

Abstract
Earlier studies of the initiating event of normal automaticity of the heart’s pacemaker cells, inspired by classical quantitative membrane theory, focused upon ion currents (IK, If) that determine the maximum diastolic potential and the early phase of the spontaneous diastolic depolarization (DD). These early DD events are caused by the prior action potential (AP) and essentially reflect a membrane recovery process. Events following the recovery process that ignite APs have not been recognized and remained a mystery until recently. These critical events are linked to rhythmic intracellular signals initiated by Ca2+ clock (i.e., sarcoplasmic reticulum [SR] cycling Ca2+). Sinoa-trial cells, regardless of size, exhibit intense ryanodine receptor (RyR), Na+/Ca2+ exchange (NCX)-1, and SR Ca2+ ATPase-2 immunolabeling and dense submembrane NCX/RyR colocalization; Ca2+ clocks generate spontaneous stochastic but roughly periodic local subsarcolemmal Ca2+ releases (LCR). LCRs generate inward currents via NCX that exponentially accelerate the late DD. The timing and amplitude of LCR/INCX-coupled events control the timing and amplitude of the nonlinear terminal DD and therefore ultimately control the chronotropic state by determining the timing of the ICaL activation that initiates the next AP. LCR period is precisely controlled by the kinetics of SR Ca2+ cycling, which, in turn, are regulated by 1) the status of protein kinase A-dependent phosphorylation of SR Ca2+ cycling proteins; and 2) membrane ion channels ensuring the Ca2+ homeostasis and therefore the Ca2+ available to Ca2+ clock. Thus, the link between early DD and next AP, missed in earlier studies, is ensured by a precisely physiologically regulated Ca2+ clock within pacemaker cells that integrates multiple Ca2+-dependent functions and rhythmically ignites APs during late DD via LCRs-INCX coupling.
Keywords: cardiac pacemaker, calcium, ryanodine receptor, Na–Ca exchanger, sarcoplasmic reticulum
Introduction
Sinoatrial nodal cell (SANC) robustness (“fail-safe” properties conserved during evolution of the animal kingdom) and flexibility (the ability to react to demands for faster or slower firing rate) require multiple interactions among intrinsic cell mechanisms. This interactive network of mechanisms intrinsic to nodal cells must also interpret and react to signals arising extrinsic to the cell (e.g., stretch or neurotransmitter or hormonal stimulation of surface membrane receptors).
During the past five decades, pacemaker theory, formulated on the basis of an ensemble of multiple voltage and time-dependent rhythms of membrane ion channels (membrane clock, Fig. 1A), has produced numerous numerical models of SANC function that are able to reproduce recurring action potentials (APs; i.e., spontaneous firing). Such membrane-delimited models, however, turned out to be fundamentally incomplete and, hence, incorrect, as they all ignored critical intracellular rhythmic signals that ensure both robustness and flexibility to SANC pacemaker function.
FIGURE 1.

Schematic representation of an ion channel membrane clock (A) and Ca2+ clock (B) in cardiac pacemaker cells. Panel C illustrates the idea that the Ca2+ clock ignites the membrane clock to entrain normal automaticity in cardiac pacemaker cells (see text for details). (In color in Annals online.)
Recent studies in SANC suggest that the missing signal comes from a rhythmic intracellular Ca2+ oscillator or Ca2+ clock (Fig. 1B) that “crosstalks” to multiple other mechanisms that govern membrane potential and, in this way, coordinates their interaction to regulate the SANC firing rate. This Ca2+ clock dispenses localized, critically timed, submembrane pulses of Ca2+ during the SANC cycle. The timing (phase) of these rhythmic Ca2+ releases and their size determine their impact on the SANC firing rate and pattern (Fig. 1C). Rhythmic APs, in turn, sustain the Ca2+ clock by maintaining cell Ca2+ homeostasis via ion fluxes of membrane ion channels. Novel numerical models demonstrate that mutual entrainment of membrane and Ca2+ clocks confers both stability and flexibility to the SANC pacemaker function. This review chronicles the evolution of thought that has led to this concept.
The Membrane-delimited Ion Channel Clock Concept
Historical Perspectives: Development of Theory and Mechanistic Studies of Cell Membrane Excitation
A major function of the heart’s pacemaker cells is to generate spontaneous APs. In 1868, Bernstein plotted a galvanometer response to nerve excitation that was, in fact, the first record of an “action potential.”1 By 1912, based on the work of Ostwald and Nernst, Bernstein developed and experimentally proved the membrane theory of bioelectric potentials,2 which linked the bioelectric potential (Vm) of nerve and muscle to ion gradients across their cell membrane. At the same time, he suggested that a mechanism of membrane excitability involved a sudden reduction of the K+ permeability of the membrane, which reduced Vm to zero.
In 1949, testing the effect of Na+ removal on Vm in large diameter squid neurons, Hodgkin and Katz3 concluded (contrary to the original Bernstein K+-driven excitability theory) that Na+ was responsible for the AP upstroke and positive membrane polarization. In the same year, Ling and Gerard invented the glass microelectrode that allowed recording activity from individual cells, and Kenneth Cole suggested placing a second electrode inside the cell in order to inject the current and to “voltage clamp” the interior of the cell. In 1952, Hodgkin and Huxley applied the voltage clamp approach to nerve cells of a giant squid and established current–voltage relationships for Na+ and K+ currents.4 Based on these data, they formulated a new quantitative theory of membrane excitation (i.e., of the AP generation). According to their theory, the movement of selective ions through the excitable membrane is determined by separate voltage-dependent “gates” with a simple kinetics described by differential equations.
Using the new microelectrode technique, Silvio Weidmann5 confirmed the application of ionic theory to the cardiac muscle and explained the differences between cardiac APs and those of nerve and skeletal muscle. His findings include the genesis of the resting membrane potential, the increased permeability to Na+ during membrane depolarization, high membrane resistance during the AP plateau, and the definition of threshold potential.
The Slow Diastolic Depolarization Concept
The unique feature of the APs of pacemaker cells is that they arise spontaneously because of events that occur prior to the rapid AP upstroke. A slow potential change preceding the discharge of cardiac impulses was first observed by Arvanitaki, who was working on the snail heart in 1937.6 Later, in 1943, Bozler used monophasic potential recording and found a similar phenomenon in sinus venosus of turtle.7 He called the slow depolarizing potential change a “prepotential” and suggested that it was “the basis underlying automaticity” including “normal rhythmicity.” Bozler worked with rhythmically active strips of cardiac muscle (turtles, rabbits, cats, and dogs) and sinus venosus (turtle, high K+)7 and noted oscillatory monophasic subthreshold potentials of relatively (compared to nerve) low frequency of approximately 1 Hz, which were enhanced by increasing bathing [Ca2+] (Cao) or by adding adrenaline. Discharging impulses on the top of oscillatory potentials were accompanied by synchronized variations in isometric force (tonus).8 Further, an important idea postulated at that time was that the observed oscillatory phenomena might be somehow linked to cardiac automaticity.8 Specifically it was postulated that:
the tonus changes and the local potentials are probably manifestations of a more fundamental process, a fluctuation in resting metabolism. … Their chief interest lies in their relation to the automaticity and rhythmicity of the muscle. It may be assumed that an increase in metabolism causes a rise in tonus and a decreased surface polarization. The decrease in polarization in turn may be considered as the last link in the chain of processes leading to the discharge of an impulse.
The prepotential (i.e., diastolic depolarization [DD]) was later observed with microelectrode techniques by Brady and Hecht9 in turtle hearts and by Coraboeuf and Weidmann10 in spontaneously active Purkinje tissue. Spontaneous APs from the sinoatrial node were first recorded using microelectrodes by West in 1955.11 Microelectrode studies of sinoatrial node identified three important factors of pacemaker potential: the DD slope, the maximum diastolic potential, and the threshold for AP upstroke.
First Models of the Membrane-delimited Pacemaker Clock
Noble modified the Hodgkin–Huxley model to simulate both muscle (triggered) and pacemaker (spontaneous) cardiac APs.12,13 AP shapes, known from microelectrode studies, were reproduced in the model by an interplay of hypothesized voltage-gated and time-dependent Na+ and K+ currents. Although specific cardiac ion currents had not yet been measured at that time, there were some clues from earlier microelectrode findings of a membrane conductance decrease during DD in pacing Purkinje fibers (first reported by Weidman14 in 1951) and then in beating sinus preparation of the rabbit’s heart (by Dudel and Trautwein15 in 1958). The membrane response with various bathing [K+] and [Na+] (Ko and Nao) in the latter study suggested that the spontaneous depolarization was a result of the decay of a specific K+ conductance (gK). The first Purkinje fiber models12,13 described the AP as a rapid membrane depolarization produced by a voltage-gated Na+ current (INa) followed by a repolarization from a delayed increase in gK. Generation of spontaneous APs in these models was thus ascribed to a slow decay of the gK (termed gK decay mechanism) so allowing any background permeability (e.g., to Na+ ions) to drive Vm away from the K+ equilibrium potential. The 1962 cardiac modification of the Hodgkin–Huxley model represented a mechanistic model based upon solid principles of thermodynamics of Bernstein’s membrane theory and the Nernst equation. The formulation of the 1962 model was a case of “theory leading experiment.”16 The membrane-delimited pacemaker mechanism, predicted by the theory, became the subject of an extensive experimental search driven by the successful application of the voltage clamp technique to various cardiac preparations.17–20 Many ion currents were discovered in cardiac pacemaker cells, and a variety of pacemaker mechanisms have been further suggested based on the characteristics of these currents.16,21,22
The Pacemaker Current
The Quest to Discover the Most Important Current that Governs Pacemaker Function
Purkinje Cells
The search for the surface membrane cardiac pacemaker mechanism was initially and mainly studied experimentally and theoretically (from about 1950 to about 1980) in Purkinje fibers and not in true pacemaker cells (i.e., SANC). A slow inward current carried by Ca2+ (Is, Isi, or ICa), activated by membrane depolarization, and increased by adrenaline was discovered in Purkinje fibers by Reuter.23 A “pure K+ selective” IK2 current (later redefined as If), a “funny,” nonselective, monovalent cation current activated by membrane hyperpolarization,24 was and continues to be acclaimed by some as the true “pacemaker current.” But the fundamental pacemaker concept formulated in the 1962 model13 and then revised in the McAllister–Noble–Tsien (MNT) model of 1975,25 based upon Purkinje fiber voltage clamp studies, did not work for all pacemakers (see review26).
Sinoatrial Nodal Cells
Following the invention of the patch clamp method in 1976 by Neher and Sakmann27 and successful isolation of single SANC by Taniguchi et al. in 1981,20 further extensive voltage clamp studies have identified abundant ion current components in SANC, and new components are still being discovered. Many previously identified currents are split into subcomponents; others are species dependent. For example, studies of single cardiac cells showed that the “slow” inward current Isi is approximately 10 times faster than previously reported in multicellular preparations (2–5 ms versus 20–100 ms) and consists of three components: T- and L-type Ca2+ currents (ICaT, ICaL) and Na+/Ca2+ exchange (NCX) current (INCX) (all three in current terminology). ICaL is responsible for AP upstroke in SANC. In most species, SANC had neither a fast Na+ current nor an inward rectifying K+ current (IK1) (see review28 for species dependency). The latter keeps the resting potential low in Purkinje fibers but not in SANC. While SANC of many species did exhibit the funny current, it plays only a minor role in pacemaker mechanism of SANC as its activation voltage is relatively low compared to maximum diastolic potential inthe cells.16,29 Furthermore, If has relatively slow (compared to DD duration in SANC) activation kinetics. It is not surprising that the block of If by Cs+ had only a minor effect on the pacemaker rhythm.30 It was also found that bullfrog sinus venosus cells lack If31. The delayed rectifier K+ current (IK) is split into slow and rapid components (IKs and IKr), with IKs being present in porcine SANC, but IKr in rat and rabbit.28 ICaT was absent in porcine SANC.32 Other currents were observed only in subpopulations of SANC (INa and Cl− current, ICl)33 or had no obvious molecular identity (the sustained current, Ist).34 As a result, net membrane current is currently viewed as being spread among numerous components and subcomponents,22 and the fundamental (and thus rather simple) pacemaker mechanism claimed in early theoretical models has been lost. Several early35 and later studies,36 which specifically compared the contribution of different ion currents to the DD, concluded that there is no single pacemaker current in the sinoatrial node.
Currently there is no consensus as to which of the SANC currents makes the major contribution to pacemaker activity. Various viewpoints on the membrane clock (Fig. 1A) include essentially combinations of previously suggested DD mechanisms: 1) original gK decay mechanism of the 1962 model (IKr in rabbit37); 2) Is (ICaL) activation38; 3) If activation39,40; 4) ICaT activation41; 5) INCX activation42–44; and 6) Ist activation.34 Numerous numerical models, based upon gating schema of ensembles of SANC membrane ion currents, have been devised, and all these models featuring different contributions of the ion currents into DD can indeed generate spontaneous APs (see review22).
However, since the idea of a unique pacemaker current was not confirmed in the extensive experimental studies across numerous species, there was a need to formulate another fundamental principle of cardiac pacemaker function. Could it be that regardless of combination or contributions of the different ion channel types, not all of the crucial mechanisms that are implicated in spontaneous excitation of pacemaker cells are embodied in the ensemble of cell surface ion channels? Moreover, and very importantly, could it be that the formal cause of spontaneous rhythmic APs in SANC (i.e., the initiating step of their normal automaticity) is an intracellular process (e.g., similar to the one that Bozler had suggested earlier in 19437)?
The Existence and the Identity of Intracellular Ca2+ Clock
Studies in Contractile Cardiac Cells Lead the Way
Heart research had been tuned in to the crucial importance of extracellular Ca2+ in the cardiac muscle duty cycle since 1883 when Sidney Ringer’s London tap water mistake illuminated the field.45 Subsequent studies identified the sarcoplasmic reticulum (SR) and its Ca2+ pump, and the idea that cyclic Ca2+ release from SR is followed by pumping of Ca2+ back into the SR was born.
Ca2+ pumping into the SR is accomplished mainly by the SR Ca2+ ATPase (SERCA). The Ca2+ pump isoform in the heart, SERCA2A, is the dominant mechanism to remove Ca2+ from the cytosol following excitation, but its relative contribution varies among species. In most mammalian ventricular myocytes (including rabbit, guinea-pig, ferret, dog, cat, and human), SERCA function accounts for about 60–75% of Ca2+ extrusion, with 25–40% attributable to the sarcolemmal NCX.46
It was shown that ryanodine, an alkaloid that makes the SR Ca2+ release channel leaky and inefficient, greatly altered the contractile state of mammalian ventricular myocardium.47,48 This extremely important pharmacological tool was used in numerous later studies to isolate, purify, and clone ryanodine receptor (RyR) permitting the assessment of SR Ca2+ release in the function of different cardiac cell types. Other studies49 demonstrated a “memory” within heart muscle that produces contractions of differing amplitudes in response to changes in the rate or pattern of stimulation. It was later inferred that staircases in contraction amplitudes prior to achieving a new steady state involved changes that occur over several heart beats in Ca2+ loading and release from the SR50 from beat-dependent net changes in cell Ca2+. Although not articulated as such at that time, such Ca2+ oscillations generated by SR Ca2+ pumping and Ca2+ release are a manifestation of an intracellular Ca2+ clock (Fig. 1B).
In mechanically skinned cardiac cell fragments, application of graded [Ca2+] induced graded Ca2+ releases from the SR to generate graded transient increases in cytosolic Ca2+ that initiate graded contractions,51,52 a process referred to as Ca2+-induced Ca2+-release.
The advent of intracellular Ca2+ indicators permitted direct demonstrations that the AP does indeed result in a release of Ca2+ from the SR, resulting in a transient increase in cytosolic Ca2+ that activates the myofilaments to drive contraction. Cytosolic Ca2+ transients evoked by APs were demonstrated in 1978 by Allen and Blinks53 in atrial muscle and in 1980 by Wier54 in Purkinje fibers by using aequorin, a Ca2+-binding photoprotein. Thus, the earlier inference that Ca2+ releases, triggered by an AP, couples membrane excitation to contraction was correct, and the concept of “excitation-induced-Ca2+-release” evolved.
It was also discovered that increases in Ca2+ beneath the cell surface membrane and the mechanical strain during contraction produced feedback on ion channels and transporters that underlie the AP.55 One crucial aspect of such tuning of excitation by Ca2+ release in ventricular myocytes occurs during the AP and involves Ca2+ removal from the cell to balance the Ca2+ influx via L-type Ca2+ channels. This is achieved, in part, by the cell surface membrane NCX, which extrudes one Ca2+ for three Na+. This process generates an inward current (INCX) that affects the AP repolarization phase shape56 as well as the diastolic membrane potential.
The Potential Importance of Intracellular Ca2+ in Generating Spontaneous APs
Evidence that an oscillatory intracellular process can initiate both normal and abnormal cardiac rhythms stems from the turn of the last century.57,58 The initial perspectives of the origin of cardiac automaticity focused not on membrane-delimited initiation of the heart beat but on myogenic mechanisms. In the early 1880s, Walter Gaskell, who conducted experiments applying heat to the tortoise heart, demonstrated that heart automaticity was myogenic in origin (rather than neuorogenic). He noted that, importantly, the automaticity could occur in all the different regions of the heart including the ventricle,59 indicating that, in contrast to skeletal muscle (which strictly followed the nerve-mediated excitation → contraction paradigm), cardiac tissues possess their own internal excitatory oscillator. The area of the turtle heart with the greatest automaticity was the sinus venosus, which emanated impulses that entrained the entire heart. Early pharmacological experimental approaches7,8,60 towards understanding normal automaticity produced “brute force” Ca2+ overload. Thus, abnormal rather than normal automaticity was being explored in early studies. Nonetheless, these studies set the stage for the later rejuvenation of the hypothesis that intracellular Ca2+ is a partner with surface membrane channels in the process of normal automaticity.
In the late 1970s to early 1980s, the idea that surface membrane-delimited ion channel rhythms were sufficient to confer automaticity to pacemaker cells dominated the research field of cardiac automaticity. During this time, however, the idea that an intracellular oscillator drives membrane excitations was temporarily revived as the importance of intracellular Ca2+ transients in cardiac cells came into focus. Unfortunately, these studies of a role for Ca2+ in pacemaker cell automaticity continued to employ brute force Ca2+ overload in their approach to the problem. Pharmacological microelectrode studies of APs in the early 1970s57 rediscovered the triggered activity described approximately 30 years earlier in the monophasic recordings.7,8,60 It was shown that the APs of Purkinje fibers exposed to ouabain or to acetylstrophanthidin develop “delayed afterdepolarizations” and aftercontractions; the amplitude of these was enhanced by driving the fiber more rapidly,61–64 by increasing Cao or by decreasing Ko, and was depressed by Mn2+.64
Based upon experimental observations that cardiac glycosides, a reduction in Nao, or an elevation in Cao cause spontaneous contractions, it was suggested that an elevation of intracellular Ca2+ drives these spontaneous contractions.57 But in contrast to the idea of a spontaneous intracellular oscillator, initial interpretations from microelectrode studies suggested that spontaneous depolarization (i.e., the membrane voltage change), per se, initiates internal Ca2+ release which drives spontaneous cardiac contraction.63–66 However, microelectrode recordings of the membrane potential do not allow assessment of the underlying ion currents, and this interpretation was disproven by voltage clamp studies that demonstrated a subcellular origin of the oscillations. It thus became apparent that intracellular Ca2+ could initiate triggered activity or abnormal automaticity. The concept was further supported by the knowledge of oscillatory Ca2+ activity in a wide variety of cells.67 Data from studies of cardiac cells, including Ca2+-dependent tension oscillations in skinned fibers (Fabiato and Fabiato68) and voltage noise analysis (De-Felice and DeHaan69) in chick heart cell aggregates led to the hypothesis that it is Ca2+ that provides the oscillatory substrate in the intracellular “metabolic” oscillator suggested earlier.8
Oscillatory Ca-dependent SR-driven Ion Current under Voltage Clamp in Cardiac Cells
Oscillatory current fluctuations under voltage clamp were observed in Purkinje fibers,70–74 single Purkinje cells,75 and sinoatrial node.76 Both current and mechanical fluctuations are enhanced when intracellular free Ca2+ is elevated by bathing solutions containing high Ca2+, low Na+, low K+, or toxic concentrations of digitalis. The power spectra of the currents showed peaks at oscillatory frequencies near 1 Hz at room temperature,77 and the current oscillations were correlated with spontaneous contractile fluctuations. These oscillatory currents in Purkinje fibers disappeared after removing extracellular Ca2+ or chelating intracellular Ca2+ with injected ethylene glycol tetraacetic acid (EGTA).74 Based on voltage clamp data, Kass and Tsien, in 1982,74 proposed that the intracellular oscillatory mechanism involves cycles of Ca2+ movement between SR and myoplasm, as previously suggested for skinned cardiac preparations by Fabiato and Fabiato.51 Since the oscillatory currents were inhibited by D600, a Ca2+ current blocker, a new and important idea was that Ca2+ oscillations represent the result of “a two-way interaction between surface membrane potential and intracellular Ca2+ stores”.74 The Ca2+ signals, including Ca2+ oscillations, were then indeed measured directly by aequorin in Purkinje fibers.78,79 An important issue that needed to be addressed was how Ca2+ oscillatory signals are transformed into respective electrical signals of cell membrane. It turned out that major ion (K+ and Ca2+) currents in cardiac cells, including SANC, were modulated by Ca2+ and cyclic nucleotides. Based on this knowledge, it was speculated that intracellular Ca2+ oscillations could be linked to membrane potential via 1) a Ca2+-dependent K+ conductance76; 2) a nonselective (Na+ and K+) cation channel74; or 3) INCX.16 Further studies of automaticity in atrial latent pacemaker cells indicated that Ca2+ release interacts with cell membrane by stimulating INCX.80
The “Abnormal” Flavor of the Initially Discovered Internal Ca2+ Oscillator
As noted, in order to demonstrate Ca2+-driven oscillatory currents by techniques available during the late 1970s, Ca2+ overload was produced in most experiments. Thus, the prevailing assumption at the time was that the oscillating current was a characteristic of abnormal function, similar to that described by Lewis, Rothberger, Scherf, Segers, and Bozler (review Ref. 57). In these earlier studies Ca2+ overload (not realized at that time by these investigators) was employed to disengage the intracellular Ca2+ clock from its entrainment by externally driven APs. Despite the fact that it had already been demonstrated that similar fluctuations can also appear quite prominently in some heart cells in the absence of overt intervention, current fluctuations were primarily interpreted as a disturbance of normal cardiac function.74 Upon testing an integration of a simple Ca2+ oscillator scheme into a pacemaker model, Noble, in 1984,16 concluded that clarification of these abnormal rhythm mechanisms required additional experiments. Other pacemaker cognoscenti of the day, although basically in agreement with this view, were somewhat more liberal in their vision. Crane-field57 reasoned that the true automaticity of the sinoa-trial node or of Purkinje fibers might have a close connection with triggered activity. Irisawa speculated that subthreshold oscillations contribute to a “safety margin to prevent the sinoatrial node cell from becoming quiescent.”76 However, since the spontaneous, localized, oscillatory Ca2+ signals in SANC could not be measured at the time, the concept that an internal Ca2+ oscillator provides the initiating signals for normal automaticity was abandoned again.
The Physiologic Intracellular Ca2+ Clock within Pacemaker Cells Is Rejuvenated
In the late 1980s, experimental investigation of the role of Ca2+ in pacemaker function began to shift from an experimental Ca2+-overload paradigm to more physiologic conditions. Afterdepolarizations and contractions were recorded in Purkinje fibers under normal Ca2+ loading conditions,81 and these fibers demonstrated localized spontaneous myofilament motion caused by spontaneous local Ca2+ oscillations.82 It was discovered that ryanodine (depleting SR content) has a profound negative chronotropic effect on automaticity of subsidiary atrial pacemakers.83 Analyses of the ryanodine effect on AP shape led to the suggestion that Ca2+ released from the SR contributes to DD, primarily during its latter half, and plays a prominent role in bringing the late pacemaker potential to the AP activation threshold. The importance of RyR-mediated Ca2+ release and INCX for normal spontaneous beating was also demonstrated in toad pacemaker cells.84
Additional studies provided compelling evidence that rhythmic Ca2+ cycling within SANC is crucial for their normal automaticity. Ca2+ cycling proteins, SERCA, RyR, and NCX previously identified in atrial or ventricular cells, were also identified in SANC.85,85a A possible importance of SR Ca2+ release and related INCX for SANC pacemaker function was demonstrated by the ryanodine effect in these cells.42–44 When it became possible to routinely measure intra-cellular Ca2+ in SANC, it was observed that a cytosolic Ca2+ transient is evoked by the AP, and that Ca2+ influx via L-type Ca2+ channels affects Ca2+ loading of the SR; chelation of intracellular Ca2+ in rabbit SANC by bis-(o-aminophenoxy)-N,N,N′,N′-tetraacetic acid (BAPTA)43,86 (but not EGTA!86,87), applied intracellularly, markedly slowed or abolished spontaneous beating of rabbit SANC. A strong, negative, chronotropic effect of EGTA, however, had been reported in SANC88 and atrioventricular node cells89 of guinea pig. Changes in the cytosolic Ca2+ transient in response to β-adrenergic stimulation were correlated with changes in the beating rate.84,85 Thus, it was becoming recognized that intracellular Ca2+ cycling within pacemaker cells is somehow involved in normal SANC pacemaker function.
A New Pacemaker Theory Based on Integration of the Intracellular Ca2+ Clock and Membrane Ion Channel Clock
Ca2+ Clocks in SANC Ignite Rhythmic APs via Activation of NCX Current
Confocal Ca2+ imaging permitted detection of a more subtle form of spontaneous Ca2+ release than those caused by experimental Ca2+ overload. Recent studies in rabbit SANC (Fig. 1C) employing confocal imaging showed that, in contrast to ventricular myocytes, the SR Ca2+ clocks are, in part, entrained by an AP but later in the cycle become “free running.”
The rhythmic local Ca2+ releases (LCRs) via RyRs occur spontaneously beneath the cell membrane in SANC during the later part of the spontaneous DD (Fig. 2A). Dense immunolabeling and clear colocalization of RyR and NCX beneath the surface membrane of SANC have been recently demonstrated (Fig. 2C–E).90 LCRs activate NCX inward currents producing miniature voltage fluctuations. The ensemble of the inward NCX currents causes the later part of the DD to increase exponentially (Fig. 2B, “Nonlinear DD”), leading to the generation of spontaneous APs.91 LCRs are a manifestation of spontaneous SR Ca2+ release that does not require a trigger mechanism, such as surface membrane depolarization or inward currents; rhythmic spontaneous LCRs occur in skinned SANC and under voltage clamp.92,93
FIGURE 2.

Diastolic submembrane LCRs via RyRs ignite rhythmic APs via activation of NCX imparting nonlinear (exponential) late DD in rabbit SANC. (A) Linescan Ca2+ image with superimposed spontaneous APs. Arrows mark LCRs. Modified from Ref. 91. (B) Ryanodine abolishes LCRs and thus abolishes the LCR-mediated AP ignition early in the cycle by inhibition of the nonlinear DD (gray area shown by arrow). Modified from Ref. 95. (C) Confocal whole cell image of cells doubly labeled for NCX and RyR. (D) Graphed pixel-by-pixel fluorescence intensities of labeling along an arbitrary line, positioned as indicated by a thick white line in C. The horizontal dashed lines report the average pixel intensity. (E) The topographical profiles of the pixel intensity levels of each antibody labeling and overlay of the small SANC. The maximum height represents the brightest possible pixel in the source image (using an 8-bit image intensity scale). Less bright pixels are accordingly scaled to a smaller height. C–E from Ref. 90. (In color in Annals online.)
An argument that is often raised in an attempt to exclude submembrane pacemaker mechanisms is that in voltage-clamped SANC the membrane current does not show cyclic changes.21,94 However, this is only true with reference to a comparison with oscillatory current scaling of more than 100 pA, as observed previously in digitalis-treated ventricular myocytes, and now interpreted as the gold standard of the oscillatory current (Fig. 1 in Ref. 94). Further, accurate perforated patch clamp studies, which preserve crucial intracellular interactions within rabbit SANC, clearly demonstrate an oscillatory nature of the net membrane current (Fig. 3).92,93 Since the scale of the oscillatory current in SANC is only 10 pA, it likely was distorted or missed in early patch clamp whole-cell experiments.21,94 However, it is the 10 pA current that has crucial importance during DD because membrane electrical resistance during this phase of cycle is very high; a tiny current change of approximately 3 pA is enough to drive the critical DD change in rabbit SANC.21,40 The rhythmic oscillatory currents in SANC under voltage clamp are produced by rhythmic LCRs, since both LCRs and the currents exhibit fluctuations of the same frequency,92,93 and both are abolished by ryanodine95 (Fig. 4).
FIGURE 3.

An example of analysis of the net membrane current (A) in a voltage-clamped rabbit SANC to detect cyclic current fluctuations. Arrows show the analysis sequence. The net current is first zoomed to a 10 pA scale and its slow component is fitted by a cubic polynomial function (solid line in (B). The slow component (likely from K+ current activation) is subtracted (C). The current fluctuations show a dominant oscillation frequency of 4.9 Hz in its power spectrum (D).
FIGURE 4.

(A,B) Simultaneous recordings of membrane potential or current (top), confocal linescan image (middle), and normalized fluo-3 fluorescent (bottom) averaged over the linescan image in a representative spontaneously beating rabbit SANC with intact sarcolemma prior to and during voltage clamp to -10 mV in control (A) and following inhibition of RyR with 3 μmol/L ryanodine (B). The traces in panels A and B under voltage clamp depict membrane current fluctuations generated by LCR occurrence. (C,D) Fast Fourier Transform (FFT) of Ca2+ (D) and membrane current (C) fluctuations during voltage clamp in control and after ryanodine. Note that the rate of these current fluctuations is the same as that of LCRs and is similarly suppressed by ryanodine. From Ref. 95. (In color in Annals online.)
The Spontaneous Cycle Length is Tightly Linked to LCR Period of SANC
The Ca2+ clock rate in cardiac cells is reflected in the LCR period observed as a delay from the AP-triggered global Ca2+ transient and the subsequent LCR occurrence (Fig. 5A). The likelihood for spontaneous SR Ca2+ release to occur increases as a function of the SR Ca2+ load determined in a physiologic content by the Ca2+ available for pumping and the phosphorylation status of the SR Ca2+ cycling proteins. Thus, the convergence of multiple factors governs the LCR period. These include the rate at which Ca2+ is pumped into the SR, the threshold SR Ca2+ load required to initiate spontaneous Ca2+ release, and the availability of SR Ca2+ release channels (i.e., RyRs).
FIGURE 5.
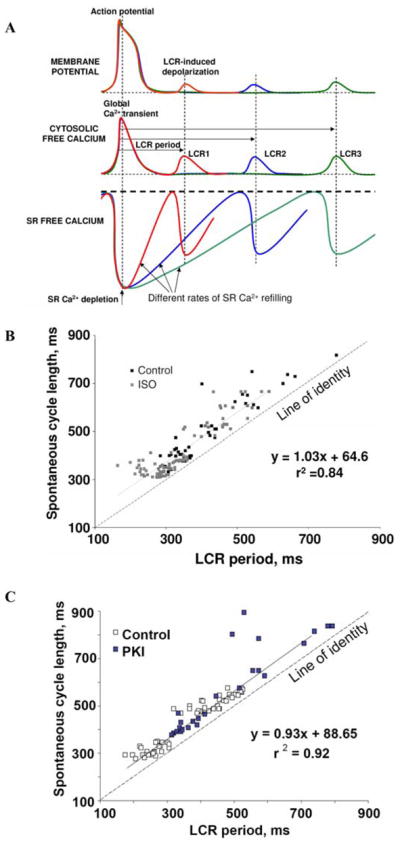
Variations in the LCR period are tightly linked to variations in the spontaneous beating. A schematically illustrates the idea that the rate of SR Ca2+ refilling controls the LCR period and the timing of the LCR-induced depolarization. (B) Relationship between LCR period and spontaneous cycle length is shifted to shorter periods by β-adrenergic stimulation with 0.1 μmol/L isoproterenol (ISO). (C) The PKI effect to increase the cycle length is linked to its effect on the LCR period. B and C are modified from Ref. 93. (In color in Annals online.)
Each spontaneous cycle of the SANC LCR clock can be envisioned to initiate the occurrence of an AP via INCX activation. Ca2+ influx via ICaL during AP triggers Ca2+-induced Ca2+-release. The resulting global SR Ca2+ depletion synchronizes SR throughout the cell in a Ca2+-depleted state. Refilling of the SR ensures that the threshold of Ca2+ load required for spontaneous release is achieved at about the time when RyR inactivation following prior activation is removed; then the spontaneous LCR occurs, activating INCX, igniting next AP, and so on.
Thus, Ca2+ refilling of the SR to the spontaneous release threshold yields the delay determining LCR period so that the LCR period is not fixed but can vary within the kinetics of SR Ca2+ reloading (Fig. 5A). Variations in the LCR period are, in turn, linked to variations in the spontaneous beating rate. Thus, an extremely tight link between the LCR period and the spontaneous cycle length has been experimentally proven (Table 1). Note that the relationship of LCR periods to the spontaneous cycle length of SANC (slope coefficient in Table 1) is nearly identical and is near unity over a wide range of conditions, and the intercept indicates that, regardless of the conditions and absolute cycle length, LCRs shortly precede (within approximately 100 ms) and ignite APs.
TABLE 1.
Variations in spontaneous cycle length are strongly correlated with variations in LCR period
| Experimental paradigm | Cycle length = a·LCR Period + b |
|---|---|
| Different SANC modified from Ref. 92 | CL = 0.86 · LCR period + 23 ms, r2 = 0.85 |
| SR Ca2+ repletion following SR Ca2+ depletion modified from Ref. 92 | CL = 0.90 · LCR period + 85 ms, r2 = 0.86 |
| Ryanodine (Vinogradova and Lakatta) | CL = 1.00 · LCR period + 110 ms, r2 = 0.90 |
| Suppression of SR Ca pump (Vinogradova and Lakatta) | CL = 0.97 · LCR period + 98 ms, r2 = 0.85 |
| PKA inhibition (PKI)93 | CL = 0.93 · LCR period + 89 ms, r2 = 0.92 |
| PKA inhibition (H89)93 | CL = 0.97 · LCR period + 86 ms, r2 = 0.85 |
| Basal PDE inhibition induced reduction in cycle length (Vinogradova and Lakatta) | CL = 0.93 · LCR period + 69 ms, r2 = 0.89 |
| β-AR stimulation induced reduction in cycle length (modified from Ref. 93) | CL = 1.03 · LCR period + 64.6 ms, r2 = 0.84 |
An extremely tight link between the LCR period and the spontaneous cycle length (CL) of SANC was identified experimentally (slope a, intercept b, and correlation r2) over a wide range of conditions (experimental paradigm).
The Crucial Role of Protein Kinase A-dependent Protein Phosphorylation in the Tightly Controlled Variability of Ca2+ Clock Rate and SANC Chronotropy
Protein kinase A (PKA)-dependent phosphorylation is obligatory for basal LCR occurrence; gradations in basal PKA-dependent phosphorylation result in gradations in the LCR period93 (Fig. 5B,C). Stimulation of β-adrenergic receptors extends the range of PKA dependence of the LCR period. The PKA dependence of the LCR period is attributable to at least three (and probably several more) potential mechanisms: augmentation of Ca2+ influx via phosphorylation of L-type Ca2+ channels; augmentation of Ca2+ pumping into the SR via phospholamban (PLB) phosphorylation; and increased availability of RyRs via their phosphorylation.
In spontaneously beating SANC, inhibition or activation of PKA results in gradations in PLB phosphorylation, paralleled by gradations in LCR period93; but graded phosphorylation of L-type channels and RyRs also likely occurs in such experiments. The dependence of LCR period on PKA inhibition or the addition of cyclic adenosine 3′,5′-phsophate (cAMP) to skinned SANC or during voltage clamp93 demonstrates the role of PKA-dependent mechanisms distinct from those of L-type Ca2+ channels (Fig. 6A). However, there have been no experiments to date that separate the roles of Ca2+ pumping from RyR availability in the generation of spontaneous LCRs. As to ICaL, its roles in SANC spontaneous firing rate regulation are 1) to provide AP upstroke; 2) to synchronize SR in a Ca2+-depleted state; and 3) to support the increased Ca2+ supply–demand balance at higher rates as ignition from lower voltages requires a larger NCX current and is thus associated with higher Ca2+ efflux by the NCX.
FIGURE 6.

(A) Schematic illustration of functional integration and regulation of membrane and submembrane Ca2+ cycling to control pacemaker function via NCX-mediated ignition of rhythmic APs. The thick line indicates spontaneous SR Ca2+ cycling. Modified from Ref. 93. (B) Spontaneous beating of rabbit SANC critically depends upon Ca2+-related mechanisms and protein phosphorylation. Bars show a decrease in the beating rate (% control) induced by different drugs that affect Ca2+ cycling (ryanodine receptors, cytosolic Ca2+), NCX (Li+ substitution for Na+), protein phosphorylation (PKI, H-89, MDL), or ion channels: If (Cs+), ICaT (Ni2+), ICaL (nifedipine * 105), IK (E-4031 ** 106). PKI and H-89 are PKA inhibitors; MDL is an adenylyl cyclase inhibitor. Modified from Ref. 95. (In color in Annals online.)
Novel Numerical Pacemaker Models Test Ca2+-driven Pacemaker Mechanism
Initial tests of the ability of LCRs during the DD to command the AP firing rate used a primary rabbit SANC model developed by Zhang et al.96 When experimentally measured waveforms of submembrane, LCRs were introduced into the model, spontaneous AP firing rate was easily entrained at a rate determined by the period of these LCRs.97 Subsequently, Kurata et al. developed a model98 that described Ca2+ concentration in submembrane space and predicted a strong negative chronotropic effect of Ca2+ chelation in this critical location. A novel pacemaker model99 included LCR in the submembrane space and reproduced, for the first time, the negative chronotropic effect of ryanodine and predicted a new powerful mechanism of rate regulation by varying Ca2+ release rate and phase; the larger release resulted in the larger INCX that, in turn, allowed wider rate regulation range by the phase of the release covering basically the entire physiologic range. In terms of the fine DD structure, the model showed that the LCR-activated NCX current imparts an exponentially rising part to the late DD that culminates in an AP upstroke.99
A subsequent upgrade of the model reproduced individual stochastic LCRs (a multicompartment SR model).91,93 Varying frequency, size, and phase of the LCRs, according to experimental data, this model predicted the wide range of chronotropic effects that were experimentally observed with graded PKA activity; that is, basal and reserve rate regulation via a stochastic ensemble of local NCX currents induced by the LCRs.93 The model also provided further details of fine DD structure, including its fluctuations and exponentially rising late DD component observed experimentally shortly before the excitation as well as modest beat-to-beat cycle variations from the stochastic occurrence of the LCRs.91 This model identified substantial benefits of interactions of the rhythmic LCRs and the membrane ion channel clock in terms of overall performance of the new pacemaker mechanism. Under conditions when ion channels operating alone in numerical models fail to generate rhythmic APs, stable and rhythmic AP firing resumes when the timely and powerful prompt to the membrane sent by SR via LCRs and NCX is introduced into the model.100
The numerical model approaches described above illustrated that the concept of Ca2+ release–ignited excitation is indeed operational in primary SANC, based on the existing knowledge of LCRs and SANC-specific ion channels. However, the Ca2+ release function in these model simulations was either substituted by the experimental waveform or approximated by a sinusoidal function. The most recent approaches to quantitatively describe Ca2+ integrated SANC function has included spontaneous Ca2+ release mechanisms101 and local Ca2+ dynamics.102 More specifically, this approach has developed a new model101 of primary rabbit SANC in which an integrated Ca2+ release mechanism is controlled by the SR Ca2+ load (modified from Shannon et al. 2004103). This more mechanistic model now predicts SR Ca2+ depletion–refilling dynamics measured by Fluo-5N101,104 in spontaneously beating SANC and exhibits spontaneous Ca2+ oscillations under voltage clamp. Varying the Ca2+ clock model parameters results in a graded control of the timing of spontaneous Ca2+ release and the pacemaker rate. This result was corroborated by the experimental finding that cyclopiazonic acid, an inhibitor of SR Ca2+ ATPase, effects about a 50% reduction of the spontaneous beating rate of rabbit SANC.104 These recent numerical model studies thus show that 1) periodic SR Ca2+ refilling and release indeed define (quantitatively rather than speculatively or phenomenologically) the Ca2+ clock within SANC and 2) beat-to-beat interaction of this Ca2+ clock with the membrane clock results in a robust pacemaker function with an effectively controlled beating rate.
The great acceleration of computational power of modern computers permits development of an extension of the presently available models to simulate local Ca2+ dynamics in the submembrane space of SANC, based on approximation of Ca2+ diffusion and stochastic activation of RyR clusters (Ca2+ release units).102 These simulations predict the occurrence of local stochastic Ca2+ wavelets with spatiotemporal properties similar to those found for LCRs in SANC. In perspective, when the above two modeling approaches converge, a rather sophisticated powerful SANC model integrating function of membrane channels, SR Ca2+ release mechanism, and local Ca2+ dynamics will likely emerge.
Mutual Entrainment of Ca2+ and Membrane Clocks
The true cardiac pacemaker mechanism is complex and includes a balanced integration of both intracellular and membrane-delimited processes (Fig. 6A). Interfering with NCX function or SR internal Ca2+ cycling directly (e.g., ryanodine) or indirectly (by inhibiting PKA-dependent phosphorylation) precludes normal pacemaker functions, just as does interfering with membrane ion channels (ICaL and IK) that generate the AP upstroke and repolarization (Fig. 6B). Note that blockade of ICaT or If has relatively minor effects on the SANC pacemaker firing rate.
While the SANC ion channel membrane clocks are necessary to effect an AP, they are not sufficient, in our opinion, to ignite rhythmic APs at variable rates. Rather, both experimental data and numerical simulations (see above) demonstrate that the intracellular Ca2+ clock and its rhythmic LCRs do not merely fine tune the normal automaticity of SANC but, by igniting excitations from within the cell, represent the formal cause of APs (Figs. 1 and 6A). The true initiation of cardiac pacemaker automaticity begins during the late part of the DD when the Ca2+ clock ignites the surface membrane excitation. Earlier events, such as the maximum diastolic potential and early linear DD, ensure recovery of the membrane clocks that are activated during the prior AP and need to be reset before the next AP. The spontaneous, but precisely controlled, rhythmic SR Ca2+ clock of SANC ensures the stability of basal rhythm and sets the basal firing rate by integrating multiple Ca2+-dependent functions and rhythmically interacting with one, igniting the ensemble of surface membrane clocks to effect APs. Variable degrees of intrinsic PKA-dependent protein phosphorylation, and its attendant variations in intracellular Ca2+ (Fig. 6A), regulate the time that the SR Ca2+ clock keeps. This regulates the LCR period, which modulates the voltage- and time-dependent kinetics of the membrane clocks, thus ensuring that they produce APs with characteristics commensurate with given rhythmic ignition rates commanded by LCRs.
The rate at which the Ca2+ clock ticks controls rhythmic spontaneous AP firing across the broad range of physiologic firing rates. In short, variations of internal Ca2+ cycling within SANC result in corresponding variations in LCRs that are tightly linked to gradations in their automaticity. In addition to AP generation, surface membrane ion channels ensure the robust function of the Ca2+ clock by keeping intracellular ion balance, especially for Ca2+. Ca2+ influx via ICaL during AP “rewinds” the Ca2+ clock by balancing Ca2+ efflux via NCX each cycle. Maintaining this balance is indeed critical for the Ca2+ clock’s long-term operation since LCRs become damped and cease within a few seconds following cessation of APs during voltage clamp near the maximum diastolic potential.92 K+ currents, in turn, regulate the AP duration commensurate with the ignition rate commanded by the Ca2+ clock.
Summary
The mutual entrainment of the Ca2+ clocks and surface membrane ion channel clocks regulate the kinetics of the SANC duty cycle. We believe that this new concept of mutually interacting clocks, governing normal automaticity of pacemaker cells, marks a new beginning of cardiac pacemaker research.
Further evidence for the crucial role of Ca2+ and rhythmic spontaneous Ca2+ releases in the initiation and regulation of normal cardiac automaticity provides the key that reunites the fields of pacemaker automaticity and cardiac muscle function.26 The separate achievements of reductionist approaches within each of these fields of research to elucidate the strength of contraction of ventricular myocytes and the spontaneous beating rate of pacemaker cells converge and a general theory of cardiac chronotropic and inotropic mechanisms emerges: SR Ca2+ cycling both initiates the heartbeat in pacemaker cells and executes the heartbeat in ventricular cells.
Acknowledgments
This research was supported by the Intramural Research Program of the National Institutes of Health, National Institute on Aging.
Footnotes
Conflicts of Interest
The authors declare no conflicts of interest.
References
- 1.Bernstein J. Ueber den zeitlichen Verlauf der negativen Schwankung des Nervenstroms. Pflugers Arch. 1868;1:173–207. (in German) [Google Scholar]
- 2.Bernstein J. Die Lehre von den elektrischen Vorgangen im Organismus auf moderner Grundlage dargestellt. Vieweg & Sohn; Braunschweig: 1912. Elektrobiologie; pp. 138–141. (in German) [Google Scholar]
- 3.Hodgkin AL, Katz B. The effect of sodium ions on the electrical activity of the giant axon of the squid. J Physiol. 1949;108:37–77. doi: 10.1113/jphysiol.1949.sp004310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kleber AG, et al. The early years of cellular cardiac electrophysiology and Silvio Weidmann (1921–2005) Heart Rhythm. 2006;3:353–359. doi: 10.1016/j.hrthm.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 6.Arvanitaki A, et al. Reactions electroniques du myocarde en function de sur tonus initial. Compt Rend Sot Biol. 1937;124:165–167. [Google Scholar]
- 7.Bozler E. The initiation of impulses in cardiac muscle. Am J Physiol. 1943;138:273–282. [Google Scholar]
- 8.Bozler E. Tonus changes in cardiac muscle and their significance for the initiation of impulses. Am J Physiol. 1943;139:477–480. [Google Scholar]
- 9.Brady AJ, Hecht HH. On the origin of the heart beat. Am J Med. 1954;17:110. [Google Scholar]
- 10.Coraboeuf E, Weidmann S. Temperature effects on the electrical activity of Purkinje fibres. Helv Physiol Pharmacol Acta. 1954;12:32–41. [PubMed] [Google Scholar]
- 11.West TC. Ultramicroelectrode recording from the cardiac pacemakers. J Pharmacol Exp Ther. 1955;115:283–290. [PubMed] [Google Scholar]
- 12.Noble D. Cardiac action and pacemaker potentials based on the Hodgkin-Huxley equations. Nature. 1960;188:495–497. doi: 10.1038/188495b0. [DOI] [PubMed] [Google Scholar]
- 13.Noble D. A modification of the Hodgkin-Huxley equations applicable to Purkinje fibre action and pacemaker potentials. J Physiol. 1962;160:317–352. doi: 10.1113/jphysiol.1962.sp006849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weidmann S. Effect of current flow on the membrane potential of cardiac muscle. J Physiol. 1951;115:227–236. doi: 10.1113/jphysiol.1951.sp004667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dudel J, Trautwein W. The mechanism of formation of automatic rhythmical impulses in heart muscle. Pflugers Arch. 1958;267:553–565. doi: 10.1007/BF00362959. [DOI] [PubMed] [Google Scholar]
- 16.Noble D. The surprising heart: a review of recent progress in cardiac electrophysiology. J Physiol. 1984;353:1–50. doi: 10.1113/jphysiol.1984.sp015320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deck KA, Kern R, Trautwein W. Voltage clamp technique in mammalian cardiac fibres. Pflugers Arch Gesamte Physiol Menschen Tiere. 1964;280:50–62. doi: 10.1007/BF00412615. [DOI] [PubMed] [Google Scholar]
- 18.Powell T, Terrar DA, Twist VW. Electrical properties of individual cells isolated from adult rat ventricular myocardium. J Physiol. 1980;302:131–153. doi: 10.1113/jphysiol.1980.sp013234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Irisawa H. Electrical activity of rabbit sino-atrial node as studies by a double sucrose gap method. In: Rijlant P, editor. Proceedings of the Satellite Symposium of the XXVth Interneational Congress of Physiological Science. The Electrical Field of the Heart. Press Academiques Europeenes; Bruxelles: 1972. pp. 242–248. [Google Scholar]
- 20.Taniguchi J, et al. Spontaneously active cells isolated from the sino-atrial and atrio-ventricular nodes of the rabbit heart. Jpn J Physiol. 1981;31:547–558. doi: 10.2170/jjphysiol.31.547. [DOI] [PubMed] [Google Scholar]
- 21.Irisawa H, Brown HF, Giles W. Cardiac pacemaking in the sinoatrial node. Physiol Rev. 1993;73:197–227. doi: 10.1152/physrev.1993.73.1.197. [DOI] [PubMed] [Google Scholar]
- 22.Wilders R. Computer modelling of the sinoatrial node. Med Biol Eng Comput. 2007;45:189–207. doi: 10.1007/s11517-006-0127-0. [DOI] [PubMed] [Google Scholar]
- 23.Reuter H. Current-tension relations of Purkinje fibers in different extracellular concentrations of calcium and under the influence of adrenaline. Pflugers Arch Gesamte Physiol Menschen Tiere. 1966;287:357–367. [PubMed] [Google Scholar]
- 24.DiFrancesco D, Ojeda C. Properties of the current if in the sino-atrial node of the rabbit compared with those of the current iK, in Purkinje fibres. J Physiol. 1980;308:353–367. doi: 10.1113/jphysiol.1980.sp013475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McAllister RE, Noble D, Tsien RW. Reconstruction of the electrical activity of cardiac Purkinje fibres. J Physiol. 1975;251:1–59. doi: 10.1113/jphysiol.1975.sp011080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maltsev VA, Vinogradova TM, Lakatta EG. The emergence of a general theory of the initiation and strength of the heartbeat. J Pharmacol Sci. 2006;100:338–369. doi: 10.1254/jphs.cr0060018. [DOI] [PubMed] [Google Scholar]
- 27.Neher E, Sakmann B. Single-channel currents recorded from membrane of denervated frog muscle fibres. Nature. 1976;260:799–802. doi: 10.1038/260799a0. [DOI] [PubMed] [Google Scholar]
- 28.Satoh H. Sino-atrial nodal cells of mammalian hearts: ionic currents and gene expression of pacemaker ionic channels. J Smooth Muscle Res. 2003;39:175–193. doi: 10.1540/jsmr.39.175. [DOI] [PubMed] [Google Scholar]
- 29.Vassalle M. The pacemaker current (If) does not play an important role in regulating SA node pacemaker activity. Cardiovasc Res. 1995;30:309–310. [PubMed] [Google Scholar]
- 30.Brown H, Kimura J, Noble S. The relative contributions of various time-dependent membrane currents to pacemaker activity in the sino atrial node. In: Bouman LN, Jongsma HJ, editors. Cardiac Rate and Rhythm. Martinus Nijhoff; The Hague: 1982. pp. 53–68. [Google Scholar]
- 31.Shibata EF, Giles WR. Ionic currents that generate the spontaneous diastolic depolarization in individual cardiac pacemaker cells. Proc Natl Acad Sci USA. 1985;82:7796–7800. doi: 10.1073/pnas.82.22.7796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ono K, Iijima T. Pathophysiological significance of T-type Ca2+ channels: properties and functional roles of T-type Ca2+ channels in cardiac pacemaking. J Pharmacol Sci. 2005;99:197–204. doi: 10.1254/jphs.fmj05002x2. [DOI] [PubMed] [Google Scholar]
- 33.Verkerk AO, et al. Ca2+-activated Cl- current in rabbit sinoatrial node cells. J Physiol. 2002;540:105–17. doi: 10.1113/jphysiol.2001.013184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo J, Ono K, Noma A. A sustained inward current activated at the diastolic potential range in rabbit sino-atrial node cells. J Physiol. 1995;483(Pt 1):1–13. doi: 10.1113/jphysiol.1995.sp020563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown HF, et al. The ionic currents underlying pacemaker activity in rabbit sino-atrial node: experimental results and computer simulations. Proc R Soc Lond B Biol Sci. 1984;222:329–347. doi: 10.1098/rspb.1984.0067. [DOI] [PubMed] [Google Scholar]
- 36.Zaza A, et al. Ionic currents during sustained pacemaker activity in rabbit sino-atrial myocytes. J Physiol. 1997;505(Pt 3):677–688. doi: 10.1111/j.1469-7793.1997.677ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ono K, Ito H. Role of rapidly activating delayed rectifier K+ current in sinoatrial node pacemaker activity. Am J Physiol. 1995;269:H453–462. doi: 10.1152/ajpheart.1995.269.2.H453. [DOI] [PubMed] [Google Scholar]
- 38.Yanagihara K, Noma A, Irisawa H. Reconstruction of sino-atrial node pacemaker potential based on the voltage clamp experiments. Jpn J Physiol. 1980;30:841–857. doi: 10.2170/jjphysiol.30.841. [DOI] [PubMed] [Google Scholar]
- 39.Maylie J, Morad M, Weiss J. A study of pacemaker potential in rabbit sino-atrial node: measurement of potassium activity under voltage-clamp conditions. J Physiol. 1981;311:161–178. doi: 10.1113/jphysiol.1981.sp013579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DiFrancesco D. The contribution of the ‘pacemaker’ current (if) to generation of spontaneous activity in rabbit sino-atrial node myocytes. J Physiol. 1991;434:23–40. doi: 10.1113/jphysiol.1991.sp018457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nilius B. Possible functional significance of a novel type of cardiac Ca channel. Biomed Biochim Acta. 1986;45:K37–45. [PubMed] [Google Scholar]
- 42.Rigg L, Terrar DA. Possible role of calcium release from the sarcoplasmic reticulum in pacemaking in guinea-pig sino-atrial node. Exp Physiol. 1996;81:877–880. doi: 10.1113/expphysiol.1996.sp003983. [DOI] [PubMed] [Google Scholar]
- 43.Li J, Qu J, Nathan RD. Ionic basis of ryanodine’s negative chronotropic effect on pacemaker cells isolated from the sinoatrial node. Am J Physiol. 1997;273:H2481–2489. doi: 10.1152/ajpheart.1997.273.5.H2481. [DOI] [PubMed] [Google Scholar]
- 44.Satoh H. Electrophysiological actions of ryanodine on single rabbit sinoatrial nodal cells. Gen Pharmacol. 1997;28:31–38. doi: 10.1016/s0306-3623(96)00182-6. [DOI] [PubMed] [Google Scholar]
- 45.Ringer S. A further contribution regarding the influence of the different constituents of the blood on the contraction of the heart. J Physiol. 1883;4:29–42. doi: 10.1113/jphysiol.1883.sp000120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. 2. Kluwer Academic Publishers; Norwell, Mass: 2001. [Google Scholar]
- 47.Sutko JL, et al. Ryanodine: its alterations of cat papillary muscle contractile state and responsiveness to inotropic interventions and a suggested mechanism of action. J Pharmacol Exp Ther. 1979;209:37–47. [PubMed] [Google Scholar]
- 48.Sutko JL, Willerson JT. Ryanodine alteration of the contractile state of rat ventricular myocardium. Comparison with dog, cat, and rabbit ventricular tissues. Circ Res. 1980;46:332–343. doi: 10.1161/01.res.46.3.332. [DOI] [PubMed] [Google Scholar]
- 49.Schaefer J, et al. Historical note on the translation of H.P. Bowditch’s paper’ Über die Eigenthümlichkeiten der Reizbarkeit, welche die Muskelfasern des Herzens zeigen’ (On the peculiarities of excitability which the fibres of cardiac muscle show) In: Noble M, Seed WA, editors. The Interval-Force Relationship of the Heart: Bowditch revisited. Cambridge University Press; Cambridge: 1992. [Google Scholar]
- 50.Wood EH, Heppner RL, Weidmann S. Inotropic effects of electric currents. I. Positive and negative effects of constant electric currents or current pulses applied during cardiac action potentials. II. Hypotheses: calcium movements, excitation-contraction coupling and inotropic effects. Circ Res. 1969;24:409–445. doi: 10.1161/01.res.24.3.409. [DOI] [PubMed] [Google Scholar]
- 51.Fabiato A, Fabiato F. Calcium-induced release of calcium from the sarcoplasmic reticulum of skinned cells from adult human, dog, cat, rabbit, rat, and frog hearts and from fetal and new-born rat ventricles. Ann NY Acad Sci. 1978;307:491–522. doi: 10.1111/j.1749-6632.1978.tb41979.x. [DOI] [PubMed] [Google Scholar]
- 52.Fabiato A, Fabiato F. Excitation-contraction coupling of isolated cardiac fibers with disrupted or closed sarcolemmas. Calcium-dependent cyclic and tonic contractions. Circ Res. 1972;31:293–307. doi: 10.1161/01.res.31.3.293. [DOI] [PubMed] [Google Scholar]
- 53.Allen DG, Blinks JR. Calcium transients in aequorin-injected frog cardiac muscle. Nature. 1978;273:509–513. doi: 10.1038/273509a0. [DOI] [PubMed] [Google Scholar]
- 54.Wier WG. Calcium transients during excitation-contraction coupling in mammalian heart: aequorin signals of canine Purkinje fibers. Science. 1980;207:1085–1087. doi: 10.1126/science.7355274. [DOI] [PubMed] [Google Scholar]
- 55.Lab MJ, Allen DG, Orchard CH. The effects of shortening on myoplasmic calcium concentration and on the action potential in mammalian ventricular muscle. Circ Res. 1984;55:825–829. doi: 10.1161/01.res.55.6.825. [DOI] [PubMed] [Google Scholar]
- 56.duBell WH, et al. The cytosolic calcium transient modulates the action potential of rat ventricular myocytes. J Physiol. 1991;436:347–369. doi: 10.1113/jphysiol.1991.sp018554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cranefield PF. Action potentials, afterpotentials, and arrhythmias. Circ Res. 1977;41:415–423. doi: 10.1161/01.res.41.4.415. [DOI] [PubMed] [Google Scholar]
- 58.Tsien RW, Kass RS, Weingart R. Cellular and subcellular mechanisms of cardiac pacemaker oscillations. J Exp Biol. 1979;81:205–215. doi: 10.1242/jeb.81.1.205. [DOI] [PubMed] [Google Scholar]
- 59.Gaskell WH. On the innervation of the heart, with special reference to the heart of the tortoise. J Physiol. 1881;4:43–127. doi: 10.1113/jphysiol.1883.sp000121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Segers M. Le rôle des potentiels tardifs du coeur. Mem Acad R Med. 1941;1:1–30. (in French) [Google Scholar]
- 61.Davis LD. Effect of changes in cycle length on diastolic depolarization produced by ouabain in canine Purkinje fibers. Circ Res. 1973;32:206–214. doi: 10.1161/01.res.32.2.206. [DOI] [PubMed] [Google Scholar]
- 62.Rosen MR, Gelband H, Hoffman BF. Correlation between effects of ouabain on the canine electrocardiogram and transmembrane potentials of isolated Purkinje fibers. Circulation. 1973;47:65–72. doi: 10.1161/01.cir.47.1.65. [DOI] [PubMed] [Google Scholar]
- 63.Ferrier GR, Saunders JH, Mendez C. A cellular mechanism for the generation of ventricular arrhythmias by acetylstrophanthidin. Circ Res. 1973;32:600–609. doi: 10.1161/01.res.32.5.600. [DOI] [PubMed] [Google Scholar]
- 64.Ferrier GR, Moe GK. Effect of calcium on acetylstrophanthidin-induced transient depolarizations in canine Purkinje tissue. Circ Res. 1973;33:508–515. doi: 10.1161/01.res.33.5.508. [DOI] [PubMed] [Google Scholar]
- 65.Kaufmann R, Fleckenstein A, Antoni H. Causes and Conditions of Release of Myocardial Contractions without a Regular Action Potential. Pflugers Arch Gesamte Physiol Menschen Tiere. 1963;278:435–446. (in German) [PubMed] [Google Scholar]
- 66.Akselrod S, et al. Electro-mechanical noise in atrial muscle fibres of the carp. Experientia. 1977;33:1058–1060. doi: 10.1007/BF01945968. [DOI] [PubMed] [Google Scholar]
- 67.Rapp PE, Berridge MJ. Oscillations in calcium-cyclic AMP control loops form the basis of pacemaker activity and other high frequency biological rhythms. J Theor Biol. 1977;66:497–525. doi: 10.1016/0022-5193(77)90299-5. [DOI] [PubMed] [Google Scholar]
- 68.Fabiato A, Fabiato F. Contractions induced by a calcium-triggered release of calcium from the sarcoplasmic reticulum of single skinned cardiac cells. J Physiol. 1975;249:469–495. doi: 10.1113/jphysiol.1975.sp011026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Defelice LJ, Dehaan RL. Voltage noise and impedance from heart cell aggregates. Biophys J. 1975;15:130a. doi: 10.1016/S0006-3495(79)85169-3. (Abstract) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lederer WJ, Tsien RW. Transient inward current underlying arrhythmogenic effects of cardiotonic steroids in Purkinje fibres. J Physiol. 1976;263:73–100. doi: 10.1113/jphysiol.1976.sp011622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Aronson RS, Gelles JM. The effect of ouabain, dinitrophenol, and lithium on the pacemaker current in sheep cardiac Purkinje fibers. Circ Res. 1977;40:517–524. doi: 10.1161/01.res.40.5.517. [DOI] [PubMed] [Google Scholar]
- 72.Kass RS, et al. Role of calcium ions in transient inward currents and aftercontractions induced by strophanthidin in cardiac Purkinje fibres. J Physiol. 1978;281:187–208. doi: 10.1113/jphysiol.1978.sp012416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eisner DA, Lederer WJ. Inotropic and arrhythmogenic effects of potassium-depleted solutions on mammalian cardiac muscle. J Physiol. 1979;294:255–277. doi: 10.1113/jphysiol.1979.sp012929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kass RS, Tsien RW. Fluctuations in membrane current driven by intracellular calcium in cardiac Purkinje fibers. Biophys J. 1982;38:259–269. doi: 10.1016/S0006-3495(82)84557-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mehdi T, Sachs F. Voltage clamp of isolated cardiac Purkinje cells. Biophys J. 1978;21:165a. (Abstract) [Google Scholar]
- 76.Irisawa H. Comparative physiology of the cardiac pacemaker mechanism. Physiol Rev. 1978;58:461–498. doi: 10.1152/physrev.1978.58.2.461. [DOI] [PubMed] [Google Scholar]
- 77.Kass RS, Lederer WJ, Tsien RW. Current fluctuations in strophanthidin-treated cardiac Purkinje fibers. Biophys J. 1976;16:25a. (Abstract) [Google Scholar]
- 78.Wier WG, et al. Cellular calcium fluctuations in mammalian heart: direct evidence from noise analysis of aequorin signals in Purkinje fibers. Proc Natl Acad Sci USA. 1983;80:7367–7371. doi: 10.1073/pnas.80.23.7367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kort AA, et al. Fluctuations in intracellular calcium concentration and their effect on tonic tension in canine cardiac Purkinje fibres. J Physiol. 1985;367:291–308. doi: 10.1113/jphysiol.1985.sp015825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhou Z, Lipsius SL. Na+-Ca2+ exchange current in latent pacemaker cells isolated from cat right atrium. J Physiol. 1993;466:263–285. [PMC free article] [PubMed] [Google Scholar]
- 81.Lipsius SL, Gibbons WR. Membrane currents, contractions, and aftercontractions in cardiac Purkinje fibers. Am J Physiol. 1982;243:H77–86. doi: 10.1152/ajpheart.1982.243.1.H77. [DOI] [PubMed] [Google Scholar]
- 82.Kort AA, Lakatta EG. Calcium-dependent mechanical oscillations occur spontaneously in unstimulated mammalian cardiac tissues. Circ Res. 1984;54:396–404. doi: 10.1161/01.res.54.4.396. [DOI] [PubMed] [Google Scholar]
- 83.Rubenstein DS, Lipsius SL. Mechanisms of automaticity in subsidiary pacemakers from cat right atrium. Circ Res. 1989;64:648–657. doi: 10.1161/01.res.64.4.648. [DOI] [PubMed] [Google Scholar]
- 84.Ju YK, Allen DG. How does beta-adrenergic stimulation increase the heart rate? The role of intra-cellular Ca2+ release in amphibian pacemaker cells. J Physiol. 1999;516(Pt 3):793–804. doi: 10.1111/j.1469-7793.1999.0793u.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rigg L, et al. Localisation and functional significance of ryanodine receptors during beta-adrenoceptor stimulation in the guinea-pig sino-atrial node. Cardiovasc Res. 2000;48:254–264. doi: 10.1016/s0008-6363(00)00153-x. [DOI] [PubMed] [Google Scholar]
- 85a.Musa H, et al. Heterogenous expression of Ca2+ handling proteins in rabbit sinoatrial node. J Histochem Cytochem. 2002;50:311–324. doi: 10.1177/002215540205000303. [DOI] [PubMed] [Google Scholar]
- 86.Vinogradova TM, et al. Sinoatrial node pacemaker activity requires Ca2+/calmodulin-dependent protein kinase II activation. Circ Res. 2000;87:760–767. doi: 10.1161/01.res.87.9.760. [DOI] [PubMed] [Google Scholar]
- 87.Cho HS, Takano M, Noma A. The electro-physiological properties of spontaneously beating pacemaker cells isolated from mouse sinoatrial node. J Physiol. 2003;550:169–180. doi: 10.1113/jphysiol.2003.040501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sanders L, et al. Fundamental importance of Na+-Ca2+ exchange for the pacemaking mechanism in guinea-pig sino-atrial node. J Physiol. 2006;571:639–649. doi: 10.1113/jphysiol.2005.100305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hancox JC, Levi AJ, Brooksby P. Intracellular calcium transients recorded with Fura-2 in spontaneously active myocytes isolated from the atrioventricular node of the rabbit heart. Proc Biol Sci. 1994;255:99–105. doi: 10.1098/rspb.1994.0014. [DOI] [PubMed] [Google Scholar]
- 90.Lyashkov AE, et al. Calcium cycling protein density and functional importance to automaticity of isolated sinoatrial nodal cells are independent of cell size. Circ Res. 2007;100:1723–1731. doi: 10.1161/CIRCRESAHA.107.153676. [DOI] [PubMed] [Google Scholar]
- 91.Bogdanov KY, et al. Membrane potential fluctuations resulting from submembrane Ca2+ releases in rabbit sinoatrial nodal cells impart an exponential phase to the late diastolic depolarization that controls their chronotropic state. Circ Res. 2006;99:979–987. doi: 10.1161/01.RES.0000247933.66532.0b. [DOI] [PubMed] [Google Scholar]
- 92.Vinogradova TM, et al. Rhythmic ryanodine receptor Ca2+ releases during diastolic depolarization of sinoatrial pacemaker cells do not require membrane depolarization. Circ Res. 2004;94:802–809. doi: 10.1161/01.RES.0000122045.55331.0F. [DOI] [PubMed] [Google Scholar]
- 93.Vinogradova TM, et al. High basal protein kinase A-dependent phosphorylation drives rhythmic internal Ca2+ store oscillations and spontaneous beating of cardiac pacemaker cells. Circ Res. 2006;98:505–514. doi: 10.1161/01.RES.0000204575.94040.d1. [DOI] [PubMed] [Google Scholar]
- 94.Noma A. Ionic mechanisms of the cardiac pacemaker potential. Jpn Heart J. 1996;37:673–682. doi: 10.1536/ihj.37.673. [DOI] [PubMed] [Google Scholar]
- 95.Lakatta EG, et al. The integration of spontaneous intracellular Ca2+ cycling and surface membrane ion channel activation entrains normal automaticity in cells of the heart’s pacemaker. Ann NY Acad Sci. 2006;1080:178–206. doi: 10.1196/annals.1380.016. [DOI] [PubMed] [Google Scholar]
- 96.Zhang H, et al. Mathematical models of action potentials in the periphery and center of the rabbit sinoatrial node. Am J Physiol. 2000;279:H397–421. doi: 10.1152/ajpheart.2000.279.1.H397. [DOI] [PubMed] [Google Scholar]
- 97.Lakatta EG, et al. Cyclic variation of intracellular calcium: a critical factor for cardiac pacemaker cell dominance. Circ Res. 2003;92:e45–50. doi: 10.1161/01.res.0000055920.64384.fb. [DOI] [PubMed] [Google Scholar]
- 98.Kurata Y, et al. Dynamical description of sinoa-trial node pacemaking: improved mathematical model for primary pacemaker cell. Am J Physiol. 2002;283:H2074–101. doi: 10.1152/ajpheart.00900.2001. [DOI] [PubMed] [Google Scholar]
- 99.Maltsev VA, et al. Diastolic calcium release controls the beating rate of rabbit sinoatrial node cells: numerical modeling of the coupling process. Biophys J. 2004;86:2596–2605. doi: 10.1016/S0006-3495(04)74314-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Maltsev VA, et al. Local subsarcolemmal Ca2+ releases within rabbit sinoatrial nodal cells not only regulate beating rate but also ensure normal rhythm during protein kinase A inhibition. Biophys J. 2005;88:89a–90a. (Abstract) [Google Scholar]
- 101.Maltsev VA, et al. A numerical model of a Ca2+ clock within sinoatrial node cells: Interactive membrane and submembrane Ca2+ cycling provides a novel mechanism of normal cardiac pacemaker function. Biophys J Supplement. 2007:77a. (Abstract) [Google Scholar]
- 102.Maltsev AV, et al. A simple stochastic mechanism of a roughly periodic Ca2+ clock within cardiac cells. Biophys J Supplement. 2007:344a. (Abstract) [Google Scholar]
- 103.Shannon TR, et al. A mathematical treatment of integrated Ca dynamics within the ventricular myocyte. Biophys J. 2004;87:3351–3371. doi: 10.1529/biophysj.104.047449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Vinogradova TM, et al. Sarcoplasmic reticulum (SR) Ca2+ refilling kinetics controls the period of local subsarcolemmal Ca2+ releases (LCR) and the spontaneous beating rate of sinoatrial node cells (SANC) Biophys J Supplement. 2007:31a. (Abstract) [Google Scholar]
- 105.Verheijck EE, et al. Contribution of L-type Ca2+ current to electrical activity in sinoatrial nodal myocytes of rabbits. Am J Physiol. 1999;276:H1064–1077. doi: 10.1152/ajpheart.1999.276.3.H1064. [DOI] [PubMed] [Google Scholar]
- 106.Verheijck EE, et al. Effects of delayed rectifier current blockade by E-4031 on impulse generation in single sinoatrial nodal myocytes of the rabbit. Circ Res. 1995;76:607–615. doi: 10.1161/01.res.76.4.607. [DOI] [PubMed] [Google Scholar]