

Abstract
Background:
The etiology of Alzheimer’s disease remains poorly understood at the mechanistic level, and genome-wide network-based genetics have the potential to provide new clues of the disease mechanisms.
Objective:
To explore the collective effects of multiple genetic association signals on an AV-45 PET measure, which is a well-known Alzheimer’s disease biomarker, by employing a network-assisted strategy.
Method:
First, we took advantage of dense module search algorithm to identify modules enriched by genetic association signals in a protein-protein interaction network. Next, we performed statistical evaluations to the modules identified by dense module search, including a normalization process to adjust the topological bias in the network, a replication test to ensure the modules were not found by chance, and a permutation test to evaluate unbiased associations between the modules and amyloid imaging phenotype. Finally, topological analysis, module similarity tests and functional enrichment analysis were performed for the identified modules.
Results:
We identified 24 consensus modules enriched by robust genetic signals in a genome-wide association analysis. The results not only validated several previously reported AD genes (APOE, APP, TOMM40, DDAH1, PARK2, ATP5C1, PVRL2, ELAVL1, ACTN1 and NRF1), but also nominated a few novel genes (ABL1, ABLIM2) that have not been studied in Alzheimer’s disease but have shown associations with other neurodegenerative diseases.
Conclusion:
The identified genes, consensus modules and enriched pathways may provide important clues to future research on the neurobiology of Alzheimer’s disease and suggest potential therapeutic targets.
Keywords: Alzheimer’s disease, amyloid imaging phenotype, genome-wide association, network analysis, pathway enrichment, consensus modules, neurodegenerative disease
1. INTRODUCTION
Alzheimer’s disease (AD) is an irreversible, neurodegenerative dementia characterized by gradual memory loss and other cognitive impairments. In 2017, AD became the sixth leading cause of 121,404 death in the US according to the record of official death certificates [1]. Currently, the number of people affected by AD is about 50 million and is expected to be over 152 million by 2050 [2]. Genetic factors play an important part in AD with heritability estimates up to 79% [3]. To identify common variants associated with AD, efforts have been focused on conducting genome-wide association (GWA) studies [4–10] in the last few years. However, most variants identified so far explain only a small proportion of the disease inherited risk [11]. A possible explanation is that real but individually moderate associations may be lost due to the stringent significance level after multiple comparison correction for a large number of tests conducted. Therefore, new strategies are required to improve the power of GWA analysis.
It has been commonly accepted that genes are not randomly distributed in regard of their biological function, but instead clustered in common biological pathways and networks [12,13]. Thus, the recognition of genes directly or indirectly interacting in pathway or complex network is gradually emerging [14,15]. Network-based approaches, which are not limited to sort the genes into not fully annotated predefined gene sets, have been proved successful in the identification of subnetworks of markers with higher reproducibility and higher predictability than individual markers [16]. They have been widely applied to the analysis of high-throughput expression data for various diseases [17]. More recent studies adopted protein-protein interaction networks (PPIN) to the analysis of GWA data to identify genome-wide enriched pathways [18–24]. In combination with the biological knowledge, they are of great help to interpret the function of the genetic variants, and to further understand the molecular mechanism influencing the traits of interest [25].
In the current study, we integrated GWA studies and PPIN to identify modules that contribute to an AD biomarker. Briefly, we aimed to (1) perform a dense module search (DMS) within the PPIN to identify modules enriched with genes associated with an amyloid imaging phenotype that is an AD biomarker; (2) estimate the association of the modules with the phenotype across another independent sample; (3) examine the role of the same module in permuted datasets which were generated by swapping the disease labels; and (4) complete similarity tests and functional enrichment analysis to assess whether the identified modules have common biological function. Our analysis yielded 24 modules including 156 genes, which formed a highly-connected and statistically significant subnetwork. The genes, modules and enriched pathways identified in this analysis are promising targets for further studies of the mechanism underlying AD.
2. MATERIALS AND METHOD
Data used in the preparation of this article were obtained from the ADNI database (http://adni.loni.usc.edu/). The ADNI was launched in 2003 as a public-private partnership, led by Principal Investigator Michael W. Weiner, MD. The primary goal of ADNI has been to test whether serial magnetic resonance imaging (MRI), positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment (MCI) and early Alzheimer’s disease (AD).
As shown in Fig. 1, we first randomly split the subjects into two groups (G1, G2) while maintaining the same gender and diagnosis distributions in each group. Then, we performed a DMS algorithm [26] on G1 to identify modules enriched with phenotype-associated genes. After that, significant modules generated by G1 were assessed whether they were significant in the independent samples G2. Next, we used a permutation-based association test on G2 to evaluate empirically whether the significant modules identified in both G1 and G2 were associated with the amyloid imaging phenotype. Finally, we evaluated the selected promising modules for centrality measurements, similarity tests and functional enrichment analysis.
Figure 1.
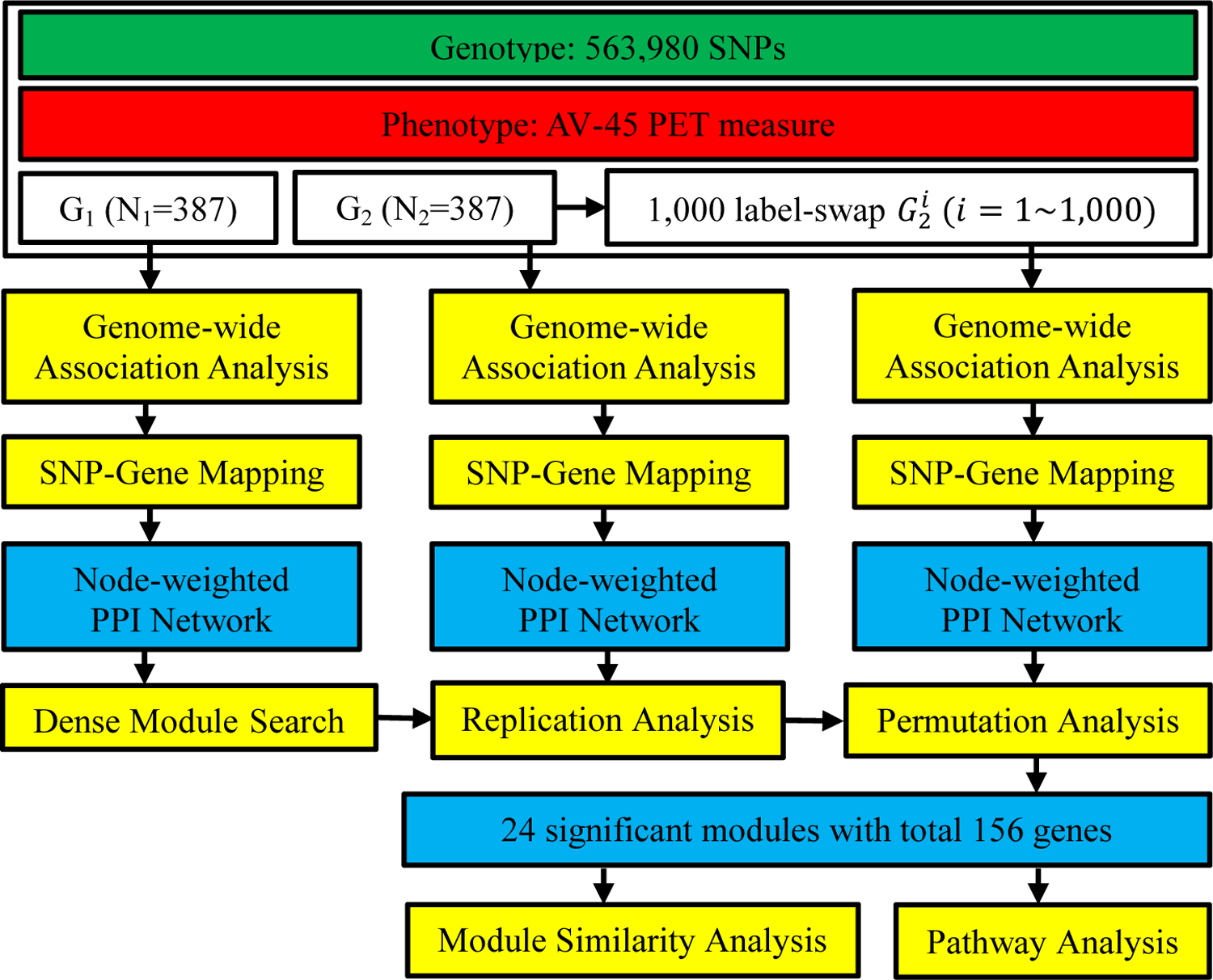
Workflow of network-assisted strategy to identify modules associated with AV-45 measure, an Alzheimer’s disease biomarker.
2.1. Genotype Data and Quality Control
The genotype data of the ADNI GO/2 cohort were collected by either the Illumina 2.5M array or the Illumina OmniQuad array [27]. The single nucleotide polymorphism (SNP) data we used were from both arrays for the present study.
A consistent quality control process described in Li et al. [28] was performed on these genotype data using the PLINK [29] (http://pngu.mgh.harvard.edu/purcell/plink/). SNPs were excluded from the GWA analysis if they could not satisfy any of the following condition: (1) no SNPs from chromosome 0 and chromosomes 22–25 (14,247 SNPs were excluded); (2) call rate per SNP ≥95%; (3) minor allele frequency ≥5% (1,845,510 SNPs were excluded based on criteria 2–3); and (4) Hardy-Weinberg equilibrium test of p ≥10−6 using control subjects only (198 SNPs were excluded). Participants were excluded from the analysis if any of the following condition was not met: (1) call rate per participant ≥90% (0 participants were excluded); (2) gender check (1 participant was excluded); and (3) identity check for related siblings (8 sibling pairs and 1 sibling triplet were identified with PI_HAT > 0.45, one participant was selected randomly from each family). Population stratification analysis was performed using complete linkage agglomerative clustering based on pairwise identity-by-state distance. We excluded 89 study participants who did not cluster with the CEU HapMap samples who are primarily of European ancestry (non-Hispanic Caucasians). After quality control, 1,079 participants each with 563,980 SNPs remained available for the subsequent analysis.
2.2. Subjects and Quantitative Traits
We focused this study on an AV-45 amyloid PET measure called SUMMARYSUVR_WHOLECEREBNORM for these ADNI-GO/2 participants, which is a florbetapir composite standardized uptake value ratio (SUVR) created by averaging across the 4 cortical regions (frontal regions, anterior/posterior cingulate regions, lateral parietal regions, and lateral temporal regions) and dividing this average by the reference region whole cerebellum [30].
For the present work, we concentrated our analyses on subjects with both genotype data and AV-45 PET baseline data available. This sample G0 (N0 = 774) included five diagnostic groups: 155 cognitively normal, 72 significant memory concern, 278 early mild cognitive impairment, 144 late mild cognitive impairment, and 125 Alzheimer’s disease subjects. To facilitate validation of the findings, we randomly split the 774 participants into two groups: G1 (N1=387) and G2 (N2=387), while maintaining the same distributions of gender and diagnosis in each group.
2.3. Integration of Genome-wide Association Analysis with Human Protein-Protein Interaction Network
SNP-trait association was evaluated by PLINK [29], using an additive genetic model with age, gender and diagnosis as covariates. SNPs from GWA analysis were further assigned to specific genes according to their genomic coordinates. A SNP was assigned to a gene if it was located within the gene using the hg19 genome build, which generated a total of 18,614 genes. Then, the most significant SNP’s P-value on a gene was used as its gene-wise P-value. Finally, after removing isolated nodes and self-interaction edges, a node-weighted PPIN with 11,960 nodes and 163,307 edges was constructed by mapping the gene-wise P-values on the PPIN. The comprehensive human PPIN was downloaded from the Protein Interaction Network Analysis platform [31] (http://cbg.garvan.unsw.edu.au/pina/).
2.4. Dense Module Search and Assessment
A DMS algorithm [26] was introduced to our analysis on the discovery set G1 to search for dense modules by the node-weighted PPIN. Briefly, a seed module only had a seed gene at the beginning and started to grew by adding the neighbor nodes with an increment of . is a module score computed for the current module. Module growth is stopped until there is no increment greater than 10% of the current module score. Specifically, , where k is the number of genes for the current module. And was computed by , where is the inverse standard normal cumulative distribution function.
To determine how likely the identified modules could be found by chance, a background distribution of module scores were created by computing through selecting the same number of genes randomly from the entire network for 100,000 times. And then the module scores were standardized by , where SD is standard deviation. Normalized module scores were transformed to P-values by , where is the standard normal cumulative density function. This analysis calculates test statistics for the modules defined in the discovery dataset G1 and then for the same modules in the replication dataset G2.
To further evaluate whether the enriched modules were significantly associated with the amyloid imaging phenotype, we performed 1,000 permutation tests of the validation dataset G2 by randomly shuffling the phenotypes and genotypes. For each permutation data set, we calculated of the same modules searched in the real data set G2. Thus 1,000 instances of were acquired to get the empirical normal distribution with mean and standard deviation . Unlike the original DMS algorithm, the empirical P-values for the association of the modules with the amyloid imaging phenotype was estimated by , where F is the normal cumulative distribution function. The final candidate modules were those modules passed the tests: (1) in both G1 and G2 dataset; and (2) after Bonferroni correction. A final subnetwork was then constructed for further functional analysis by merging candidate modules.
2.5. Module Similarity Tests and Functional Enrichment Analysis
To understand the topological property of the final identified subnetwork, genes in the subnetwork were ranked by some centrality measures, including degree centrality, betweenness centrality, closeness centrality, and page-rank centrality.
A biological process is a cooperation of a set of genes. If two or more modules consist of similar sets of genes, those modules might have a similar biological function or be related in the biological process somehow. We adopted Jaccard similarity coefficient [32] (JSC) to quantitatively measure the similarity between two different modules. , where represents the number of shared genes between module A and module B, represents the number of total genes in module A and module B. All pairs of significant modules were scanned.
We performed KEGG [33] pathway enrichment analysis for each of the identified significant modules by using the web-based tool Enrichr [34] (http://amp.pharm.mssm.edu/Enrichr). The enrichment significance for each KEGG term was computed by ‘fuzzy’ enrichment analysis and only terms with an adjusted P-value of less than 0.01 in at least one module were retained.
3. RESULTS
3.1. Significant Signals Identified by Genome-wide and Gene-based Association Analysis
GWA analysis detected some significant SNPs associated with the amyloid imaging phenotype. As we can see from Fig. 2, four SNPs (rs4420638, rs769449, rs2075650, rs157582) in G1 and one SNP (rs4420638) in G2 reached our genome-wide significance (P<8.87×10−8) after Bonferroni correction. In particular, rs4420638 was replicated in these two independent datasets. After GWA analysis, we conducted the gene-based association analysis for G1 and G2 separately using gene’s smallest P-value. Overall, 3 genes (APOE, TOMM40, PVRL2) and 2 genes (APOE, TOMM40) were identified to reach the significance level (P<2.69×10−6) after Bonferroni correction in G1 and G2 respectively.
Figure 2. Manhattan plot of the genome-wide association analysis (GWA) of AV-45 PET measure.
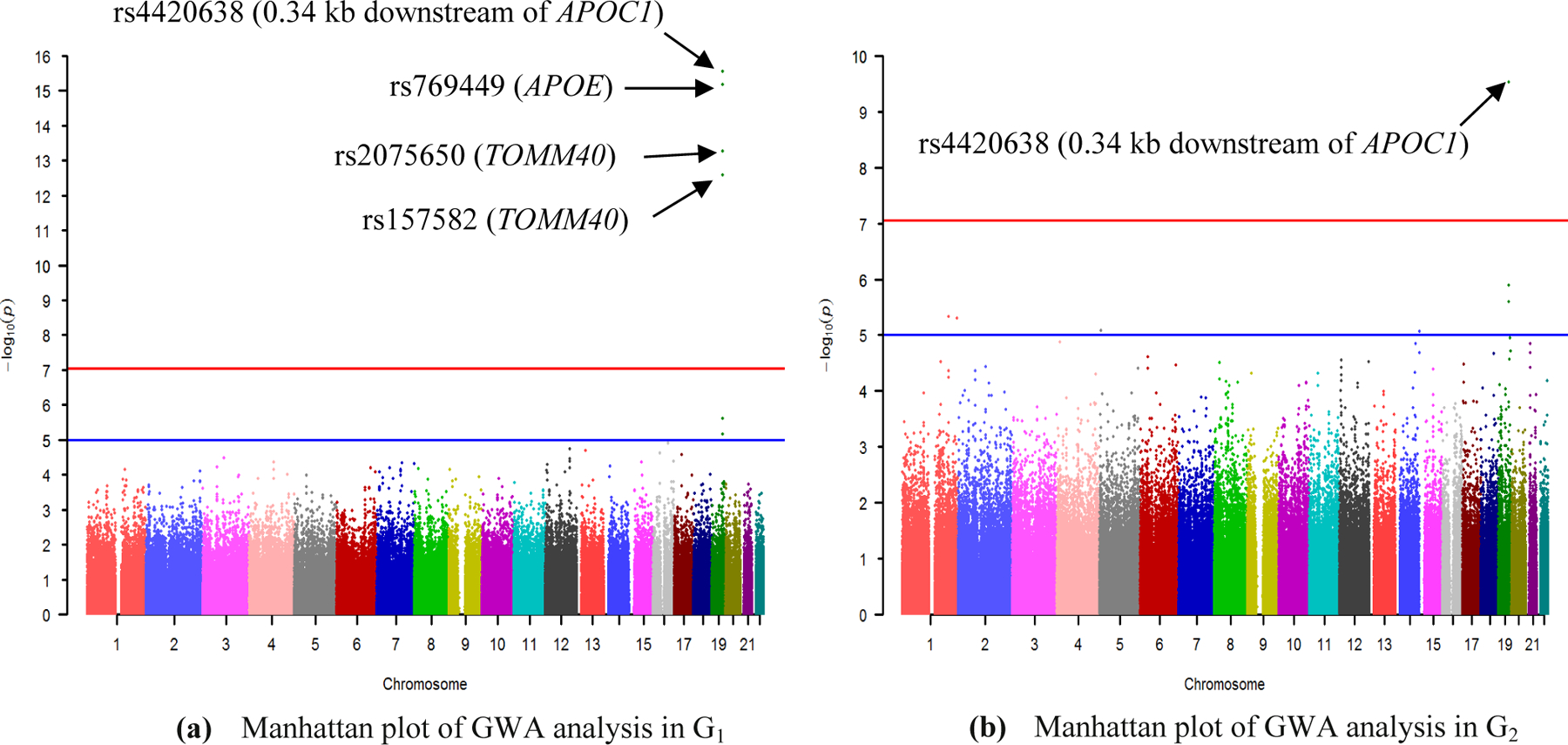
(a) Manhattan plot of GWA in G1; (b) Manhattan plot of GWA in G2. 563,980 SNPs were tested for association with Aβ burden of participants from group G1 and G2 separately under an additive model, with age, gender, and diagnosis as covariates. The blue line indicates the significance level P = 10−5 and the red line is the genome-wide significance level P = 8.87×10−8 (0.05/563,980) after Bonferroni correction in our analysis.
3.2. Modules Associated with Amyloid Imaging Phenotype
By performing DMS with each gene as a seed in the node-weighted PPIN, a total of 11,782 modules were initially obtained for the discovery dataset G1 after removing the replicated modules. For each module in G1, we evaluated whether the module contains a proportion of significant genes rather than randomly expected and whether the module can be found in another independent dataset G2. Of the 11,782 modules identified in G1, 8,630 ones were significant; and 94 of those were also significant in G2 after Bonferroni correction ( in both G1 and G2). Then, a permutation test (N =1000) was conducted to evaluate whether the enriched modules were highly related to the amyloid imaging phenotype. 24 modules with 156 non-redundant genes (shown in Table 1) were enriched for phenotype-associated signals after Bonferroni correction (. Therefore, these results provided an independent replication for our module identification results and showed significant enrichment with association signals to the amyloid imaging phenotype.
Table 1.
24 significant modules enriched for amyloid imaging phenotype-associated signals.
| No. | #G | Pz1 | Pz2 | P | Start Node | Module Genes |
|---|---|---|---|---|---|---|
| 1 | 13 | 1.19E-13 | 2.61E-09 | 3.46E-08 | TAX1BP1 | TAX1BP1;ARHGAP1;CALCOCO2;CNTN2;GABRR1;IKBKE;PARK2; STARD13;APP;ATP5O;ITPR1;TOMM40;APOE |
| 2 | 13 | 2.89E-15 | 7.87E-09 | 3.78E-08 | HSDL2 | HSDL2;EEF2K;IKBKE;NRF1;PARK2;ABCA3;ABLIM2;ACTG1; ADCY9;DAGLB;DDAH1;TOMM40;APOE |
| 3 | 11 | 8.91E-11 | 3.57E-07 | 4.78E-08 | RNF141 | RNF141;SMURF1;TRIM24;ACAP2;ACTR1A;APP;ARHGAP15; CBX1;CDK14;U2AF1;APOE |
| 4 | 15 | 4.78E-12 | 3.38E-07 | 7.13E-08 | PSKH1 | PSKH1;CDC6;HSP90AA1;RDH13;SNTA1;AARS;ABCA1;ABCC4; ACTG1;ACTN1;APP;CDK14;FBXO18;TOMM40;APOE |
| 5 | 11 | 3.57E-10 | 1.32E-06 | 1.29E-07 | RET | RET;CASP3;CBL;CBLC;DOK5;DOK6;PTPRF;APP; CASP10;SREBF2;APOE |
| 6 | 12 | 2.97E-10 | 7.85E-07 | 1.92E-07 | DPPA4 | DPPA4;BTBD2;MAPK6;PLEKHF2;PTPRO;SH3GL2;SH3GL3; ACTG1;AIDA;AMPH;PVRL2;APOE |
| 7 | 16 | 8.36E-12 | 9.00E-07 | 3.55E-07 | CISD1 | CISD1;ATP6V1C1;NR4A2;NRF1;PARK2;PRKAB1;RNF2;ABCA3; ABCF1;ABLIM2;ACTG1;ADCY9;DAGLB;DDAH1;TOMM40;APOE |
| 8 | 14 | 3.36E-12 | 2.08E-08 | 3.80E-07 | PSME2 | PSME2;APP;BAG3;CFTR;EPHA2;IKBKE;MGMT;PARD3; PARK2;PSMA1;ABHD10;ANKS1B;APOE;TOMM40 |
| 9 | 14 | 5.53E-13 | 3.20E-07 | 5.37E-07 | KLHL6 | KLHL6;HSP90AA1;AARS;ABL1;ACTG1;ACTN1;AHR;AHSA2; ALK;APP;CDK14;FBXO18;TOMM40;APOE |
| 10 | 14 | 4.85E-13 | 2.31E-07 | 5.78E-07 | MYLK4 | MYLK4;HSP90AA1;AARS;ABL1;ACTG1;ACTN1;AHR;AHSA2; ALK;APP;CDK14;FBXO18;TOMM40;APOE |
| 11 | 16 | 1.13E-09 | 2.38E-07 | 6.31E-07 | SUCLG1 | SUCLG1;CHMP1B;ECT2;OXCT1;SEPT11;SEPT9;SUCLA2;SUCLG2; ABR;AGGF1;APP;CUX1;ELAVL1;MYO1D;TOMM40;APOE |
| 12 | 12 | 0 | 1.23E-07 | 6.75E-07 | TPM4 | TPM4;ARRB1;CDH2;DBN1;ILF3;PARK2;APP;ATP5O; CDH4;PVRL2;TOMM40;APOE |
| 13 | 11 | 6.53E-14 | 2.92E-06 | 7.37E-07 | AMY2B | AMY2B;FOS;PARK2;ACSL1;ACTA2;APP;ATF7;ATP5C1; ATP5O;TOMM40;APOE |
| 14 | 11 | 3.33E-16 | 5.63E-08 | 9.93E-07 | SLC11A2 | SLC11A2;NDFIP1;NEDD4;NEDD4L;PARK2;ANXA13;APP;ATP5O; GRIA1;TOMM40;APOE |
| 15 | 10 | 0 | 9.59E-08 | 1.10E-06 | PARK2 | PARK2;ACTA2;ANXA2;APP;ATP5C1;ATP5O;CUL1;ESRRG; TOMM40;APOE |
| 16 | 14 | 5.13E-13 | 2.89E-06 | 1.20E-06 | DMRTA1 | DMRTA1;HSP90AA1;ORC5;STAT5B;AARS;ACTG1;ACTN1;AHSA2; ALK;APP;CDK14;FBXO18;TOMM40;APOE |
| 17 | 13 | 2.29E-12 | 1.46E-06 | 1.27E-06 | HTRA2 | HTRA2;ALDH1A2;APC;APP;BIRC2;CDC40;MAPK3;PAG1; PARK2;ABHD10;ANKS1B;APOE;TOMM40 |
| 18 | 11 | 5.53E-13 | 3.12E-07 | 1.77E-06 | SLC22A4 | SLC22A4;PDZK1;SLC9A3R2;ABCA1;ABCC4;ADORA2B;APP; ATP2B2;SLCO3A1;TOMM40;APOE |
| 19 | 11 | 3.57E-14 | 1.86E-06 | 1.81E-06 | GPD2 | GPD2;ALDOA;PARK2;ACSL1;ACTA2;APP;ATP5C1;ATP5O; CUL1;TOMM40;APOE |
| 20 | 13 | 1.97E-13 | 1.48E-07 | 2.48E-06 | SCYL1 | SCYL1;CD93;CHD1L;APLF;ARHGAP15;CBX1;COL4A1;COL4A2; APP;PARK2;SMURF1;APOE;TOMM40 |
| 21 | 12 | 0 | 9.46E-07 | 2.58E-06 | TGM3 | TGM3;ATG12;PARK2;PRKAB1;USP53;WWOX;APP;ATP5C1; ATP5O;PVRL2;TOMM40;APOE |
| 22 | 16 | 8.86E-14 | 2.32E-06 | 2.64E-06 | ZNF787 | ZNF787;FMNL1;IL7R;NRF1;PTEN;RNF2;ABCA3;ABCF1; ABLIM2;ACTG1;ADCY9;ANK3;DDAH1;INO80C;TOMM40;APOE |
| 23 | 8 | 0 | 2.06E-06 | 2.69E-06 | ESRRB | ESRRB;NCOA3;PARK2;APP;ATP5C1;ATP5O;TOMM40;APOE |
| 24 | 16 | 6.51E-11 | 2.15E-06 | 2.84E-06 | STYK1 | STYK1;GSK3B;HSP90AA1;AARS;ABL1;ACACA;ACLY;ACTG1; ACTN1;AHSA2;ALK;APP;BZW2;FBXO18;TOMM40;APOE |
No.: the identifier of the module; #G: number of genes in the module; Pz1: P-value transferred from z-score of modules identified in G1; Pz2: P-value transferred from z-score of modules identified in G2; P: Peval, the empirical P-value of the module; Start Node: source node from which the module search started; Module Genes: genes enriched in the module.
Table 2 shows 23 genes appearing in at least three modules. Detail about all 156 genes are shown in Table S1. Significant genes (APOE, TOMM40, PVRL2) identified by gene-base analysis approach were also included in several significant modules, and APOE, TOMM40, APP enriched in more than 20 modules. In addition, links between these genes were overrepresented in several modules, such as ATP50-PARK2 (8 modules), ATPG1-APOE (9 modules), APOE-APP (20 modules), APOE-APP-PARK2-TOMM40 (13 modules).
Table 2. Genes appeared in more than three modules.
P-values smaller than 0.001 were in bold.
| Genes | Degree | PCT (%) | P-G1 | P-G2 | #M |
|---|---|---|---|---|---|
| APOE | 55 | 7.35 | 6.86E-16 | 1.28E-06 | 24 |
| TOMM40 | 41 | 10.54 | 5.39E-14 | 2.49E-06 | 21 |
| APP | 1649 | 0.01 | 5.27E-03 | 3.73E-02 | 20 |
| PARK2 | 314 | 0.33 | 3.66E-03 | 1.21E-03 | 13 |
| ACTG1 | 111 | 2.48 | 1.12E-02 | 1.90E-01 | 9 |
| ATP5O | 25 | 18.32 | 2.86E-03 | 4.25E-03 | 8 |
| AARS | 40 | 10.84 | 6.93E-02 | 3.31E-02 | 5 |
| ACTN1 | 95 | 3.23 | 1.19E-02 | 1.20E-01 | 5 |
| ATP5C1 | 44 | 9.77 | 5.34E-03 | 2.80E-01 | 5 |
| CDK14 | 18 | 24.82 | 1.35E-03 | 4.63E-03 | 5 |
| FBXO18 | 11 | 36.21 | 3.62E-04 | 9.14E-02 | 5 |
| HSP90AA1 | 630 | 0.06 | 2.92E-01 | 9.35E-02 | 5 |
| AHSA2 | 1 | 83.60 | 1.60E-02 | 2.28E-02 | 4 |
| ALK | 52 | 7.92 | 1.30E-02 | 9.71E-05 | 4 |
| ABCA3 | 2 | 73.34 | 2.74E-02 | 1.07E-01 | 3 |
| ABL1 | 287 | 0.42 | 1.24E-01 | 8.14E-02 | 3 |
| ABLIM2 | 4 | 59.37 | 1.80E-02 | 5.08E-03 | 3 |
| ACTA2 | 55 | 7.35 | 5.59E-02 | 2.43E-03 | 3 |
| ADCY9 | 1 | 83.60 | 3.32E-03 | 2.67E-03 | 3 |
| DDAH1 | 4 | 59.37 | 4.54E-04 | 4.40E-03 | 3 |
| IKBKE | 299 | 0.38 | 2.68E-02 | 4.44E-03 | 3 |
| NRF1 | 2053 | 0 | 9.09E-02 | 2.51E-02 | 3 |
| PVRL2 | 11 | 36.21 | 2.46E-06 | 1.92E-04 | 3 |
Degree: degree of the gene in PPIN; PCT: percentage, number of genes with degree larger than the gene divided by the number of all genes; P-G1: P-value of the gene from GWA analysis of G1 dataset; P-G2: P-value of the gene from GWA analysis of G2 dataset; #M: number of identified modules containing the gene.
3.3. Statistical and Functional Analysis of the Significantly Enriched Modules
Table 3 shows the top 10 % genes ranked according to their topological centralities in the subnetwork, four of which were also found in more than three identified modules. Please see Table S1 for the topological centralities of all 156 genes.
Table 3.
Top 10% genes in all four topological centralities.
| Gene | Betweenness | Closeness | PageRank | Degree |
|---|---|---|---|---|
| APP | 0.28 | 0.50 | 0.04 | 0.22 |
| HSP90AA1 | 0.23 | 0.47 | 0.04 | 0.19 |
| PARK2 | 0.19 | 0.49 | 0.03 | 0.18 |
| NRF1 | 0.12 | 0.41 | 0.03 | 0.15 |
| ELAVL1 | 0.10 | 0.42 | 0.02 | 0.14 |
| RNF2 | 0.05 | 0.40 | 0.02 | 0.10 |
Betweenness: betweenness centrality, denotes the number of short communication paths that a node participates in; Closeness: closeness centrality, corresponds to the average distance (the length of the shortest paths) between a given node and the rest of the network; PageRank: PageRank centrality, takes into account the quantity and quality of connections to a given node; Degree: degree centrality, counts the number of interactions with a given node.
A crosstalk analysis was performed for these 24 enriched modules. when there are no shared genes between module A and module B. JSC(A,B)=1 when module A and module B share exactly the same genes. Thus the higher the JSC is, the more similar the two modules are. As shown in Fig. 3, there are three large clusters (S1 = M9,10,16,24,4; S2 = M13,15,19,23,21; S3 = M2,7,22), each of which consists of at least a pair of modules with JSC more than 0.5.
Figure 3. Heat map of Jaccard similarity coefficients (JSC) of 24 significant modules.
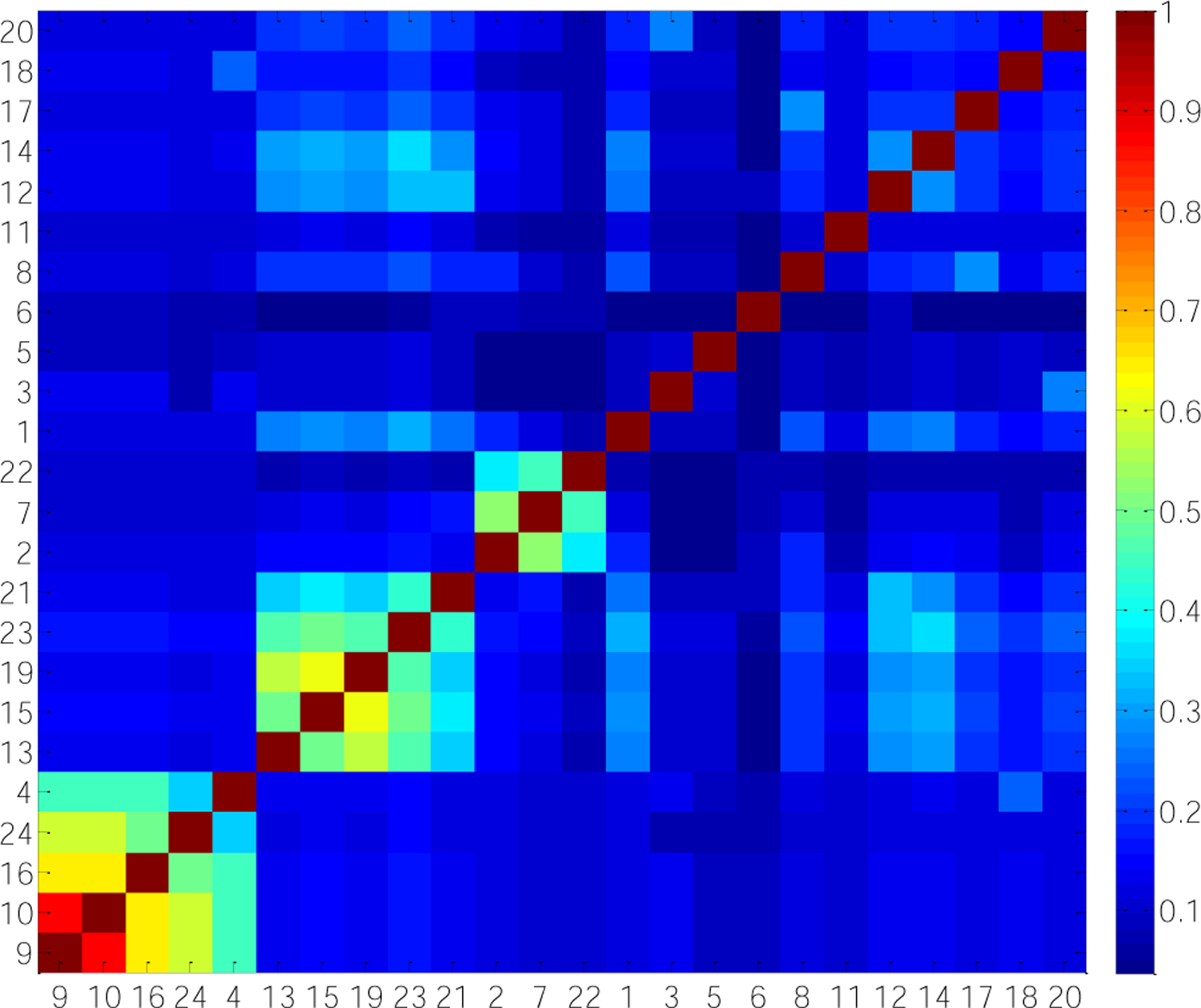
JSC ranges from 0 to 1. JSC(A,B)=0 when there are no shared genes between module A and module B. JSC(A,B)=1 when module A and module B share exactly the same genes. Thus the larger the JSC is, the stronger agreement the modules have. In order to make an intuitive understanding of JSC among 24 modules, the clusters were arranged according to hierarchical clustering algorithm performed in Matlab.
Fig. 4 shows the most significant module M1 and three subnetworks composed of those clustered modules separately. All significant modules included about 58.3% nominally significant (gene-wise P-value, 0.05) genes in G1 and 57.1% nominal significant genes in G2, while 122 genes (78.2%) reached the nominal significance in either dataset. These modules were highly significant in terms of not only nominal P-value but also topological structure measurements. 23.1% genes were with large degree — the degree of at least 95% genes in the network was smaller than that of these genes. Some non-significant genes were included in these modules, mainly because they were surrounded by low-P-value genes, such as TAX1BP1 in Fig. 4(a), HSP90AA1 in Fig. 4(b), NRF1 in Fig. 4(d). This further demonstrated the capability of the network-assisted strategy to identify collective effect of multiple genes, although they may not show significant association with the amyloid imaging phenotype on their own. The subnetwork with genes appearing in more than three modules were shown in Fig. 4(e).
Figure 4. Subnetworks composed of clustered modules with the Jaccard similarity coefficients larger than 0.5.
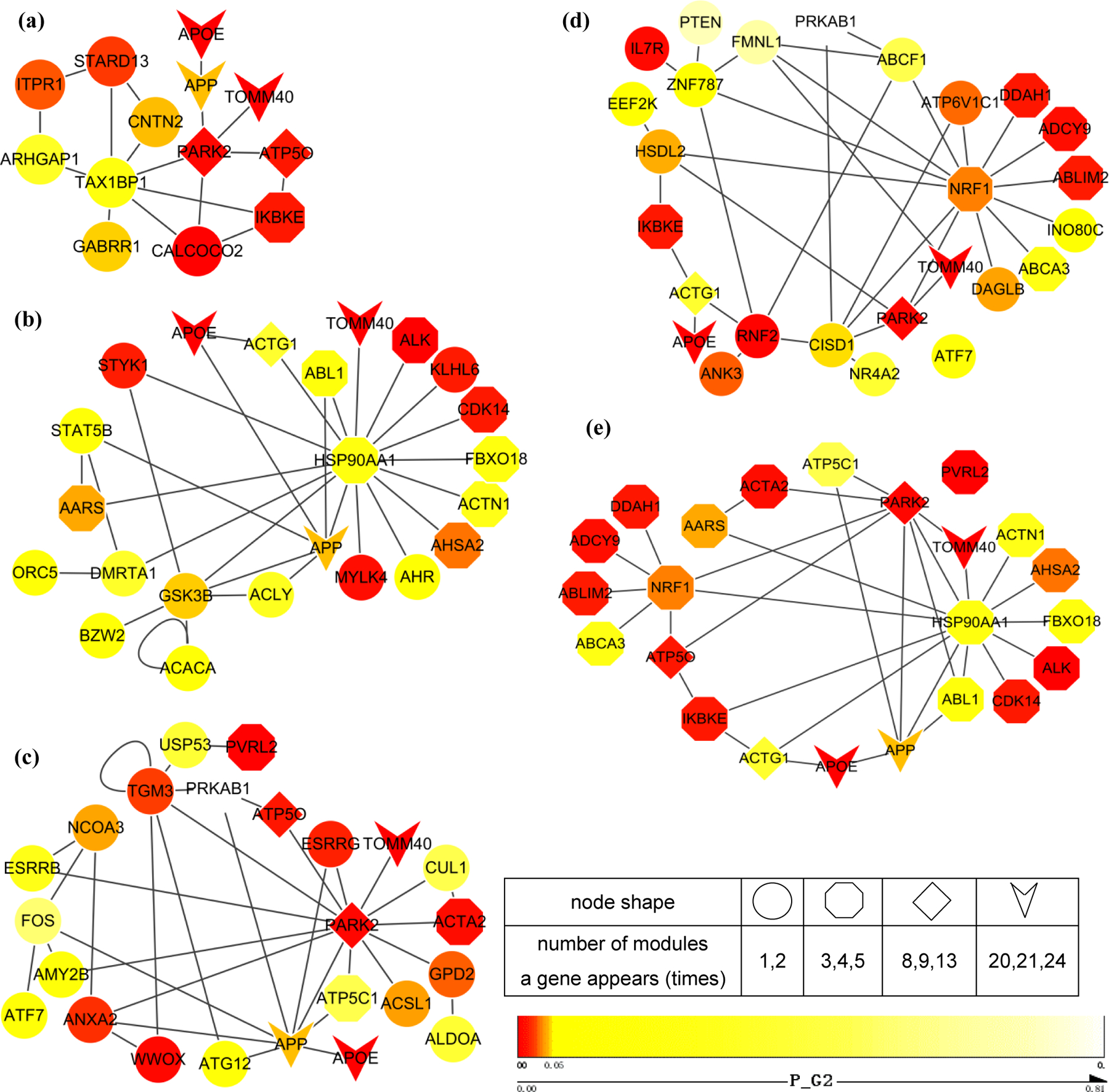
(a) The most significant module; (b) Subnetwork composed of M4,M9,M10,M16,M24; (c) Subnetwork composed of M13,M15,M19,M21,M23; (d) Subnetwork composed of M2,M7,M22; (e) Subnetwork with genes appearing in more than 3 modules. P-values from genome-wide association analysis of G2 are color-mapped. Node shape represents the number of modules a gene appears in.
Pathway enrichment analyses were conducted for the 24 significant modules respectively to explore their biological functions. 16 modules with at least one significantly (adjusted P-value < 0.01) enriched KEGG term were shown in Fig. 5. We observed the following. (1) Alzheimer’s disease pathway was enriched by 10 modules. (2) The most significant module M1 only enriched the Alzheimer’s disease pathway. (3) Six pathways with adjusted P-value < 0.001 were enriched by M1,7,11,13,15,19,21,23, including three metabolism-related pathways (Citrate cycle, Propanoate metabolism and Carbon metabolism), two neurodegenerative diseases (Parkinson’s disease and Alzheimer’s disease) and Vibrio cholerae infection pathway. (4) The metabolism-related pathways in (3) were enriched by M11. (5) The neurodegenerative diseases in (3) were commonly enriched by M13,15,19,21,23. This result is consistent with the crosstalk-analysis results, suggesting M13,15,19,21,23 may contribute to Parkinson’s disease and Alzheimer’s disease. (6) Besides two neurodegenerative diseases mentioned in (3), there are five other pathways enriched by two or more modules. They are Ubiquitin mediated proteolysis, Apoptosis, Bacterial invasion of epithelial cells, Oxytocin signaling pathway and Oxidative phosphorylation.
Figure 5. Heat map of pathway enrichment analysis at the significance level of P<0.01.
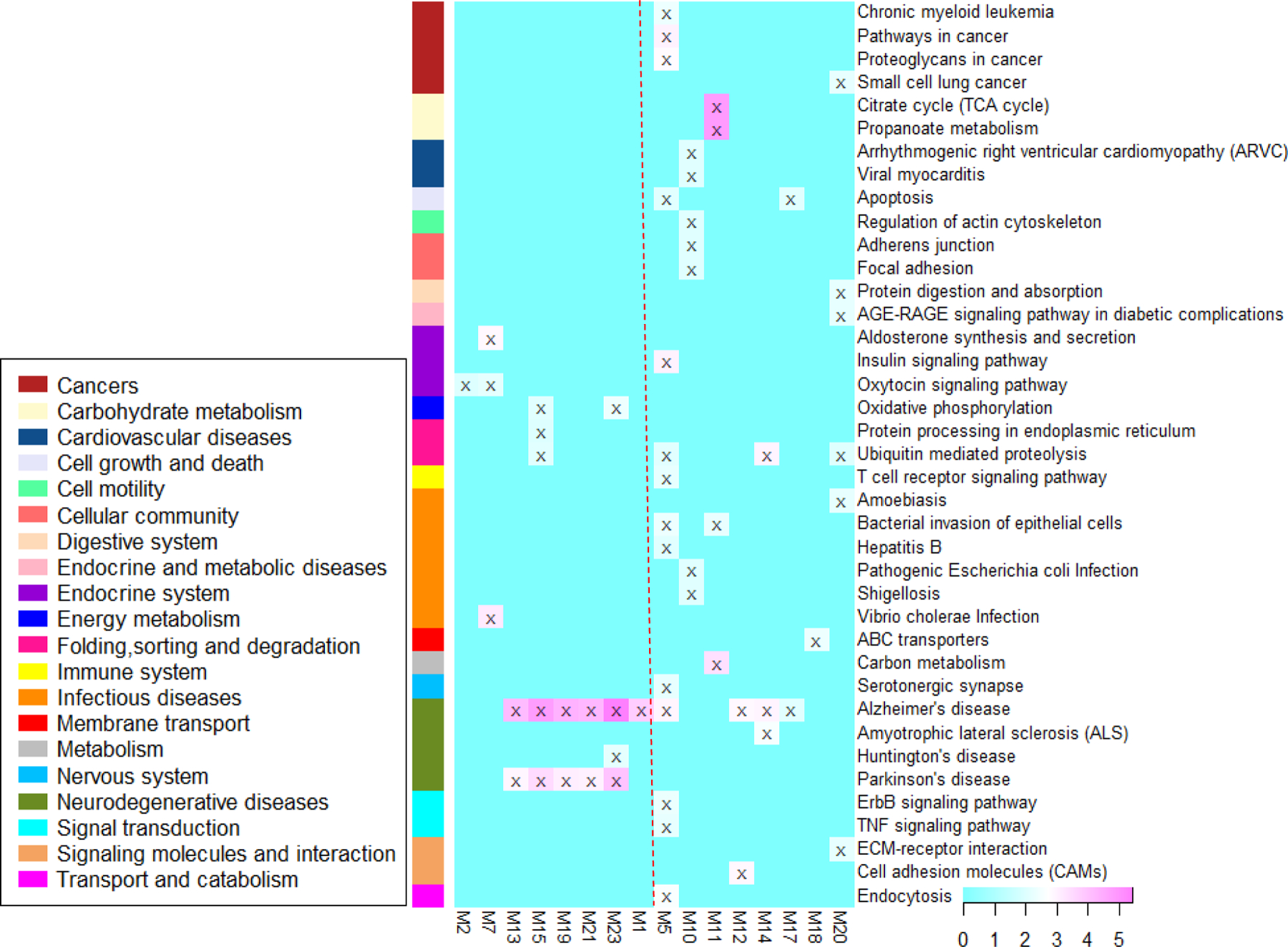
–log10(P-values) from each analysis are color-mapped in the heat map. Heat map blocks labeled with “x” reach the adjusted significance level of P<0.01. Each row (pathways) and column (modules) with at least one “x” block are included in the heat map. Modules on the left of the red dotted line are clustered modules. The remaining modules are on the right.
4. DISCUSSION
In the 24 identified modules, 23 genes appeared in more than three modules (Table 2). Six genes were among the top 10% genes ranked by their topological centralities (Table 3), four of which also appeared in Table 2 (PARK2, APP, NRF1, HSP90AA1). Among those 25 (23+6–4) genes, associations of several genes with Aβ were previously well-studied.
The amyloid plaques in the brain due to the imbalance between production and clearance of amyloid-β (Aβ) peptides is one major hallmark of amyloid pathology in AD brains [35]. It damages synapses and eventually leads to neurodegeneration and dementia [36]. As shown in Fig. 2, SNPs downstream of APOC1 and SNPs in APOE and TOMM40 genes displayed genome-wide significant associations with Aβ in GWA analysis. The recent cell type-specific and inducible mouse models support that APOE has the greatest influence on amyloid during the initial stage by affecting the clearance and aggregation of Aβ [37]. Genetic variants in the TOMM40 gene have been reported to greatly increased the precision in the estimation of late-onset AD age in APOE ε3 carriers [38]. Apolipoprotein C-I, the increased expression of which induced by the H2 allele of APOC1, co-localizes in plaques with Aβ and delays its aggregation in vitro [39]. Mutations in the APP gene could cause the excessive secretion of Aβ, which is a primary cause of early-onset familial AD [35,36]. The overexpression of PARK2 induces the clearance of Aβ by enhancing autophagy, increases tricarboxylic acid cycle activity, and improves mitochondrial function as well as a synaptic restoration function in a triple transgenic AD mice model [40,41]. ABL1 inhibition has an effect on Aβ deposits in the brain [42]. Overexpression of DDAH1 significantly decreases Aβ secretion in Aβ precursor protein cells, whereas knockdown of endogenous DDAH1 exacerbates cellular Aβ secretion [43].
Besides those gene related with Aβ, we have highlighted a panel of genes have already been implicated in the etiology of AD. ACTN1 has been identified as one of high ranking genes being associated with AD related gene expression changes in the hippocampus [44]. ATP5C1 has been identified to be susceptible to AD by the analysis of the differentially expressed genes in entorhinal cortex with AD [45]. Increased mRNA expressions and protein levels of NRF1 have been found in 6-month-old in double mutant APPXDrp1+/− mice relative to APP mice [46]. PVRL2 has exhibited significant association with AD in late onset AD family study data, through GWA studies using both logistic and Cox’s regression [47]. Amadio et al have found that neuronal ELAV proteins were decreased in postmortem patients along with AD progression, but the ELAVL1 levels did not change, thus indicating that only neuronal ELAV proteins are affected in AD pathology [48]. ABCA1 has been identified one of the hub genes associated with late-onset AD APOE4 non-carriers [49].
Among the genes appeared in more than three modules, there were also some novel genes that had not been studied in AD, yet have associations with other types of neurodegenerative diseases. A novel mutation in AARS has been reported causing a progressive, mild myeloneuropathy [50]. ABLIM2 has been found to take part in the neuron guidance processes in human, rat and mouse [51]. Other genes in those 25 genes require more evidences to demonstrate the role they played in biology process. In conclusion, those 25 genes could be promising novel candidate genes for AD risk.
In the 24 identified modules, five links between genes (ATP50-PARK2, ATPG1-APOE, APOE-APP-PARK2-TOMM40) were found for at least more than eight times, two of which (APOE-APP and PARK2-TOMM40) have been reported by in vivo and/or vitro experiments. It has been announced that Apolipoprotein E (ApoE) activates a non-canonical MAP-kinase cascade stimulating APP transcription and Aβ synthesis [52]. Mitochondrial clearance triggered by PARK2 are reversed by the overproduction of TOMM40 after mitochondrial transmembrane potential loss, and the molecular interaction between PARK2 and TOMM40 are weakened or disrupted by PARK2 mutations [53].
Pathway enrichment analyses were conducted for the 24 significant modules respectively to explore their biological function (Fig. 5). Please see Section 3.3 for detailed information of the observation. Briefly, several modules enriched in two significant neurodegenerative disease pathways (Alzheimer’s disease, Parkinson’s disease) and Ubiquitin mediated proteolysis pathway. Parkinson’s disease has been previously found to be associated with AD [40,41]. The dysfunction of ubiquitin proteasome system has been reported associated with the accumulation of hyper-phosphorylated tau oligomers at human AD synapses [54]. Another highly enriched pathway is carbohydrate metabolism-related Citrate cycle (TCA cycle) pathway. A lower conversion of glucose into TCA cycle-related metabolites is shown in the brain of 13-month-old triple transgenic mice (a mouse model of AD) [55]. Another two pathways are Apoptosis pathway and Oxidative phosphorylation pathway. It has been reported that genetic variation within oxidative phosphorylation genes have associations with AD clinical status and neuroimaging measures [56]. The increased levels of apoptosis proteins in amnestic MCI have been found between AD and normal controls [57]. In conclusion, the enrichment results may provide valuable information for the subsequent finding of functional similar signals associated with AD.
CONCLUSION
For the current study, we performed a genome-wide network-assisted association and enrichment study to explore the collective effects of multiple genetic association signals on an AV-45 PET measure. First, we took advantage of the DMS algorithm [26] to identify modules enriched by the genetic association signals in PPIN. Next, we employed statistical evaluations to the identified modules, including a normalization process to adjust the topological bias in network (e.g., nodes with high degree), a replication test to ensure the modules were not found by chance (i.e. high probability found in other AD-related datasets), and a permutation test to evaluate unbiased association between the modules and AV-45 PET measure. At last, 24 modules (156 genes in total) enriched by robust genetic signals in GWA analysis datasets were found. Several centrality measures were computed to rank those 156 genes, and module similarity tests and functional enrichment analysis were performed subsequently for those 24 modules. Those highly reproducible and biologically meaningful modules not only confirmed previously reported AD genes, but also highlight several new candidate genes, gene-gene functional relationship and molecular pathways underlying AD or other types of neurodegenerative diseases.
Our results derived from computational analysis in this study need to be validated by additional biological experiments more than literature survey. The identified genes and network modules may provide more insights into the neurobiology of AD and suggest potential therapeutic targets.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported in part by grants from National Natural Science Foundation of China (61773134, 61803117), Fundamental Research Funds for the Central Universities (3072019CFM0403, HEUCFG201824) and Natural Science Foundation of Heilongjiang Province of China (YQ2019F003) at Harbin Engineering University; and MOE (Ministry of Education in China) Project of Humanities and Social Sciences (Project No.19YJCZH120) at Changzhou Institute of Technology; and by NIH R01 EB022574, R01 LM011360, U01 AG024904, RC2 AG036535, R01 AG19771, and P30 AG10133 at Indiana University and University of Pennsylvania.
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12–2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson& Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Footnotes
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No Animals/Humans were used for the studies that are base of this research.
CONCENT FOR PUBLICATION
Not applicable.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- [1].As Association. 2019 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia 15: 321–87 (2019). [Google Scholar]
- [2].Patterson C World Alzheimer Report 2018 https://www.alz.co.uk/research/WorldAlzheimerReport2018.pdf (2018).
- [3].Wingo TS, Lah JJ, Levey AI,Cutler DJ. Autosomal Recessive Causes Likely in Early-Onset Alzheimer Disease. JAMA Neurology 69: 59–64 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Nussbaum RL. Genome-Wide Association Studies, Alzheimer Disease, and Understudied PopulationsGenome-Wide Association Studies in Alzheimer Disease. Jama 309: 1527–8 (2013). [DOI] [PubMed] [Google Scholar]
- [5].Naj AC, Jun G, Reitz C, Kunkle BW, Perry W, Park YS et al. Effects of Multiple Genetic Loci on Age at Onset in Late-Onset Alzheimer Disease: A Genome-Wide Association StudyLoci Modifying Age at Onset in Late-Onset ADLoci Modifying Age at Onset in Late-Onset AD. JAMA Neurology 71: 1394–404 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ramanan VK, Risacher SL, Nho K, Kim S, Shen L, McDonald BC et al. GWAS of longitudinal amyloid accumulation on 18F-florbetapir PET in Alzheimer’s disease implicates microglial activation gene IL1RAP. Brain : a journal of neurology 138: 3076–88 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Vardarajan BN, Ghani M, Kahn A, Sheikh S, Sato C, Barral S et al. Rare coding mutations identified by sequencing of Alzheimer disease genome-wide association studies loci. Annals of neurology 78: 487–98 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cuyvers E,Sleegers K. Genetic variations underlying Alzheimer’s disease: evidence from genome-wide association studies and beyond. The Lancet Neurology 15: 857–68 (2016). [DOI] [PubMed] [Google Scholar]
- [9].Deming Y, Li Z, Kapoor M, Harari O, Del-Aguila JL, Black K et al. Genome-wide association study identifies four novel loci associated with Alzheimer’s endophenotypes and disease modifiers. Acta Neuropathologica 133: 839–56 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Young AL, Scelsi MA, Marinescu RV, Schott JM, Ourselin S, Alexander DC et al. GENOMEWIDE ASSOCIATION STUDY OF DATA-DRIVEN ALZHEIMER’S DISEASE SUBTYPES. Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association 14: P1042–P3 (2018). [Google Scholar]
- [11].Chen X, Kuja-Halkola R, Rahman I, Arpegård J, Viktorin A, Karlsson R et al. Dominant Genetic Variation and Missing Heritability for Human Complex Traits: Insights from Twin versus Genome-wide Common SNP Models. The American Journal of Human Genetics 97: 708–14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 467: 832–8 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cho DY, Kim YA,Przytycka TM. Chapter 5: Network biology approach to complex diseases. PLoS computational biology 8: e1002820 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yan J, Risacher SL, Shen L,Saykin AJ. Network approaches to systems biology analysis of complex disease: integrative methods for multi-omics data. Briefings in Bioinformatics 19: 1370–81 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gosak M, Markovič R, Dolenšek J, Slak Rupnik M, Marhl M, Stožer A et al. Network science of biological systems at different scales: A review. Physics of Life Reviews 24: 118–35 (2018). [DOI] [PubMed] [Google Scholar]
- [16].Chuang HY, Lee E, Liu YT, Lee D,Ideker T. Network-based classification of breast cancer metastasis. Molecular systems biology 3: 140 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hu JX, Thomas CE,Brunak S. Network biology concepts in complex disease comorbidities. Nature Reviews Genetics 17: 615 (2016). [DOI] [PubMed] [Google Scholar]
- [18].International Multiple Sclerosis Genetics C. Network-based multiple sclerosis pathway analysis with GWAS data from 15,000 cases and 30,000 controls. American journal of human genetics 92: 854–65 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yu H, Bi W, Liu C, Zhao Y, Zhang JF, Zhang D et al. Protein-interaction-network-based analysis for genome-wide association analysis of schizophrenia in Han Chinese population. Journal of psychiatric research 50: 73–8 (2014). [DOI] [PubMed] [Google Scholar]
- [20].Chang S, Fang K, Zhang K,Wang J. Network-Based Analysis of Schizophrenia Genome-Wide Association Data to Detect the Joint Functional Association Signals. PloS one 10: e0133404 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kar SP, Tyrer JP, Li Q, Lawrenson K, Aben KKH, Anton-Culver H et al. Network-Based Integration of GWAS and Gene Expression Identifies a <em>HOX</em>-Centric Network Associated with Serous Ovarian Cancer Risk. Cancer Epidemiology Biomarkers & Prevention 24: 1574 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Heiland DH, Mader I, Schlosser P, Pfeifer D, Carro MS, Lange T et al. Integrative Network-based Analysis of Magnetic Resonance Spectroscopy and Genome Wide Expression in Glioblastoma multiforme. Scientific Reports 6: 29052 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yao X, Yan J, Liu K, Kim S, Nho K, Risacher SL et al. Tissue-specific network-based genome wide study of amygdala imaging phenotypes to identify functional interaction modules. Bioinformatics 33: 3250–7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lee T,Lee I. araGWAB: Network-based boosting of genome-wide association studies in Arabidopsis thaliana. Scientific Reports 8: 2925 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sun YV. Integration of biological networks and pathways with genetic association studies. Human genetics 131: 1677–86 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Jia P, Zheng S, Long J, Zheng W,Zhao Z. dmGWAS: dense module searching for genome-wide association studies in protein-protein interaction networks. Bioinformatics 27: 95–102 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Shen L, Thompson PM, Potkin SG, Bertram L, Farrer LA, Foroud TM et al. Genetic analysis of quantitative phenotypes in AD and MCI: imaging, cognition and biomarkers. Brain imaging and behavior 8: 183–207 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Li J, Zhang Q, Chen F, Yan J, Kim S, Wang L et al. Genetic Interactions Explain Variance in Cingulate Amyloid Burden: An AV-45 PET Genome-Wide Association and Interaction Study in the ADNI Cohort. BioMed research international 2015: 647389 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. American journal of human genetics 81: 559–75 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Initiative AsDN. Florbetapir processing methods https://adni.bitbucket.io/reference/docs/UCBERKELEYAV45/ADNI_AV45_Methods_JagustLab_06.25.15.pdf (2012).
- [31].Cowley MJ, Pinese M, Kassahn KS, Waddell N, Pearson JV, Grimmond SM et al. PINA v2.0: mining interactome modules. Nucleic acids research 40: D862–D5 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Levandowsky M,Winter D. Distance between Sets. Nature 234: 34–5 (1971). [Google Scholar]
- [33].Kanehisa M, Sato Y, Furumichi M, Morishima K,Tanabe M. New approach for understanding genome variations in KEGG. Nucleic acids research 47: D590–D5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic acids research 44: W90–7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Blennow K, de Leon MJ,Zetterberg H. Alzheimer’s disease. Lancet 368: 387–403 (2006). [DOI] [PubMed] [Google Scholar]
- [36].Hardy J,Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297: 353–6 (2002). [DOI] [PubMed] [Google Scholar]
- [37].Liu CC, Zhao N, Fu Y, Wang N, Linares C, Tsai CW et al. ApoE4 Accelerates Early Seeding of Amyloid Pathology. Neuron 96: 1024–32e3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Roses AD, Lutz MW, Amrine-Madsen H, Saunders AM, Crenshaw DG, Sundseth SS et al. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. The pharmacogenomics journal 10: 375–84 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Abildayeva K, Berbée JF, Blokland A, Jansen PJ, Hoek FJ, Meijer O et al. Human apolipoprotein CI expression in mice impairs learning and memory functions. Journal of lipid research 49: 856–69 (2008). [DOI] [PubMed] [Google Scholar]
- [40].Khandelwal PJ, Herman AM, Hoe HS, Rebeck GW,Moussa CE. Parkin mediates beclin-dependent autophagic clearance of defective mitochondria and ubiquitinated Abeta in AD models. Human molecular genetics 20: 2091–102 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lonskaya I, Shekoyan AR, Hebron ML, Desforges N, Algarzae NK,Moussa CE. Diminished parkin solubility and co-localization with intraneuronal amyloid-β are associated with autophagic defects in Alzheimer’s disease. Journal of Alzheimer’s disease : JAD 33: 231–47 (2013). [DOI] [PubMed] [Google Scholar]
- [42].Estrada LD, Chamorro D, Yañez MJ, Gonzalez M, Leal N, von Bernhardi R et al. Reduction of Blood Amyloid-β Oligomers in Alzheimer’s Disease Transgenic Mice by c-Abl Kinase Inhibition. Journal of Alzheimer’s Disease 54: 1193–205 (2016). [DOI] [PubMed] [Google Scholar]
- [43].Luo Y, Yue W, Quan X, Wang Y, Zhao B,Lu Z. Asymmetric dimethylarginine exacerbates Abeta-induced toxicity and oxidative stress in human cell and Caenorhabditis elegans models of Alzheimer disease. Free radical biology & medicine 79: 117–26 (2015). [DOI] [PubMed] [Google Scholar]
- [44].Silver M, Janousova E, Hua X, Thompson PM, Montana G,Neuroimaging AsD. Identification of gene pathways implicated in Alzheimer’s disease using longitudinal imaging phenotypes with sparse regression. NeuroImage 63: 1681–94 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ding B, Xi Y, Gao M, Li Z, Xu C, Fan S et al. Gene expression profiles of entorhinal cortex in Alzheimer’s disease. American journal of Alzheimer’s disease and other dementias 29: 526–32 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Manczak M, Kandimalla R, Fry D, Sesaki H,Reddy PH. Protective effects of reduced dynamin-related protein 1 against amyloid beta-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Human molecular genetics 25: 5148–66 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Yashin AI, Fang F, Kovtun M, Wu D, Duan M, Arbeev K et al. Hidden heterogeneity in Alzheimer’s disease: Insights from genetic association studies and other analyses. Exp Gerontol 107: 148–60 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Amadio M, Pascale A, Wang J, Ho L, Quattrone A, Gandy S et al. nELAV proteins alteration in Alzheimer’s disease brain: a novel putative target for amyloid-beta reverberating on AbetaPP processing. Journal of Alzheimer’s disease : JAD 16: 409–19 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Jiang S, Zhang CY, Tang L, Zhao LX, Chen HZ,Qiu Y. Integrated Genomic Analysis Revealed Associated Genes for Alzheimer’s Disease in APOE4 Non-Carriers. Curr Alzheimer Res 16: 753–63 (2019). [DOI] [PubMed] [Google Scholar]
- [50].Motley WW, Griffin LB, Mademan I, Baets J, De Vriendt E, De Jonghe P et al. A novel AARS mutation in a family with dominant myeloneuropathy. Neurology 84: 2040–7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Klimov E, Rud’ko O, Rakhmanaliev E,Sulimova G. Genomic organisation and tissue specific expression of ABLIM2 gene in human, mouse and rat. Biochimica et biophysica acta 1730: 1–9 (2005). [DOI] [PubMed] [Google Scholar]
- [52].Huang Y-WA, Zhou B, Wernig M,Südhof TC. ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Aβ Secretion. Cell 168: 427–41.e21 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Bertolin G, Ferrando-Miguel R, Jacoupy M, Traver S, Grenier K, Greene AW et al. The TOMM machinery is a molecular switch in PINK1 and PARK2/PARKIN-dependent mitochondrial clearance. Autophagy 9: 1801–17 (2013). [DOI] [PubMed] [Google Scholar]
- [54].Tai H-C, Serrano-Pozo A, Hashimoto T, Frosch MP, Spires-Jones TL,Hyman BT. The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am J Pathol 181: 1426–35 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Sancheti H, Kanamori K, Patil I, Díaz Brinton R, Ross BD,Cadenas E. Reversal of metabolic deficits by lipoic acid in a triple transgenic mouse model of Alzheimer’s disease: a 13C NMR study. J Cereb Blood Flow Metab 34: 288–96 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Biffi A, Sabuncu MR, Desikan RS, Schmansky N, Salat DH, Rosand J et al. Genetic variation of oxidative phosphorylation genes in stroke and Alzheimer’s disease. Neurobiology of aging 35: 1956.e1-.e19568 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zhao S, Zhao J, Zhang T,Guo C. Increased apoptosis in the platelets of patients with Alzheimer’s disease and amnestic mild cognitive impairment. Clinical neurology and neurosurgery 143: 46–50 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.