

ABSTRACT
Treponema pallidum, an obligate human pathogen, has an outer membrane (OM) whose physical properties, ultrastructure, and composition differ markedly from those of phylogenetically distant Gram-negative bacteria. We developed structural models for the outer membrane protein (OMP) repertoire (OMPeome) of T. pallidum Nichols using solved Gram-negative structures, computational tools, and small-angle X-ray scattering (SAXS) of selected recombinant periplasmic domains. The T. pallidum “OMPeome” harbors two “stand-alone” proteins (BamA and LptD) involved in OM biogenesis and four paralogous families involved in the influx/efflux of small molecules: 8-stranded β-barrels, long-chain-fatty-acid transporters (FadLs), OM factors (OMFs) for efflux pumps, and T. pallidum repeat proteins (Tprs). BamA (TP0326), the central component of a β-barrel assembly machine (BAM)/translocation and assembly module (TAM) hybrid, possesses a highly flexible polypeptide-transport-associated (POTRA) 1-5 arm predicted to interact with TamB (TP0325). TP0515, an LptD ortholog, contains a novel, unstructured C-terminal domain that models inside the β-barrel. T. pallidum has four 8-stranded β-barrels, each containing positively charged extracellular loops that could contribute to pathogenesis. Three of five FadL-like orthologs have a novel α-helical, presumptively periplasmic C-terminal extension. SAXS and structural modeling further supported the bipartite membrane topology and tridomain architecture of full-length members of the Tpr family. T. pallidum’s two efflux pumps presumably extrude noxious small molecules via four coexpressed OMFs with variably charged tunnels. For BamA, LptD, and OMFs, we modeled the molecular machines that deliver their substrates into the OM or external milieu. The spirochete’s extended families of OM transporters collectively confer a broad capacity for nutrient uptake. The models also furnish a structural road map for vaccine development.
IMPORTANCE The unusual outer membrane (OM) of T. pallidum, the syphilis spirochete, is the ultrastructural basis for its well-recognized capacity for invasiveness, immune evasion, and persistence. In recent years, we have made considerable progress in identifying T. pallidum’s repertoire of OMPs. Here, we developed three-dimensional (3D) models for the T. pallidum Nichols OMPeome using structural modeling, bioinformatics, and solution scattering. The OM contains three families of OMP transporters, an OMP family involved in the extrusion of noxious molecules, and two “stand-alone” proteins involved in OM biogenesis. This work represents a major advance toward elucidating host-pathogen interactions during syphilis; understanding how T. pallidum, an extreme auxotroph, obtains a wide array of biomolecules from its obligate human host; and developing a vaccine with global efficacy.
KEYWORDS: syphilis, Treponema pallidum, bioinformatics, outer membrane proteins, structural biology, vaccines
INTRODUCTION
Syphilis is a multistage, sexually transmitted infection renowned for its protean clinical manifestations and protracted course (1, 2). Since the start of the new millennium, the disease has undergone a dramatic resurgence in the United States, particularly among men who have sex with men (3), in addition to posing an ongoing global health threat to at-risk populations in resource-poor nations (4). These alarming trends underscore the urgent need for a vaccine with global efficacy (4). The complex natural history of syphilis reflects the invasiveness, immunoevasiveness, and inflammatory potential of its etiological agent, the pathogenic spirochete Treponema pallidum subsp. pallidum (5, 6). As an extreme auxotroph, T. pallidum must obtain a vast array of biomolecules from its obligate human host (7, 8). To support its distinct parasitic lifestyle (6), T. pallidum has evolved an outer membrane (OM) (see Table S1 in the supplemental material, which defines all abbreviations used in this document) whose physical properties, ultrastructure, and composition differ markedly from those of phylogenetically distant Gram-negative bacteria (9–12). The T. pallidum OM is a fragile lipid bilayer lacking lipopolysaccharide (LPS) (7) with a much lower density of membrane-spanning proteins than its Gram-negative counterparts (10, 11). The OM’s paucity of surface-exposed pathogen-associated molecular patterns and membrane-spanning proteins is the ultrastructural basis for the syphilis spirochete’s remarkable capacity for persistence and immune evasion, attributes that have earned it the designation “stealth pathogen” (6).
The Gram-negative OM is an asymmetric lipid bilayer consisting of an inner (periplasmic) leaflet of glycerophospholipids and an outer leaflet of LPS, the highly inflammatory glycolipid that establishes the permeability barrier (13). In disease-causing organisms, the OM constitutes the host-pathogen interface, serving adhesive, invasive, and immunoevasive functions integral to a bacterium’s parasitic program (14–16). The structure also houses the surface-exposed targets for protective antibodies elicited during infection or by immunization (17–20) and, therefore, is the focus of vaccine development. A characteristic feature of this unusual bilayer is its high density of integral membrane proteins that, with rare exceptions (21), adopt the versatile β-barrel conformation in which an even number of antiparallel, amphipathic β-strands circularize to form a closed structure, typically with a central channel (22, 23). Bridging adjacent strands on the external side of the OM are large extracellular loops (ECLs) that mediate interactions with host cells and tissue components (24–26) and contain B-cell epitopes (BCEs) subject to sequence/antigenic variation due to host immune pressure (27–30). Gram-negative β-barrel proteins serve transport-related functions that contribute to the maintenance of cellular homeostasis. General porins are abundant, trimeric aqueous channels for the passive diffusion of small, water-soluble molecules (31, 32), while low-abundance, energy-coupled, monomeric transporters serve as substrate-specific, ligand-gated channels for the uptake of micronutrients such as iron, nickel, and vitamin B12 (33). Gram-negative bacteria also express OM transporters that enable growth on alternative carbon sources, such as maltodextrins (LamB) and fatty acids (FadL) (34, 35). BamA and LptD are the terminal β-barrel components of the molecular machines that insert nascent outer membrane proteins (OMPs) and LPS, respectively, into the OM bilayer (36–38). The OMs of Gram-negative bacteria also contain a considerable number of proteins that form 8- to 10-stranded monomeric β-barrel structures that are implicated in the transport of small molecules and cell adhesion (39, 40). In addition to shuttling molecules into the cell, Gram-negative bacteria need to expel exogenous and endogenous toxins. They achieve this by coupling cytoplasmic membrane (CM)-associated efflux pumps to an interesting variation on the β-barrel theme: trimeric outer membrane factors (OMFs) in which each monomer contributes 4 β-strands to complete a single 12-stranded β-barrel (41, 42).
Among the many factors hampering the molecular characterization of T. pallidum strain Nichols rare OMPs was their lack of sequence relatedness to prototypical OMPs of Gram-negative bacteria (11). To overcome this hurdle, we mined the T. pallidum genome for proteins predicted to form β-barrels (6, 43), the hallmark conformation of OMPs in double-membrane organisms (22, 23), and subsequently demonstrated that selected candidate OMPs, in fact, adopt this conformation in vitro and are surface exposed in vivo (44, 45). The expansion of OMP structures in the RCSB PDB database (www.rcsb.org), coupled with newly developed structural algorithms (46–48), has enabled us to begin to generate three-dimensional (3D) models for candidate OMPs previously identified by secondary structure predictions (11).
For the iteration of the T. pallidum (Nichols) OMP repertoire (OMPeome) described here, we refined our structural models using solved structures as the templates, an expanded battery of computational and bioinformatic tools, recently published experimental data, and small-angle X-ray scattering (SAXS) analysis of selected recombinant aqueous soluble periplasmic domains. Our analyses revealed that, in addition to the BamA and LptD “stand-alone” proteins with novel structural features, the spirochete’s OM contains extended families of transporters and efflux pump OMFs that presumably endow the organism with a broad capacity for nutrient uptake and molecule extrusion. For BamA, LptD, and efflux pump OMFs, we identified and modeled the components of the cognate molecular machines that enable these OMPs to deliver their unique cargos into the OM bilayer or the external milieu. SAXS provided new insight into the function of T. pallidum’s β-barrel assembly machine (BAM) apparatus and additional biophysical evidence for the bipartite membrane topology of full-length members of the T. pallidum repeat (Tpr) family. Collectively, the 3D models reveal that T. pallidum has evolved a unique repertoire of OMPs to address its physiological needs while meeting the demands of stealth pathogenicity. Strategies to elicit protective antibodies by immunization with recombinant treponemal proteins require detailed knowledge of the molecular architecture of the spirochete’s OM as well as the membrane topology and structure of candidate vaccinogens. Thus, the models described here also provide a structural road map for the development of a broadly protective vaccine for a disease that has afflicted humankind for centuries.
RESULTS AND DISCUSSION
T. pallidum BamA (TP0326) possesses a divergent β-barrel domain and a highly flexible POTRA arm.
BamA is the central catalyst of the BAM apparatus, the molecular machine that inserts newly synthesized OMPs into the OMs of diderm bacteria (49). In Escherichia coli, BamA consists of a C-terminal β-barrel and an extended N-terminal periplasmic region containing five polypeptide-transport-associated (POTRA) domains (50) (Fig. 1A). Following export across the CM by the Sec translocon, nascent OMPs are transported to the POTRA arm of BamA by the periplasmic chaperones Skp and SurA (51). The POTRA arm then works in concert with the accessory lipoproteins BamBCDE to thread unfolded OMPs into the lumen of the β-barrel (52). To accept its cargo, the BamA β-barrel adopts a conformational state in which the periplasmic aqueous pore is open and the lateral gate is closed (53).
FIG 1.

The highly flexible POTRA1-5 of T. pallidum BamA lack BamBCDE-interacting residues. (A and B) Ribbon diagrams for the crystal structure of E. coli BamA (PDB accession number 5D0Q) and the structural model of TP0326. Both proteins are in the same orientation. The arrows indicate ECL4 in both proteins. The β-strands (β1 and β16) forming the lateral gate are shown in magenta. The polyserine tract in ECL7 of TP0326 is displayed as green spheres. A nonconservative amino acid substitution (L593Q) in ECL4 of TP0326 is depicted as an orange sphere with the BCE (shown as a blue ribbon). (C and D) The LptD interaction sites (59) in the β-barrel domain of E. coli BamA (PDB accession number 5D0Q) and their equivalent residues in the 3D model of TP0326 are labeled and shown as sticks. (E and F) BamBCDE-interacting residues of the E. coli POTRA arm (PDB accession number 5D0Q) and their equivalent residues in the 3D model of TP0326 POTRA1-5 are depicted as blue, green, cyan, and red surfaces, respectively. Both POTRA arms (E and F) are in the same orientation. (G) Rg distribution of EOM-generated major conformations for the SAXS data of TP0326 POTRA1-5. Three major conformations (compact, intermediate, and extended) for TP0326 POTRA1-5 are shown in the 3D models. (H) The SAXS envelope (gray surface) of TP0326 POTRA1-5 overlaid with the 3D model of its compact conformation (green 3D model in panel G).
The discovery that T. pallidum contains a BamA ortholog (TP0326), its first characterized rare OMP (54), provided strong support for the assumption that, as in Gram-negative bacteria, the spirochete’s OMPs adopt a β-barrel conformation. Here and elsewhere, we evaluated the integrity of our structural models using MolProbity (55) and WinCoot (56). Our 3D structural model of the BamA β-barrel (Table 1) revealed features typical of Gram-negative BamAs, such as a lateral gate formed by β-strands 1 and 16; a dome comprised of ECL4, -6, and -7 that occludes the barrel’s extracellular opening; and the projection of ECL6 into the barrel interior (44). However, the model also exhibited features suggesting that evolution has tailored T. pallidum’s BamA to catalyze the OM insertion of a distinctive repertoire of OMPs while contending with host-derived immune selection pressures (30, 44). In addition to having a positively charged lumen and different anchoring residues within the barrel for ECL6 (44), TP0326 also harbors an unusual ECL (ECL7) with a two-tiered polyserine-rich tract found only in pathogenic treponemes (44, 57) and a large ECL (ECL4) harboring an antigenically variable BCE (Fig. 1B), which can be targeted by opsonic antibodies (30, 44). Striking molecular differences were also noted between the BAM complex of T. pallidum and its E. coli prototype. The T. pallidum BAM complex is larger than that of E. coli (∼300 to 400 kDa versus 232 kDa), lacks BamBCDE subunits based on genome mining for structural as well as sequence orthologs, and, notably, dissociates in relatively low concentrations of the detergent n-dodecyl-β-d-maltoside (DDM) (54, 58).
TABLE 1.
Structural modeling of the T. pallidum OMPeome
| T. pallidum protein | Modeling softwarea | Identified structural ortholog; organism; PDB accession no. | % sequence identity with orthologb |
|---|---|---|---|
| Components of the BAM complex | |||
| TP0326 | ModWeb | BamA; E. coli; 4K3B | 19 |
| DUF domain of TP0325 | SWISS-MODEL | DUF490 domain of TamB; E. coli; 5VTG | 22 |
| Components of the Lpt complex | |||
| TP0515 | I-TASSER | LptD; Shigella flexneri; 4Q35 | 20 |
| TP0784 | Phyre2 | LptC; Vibrio cholerae; 6MJP | 18 |
| TP0785 | Phyre2 | LptA; E. coli; 2R1A | 18 |
| TP0786 | Phyre2 | LptB; Burkholderia phymatum; 4WBS | 50 |
| TP0883 | Phyre2 | LptF; Vibrio cholerae; 6MJP | 15 |
| TP0884 | Phyre2 | LptG; Vibrio cholerae; 6MJP | 17 |
| 8-stranded β-barrels | |||
| TP0126 | Phyre2, SWISS-MODEL | OmpW; E. coli; 2MHL (NMR structure), 2F1T (crystal structure) | 19 |
| TP0733 | Phyre2, SWISS-MODEL | 23 | |
| TP0479 | I-TASSER, SWISS-MODEL | OprG; Pseudomonas aeruginosa; 2N6L (NMR structure), 2X27 (crystal structure) | 16 |
| TP0698 | I-TASSER, SWISS-MODEL | 18 | |
| FadL-like proteins | |||
| TP0548 | trRosetta | Long-chain-fatty-acid transporter FadL; E. coli; 1T1L | 17 |
| TP0856 | trRosetta | 19 | |
| TP0858 | trRosetta | 18 | |
| TP0859 | trRosetta | 17 | |
| TP0865 | trRosetta | 18 | |
| Components of efflux systems | |||
| TP0966 | trRosetta | OMF (TolC); E. coli; 1TQQ | 15 |
| TP0967 | trRosetta | 16 | |
| TP0968 | trRosetta | 16 | |
| TP0969 | trRosetta | 16 | |
| TP0962 | Phyre2 | IM-spanning domain of MacB; Acinetobacter baumannii; 5GKO | 25 |
| TP0963 | Phyre2 | IM-spanning domain of MacB; Acinetobacter baumannii; 5GKO | 27 |
| TP0964 | Phyre2 | Cytoplasmic domain of MacB; Acinetobacter baumannii; 5GKO | 51 |
| TP0965 | Phyre2 | MacA; E. coli; 5NIK | 19 |
| TP0790 | Phyre2 | AcrB; E. coli; 5V5S | 21 |
| T. pallidum repeat proteins: all members of Tprs | trRosetta | No structural template was identified | NA |
Recent biochemical and structural studies in E. coli have established that folding of nascent OMPs occurs within the lumen of the BamA β-barrel and, for one substrate, LptD, have identified residues vital to this process (59). To determine whether these essential residues are conserved in T. pallidum BamA, we performed structure-based sequence alignment of the TP0326 3D model with the crystal structures of E. coli BamA (PDB accession numbers 5D0Q and 5D0O) (60). The presence of an LptD ortholog (TP0515 [see below]) in T. pallidum (11) supports the validity of this comparison. Figure 1C and D show that E470, R488, and N666, hydrophilic residues in E. coli BamA essential for LptD folding (59), have been replaced by K484, D502, and D718 in TP0326. In separate study, Hart et al. (61) demonstrated that an E470K mutation in E. coli BamA bypasses the BAM machinery’s requirement for BamBCDE albeit with decreased efficiency. K484, located in the equivalent position in TP0326 (Fig. 1C and D; see also Fig. S1A in the supplemental material), could serve as a comparable “gain-of-function” replacement. While TP0326 maintains a lateral gate for the egress of OMPs, the positions of critical gate residues are swapped in E. coli and T. pallidum (Fig. 1C and D; Fig. S1A): N427 and Q803 on β-strands 1 and 16, respectively, in E. coli BamA (59) have been replaced by Q436 and N827 in TP0326. Thus, whereas the overall mechanism for folding nascent OMPs appears to be preserved in T. pallidum, substitutions in the BamA barrel collectively imply that specific residue interactions differ substantially. Given the much lower replication rate of T. pallidum (30 h in vivo versus 20 min for E. coli) (62), these divergences could also serve as an allosteric mechanism to coordinate the kinetics of OMP folding and OM insertion with the growth rate (63). Other factors, such as the difference in lipid compositions between the T. pallidum and Gram-negative OMs (64), particularly the absence of LPS in the former, may also be important in regulating the rate at which the β-barrel of BamA inserts nascent OMPs into the OM bilayer (65).
As shown in Fig. 1E, the contact residues of the five POTRA domains with BamBCDE have been extensively mapped from the crystal structures of the E. coli BAM complex (PDB accession numbers 5D0Q, 5D0O, and 5EKQ) (60, 66). Not surprisingly, most of these interacting residues are absent in the POTRA arm of TP0326 (Fig. 1F). Although T. pallidum contains Skp (TP0327) (54) and a recently identified SurA (TP1016) ortholog (67), the differences in the POTRA arms and the lack of BAM accessory subunits in T. pallidum strongly imply that the syphilis spirochete employs a divergent mechanism for the transfer of nascent OMPs to its BamA β-barrel.
In addition to the BAM complex, E. coli contains a translocation and assembly module (TAM) required for the export of a subset of OMPs, particularly autotransporters, to the cell surface (68, 69). The TAM complex consists of TamA, also a member of the OMP85 superfamily, and a CM protein, TamB, containing a periplasmic DUF490 domain that interacts with the POTRA arm of TamA. tp0325, the gene immediately upstream of tp0326 (Fig. S1B) and likely cotranscribed with it, encodes a large (108-kDa) TamB ortholog with a C-terminal DUF490 domain (68). Webb et al. (70) proposed that T. pallidum harbors a chimeric BAM/TAM OM biogenesis machine. In support of this conjecture, Iqbal and colleagues (71) demonstrated that the TamB ortholog (bb0794) of Borrelia burgdorferi, also upstream of bamA (bb0795), is part of the Lyme disease spirochete’s BAM complex. Homology modeling reveals that, like the TamA interaction partner in E. coli (72), the DUF490 domain of TP0325 is an elongated taco-shaped molecule composed entirely of β-sheets and random coils (Table 1 and Fig. S1C).
Using SAXS, we solved the solution structure of aqueous soluble, monomeric (∼45-kDa), recombinant POTRA1 to -5 (POTRA1-5) of TP0326 (Fig. S2A) to gain insights into the mechanism of transfer of nascent OMPs. The structural model of POTRA1-5 (Fig. 1F) poorly matched the experimental scattering data (χ2 = 9.0) (Fig. S2B). Consequently, we assessed whether the SAXS data represent a mixture of conformations by employing an ensemble optimization method (EOM) (73). The EOM generated a three-state ensemble (compact, intermediate, and extended conformations), which dramatically improved the χ2 value from 9.0 to 1.0 (Fig. 1G and Fig. S2B). Ab initio shape reconstruction from the SAXS data, using 10 independent bead models (see Materials and Methods), yielded a multidomain envelope. The EOM-generated structural model of the compact conformation of POTRA1-5 (Fig. 1G) shows an excellent fit for the ab initio molecular envelope (Fig. 1H). Based on the flexibility of the T. pallidum POTRA1-5 solution structure (Fig. 1G and H) and previously solved E. coli BamA structures in multiple conformations (37), we can propose a working model for POTRA-mediated transfer to the BamA channel of nascent treponemal OMPs delivered by the periplasmic chaperones SurA and Skp (Fig. S3). Interaction with the DUF490 domain of TP0325 with TP0326 occurs when the POTRA arm is extended. In this conformation, the periplasmic pore of TP0326 is closed, and the lateral gate is open, permitting the egress of substrates folding within the barrel. The receipt of unfolded, newly exported OMPs from periplasmic chaperones results in the detachment of the POTRA from TP0325, enabling it to assume the bent, “transfer” conformation, which opens the periplasmic pore and closes the lateral gate. This scenario potentially explains the previously observed dissociable nature of the T. pallidum BAM complex (54).
T. pallidum harbors an LptD ortholog and an Lpt apparatus for transport of an uncharacterized substrate other than LPS.
The LPS transport (Lpt) machinery consists of seven essential proteins (LptABCDEFG) that mediate the transfer of newly synthesized LPS from the periplasmic side of the CM to the OM (74). The inner membrane (IM) components LptB2FG form an ATP binding cassette (ABC) transporter that uses the energy from the hydrolysis of ATP to extract LPS from the CM and push it along the periplasmic jelly roll bridge formed by LptCAD (the so-called “Pez model” for LPS transport) (74). A translocon, consisting of the integral OMP LptD and the LptE lipoprotein “plug” within the lumen of the LptD β-barrel, transfers LPS from the periplasmic bridge to its final destination: the outer leaflet of the OM (74). LptD is a 26-stranded β-barrel with a hydrophilic interior, ECLs that close off the external opening of the barrel and fold into the lumen, a lateral gate between β-strands 1 and 26, and an N-terminal β-jelly roll domain that completes the periplasmic bridge (Fig. 2). A hydrophobic slide within the β-jelly roll domain of LptD guides LPS through a hydrophobic intramembrane hole situated within the bilayer and contiguous with the lateral gate and negatively charged exit pore (Fig. 2) (38, 75). Following transfer from the periplasmic bridge, the lipid A moiety of LPS inserts into the OM through the hydrophobic hole, while the O-antigen is delivered to the extracellular milieu through the lateral gate and exit pore with the assistance of LptE (38, 75). A functional Lpt pathway depends upon the proper folding of LptD, which requires LptE and involves a complex cycle of disulfide bond rearrangements between four conserved cysteines in the β-jelly roll domain and the β-barrel (76).
FIG 2.

Structural model of T. pallidum LptD (TP0515). Shown are cartoon diagrams of the LptD-LptE complex crystal structure (PDB accession number 4Q35) from Shigella flexneri (left) and the 3D structure model of the T. pallidum LptD ortholog (TP0515) (right). Both proteins are in the same orientation. The β-jelly roll N-terminal domain and 26-stranded β-barrel are shown in green and cyan, respectively. LptE of S. flexneri and the C-terminal extension of TP0515 are depicted as orange ribbons. With both proteins, the β-strands (β1 and β26) of the lateral gate and β2 are shown in magenta. Essential amino acids of the lateral gate (N232, P231, and P246) (38) and the intramembrane hole (W180, F203, Y205, V208, F211, and Y212) (75) and their T. pallidum TP0515 equivalents are displayed as sticks. Residues of intramembrane holes are zoomed-in for clarity. The four Cys residues required for the folding of S. flexneri LptD (76) are shown in red. Cys residues in T. pallidum LptD are shown as dark gray sticks.
Because T. pallidum lacks an LPS biosynthetic pathway (6), we were surprised to discover that TP0515, originally identified as a candidate OMP based on secondary structure predictions (43), is a structural ortholog of LptD (11). Moreover, as shown in Fig. S4, with the exception of LptE, T. pallidum contains all the components of the Lpt pathway (Table 1) (74). The 3D model of TP0515, built by an ab initio modeling program, I-TASSER (Table 1) (46), contains the hallmark features of Gram-negative LptD described above (Fig. 2). DiscoTope 2.0 analysis of TP0515 predicts that several large ECLs (ECL3, -5, -6, -7, and -12) in its β-barrel domain contain strong BCEs (Fig. S4 and Table S2), which could serve as antigenic targets for host recognition. The likely cargo for the Lpt pathway in T. pallidum is its uncharacterized glycolipids (77), and this probably accounts for structural differences between Gram-negative LptD and TP0515 (Fig. 2). Moreover, it seems reasonable to presume that the putative glycolipid cargo is delivered to the outer leaflet of the OM where it would serve a barrier function. Most notable is an unstructured hydrophilic C-terminal extension in TP0515 that I-TASSER positioned inside the hydrophilic lumen of the β-barrel (Fig. 2); it is tempting to speculate that this region fulfills the functional roles of LptE. The essential hydrophobic residues (75) of the intramembrane hole in TP0515 are well conserved (Fig. 2). Although TP0515 maintains asparagine residues in its lateral gate (N227 and N848 in β1 and β26, respectively), it lacks one of the two essential prolines in the β2 strand (Fig. 2) needed for lateral gate function in Gram-negative LptDs (38), implying somewhat different mechanics of lateral gate function. Whereas Gram-negative LptD has a negatively charged exit pore to expel negatively charged LPS into the outer leaflet of the OM, the exit pore of TP0515 is positively charged (Fig. S5A) and presumably exports an uncharacterized, positively charged glycolipid (77). Interestingly, the exit pore of LptD (LIC11458) from Leptospira interrogans, a spirochete that contains LPS, is negatively charged (Fig. S5A). Finally, TP0515 has four cysteines, but their positions are not conserved with respect to the cysteines in Gram-negative LptD (Fig. 2 and Fig. S5B).
Eight-stranded β-barrels.
Eight-stranded β-barrels comprise a group of functionally versatile OMPs that are ubiquitous in Gram-negative bacteria (78). Members of this group have either a single-domain β-barrel architecture (e.g., OmpW, Opr, and CarO) (79–81) or a bipartite topology (OmpA) (82) in which the N-terminal domain forms a β-barrel and the periplasmic C-terminal domain interacts with peptidoglycan. Despite their conserved β-barrel architecture, members of this group have low overall sequence similarity, with the ECLs, in particular, showing extensive diversity (83). E. coli OmpW, Pseudomonas aeruginosa OprG, and Acinetobacter baumannii CarO are the most extensively studied representatives of the single-domain subgroup. The crystal structures of OmpW and OprG revealed a narrow hydrophobic channel leading to a lateral opening in the barrel wall (79, 84), while CarO has a hydrophilic channel and lacks a lateral gate (85). It is often stated that OmpW transports small nonpolar substrates (86), OprG transports small neutral amino acids (87), and CarO mediates the uptake of glycine and ornithine (85). However, there is considerable disagreement about the cargos that these proteins handle and their mechanisms of transport (86–88). The dynamic structures of OmpW (PDB accession number 2MHL), OprG (PDB accession number 2N6L), and AlkL of Pseudomonas putida (transports alkanes) (PDB accession number 6QAM) solved by nuclear magnetic resonance (NMR), and their electrostatic potentials, appear to provide a more uniform picture of structure-function relationships in the 8-stranded β-barrels (Fig. S6). The NMR structures revealed that each has a narrow channel, long flexible ECLs, and short β-strands with lengths closely matching the thickness of the OM bilayer (87, 89, 90). These studies, bolstered by molecular simulations, yield a mechanistic model in which substrate transport occurs via dynamic mobility of the ECLs and transient strand separation (89). Eight-stranded β-barrels also contribute to bacterial virulence by virtue of their ECLs (78). As an example, Ail of Yersinia pestis binds to negatively charged extracellular matrix (ECM) components via its positively charged ECLs (83).
The T. pallidum Nichols genome encodes two proteins (TP0624 and TP0292) with OmpA-like domains containing peptidoglycan recognition motifs; neither, however, contains a β-barrel domain (91–93). The genome encodes four single-domain 8-stranded β-barrels (TP0126, TP0479, TP0698, and TP0733). TP0126 and TP0733 were identified by ab initio modeling using the OmpW crystal structure (PDB accession number 2F1T) (11, 94). TP0479 and TP0698, discovered here from a reanalysis of T. pallidum’s potential β-barrel-forming proteins (43), best fit the crystal structure of OprG (PDB accession number 2X27). Knowing that the NMR and crystal structures for 8-stranded barrels can be at variance, we also used the NMR structures of OmpW (PDB accession number 2MHL) (87) and OprG (PDB accession number 2N6L) (90) as the templates to construct homology models (Table 1). We consider the NMR-based models to be preferable because they provide greater coverage of the TP0126 and TP0733 sequences and yield barrels and ECLs with more open conformations (Fig. 3A). Calculations done using MOLEonline (95) predict that all four proteins have a narrow, continuous channel (Fig. 3B) consistent with possible transport functions. Typical of 8-stranded barrels, structure-based sequence alignments show that the ECLs vary markedly in amino acid composition and length (Fig. S7), with some (e.g., ECL4 of TP0479) likely being highly flexible. As a consequence of this diversity, the ECLs vary in both surface charge (Fig. 3C) and density/distribution of BCEs (Fig. 3A and Table S3). These models support the notion that the syphilis spirochete’s 8-stranded β-barrels transport a variety of small-molecule substrates and engage in diverse interactions at the host-pathogen interface. In other bacteria, 8-stranded β-barrels have shown promise as vaccine candidates (16, 78). The presence of multiple surface-exposed BCEs in the T. pallidum 8-stranded barrels (Fig. 3A) suggests that these proteins could also be targets for a protective immune response. It is noteworthy, therefore, that recombinant TP0126 failed to protect in the rabbit model of experimental syphilis (96). As Haynes et al. noted (96), it remains to be determined whether this negative result reflects the biology of the native protein, which has been proposed to undergo phase variation (94), and/or deficiencies in their immunization/challenge protocol.
FIG 3.

Eight-stranded β-barrels in T. pallidum. Shown are structural models with BCEs (A), predicted luminal cavities (B), and comparative electrostatics (C) for TP0126, TP0733, TP0479, and TP0698. BCEs are shown as transparent surfaces in panel A. The luminal cavities are shown as gray surfaces in panel B.
FadL-like 14-stranded β-barrels.
Although biomembranes typically are highly permeable to long-chain fatty acids (LCFAs), Gram-negative OMs are a notable exception because of the permeability barrier created by an outer leaflet composed of LPS (31). Consequently, the transport of LCFAs across the OM of Gram-negative bacteria requires a dedicated transporter (34, 97). Based on the crystal structure of E. coli FadL (PDB accession number 1T1L), the proposed pathway for fatty acid uptake involves two low-affinity binding sites within a hydrophobic groove formed by ECL3 and -4 and a conserved high-affinity site at the entrance of the barrel (Fig. S8A) (34). Once inside the barrel, the substrate disrupts a hydrogen bond between a conserved glycine (G103) in β-strand 3 and a phenylalanine (F3) in a hydrophilic, N-terminal “hatch” region that plugs the lumen (34). Disruption of this bond induces a transient lateral opening at the kink formed by residues SNYG in β-strand 3 (S3 kink); a conserved Asn-Pro-Ala (NPA) motif in the hatch is essential for this substrate-induced conformational change (98). To date, only two other FadL-like OMPs, TbuX of Ralstonia pickettii (PDB accession number 3BRY) and TodX of Pseudomonas putida (PDB accession number 3BRZ), which transport aromatic hydrocarbons, have been structurally characterized (99). Compared to E. coli FadL, both structures have similar LCFA high-affinity binding sites but less pronounced S3 kinks and continuous channels extending through their barrels.
Although a previous study proposed that hydrocarbon substrates traverse the channels of TbuX and TodX to reach the periplasm, a recent investigation provided evidence that traversal of the lateral gate would be the preferred mechanism. Although a previous study (99) proposed that hydrocarbon substrates traverse the TbuX and TodX channels to reach the periplasm, a recent investigation (169) provided evidence that aromatic compounds exit their barrels via a lateral gate.
How T. pallidum obtains LCFAs, which it cannot synthesize (7), has been a long-standing question (77). Although the syphilis spirochete’s OM is more permeable to LCFAs than that of E. coli (100), the discovery that it harbors five FadL orthologs (TP0548, TP0856, TP0858, TP0859, and TP0865) (Fig. S8B) (11) argues that the diffusion of LCFAs alone is not sufficient to meet the bacterium’s needs. Moreover, the presence of five paralogous FadL-like OMPs suggests that this family imports a wider range of essential hydrophobic nutrients. Flavins might be one such substrate. In addition to being a fatty acid auxotroph, recent studies by Deka and colleagues (101) have called attention to the spirochete’s “flavin-centric” lifestyle as a basis for many redox biochemical reactions. Flavins have limited water solubility (102), and T. pallidum lacks the ability to synthesis them (6). An alignment percentage matrix (Table S4) shows that T. pallidum FadL-like proteins fall into three subgroups: TP0548 (subgroup I), TP0856 and TP0858 (subgroup II), and TP0859 and TP0865 (subgroup III). To better understand the evolutionary and functional relationships between T. pallidum FadL-like proteins and other bacterial orthologs, we performed a sequence-based phylogenetic analysis of 415 unique FadLs from the Pfam database (http://pfam.xfam.org) (Fig. S9). Interestingly, the T. pallidum FadL-like proteins are much more closely related to R. pickettii TbuX and P. putida TodX than to E. coli FadL and cluster on the same branch of the tree, suggesting evolution from a single precursor.
Three-dimensional ab initio modeling using either Phyre2 or I-TASSER predicts that all five FadL-like proteins form 14-stranded β-barrels with an unstructured N-terminal hatch within the barrel lumen. However, neither program provided complete coverage for TP0548, TP0859, or TP0865. More specifically, TP0859 and TP0865 have unmodeled stretches at their N and C termini, while TP0548 has an unmodeled stretch at its C terminus (data not shown). Additionally, ECL4 of TP0858 was incomplete. We then used trRosetta (48), an advanced computational program based on direct energy minimization, in an attempt to obtain complete structural models (Fig. 4, Table 1, and Fig. S8B). The complete trRosetta models predict that N- and C-terminal regions of TP0548, TP0859, and TP0865 (subgroups I and III) possess two structural features not seen in the solved crystal structures (34, 99): (i) the N-terminal hatch regions extend from within the barrel to the external surface, where they form α-helices that overlay the entrances (Fig. 4), and (ii) the C-terminal stretches form three α-helical bundle domains attached to the β-barrel by short linkers. A search of all FadL members in the Pfam database failed to identify any other orthologs containing a similar C-terminal domain. Based on its location distal to the β-barrel and predicted surface hydrophilicity, we assigned the C-terminal α-helical bundles to the periplasmic compartment (Fig. 4). MOTIF Search (www.genome.jp/tools/motif) identified a tetratricopeptide repeat (TPR) motif distributed over two antiparallel α-helices of each helical bundle (Fig. 4 and Fig. S8B). Such motifs typically form a hydrophobic groove between the α-helices that mediates protein-protein interactions (103); analysis using YRB (104) revealed a potential hydrophobic cleft in each C-terminal domain (Fig. S10). Phyre2, I-TASSER, and trRosetta all predict that the five FadL orthologs lack the LCFA high-affinity binding site; however, the luminal plug portions of the hatch regions contain either a highly conserved NPA motif (TP0548, TP0856, and TP0858) or an NAA variant (TP0859 and TP0865) (Fig. S8B). Hydrophobicity analysis by the YRB program (104) revealed that each of the treponemal FadL-like proteins contains hydrophobic patches extending from the rim through the barrel (Fig. S11A), providing a potential passageway for hydrophobic substrates into the periplasm. trRosetta also predicts that ECL4 of TP0858 is a long cystine-rich loop comparable to ECL4 of TP0856 (Fig. 4); both could form rigid, disulfide-bonded scaffolds to facilitate the capture of the substrate from the extracellular milieu. In this regard, the extended hydrophobic cleft in ECL4 of TP0858 is particularly noteworthy (Fig. S11B). All five T. pallidum FadL-like proteins harbor one or more predicted conformational BCEs (Fig. 4 and Table S5). Members of subfamilies I and III contain predicted BCEs in their hatch regions, ECLs, and C-terminal extensions, while TP0856 and TP0858 have predicted BCEs only in their ECLs or hatch domains, respectively. These predictions suggest that family members are subject to various degrees of host immunological pressure. Three (tp0856, tp0858, and tp0859) of the five genes are cotranscribed (Fig. S11B and C), suggesting that their gene produces function in a coordinated fashion. It is widely assumed that an effective syphilis vaccine has to target multiple OMPs (105). The coexpression of these three OMPs makes them attractive vaccine candidates since their ECLs would be accessible to antibodies at the same time.
FIG 4.

Three T. pallidum FadL-like proteins have a bipartite membrane topology. β-Barrel domains are shown in various cyan shades. In all models, the N-terminal hatch regions are shown in magenta. BCEs are depicted as transparent surfaces. Cysteine residues in the cysteine-rich ECL4 of TP0856 and TP0858 are represented as sticks. The C-terminal helical domains of TP0548, TP0859, and TP0865 are shown in orange and indicated by arrowheads. The TPR motifs in the C-terminal domains are shown in black.
It remains to be determined how T. pallidum FadL-like proteins transfer hydrocarbon substrates to the periplasm. Once in the periplasm, however, the transport pathways might diverge based on the physical properties of the substrates, particularly solubility. Conceivably, TP0548, TP0859, and TP0865 transport more hydrophobic substrates. The interaction between their C-terminal α-helical domains and the TRAP transporter (TP0956/TP0957) described by Norgard and colleagues (106, 107) would establish a mechanism to shuttle less-water-soluble nutrients across the periplasm, delivering them to a dedicated CM permease (TP0958). TP0856 and TP0858 would handle more-water-soluble cargo discharged directly into the periplasmic compartment. The periplasmic lipocalin TP0751, which we recently demonstrated has a hydrophobic rim capable of binding small molecules (108), might also serve as a conduit of cargo(s) imported by one or more members of the FadL-like family.
Treponema pallidum repeat proteins.
Arguably, the most important initial discovery from the T. pallidum Nichols genomic sequence was the Treponema pallidum repeat (Tpr) family of proteins, whose members have sequence homology to the major outer sheath protein (MOSP) of Treponema denticola (7, 109), a known trimeric pore-forming adhesin (110–113) and a major virulence determinant (114). Centurion-Lara and coworkers divided the Tprs into three subfamilies based on sequence relationships (109) (Fig. S12). In the Nichols strain, TprA and TprF are truncated at their C-terminal ends.
Full-length Tprs consist of an extreme amino-terminal stretch (∼50 amino acids [aa]), a conserved N-terminal domain related to the corresponding domain in the N-terminal portion of T. denticola MOSP (MOSPN), a central region that varies in both length and sequence among family members (central variable region [CVR]), and a conserved C-terminal domain related to the corresponding domain in MOSP (MOSPC) (6, 111). Previous examinations of subfamily I Tprs (TprC/D and TprI) revealed that their MOSPC domains form amphiphilic, trimeric β-barrels with porin activity in vitro and are surface exposed in T. pallidum (45, 115). In contrast, the Tpr MOSPN domains and CVRs are water soluble in vitro and periplasmic in T. pallidum. The truncated TprF is water soluble in vitro and periplasmic in T. pallidum. We undertook further analyses to expand our knowledge of Tpr domain architecture beyond subfamily I to the entire family.
(i) Domain analysis and modeling predict a tridomain architecture of full-length Tprs.
According to the updated Pfam database (116), all full-length Tprs possess MOSPN and MOSPC domains (Fig. S12). A comparative percentage alignment matrix of all Tpr MOSPN and MOSPC domains revealed less than 23% similarity between the two domains (Table S6). This comparison of amino acid sequences between the two domains suggests that they have distinct evolutionary origins. While the percent similarities within the MOSPN and MOSPC domains are similar (Table S7), their stretches of amino acid similarity differ in distribution (Fig. S13). With both domains, diversity is generated by block insertions as well as amino acid substitutions. Particularly noteworthy in comparisons with the MOSPN and MOSPC domains are the marked differences in the lengths and amino acid compositions of the CVRs (Fig. S12 and S13). Subfamily II CVRs are considerably larger than those in the other subfamilies, also as a result of block insertions, while the CVR of TprK is the smallest (Fig. S13). Collectively, these results imply that the variable length of CVRs, resulting from the wholesale insertion of sequence cassettes, was a major factor in the evolution of the Tprs (111). Previous unsuccessful efforts to predict secondary structures for the CVRs prompted us to consider the possibility that they are unstructured. A search for disorder-based binding regions using the ANCHOR program (117) predicts that several Tprs have short molecular recognition features (MoRFs) (∼6 to 18 aa in length) (Fig. S12). MoRFs are expected to undergo disorder-to-order transitions upon interaction with specific protein partners (118).
In the absence of sequence or structural orthologs of the Tprs, we previously used the TMBpro Web server to transform the MOSPC domain of TprC/D into a 10-stranded β-barrel topology (30); this conformation is supported by our biophysical data (45). Given that the TMBpro Web server (119) lacks a modeling module to predict secondary and tertiary structures unbiasedly, we used the more accurate ab initio modeling program trRosetta for the full-length Tprs (Table 1). trRosetta was unable to generate a β-barrel model for either the full-length polypeptides or individual domains, presumably reflecting the lack of structural templates in the databases. However, it predicted that all nine full-length Tprs have a tridomain architecture with domain boundaries (Fig. 5A) corresponding closely to those predicted by Pfam (Fig. S12). The MOSPN and MOSPC domains are predominantly β-stranded, in agreement with secondary structure analyses previously obtained by circular dichroism (CD) spectroscopy (45, 115). Importantly, trRosetta predicts that all CVRs, including the diminutive CVR of TprK, consist exclusively of α-helices (Fig. 5A). The predicted conservation of secondary structure for the CVRs is noteworthy given their low degree of sequence similarity (Fig. S13 and Table S7).
FIG 5.
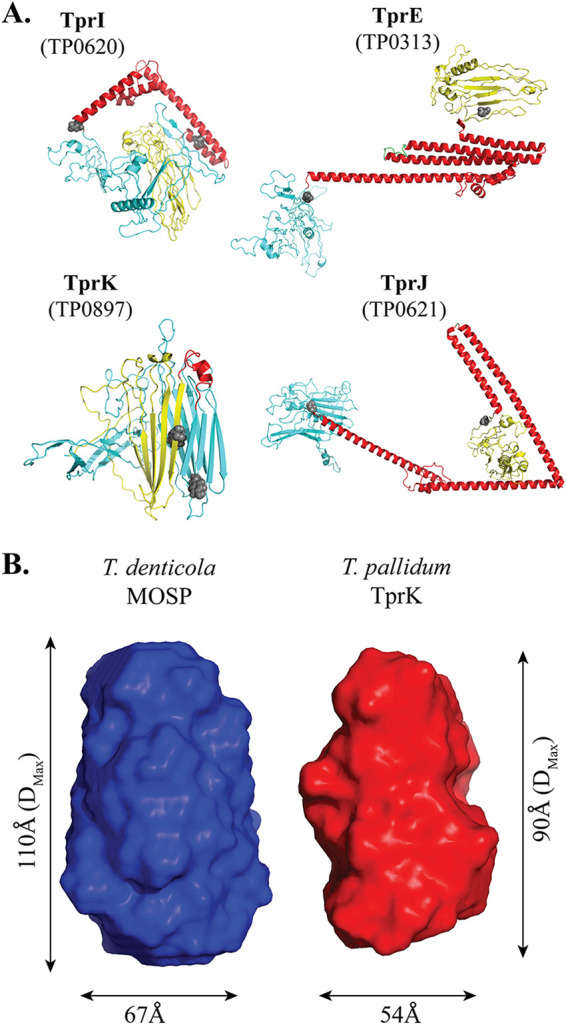
trRosetta modeling of full-length Tprs and solution structures of MOSPN domains of T. denticola MOSP and T. pallidum TprK. (A) MOSPN and MOSPC domains of all Tprs, shown in cyan and yellow, respectively. α-Helices of CVRs are red. Residues corresponding to predicted CVR domain boundaries from Pfam are shown as spheres. (B) Ab initio reconstruction of the low-resolution molecular envelopes of T. denticola MOSP and T. pallidum TprK MOSPN domains (blue and red, respectively) calculated using DAMMIN (166) and DAMAVER (167). Both envelopes were generated without enforcing any symmetry. The maximum dimensions (Dmax) and the widths of both envelopes are also labeled.
(ii) SAXS supports the bipartite membrane topology of all Tprs.
Taken together, the above-described findings indicate that full-length Tprs have a tridomain architecture and a bipartite membrane topology. We used SAXS to garner additional experimental evidence for the bipartite membrane topology of Tprs. For these investigations, we chose MOSPN of T. denticola MOSP as a structural surrogate for the corresponding Tpr domains and MOSPN of TprK given the widespread belief that all seven of its variable regions occur within a β-barrel formed by the full-length polypeptide (120–122). The MOSPN domains of both proteins could be purified by Ni-nitrilotriacetic acid (NTA) chromatography without denaturants or detergents and, by size exclusion chromatography (SEC), eluted as monomers with molecular weights of ∼25 kDa (Fig. S14A). In SAXS experiments, the scattering curves and intraparticle distance distribution functions [P(r)] of both domains showed correct folding without aggregation (Fig. S14B and C). As with TprF (45), ab initio envelope reconstructions from the SAXS data reveal rigid, nonglobular shapes (Fig. 5B).
T. pallidum efflux systems and outer membrane factors.
Bacteria not only need to import nutrients but also need to expel toxigenic molecules of endogenous or exogenous origin (123). The capacity of numerous bacterial species to tolerate antibiotics and other toxic compounds elaborated by the commensal flora and/or host defenses arises in large part from the activity of efflux systems (124). Additionally, efflux systems enhance the virulence potential of bacterial pathogens by protecting against reactive oxygen species (125).
In Gram-negative bacteria, multicomponent efflux systems span both the inner and outer membranes and consist of an OMF, an inner membrane-spanning transporter (pump), and a periplasmic adaptor that connects the two (41). The pumps for these multicomponent systems fall into two families: ATP binding cassette (ABC) and resistance-nodulation-cell division (RND) transporters. ABC transporters (e.g., E. coli MacB) consist of an N-terminal nucleotide binding domain (NBD) and four transmembrane (TM) helices and hydrolyze ATP to drive transport (126). RND transporters (e.g., E. coli AcrB) consist of a TM domain with 12 α-helices within the CM, a porter domain in the periplasm containing the substrate binding pocket, and a docking domain that connects the pump to the adaptor; RND transporters are powered by the proton motive force (126). In both systems, OMF monomers (e.g., E. coli TolC) form a trimer with 3-fold symmetry in which 4 β-strands and 6 α-helices from each subunit associate to form a 12-stranded β-barrel within the OM and an α-helical barrel within the periplasm, which interacts with a hexameric adaptor (e.g., AcrA and MacA) (127, 128). The periplasmic adaptor and the IM transporter maintain a stable association, while the OMF interacts transiently with the periplasmic adaptor. As a result, in E. coli, TolC can associate with adaptors for both pumps (129).
An investigation by Brautigam and colleagues of Tp34 (TP0971), a T. pallidum lipoprotein involved in metal homeostasis, resulted in the serendipitous discovery of a large operon (tp0959 to tp0972) encoding a MacB transporter (TP0962, TP0963, and TP0964), a periplasmic adaptor (TP0965), and four OMFs (TP0966 to TP0969) (130). In contrast to conventional MacB (128, 131), which is a homodimer, the MacB transporter in T. pallidum consists of a heterodimeric TM component (TP0962/TP0963) and two identical NBDs (TP0964) (130). We identified an ortholog (TP0790) of the RND transporter AcrB elsewhere in the T. pallidum genome (11). Multiple sequence alignment (MSA) (Table S8) and phylogenetic analysis (Fig. S15A) revealed that the four T. pallidum OMFs share 30% to 40% sequence similarity and cluster on a single branch close to E. coli TolC and Salmonella enterica serovar Typhi St50. 3D models of the TP0966, TP0967, TP0968, and TP0969 protomers (Fig. 6A, Table 1, and Fig. S15B) have a typical TolC-like topology consisting of four β-strands with two large ECLs and six α-helices. Threefold symmetry was applied to assemble trimers for the four OMFs. Whereas the predicted topologies of the T. pallidum OMFs closely overlap that of TolC (PDB accession number 1TQQ) (132) (Fig. 6A and Table S9), the electrostatics of the channels differ greatly from that of TolC as well as each other. Notably, the TolC channel is entirely negative, while the T. pallidum OMF channels contain variable patches of basic and acidic residues (Fig. 6B). Interestingly, the TP0967 channel, the most negatively charged of the four, also contains an aspartate ring (Fig. 6C), which, in TolC, determines ion selectivity (133). As with the genes encoding the FadL-like proteins, the cotranscription of OMF genes (tp0966 to tp0969) implies that they function in parallel (130). Also, as with the fadL-like genes, cotranscription makes them attractive vaccine candidates. DiscoTope 2.0 predicts that all four trimers, most notably TP0969, contain surface-exposed BCEs (Fig. 6A and Table S10).
FIG 6.

Channels of T. pallidum OMFs have dissimilar electrostatic potentials. (A) Ribbon diagrams for trimeric structural models of T. pallidum OMFs and the crystal structure of E. coli TolC (PDB accession number 1TQQ). One protomer in all four OMFs is shown in various shades of gold, while the TolC protomer is blue. BCEs are displayed as surfaces. All proteins are in the same orientation, with β-strands labeled. (B) Comparative electrostatics (same orientation) of T. pallidum OMFs and TolC. (C) View from the periplasmic entrance showing the aspartate ring (red spheres) of TP0967 and E. coli TolC. Both proteins are in the same orientation.
We used Phyre2 and PyMOL to construct a hexameric assembly of the periplasmic adaptor (TP0965), which exhibits a typical MacA-like funnel but is shorter than E. coli MacA (∼121 Å versus ∼152 Å, respectively) (Fig. 7A and B). Because our mining of the T. pallidum genome revealed only one adaptor, we hypothesize that it functions in both efflux systems. This conjecture is based on the mixed electrostatic charges at the interface of TP0965 with the two efflux pumps (TP0962/TP0963/TP0964 and TP0790) (Fig. S16A). In E. coli, the positively charged periplasmic interface of MacB interacts with the negatively charged projections of MacA, while the converse is true for AcrB and AcrA (Fig. S16B). We next assembled the complete Mac and Acr efflux system in T. pallidum (Fig. 7B) using the cryo-electron microscopy (cryo-EM) structures of E. coli Mac and Acr (PDB accession numbers 5NIK and 5V5S, respectively) (127, 128) (Fig. S16B).
FIG 7.

Modeling of T. pallidum periplasmic adaptors and TM transporters. (A) Structural models of the T. pallidum periplasmic adaptor (TP0965), the Mac-like TM transporter (TP0962/TP0963/TP964), and the RND-like TM transporter (TP0790) and OMF (TP0969). (B) Cryo-EM structures of E. coli Mac-type (PDB accession number 5NIK) and Acr-type (PDB accession number 5V5S) efflux systems.
In contrast to E. coli, in which one OMF (TolC) interacts with multiple adaptors (129), T. pallidum appears to have taken a different interactome approach to achieve substrate versatility of its efflux systems. The spirochete’s use of four OMFs, one periplasmic adaptor, and two TM exporters theoretically enables it to contend with a wide range of biochemical threats encountered in different tissues during the protracted course of this chronic disease. Because of its extraordinary invasiveness, T. pallidum is usually conceived of as a “loner” pathogen (6). However, during sexual activity, syphilis spirochetes are often inoculated into sites that teem with commensal microorganisms (4). The efflux pumps are likely a critical component of the spirochete’s early survival strategy, particularly in sites such as the female genital tract and the rectal mucosa, which have complex microbial communities that produce a variety of bactericidal small molecules and peptides. In addition, one can assume that mucosal epithelial cells respond to the invader by secreting antimicrobial peptides (AMPs) and sequestering transition metals (“nutritional immunity”) (134). A mouse model of lower female genital tract infection with Neisseria gonorrhoeae provides precedent for this notion; the absence of the MtrC-MtrD-MtrE efflux system decreases the survival of gonococci (135). Given the complex cellular inflammatory response that T. pallidum elicits in all stages of syphilis, the spirochete’s need to deal with toxigenic and redox-active molecules of exogenous origins likely extends beyond early infection (6). T. pallidum’s ability to evade host defenses and establish persistence due to its OM molecular architecture is the crux of its designation as the stealth pathogen (6). The potential ability to extrude different combinations of noxious molecules in diverse milieus, supported by its OMF repertoire, likely adds a new structure-function dimension to the concept of stealth pathogenicity.
Concluding remarks.
The OM is a durable, adaptable permeability barrier that enables Gram-negative bacteria to thrive in unpredictable, and often hostile, environments as free-living organisms, pathogens, or both (31). After more than 50 years of intensive investigation, the ultrastructure, constituents, and biogenesis of the Gram-negative OM are reasonably well understood (23, 52). The T. pallidum OM must also support homeostatic, protective, and virulence-related functions but for a bacterium with a markedly limited biosynthetic capacity and an exclusively pathogenic, invasive lifestyle capable of causing protracted, and even lifelong, infection in humans (2, 136). We have known for years that the properties, composition, and molecular architecture of the T. pallidum OM differ greatly from those of its Gram-negative counterparts (6, 11, 137). Cryo-EM has revealed that spatial relationships between the OM of T. pallidum and the other compartments of the cell envelope differ greatly from those of Gram-negative bacteria (9, 10). Previously, we identified candidate OMPs in the spirochete based on their predicted ability to adopt a β-barrel conformation (43). The characterization of these molecules and their interaction partners using structure-based approaches (30, 44) represents an important step toward deconvoluting the spirochete’s enigmatic mechanisms for persistence and devising strategies to elicit protective immunity.
How T. pallidum obtains the vast array of nutrients required to sustain infection of its obligate human host is a long-standing conundrum of syphilis pathogenesis, well predating the sequencing of the spirochete’s minimalist genome (7, 8, 138, 139). The analyses presented here reveal that much of the answer to the enigma resides in the previously unsuspected diversity and complexity of its OMPeome. T. pallidum differs from many (although not all) Gram-negative bacteria in that it contains multiple paralogous OMP families. We postulate that each family enables the bacterium to import a different “class” of substrate and that members within a family have different substrate specificities within that class. With the important exception of the Tprs, T. pallidum has achieved this functional diversity by utilizing known structural orthologs as evolutionary templates. The multidomain Tprs, on the other hand, which have no Gram-negative counterpart, appear to have evolved under a combination of pressures related to the spirochete’s architectural plan (i.e., the need to stabilize the OM by tethering it to the peptidoglycan sacculus [9, 10]) in addition to its metabolic requirements (i.e., the need for porin-like proteins to passively import numerous small molecules). While the bacterium requires an OMP repertoire with broad importation capabilities, it does not need high copy numbers of individual importers given its dramatically slower replication time (∼30 h) than that of Gram-negative bacteria (62, 140, 141). From the standpoint of immune evasion, the benefits of this evolutionary path seem readily apparent. T. pallidum appears to contend with a less obvious adverse consequence of this broad importation strategy, the incidental uptake of cytotoxic small molecules, by deploying a robust combinatorial efflux system to function in concert with its importers.
As an obligate human pathogen with no known environmental or zoonotic reservoir that has remained exquisitely sensitive to penicillin G for the past 75 years (2, 142, 143), one would expect that eradication of T. pallidum could be accomplished by aggressive public health measures. The explosive increase in new syphilis cases in the United States and globally during the past 20 years clearly argues otherwise (1, 4, 144). Consequently, there is a growing sense of urgency about the need for a vaccine as a cornerstone of a global containment strategy (105, 145). Because T. pallidum is an extracellular bacterium (6, 8), an effective vaccine must elicit antibodies against antigens on the bacterial surface (105, 145). Years of investigation of the molecular architecture of the T. pallidum outer membrane have shown that the principal candidates for syphilis vaccine design are contained within the spirochete’s OMPeome (6, 11, 108, 137). The structural models described here provide investigators, for the first time, with a platform for a rational, systematic investigation of the combination of OMPs needed to elicit long-lived protection. With β-barrel proteins, protective antibodies must be directed against BCEs located in ECLs. Applying the principles of structural vaccinology will enable investigators to ascertain that antibodies elicited in syphilis vaccine trials with animal models precisely target the Achilles’ heels of candidate OMP vaccinogens: their ECLs.
MATERIALS AND METHODS
Ethics statement.
All animal experimentation was conducted according to the Guide for the Care and Use of Laboratory Animals, 8th ed. (146), and in accordance with protocols reviewed and approved by the UConn Health Institutional Animal Care and Use Committee under the auspices of Animal Welfare Assurance A3471-01.
Propagation of T. pallidum.
T. pallidum (Nichols strain) was propagated by intratesticular inoculation of adult male New Zealand White rabbits and harvested in CMRL medium (Gibco) supplemented with 20% heat-inactivated normal rabbit serum at peak orchitis as previously described (54). Spirochetes were enumerated by dark-field microscopy using a Petroff-Hausser counting chamber (Hausser Scientific).
Cloning procedures.
Recombinant constructs and oligonucleotide primers used in this study are listed in Table S10 in the supplemental material. All cloned constructs are based on the T. pallidum Nichols strain. Constructs for POTRA1-5 of BamA (TP0326) and the MOSPN domain of T. denticola strain ATCC 35405 without its signal sequence were described previously (44, 67). DNA encoding the MOSPN domain (amino acid residues 29 through 259) of TprK (TP0897) (GenBank accession number WP_010882340) without its signal sequence was PCR amplified from the pUK57 plasmid harboring an E. coli codon-optimized version of tprK (synthesized by GenScript). The resulting amplicon was cloned into linearized pET28a in frame with the N-terminal His6 tag using in-fusion cloning (147) (TaKaRa).
Expression and purification.
All constructs in this study were overexpressed in E. coli OverExpress C41(DE3) (Lucigen) at 20°C. Expression was induced at an optical density at 600 nm (OD600) of ∼0.4 by the addition of isopropyl-β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 0.5 mM, followed by 12 to 44 h of growth. Following induction, cells were harvested at 7,000 × g for 20 min at 4°C. The pellet was lysed in Tris-NaCl buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl) supplemented with a protease inhibitor cocktail (PIC; Sigma-Aldrich). The lysate was centrifuged at 14,000 rpm for 40 min at 4°C to remove insoluble proteins and cell debris. The supernatant was mixed with Ni-NTA resin (Qiagen, USA) for 1 h and washed with Tris-NaCl buffer, followed by another wash with the same buffer containing 40 mM imidazole. Elution was carried out in Tris-NaCl buffer with 400 mM imidazole at pH 7.5. Elution fractions were pooled and subjected to size exclusion chromatography (SEC) using a Superdex-200 HR 16/60 column (GE Healthcare Life Sciences) preequilibrated with a solution containing 50 mM Tris (pH 7.5), 100 mM NaCl, and 1 mM dithiothreitol (DTT). The purity of all proteins was verified by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The final proteins (POTRA arm and the MOSPN domains of TprK and MOSP) were concentrated to 1 to 3 mg/ml using an Amicon filtration device, flash-frozen in liquid nitrogen, and stored at −80°C until use.
3D modeling of T. pallidum OMPs.
To predict the 3D structures of recently identified T. pallidum OMPs (11), amino acid sequences were submitted to the Phyre2 server (http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index) (47). A confidence score of 90% or higher (47) between the template model and the target match was set as a cutoff value (47). The high confidence level (90%) indicates that the query protein is likely to adopt the overall predicted β-barrel at high accuracy (47). Proteins that did not meet the 90% threshold or had unmodeled regions in Phyre2 were submitted to I-TASSER and/or trRosetta (46). The accuracy of 3D models from I-TASSER was quantitatively measured by the C score (148). The structural model of BamA was made, as described previously by Luthra et al. (44), using the solved structure of full-length BamA (PDB accession number 3KGP) from N. gonorrhoeae (149). The quality of all models was evaluated with MolProbity (55). All Tpr models were made using trRosetta (46). Steric clashes in each model were evaluated using WinCoot (56). Structural modeling and sequence identities are summarized in Table 1.
3D modeling data availability.
Atomic coordinates for all structural models of the T. pallidum OMPeome are downloadable from https://drive.google.com/file/d/1EurEnlwAiqtsUm8t-jC3Xuz5e7nV45mT/view?usp=sharing&export=download.
Modeling and assembly of the T. pallidum Lpt complex.
The Phyre2 server (47) was used to build the initial 3D structural model of T. pallidum LptB (TP0786), LptF (TP0883), LptG (TP0884), LptC (TP0784), and LptA (TP0785) orthologs. Next, the crystal structure of Vibrio cholerae LptB2FGC (PDB accession number 6MJP) (150) was applied as a template to assemble a structural model for T. pallidum LptB2FGC (TP07862-TP0883-TP0884-TP0784). Finally, T. pallidum LptA (TP0785) was manually aligned between the N-terminal domain of LptD (TP0515) and LptC (TP0784).
Modeling and assembly of T. pallidum efflux systems.
The crystal structure of trimeric E. coli TolC (PDB accession number 1TQQ) (132) was used to build trimers of T. pallidum OMFs TP0966, TP0967, TP0968, and TP0969 using WinCoot (56). Structural models of TP0790, TP0962/TP0963, TP0964, and TP0965 were made in Phyre2 (47). The cryo-EM structures of the MacAB-TolC (PDB accession number 5NIK) (128) and AcrAB-TolC (PDB accession number 5V5S) (127) efflux pump-OMF complexes were used as the templates to assemble T. pallidum ABC-type and RND-based pumps with the periplasmic adaptor (TP0965) and OMFs (TP0966, TP0967, TP0968, and TP0969).
Bioinformatics.
DOG 2.0 (151) was used to delineate domains and extracellular loops of T. pallidum OMPs on the primary (one-dimensional [1D]) protein structures. Structures were aligned using the secondary structure matching (SSM) superimposition algorithm in WinCoot (56). All structural figures were rendered in PyMOL (https://pymol.org/) (152). MSA was performed using ClustalW with the GONNET matrix in MacVector. Sequence identity matrices were calculated in Clustal Omega using the default parameters (153). PROMALS3D (154) and ESPript3.0 (155) were used to generate structure-based sequence alignments. The electrostatic potential of 3D structures was calculated using the adaptive Poisson-Boltzmann solver (APBS) (156). Discontinuous BCEs were predicted from each 3D model using a threshold of −3.7 with the DiscoTope 2.0 server (157). The score for BCEs is calculated as a combination of propensity scores of amino acids in spatial proximity and contact numbers. The residue contact number is the number of Cα atoms in the antigen within a distance of 10 Å (158). All phylogenetic analyses were performed using Clustal Omega (153) and visualized using Interactive Tree of Life (iTOL) (V4) (159).
Acquisition of small-angle X-ray scattering data.
Online SEC–small-angle X-ray scattering (SAXS) at the 16 LiX beamline (160) of National Synchrotron Light Source II (Brookhaven National Laboratory) was used to collect scattering data for the MOSPN domain of TprK. One hundred fifty microliters (∼200 μM) of a protein sample (TprK) was injected into a Superdex Increase 200 10/300 GL column (GE Healthcare) at a flow rate of 0.45 ml/min. Scattering images were collected continuously at 1-s exposures per frame. The data processing program pyXS (161) was used for scaling, integration, and averaging of individual scattering images after inspection for aggregation. SAXS data of MOSPN of T. denticola MOSP were also collected at the 16 LiX beamline (160) under static data collection conditions using a 1.0-mg/ml protein concentration. pyXS (161) was used to determine the scattering contribution of the MOSPN domain of T. denticola MOSP by subtracting the background scattering of the buffer.
The SAXS data of TP0326 POTRA1-5 were collected at the Cornell High-Energy Synchrotron Source (CHESS) beamline using a dual Pilatus 100 K-S SAXS/wide-angle X-ray scattering (WAXS) detector, and background subtraction of SAXS buffer was performed using the open-source software RAW (162).
SAXS analysis and model building.
The ATSAS software package (163) was used to generate 3D ab initio envelopes of BamA POTRA1-5, T. denticola MOSPN, and TprK MOSPN from one-dimensional SAXS profiles. Radii of gyration (Rg), deduced from the Guinier region of the Guinier plots, were computed using PRIMUS (164). GNOM (165) was used to generate a well-behaved P(r) curve. Ab initio envelope reconstructions were performed using DAMMIN (166) and DAMAVER (167). Theoretical scattering curves were compared against experimental scattering data using FoXS (168). The predicted flexibility of POTRA1-5 was determined using the ensemble optimization method (EOM) (73).
Operon prediction of FadL-like genes in T. pallidum.
The FGENESB server (http://www.softberry.com/berry.phtml?topic=fgenesb&group=programs&subgroup=gfindb) was used for operon prediction for the region encompassing nucleotide positions 937198 to 932966 of the T. pallidum Nichols strain (NCBI accession number NC_021490.2) genome carrying tp0856, tp0858, and tp0859, including intergenic regions between tp0856 and tp0858 and between tp0858 and tp0859.
Expression analysis of fadL-like genes.
RNA was extracted from testicular tissue of four rabbits inoculated with the T. pallidum Nichols strain as previously described (54). The RNA concentration was measured using a Nanodrop spectrophotometer (Thermo Scientific). cDNA synthesis was performed using the high-capacity cDNA archive kit (Applied Biosystems) according to the manufacturer’s instructions. Primers used to amply transcripts for tp0856, tp0858, tp0859, and the intergenic regions between tp0856, tp0858, and tp0859 are listed in Table S10. Amplification reactions were performed in a GeneAmp 9700 PCR system (Applied Biosystems) as follows: 1 cycle at 98°C for 3 min, followed by 35 cycles of 98°C for 10 s, 55°C for 15 s, and 72°C for 10 s and 1 cycle of 72°C for 3 min.
ACKNOWLEDGMENTS
We thank Carson J. La Vake and Morgan LeDoyt (UConn Health) for their technical support. We thank Shirish Chodankar and Lin Yang (Brookhaven National Laboratory) for assistance with SAXS measurements at beamline 16-ID (LiX).
This work was supported by NIAID grants R01 AI26756 (J.D.R.) and U19 AI144177 (J.D.R. and M.J.C.) and research funds generously provided by Connecticut Children’s (K.L.H., M.J.C., and J.D.R.).
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
K.L.H., V.N.U., M.J.C., J.D.R., and A.L. contributed to the design of experiments. K.L.H., J.M.M.-R., K.N.D., N.S., V.N.U., M.J.C., and A.L. performed the experiments. K.L.H., J.D.R., and A.L. analyzed the data. K.L.H., J.D.R., and A.L. wrote the manuscript. All authors reviewed and approved the manuscript.
Footnotes
Supplemental material is available online only.
Contributor Information
Amit Luthra, Email: amluthra@uchc.edu.
Michael Y. Galperin, NCBI, NLM, National Institutes of Health
REFERENCES
- 1.Hook EWR. 2017. Syphilis. Lancet 389:1550–1557. 10.1016/S0140-6736(16)32411-4. [DOI] [PubMed] [Google Scholar]
- 2.Radolf JD, Tramont EC, Salazar JC. 2019. Syphilis (Treponema pallidum), p 2865–2892. In Bennett JE, Dolin R, Blaser MJ (ed), Mandell, Douglas, and Bennett’s principles and practice of infectious diseases, 9th ed. Elsevier, Philadelphia, PA. [Google Scholar]
- 3.Patton ME, Su JR, Nelson R, Weinstock H, Centers for Disease Control and Prevention . 2014. Primary and secondary syphilis—United States, 2005-2013. MMWR Morb Mortal Wkly Rep 63:402–406. [PMC free article] [PubMed] [Google Scholar]
- 4.Peeling RW, Mabey D, Kamb ML, Chen XS, Radolf JD, Benzaken AS. 2017. Syphilis. Nat Rev Dis Primers 3:17073. 10.1038/nrdp.2017.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salazar JC, Hazlett KR, Radolf JD. 2002. The immune response to infection with Treponema pallidum, the stealth pathogen. Microbes Infect 4:1133–1140. 10.1016/S1286-4579(02)01638-6. [DOI] [PubMed] [Google Scholar]
- 6.Radolf JD, Deka RK, Anand A, Smajs D, Norgard MV, Yang XF. 2016. Treponema pallidum, the syphilis spirochete: making a living as a stealth pathogen. Nat Rev Microbiol 14:744–759. 10.1038/nrmicro.2016.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fraser CM, Norris SJ, Weinstock GM, White O, Sutton GG, Dodson R, Gwinn M, Hickey EK, Clayton R, Ketchum KA, Sodergren E, Hardham JM, McLeod MP, Salzberg S, Peterson J, Khalak H, Richardson D, Howell JK, Chidambaram M, Utterback T, McDonald L, Artiach P, Bowman C, Cotton MD, Fujii C, Garland S, Hatch B, Horst K, Roberts K, Sandusky M, Weidman J, Smith HO, Venter JC. 1998. Complete genome sequence of Treponema pallidum, the syphilis spirochete. Science 281:375–388. 10.1126/science.281.5375.375. [DOI] [PubMed] [Google Scholar]
- 8.Norris SJ, Cox DL, Weinstock GM. 2001. Biology of Treponema pallidum: correlation of functional activities with genome sequence data. J Mol Microbiol Biotechnol 3:37–62. [PubMed] [Google Scholar]
- 9.Izard J, Renken C, Hsieh CE, Desrosiers DC, Dunham-Ems S, La Vake C, Gebhardt LL, Limberger RJ, Cox DL, Marko M, Radolf JD. 2009. Cryo-electron tomography elucidates the molecular architecture of Treponema pallidum, the syphilis spirochete. J Bacteriol 191:7566–7580. 10.1128/JB.01031-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu J, Howell JK, Bradley SD, Zheng Y, Zhou ZH, Norris SJ. 2010. Cellular architecture of Treponema pallidum: novel flagellum, periplasmic cone, and cell envelope as revealed by cryo electron tomography. J Mol Biol 403:546–561. 10.1016/j.jmb.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Radolf JD, Kumar S. 2018. The Treponema pallidum outer membrane. Curr Top Microbiol Immunol 415:1–38. 10.1007/82_2017_44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forterre P. 2015. The universal tree of life: an update. Front Microbiol 6:717. 10.3389/fmicb.2015.00717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henderson JC, Zimmerman SM, Crofts AA, Boll JM, Kuhns LG, Herrera CM, Trent MS. 2016. The power of asymmetry: architecture and assembly of the Gram-negative outer membrane lipid bilayer. Annu Rev Microbiol 70:255–278. 10.1146/annurev-micro-102215-095308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krishnan S, Prasadarao NV. 2012. Outer membrane protein A and OprF: versatile roles in Gram-negative bacterial infections. FEBS J 279:919–931. 10.1111/j.1742-4658.2012.08482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oleastro M, Menard A. 2013. The role of Helicobacter pylori outer membrane proteins in adherence and pathogenesis. Biology (Basel) 2:1110–1134. 10.3390/biology2031110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fairman JW, Dautin N, Wojtowicz D, Liu W, Noinaj N, Barnard TJ, Udho E, Przytycka TM, Cherezov V, Buchanan SK. 2012. Crystal structures of the outer membrane domain of intimin and invasin from enterohemorrhagic E. coli and enteropathogenic Y. pseudotuberculosis. Structure 20:1233–1243. 10.1016/j.str.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McMichael JC. 2000. Vaccines for Moraxella catarrhalis. Vaccine 19(Suppl 1):S101–S107. 10.1016/S0264-410X(00)00287-5. [DOI] [PubMed] [Google Scholar]
- 18.Wright JC, Williams JN, Christodoulides M, Heckels JE. 2002. Immunization with the recombinant PorB outer membrane protein induces a bactericidal immune response against Neisseria meningitidis. Infect Immun 70:4028–4034. 10.1128/iai.70.8.4028-4034.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stork M, Bos MP, Jongerius I, de Kok N, Schilders I, Weynants VE, Poolman JT, Tommassen J. 2010. An outer membrane receptor of Neisseria meningitidis involved in zinc acquisition with vaccine potential. PLoS Pathog 6:e1000969. 10.1371/journal.ppat.1000969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lun J, Xia C, Yuan C, Zhang Y, Zhong M, Huang T, Hu Z. 2014. The outer membrane protein, LamB (maltoporin), is a versatile vaccine candidate among the Vibrio species. Vaccine 32:809–815. 10.1016/j.vaccine.2013.12.035. [DOI] [PubMed] [Google Scholar]
- 21.Dong C, Beis K, Nesper J, Brunkan-Lamontagne AL, Clarke BR, Whitfield C, Naismith JH. 2006. Wza the translocon for E. coli capsular polysaccharides defines a new class of membrane protein. Nature 444:226–229. 10.1038/nature05267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wimley WC. 2003. The versatile β-barrel membrane protein. Curr Opin Struct Biol 13:404–411. 10.1016/s0959-440x(03)00099-x. [DOI] [PubMed] [Google Scholar]
- 23.Chaturvedi D, Mahalakshmi R. 2017. Transmembrane β-barrels: evolution, folding and energetics. Biochim Biophys Acta 1859:2467–2482. 10.1016/j.bbamem.2017.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin J, Huang S, Zhang Q. 2002. Outer membrane proteins: key players for bacterial adaptation in host niches. Microbes Infect 4:325–331. 10.1016/S1286-4579(02)01545-9. [DOI] [PubMed] [Google Scholar]
- 25.Mikula KM, Kolodziejczyk R, Goldman A. 2012. Yersinia infection tools—characterization of structure and function of adhesins. Front Cell Infect Microbiol 2:169. 10.3389/fcimb.2012.00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu Z, Periaswamy B, Sahin O, Yaeger M, Plummer P, Zhai W, Shen Z, Dai L, Chen SL, Zhang Q. 2016. Point mutations in the major outer membrane protein drive hypervirulence of a rapidly expanding clone of Campylobacter jejuni. Proc Natl Acad Sci U S A 113:10690–10695. 10.1073/pnas.1605869113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hobbs MM, Alcorn TM, Davis RH, Fischer W, Thomas JC, Martin I, Ison C, Sparling PF, Cohen MS. 1999. Molecular typing of Neisseria gonorrhoeae causing repeated infections: evolution of porin during passage within a community. J Infect Dis 179:371–381. 10.1086/314608. [DOI] [PubMed] [Google Scholar]
- 28.Haake DA, Suchard MA, Kelley MM, Dundoo M, Alt DP, Zuerner RL. 2004. Molecular evolution and mosaicism of leptospiral outer membrane proteins involves horizontal DNA transfer. J Bacteriol 186:2818–2828. 10.1128/jb.186.9.2818-2828.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stenkova AM, Isaeva MP, Shubin FN, Rasskazov VA, Rakin AV. 2011. Trends of the major porin gene (ompF) evolution: insight from the genus Yersinia. PLoS One 6:e20546. 10.1371/journal.pone.0020546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumar S, Caimano MJ, Anand A, Dey A, Hawley KL, LeDoyt ME, La Vake CJ, Cruz AR, Ramirez LG, Paštěková L, Bezsonova I, Šmajs D, Salazar JC, Radolf JD. 2018. Sequence variation of rare outer membrane protein β-barrel domains in clinical strains provides insights into the evolution of Treponema pallidum subsp. pallidum, the syphilis spirochete. mBio 9:e01006-18. 10.1128/mBio.01006-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nikaido H. 2003. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev 67:593–656. 10.1128/mmbr.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vergalli J, Bodrenko IV, Masi M, Moynie L, Acosta-Gutierrez S, Naismith JH, Davin-Regli A, Ceccarelli M, van den Berg B, Winterhalter M, Pages JM. 2020. Porins and small-molecule translocation across the outer membrane of Gram-negative bacteria. Nat Rev Microbiol 18:164–176. 10.1038/s41579-019-0294-2. [DOI] [PubMed] [Google Scholar]
- 33.Celia H, Noinaj N, Buchanan SK. 2020. Structure and stoichiometry of the Ton molecular motor. Int J Mol Sci 21:375. 10.3390/ijms21020375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van den Berg B, Black PN, ClemonsWM, Jr, Rapoport TA. 2004. Crystal structure of the long-chain fatty acid transporter FadL. Science 304:1506–1509. 10.1126/science.1097524. [DOI] [PubMed] [Google Scholar]
- 35.Meyer JEW, Hofnung M, Schulz GE. 1997. Structure of maltoporin from Salmonella typhimurium ligated with a nitrophenyl-maltotrioside. J Mol Biol 266:761–775. 10.1006/jmbi.1996.0823. [DOI] [PubMed] [Google Scholar]
- 36.Okuda S, Sherman DJ, Silhavy TJ, Ruiz N, Kahne D. 2016. Lipopolysaccharide transport and assembly at the outer membrane: the PEZ model. Nat Rev Microbiol 14:337–345. 10.1038/nrmicro.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Noinaj N, Gumbart JC, Buchanan SK. 2017. The β-barrel assembly machinery in motion. Nat Rev Microbiol 15:197–204. 10.1038/nrmicro.2016.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Botos I, Majdalani N, Mayclin SJ, McCarthy JG, Lundquist K, Wojtowicz D, Barnard TJ, Gumbart JC, Buchanan SK. 2016. Structural and functional characterization of the LPS transporter LptDE from Gram-negative pathogens. Structure 24:965–976. 10.1016/j.str.2016.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chevalier S, Bouffartigues E, Bodilis J, Maillot O, Lesouhaitier O, Feuilloley MGJ, Orange N, Dufour A, Cornelis P. 2017. Structure, function and regulation of Pseudomonas aeruginosa porins. FEMS Microbiol Rev 41:698–722. 10.1093/femsre/fux020. [DOI] [PubMed] [Google Scholar]
- 40.Catel-Ferreira M, Marti S, Guillon L, Jara L, Coadou G, Molle V, Bouffartigues E, Bou G, Shalk I, Jouenne T, Vila-Farres X, De E. 2016. The outer membrane porin OmpW of Acinetobacter baumannii is involved in iron uptake and colistin binding. FEBS Lett 590:224–231. 10.1002/1873-3468.12050. [DOI] [PubMed] [Google Scholar]
- 41.Du D, van Veen HW, Luisi BF. 2015. Assembly and operation of bacterial tripartite multidrug efflux pumps. Trends Microbiol 23:311–319. 10.1016/j.tim.2015.01.010. [DOI] [PubMed] [Google Scholar]
- 42.Franklin MW, Nepomnyachiy S, Feehan R, Ben-Tal N, Kolodny R, Slusky JSG. 2018. Efflux pumps represent possible evolutionary convergence onto the β-barrel fold. Structure 26:1266–1274.e2. 10.1016/j.str.2018.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cox DL, Luthra A, Dunham-Ems S, Desrosiers DC, Salazar JC, Caimano MJ, Radolf JD. 2010. Surface immunolabeling and consensus computational framework to identify candidate rare outer membrane proteins of Treponema pallidum. Infect Immun 78:5178–5194. 10.1128/IAI.00834-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luthra A, Anand A, Hawley KL, LeDoyt M, La Vake CJ, Caimano MJ, Cruz AR, Salazar JC, Radolf JD. 2015. A homology model reveals novel structural features and an immunodominant surface loop/opsonic target in the Treponema pallidum BamA ortholog TP_0326. J Bacteriol 197:1906–1920. 10.1128/JB.00086-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anand A, LeDoyt M, Karanian C, Luthra A, Koszelak-Rosenblum M, Malkowski MG, Puthenveetil R, Vinogradova O, Radolf JD. 2015. Bipartite topology of Treponema pallidum repeat proteins C/D and I: outer membrane insertion, trimerization, and porin function require a C-terminal β-barrel domain. J Biol Chem 290:12313–12331. 10.1074/jbc.M114.629188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. 2015. The I-TASSER suite: protein structure and function prediction. Nat Methods 12:7–8. 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. 2015. The Phyre2 Web portal for protein modeling, prediction and analysis. Nat Protoc 10:845–858. 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang J, Anishchenko I, Park H, Peng Z, Ovchinnikov S, Baker D. 2020. Improved protein structure prediction using predicted interresidue orientations. Proc Natl Acad Sci U S A 117:1496–1503. 10.1073/pnas.1914677117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hagan CL, Silhavy TJ, Kahne D. 2011. β-Barrel membrane protein assembly by the Bam complex. Annu Rev Biochem 80:189–210. 10.1146/annurev-biochem-061408-144611. [DOI] [PubMed] [Google Scholar]
- 50.Ricci DP, Silhavy TJ. 2012. The Bam machine: a molecular cooper. Biochim Biophys Acta 1818:1067–1084. 10.1016/j.bbamem.2011.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rollauer SE, Sooreshjani MA, Noinaj N, Buchanan SK. 2015. Outer membrane protein biogenesis in Gram-negative bacteria. Philos Trans R Soc Lond B Biol Sci 370:20150023. 10.1098/rstb.2015.0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Konovalova A, Kahne DE, Silhavy TJ. 2017. Outer membrane biogenesis. Annu Rev Microbiol 71:539–556. 10.1146/annurev-micro-090816-093754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu R, Stephenson R, Gichaba A, Noinaj N. 2020. The big BAM theory: an open and closed case? Biochim Biophys Acta 1862:183062. 10.1016/j.bbamem.2019.183062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Desrosiers DC, Anand A, Luthra A, Dunham-Ems SM, LeDoyt M, Cummings MA, Eshghi A, Cameron CE, Cruz AR, Salazar JC, Caimano MJ, Radolf JD. 2011. TP0326, a Treponema pallidum β-barrel assembly machinery A (BamA) orthologue and rare outer membrane protein. Mol Microbiol 80:1496–1515. 10.1111/j.1365-2958.2011.07662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, ArendallWB, III, Snoeyink J, Richardson JS, Richardson DC. 2007. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35:W375–W383. 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pětrošová H, Zobaníková M, Čejková D, Mikalová L, Pospíšilová P, Strouhal M, Chen L, Qin X, Muzny DM, Weinstock GM, Šmajs D. 2012. Whole genome sequence of Treponema pallidum ssp. pallidum, strain Mexico A, suggests recombination between yaws and syphilis strains. PLoS Negl Trop Dis 6:e1832. 10.1371/journal.pntd.0001832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hagan CL, Kim S, Kahne D. 2010. Reconstitution of outer membrane protein assembly from purified components. Science 328:890–892. 10.1126/science.1188919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee J, Tomasek D, Santos TM, May MD, Meuskens I, Kahne D. 2019. Formation of a β-barrel membrane protein is catalyzed by the interior surface of the assembly machine protein BamA. Elife 8:e49787. 10.7554/eLife.49787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gu Y, Li H, Dong H, Zeng Y, Zhang Z, Paterson NG, Stansfeld PJ, Wang Z, Zhang Y, Wang W, Dong C. 2016. Structural basis of outer membrane protein insertion by the BAM complex. Nature 531:64–69. 10.1038/nature17199. [DOI] [PubMed] [Google Scholar]
- 61.Hart EM, Gupta M, Wuhr M, Silhavy TJ. 2020. The gain-of-function allele bamA E470K bypasses the essential requirement for BamD in β-barrel outer membrane protein assembly. Proc Natl Acad Sci U S A 117:18737–18743. 10.1073/pnas.2007696117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cumberland MC, Turner TB. 1949. The rate of multiplication of Treponema pallidum in normal and immune rabbits. Am J Syph Gonorrhea Vener Dis 33:201–212. [PubMed] [Google Scholar]
- 63.Warner LR, Gatzeva-Topalova PZ, Doerner PA, Pardi A, Sousa MC. 2017. Flexibility in the periplasmic domain of BamA is important for function. Structure 25:94–106. 10.1016/j.str.2016.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Radolf JD, Robinson EJ, Bourell KW, Akins DR, Porcella SF, Weigel LM, Jones JD, Norgard MV. 1995. Characterization of outer membranes isolated from Treponema pallidum, the syphilis spirochete. Infect Immun 63:4244–4252. 10.1128/IAI.63.11.4244-4252.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Horne JE, Brockwell DJ, Radford SE. 2020. Role of the lipid bilayer in outer membrane protein folding in Gram-negative bacteria. J Biol Chem 295:10340–10367. 10.1074/jbc.REV120.011473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bakelar J, Buchanan SK, Noinaj N. 2016. The structure of the β-barrel assembly machinery complex. Science 351:180–186. 10.1126/science.aad3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Puthenveetil R, Kumar S, Caimano MJ, Dey A, Anand A, Vinogradova O, Radolf JD. 2017. The major outer sheath protein forms distinct conformers and multimeric complexes in the outer membrane and periplasm of Treponema denticola. Sci Rep 7:13260. 10.1038/s41598-017-13550-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Selkrig J, Mosbahi K, Webb CT, Belousoff MJ, Perry AJ, Wells TJ, Morris F, Leyton DL, Totsika M, Phan M-D, Celik N, Kelly M, Oates C, Hartland EL, Robins-Browne RM, Ramarathinam SH, Purcell AW, Schembri MA, Strugnell RA, Henderson IR, Walker D, Lithgow T. 2012. Discovery of an archetypal protein transport system in bacterial outer membranes. Nat Struct Mol Biol 19:506–510, S1. 10.1038/nsmb.2261. [DOI] [PubMed] [Google Scholar]
- 69.Shen HH, Leyton DL, Shiota T, Belousoff MJ, Noinaj N, Lu J, Holt SA, Tan K, Selkrig J, Webb CT, Buchanan SK, Martin LL, Lithgow T. 2014. Reconstitution of a nanomachine driving the assembly of proteins into bacterial outer membranes. Nat Commun 5:5078. 10.1038/ncomms6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Webb CT, Heinz E, Lithgow T. 2012. Evolution of the β-barrel assembly machinery. Trends Microbiol 20:612–620. 10.1016/j.tim.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 71.Iqbal H, Kenedy MR, Lybecker M, Akins DR. 2016. The TamB ortholog of Borrelia burgdorferi interacts with the beta-barrel assembly machine (BAM) complex protein BamA. Mol Microbiol 102:757–774. 10.1111/mmi.13492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Josts I, Stubenrauch CJ, Vadlamani G, Mosbahi K, Walker D, Lithgow T, Grinter R. 2017. The structure of a conserved domain of TamB reveals a hydrophobic β taco fold. Structure 25:1898–1906.e5. 10.1016/j.str.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bernado P, Mylonas E, Petoukhov MV, Blackledge M, Svergun DI. 2007. Structural characterization of flexible proteins using small-angle X-ray scattering. J Am Chem Soc 129:5656–5664. 10.1021/ja069124n. [DOI] [PubMed] [Google Scholar]
- 74.Lundstedt E, Kahne D, Ruiz N. 2021. Assembly and maintenance of lipids at the bacterial outer membrane. Chem Rev 121:5098–5123. 10.1021/acs.chemrev.0c00587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gu Y, Stansfeld PJ, Zeng Y, Dong H, Wang W, Dong C. 2015. Lipopolysaccharide is inserted into the outer membrane through an intramembrane hole, a lumen gate, and the lateral opening of LptD. Structure 23:496–504. 10.1016/j.str.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chng SS, Xue M, Garner RA, Kadokura H, Boyd D, Beckwith J, Kahne D. 2012. Disulfide rearrangement triggered by translocon assembly controls lipopolysaccharide export. Science 337:1665–1668. 10.1126/science.1227215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Belisle JT, Brandt ME, Radolf JD, Norgard MV. 1994. Fatty acids of Treponema pallidum and Borrelia burgdorferi lipoproteins. J Bacteriol 176:2151–2157. 10.1128/jb.176.8.2151-2157.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McClean S. 2012. Eight stranded β-barrel and related outer membrane proteins: role in bacterial pathogenesis. Protein Pept Lett 19:1013–1025. 10.2174/092986612802762688. [DOI] [PubMed] [Google Scholar]
- 79.Hong H, Patel DR, Tamm LK, van den Berg B. 2006. The outer membrane protein OmpW forms an eight-stranded β-barrel with a hydrophobic channel. J Biol Chem 281:7568–7577. 10.1074/jbc.M512365200. [DOI] [PubMed] [Google Scholar]
- 80.Mussi MA, Relling VM, Limansky AS, Viale AM. 2007. CarO, an Acinetobacter baumannii outer membrane protein involved in carbapenem resistance, is essential for L-ornithine uptake. FEBS Lett 581:5573–5578. 10.1016/j.febslet.2007.10.063. [DOI] [PubMed] [Google Scholar]
- 81.McPhee JB, Tamber S, Bains M, Maier E, Gellatly S, Lo A, Benz R, Hancock RE. 2009. The major outer membrane protein OprG of Pseudomonas aeruginosa contributes to cytotoxicity and forms an anaerobically regulated, cation-selective channel. FEMS Microbiol Lett 296:241–247. 10.1111/j.1574-6968.2009.01651.x. [DOI] [PubMed] [Google Scholar]
- 82.Smith SG, Mahon V, Lambert MA, Fagan RP. 2007. A molecular Swiss army knife: OmpA structure, function and expression. FEMS Microbiol Lett 273:1–11. 10.1111/j.1574-6968.2007.00778.x. [DOI] [PubMed] [Google Scholar]
- 83.Yamashita S, Lukacik P, Barnard TJ, Noinaj N, Felek S, Tsang TM, Krukonis ES, Hinnebusch BJ, Buchanan SK. 2011. Structural insights into Ail-mediated adhesion in Yersinia pestis. Structure 19:1672–1682. 10.1016/j.str.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Touw DS, Patel DR, van den Berg B. 2010. The crystal structure of OprG from Pseudomonas aeruginosa, a potential channel for transport of hydrophobic molecules across the outer membrane. PLoS One 5:e15016. 10.1371/journal.pone.0015016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zahn M, D’Agostino T, Eren E, Basle A, Ceccarelli M, van den Berg B. 2015. Small-molecule transport by CarO, an abundant eight-stranded β-barrel outer membrane protein from Acinetobacter baumannii. J Mol Biol 427:2329–2339. 10.1016/j.jmb.2015.03.016. [DOI] [PubMed] [Google Scholar]
- 86.Xiao M, Lai Y, Sun J, Chen G, Yan A. 2016. Transcriptional regulation of the outer membrane porin gene ompW reveals its physiological role during the transition from the aerobic to the anaerobic lifestyle of Escherichia coli. Front Microbiol 7:799. 10.3389/fmicb.2016.00799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Horst R, Stanczak P, Wuthrich K. 2014. NMR polypeptide backbone conformation of the E. coli outer membrane protein W. Structure 22:1204–1209. 10.1016/j.str.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sanganna Gari RR, Seelheim P, Marsh B, Kiessling V, Creutz CE, Tamm LK. 2018. Quaternary structure of the small amino acid transporter OprG from Pseudomonas aeruginosa. J Biol Chem 293:17267–17277. 10.1074/jbc.RA118.004461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schubeis T, Le Marchand T, Daday C, Kopec W, Tekwani Movellan K, Stanek J, Schwarzer TS, Castiglione K, de Groot BL, Pintacuda G, Andreas LB. 2020. A β-barrel for oil transport through lipid membranes: dynamic NMR structures of AlkL. Proc Natl Acad Sci U S A 117:21014–21021. 10.1073/pnas.2002598117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kucharska I, Seelheim P, Edrington T, Liang B, Tamm LK. 2015. OprG harnesses the dynamics of its extracellular loops to transport small amino acids across the outer membrane of Pseudomonas aeruginosa. Structure 23:2234–2245. 10.1016/j.str.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cox DL, Akins DR, Porcella SF, Norgard MV, Radolf JD. 1995. Treponema pallidum in gel microdroplets: a novel strategy for investigation of treponemal molecular architecture. Mol Microbiol 15:1151–1164. 10.1111/j.1365-2958.1995.tb02288.x. [DOI] [PubMed] [Google Scholar]
- 92.Hardham JM, Stamm LV. 1994. Identification and characterization of the Treponema pallidum tpn50 gene, an ompA homolog. Infect Immun 62:1015–1025. 10.1128/IAI.62.3.1015-1025.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Parker ML, Houston S, Wetherell C, Cameron CE, Boulanger MJ. 2016. The structure of Treponema pallidum Tp0624 reveals a modular assembly of divergently functionalized and previously uncharacterized domains. PLoS One 11:e0166274. 10.1371/journal.pone.0166274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Giacani L, Brandt SL, Ke W, Reid TB, Molini BJ, Iverson-Cabral S, Ciccarese G, Drago F, Lukehart SA, Centurion-Lara A. 2015. Transcription of TP0126, Treponema pallidum putative OmpW homolog, is regulated by the length of a homopolymeric guanosine repeat. Infect Immun 83:2275–2289. 10.1128/IAI.00360-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pravda L, Sehnal D, Tousek D, Navratilova V, Bazgier V, Berka K, Svobodova Varekova R, Koca J, Otyepka M. 2018. MOLEonline: a Web-based tool for analyzing channels, tunnels and pores (2018 update). Nucleic Acids Res 46:W368–W373. 10.1093/nar/gky309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Haynes AM, Godornes C, Ke W, Giacani L. 2019. Evaluation of the protective ability of the Treponema pallidum subsp. pallidum Tp0126 OmpW homolog in the rabbit model of syphilis. Infect Immun 87:e00323-19. 10.1128/IAI.00323-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.van den Berg B. 2005. The FadL family: unusual transporters for unusual substrates. Curr Opin Struct Biol 15:401–407. 10.1016/j.sbi.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 98.Hearn EM, Patel DR, Lepore BW, Indic M, van den Berg B. 2009. Transmembrane passage of hydrophobic compounds through a protein channel wall. Nature 458:367–370. 10.1038/nature07678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hearn EM, Patel DR, van den Berg B. 2008. Outer-membrane transport of aromatic hydrocarbons as a first step in biodegradation. Proc Natl Acad Sci U S A 105:8601–8606. 10.1073/pnas.0801264105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cox DL, Radolf JD. 2001. Insertion of fluorescent fatty acid probes into the outer membranes of the pathogenic spirochaetes Treponema pallidum and Borrelia burgdorferi. Microbiology (Reading) 147:1161–1169. 10.1099/00221287-147-5-1161. [DOI] [PubMed] [Google Scholar]
- 101.Deka RK, Brautigam CA, Liu WZ, Tomchick DR, Norgard MV. 2016. Molecular insights into the enzymatic diversity of flavin-trafficking protein (Ftp; formerly ApbE) in flavoprotein biogenesis in the bacterial periplasm. Microbiologyopen 5:21–38. 10.1002/mbo3.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Edwards AM. 2014. Structure and general properties of flavins. Methods Mol Biol 1146:3–13. 10.1007/978-1-4939-0452-5_1. [DOI] [PubMed] [Google Scholar]
- 103.D’Andrea LD, Regan L. 2003. TPR proteins: the versatile helix. Trends Biochem Sci 28:655–662. 10.1016/j.tibs.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 104.Hagemans D, van Belzen IA, Moran Luengo T, Rudiger SG. 2015. A script to highlight hydrophobicity and charge on protein surfaces. Front Mol Biosci 2:56. 10.3389/fmolb.2015.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lithgow KV, Cameron CE. 2017. Vaccine development for syphilis. Expert Rev Vaccines 16:37–44. 10.1080/14760584.2016.1203262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Brautigam CA, Deka RK, Schuck P, Tomchick DR, Norgard MV. 2012. Structural and thermodynamic characterization of the interaction between two periplasmic Treponema pallidum lipoproteins that are components of a TPR-protein-associated TRAP transporter (TPAT). J Mol Biol 420:70–86. 10.1016/j.jmb.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Deka RK, Brautigam CA, Goldberg M, Schuck P, Tomchick DR, Norgard MV. 2012. Structural, bioinformatic, and in vivo analyses of two Treponema pallidum lipoproteins reveal a unique TRAP transporter. J Mol Biol 416:678–696. 10.1016/j.jmb.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Luthra A, Montezuma-Rusca JM, La Vake CJ, LeDoyt M, Delgado KN, Davenport TC, Fiel-Gan M, Caimano MJ, Radolf JD, Hawley KL. 2020. Evidence that immunization with TP0751, a bipartite Treponema pallidum lipoprotein with an intrinsically disordered region and lipocalin fold, fails to protect in the rabbit model of experimental syphilis. PLoS Pathog 16:e1008871. 10.1371/journal.ppat.1008871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Centurion-Lara A, Castro C, Barrett L, Cameron C, Mostowfi M, Van Voorhis WC, Lukehart SA. 1999. Treponema pallidum major sheath protein homologue Tpr K is a target of opsonic antibody and the protective immune response. J Exp Med 189:647–656. 10.1084/jem.189.4.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Egli C, Leung WK, Muller KH, Hancock RE, McBride BC. 1993. Pore-forming properties of the major 53-kilodalton surface antigen from the outer sheath of Treponema denticola. Infect Immun 61:1694–1699. 10.1128/IAI.61.5.1694-1699.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Anand A, Luthra A, Edmond ME, LeDoyt M, Caimano MJ, Radolf JD. 2013. The major outer sheath protein (Msp) of Treponema denticola has a bipartite domain architecture and exists as periplasmic and outer membrane-spanning conformers. J Bacteriol 195:2060–2071. 10.1128/JB.00078-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Visser MB, Ellen RP. 2011. New insights into the emerging role of oral spirochaetes in periodontal disease. Clin Microbiol Infect 17:502–512. 10.1111/j.1469-0691.2011.03460.x. [DOI] [PubMed] [Google Scholar]
- 113.Fenno JC, Muller KH, McBride BC. 1996. Sequence analysis, expression, and binding activity of recombinant major outer sheath protein (Msp) of Treponema denticola. J Bacteriol 178:2489–2497. 10.1128/jb.178.9.2489-2497.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ishihara K. 2010. Virulence factors of Treponema denticola. Periodontol 2000 54:117–135. 10.1111/j.1600-0757.2009.00345.x. [DOI] [PubMed] [Google Scholar]
- 115.Anand A, Luthra A, Dunham-Ems S, Caimano MJ, Karanian C, LeDoyt M, Cruz AR, Salazar JC, Radolf JD. 2012. TprC/D (Tp0117/131), a trimeric, pore-forming rare outer membrane protein of Treponema pallidum, has a bipartite domain structure. J Bacteriol 194:2321–2333. 10.1128/JB.00101-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mistry J, Chuguransky S, Williams L, Qureshi M, Salazar GA, Sonnhammer ELL, Tosatto SCE, Paladin L, Raj S, Richardson LJ, Finn RD, Bateman A. 2021. Pfam: the protein families database in 2021. Nucleic Acids Res 49:D412–D419. 10.1093/nar/gkaa913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dosztanyi Z, Meszaros B, Simon I. 2009. ANCHOR: Web server for predicting protein binding regions in disordered proteins. Bioinformatics 25:2745–2746. 10.1093/bioinformatics/btp518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Vacic V, Oldfield CJ, Mohan A, Radivojac P, Cortese MS, Uversky VN, Dunker AK. 2007. Characterization of molecular recognition features, MoRFs, and their binding partners. J Proteome Res 6:2351–2366. 10.1021/pr0701411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Randall A, Cheng J, Sweredoski M, Baldi P. 2008. TMBpro: secondary structure, beta-contact and tertiary structure prediction of transmembrane beta-barrel proteins. Bioinformatics 24:513–520. 10.1093/bioinformatics/btm548. [DOI] [PubMed] [Google Scholar]
- 120.Ho EL, Lukehart SA. 2011. Syphilis: using modern approaches to understand an old disease. J Clin Invest 121:4584–4592. 10.1172/JCI57173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pinto M, Borges V, Antelo M, Pinheiro M, Nunes A, Azevedo J, Borrego MJ, Mendonca J, Carpinteiro D, Vieira L, Gomes JP. 2016. Genome-scale analysis of the non-cultivable Treponema pallidum reveals extensive within-patient genetic variation. Nat Microbiol 2:16190. 10.1038/nmicrobiol.2016.190. [DOI] [PubMed] [Google Scholar]
- 122.Centurion-Lara A, Giacani L, Godornes C, Molini BJ, Brinck Reid T, Lukehart SA. 2013. Fine analysis of genetic diversity of the tpr gene family among treponemal species, subspecies and strains. PLoS Negl Trop Dis 7:e2222. 10.1371/journal.pntd.0002222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Piddock LJ. 2006. Multidrug-resistance efflux pumps—not just for resistance. Nat Rev Microbiol 4:629–636. 10.1038/nrmicro1464. [DOI] [PubMed] [Google Scholar]
- 124.Baranova N. 2016. Involvement of antimicrobial drug efflux systems in bacterial fitness and virulence, p 701–727. In Li X-Z, Elkins CA, Zgurskaya HI (ed), Efflux-mediated antimicrobial resistance in bacteria: mechanisms, regulation and clinical implications. Springer International Publishing, Cham, Switzerland. 10.1007/978-3-319-39658-3_27. [DOI] [Google Scholar]
- 125.Alcalde-Rico M, Hernando-Amado S, Blanco P, Martinez JL. 2016. Multidrug efflux pumps at the crossroad between antibiotic resistance and bacterial virulence. Front Microbiol 7:1483. 10.3389/fmicb.2016.01483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Du D, Wang-Kan X, Neuberger A, van Veen HW, Pos KM, Piddock LJV, Luisi BF. 2018. Multidrug efflux pumps: structure, function and regulation. Nat Rev Microbiol 16:523–539. 10.1038/s41579-018-0048-6. [DOI] [PubMed] [Google Scholar]
- 127.Wang Z, Fan G, Hryc CF, Blaza JN, Serysheva II, Schmid MF, Chiu W, Luisi BF, Du D. 2017. An allosteric transport mechanism for the AcrAB-TolC multidrug efflux pump. Elife 6:e24905. 10.7554/eLife.24905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fitzpatrick AWP, Llabres S, Neuberger A, Blaza JN, Bai XC, Okada U, Murakami S, van Veen HW, Zachariae U, Scheres SHW, Luisi BF, Du D. 2017. Structure of the MacAB-TolC ABC-type tripartite multidrug efflux pump. Nat Microbiol 2:17070. 10.1038/nmicrobiol.2017.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Du D, van Veen HW, Murakami S, Pos KM, Luisi BF. 2015. Structure, mechanism and cooperation of bacterial multidrug transporters. Curr Opin Struct Biol 33:76–91. 10.1016/j.sbi.2015.07.015. [DOI] [PubMed] [Google Scholar]
- 130.Brautigam CA, Deka RK, Ouyang Z, Machius M, Knutsen G, Tomchick DR, Norgard MV. 2012. Biophysical and bioinformatic analyses implicate the Treponema pallidum Tp34 lipoprotein (Tp0971) in transition metal homeostasis. J Bacteriol 194:6771–6781. 10.1128/JB.01494-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Crow A, Greene NP, Kaplan E, Koronakis V. 2017. Structure and mechanotransmission mechanism of the MacB ABC transporter superfamily. Proc Natl Acad Sci U S A 114:12572–12577. 10.1073/pnas.1712153114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Higgins MK, Eswaran J, Edwards P, Schertler GF, Hughes C, Koronakis V. 2004. Structure of the ligand-blocked periplasmic entrance of the bacterial multidrug efflux protein TolC. J Mol Biol 342:697–702. 10.1016/j.jmb.2004.07.088. [DOI] [PubMed] [Google Scholar]
- 133.Andersen C, Koronakis E, Hughes C, Koronakis V. 2002. An aspartate ring at the TolC tunnel entrance determines ion selectivity and presents a target for blocking by large cations. Mol Microbiol 44:1131–1139. 10.1046/j.1365-2958.2002.02898.x. [DOI] [PubMed] [Google Scholar]
- 134.Hood MI, Skaar EP. 2012. Nutritional immunity: transition metals at the pathogen-host interface. Nat Rev Microbiol 10:525–537. 10.1038/nrmicro2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jerse AE, Sharma ND, Simms AN, Crow ET, Snyder LA, Shafer WM. 2003. A gonococcal efflux pump system enhances bacterial survival in a female mouse model of genital tract infection. Infect Immun 71:5576–5582. 10.1128/iai.71.10.5576-5582.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lafond RE, Lukehart SA. 2006. Biological basis for syphilis. Clin Microbiol Rev 19:29–49. 10.1128/CMR.19.1.29-49.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Radolf JD. 1995. Treponema pallidum and the quest for outer membrane proteins. Mol Microbiol 16:1067–1073. 10.1111/j.1365-2958.1995.tb02332.x. [DOI] [PubMed] [Google Scholar]
- 138.Turner TB, Hollander DH. 1957. Biology of the treponematoses based on studies carried out at the International Treponematosis Laboratory Center of the Johns Hopkins University under the auspices of the World Health Organization. Monogr Ser World Health Organ 1957:3–266. [PubMed] [Google Scholar]
- 139.Fieldsteel AH, Cox DL, Moeckli RA. 1981. Cultivation of virulent Treponema pallidum in tissue culture. Infect Immun 32:908–915. 10.1128/IAI.32.2.908-915.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Magnuson HJ, Eagle H, Fleischman R. 1948. The minimal infectious inoculum of Spirochaeta pallida (Nichols strain) and a consideration of its rate of multiplication in vivo. Am J Syph Gonorrhea Vener Dis 32:1–18. [PubMed] [Google Scholar]
- 141.Edmondson DG, Hu B, Norris SJ. 2018. Long-term in vitro culture of the syphilis spirochete Treponema pallidum subsp. pallidum. mBio 9:e01153-18. 10.1128/mBio.01153-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rein MF. 1976. Biopharmacology of syphilotherapy. J Am Vener Dis Assoc 3:109–127. [PubMed] [Google Scholar]
- 143.Galvao TF, Silva MT, Serruya SJ, Newman LM, Klausner JD, Pereira MG, Fescina R. 2013. Safety of benzathine penicillin for preventing congenital syphilis: a systematic review. PLoS One 8:e56463. 10.1371/journal.pone.0056463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ghanem KG, Ram S, Rice PA. 2020. The modern epidemic of syphilis. N Engl J Med 382:845–854. 10.1056/NEJMra1901593. [DOI] [PubMed] [Google Scholar]
- 145.Gottlieb SL, Deal CD, Giersing B, Rees H, Bolan G, Johnston C, Timms P, Gray-Owen SD, Jerse AE, Cameron CE, Moorthy VS, Kiarie J, Broutet N. 2016. The global roadmap for advancing development of vaccines against sexually transmitted infections: update and next steps. Vaccine 34:2939–2947. 10.1016/j.vaccine.2016.03.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington, DC. [Google Scholar]
- 147.Spiliotis M. 2012. Inverse fusion PCR cloning. PLoS One 7:e35407. 10.1371/journal.pone.0035407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Roy A, Kucukural A, Zhang Y. 2010. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5:725–738. 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Noinaj N, Kuszak AJ, Gumbart JC, Lukacik P, Chang H, Easley NC, Lithgow T, Buchanan SK. 2013. Structural insight into the biogenesis of β-barrel membrane proteins. Nature 501:385–390. 10.1038/nature12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Owens TW, Taylor RJ, Pahil KS, Bertani BR, Ruiz N, Kruse AC, Kahne D. 2019. Structural basis of unidirectional export of lipopolysaccharide to the cell surface. Nature 567:550–553. 10.1038/s41586-019-1039-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ren J, Wen L, Gao X, Jin C, Xue Y, Yao X. 2009. DOG 1.0: illustrator of protein domain structures. Cell Res 19:271–273. 10.1038/cr.2009.6. [DOI] [PubMed] [Google Scholar]
- 152.Janson G, Zhang C, Prado MG, Paiardini A. 2017. PyMod 2.0: improvements in protein sequence-structure analysis and homology modeling within PyMOL. Bioinformatics 33:444–446. 10.1093/bioinformatics/btw638. [DOI] [PubMed] [Google Scholar]
- 153.Sievers F, Higgins DG. 2018. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci 27:135–145. 10.1002/pro.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Pei J, Kim BH, Grishin NV. 2008. PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res 36:2295–2300. 10.1093/nar/gkn072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gouet P, Robert X, Courcelle E. 2003. ESPript/ENDscript: extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res 31:3320–3323. 10.1093/nar/gkg556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Jurrus E, Engel D, Star K, Monson K, Brandi J, Felberg LE, Brookes DH, Wilson L, Chen J, Liles K, Chun M, Li P, Gohara DW, Dolinsky T, Konecny R, Koes DR, Nielsen JE, Head-Gordon T, Geng W, Krasny R, Wei GW, Holst MJ, McCammon JA, Baker NA. 2018. Improvements to the APBS biomolecular solvation software suite. Protein Sci 27:112–128. 10.1002/pro.3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kringelum JV, Lundegaard C, Lund O, Nielsen M. 2012. Reliable B cell epitope predictions: impacts of method development and improved benchmarking. PLoS Comput Biol 8:e1002829. 10.1371/journal.pcbi.1002829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Nishikawa K, Ooi T. 1980. Prediction of the surface-interior diagram of globular proteins by an empirical method. Int J Pept Protein Res 16:19–32. 10.1111/j.1399-3011.1980.tb02931.x. [DOI] [PubMed] [Google Scholar]
- 159.Letunic I, Bork P. 2019. Interactive Tree of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res 47:W256–W259. 10.1093/nar/gkz239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Yang L, Antonelli S, Chodankar S, Byrnes J, Lazo E, Qian K. 2020. Solution scattering at the Life Science X-Ray Scattering (LiX) beamline. J Synchrotron Radiat 27:804–812. 10.1107/S1600577520002362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Allaire M, Yang L. 2011. Biomolecular solution X-ray scattering at the National Synchrotron Light Source. J Synchrotron Radiat 18:41–44. 10.1107/S0909049510036022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hopkins JB, Gillilan RE, Skou S. 2017. BioXTAS RAW: improvements to a free open-source program for small-angle X-ray scattering data reduction and analysis. J Appl Crystallogr 50:1545–1553. 10.1107/S1600576717011438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Petoukhov MV, Franke D, Shkumatov AV, Tria G, Kikhney AG, Gajda M, Gorba C, Mertens HD, Konarev PV, Svergun DI. 2012. New developments in the ATSAS program package for small-angle scattering data analysis. J Appl Crystallogr 45:342–350. 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Konarev PV, Volkov VV, Sokolova AV, Koch MH, Svergun DI. 2003. Primus: a Windows PC-based system for small-angle scattering data analysis. J Appl Crystallogr 36:1277–1282. 10.1107/S0021889803012779. [DOI] [Google Scholar]
- 165.Svergun DI. 1992. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Crystallogr 25:495–503. 10.1107/S0021889892001663. [DOI] [Google Scholar]
- 166.Svergun DI. 1999. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys J 76:2879–2886. 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Volkov VV, Svergun DI. 2003. Uniqueness of ab initio shape determination in small-angle scattering. J Appl Crystallogr 36:860–864. 10.1107/S0021889803000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Schneidman-Duhovny D, Hammel M, Tainer JA, Sali A. 2016. FoXS, FoXSDock and MultiFoXS: single-state and multi-state structural modeling of proteins and their complexes based on SAXS profiles. Nucleic Acids Res 44:W424–W429. 10.1093/nar/gkw389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Somboon K, Doble A, Bulmer D, Basle A, Khalid S, van den Berg B. 2020. Uptake of monoaromatic hydrocarbons during biodegradation by FadL channel-mediated lateral diffusion. Nat Commun 11:6331. 10.1038/s41467-020-20126-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S16 and Tables S1 to S10. Download JB00082-21-s0001.pdf, PDF file, 1.39 MB (5.5MB, pdf)