] [Google Scholar]
Oran 2020
- Oran DP, Topol EJ. Prevalence of asymptomatic SARS-CoV-2 infection: a narrative review. Annals of Internal Medicine 2020;173(5):362-7. [DOI: 10.7326/M20-3012] [DOI] [PMC free article] [PubMed] [Google Scholar]
Pan 2020
- Pan A, Liu L, Wang C, Guo H, Hao X, Wang Q, et al. Association of public health interventions with the epidemiology of the COVID-19 outbreak in Wuhan, China. JAMA 2020;323(19):1915-23. [DOI: 10.1001/jama.2020.6130] [DOI] [PMC free article] [PubMed] [Google Scholar]
Parmar 1998
- Parmar MK, Torri V, Stewart L. Extracting summary statistics to perform metaanalyses of the published literature for survival endpoints. Statistics in Medicine 1998;17(24):2815-34. [DOI: ] [DOI] [PubMed] [Google Scholar]
Petrilli 2020
- Petrilli CM, Jones SA, Yang J, Rajagopalan H, O'Donnel L, Chernyak Y, et al. Factors associated with hospital admission and critical illness among 5279 people with coronavirus disease 2019 in New York City: prospective cohort study. BMJ 2020;369:m1966. [DOI: 10.1136/bmj.m1966] [DOI] [PMC free article] [PubMed] [Google Scholar]
Piechotta 2020
- Piechotta V, Valk SJ, Chai KL, Wood EM, Lamikanra A, Kimber C, et al. Safety and effectiveness of convalescent plasma or hyperimmune globulin for people with COVID-19: a rapid review. OSF Registries 2020. [DOI: 10.17605/OSF.IO/DWF53] [DOI] [PMC free article] [PubMed] [Google Scholar]
Piscoya 2020
- Piscoya A, Ng-Sueng LF, Del Riego AP, Cerna-Viacava R, Pasupuleti V, Roman YM, et al. Efficacy and harms of remdesivir for the treatment of COVID-19: a systematic review and meta-analysis. PLOS ONE 2020;15(12):e0243705. [DOI: 10.1371/journal.pone.0243705] [DOI] [PMC free article] [PubMed] [Google Scholar]
Potere 2020
- Potere N, Valeriani E, Candeloro M, Tana M, Porreca E, Abbate A, et al. A higher mortality rate and long ventilation times differentiate COVID-19 from severe respiratory infections in flu waves [Eine höhere Letalität und lange Beatmungsdauer unterscheiden COVID-19 von schwer verlaufenden Atemwegsinfektionen in Grippewellen]. Critical Care 2020;24(1):389. [DOI: 10.25646/7111] [DOI] [PMC free article] [PubMed] [Google Scholar]
RevMan Web 2021 [Computer program]
- The Cochrane Collaboration Review Manager Web (RevMan Web). Version 3.4.0. The Cochrane Collaboration, 2021. Available at revman.cochrane.org.
Richardson 2020
- Richardson S, Hirsch JS, Narasimhan M, Crawford JM, McGinn T, Davidson KW. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City area. JAMA 2020;323(20):2052-9. [DOI: 10.1001/jama.2020.6775] [DOI] [PMC free article] [PubMed] [Google Scholar]
Roshanshad 2020
- Roshanshad A, Kamalipour A, Ashraf MA, Roshanshad R, Jafari S, Nazemi P, et al. The efficacy of remdesivir in coronavirus disease 2019 (COVID-19): a systematic review. Iranian Journal of Microbiology 2020;12(5):376-87. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Santesso 2020
- Santesso N, Glenton C, Dahm P, Garner P, Akl A, Alper B, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. Journal of Clinical Epidemiology 2020;119:126-35. [DOI: 10.1016/j.jclinepi.2019.10.014] [DOI] [PubMed] [Google Scholar]
Schilling 2020
- Schilling J, Lehfeld AS, Schumacher D, Ullrich A, Diercke M. Disease severity of the first COVID-19 wave in Germany using reporting data from the national notification system. Journal of Health Monitoring 2020;5(S11):2-20. [DOI: 10.25646/7170] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schünemann 2021
- Schünemann HJ, Higgins JP, Vist GE, Glasziou P, Akl EA, Skoetz N, et al. Chapter 14: Completing ‘Summary of findings’ tables and grading the certainty of the evidence. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from training.cochrane.org/handbook.
Schwarz 2021
- Schwarz T, Tober-Lau P, Hillus D, Helbig ET, Lippert LJ, Thibeault C, et al. Delayed antibody and T-cell response to BNT162b2 vaccination in the elderly, Germany. Emerging Infectious Diseases 08 June 2021;27 (early release). [DOI: 10.3201/eid2708.211145] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sheahan 2017
- Sheahan TP, Sims AC, Graham RL, Menachery VD, Gralinski LE, Case JB, et al. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Science Translational Medicine 2017;9(396):eaal3653. [DOI: 10.1126/scitranslmed.aal3653] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sheahan 2020
- Sheahan TP, Sims AC, Leist SR, Schäfer A, Won J, Brown AJ, et al. Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir and interferon beta against MERS-CoV. Nature Communications 2020;11(1):222. [DOI: 10.1038/s41467-019-13940-6] [DOI] [PMC free article] [PubMed] [Google Scholar]
Siegel 2017
- Siegel D, Hui HC, Doerffler E, Clarke MO, Chun K, Zhang L, et al. Discovery and synthesis of a phosphoramidate prodrug of a pyrrolo[2,1-f][triazin-4-amino] Adenine C-Nucleoside (GS-5734) for the treatment of ebola and emerging viruses. Journal of Medicinal Chemistry 2017;60(5):1648-61. [DOI: 10.1021/acs.jmedchem.6b01594] [DOI] [PubMed] [Google Scholar]
Siemieniuk 2020
- Siemieniuk RA, Bartoszko JJ, Ge L, Zeraatkar D, Izcovich A, Kum E, et al. Drug treatments for COVID-19: living systematic review and network meta-analysis. BMJ 2020;370:m2980. [DOI: 10.1136/bmj.m2980] [DOI] [PMC free article] [PubMed] [Google Scholar]
Skoetz 2020
- Skoetz N, Goldkuhle M, Van Dalen EC, Akl EA, Trivella M, Mustafa RA, et al. GRADE guidelines 27: how to calculate absolute effects for time-to-event outcomes in summary of findings tables and evidence profiles. Journal of Clinical Epidemiology 2020;118:124-31. [DOI: 10.1016/j.jclinepi.2019.10.015] [DOI] [PubMed] [Google Scholar]
Sterne 2019
- Sterne JA, Savovic J, Page MJ, Elbers RG, Blencowe N, Boutron I, et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ 2019;366:l4898. [DOI: 10.1136/bmj.l4898] [DOI] [PubMed] [Google Scholar]
Struyf 2021
- Struyf T, Deeks JJ, Dinnes J, Takwoingi Y, Davenport C, Leeflang MM, et al. Signs and symptoms to determine if a patient presenting in primary care or hospital outpatient settings has COVID-19 disease. Cochrane Database of Systematic Reviews 2021, Issue 2. Art. No: CD013665. [DOI: 10.1002/14651858.CD013665.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tchesnokov 2019
- Tchesnokov E, Feng J, Porter D, Götte M. Mechanism of inhibition of ebola virus RNA-dependent RNA polymerase by remdesivir. Viruses 2019;11(4):326. [DOI: 10.3390/v11040326] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tierney 2007
- Tierney JF, Stewart LA, Ghersi D, Burdett S, Sydes MR. Practical methods for incorporating summary time-to-event data into meta-analysis. Trials 2007;8:16. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tolksdorf 2020
- Tolksdorf K, Buda S, Schuler E, Wieler LH, Haas W. Higher lethality and long duration of ventilation distinguish COVID-19 from severe respiratory tract infections in influenza waves. [Eine höhere Letalität und lange Beatmungsdauer unterscheiden COVID-19 von schwer verlaufenden Atemwegsinfektionen in Grippewellen]. Epidemiologisches Bulletin 2020;41:3-10. [DOI: 10.25646/7111] [DOI] [Google Scholar]
Vegivinti 2021
- Vegivinti CT, Pederson JM, Saravu K, Gupta N, Barrett A, Davis AR, et al. Remdesivir therapy in patients with COVID-19: a systematic review and meta-analysis of randomised controlled trials. Annals of Medicine and Surgery 2021;62:43-8. [DOI: 10.1016/j.amsu.2020.12.051] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wang 2020
- Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Research 2020;30(3):269-71. [DOI: 10.1038/s41422-020-0282-0] [DOI] [PMC free article] [PubMed] [Google Scholar]
Warren 2016
- Warren TK, Jordan R, Lo MK, Ray AS, Mackman RL, Soloveva V, et al. Therapeutic efficacy of the small molecule GS-5734 against ebola virus in rhesus monkeys. Nature 2016;531(7594):381-5. [DOI: 10.1038/nature17180] [DOI] [PMC free article] [PubMed] [Google Scholar]
Welte 2021
- Welte T, Ambrose LJ, Sibbring GC, Sheikh S, Müllerová H, Sabir I. Current evidence for COVID-19 therapies: a systematic literature review. European Respiratory Review 2021;30:200384. [DOI: 10.1183/16000617.0384-2020] [DOI] [PMC free article] [PubMed] [Google Scholar]
WHO 2003
- World Health Organization. Cumulative number of reported probable cases of SARS. www.who.int/csr/sars/country/2003_07_11/en (accessed 13 April 2020).
WHO 2019
- World Health Organization. Middle East respiratory syndrome coronavirus (MERSCoV). www.who.int/emergencies/mers-cov/en (accessed 13 April 2020).
WHO 2020a
- World Health Organization. Timeline: WHO's COVID-19 response. www.who.int/emergencies/diseases/novel-coronavirus-2019/interactive-timeline (accessed 17 March 2021).
WHO 2020b
- World Health Organization. Estimating mortality from COVID-19 - scientific brief. www.who.int/publications/i/item/WHO-2019-nCoV-Sci-Brief-Mortality-2020.1 (accessed 2 November 2020).
WHO 2020c
- World Health Organization. Report of the WHO‐China joint mission on coronavirus disease 2019 (COVID‐19). www.who.int/docs/default-source/coronaviruse/who-china-joint-mission-on-covid-19-final-report (accessed 6 November 2020).
WHO 2020d
- WHO Working Group on the Clinical Characterisation and Management of COVID-19 infection. A minimal common outcome measure set for COVID-19 clinical research. Lancet Infectious Diseases 2020;20(8):e192-7. [DOI: 10.1016/S1473-3099(20)30483-7] [DOI] [PMC free article] [PubMed] [Google Scholar]
WHO 2020e
- World Health Organization. SARS-CoV-2 mink-associated variant strain – Denmark. www.who.int/csr/don/06-november-2020-mink-associated-sars-cov2-denmark/en (accessed 9 November 2020).
WHO 2021
- World Health Organization. Therapeutics and COVID-19: living guideline. www.who.int/publications/i/item/therapeutics-and-covid-19-living-guideline (accessed 8 March 2021). [PubMed]
WHO 2021a
- World Health Organization. WHO Coronavirus Disease (COVID-19) Dashboard. covid19.who.int (accessed 09 June 2021).
WHO 2021b
- World Health Organization. WHO Coronavirus (COVID-19) Dashboard. covid19.who.int/ (accessed 02 August 2021).
WHO 2021c
- World Health Organization. Origins of the SARS-CoV-2 virus. www.who.int/health-topics/coronavirus/origins-of-the-virus (accessed 30 March 2021).
WHO 2021d
- World Health Organization. Weekly epidemiological update - 16 March 2021. https://www.who.int/publications/m/item/weekly-epidemiological-update---16-march-2021 (accessed 20 March 2021).
Williamson 2020a
- Williamson E, Walker AJ, Bhaskaran KJ, Bacon S, Bates C, Morton CE, et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 2020;584:430-6. [DOI: 10.1038/s41586-020-2521-4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Williamson 2020b
- Williamson BN, Feldmann F, Schwarz B, Meade-White K, Porter DP, Schulz J, et al. Clinical benefit of remdesivir in rhesus macaques infected with SARS-CoV-2. Nature 2020;585:7824. [DOI: 10.1038/s41586-020-2423-5] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wilt 2021
- Wilt TJ, Kaka AS, MacDonald R, Greer N, Obley A, Duan-Porter W. Remdesivir for adults with COVID-19: a living systematic review for an American College of Physicians Practice Point. Annals of Internal Medicine 2021;174(2):209-20. [DOI: 10.7326/M20-5752] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wu 2020
- Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72314 cases from the Chinese Center for Disease Control and Prevention. JAMA 2020;323(13):1239-42. [DOI: 10.1001/jama.2020.2648] [DOI] [PubMed] [Google Scholar]
Yan 2020
- Yan R, Zhang Y, Li Y, Guo Y, Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020;367(6485):1444-8. [DOI: 10.1126/science.abb2762] [DOI] [PMC free article] [PubMed] [Google Scholar]
Yokoyama 2020
- Yokoyama Y, Briasoulis A, Takagi H, Kuno T. Effect of remdesivir on patients with COVID-19: a network meta-analysis of randomised control trials. Virus Research 2020;288:198137. [DOI: 10.1016/j.virusres.2020.198137] [DOI] [PMC free article] [PubMed] [Google Scholar]