

Abstract
Background
Botulinum toxin type A (BontA) is the most frequent treatment for facial wrinkles, but its effectiveness and safety have not previously been assessed in a Cochrane Review.
Objectives
To assess the effects of all commercially available botulinum toxin type A products for the treatment of any type of facial wrinkles.
Search methods
We searched the following databases up to May 2020: the Cochrane Skin Specialised Register, CENTRAL, MEDLINE, Embase, and LILACS. We also searched five trials registers, and checked the reference lists of included studies for further references to relevant randomised controlled trials (RCTs).
Selection criteria
We included RCTs with over 50 participants, comparing BontA versus placebo, other types of BontA, or fillers (hyaluronic acid), for treating facial wrinkles in adults.
Data collection and analysis
We used standard methodological procedures expected by Cochrane. Primary outcomes were participant assessment of success and major adverse events (AEs) (eyelid ptosis, eyelid sensory disorder, strabismus). Secondary outcomes included physician assessment of success; proportion of participants with at least one AE and duration of treatment effect. We used GRADE to assess the certainty of the evidence for each outcome.
Main results
We included 65 RCTs, involving 14,919 randomised participants. Most participants were female, aged 18 to 65 years. All participants were outpatients (private office or day clinic). Study duration was between one week and one year. No studies were assessed as low risk of bias in all domains; the overall risk of bias was unclear for most studies.
The most common comparator was placebo (36 studies). An active control was used in 19 studies. There were eight dose‐ranging studies of onabotulinumtoxinA, and a small number of studies compared against fillers. Treatment was given in one cycle (54 studies), two cycles (three studies), or three or more cycles (eight studies).
The treated regions were glabella (43 studies), crow's feet (seven studies), forehead (two studies), perioral (two studies), full face (one study), or more than two regions (nine studies). Most studies analysed moderate to severe wrinkles; mean duration of treatment was 20 weeks.
The following results summarise the main comparisons, based on studies of one treatment cycle for the glabella. AEs were collected over the duration of these studies (over four to 24 weeks).
Compared to placebo, onabotulinumtoxinA‐20 U probably has a higher success rate when assessed by participants (risk ratio (RR) 19.45, 95% confidence interval (CI) 8.60 to 43.99; 575 participants; 4 studies; moderate‐certainty evidence) or physicians (RR 17.10, 95% CI 10.07 to 29.05; 1339 participants; 7 studies; moderate‐certainty evidence) at week four. Major AEs are probably higher with onabotulinumtoxinA‐20 U (Peto OR 3.62, 95% CI 1.50 to 8.74; 1390 participants; 8 studies; moderate‐certainty evidence), but there may be no difference in any AEs (RR 1.14, 95% CI 0.89 to 1.45; 1388 participants; 8 studies; low‐certainty evidence).
Compared to placebo, abobotulinumtoxinA‐50 U has a higher participant‐assessed success rate at week four (RR 21.22, 95% CI 7.40 to 60.56; 915 participants; 6 studies; high‐certainty evidence); and probably has a higher physician‐assessed success rate (RR 14.93, 95% CI 8.09 to 27.55; 1059 participants; 7 studies; moderate‐certainty evidence). There are probably more major AEs with abobotulinumtoxinA‐50 U (Peto OR 3.36, 95% CI 0.88 to 12.87; 1294 participants; 7 studies; moderate‐certainty evidence). Any AE may be more common with abobotulinumtoxinA‐50 U (RR 1.25, 95% CI 1.05 to 1.49; 1471 participants; 8 studies; low‐certainty evidence).
Compared to placebo, incobotulinumtoxinA‐20 U probably has a higher participant‐assessed success rate at week four (RR 66.57, 95% CI 13.50 to 328.28; 547 participants; 2 studies; moderate‐certainty evidence), and physician‐assessed success rate (RR 134.62, 95% CI 19.05 to 951.45; 547 participants; 2 studies; moderate‐certainty evidence). Major AEs were not observed (547 participants; 2 studies; moderate‐certainty evidence). There may be no difference between groups in any AEs (RR 1.17, 95% CI 0.90 to 1.53; 547 participants; 2 studies; low‐certainty evidence).
AbobotulinumtoxinA‐50 U is no different to onabotulinumtoxinA‐20 U in participant‐assessed success rate (RR 1.00, 95% CI 0.92 to 1.08, 388 participants, 1 study, high‐certainty evidence) and physician‐assessed success rate (RR 1.01, 95% CI 0.95 to 1.06; 388 participants; 1 study; high‐certainty evidence) at week four. Major AEs are probably more likely in the abobotulinumtoxinA‐50 U group than the onabotulinumtoxinA‐20 U group (Peto OR 2.65, 95% CI 0.77 to 9.09; 433 participants; 1 study; moderate‐certainty evidence). There is probably no difference in any AE (RR 1.02, 95% CI 0.67 to 1.54; 492 participants; 2 studies; moderate‐certainty evidence).
IncobotulinumtoxinA‐24 U may be no different to onabotulinumtoxinA‐24 U in physician‐assessed success rate at week four (RR 1.01, 95% CI 0.96 to 1.05; 381 participants; 1 study; low‐certainty evidence) (participant assessment was not measured). One participant reported ptosis with onabotulinumtoxinA, but we are uncertain of the risk of AEs (Peto OR 0.02, 95% CI 0.00 to 1.77; 381 participants; 1 study; very low‐certainty evidence).
Compared to placebo, daxibotulinumtoxinA‐40 U probably has a higher participant‐assessed success rate (RR 21.10, 95% CI 11.31 to 39.34; 683 participants; 2 studies; moderate‐certainty evidence) and physician‐assessed success rate (RR 23.40, 95% CI 12.56 to 43.61; 683 participants; 2 studies; moderate‐certainty evidence) at week four. Major AEs were not observed (716 participants; 2 studies; moderate‐certainty evidence). There may be an increase in any AE with daxibotulinumtoxinA compared to placebo (RR 2.23, 95% CI 1.46 to 3.40; 716 participants; 2 studies; moderate‐certainty evidence).
Major AEs reported were mainly ptosis; BontA is also known to carry a risk of strabismus or eyelid sensory disorders.
Authors' conclusions
BontA treatment reduces wrinkles within four weeks of treatment, but probably increases risk of ptosis. We found several heterogeneous studies (different types or doses of BontA, number of cycles, and different facial regions) hindering meta‐analyses. The certainty of the evidence for effectiveness outcomes was high, low or moderate; for AEs, very low to moderate. Future RCTs should compare the most common BontA (onabotulinumtoxinA, abobotulinumtoxinA, incobotulinumtoxinA, daxibotulinumtoxinA, prabotulinumtoxinA) and evaluate long‐term outcomes. There is a lack of evidence about the effects of multiple cycles of BontA, frequency of major AEs, duration of effect, efficacy of recently‐approved BontA and comparisons with other treatments.
Plain language summary
How well does botulinum toxin (type A; often called ‘Botox’) treat wrinkles on the face?
Key messages
Injecting botulinum toxin type A (a Botox‐like treatment) reduces wrinkles between the eyebrows, and is relatively safe to use. The effects on wrinkles were seen when measured at four weeks after the injection. Injecting botulinum toxin type A probably increases the risk of eyelid drooping. More studies are needed to assess the longer‐term benefits and harms of repeated treatment with botulinum toxin.
Treating facial wrinkles
Continuous movement of muscles in the face can cause the skin to wrinkle as it ages and becomes less elastic. Botulinum toxin type A is a chemical that relaxes muscles; it is produced by a type of bacteria. It is commonly used to smooth out lines and wrinkles by injecting it into the muscles of the face to stop their movement for a short time. Muscle activity usually stops completely within five to 15 days after the injection. The effects on the muscles are temporary and usually last for around four to six months.
What did we want to find out?
We wanted to find out how well botulinum toxin could treat wrinkles on the face, and if it causes any unwanted effects.
What did we do?
We searched for studies that tested the effects of botulinum toxin to treat wrinkles on the face.
What did we find?
We found 65 studies in 14,919 people (mostly women) who went to a day clinic or private office for treatment. The studies lasted from one week to one year; the average length of treatment was 20 weeks. The studies compared one type of botulinum toxin against another type, against a placebo (an injection that did not contain any botulinum toxin), or against an alternative treatment. Several studies were funded by pharmaceutical companies.
The studies tested four types of botulinum toxin that were licensed for use and some other types that were not yet licensed.
All studies assessed the success of treatment by measuring wrinkles and lines when facial muscles were at their most tense. Most studies treated wrinkles that develop between the eyebrows, known as 'glabellar lines'.
What are the main results of our review?
At four weeks after injection, all types of botulinum toxin reduced glabellar lines more than a placebo. This effect was seen whether the wrinkles were assessed by doctors or by the people who had the injections.
Unwanted effects are probably more common with botulinum toxin than with placebo injections. The most commonly reported unwanted effects are drooping eyelids, squinting (when the eyes point in different directions) and numbness of the eyelid.
Two studies compared two different types of botulinum toxin and found no difference between the types for how well they reduced glabellar lines.
What are the limitations of the evidence?
Our confidence in the evidence is moderate to high that botulinum toxin reduces wrinkles between the eyebrows better than a placebo. We are less confident in some of the evidence for other comparisons or studies, because some studies enrolled only a small number of people, and in some studies it was unclear how people were assigned to different treatment groups or if people knew which treatment they received. Further research is likely to increase our confidence in the evidence.
How up to date is this evidence?
The evidence is current up to May 2020.
Summary of findings
Background
Please note that unfamiliar terms may be listed in the Glossary in Appendix 1.
Description of the condition
Aging is a biological process; however, it is not well accepted by all in western cultures, who desire to retain a youthful appearance and optimal level of beauty, equating it with increased socialisation, power, success, and happiness. Preventing and treating the consequences of aging in the body has become almost a fixation (Garnham 2013).
Facial ageing depends on intrinsic factors, which include genetics (heredity) and ethnicity, and extrinsic factors, such as environmental conditions (e.g. sun exposure, smoking habits, and nutritional status). All of these factors contribute towards the appearance of ageing signs: fat absorption, flaccidity, and wrinkles (Sveikata 2011). The aging process turns the skin thinner, drier, and less elastic, and less able to protect itself from internal and external aggressions. Due to these factors, the continuous muscle movement (facial expression) can lead to wrinkles.One of the first stages of facial aging includes the appearance of dynamic wrinkles. Additionally, the appearance of dynamic wrinkles occurs through increased muscle tone, as shown by electromyographic alterations Le Louarn 2007. Over an individual's lifetime, however, resting muscle tone increases and creases the skin causing fine wrinkles and lines in the skin surface (hyperdynamic wrinkles). If these wrinkles do not receive any treatment, the skin shows a permanent mark (static wrinkles) (Carruthers 2008a). Due to this fact, the dynamic rhytides treatment is more indicated in the clinical practice. Facial wrinkles can be classified in glabellar lines (vertical lines or furrows in the region between the eyebrows, above the nose); forehead lines (vertical or diagonal lines in the forehead region), crow's feet lines (lines or furrows in the periorbicular region, around the eyes).
Several surgical and non‐surgical procedures for dynamic wrinkles are available. Amongst all therapies, botulinum toxin type A (BontA) injections are considered the most frequent treatment for this condition. According to the American Society of aesthetic surgery statistics, 4,597,886 injections of BontA were performed in 2016 (ASAPS 2016). The reason for BontA success can be attributed to low cost, no recovery time and temporary effect (Glogau 2012). The BontA average cost for wrinkle treatment is USD 385 per session (ASAPS 2017). This treatment is performed in outpatients, during daily activities. The temporary effect ranges from four to six months.
Description of the intervention
Botulinum toxin has been used since the 1980s; there are eight subtypes available (A, B, C1, C2, D, E, F, and G). Serotypes A and B are commercially available (Berry 2012). Botulinum toxinA is the most used in clinical practice due to its duration effect. Moreover, several brands are available in the market. Although all these toxins are type A, all companies have their particular strains. Due to this fact, each brand has specific biological characteristics: units equivalence (ratio) and dermal diffusion (Glogau 2012).
For this reason, there is a conversion ratio.
OnabotulinumtoxinA: AbobotulinumtoxinA, ratio = 1 unit (U) : 2.5 U or 3U
OnabotulinumtoxinA: IncobobotulinintoxinA, ratio = 1 U:1 U
The other BontA brands (daxibotulintoxinA (DWP450), PraxibotulinumtoxinA, HBTX‐A, Prosigne®, CBFC26, MT10109L, Medytox®, Neuronox®) follow the conversion ratio of 1:1
Despite these biological properties differences, the medical community recommend guidelines to treat facial wrinkles.
In 2008 and 2016, an American committee discussed the dose treatment related to onabotulinumtoxinA.
Glabellar region, the therapeutical dose range from 12 U to 40 U, 2 U to 4 U per injection, distributed in three to seven intramuscular injections points (procerus muscle, corrugator supercilii muscle, orbicularis oculi, muscle depressor supercilii muscle)frontal lines, the therapeutical dose range from 8 U to 25 U distributed in four to eight points of intramuscular injection along the frontal muscle wrinkles with a 2.0 cm above the eyebrows; and crow's feet, the therapeutical dose range from 6 U to 15 U distributed in two to five subcutaneous injections per side (one injection at least 1.5 cm to 2.0 cm from lateral canthus, one injection in the orbital rim next to the eyebrow extremity, and one injection near the zygomatic process in the orbital rim, the other injections along the crow’s feet lines laterally to the previous injections) (Sundaram 2016) (Carruthers 2008a).
In 2010, a European committee addressed the same issues and created an equivalent botulinum toxin type A guideline based on the other brand of BontA, AbobotulinumtoxinA biological properties:
for the glabellar region, 50U (Speywood units) in five points;
for frontal lines, 20 U to 60 U in four to six points; and
for crow's feet, 30 U to 60 U in three points per side (Ascher 2010).
The guidelines shown above studied the most common BontA used in the clinical practice.
Until now, the medical community does not know if these brands behave differently regarding effectiveness, duration of treatment and adverse events.
How the intervention might work
After injection into the muscle, botulinum toxin diffuses to the nerve terminal, where it binds, preventing the release of the neurotransmitter, acetylcholine, from the nerve synapse; thus, preventing its effect on the neuromuscular junction and consequently the muscle does not contract and does not crease the skin (no hyperdynamic wrinkle). Complete lack of muscle activity occurs after approximately five to 15 days (Berry 2012).
It is perceived that there are fewer wrinkles due to the non‐contraction of specific facial muscles (Fagien 2003). However, this muscle atrophy due to chemical effect provokes regeneration at the nerve terminal known as 'sprouting'. This process, which lasts for 120 days, will originate in a new terminal at the neuromuscular junction, which will bring back muscle activity (Berry 2012). Because of this, the duration of clinical treatment is in the range of three to six months.
It is important that during the consultation prior to the botulinum toxin procedure, the medical professional establishes the expectations of the person and whether these will be achieved, explains all possible outcomes, safety issues, duration of treatment, and potential adverse effects, and examines the anatomic regions, in rest and contraction, and any pre‐existing asymmetry. Otherwise, the botulinum toxin treatment may cause frustration and disappointment.
Moreover, for an optimal result, all medical professionals (dermatologists, plastic surgeons) should have a complete knowledge of functional muscle anatomy (Carruthers 2008a). This injection attenuates wrinkle appearance progressively (within 15 days), but the effect is temporary (four to six months) (Berry 2012; Carruthers 2008a).
Why it is important to do this review
Botulinum toxin has been used to reduce hyperdynamic facial wrinkles for more than 20 years. During this period, several formulations have appeared on the market. Although these neurotoxins are not comparable, the U.S. Food and Drug Administration (FDA), European Medicines Agency (EMA), and other drugs evaluation boards have been attempting to organise and classify them. Currently, there are several botulinum toxin type A products on the market: Botox®/Vistabel®/Vistabex® (Allergan); Dysport®/Disport® (Ipsen); Azzulure® (Galderma); XEOMIN®/Bocouture®/Xeomeen® (Merz Aesthetics); Neuronox®/Siax® (Medytox); Prosigne® (Cristalia); Lantox® (Dermacare), also known as BTXA™ (Lanzhou Institute of Biological Products (LIBP)® ‐ Hong Kong); and Lanzox® (Kalbe ‐ Indonesia) (Brandt 2009; Nettar 2011; Won 2013).
A Cochrane Review that assessed treatments for wrinkles and other skin changes provoked by photoageing included an evaluation of topical treatments (tretinoin, lactic, glycolic acids, moisturiser), and oral and topical polysaccharides and surgical procedures (CO2 laser, YAG laser, dermabrasion), but did not assess botulinum toxin for facial wrinkles (Samuel 2005).
It is important to compare the efficacy of BontA versus different BontA brands, filler (hyaluronic acid, methacrylate, calcium hydroxyapatite, Polyalkylimide, Polylactic acid), and surgery. Also, it is crucial to analyse BontA safety, for example, the major adverse effects are: blepharoptosis (abnormal low‐lying upper eyelid margin with the eye in primary gaze) and strabismus (inability of one eye to attain binocular vision with the other because of imbalance of the muscles of the eyeball).
So far, no systematic reviews have been conducted on the effectiveness and safety of botulinum toxin type A in cosmetic procedures. As a consequence of the lack of robust clinical evidence, decisions about the use of different therapies for facial wrinkles are made at the discretion of plastic surgeons or dermatologists working with the person concerned.
The aim of the present systematic review is to determine the effectiveness of botulinum toxin for the treatment of any type of facial wrinkle (dynamic or static).
The methods for this review were published as a protocol 'Botulinum toxin for facial wrinkles' (Camargo 2014).
Objectives
To assess the effects of all commercially available botulinum toxin type A products for the treatment of any type of facial wrinkles.
Methods
Criteria for considering studies for this review
Types of studies
We included, randomised controlled trials (RCTs). Additionally, we also included split‐face designs (studies that compared two different treatments, each one applied to one side of the face). We did not include cluster‐ and cross‐over trials. All the studies had to have 50 or more participants.
Types of participants
Individuals of either gender, aged 18 years and above, with a diagnosis of dynamic or static facial wrinkles (glabellar, forehead, or crow's feet).
Types of interventions
All types of botulinum toxin type A in any dose, single or multiple treatments, compared to placebo, other types of botulinum toxin type A, and fillers (hyaluronic acid).
Types of outcome measures
We included studies assessing at least one of the outcomes below.
Primary outcomes
1. Participant assessment of success, measured by validated scores or scales (Bertucci 2020; Carruthers 2003; Honeck 2003; Hund 2006; Rzany 2006). We considered wrinkles and lines at maximum contraction assessed by the following tools.
Four‐point scale (Carruthers 2003)
Patient Frown Wrinkle Severity (PFWS) scale (Bertucci 2020)
Facial Line Treatment Satisfaction (FTS) Questionnaire (14‐item) (Cox 2003)
Facial Line Outcomes Questionnaire (FLO‐7) (Cox 2003; Fagien 2007b)
Self perception of age (SPA) (Fagien 2007b; Fagien 2008)
5‐point Merz Aesthetic Scale (Rzany 2006)
2. Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus).
Secondary outcomes
1. Physician assessment of success, measured by validated scores or scales (Bertucci 2020; Carruthers 2012; Flynn 2012; Kane 2012; Narins 2012; Rzany 2012; Sattler 2012). We considered wrinkles and lines at maximum contraction assessed by the following tools.
Five‐point scale (Flynn 2012)
Investigator Global AssessmenteFrown Wrinkle Severity (IGA‐FWS) (Bertucci 2020)
Facial Wrinkle Scale (FWS) (Carruthers 2003)
Brow Positioning Grading Scale (five‐point scale) (Carruthers 2008b)
Forehead Lines Grading Scale (five‐point scale) (Carruthers 2008c)
Crow's Feet Grading Scale (Carruthers 2008d)
5‐point Merz Aesthetic Scale (Rzany 2006)
2. Any adverse event, measured by the proportion of participants presenting at least one adverse event.
3. Duration of treatment effect
Timing of outcome measurement
We assessed these outcome measures before and after treatment (predominantly focusing on 4, 8, 12, 16 weeks, or more).
Search methods for identification of studies
We aimed to identify all relevant RCTs regardless of language or publication status (published, unpublished, in press, or in progress).
Electronic searches
The Cochrane Skin Information Specialist searched the following databases up to 5 May 2020 using strategies based on the draft strategy for MEDLINE in our published protocol (Camargo 2014):
the Cochrane Skin Specialised Register using the search strategy listed in Appendix 2;
the Cochrane Central Register of Controlled Trials (CENTRAL) 2020, Issue 5 in the Cochrane Library using the strategy listed in Appendix 3
MEDLINE via Ovid (from 1946) using the strategy listed in Appendix 4;
Embase via Ovid (from 1974) using the strategy listed in Appendix 5; and
LILACS (Latin American and Caribbean Health Science Information database, from 1982) using the strategy listed in Appendix 6.
Trials registers
We (CPC, RR) searched the following trials registers up to 5 May 2020 using the search terms in Appendix 7:
the ISRCTN registry (www.isrctn.com);
ClinicalTrials.gov (www.clinicaltrials.gov);
the Australian New Zealand Clinical Trials Registry (www.anzctr.org.au);
the World Health Organization International Clinical Trials Registry Platform (ICTRP) (apps.who.int/trialsearch/); and
the EU Clinical Trials Register (www.clinicaltrialsregister.eu).
Searching other resources
References from included studies
We checked the bibliographies of included studies for further references to relevant trials.
Unpublished literature
We contacted specialists in the field, authors of the included trials, and pharmaceutical companies, to request relevant unpublished data.
We handsearched the following plastic and dermatological conference proceedings for further references to relevant RCTs:
AAD Annual Meeting (2013‐2016); and
Brazilian Congress of Dermatologic Society (2013‐2016).
Adverse events
We did not perform a separate search for adverse effects of botulinum toxin. However, we examined data on adverse effects from the included studies we identified.
Data collection and analysis
Selection of studies
Two review authors (CPC and RG) independently assessed and selected studies. We checked the full text of studies for inclusion or exclusion. We recorded reasons for exclusion in the 'Characteristics of excluded studies' tables in the review.
We referred to a third review (CSC) in any case of disagreement.
Data extraction and management
Two review authors (CPC and RG) created, piloted, and managed data extraction forms. They independently extracted data from the full text of the included studies, and a third review author (RR) resolved any discrepancies.
We considered the following data and inserted into the data extraction form:
publication information (e.g. journal, year, and authors);
study design, including details of randomisation methods and blinding of treatments;
methodology, such as inclusion and exclusion criteria and risk of bias (e.g. selection, performance, detection, and attrition);
population;
outcome measures of the study (we will indicate where these are our prespecified outcomes for this review);
dropouts; and
treatment (e.g. total units, duration, and number of treatments).
Assessment of risk of bias in included studies
Two review authors (CPC and RG) independently applied Cochrane's risk of bias tool (Higgins 2017). We referred to a third review author (RR) in any case of disagreement.
We assessed the following domains to evaluate risk of bias (low, high, or unclear):
(a) random sequence generation; (b) allocation concealment; (c) blinding (e.g. blinding of participants, personnel, and outcome assessment); (d) attrition (i.e. incomplete outcome reporting); (e) selective reporting bias; and (f) other risks of bias.
Measures of treatment effect
Considering treatment effects with 95% confidence intervals (CIs), we reported dichotomous outcomes as risk ratios (RR) and continuous outcomes as standardised mean difference (SMD) when studies used different scales. In rare events (any major adverse event) we reported dichotomous outcomes as Peto odds ratio (OR), since when there is a low number of events, OR is similar to RR.
| Score scale | Method | Assessment of outcomes | Type of variable |
| 4‐point scale | 0 = none 1 = mild 2 = moderate 3 = severe |
Mean/median | Ordinal |
| 9‐point scale | +4 = complete improvement (100%) 0 = no change ‐4 = 100% worse |
Mean/median | Ordinal |
| Participant satisfaction | 0 to 7 | Categorical | |
| FLO | Age perception | Ordinal | |
| FTLS | 7‐point scale | Mean/median | Ordinal |
We considered an 'event' for a dichotomous variable (success) when the patients showed 2‐points of improvement in the wrinkles scale.
Unit of analysis issues
We considered the individual participant as the unit of analysis. We also considered each side or region of the face as the unit of analysis for split‐face studies, and described these studies narratively.
Dealing with missing data
In case of missing data, we contacted study authors for more information. When the authors did not respond satisfactorily, we did not use the study for quantitative analysis, and we used intention‐to‐treat (ITT) analysis. We utilised dropouts as ITT analysis.
We considered outcome data complete if the analysis included more than 80% of participants. We applied these criteria to all trials. When data were missing and the study was not included in a meta‐analysis, we discussed it in the text of this review.
Assessment of heterogeneity
We used the I² statistic to quantify the level of statistical heterogeneity for each outcome. According to the I² statistic, we classified heterogeneity as follows: low heterogeneity (0% to 25%), moderate (25% to 75%), or substantial (more than 75%) as suggested in the Cochrane Handbook for Systematic Review of Interventions (Higgins 2020).
We performed a random‐effect meta‐analysis by default, since regardless of statistical heterogeneity, we expected a significant clinical and/or methodological heterogeneity among included RCTs.
Assessment of reporting biases
We contacted study authors to clarify non‐reporting of their outcomes. We did not perform a funnel plot because there was less than 10 papers in each analysis.
Data synthesis
We summarised data using the Review Manager 5 software (RevMan). When pooling data was not appropriate or possible (lack of data), we described the results in the main text.
Subgroup analysis and investigation of heterogeneity
We planned the following subgroups analyses:
age;
gender;
ethnic group;
type of wrinkles: static or dynamic; and
total doses per area of the face (e.g. glabellar, forehead, periorbicular).
We also planned to undertaken sensitivity analyses by removing studies at high risk of bias.
However, we did not carry out any subgroup analyses due to lack of available data.
Sensitivity analysis
We planned to perform sensitivity analysis considering studies with high risk of bias (allocation) and comparing the results with the overall findings. However, due to the low number of included studies in quantitative synthesis, we deemed this analysis inappropriate.
Summary of findings and assessment of the certainty of the evidence
We created six summary of findings tables for the comparisons below (chosen based on clinical relevance considering type of toxin and face region):
OnabotulinumtoxinA 20 units compared to placebo, one cycle of treatment in glabellar lines for facial wrinkles
AbobotulinumtoxinA 50 units compared to placebo, one cycle of treatment in glabellar lines for facial wrinkles
IncobotulinumtoxinA 20 units compared to placebo, one cycle of treatment in glabellar lines for facial wrinkles
AbobotulinumtoxinA 50 units compared to onabotulinumtoxinA 20 units, one cycle of treatment in glabellar lines for facial wrinkles
OnabotulinumtoxinA 24 units compared to incobotulinumtoxinA 24 units, one cycle of treatment in glabellar lines for facial wrinkles
DaxibotulinumtoxinA 40 units compared to placebo, one cycle of treatment in glabellar lines for facial wrinkles
We included both our primary and secondary outcomes in each table. For participant‐ and physician‐assessment of success we chose the time point closest to four weeks to include in our summary of findings tables. For the major adverse events and treatment duration, we used the longer time point reported. We used the five GRADE criteria (study limitations, consistency of effect, imprecision, indirectness, and publication bias) to assess the certainty of the evidence related with each prespecified primary outcomes. We used methods described in Chapter 14 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2020), and used the platform GRADEpro GDT (GRADEpro GDT). We explained each decisions for down‐ or upgrading the criteria using footnotes, and added comments where necessary.
Results
Description of studies
Results of the search
The Electronic searches retrieved 441 records and our handsearches retrieved 15 further records. We had a total of 456 records. After removing duplicates, 425 records were screened. We excluded 325 records based on titles and abstracts. We obtained the full text of the remaining 100 records for further scrutiny against our inclusion criteria. We excluded 11 studies (see Characteristics of excluded studies). Twenty‐four studies are awaiting classification (see Characteristics of studies awaiting classification ). We included 65 studies reported in 75 references (see Characteristics of included studies). Twenty‐seven studies were included in the quantitative synthesis.
For a further description of our screening process, see the study flow diagram Figure 1.
1.
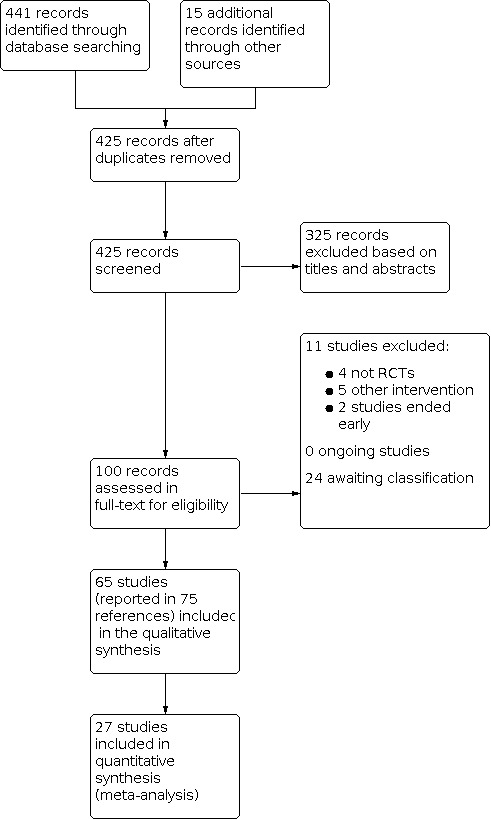
Study flow diagram.
Included studies
We included 65 studies (reported in 75 references), which randomised a total of 14,919 participants. Four were published only as abstracts (Ascher 2018; Firoz 2012; Lee 2013; Solish 2018), and 61 were published as full text (see Characteristics of included studies).
We sent 21 emails to the authors of the following studies asking for additional data (Ascher 2004; Ascher 2005; Ascher 2009; Beer 2006; Beer 2019a; Brandt 2009; Carruthers 2003a; Carruthers 2004; Carruthers 2005a; Cohen 2012; Dayan 2010; Feng 2015; Hexsel 2013; Kane 2009; Kassir 2013; Michaels 2012; Moers‐Carpi 2015; Monheit 2007; Rappl 2013; Rzany 2006; Won 2015).
We did not receive any answer from authors of 12 studies (Ascher 2004; Ascher 2005; Ascher 2009; Beer 2006; Beer 2019a; Brandt 2009; Dayan 2010; Feng 2015; Kane 2009; Michaels 2012; Moers‐Carpi 2015; Won 2015).
Nine authors answered our emails.
Four authors answered that data were no longer available because the studies were carried out too long ago (Carruthers 2003a; Carruthers 2004; Carruthers 2005a; Cohen 2012).
Hexsel 2013 provided a SPSS file, but did not provide any information to clarify our questions.
Kassir 2013 provided the full paper, but there was no additional information about missing data.
Monheit 2007 answered the following: “The assessment for primary response was at maximal contraction”
Rappl 2013 clarified a discrepancy confirming that 21U was the correct dose used.
Rzany 2006 answered the following: “Concerning the data. This was an IPSEN initiated trial. All analysis was done through IPSEN. I would suggest that you contact IPSEN directly”.
Despite our best efforts, we could not find a validated email for the authors of two studies (Harii 2008; Lee 2013).
Design
Five studies were split‐face design (Firoz 2012; Michaels 2012; Kassir 2013; Nettar 2011; Park 2014). The remaining studies were randomised controlled trials (RCTs) with parallel design.
Setting
In total, 51 RCTs were multicentre studies.
USA (n = 8) (Brandt 2009; Carruthers 2002; Hanke 2013; Kane 2009; Monheit 2019; Beer 2019a; Beer 2019b; Rubin 2009)
Canada (n = 3) (Carruthers 2017; Rivers 2015; Carruthers 2017)
Europe, single country (n = 4) (Ascher 2004; Ascher 2005; Ascher 2009; Rzany 2006)
Europe, two or more countries (n = 5) (Ascher 2018; Ascher 2020; Kerscher 2015; NCT02493946; Satler 2010)
North America, two or more countries (n = 8) (Bertucci 2020; Carruthers 2004; Carruthers 2010; Carruthers 2013; Carruthers 2003b; Carruthers 2015; Monheit 2007; Solish 2016)
Transcontinental (n = 8) (Carruthers 2014; Kane 2015; Lowe 2006; Moers‐Carpi 2012; Moers‐Carpi 2015; De Boulle 2018; Ogilvie 2019; Rzany 2019)
Asia, single country (n = 11) (Cheon 2019; Feng 2015; Harii 2008; Harii 2017; Kim 2014; Kim 2015; NCT02450526; Won 2013; Won 2015; Wu 2010; Wu 2019)
South America (n = 1) (Costa 2016)
No information (n = 3) (Cohen 2012; Dayan 2010; Solish 2018)
Eight RCTs were developed in a single centre: USA (Beer 2006; Michaels 2012; Kassir 2013), Canada (Carruthers 2005a; Carruthers 2005b; Carruthers 2009), Brazil (Hexsel 2013), Austria (Rappl 2013).
Seven studies did not provide any information about setting (Carruthers 2003a; Fagien 2007a; Firoz 2012; Lee 2013; Nettar 2011; Park 2014; Patel 2004).
All patients were outpatients (private office or day clinic).
Study duration
The mean duration of studies was 20.75 weeks ± 11.7 (Nettar 2011) (range 1 to 52 weeks) (Carruthers 2004). The interval between treatments was 24 weeks (six months). Most of the studies analysed the effects from 16 weeks (when the muscle begins to work again) to 24 weeks (the new treatment interval).
Funding
Most of the studies were reported to have received pharmaceutical industry funding.
Participants
The total study population was 14,919 participants. Participants of Carruthers 2015 partially overlapped with participants of Moers‐Carpi 2015.
Age
Apart from four studies, which reported age by range (Ascher 2015; Beer 2006; Carruthers A 2003;NCT02493946 Solish 2018), the majority of the studies (58 studies) reported mean age either by treatment group or by total study population. The mean age of study participants ranged from 18 to 65 years in the majority of studies. Two studies did not report the age, gender, or other demographic data (Lee 2013; Patel 2004).
Gender
Seven studies did not provide any information about gender (Fagien 2007a; Feng 2015; Firoz 2012; Lee 2013; Nettar 2011; Patel 2004; Solish 2018). Ten studies included only female participants (Ascher 2018; Beer 2006; Carruthers 2005b; Carruthers 2009; Carruthers 2003a; Carruthers 2010; Cohen 2012; Costa 2016; Kane 2015; Satler 2010). One paper studied only men (Carruthers 2005a).
The majority of the studies included more than 80% female participants (Ascher 2004; Ascher 2005; Ascher 2009; Ascher 2020; Bertucci 2020; Carruthers 2004; Carruthers 2005b; Carruthers 2014; Carruthers 2003b; Carruthers 2015; Carruthers 2017; Cheon 2019; Dayan 2010; De Boulle 2018; Feng 2015; Hanke 2013; Harii 2008; Harii 2017; Hexsel 2013; Kane 2009; Kane 2015; Kassir 2013; Kerscher 2015; Kim 2014; Kim 2015; Lowe 2006; Michaels 2012; Moers‐Carpi 2012; Moers‐Carpi 2015; Monheit 2007; Monheit 2019; NCT02450526; NCT02493946; Beer 2019a; Beer 2019b; Ogilvie 2019; Park 2014; Rappl 2013; Rivers 2015; Rubin 2009; Rzany 2006; Rzany 2019; Solish 2016; Won 2013; Won 2015; Wu 2010; Wu 2019).
Facial region
Glabellar lines (GL): 43 RCTs (Ascher 2004; Ascher 2005; Ascher 2018; Ascher 2020; Beer 2006; Bertucci 2020; Brandt 2009; Carruthers 2004; Carruthers 2005a; Carruthers 2005b; Carruthers 2013; Carruthers 2002; Carruthers 2003b; Carruthers 2017; Costa 2016; Fagien 2007a; Feng 2015; Hanke 2013; Harii 2008; Kane 2009; Kane 2015; Kassir 2013; Kim 2014; Kim 2015; Lee 2013; Lowe 2006; Moers‐Carpi 2012; Monheit 2007; Monheit 2019; NCT02450526; NCT02493946; Beer 2019a; Beer 2019b; Patel 2004; Rappl 2013; Rzany 2019; Rubin 2009; Rzany 2006; Satler 2010; Solish 2018; Won 2013; Won 2015; Wu 2010).
Crow's feet lines: 7 RCTs (Ascher 2009; Carruthers 2014; Cheon 2019; Harii 2017; Nettar 2011; Park 2014; Wu 2019).
Perioral lines: 2 RCTs (Carruthers 2010; Cohen 2012).
Forehead line: 2 RCTs (Carruthers 2003a; Solish 2016).
Forehead lines and crow's feet line: one RCT (Michaels 2012).
Upper lines (glabellar lines, crow's feet lines, forehead lines): 3 RCTs (Carruthers 2009; Dayan 2010; De Boulle 2018).
Forehead lines and glabellar lines: 3 RCTs (Firoz 2012; Kerscher 2015; Ogilvie 2019).
Crow's feet lines and glabellar lines: 3 RCTs (Carruthers 2015; Moers‐Carpi 2015; Rivers 2015).
Full face: one RCT (Hexsel 2013).
Severity of the wrinkles
Most of the papers which treated glabellar lines included moderate‐to‐severe glabellar lines according to Facial Wrinkle Scale score or Glabellar Lines Severity Scale. The others regions did not present details about severity.
Sample size
The sample size of the studies ranged from 56 to 917 participants (mean = 230.14).
Unit of analysis
Five studies were split‐face design (Firoz 2012; Michaels 2012; Kassir 2013; Nettar 2011; Park 2014). In the remaining studies, the unit of analysis was the individual. In meta‐analysis we only compared parallel study groups.
Intervention
Types of botulinum toxin type A (BontA)
11 commercial types of BontA (produced from different strains of BontA with unique biological behaviour) were addressed in RCTS:
Botox®, Vistabel®, Vistabex® (Allergan ‐ onabotulinumtoxinA (Beer 2006; Carruthers 2003a; Carruthers 2004; Carruthers 2005a; Carruthers 2005b; Carruthers 2009; Carruthers 2010; Carruthers 2014; Carruthers 2002; Carruthers 2003b; Carruthers 2015; Carruthers 2017; Cohen 2012; Dayan 2010; De Boulle 2018; Fagien 2007a; Firoz 2012; Harii 2008; Harii 2017; Kane 2009; Kane 2015; Kassir 2013; Lowe 2006; Michaels 2012; Moers‐Carpi 2012; Moers‐Carpi 2015; Nettar 2011; Ogilvie 2019; Park 2014; Park 2014; Patel 2004; Rappl 2013; Rzany 2019; Rivers 2015; Satler 2010; Solish 2016; Won 2015; Wu 2010; Wu 2019)
Dysport® (Ipsen); Azzulure® (Galderma ‐ abobotulinumtoxinA (Ascher 2004; Ascher 2005; Ascher 2009; Ascher 2018; Brandt 2009; Hexsel 2013; Kane 2009; Kassir 2013; Lowe 2006; Michaels 2012; Monheit 2007; Monheit 2019; Nettar 2011; Rappl 2013; Rubin 2009; Rzany 2006)
Xeomeen®, Xeomin®, Bocouture® (Merz Aesthetics ‐ incobotulinumtoxinA (Carruthers 2013; Dayan 2010; Hanke 2013; Kane 2009; Kane 2015; Kerscher 2015; Moers‐Carpi 2012; Park 2014; Rappl 2013; Satler 2010)
HBTX‐A (Feng 2015; NCT02493946)
Neuronox®, Botulift®, Siax®, Medytox® (Medytox, Inc., Cheonwon‐gun, Republic of Korea) (Cheon 2019; Lee 2013; Won 2013)
Liquid BontA (MT10109L) (Kim 2015)
DaxibotulinumtoxinA (DWP450) (Daewoong Pharmaceutical, Seoul, Korea) (Bertucci 2020; Carruthers 2017; Won 2015)
Liquid BontA (Ipsen) (Ascher 2018; Ascher 2020; NCT02450526)
CBFC26 (SNUH) (Kim 2014)
Prosigne® (Lanzhou Institute of Biological Products) (Costa 2016)
PrabotulinumtoxinA (Beer 2019a; Beer 2019b; Rzany 2019; Solish 2018)
Number of cycles
One single cycle of treatment: 54 RCTs (Ascher 2004; Ascher 2009; Ascher 2018; Beer 2006; Beer 2019a; Bertucci 2020; Brandt 2009; Carruthers 2003a; Carruthers 2005a; Carruthers 2005b; Carruthers 2009; Carruthers 2010; Carruthers 2013; Carruthers 2014; Carruthers 2002; Carruthers 2003b; Carruthers 2010; Carruthers 2017; Cheon 2019; Cohen 2012; Costa 2016; Dayan 2010; Fagien 2007a; Feng 2015; Firoz 2012; Hanke 2013; Hexsel 2013; Kane 2009; Kane 2015; Kassir 2013; Kerscher 2015; Kim 2014; Kim 2015; Lee 2013; Lowe 2006; Michaels 2012; Moers‐Carpi 2012; Monheit 2007; Monheit 2019; Nettar 2011; NCT02493946; Park 2014; Patel 2004; Rappl 2013; Rivers 2015; Rzany 2006; Rzany 2019; Satler 2010; Solish 2016; Solish 2018 ; Won 2013; Won 2015; Wu 2010; Wu 2019)
Two cycles of treatment: 3 RCTs (Ascher 2005; Moers‐Carpi 2015; Ogilvie 2019)
Three or more cycles treatments: 8 RCTs (Ascher 2020; Carruthers 2004; Carruthers 2015; De Boulle 2018; Harii 2008; Harii 2017; Rubin 2009; NCT02450526)
Dose of the treatment
The dose ranged according to the facial region and BontA's brand.
Glabellar lines
OnabotulinumtoxinA, from 8 U to 80 U (Beer 2006; Carruthers 2003a; Carruthers 2004; Carruthers 2005a; Carruthers 2005b; Carruthers 2009; Carruthers 2010; Carruthers 2014; Carruthers 2015; Carruthers 2002; Carruthers 2003b; Carruthers 2017; Cohen 2012; Dayan 2010; De Boulle 2018; Fagien 2007a; Firoz 2012; Harii 2008; Harii 2017; Kane 2009; Kane 2015; Kassir 2013; Lowe 2006; Michaels 2012; Moers‐Carpi 2012; Moers‐Carpi 2015; Nettar 2011; Ogilvie 2019; Park 2014; Patel 2004; Rappl 2013; Rzany 2019; Rivers 2015; Satler 2010; Solish 2018; Won 2015; Wu 2010)
AbobotulinumtoxinA, from 20 U to 75 U (Ascher 2004; Ascher 2005; Ascher 2009; Ascher 2018; Brandt 2009; Hexsel 2013; Kane 2009; Kassir 2013; Lowe 2006; Michaels 2012; Monheit 2007; Monheit 2019; Nettar 2011; Rappl 2013; Rubin 2009; Rzany 2006)
IncobotulinumtoxinA, from 20 U to 24 U (Carruthers 2013; Hanke 2013; Kane 2009; Kane 2015; Kerscher 2015; Moers‐Carpi 2012; Rappl 2013; Satler 2010)
HBTX‐A, 20U (Feng 2015; NCT02493946)
NewBontA [Medytox®], 20 U (Lee 2013)
NewBontA [Neuronox®], 20 U (Won 2013)
DaxibotulinumtoxinA (DWP450), 20 U to 60 U (Bertucci 2020; Carruthers 2017; Won 2015)
MT10109L, 20 U (Kim 2015)
LiquidBontA 20 U to 75 U (Ascher 2018; Ascher 2020; NCT02450526)
CBFC26, 20 U (Kim 2014)
NewBontA [Prosigne®], 20 U (Costa 2016)
PrabotulinumtoxinA 20U to 60 U (Beer 2019a; Beer 2019b; Rzany 2019; Solish 2018)
Forehead lines
OnabotulinumtoxinA, 10U to 48 U (Carruthers 2003a; Carruthers 2009; Dayan 2010)
IncobotulinumtoxinA, 10 U to 20 U (Kerscher 2015)
Crow's feet lines
OnabotulinumtoxinA 7.5 U to 24 U (Harii 2017; Kassir 2013; Moers‐Carpi 2012; Moers‐Carpi 2015; Nettar 2011; Park 2014; Rivers 2015; Wu 2019)
AbobotulinumtoxinA 30 U (Kassir 2013; Nettar 2011)
IncobotulinumtoxinA 7.5 U to 12 U (Dayan 2010; Kerscher 2015; Park 2014)
Neuronox® 24U‐ (Cheon 2019)
Perioral lines
OnabotulinumtoxinA, from7.5 U to 12 U (Carruthers 2010; Cohen 2012)
Distribution of the injection points
The distribution of all injections points followed the American and European consensus.
Glabellar lines‐ three to seven intramuscular injections points (procerus muscle, corrugator supercilii muscle, orbicularis oculi, muscle depressor supercilii muscle) (Figure 2)
Forehead lines‐ four to eight points of intramuscular injection along the frontal muscle wrinkles with a 2.0 cm above the eyebrows; and
Crow's feet‐ one injection at least 1.5 cm to 2.0 cm from lateral canthus, one injection in the orbital rim next to the eyebrow caudal extremity, and one injection near the zygomatic process in the orbital rim, the other injections along the crow’s feet lines laterally to the previous injections per side (Carruthers 2008a; Ascher 2010; Sundaram 2016)
Perioral lines‐ four injections, two symmetric injections per lip (lower and upper lip) (Cohen 2012)
2.

Illustration of injection sites in glabellar region. Copyright [2020] [Cristina Pires Camargo]Reproduced with permission
Comparisons
BontA versus placebo, at least one cycle of treatment (36 studies); BontA at different doses, one cycle of treatment (21 studies); BontA versus placebo, at least two cycles of treatment (11 studies); BontA versus facial cream (one study); BontA associated to fillers (2 studies)
The studies tested the effect of different types of BontA in facial wrinkles through the following comparisons.
OnabotulinumtoxinA versus placebo
OnabotulinumtoxinA versus placebo, glabellar lines, one cycle of treatment, both genders (Carruthers 2002; Carruthers 2003b; Carruthers 2017; Fagien 2007a; Rzany 2019; Solish 2018; Wu 2010)
OnabotulinumtoxinA versus placebo, crow's feet lines, one cycle of treatment, both genders (Carruthers 2014; Wu 2019)
OnabotulinumtoxinA versus placebo, glabellar lines, and crow's feet lines, one cycle of treatment, both genders (Rivers 2015)
OnabotulinumtoxinA versus placebo, glabellar lines, forehead lines, and crow's feet lines, one cycle of treatment, both genders (De Boulle 2018)
OnabotulinumtoxinA versus placebo, glabellar lines, five cycles of treatment (Harii 2017)
OnabotulinumtoxinA, different doses
OnabotulinumtoxinA, different doses, one cycle of treatment, glabellar lines in men (Carruthers 2005a)
OnabotulinumtoxinA, different doses, one cycle of treatment, glabellar lines in women (Carruthers 2005b)
OnabotulinumtoxinA, different doses, one cycle of treatment, upper wrinkles (forehead lines, glabellar lines, crow's feet lines) in women (Carruthers 2009; Dayan 2010)
OnabotulinumtoxinA, different doses, one cycle of treatment, forehead lines, dose‐ranging in women (Carruthers 2003a; Solish 2016)
OnabotulinumtoxinA, different doses, one cycle of treatment, forehead lines and glabella lines, dose‐ranging, both genders (Ogilvie 2019)
OnabotulinumtoxinA, different doses, one cycle of treatment, perioral lines in women (Cohen 2012)
OnabotulinumtoxinA versus placebo
OnabotulinumtoxinA versus placebo, glabellar lines, two‐three cycles of treatment, both genders (Carruthers 2004)
OnabotulinumtoxinA versus placebo, glabellar lines and crow's feet lines, two cycles of treatment, both genders (Moers‐Carpi 2015; Carruthers 2015)
OnabotulinumtoxinA versus placebo, glabellar lines, five cycles of treatment, both genders (Harii 2017). We only use double‐blind data.
AbobotulinumtoxinA versus placebo
AbobotulinumtoxinA versus placebo, one cycle of treatment, glabellar lines (Ascher 2005)
AbobotulinumtoxinA versus placebo, one cycle of treatment, glabellar lines, both gender (Brandt 2009)
AbobotulinumtoxinA, different doses, versus placebo
AbobotulinumtoxinA versus placebo, different doses, glabellar lines, both genders (Ascher 2004; Monheit 2019; Kane 2009; Monheit 2007; Rzany 2006)
AbobotulinumtoxinA versus placebo, different doses, crow's feet, both genders (Ascher 2009)
AbobotulinumtoxinA versus placebo, multiple cycles of treatment, glabellar lines, both genders (Ascher 2020; NCT02450526)
AbobotulinumtoxinA versus placebo, two cycles of treatment, glabellar lines, both genders (Ascher 2005)
AbobotulinumtoxinA versus placebo, three cycles of treatment, glabellar lines, both genders (Rubin 2009)
AbobotulinumtoxinA, different doses
AbobotulinumtoxinA, three different doses in full‐face treatment, both genders (Hexsel 2013)
IncobotulinumtoxinA versus placebo
IncobotulinumtoxinA versus placebo, glabellar lines, both genders (Carruthers 2013; Hanke 2013)
IncobotulinumtoxinA versus placebo, forehead lines, glabellar lines, and crow's feet lines, both genders (Kerscher 2015)
HBTX‐A versus placebo
HBTX‐A versus placebo, glabellar lines, one cycle of treatment, both genders (Feng 2015; NCT02493946)
Neuronox® versus placebo
Neuronox® versus placebo, crow's feet lines, one cycle of treatment, both genders (Cheon 2019)
Liquid BontA (Ipsen®), different doses, versus placebo
Liquid BontA (Ipsen®) different doses versus placebo versus abobotulinumtoxinA, glabellar lines, both genders (Ascher 2018)
Liquid BontA (Ipsen®) versus placebo, glabellar lines, both genders (Ascher 2020)
DaxibotulinumtoxinA versus placebo
DaxibotulinumtoxinA versus placebo from one cycle to 5 cycles of treatment in crow's feet lines, both genders (Bertucci 2020; Harii 2017; Solish 2018)
DaxibotulinumtoxinA, dose‐ranging, versus onabotulinumtoxinA, one cycle of treatment, both genders (Carruthers 2017)
PrabotulinimtoxinA versus placebo
PrabotulinimtoxinA versus placebo, one cycle of treatment in glabellar lines, both genders (Beer 2019a; Beer 2019b; Rzany 2019)
BontA versus active control
OnabotulinumtoxinA versus AbobotulinumtoxinA, one cycle of treatment, glabellar lines and forehead lines, both genders (Firoz 2012)
OnabotulinumtoxinA versus AbobotulinumtoxinA, one cycle of treatment, glabellar lines, both genders (Kassir 2013; Lowe 2006)
OnabotulinumtoxinA versus AbobotulinumtoxinA, one cycle of treatment, glabellar lines, crow's feet lines, and forehead lines, both genders (Michaels 2012)
OnabotulinumtoxinA versus AbobotulinumtoxinA, one cycle of treatment, in crow's feet lines, both genders (Nettar 2011)
OnabotulinumtoxinA versus IncobotulinumtoxinA, one cycle of treatment, glabellar lines, both genders (Kane 2015; Moers‐Carpi 2012; Satler 2010)
OnabotulinumtoxinA versus IncobotulinumtoxinA, one cycle of treatment, crow's feet lines, both genders (Park 2014)
OnabotulinumtoxinA versus AbobotulinumtoxinA versus IncobotulinumtoxinA, one cycle of treatment, glabellar lines, both genders (Rappl 2013)
Neuronox® versus onabotulinumtoxinA, one cycle of treatment, glabellar lines, both genders (Won 2013)
Liquid BontA (MT10109L) versus onabotulinumtoxinA, one cycle of treatment, glabellar lines, both genders (Kim 2015)
New BontA (Medytox®) versus onabotulinumtoxinA, one cycle of treatment, glabellar lines, both genders (Lee 2013)
DaxibotulinumtoxinA versus onabotulinumtoxinA, one cycle of treatment, glabellar lines, both genders (Won 2015)
CBFC26 versus onabotulinumtoxinA, one cycle of treatment, glabellar lines, both genders (Kim 2014)
Liquid BontA (Ipsen®) different doses versus AbobotulinumtoxinA, glabellar lines, both genders (Ascher 2018)
PrabotulinimtoxinA versus placebo, one cycle of treatment in glabellar lines, both genders (Beer 2019a; Beer 2019b; Rzany 2019)
PrabotulinimtoxinA versus onabotulinumtoxinA, one cycle of treatment in glabellar lines, both genders (Rzany 2019)
BontA associated with creams
OnabotulinumtoxinA, one cycle of treatment, versus facial cream in glabellar lines in women (Beer 2006)
BontA associated with fillers
OnabotulinumtoxinA associated with fillers (collagen), one cycle of treatment, versus onabotulinumtoxinA, glabellar lines (Patel 2004)
OnabotulinumtoxinA associated with collagen, one cycle of treatment, versus collagen, glabellar lines, no information about genders (Patel 2004)
OnabotulinumtoxinA associated with Hyaluronic acid, one cycle of treatment, versus onabotulinumtoxinA, lips and perioral lines, in women (Carruthers 2010)
OnabotulinumtoxinA associated with Hyaluronic acid, one cycle of treatment, versus Hyaluronic acid, lips and perioral lines, in women (Carruthers 2010)
Outcomes
Primary outcomes
Thirty‐five studies evaluated participant assessment of success by analysing scores and scales (the responder rate at maximum contraction): Ascher 2020; Ascher 2018; Beer 2019a; Beer 2019b; Brandt 2009; Bertucci 2020; Carruthers 2003a; Carruthers 2005a; Carruthers 2005b; Carruthers 2009; Carruthers 2013; Carruthers 2014; Carruthers 2015; Carruthers 2017; Cheon 2019; De Boulle 2018; Feng 2015; Hanke 2013; Harii 2008; Kane 2009; Kim 2014; Kim 2015; Moers‐Carpi 2012; Moers‐Carpi 2015; Monheit 2019; NCT02493946; Nettar 2011; Ogilvie 2019; Rzany 2019; Rubin 2009; Solish 2016; Solish 2018; Won 2013; Won 2015; Wu 2019. The most common scales used by studies were Facial Wrinkle Scale and 5‐Point Scale.
Forty‐six studies evaluated any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus): Ascher 2020; Ascher 2004; Ascher 2005; Ascher 2009; Ascher 2018; Beer 2006; Beer 2019a; Beer 2019b; Bertucci 2020; Brandt 2009; Carruthers 2003a; Carruthers 2004; Carruthers 2005b; Carruthers 2003b; Carruthers 2002; Carruthers 2010; Carruthers 2013; Carruthers 2015; Carruthers 2017; Cheon 2019; De Boulle 2018; Feng 2015; Hanke 2013; Harii 2008; Harii 2017; Kane 2015; Kassir 2013; Kerscher 2015; Kim 2014; Kim 2015; Moers‐Carpi 2015; Monheit 2019; NCT02450526 NCT02493946; Patel 2004; Rivers 2015; Rubin 2009; Rzany 2006; Rzany 2019; Satler 2010; Solish 2016; Solish 2018; Won 2013; Won 2015; Wu 2010; Wu 2019.
Secondary outcomes
Forty‐nine studies evaluated an assessment of the physician assessment of success by analysing scores and scales (responder rate at maximum contraction): Ascher 2020; Ascher 2004; Ascher 2005; Ascher 2009; Ascher 2018; Beer 2019a; Beer 2019b; Bertucci 2020; Carruthers 2003a; Carruthers 2005a; Carruthers 2005b; Carruthers 2009; Carruthers 2010; Carruthers 2013; Carruthers 2014; Carruthers 2003b; Carruthers 2002; Carruthers 2015; Carruthers 2017; Cheon 2019; Cohen 2012; Costa 2016; De Boulle 2018; Hanke 2013; Harii 2008; Harii 2017 Kane 2009; Kane 2015; Kassir 2013; Kerscher 2015; Kim 2014; Kim 2015; Lee 2013; Lowe 2006; Moers‐Carpi 2012; Moers‐Carpi 2015; Monheit 2019; NCT02450526; NCT02493946;; Rappl 2013; Rivers 2015; Rubin 2009; Rzany 2006; Satler 2010; Solish 2016; Won 2013; Won 2015; Wu 2010; Wu 2019. The most common scale used by studies were Facial Wrinkle Scale and 5‐Point Scale.
Fifty‐one studies evaluated the occurrence of any adverse event: Ascher 2020; Ascher 2004; Ascher 2005; Ascher 2009; Ascher 2018; Beer 2006; Beer 2019a; Beer 2019b; Bertucci 2020; Brandt 2009; Carruthers 2003a; Carruthers 2004; Carruthers 2005a; Carruthers 2005b; Carruthers 2009; Carruthers 2010; Carruthers 2013; Carruthers 2002; Carruthers 2003b; Carruthers 2015; Carruthers 2017; Cheon 2019; Cohen 2012; De Boulle 2018; Feng 2015; Hanke 2013; Harii 2008; Harii 2017; Kane 2015; Kerscher 2015; Kim 2014; Kim 2015; Lowe 2006; Moers‐Carpi 2012; Moers‐Carpi 2015; Monheit 2007; Monheit 2019; NCT02450526; NCT02493946; Rappl 2013; Rivers 2015; Rubin 2009; Rzany 2006; Rzany 2019; Satler 2010; Solish 2016; Solish 2018; Won 2013; Won 2015; Wu 2010; Wu 2019.
Twenty‐one studies evaluated the duration of treatment effect (weeks): Ascher 2005; Bertucci 2020; Brandt 2009; Carruthers 2005a; Carruthers 2009; Carruthers 2010; Carruthers 2014; Carruthers 2017; Cheon 2019; Costa 2016; Feng 2015; Harii 2008; Harii 2017; Kane 2009; Monheit 2019; Beer 2019a; Beer 2019b; Rappl 2013; Rzany 2019; Solish 2016; Wu 2019)
One RCT (NCT02493946) evaluated only HBTX‐A duration in days.
Excluded studies
We excluded 11 studies. Four studies were not randomised clinical trials (Hexsel 2018; Mahmoud 2016; Rzany 2013; 2014‐003770‐16). Five studies analysed interventions outside the scope of this review (Cartier 2020; NCT02297516; Punga 2016; Wilson 2016; Zhang 2018). Two studies ended when the company involved (Johnson & Johnson) changed their plans to produce the BontA (NCT00752050; NCT00752297) (see Characteristics of excluded studies).
Ongoing studies
No studies were identified as ongoing.
Studies awaiting classification
We identified 24 studies awaiting classification. These studies were listed as completed on the clinical trial register, but no relevant results are available yet. See Characteristics of studies awaiting classification for more details.
Risk of bias in included studies
The risk of bias of each study is detailed in the Characteristics of included studies table. Figure 3 and Figure 4 present the risk of bias summary along with review authors' judgements about each risk of bias item for an individual study. The overall risk of bias of the studies was unclear in all of them as we categorised at least one of the domains as having an unclear risk of bias.
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
4.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
Random sequence generation
Twenty‐five studies described randomisation sequence adequately and were considered as low risk of bias. One study used 'tossing a coin' to generate random sequence (Michaels 2012) and the remaining 24 studies used computer‐generated random numbers (Ascher 2004; Ascher 2005; Ascher 2009; Ascher 2018;Ascher 2020 ; Beer 2019a; Beer 2019b; Bertucci 2020; Brandt 2009; Costa 2016; Firoz 2012; Harii 2008; Hexsel 2013; Kassir 2013; Kerscher 2015; Kim 2014; Lowe 2006; Moers‐Carpi 2012; Nettar 2011; Ogilvie 2019 ; Rappl 2013; Rzany 2019; Won 2015; Wu 2019).
Thirty‐nine studies studies were reported as being randomised, but further description of sequence generation was not reported; hence, we classified these as unclear risk of bias (Beer 2006; Carruthers 2003a; Carruthers 2004; Carruthers 2005a; Carruthers 2005b; Carruthers 2009; Carruthers 2010; Carruthers 2013; Carruthers 2002; Carruthers 2003b; Carruthers 2014; Carruthers 2015; Carruthers 2017; Cheon 2019; Cohen 2012; Dayan 2010; De Boulle 2018; Fagien 2007a; Feng 2015; Hanke 2013; Harii 2017; Kane 2009; Kane 2015; Kim 2015; Lee 2013; Moers‐Carpi 2015; Monheit 2007; Monheit 2019;NCT02450526; NCT02493946; Park 2014; Patel 2004; Rivers 2015; Rzany 2006; Satler 2010; Solish 2016; Solish 2018; Won 2013; Wu 2010).
Rubin 2009 added an amendment to a supplementary randomisation for the third cycle (C), but no further information about the method was reported. We considered this study as presenting high risk of bias.
Allocation sequence concealment
Nine studies described allocation concealment and were considered as low risk of bias (Ascher 2018; Ascher 2020; Bertucci 2020; Harii 2017; Kassir 2013; Kerscher 2015; Kim 2014; Lowe 2006; Ogilvie 2019).
Fifty‐six studies had reported allocation, but the authors did not show the methods used for maintaining the allocation concealment, and we considered them as presenting unclear risk of bias (Ascher 2004; Ascher 2005; Ascher 2009; Beer 2006; Beer 2019a; Beer 2019b; Brandt 2009; Carruthers 2003a; Carruthers 2004; Carruthers 2005a; Carruthers 2005b; Carruthers 2009; Carruthers 2010; Carruthers 2013; Carruthers 2002; Carruthers 2003b; Carruthers 2014; Carruthers 2015; Carruthers 2017; Cohen 2012; Cheon 2019; Costa 2016; Dayan 2010; De Boulle 2018; Fagien 2007a; Feng 2015; Firoz 2012; Hanke 2013; Harii 2008; Hexsel 2013; Kane 2009; Kane 2015; Kim 2015; Lee 2013; Michaels 2012; Moers‐Carpi 2012; Moers‐Carpi 2015; Monheit 2007; Monheit 2019; NCT02450526; NCT02493946; Nettar 2011; Park 2014; Patel 2004; Rappl 2013; Rivers 2015; Rubin 2009; Rzany 2006; Rzany 2019; Satler 2010; Solish 2016; Solish 2018; Won 2013; Won 2015; Wu 2010; Wu 2019).
Blinding
Performance bias
Twenty‐six studies presented low risk of bias related to performance (Ascher 2004; Ascher 2009; Ascher 2018; Beer 2006; Beer 2019a; Beer 2019b, Bertucci 2020; Brandt 2009; Carruthers 2004; Carruthers 2005b; Carruthers 2009; Carruthers 2003b; Carruthers 2017; Cheon 2019; Cohen 2012; Fagien 2007a; Firoz 2012; Hanke 2013; Kane 2015; Kerscher 2015; Lowe 2006; Moers‐Carpi 2012; Monheit 2007; Rappl 2013; Rzany 2019; Wu 2010). In these studies, the authors reported that the person responsible for blinding process was not directly involved in the research.
Thirty‐seven studies did not mention the details of how they blinded the participants, and we considered this as an unclear risk of bias (Ascher 2005; Ascher 2020 ; Carruthers 2003a; Carruthers 2005a; Carruthers 2013; Carruthers 2014; Carruthers 2002; Carruthers 2015; Costa 2016; Dayan 2010; De Boulle 2018; Feng 2015; Harii 2008; Harii 2017; Kane 2009; Kassir 2013; Kim 2014; Kim 2015; Lee 2013; Michaels 2012; Moers‐Carpi 2015; Monheit 2019; NCT02493946; NCT02450526 ; Nettar 2011; Ogilvie 2019; Park 2014; Patel 2004; Rivers 2015; Rubin 2009; Rzany 2006; Satler 2010; Solish 2016; Solish 2018; Won 2013; Won 2015; Wu 2019).
Two RCTs were considered high risk of bias (Carruthers 2010; Hexsel 2013). Carruthers 2010 was a single‐blinded study, and Hexsel 2013 was an open‐label trial.
Detection bias
We judged 33 studies as low risk of bias because they provided information about blinding of outcome assessment (Ascher 2004; Ascher 2009; Beer 2006; Beer 2019a; Beer 2019b, Bertucci 2020; Brandt 2009; Carruthers 2004; Carruthers 2005b; Carruthers 2009; Carruthers 2010; Carruthers 2002; Carruthers 2003b; Carruthers 2017;Cheon 2019; Cohen 2012; Fagien 2007a; Hanke 2013; Kane 2009; Kane 2015; Kerscher 2015; Kim 2014; Kim 2015; Lowe 2006; Moers‐Carpi 2012; Moers‐Carpi 2015; Monheit 2007; Nettar 2011; Patel 2004; Rappl 2013; Rzany 2019; Won 2013; Won 2015). The authors reported that the person responsible for blinding maintenance was not involved in the research
Thirty‐one studies did not describe detection bias, and we considered them as presenting unclear risk of bias (Ascher 2005; Ascher 2018; Ascher 2020; Carruthers 2003a; Carruthers 2005a; Carruthers 2013; Carruthers 2014; Carruthers 2015; Costa 2016; Dayan 2010;De Boulle 2018, Feng 2015; Firoz 2012; Harii 2008; Harii 2017; Kassir 2013; Lee 2013; Michaels 2012; Monheit 2019; NCT02450526 ; NCT02493946; Ogilvie 2019; Park 2014; Rivers 2015; Rubin 2009; Rzany 2006; Satler 2010; Solish 2016; Solish 2018; Wu 2010; Wu 2019).
One study was judged as presenting a high‐risk of bias due to open‐label design (Hexsel 2013).
Incomplete outcome data
Low risk studies were defined as low dropout rate, comparable drop‐out rate between groups, and/or comparable reasons for dropout between groups.
Thirty‐nine trials were considered low risk of bias (Ascher 2005; Ascher 2020 ; Beer 2019a; Beer 2019b; Brandt 2009; Carruthers 2005b; Carruthers 2010; Carruthers 2013; Carruthers 2014; Carruthers 2002; Carruthers 2003b; Carruthers 2015; Carruthers 2017; Cheon 2019; Costa 2016; De Boulle 2018; Feng 2015; Hanke 2013; Hexsel 2013; Kane 2009; Kane 2015; Kassir 2013; Kerscher 2015; Kim 2014; Kim 2015; Lowe 2006; Moers‐Carpi 2012; Moers‐Carpi 2015; Monheit 2007; Monheit 2019; Nettar 2011; NCT02450526; Patel 2004; Rappl 2013; Rivers 2015; Rzany 2019; Satler 2010; Won 2015; Wu 2010). We consider low risk of bias if the authors reported the reasons for dropout.
Twenty‐five RCTs were considered unclear risk of bias because they did not provide any reason of dropouts (Ascher 2004; Ascher 2018; Beer 2006; Bertucci 2020; Carruthers 2003a; Carruthers 2004; Carruthers 2005a; Carruthers 2009; Cohen 2012; Dayan 2010; Fagien 2007a; Firoz 2012; Harii 2008; Harii 2017; Lee 2013; Michaels 2012; NCT02493946; Ogilvie 2019; Park 2014; Rubin 2009; Rzany 2006; Solish 2016; Solish 2018; Won 2013; Wu 2019).
One study was consider as a high risk of bias. Ascher 2009 reported protocol violation.
Selective reporting
46 RCTs were considered low risk of bias (Ascher 2004; Ascher 2018; Ascher 2020; Beer 2019a; Beer 2019b; Bertucci 2020; Carruthers 2002; Carruthers 2004; Carruthers 2005b; Carruthers 2009; Carruthers 2010; Carruthers 2013; Carruthers 2014; Carruthers 2015; Carruthers 2017; Cohen 2012; Cheon 2019; Costa 2016; Fagien 2007a; Firoz 2012; Hanke 2013; Harii 2008; Harii 2017; Kane 2009; Kane 2015; Kim 2014; Kim 2015; Lowe 2006; Moers‐Carpi 2012; Monheit 2007; Monheit 2019; NCT02450526; NCT02493946; Nettar 2011; Ogilvie 2019; Park 2014; Patel 2004; Rappl 2013; Rivers 2015; Rubin 2009; Rzany 2019; Satler 2010; Solish 2016; Won 2013; Wu 2010; Wu 2019).
We consider low risk of bias if the authors presented all prespecified outcomes.
Five studies were considered as presenting unclear risk of bias (Ascher 2005; Ascher 2009; De Boulle 2018; Hexsel 2013; Solish 2018).
Fourteen RCTs were considered as presenting high risk of bias (Beer 2006; Brandt 2009; Carruthers 2003a; Carruthers 2005a; Carruthers 2003b; Dayan 2010; Feng 2015; Kassir 2013; Kerscher 2015; Lee 2013; Michaels 2012; Moers‐Carpi 2015; Rzany 2006; Won 2015). Beer 2006 did not report raw data for patient satisfaction, only P values; Brandt 2009 reported different data in the text compared to the graphic; Carruthers 2003a reported an imbalance in baseline data and missing data; Carruthers 2005a did not report participant satisfaction, only P values; Carruthers 2003b reported better results were seen in the subgroup analysis by age (younger than 50 years old), but no data were shown; Dayan 2010 reported only P values; Feng 2015 did not mention if the investigator assessment was done at rest or at contraction; Kassir 2013 reported inconsistencies in the number of participants included in the study; Kerscher 2015 and Solish 2018 did not report the following outcomes: the response rate at rest by investigator assessment, the proportion of one‐point responders based on the investigator’s rating of glabellar lines and forehead at rest, and investigator‐assessed and participant‐assessed outcomes; Lee 2013 reported only the outcomes assessed at week 4; Michaels 2012 only reported P values; Moers‐Carpi 2015 reported only P values for investigator‐assessed responder rates on crow's feet lines (FWS), participant's global assessment of change in crow's feet lines, patient‐reported outcomes; Rzany 2006 did not report the following outcomes: the scores at maximum frown and at rest, by the investigator assessment, at weeks 0, 2, 4, 12, and 16; the subjective assessment of improvement since the first visit by the participant assessment at weeks 2, 4, 12, and 16; Won 2015 did not report patient satisfaction.
Eight studies showed reported clinical trial register numbers (Ascher 2018 (NCT01333397); Carruthers 2014 (NCT01189747); Kane 2015 (NCT02096081); Moers‐Carpi 2012 (NCT01271452); Moers‐Carpi 2015 (NCT01189760); Rivers 2015 (NCT01777620); Satler 2010 (NCT00777803); Won 2013 (NCT01237977). Of these studies, only Moers‐Carpi 2015 had a high risk of bias (Reported Outcomes, no data shown).
Other potential sources of bias
Thirty‐six studies were considered low risk of other sources of bias (Ascher 2009; Ascher 2020; Beer 2006; Bertucci 2020; Brandt 2009; Carruthers 2003a; Carruthers 2004; Carruthers 2005a; Carruthers 2009; Carruthers 2010; Carruthers 2014; Carruthers 2002; Carruthers 2003b; Cheon 2019; Cohen 2012; Dayan 2010; Feng 2015; Firoz 2012; Harii 2008; Hexsel 2013; Kane 2015; Kassir 2013; Kerscher 2015; Kim 2014; Kim 2015; Michaels 2012; Monheit 2007;Monheit 2019; Beer 2019a; Beer 2019b; Park 2014; Patel 2004; Rubin 2009; Satler 2010; Wu 2010; Wu 2019).
In 20 studies, at least one of the authors was a sponsor employee, so we consider unclear risk of bias (Ascher 2005; Ascher 2018; Carruthers 2013; Carruthers 2017; De Boulle 2018; Fagien 2007a; Hanke 2013; Harii 2017; Lee 2013; Lowe 2006; Moers‐Carpi 2012; Moers‐Carpi 2015; NCT02450526 ; Ogilvie 2019; Rappl 2013;Rzany 2019; Solish 2016; Solish 2018, Won 2013; Won 2015). One study was considered as unclear risk of bias because some parts of the text showed discrepancies (Rappl 2013), and another was judged as unclear due to limited information provided about the trial (NCT02493946).
Besides investigators bias, four studies had baseline imbalances that could influence the outcomes, so we consider them as unclear (Ascher 2018; Carruthers 2017; De Boulle 2018; Ogilvie 2019).
Eight studies were considered as high risk of bias (Ascher 2004; Carruthers 2005b; Carruthers 2015; Costa 2016; Kane 2009; Nettar 2011; Rivers 2015; Rzany 2006). Costa 2016 reported protocol violations. In another study rated as high risk of bias, the sponsor was involved in data analysis of the trial (Rzany 2006). Three studies reported at least one of the authors wa ae sponsor stockholder, so we considered this a high risk of bias (Carruthers 2015; Kane 2009; Nettar 2011).
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6
Summary of findings 1. Summary of Findings Table ‐ OnabotulinumtoxinA 20U compared to placebo in glabellar lines.
| OnabotulinumtoxinA 20U compared to placebo in glabellar lines | ||||||
| Patient or population: glabellar lines Setting: Outpatient Intervention: OnabotulinumtoxinA 20U Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with OnabotulinumtoxinA 20U | |||||
| Partcipant assessment of success assessed with: Validated tools, considering wrinkles and lines at maximum contraction follow up: 4 weeks | 3 per 100 | 65 per 100 (29 to 100) | RR 19.45 (8.60 to 43.99) | 575 (4 RCTs) | ⊕⊕⊕⊝ MODERATE a | |
| Major adverse events follow up: range 4 weeks to 24 weeks | 1 per 100 | 2 per 100 (1 to 5) | OR 3.62 (1.50 to 8.74) | 1390 (8 RCTs) | ⊕⊕⊕⊝ MODERATE a | |
| Physician assessment of success assessed with: Validate tools, wrinkles and lines at maximum contraction follow up: 4 weeks | 4 per 100 | 61 per 100 (36 to 100) | RR 17.10 (10.07 to 29.05) | 1339 (7 RCTs) | ⊕⊕⊕⊝ MODERATE a | |
| Total adverse events follow up: range 4 weeks to 24 weeks | 27 per 100 | 31 per 100 (24 to 39) | RR 1.14 (0.89 to 1.45) | 1388 (8 RCTs) | ⊕⊕⊝⊝ LOW a,b | |
| Duration of treatment effect assessed with: weeks | The mean duration of treatment effect was 0.4 | MD 18.4 higher (16.17 higher to 20.63 higher) | ‐ | 77 (1 RCT) | ⊕⊕⊕⊝ MODERATE a | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio; MD: Mean difference | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
| See interactive version of this table: https://gdt.gradepro.org/presentations/#/isof/isof_question_revman_web_423103355840383514. | ||||||
a. Downgraded one level due to serious risk of bias:, unclear risk of bias from blinding of participants, personnel, and outcome assessors. b. Downgraded one level due to serious imprecision: wide 95% CI, crossing the null.
Summary of findings 2. Summary of Findings Table ‐ AbobotulinumtoxinA 50U compared to placebo for glabellar lines.
| AbobotulinumtoxinA 50U compared to placebo for glabellar lines | ||||||
| Patient or population: glabellar lines Setting: Outpatient Intervention: AbobotulinumtoxinA 50U Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with AbobotulinumtoxinA 50U | |||||
| Participant assessment of success by analysing scores and scales ‐ 20 weeks (Validated tools, wrinkles and lines at maximum contraction) follow up: 4 weeks | 3 per 100 | 16 per 100 (5 to 51) | RR 5.33 (1.67 to 16.99) | 300 (1 RCT) | ⊕⊕⊕⊕ HIGH | |
| Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) follow up: range 4 weeks to 12 weeks | 0 per 100 | 0 per 100 (0 to 0) | RR 3.36 (0.88 to 12.87) | 1294 (7 RCTs) | ⊕⊕⊕⊝ MODERATE a | Unable to calculate the risk with the intervention as no major adverse events occurred in the placebo group. |
| Physician assessment of success by analysing scores and scales ‐ 4 weeks assessed with: Validated tools, wrinkles and lines at maximum contraction follow up: 4 weeks | 3 per 100 | 53 per 100 (30 to 96) | RR 15.78 (8.75 to 28.45) | 1060 (7 RCTs) | ⊕⊕⊕⊝ MODERATE b | |
| Total adverse events follow up: range 4 weeks to 16 weeks | 20 per 100 | 25 per 100 (21 to 30) | RR 1.25 (1.05 to 1.49) | 1471 (8 RCTs) | ⊕⊕⊝⊝ LOW a,b | |
| Duration of treatment effect assessed with: weeks | The mean duration of treatment effect was 99.7 days | MD 17.3 days higher (15.82 higher to 18.78 higher) | ‐ | 100 (1 RCT) | ⊕⊕⊕⊝ MODERATE b | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; MD: Mean difference | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
| See interactive version of this table: https://gdt.gradepro.org/presentations/#/isof/isof_question_revman_web_423103454361439144. | ||||||
a. Downgraded one level due to serious imprecision: wide 95% CI, crossing the null. b. Downgraded one level due to serious risk of bias: unclear risk of bias from blinding of participants, personnel, and assessors.
Summary of findings 3. Summary of Findings Table ‐ IncobotulinumtoxinA 20U compared to placebo for glabellar lines.
| IncobotulinumtoxinA 20U compared to placebo for glabellar lines | ||||||
| Patient or population: glabellar lines Setting: Outpatient Intervention: IncobotulinumtoxinA 20U Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with IncobotulinumtoxinA 20U | |||||
| Participant assessment of success by analysing scores and scales ‐ 4 weeks | 1 per 100 | 37 per 100 (7 to 100) | RR 66.57 (13.50 to 328.28) | 547 (2 RCTs) | ⊕⊕⊕⊝ MODERATE a | |
| Major adverse events follow up: range 4 weeks to 16 weeks | No major adverse events were observed | 547 (2 RCTs) | ⊕⊕⊕⊝ MODERATE a | |||
| Physician assessment of success by analysing scores and scales assessed with: Validated tools, wrinkles and lines at maximum contraction follow up: 4 weeks | 0 per 100 | 0 per 100 (0 to 0) | RR 134.62 (19.05 to 951.45) | 547 (2 RCTs) | ⊕⊕⊕⊝ MODERATE a | Unable to calculate the risk with the intervention as no events occurred in the placebo group. |
| Total adverse events follow up: range 4 weeks to 16 weeks | 29 per 100 | 34 per 100 (26 to 45) | RR 1.17 (0.90 to 1.53) | 547 (2 RCTs) | ⊕⊕⊝⊝ LOW a,b | |
| Duration of treatment effect ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
| See interactive version of this table: https://gdt.gradepro.org/presentations/#/isof/isof_question_revman_web_423103474800544813. | ||||||
a. Downgraded one level due to serious risk of bias: unclear risk of bias from blinding of participants, personnel, and assessors. b. Downgraded one level due to serious imprecision: wide 95% CI, crossing the null
Summary of findings 4. Summary of Findings Table ‐ AbobotulinumtoxinA 50U compared to OnabotulinumtoxinA 20U in glabellar lines.
| AbobotulinumtoxinA 50U compared to OnabotulinumtoxinA 20U in glabellar lines | ||||||
| Patient or population: glabellar lines Setting: Outpatient Intervention: AbobotulinumtoxinA 50U Comparison: OnabotulinumtoxinA 20U | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with OnabotulinumtoxinA 20U | Risk with AbobotulinumtoxinA 50U | |||||
| Participant assessment success by analysing scores and scales ‐ 4 weeks assessed with: Validated tools, wrinkles and lines at maximum contraction follow up: 4 weeks | 894 per 1000 | 894 per 1000 (822 to 965) | RR 1.00 (0.92 to 1.08) | 388 (1 RCT) | ⊕⊕⊕⊕ HIGH | |
| Major adverse events ‐ Any major adverse events (eyelid ptosis, eyelid sensory disorder, s follow up: range 4 weeks to 12 weeks | 1 per 100 | 2 per 100 (1 to 8) | OR 2.65 (0.77 to 9.09) | 433 (1 RCT) | ⊕⊕⊕⊝ MODERATE a | |
| Physician assessment of success by analysing scores and scales ‐ 4 weeks (Validated tools, wrinkles and lines at maximum contraction) follow up: 4 weeks | 95 per 100 | 96 per 100 (90 to 100) | RR 1.01 (0.95 to 1.06) | 388 (1 RCT) | ⊕⊕⊕⊕ HIGH | |
| Total adverse events follow up: range 4 weeks to 12 weeks | 18 per 100 | 19 per 100 (12 to 28) | RR 1.02 (0.67 to 1.54) | 492 (2 RCTs) | ⊕⊕⊕⊝ MODERATE a | |
| Duration of treatment effect ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
| See interactive version of this table: https://gdt.gradepro.org/presentations/#/isof/isof_question_revman_web_423103510517664820. | ||||||
a. Downgraded one level due to serious imprecision: wide 95% CI, crossing the null.
Summary of findings 5. Summary of Findings Table ‐ IncobotulinumtoxinA 24U compared to OnabotulinumtoxinA 24U in glabellar lines.
| IncobotulinumtoxinA 24U compared to OnabotulinumtoxinA 24U in glabellar lines | ||||||
| Patient or population: glabellar lines Setting: Outpatient Intervention: IncobotulinumtoxinA 24U Comparison: OnabotulinumtoxinA 24U | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with OnabotulinumtoxinA 24U | Risk with IncobotulinumtoxinA 24U | |||||
| Participant assessment of success by analysing scores and scales ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ | |
| Major adverse events follow up: range 4 weeks to 12 weeks | 1 per 100 | 0 per 100 (0 to 2) | OR 0.02 (0.00 to 1.77) | 381 (1 RCT) | ⊕⊝⊝⊝ VERY LOW a,b,c | Ptosis was reported in one participant in onabotulinumtoxinA group |
| Physician assessment of success by analysing scores and scales ‐ 4 weeks (Validated tools, wrinkles and lines at maximum contraction) follow up: 4 weeks | 96 per 100 | 97 per 100 (92 to 100) | RR 1.01 (0.96 to 1.05) | 381 (1 RCT) | ⊕⊕⊝⊝ LOW a,b | |
| Total adverse events follow up: range 4 weeks to 12 weeks | 1 per 100 | 0 per 100 (0 to 2) | OR 0.02 (0.00 to 1.77) | 381 (1 RCT) | ⊕⊝⊝⊝ VERY LOW a,b,c | |
| Duration of treatment effect ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
| See interactive version of this table: https://gdt.gradepro.org/presentations/#/isof/isof_question_revman_web_423103538603772987. | ||||||
a. Downgraded one level due to serious risk of bias: unclear risk of bias from randomisation, allocation concealment and blinding. b. Downgraded one level due to serious imprecision: wide 95% CI, crossing the null. c. Downgraded one level due to serious imprecision: low number of events.
Summary of findings 6. Summary of Findings Table ‐ DaxibotulinumtoxinA 40U compared to placebo in glabellar lines.
| DaxibotulinumtoxinA 40U compared to placebo in glabellar lines | ||||||
| Patient or population: glabellar lines Setting: Outpatient Intervention: DaxibotulinumtoxinA 40U Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with DaxibotulinumtoxinA 40U | |||||
| Participant assessment of success by analysing scores and scales ‐ 4 weeks (Validated tools, wrinkles and lines at maximum contraction) follow up: 4 weeks | 4 per 100 | 79 per 100 (43 to 100) | RR 21.10 (11.31 to 39.34) | 683 (2 RCTs) | ⊕⊕⊕⊝ MODERATE a | |
| Major adverse events follow up: range 4 weeks to 24 weeks | No major adverse events were observed. | 716 (2 RCTs) | ⊕⊕⊕⊝ MODERATE a | |||
| Physician assessment of success by analysing scores and scales ‐ 4 weeks (Validated tools, wrinkles and lines at maximum contraction) follow up: 4 weeks | 4 per 100 | 88 per 100 (47 to 100) | RR 23.40 (12.56 to 43.61) | 683 (2 RCTs) | ⊕⊕⊕⊝ MODERATE a | |
| Total adverse events follow up: range 4 weeks to 24 weeks | 9 per 100 | 21 per 100 (14 to 32) | RR 2.23 (1.46 to 3.40) | 716 (2 RCTs) | ⊕⊕⊕⊝ MODERATE a | |
| Duration of treatment effect | The mean duration of treatment effect was 0.4 weeks | MD 22.8 weeks higher (20.74 higher to 24.8 higher) | ‐ | 74 (1 RCT) | ⊕⊕⊕⊝ MODERATE a | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; MD: Mean difference | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
| See interactive version of this table: https://gdt.gradepro.org/presentations/#/isof/isof_question_revman_web_423103545427906155. | ||||||
a. Downgraded one level due to serious risk of bias: unclear allocation concealment.
COMPARISON 1. OnabotulinumtoxinmtoxinA 10 units versus placebo for glabellar lines, one cycle of treatment
One randomised controlled trial (RCT) (n = 94 participants) assessed this comparison (Harii 2008).
For this reason (one study), we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate, by participant assessment, was higher with onabotulinumtoxinA 10 U than placebo; however, because of the wide confidence intervals the results are very uncertain; at week 4 (risk ratio (RR) 40.36, 95% confidence interval (CI) 5.78 to 281.92; participants = 92; studies = 1); week 8 (RR 67.93, 95% CI 4.28 to 1078.29; participants = 91; studies = 1); week 12 (RR 26.29, 95% CI 3.71 to 186.39; participants = 90; studies = 1); and week 16 (RR 35.33, 95% CI 2.18 to 572.97; participants = 90; studies = 1) (Analysis 1.1).
1.1. Analysis.

Comparison 1: OnabotulinumtoxinA 10units versus placebo, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major adverse events.
Secondary outcomes
Physician assessment of success by analysing scores or scales
The responder rate was higher with onabotulinumtoxinA 10 U than placebo; however, because of the wide confidence intervals the results are very uncertain; at week 4 (RR 83.84, 95% CI 5.31 to 1325.05; participants = 92; studies = 1); at week 8 (RR 32.37, 95% CI 4.60 to 227.65; participants = 91; studies = 1); at week 12 (RR 46.72, 95% CI 2.91 to 749.60; participants = 90; studies = 1); and at week 16 (RR 23.93, 95% CI 1.44 to 396.41; participants = 90; studies = 1) (Analysis 1.2).
1.2. Analysis.

Comparison 1: OnabotulinumtoxinA 10units versus placebo, Outcome 2: Physician assessment of success by analysing scores and scales
Total adverse events
No difference between groups was found (RR 1.14, 95% CI 0.84 to 1.55, participants = 95; studies = 1) (Analysis 1.3).
1.3. Analysis.

Comparison 1: OnabotulinumtoxinA 10units versus placebo, Outcome 3: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 2. OnabotulinumtoxinA 20 units versus placebo glabellar lines, one cycle of treatment
Eight RCTs (n = 1390 participants) assessed this comparison (Beer 2006; Carruthers 2003b; Carruthers 2002; Carruthers 2017; Harii 2008; NCT02450526; Rzany 2019; Wu 2010), and the results are included in Table 1.
Primary outcomes
Participant assessment of success by analysing scores and scales
Four RCTs assessed this outcome (Harii 2008 n = 94; Carruthers 2017 n = 77; NCT02450526 n = 113; Rzany 2019 n = 292).
The responder rate, by participant assessment, was higher with onabotulinumtoxinA 20 U than placebo at week 4 (RR 19.45, 95% CI 8.60 to 43.99; participants = 575; studies = 4; I2 = 0%; moderate‐certainty evidence); at week 8 (RR 28.45, 95% CI 5.92 to 136.74; participants = 204; studies = 2, I2 = 0%); at week 12 (RR 12.77, 95% CI 2.78 to 58.72; participants = 203; studies = 2, I2 = 43%); at week 16 (RR 20.71, 95% CI 2.82 to 151.91; participants = 167; studies = 2; I2 = 0%); at week 24 (RR 4.19, 95% CI 0.21 to 84.41; participants = 77; studies = 1) (Analysis 2.1).
2.1. Analysis.

Comparison 2: OnabotulinumtoxinA 20 units versus placebo one cycle of treatment in glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Eight studies assessed this outcome (n = 1390) (Beer 2006; Carruthers 2003b; Carruthers 2002; Carruthers 2017; Harii 2008; Rzany 2019; Wu 2010; NCT02450526).
The frequency of adverse events was higher with onabotulinumtoxinA 20 U when compared to placebo (Peto OR 3.62, 95% CI 1.50 to 8.74, participants = 1390; studies = 8; moderate‐certainty evidence) (Analysis 2.2).
2.2. Analysis.

Comparison 2: OnabotulinumtoxinA 20 units versus placebo one cycle of treatment in glabellar lines, Outcome 2: Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
Seven RCTs reported this outcome (n = 1339) (Carruthers 2003b; Carruthers 2002; Carruthers 2017; Harii 2008; Rzany 2019; Wu 2010; NCT02450526).
The responder rate was higher with onabotulinumtoxinA 20 U when compared to placebo at week 4 (RR 17.10, 95% CI 10.07 to 29.05; participants = 1339; studies = 7; I2 = 0%; moderate‐certainty evidence); at week 8 (RR 21.50, 95% CI 9.68 to 47.75; participants = 1046; studies = 6; I2 = 15%); at week 12 (RR 10.81, 95% CI 5.79 to 20.16; participants = 1046; studies = 6; I2 = 10%); at week 16 (RR 15.13, 95% CI 5.98 to 38.27; participants = 933; studies = 5; I2 = 0%); at week 20 (RR 5.86, 95% CI 0.31 to 109.74; participants = 77; studies = 1); at week 24 (RR 4.19, 95% CI 0.21 to 84.41; participants = 77; studies = 1) (Analysis 2.3).
2.3. Analysis.

Comparison 2: OnabotulinumtoxinA 20 units versus placebo one cycle of treatment in glabellar lines, Outcome 3: Physician assessment of success by analysing scores and scales
Total adverse events
Eight RCTs (n = 1388 participants) assessed this comparison (Beer 2006; Carruthers 2003b; Carruthers 2002; Carruthers 2017; Harii 2008; Rzany 2019; Wu 2010; NCT02450526). No difference between groups was found (RR 1.14, 95% CI 0.89 to 1.45; participants = 1388; studies = 8; I2 = 28%, low‐certainty evidence) (Analysis 2.4).
2.4. Analysis.

Comparison 2: OnabotulinumtoxinA 20 units versus placebo one cycle of treatment in glabellar lines, Outcome 4: Total adverse events
Duration of treatment effect
Only one RCT assessed this outcome (Carruthers 2017); for this reason we were not able to undertake a meta‐analysis and only a visual representation of the results is shown in the forest plots. The duration of treatment effect of onabotulinumtoxinA 20 U was higher than in the placebo group (mean difference (MD) 18.40, 95% CI 16.17 to 20.63; participants = 77; studies = 1; low‐certainty evidence) (Analysis 2.5).
2.5. Analysis.

Comparison 2: OnabotulinumtoxinA 20 units versus placebo one cycle of treatment in glabellar lines, Outcome 5: Duration of treatment (weeks)
COMPARISON 3. OnabotulinumtoxinA 20 units versus 10 units for glabellar lines, one cycle of treatment
Two RCTs (n = 131 participants) assessed this comparison (Carruthers 2005b; Harii 2008).
Primary outcomes
Participants assessment of success by analysing scores and scales
There were two studies (Carruthers 2005b; Harii 2008) which assessed this outcome. The meta‐analysis showed no clear or substantial difference between these doses at 4 weeks (RR 1.11, 95% CI 0.96 to 1.29; participants = 131; studies = 2; I² = 0%); 8 weeks (RR 1.24, 95% CI 1.01 to 1.52; participants = 131; studies = 2; I² = 0%); and 16 weeks (RR 1.23, 95% CI 0.75 to 2.01; participants = 131; studies = 2; I² = 0%) (Analysis 3.1).
3.1. Analysis.

Comparison 3: OnabotulinumtoxinA 20units versus 10 units one treatment glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events
The meta‐analysis showed no clear difference between these doses as the result was imprecise (Peto OR 1.93, 95% CI 0.20 to 18.70; participants = 131; studies = 2) (Analysis 3.2).
3.2. Analysis.

Comparison 3: OnabotulinumtoxinA 20units versus 10 units one treatment glabellar lines, Outcome 2: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scale scores or scales
The meta‐analysis showed no clear difference between doses at 4 weeks (RR 1.05, 95% CI 0.91 to 1.20; n = 131; studies = 2; I² = 0%); at 8 weeks (RR 1.53, 95% CI 0.76 to 3.10; n = 131; studies = 2; I² = 65%); and 16 weeks (RR 1.43, 95% CI 0.76 to 2.69; n = 131; studies = 2; I² = 0%) (Analysis 3.3).
3.3. Analysis.

Comparison 3: OnabotulinumtoxinA 20units versus 10 units one treatment glabellar lines, Outcome 3: Physician assessment of success by analysing scores and scales
Total adverse events
There was no difference in number of events between doses (RR 1.08, 95% CI 0.49 to 2.37; n = 131; studies = 2; I² = 0%) (Analysis 3.4).
3.4. Analysis.

Comparison 3: OnabotulinumtoxinA 20units versus 10 units one treatment glabellar lines, Outcome 4: Total adverse events
Duration of treatment effect
Harii 2008 was the only study in this comparison to report this outcome. The mean duration of effect of treatment was 9.4 weeks in the OnabotulinumtoxinA 20U group and 7.9 weeks in the onabotulinumtoxinA 10 U group (no additional data were provided; therefore, we were unable to create a forest plot).
COMPARISION 4. OnabotulinumtoxinA 64 units versus placebo for upper wrinkles, one cycle of treatment.
Only one RCT (n = 469 participants) assessed this comparison (De Boulle 2018). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate (participant assessment) was higher in onabotulinumtoxinA 64 U than placebo at week 4 (RR 8.32, 95% CI 4.53 to 15.30; participants = 469; studies = 1); at week 8 (RR 106.50, 95% CI 6.66 to 1702.48; participants = 469; studies = 1); at week 12 (RR 44.50, 95% CI 2.76 to 717.83; participants = 469; studies = 1); at week 16 (RR 26.50, 95% CI 1.63 to 431.99; participants = 469; studies = 1); at week 20 (RR 5.50, 95% CI 0.31 to 98.84; participants = 469; studies = 1); and at week 24 (RR 1.50, 95% CI 0.06 to 36.61; participants = 469; studies = 1) (Analysis 4.1).
4.1. Analysis.

Comparison 4: OnabotulinumtoxinA 64units versus placebo one cycle of treatment, upper wrinkles, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
The major adverse events was similar with onabotulinumtoxinA 64 U and placebo (RR 1.00, 95% CI 0.09 to 10.91; participants = 469; studies = 1). (Analysis 4.2).
4.2. Analysis.

Comparison 4: OnabotulinumtoxinA 64units versus placebo one cycle of treatment, upper wrinkles, Outcome 2: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by physician assessment was higher in onabotulinumtoxinA 64 U than placebo at week 4 (RR 8.32, 95% CI 4.53 to 15.30; participants = 469; studies = 1); at week 8 (RR 106.50, 95% CI 6.66 to 1702.48; participants = 469; studies = 1), at week 12 (RR 44.50, 95% CI 2.76 to 717.83; participants = 469; studies = 1), at week 16 (RR 26.50, 95% CI 1.63 to 431.99; participants = 469; studies = 1); at week 20 (RR 5.50, 95% CI 0.31 to 98.84; participants = 469; studies = 1); and at week 24 (RR 1.50, 95% CI 0.06 to 36.61; participants = 469; studies = 1) (Analysis 4.3).
4.3. Analysis.

Comparison 4: OnabotulinumtoxinA 64units versus placebo one cycle of treatment, upper wrinkles, Outcome 3: Physician assessment of success by analysing scores and scales
Total adverse events
There was higher rate of any adverse event with onabotulinumtoxinA 64 U (RR 1.32, 95% CI 1.03 to 1.71; participants = 469; studies = 1) (Analysis 4.4).
4.4. Analysis.

Comparison 4: OnabotulinumtoxinA 64units versus placebo one cycle of treatment, upper wrinkles, Outcome 4: Total adverse events
Duration of treatment effect
The RCT did not report this outcome.
COMPARISION 5. OnabotulinumtoxinA 40 units versus placebo for upper wrinkles, one cycle of treatment.
Only one RCT (n = 474 participants) assessed this comparison (De Boulle 2018). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate (participant assessment) was higher in onabotulinumtoxinA 40 U than placebo at week 4 (RR 7.41, 95% CI 4.02 to 13.64; participants = 474; studies = 1); at week 8 (RR 86.13, 95% CI 5.38 to 1378.82; participants = 474; studies = 1); at week 12 (RR 39.87, 95% CI 2.47 to 644.08; participants = 474; studies = 1; I2 = 0%); at week 16 (RR 11.32, 95% CI 0.67 to 190.86; participants = 474; studies = 1); at week 20 (RR 2.26, 95% CI 0.12 to 50.95; participants = 474; studies = 1); and at week 24 (RR 1.48, 95% CI 0.06 to 36.04; participants = 474; studies = 1) (Analysis 5.1).
5.1. Analysis.

Comparison 5: OnabotulinumtoxinA 40units versus placebo one cycle of treatment, upper wrinkles, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
The major adverse events was higher in onabotulinumtoxinA 40 U than placebo (RR 2.45, 95% CI 0.29 to 20.82; participants = 474; studies = 1). (Analysis 5.2).
5.2. Analysis.

Comparison 5: OnabotulinumtoxinA 40units versus placebo one cycle of treatment, upper wrinkles, Outcome 2: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by physician assessment was higher in onabotulinumtoxinA 40U than placebo at week 4 (RR 7.41, 95% CI 4.02 to 13.64; participants = 474; studies = 1); at week 8 (RR 86.13, 95% CI 5.38 to 1378.82; participants = 474; studies = 1); at week 12 (RR 39.87, 95% CI 2.47 to 644.08; participants = 474; studies = 1); at week 16 (RR 11.32, 95% CI 0.67 to 190.86; participants = 474; studies = 1); at week 20 (RR 2.46, 95% CI 0.12 to 50.95; participants = 474; studies = 1); and at week 24 (RR 1.48, 95% CI 0.06 to 36.04; participants = 474; studies = 1) (Analysis 5.3).
5.3. Analysis.

Comparison 5: OnabotulinumtoxinA 40units versus placebo one cycle of treatment, upper wrinkles, Outcome 3: Physician assessment of success by analysing scores and scales
Total adverse event
There was higher in onabotulinumtoxinA 40 U than placebo (RR 1.45, 95% CI 1.13 to 1.86; participants = 474; studies = 1) (Analysis 5.4).
5.4. Analysis.

Comparison 5: OnabotulinumtoxinA 40units versus placebo one cycle of treatment, upper wrinkles, Outcome 4: Total adverse events
Duration of treatment effect
The RCT did not report this outcome.
COMPARISION 6. OnabotulinumtoxinA 64 units versus onabotulinumtoxinA 40 units for upper wrinkles, one cycle of treatment.
Only one RCT (n = 631participants) assessed this comparison (De Boulle 2018). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate (participant assessment) was higher in onabotulinumtoxinA 64 U than OnabotulinumtoxinA 40 U at week 4 (RR 1.12, 95% CI 0.96 to 1.31; participants = 631; studies = 1); at week 8 (RR 1.24, 95% CI 0.98 to 1.57; participants = 631; studies = 1); at week 12 (RR 1.12, 95% CI 0.75 to 1.67; participants = 631; studies = 1); at week 16 (RR 2.40, 95% CI 1.21 to 4.78; participants = 631; studies = 1); at week 20 (RR 2.54, 95% CI 0.50 to 12.99; participants = 631; studies = 1).
However, the responder rate (participant assessment) was similar with onabotulinumtoxinA 64 U and onabotulinumtoxinA 40 U at week 24 (RR 1.02, 95% CI 0.06 to 16.17; participants = 631; studies = 1) (Analysis 6.1).
6.1. Analysis.

Comparison 6: OnabotulinumtoxinA 64units versus OnabotulinumoxinA 40U one cycle of treatment, upper wrinkles, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
No difference was found (RR 0.41, 95% CI 0.08 to 2.08; participants = 631; studies = 1) (Analysis 6.2).
6.2. Analysis.

Comparison 6: OnabotulinumtoxinA 64units versus OnabotulinumoxinA 40U one cycle of treatment, upper wrinkles, Outcome 2: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by physician assessment was higher in onabotulinumtoxinA 64 units than onabotulinumtoxinA 40 units at week 4 (RR 1.12, 95% CI 0.96 to 1.31; participants = 631; studies = 1); at week 8 (RR 1.24, 95% CI 0.98 to 1.57; participants = 631; studies = 1); at week 12 (RR 1.12, 95% CI 0.75 to 1.67; participants = 631; studies = 1); at week 16(RR 2.40, 95% CI 1.21 to 4.78; participants = 631; studies = 1); at week 20 (RR 2.54, 95% CI 0.50 to 12.99; participants = 631; studies = 1).
However, the responder rate (participant assessment) was similar with onabotulinumtoxinA 64 U and OnabotulinumtoxinA 40 U at week 24 (RR 1.02, 95% CI 0.06 to 16.17; participants = 631; studies = 1) (Analysis 6.3).
6.3. Analysis.

Comparison 6: OnabotulinumtoxinA 64units versus OnabotulinumoxinA 40U one cycle of treatment, upper wrinkles, Outcome 3: Physician assessment of success by analysing scores and scales
Total adverse events
No difference between groups was found (RR 0.91, 95% CI 0.77 to 1.08; participants = 631; studies = 1) (Analysis 6.4).
6.4. Analysis.

Comparison 6: OnabotulinumtoxinA 64units versus OnabotulinumoxinA 40U one cycle of treatment, upper wrinkles, Outcome 4: Total adverse events
Duration of treatment effect
The RCT did not report this outcome.
COMPARISON 7. OnabotulinumtoxinA 64 units versus 32 units for upper wrinkles, one cycle of treatment
Only one RCT (n = 40 participants) assessed this comparison (Carruthers 2009). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate (participant assessment) was higher in onabotulinumtoxinA 64 U than in onabotulinumtoxinA 32 U at week 12 (RR 1.32, 95% CI 1.02 to 1.72; participants = 40; studies = 1) and week 16 (RR 1.73, 95% CI 1.15 to 2.60; participants = 40; studies = 1). However, there was no clear or substantial difference between groups at week 4 (RR 1.17, 95% CI 0.96 to 1.43; participants = 40; studies = 1); at week 8 (RR 1.12, 95% CI 0.91 to 1.38; participants = 40; studies = 1); week 20 (RR 4.00, 95% CI 0.97 to 16.55; participants = 40; studies = 1); and week 24 (RR 1.00, 95% CI 0.16 to 6.42; participants = 40; studies = 1) (Analysis 7.1).
7.1. Analysis.

Comparison 7: OnabotulinumtoxinA 64units versus 32 units one cycle of treatment upper wrinkles, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by physician assessment, was not different between onabotulinumtoxinA 32 U and onabotulinumtoxinA 64 U at weeks 4 and 8 (RR 1.00, 95% CI 0.91 to 1.10; participants = 40; studies = 1); at week 12 (RR 0.95, 95% CI 0.83 to 1.09; participants = 40; studies = 1). At weeks 16 and week 20 there was no clear difference between groups (RR 0.62, 95% CI 0.33 to 1.15; participants = 40; studies = 1) (RR 0.40, 95% CI 0.09 to 1.83; participants = 40; studies = 1). At week 24, both groups were less than 2/20(10%) (Analysis 7.2).
7.2. Analysis.

Comparison 7: OnabotulinumtoxinA 64units versus 32 units one cycle of treatment upper wrinkles, Outcome 2: Physician assessment of success by analysing scores and scales
Total adverse events
No difference between groups was found (RR 0.75, 95% CI 0.49 to 1.14; participants = 40; studies = 1) (Analysis 7.3).
7.3. Analysis.

Comparison 7: OnabotulinumtoxinA 64units versus 32 units one cycle of treatment upper wrinkles, Outcome 3: Total adverse events
Duration of treatment effect
The RCT assessed this outcome, but did not report the results.
COMPARISON 8. OnabotulinumtoxinA 96 units versus 32 units for upper wrinkles, one cycle of treatment
Only one RCT (n = 40 participants) assessed this outcome (Carruthers 2009). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate, by participant assessment, was similar between onabotulinumtoxinA 96U and onabotulinumtoxinA 32 U at weeks 4 and 8 (for both time points: RR 1.17, 95% CI 0.96 to 1.43; participants = 40; studies = 1); and there was no clear difference at week 24 (RR 3.50, 95% CI 0.83 to 14.83; participants = 40; studies = 1); and week 32 (RR 3.50, 95% CI 0.83 to 14.83; participants = 40; studies = 1). The responder rate, by participant assessment, was higher with onabotulinumtoxinA 96 U than onabotulinumtoxinA 32 U at week 12 (RR 1.32, 95% CI 1.02 to 1.72; participants = 40; studies = 1); week 16 (RR 1.64, 95% CI 1.07 to 2.50; participants = 40; studies = 1); week 20 (RR 6.00, 95% CI 1.54 to 23.44; participants = 40; studies = 1); and at week 28 (RR 4.50, 95% CI 1.11 to 18.27; participants = 40; studies = 1) (Analysis 8.1).
8.1. Analysis.

Comparison 8: OnabotulinumtoxinA 96 units versus 32 units one cycle of treatment upper wrinkles, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by physician assessment, was similar between onabotulinumtoxinA 96 U and onabotulinumtoxinA 32 U at weeks 4 and 8 (RR 1.00, 95% CI 0.91 to 1.10; participants = 40; studies = 1) and at week 12 (RR 1.05, 95% CI 0.92 to 1.20; participants = 40; studies = 1). But at week 28 and week 32 we are uncertain which group is favoured due to the wide confidence intervals obtained: (RR 13.00, 95% CI 0.78 to 216.39; participants = 40; studies = 1), (RR 5.00, 95% CI 0.26 to 98.00; participants = 40; studies = 1). The responder rate, by physician assessment, was higher with onabotulinumtoxinA 96u than onabotulinumtoxinA 32 U at week 16 (RR 2.25, 95% CI 1.29 to 3.92; participants = 40; studies = 1); week 20 (RR 7.50, 95% CI 1.97 to 28.61; participants = 40; studies = 1); and week 24 (RR 27.00, 95% CI 1.71 to 425.36; participants = 40; studies = 1) (Analysis 8.2).
8.2. Analysis.

Comparison 8: OnabotulinumtoxinA 96 units versus 32 units one cycle of treatment upper wrinkles, Outcome 2: Physician assessment of success by analysing scores and scales
Total adverse events
No difference between groups was found (RR 1.25, 95% CI 0.81 to 1.94; participants = 40; studies = 1) (Analysis 8.3).
8.3. Analysis.

Comparison 8: OnabotulinumtoxinA 96 units versus 32 units one cycle of treatment upper wrinkles, Outcome 3: Total adverse events
Duration of treatment effect
This RCT did not assessed this outcome.
COMPARISON 9. OnabotulinumtoxinA 96 units versus 64 units for upper wrinkles, one cycle of treatment.
Only one RCT (n = 40 participants) assessed this comparison (Carruthers 2009). For this reason we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate, by participant assessment, was similar between onabotulinumtoxinA 96U and onabotulinumtoxinA 64U, at week 4 (RR 1.00, 95% CI 0.91 to 1.10; participants = 40; studies = 1), at week 8 (RR 1.05, 95% CI 0.92 to 1.20; participants = 40; studies = 1), at week 12 (RR 1.00, 95% CI 0.91 to 1.10; participants = 40; studies = 1), and at week 16 (RR 0.95, 95% CI 0.79 to 1.13; participants = 40; studies = 1). We are less certain of the effect at week 20 (RR 1.50, 95% CI 0.79 to 2.86; participants = 40; studies = 1), and at week 24 (RR 3.50, 95% CI 0.83 to 14.83; participants = 40; studies = 1). The responder rate, by participant assessment, were higher with onabotulinumtoxinA 96U than onabotulinumtoxinA 64U at week 28 and week 32; however, because of the very wide confidence interval the result is very uncertain: at week 28 (RR 19.00, 95% CI 1.18 to 305.88; participants = 40; studies = 1), and at week 32 (RR 17.00, 95% CI 1.05 to 276.03; participants = 40; studies = 1) (Analysis 9.1).
9.1. Analysis.

Comparison 9: OnabotulinumtoxinA 96 units versus 64 units one cycle of treatment upper wrinkles, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by participant assessment, was similar with onabotulinumtoxinA 96U versus onabotulinumtoxinA 64U, at week 4 (RR 0.95, 95% CI 0.83 to 1.09; participants = 40; studies = 1), at week 8 (RR 1.00, 95% CI 0.91 to 1.10; participants = 40; studies = 1), and at week 12 (RR 1.00, 95% CI 0.91 to 1.10; participants = 40; studies = 1). But we are less certain about the effect size at week 16 (RR 1.38, 95% CI 0.97 to 1.97; participants = 40; studies = 1). The responder rate was higher in the 96 U group at week 20 (RR 3.00, 95% CI 1.35 to 6.68; participants = 40; studies = 1), and at week 24 (RR 6.50, 95% CI 1.68 to 25.16; participants = 40; studies = 1). The responder rate, by participant assessment, were also higher with onabotulinumtoxinA 96 units than onabotulinumtoxinA 64 units at week 24 and week 32; however, because of the confidence interval the results are very uncertain: at week 28 (RR 13.00, 95% CI 0.78 to 216.39; participants = 40; studies = 1), and at week 32 (RR 5.00, 95% CI 0.26 to 98.00; participants = 40; studies = 1) (Analysis 9.2).
9.2. Analysis.

Comparison 9: OnabotulinumtoxinA 96 units versus 64 units one cycle of treatment upper wrinkles, Outcome 2: Physician assessment of success by analysing scores and scales
Any adverse event
No difference between groups was found but the confidence interval was wide (RR 0.94, 95% CI 0.67 to 1.31; participants = 40; studies = 1) (Analysis 9.3).
9.3. Analysis.

Comparison 9: OnabotulinumtoxinA 96 units versus 64 units one cycle of treatment upper wrinkles, Outcome 3: Total adverse events
Duration of treatment effect
This RCT studied this outcome as survival analysis. The authors showed a better duration of treatment effect in onabotulinumtoxinA 96 units group (61.3% of responders at week 24).
COMPARISON 10. OnabotulinumtoxinA 32 units versus 16 units for forehead lines, one cycle of treatment
Only one RCT (n = 39 participants) assessed this comparison (Carruthers 2003a). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate, by participant assessment was similar with onabotulinumtoxinA 32U versus onabotulinumtoxinA 16 U at week 8 (RR 1.05, 95% CI 0.36 to 3.07; participants = 39; studies = 1) (Analysis 10.1).
10.1. Analysis.

Comparison 10: OnabotulinumtoxinA 32 units versus 16 units one cycle of treatment forehead lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Eyelid swelling was similar with onabotulinumtoxinA 32 U and onabotulinumtoxinA 16U (RR 1.05; 95% CI 0.24 to 4.59, n = 39; studies = 1) (Analysis 10.2).
10.2. Analysis.

Comparison 10: OnabotulinumtoxinA 32 units versus 16 units one cycle of treatment forehead lines, Outcome 2: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by physician assessment was similar with onabotulinumtoxinA 32U versus onabotulinumtoxinA 16 U at week 8 (RR 0.73; 95% CI 0.41 to 1.29, participants = 39; studies = 1) (Analysis 10.3).
10.3. Analysis.

Comparison 10: OnabotulinumtoxinA 32 units versus 16 units one cycle of treatment forehead lines, Outcome 3: Physician assessment of success by analysing scores and scales
Any adverse event
This RCT did not assess this outcome.
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 11. OnabotulinumtoxinA 48 units versus 16 units for forehead lines, one cycle of treatment
Only one RCT (n = 40 participants) assessed this comparison (Carruthers 2003a). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate, by participant assessment, was higher with onabotulinumtoxinA 48 U then onabotulinumtoxinA 16 U at week 8, but the results are uncertain due to the confidence interval including 1 (RR 1.60, 95% CI 0.63 to 4.05; participants = 40; studies = 1) (Analysis 17.1).
17.1. Analysis.

Comparison 17: OnabotulinumtoxinA 48 units versus 16 units one cycle of treatment forehead lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
No difference between groups was found, but the confidence interval was wide (RR 2.00; 95% CI 0.58 to 6.91, n = 40; studies = 1) (Analysis 17.2).
17.2. Analysis.

Comparison 17: OnabotulinumtoxinA 48 units versus 16 units one cycle of treatment forehead lines, Outcome 2: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by physician's assessment, was similar with onabotulinumtoxinA 48 U and onabotulinumtoxinA 16 U at week 8 (RR 1.00; 95% CI 0.63 to 1.58, n = 40; studies = 1) (Analysis 17.3).
17.3. Analysis.

Comparison 17: OnabotulinumtoxinA 48 units versus 16 units one cycle of treatment forehead lines, Outcome 3: Physician assessment of success by analysing scores and scales
Total adverse events
This RCT did not assess this outcome.
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 12. OnabotulinumtoxinA 48 units versus 32 units for forehead lines, one cycle of treatment
Only one RCT (n = 39 participants) assessed this comparison (Carruthers 2003a). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
There was no clear difference for responder rate, by participant assessment, between doses at week 8 (RR 1.52; 95% CI 0.60 to 3.83, n = 39; studies = 1) (Analysis 12.1).
12.1. Analysis.

Comparison 12: OnabotulinumtoxinA 48 units versus 32 units one cycle of treatment forehead lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
No difference between groups was found but the confidence interval was wide (RR 0.47, 95% CI 0.10 to 2.30; participants = 39; studies = 1) (Analysis 12.2)
12.2. Analysis.

Comparison 12: OnabotulinumtoxinA 48 units versus 32 units one cycle of treatment forehead lines, Outcome 2: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
One RCT assessed this comparison (Carruthers 2003a). The responder rate, by physician assessment, were similar with OnabotulinumtoxinA 48 U versus OnabotulinumtoxinA 32 U at week 8 (RR 1.37; 95% CI 0.77 to 2.43, n = 39; studies = 1) (Analysis 12.3).
12.3. Analysis.

Comparison 12: OnabotulinumtoxinA 48 units versus 32 units one cycle of treatment forehead lines, Outcome 3: Physician assessment of success by analysing scores and scales
Total adverse events
This RCT did not assess this outcome.
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 13. OnabotulinumtoxinA 40 units versus placebo for forehead lines and glabellar lines, one cycle of treatment
Only one RCT (n = 116 participants) assessed this comparison (Solish 2016).
For this reason (one study), we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate, by participant assessment, was higher with onabotulinumtoxinA 40 U than placebo at week 4 (RR 6.60, 95% CI 3.44 to 12.64; participants = 116; studies = 1); at week 8 (RR 16.91, 95% CI 5.59 to 51.17; participants = 116; studies = 1); at week 12 (RR 4.73, 95% CI 2.27 to 9.84; participants = 116; studies = 1); and at week 20 (RR 6.90, 95% CI 2.17 to 21.96; participants = 116; studies = 1).
Although, the responder rate, by participant assessment, was higher with onabotulinumtoxinA 40 U than placebo, the results are uncertain due to the wide confidence intervals: at week 16 (RR 32.09, 95% CI 4.53 to 227.29; participants = 116; studies = 1), and at week 24 (RR 15.53, 95% CI 2.12 to 113.72; participants = 116; studies = 1) (Analysis 13.1).
13.1. Analysis.

Comparison 13: OnabotulinumtoxinA 40 units versus placebo one cycle of treatment for forehead lines and glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by physician assessment, was higher with onabotulinumtoxinA 40 U than placebo at week 16 (RR 4.49, 95% CI 2.00 to 10.08; participants = 116; studies = 1).
The responder rate, by physician assessment, was higher with onabotulinumtoxinA 40 U than placebo at week 4 (RR 26.91, 95% CI 6.88 to 105.34; participants = 116; studies = 1); at week 8 (RR 48.65, 95% CI 6.94 to 340.90; participants = 116; studies = 1); at week 12 (RR 10.70, 95% CI 3.46 to 33.04; participants = 116; studies = 1); at week 20 (RR 20.70, 95% CI 2.87 to 149.20; participants = 116; studies = 1); and at week 24 (RR 11.39, 95% CI 1.52 to 85.36; participants = 116; studies = 1) (Analysis 13.2). However, because of the wide confidence intervals the results are very uncertain.
13.2. Analysis.

Comparison 13: OnabotulinumtoxinA 40 units versus placebo one cycle of treatment for forehead lines and glabellar lines, Outcome 2: Physician assessment of success by analysing scores and scales
Total adverse events
No difference between groups was found, but the confidence interval was wide (RR 1.38; 95% CI 0.79 to 2.42; participants = 116; studies = 1) (Analysis 13.3).
13.3. Analysis.

Comparison 13: OnabotulinumtoxinA 40 units versus placebo one cycle of treatment for forehead lines and glabellar lines, Outcome 3: Total adverse events
Duration of treatment effect
The duration of the treatment effect was 16.9 weeks (118.5 days) by physician assessment and 17.8 weeks (125 days) by participant assessment in onabotulinumtoxinA 40U group.
COMPARISON 14. OnabotulinumtoxinA 30 units versus placebo, one cycle of treatment, forehead lines and glabellar lines
Only one RCT (n = 118 participants) assessed this comparison (Solish 2016). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate, by participant assessment was higher with onabotulinumtoxinA 30 U group than placebo at week 4 (RR 6.25, 95% CI 3.25 to 12.01; participants = 118; studies = 1); at week 8 (RR 14.00, 95% CI 4.59 to 42.67; participants = 118; studies = 1); at week 12 (RR 4.29, 95% CI 2.05 to 8.98; participants = 118; studies = 1); at week 20 (RR 5.33, 95% CI 1.64 to 17.34; participants = 118; studies = 1); and at week 24 (RR 10.00, 95% CI 1.32 to 75.66; participants = 118; studies = 1) (Analysis 14.1).
14.1. Analysis.

Comparison 14: OnabotulinumtoxinA 30 units versus placebo one cycle of treatment for forehead lines and glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Although the responder rate, by participant assessment, was higher with onabotulinumtoxinA 30U group than placebo at week 16, the result is uncertain due to a large confidence interval (RR 22.00, 95% CI 3.06 to 157.95; participants = 118; studies = 1).
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
No difference between groups was found but the confidence interval was wide (RR 5.00, 95% CI 0.25 to 101.97; participants = 118; studies = 1) (Analysis 14.2)
14.2. Analysis.

Comparison 14: OnabotulinumtoxinA 30 units versus placebo one cycle of treatment for forehead lines and glabellar lines, Outcome 2: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by physician assessment, was higher with onabotulinumtoxinA 30U group than placebo; however, because of the confidence interval the results is very uncertain; at week 4 (RR 24.00, 95% CI 6.11 to 94.23; participants = 118; studies = 1); at week 8 (RR 43.00, 95% CI 6.12 to 302.08; participants = 118; studies = 1); and at week 24 (RR 8.00, 95% CI 1.03 to 61.98; participants = 118; studies = 1) (Analysis 14.3).
14.3. Analysis.

Comparison 14: OnabotulinumtoxinA 30 units versus placebo one cycle of treatment for forehead lines and glabellar lines, Outcome 3: Physician assessment of success by analysing scores and scales
The responder rate, by physician assessment, was higher with onabotulinumtoxinA 30 U group than placebo at week 12 (RR 8.67, 95% CI 2.77 to 27.08; participants = 118; studies = 1); and at week 16 (RR 3.33, 95% CI 1.44 to 7.71; participants = 118; studies = 1) (Analysis 14.3).
Total adverse events
No difference between groups was found, but the confidence interval was wide(RR 1.86, 95% CI 0.80 to 4.32; participants = 118; studies = 1) (Analysis 14.4).
14.4. Analysis.

Comparison 14: OnabotulinumtoxinA 30 units versus placebo one cycle of treatment for forehead lines and glabellar lines, Outcome 4: Total adverse events
Duration of treatment effect
The duration of treatment effect was 16 weeks (113.0 days) by physician assessment and 16.5 weeks (115.5 days) by participant assessment in onabotulinumtoxinA 30u group.
COMPARISON 15. OnabotulinumtoxinA 40 units versus onabotulinumtoxinA 30 units one cycle of treatment, forehead lines and glabellar lines
Only one RCT (n = 116 participants) assessed this comparison (Solish 2016). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate, by participant assessment, were similar with onabotulinumtoxinA 40 U versus onabotulinumtoxinA 30U at week 4 (RR 1.06, 95% CI 0.92 to 1.21; participants = 116; studies = 1); at week 8 (RR 1.21, 95% CI 1.00 to 1.47; participants = 116; studies = 1); at week 12 (RR 1.10, 95% CI 0.79 to 1.55; participants = 116; studies = 1); at week 16 (RR 1.46, 95% CI 0.97 to 2.19; participants = 116; studies = 1); at week 20 (RR 1.29, 95% CI 0.75 to 2.24; participants = 116; studies = 1); and at week 24 (RR 1.55, 95% CI 0.76 to 3.17; participants = 116; studies = 1) (Analysis 15.1).
15.1. Analysis.

Comparison 15: OnabotulinumtoxinA 40 units versus OnabotulinumonabotulinumtoxinA 30 units one cycle of treatment for forehead lines and glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
No difference between groups was found, but the confidence interval was wide (RR 0.21, 95% CI 0.01 to 4.22; participants = 116; studies = 1) (Analysis 15.2).
15.2. Analysis.

Comparison 15: OnabotulinumtoxinA 40 units versus OnabotulinumonabotulinumtoxinA 30 units one cycle of treatment for forehead lines and glabellar lines, Outcome 2: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by physician assessment, were similar with onabotulinumtoxinA 40 U versus onabotulinumtoxinA 30 U at week 4 (RR 1.12, 95% CI 0.97 to 1.30; participants = 116; studies = 1); at week 8 (RR 1.13, 95% CI 0.93 to 1.38; participants = 116; studies = 1); at week 12 (RR 1.23, 95% CI 0.85 to 1.79; participants = 116; studies = 1); at week 16 (RR 1.35, 95% CI 0.85 to 2.12; participants = 116; studies = 1); at week 20 (RR 1.38, 95% CI 0.79 to 2.42; participants = 116; studies = 1); and at week 24 (RR 1.42, 95% CI 0.62 to 3.28; participants = 116; studies = 1) (Analysis 15.3).
15.3. Analysis.

Comparison 15: OnabotulinumtoxinA 40 units versus OnabotulinumonabotulinumtoxinA 30 units one cycle of treatment for forehead lines and glabellar lines, Outcome 3: Physician assessment of success by analysing scores and scales
Total adverse events
No difference between groups was found, but the confidence interval was wide (RR 1.04, 95% CI 0.63 to 1.71; participants = 116; studies = 1) (Analysis 15.4).
15.4. Analysis.

Comparison 15: OnabotulinumtoxinA 40 units versus OnabotulinumonabotulinumtoxinA 30 units one cycle of treatment for forehead lines and glabellar lines, Outcome 4: Total adverse events
Duration of treatment effect
The duration of treatment effect was 118.5 days by investigator assessment and 125 days by participant assessment in OnbotulinumtoxinA 40 U group. The duration of treatment effect was 113.0 days by investigator assessment and 115.5 days by participant assessment in OnabotulinumtoxinA 30 U group.
COMPARISON 16. OnabotulinumtoxinA 40 units versus 20 units one cycle of treatment, glabellar lines
Two RCTs assessed (n = 80 participants) this comparison (Carruthers 2005a; Carruthers 2005b).
Primary outcomes
Participant assessment of success by analysing scores and scales
The meta‐analysis showed benefit in favour of onabotulinumtoxinA 40 U group at 4 weeks (RR 1.63, 95% CI 1.13 to 2.35; participants = 80; studies = 2; I² = 0%), but not at week 8 (RR 1.52, 95% CI 0.77 to 2.97; participants = 80; studies = 2; I² = 44%), or week 16 (RR 2.51, 95% CI 0.88 to 7.16; participants = 80; studies = 2; I² = 0%) (Analysis 16.1) (Figure 5).
16.1. Analysis.

Comparison 16: OnabotulinumtoxinA 40 units versus 20 units one treatment glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Only Carruthers 2005b assessed this outcome. The rate of eyebrow ptosis was 1/20 (5%) for both doses.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The meta‐analysis showed a potential benefit in favour of onabotulinumtoxinA 40 U group at 4 weeks (RR 1.48, 95% CI 0.93 to 2.36; participants = 80; studies = 2; I² = 71%); at week 8 (RR 1.35, 95% CI 0.82 to 2.23; participants = 80; studies = 2; I² = 21%); and at 16 weeks (RR 1.34, 95% CI 0.51 to 3.53; participants = 80; studies = 2; I² = 0%) (Analysis 16.2) (Figure 5), but the difference was small and the result were not very precise.
16.2. Analysis.

Comparison 16: OnabotulinumtoxinA 40 units versus 20 units one treatment glabellar lines, Outcome 2: Physician assessment of success by analysing scores and scales
Total adverse events
No difference between groups was found, but the confidence interval was wide (RR 1.93, 95% CI 0.53 to 7.05; participants = 80; studies = 2; I² = 28%) (Analysis 16.3) (Figure 5).
16.3. Analysis.

Comparison 16: OnabotulinumtoxinA 40 units versus 20 units one treatment glabellar lines, Outcome 3: Total adverse events
Duration of treatment effect
Only one RCT assessed this outcome (Carruthers 2005b). The mean time of duration of treatment effect for onabotulinumtoxinA 20 U group was 17.6 weeks and 21.7 weeks for onabotulinumtoxinA 40 U group (no standard deviation (SD), P value, or 95% CIs were provided; hence, results were not provided in a forest plot).
COMPARISON 17. OnabotulinumtoxinA 60 units versus 20 units one cycle of treatment, glabellar lines
Only one RCT (n = 40 participants) assessed this comparison (Carruthers 2005a). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
This RCT did not assess this outcome.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by participant assessment, were higher with onabotulinumtoxinA 60 U than onabotulinumtoxinA 20 U at week 4 (RR 1.90, 95% CI 1.21 to 2.98; participants = 40; studies = 1), and at week 8 (RR 3.80, 95% CI 1.77 to 8.17; participants = 40; studies = 1). However, there was no clear or substantial difference in responder rate with onabotulinumtoxinA 60 U and onabotulinumtoxinA 20 U at week 12 (RR 2.00, 95% CI 0.72 to 5.59; participants = 40; studies = 1) at week 16 (RR 1.00, 95% CI 0.29 to 3.45; participants = 40; studies = 1); and at week 20 (RR 2.00, 95% CI 0.41 to 9.71; participants = 40; studies = 1) (Analysis 11.1).
11.1. Analysis.

Comparison 11: OnabotulinumtoxinA 60 units versus 20 units one cycle of treatment glabellar lines, Outcome 1: Physician assessment of success by analysing scores and scales
Total adverse events
No difference between groups was found, but the confidence interval was wide (RR 1.00, 95% CI 0.07 to 14.90; participants = 40; studies = 1) (Analysis 11.2).
11.2. Analysis.

Comparison 11: OnabotulinumtoxinA 60 units versus 20 units one cycle of treatment glabellar lines, Outcome 2: Total adverse events
Duration of treatment effect
The duration of treatment effect was 17.6 weeks in onabotulinumtoxinA 20U group, and 22.8 weeks in onabotulinumtoxinA 60 U group.
COMPARISON 18. OnabotulinumtoxinA 80 units versus 2 0units one cycle of treatment, glabellar lines
Only one RCT (n = 40 participants) assessed this comparison (Carruthers 2005a). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate, by participant assessment, were higher with onabotulinumtoxinA 80 U than onabotulinumtoxinA 20U at week 4 (RR 3.75, 95% CI 1.51 to 9.34; participants = 40; studies = 1) (Analysis 18.1).
18.1. Analysis.

Comparison 18: OnabotulinumtoxinA 80 units versus 20 units one cycle of treatment glabellar lines, Outcome 1: Participant assessment, maximum contraction (responder rate)
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by physician assessment, were higher with onabotulinumtoxinA 80 U than onabotulinumtoxinA 20 U at week 4 (RR 1.95, 95% CI 1.27 to 3.01; participants = 40; studies = 1); at week 8 (RR 3.20, 95% CI 1.45 to 7.05; participants = 40; studies = 1); and at week 12 (RR 3.50, 95% CI 1.39 to 8.80; participants = 40; studies = 1). However, the responder rate was similar with onabotulinumtoxinA 80 U and onabotulinumtoxinA 20 U at week 16 (RR 1.00, 95% CI 0.29 to 3.45; participants = 40; studies = 1), and at week 20 (RR 1.00, 95% CI 0.16 to 6.42; participants = 40; studies = 1). At week 24, the result was very uncertain (RR 5.00, 95% CI 0.26 to 98.00; participants = 40; studies = 1) (Analysis 18.2).
18.2. Analysis.

Comparison 18: OnabotulinumtoxinA 80 units versus 20 units one cycle of treatment glabellar lines, Outcome 2: Physician assessment of success by analysing scores and scales
Total adverse events
No difference between groups was found, but the confidence interval was wide (RR 4.00, 95% CI 0.49 to 32.72; participants = 40; studies = 1) (Analysis 18.3).
18.3. Analysis.

Comparison 18: OnabotulinumtoxinA 80 units versus 20 units one cycle of treatment glabellar lines, Outcome 3: Total adverse events
Duration of treatment effect
The duration of treatment effect was 17.6 weeks in onabotulinumtoxinA 20U group and 24.2 weeks in onabotulinumtoxinA 80U.
COMPARISON 19. OnabotulinumtoxinA 80 units versus 60 units one cycle of treatment, glabellar lines
Only one RCT (n = 20 participants) assessed this comparison (Carruthers 2005a). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
This RCT did not assess this outcome.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by physician assessment were similar with onabotulinumtoxinA 80U and onabotulinumtoxinA 60 U at week 4 (RR 1.05, 95% CI 0.92 to 1.20; participants = 40; studies = 1); at week 8 (RR 0.84, 95% CI 0.66 to 1.07; participants = 40; studies = 1); at week 12 (RR 1.75, 95% CI 0.95 to 3.22; participants = 40; studies = 1); and at week 16 (RR 1.00, 95% CI 0.29 to 3.45; participants = 40; studies = 1). At week 20 and 24, the effect sizes were less certain: (RR 0.50, 95% CI 0.10 to 2.43; participants = 40; studies = 1) (RR 5.00, 95% CI 0.26 to 98.00; participants = 40; studies = 1) (Analysis 19.1).
19.1. Analysis.

Comparison 19: OnabotulinumtoxinA 80 units versus 60 units one cycle of treatment glabellar lines, Outcome 1: Physician assessment of success by analysing scores and scales
Total adverse events
There was no difference between groups, but the confidence interval was wide and included both an increase or a reduction at the risk of total adverse events (RR 2.00, 95% CI 0.41 to 9.71; participants = 40; studies = 1) (Analysis 19.2).
19.2. Analysis.

Comparison 19: OnabotulinumtoxinA 80 units versus 60 units one cycle of treatment glabellar lines, Outcome 2: Total adverse events
Duration of treatment effect
The duration of treatment effect was 22.8 weeks in onabotulinumtoxinA 60U group, and 24.2 weeks in onabotulinumtoxinA 80 U.
COMPARISON 20. OnabotulinumtoxinA 30 units versus 10 units one cycle of treatment, glabellar lines
One RCT (n = 40 participants) assessed this comparison (Carruthers 2005a).
For this reason (one study), we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
This RCT did not assess this outcome.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by physician assessment, were similar with onabotulinumtoxinA 30 U and onabotulinumtoxinA 10 U at week 4 (RR 1.12, 95% CI 0.91 to 1.38; participants = 40; studies = 1), but favoured the 30 U group at week 16, although with some uncertainty (RR 5.00, 95% CI 0.64 to 39.06; participants = 40; studies = 1). The responder rate, by physician assessment, were higher with onabotulinumtoxinA 30 U than onabotulinumtoxinA 10 U at week 8 (RR 2.40, 95% CI 1.04 to 5.55; participants = 40; studies = 1).
Although the responder rate, by physician assessment, were higher with onabotulinumtoxinA 30 U than onabotulinumtoxinA 10 U at week 12, the result is uncertain because of the confidence interval (RR 9.00, 95% CI 1.25 to 64.59; participants = 40; studies = 1) (Analysis 20.1).
20.1. Analysis.

Comparison 20: OnabotulinumtoxinA 30 units versus 10 units one cycle of treatment glabellar lines, Outcome 1: Physician assessment of success by analysing scores and scales
Total adverse events
The was no difference between groups (RR 0.25; 95% CI 0.03 to 2.05; participants = 40; studies = 1) (Analysis 20.2).
20.2. Analysis.

Comparison 20: OnabotulinumtoxinA 30 units versus 10 units one cycle of treatment glabellar lines, Outcome 2: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 21. OnabotulinumtoxinA 40 units versus 10 units one cycle of treatment, glabellar lines
Only one RCT (n = 40 participants) assessed this comparison (Carruthers 2005b). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
There was no clear or substantial difference in responder rate, by participant assessment, between onabotulinumtoxinA 40 U and onabotulinumtoxinA 10 U at week 4 (RR 1.86, 95% CI 0.94 to 3.66; participants = 40; studies = 1); at week 8 (RR 1.00, 95% CI 0.47 to 2.14; participants = 40; studies = 1); at week 12 (RR 1.75, 95% CI 0.61 to 5.05; participants = 40; studies = 1); and at week 16 (RR 1.00, 95% CI 0.16 to 6.42; participants = 40; studies = 1) (Analysis 21.1).
21.1. Analysis.

Comparison 21: OnabotulinumtoxinA 40 units versus 10 units one cycle of treatment glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by physician assessment, were similar with onabotulinumtoxinA 40 U and onabotulinumtoxinA 10 U at week 4 (RR 1.17, 95% CI 0.96 to 1.43; participants = 40; studies = 1) but the result was more uncertain at week 16 (RR 5.00, 95% CI 0.64 to 39.06; participants = 40; studies = 1). The responder rate, by physician assessment, were higher with onabotulinumtoxinA 40 U than onabotulinumtoxinA 10 U at week 8 (RR 2.80, 95% CI 1.24 to 6.30; participants = 40; studies = 1).
Although the responder rate, by physician assessment, were higher with onabotulinumtoxinA 30u than onabotulinumtoxinA 10u at week 12, the RR is uncertain because of the wide confidence interval (RR 8.00, 95% CI 1.10 to 58.19; participants = 40; studies = 1) (Analysis 21.2).
21.2. Analysis.

Comparison 21: OnabotulinumtoxinA 40 units versus 10 units one cycle of treatment glabellar lines, Outcome 2: Physician assessment of success by analysing scores and scales
Total adverse events
No difference between groups was found, but the confidence interval was wide (RR 1.25, 95% CI 0.39 to 3.99; participants = 40; studies = 1) (Analysis 21.3).
21.3. Analysis.

Comparison 21: OnabotulinumtoxinA 40 units versus 10 units one cycle of treatment glabellar lines, Outcome 3: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 22. OnabotulinumtoxinA 40 units versus 30 units one cycle of treatment, glabellar lines
Only one RCT (n = 40 participants) assessed this comparison (Carruthers 2005b). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate, by participant assessment, were similar with onabotulinumtoxinA 40 U and onabotulinumtoxinA 30 U at week 4 (RR 1.30, 95% CI 0.75 to 2.24; participants = 40; studies = 1); at week 8 (RR 1.00, 95% CI 0.47 to 2.14; participants = 40; studies = 1); at week 12 (RR 1.17, 95% CI 0.48 to 2.86; participants = 40; studies = 1); and at week 16 (RR 1.00, 95% CI 0.16 to 6.42; participants = 40; studies = 1) (Analysis 22.1).
22.1. Analysis.

Comparison 22: OnabotulinumtoxinA 40 units versus 30 units one cycle of treatment glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by physician assessment were similar with onabotulinumtoxinA 40 U and onabotulinumtoxinA 3 0U at week 4 (RR 1.05, 95% CI 0.92 to 1.20; participants = 40; studies = 1;, at week 8 (RR 1.17, 95% CI 0.74 to 1.85; participants = 40; studies = 1); at week 12 (RR 0.89, 95% CI 0.43 to 1.83; participants = 40; studies = 1), and at week 16 (RR 1.00, 95% CI 0.34 to 2.93; participants = 40; studies = 1) (Analysis 22.2).
22.2. Analysis.

Comparison 22: OnabotulinumtoxinA 40 units versus 30 units one cycle of treatment glabellar lines, Outcome 2: Physician assessment of success by analysing scores and scales
Total adverse event
No difference between groups was found, but the confidence interval was wide (RR 5.00, 95% CI 0.64 to 39.06; participants = 40; studies = 1) (Analysis 22.3).
22.3. Analysis.

Comparison 22: OnabotulinumtoxinA 40 units versus 30 units one cycle of treatment glabellar lines, Outcome 3: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 23. OnabotulinumtoxinA 30 units versus 20 units one cycle of treatment, glabellar lines
Only one RCT (n = 40 participants) assessed this comparison (Carruthers 2005b). For this reason (one study), we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate, by participant assessment were similar with onabotulinumtoxinA 30 U and onabotulinumtoxinA 2 0U at week 4 (RR 1.11, 95% CI 0.58 to 2.14; participants = 40; studies = 1); at week 8 (RR 1.00, 95% CI 0.47 to 2.14; participants = 40; studies = 1); at week 12 (RR 1.50, 95% CI 0.50 to 4.52; participants = 40; studies = 1); and at week 16 (RR 1.00, 95% CI 0.16 to 6.42; participants = 40; studies = 1) (Analysis 23.1).
23.1. Analysis.

Comparison 23: OnabotulinumtoxinA 30 units versus 20 units one cycle of treatment glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by physician assessment, were similar with onabotulinumtoxinA 30 U and onabotulinumtoxinA 20 U at week 4 (RR 1.19, 95% CI 0.93 to 1.51; participants = 40; studies = 1); at week 8 (RR 1.00, 95% CI 0.60 to 1.66; participants = 40; studies = 1); at week 12 (RR 1.29, 95% CI 0.60 to 2.77; participants = 40; studies = 1); and at week 16 (RR 1.67, 95% CI 0.46 to 6.06; participants = 40; studies = 1) (Analysis 23.2).
23.2. Analysis.

Comparison 23: OnabotulinumtoxinA 30 units versus 20 units one cycle of treatment glabellar lines, Outcome 2: Physician assessment of success by analysing scores and scales
Total adverse events
No difference between groups was found, but the confidence interval was wide (RR 0.25, 95% CI 0.03 to 2.05; participants = 40; studies = 1) (Analysis 23.3).
23.3. Analysis.

Comparison 23: OnabotulinumtoxinA 30 units versus 20 units one cycle of treatment glabellar lines, Outcome 3: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 24. OnabotulinumtoxinA 60 units versus 40 units one cycle of treatment, glabellar lines
Only one RCT (n = 40 participants) assessed this comparison (Carruthers 2005a). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
This RCT did not assess this outcome.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by physician assessment, were similar with onabotulinumtoxinA 60U and onabotulinumtoxinA 40 U at week 4 (RR 1.12, 95% CI 0.91 to 1.38; participants = 40; studies = 1); at week 12 (RR 1.00, 95% CI 0.47 to 2.14; participants = 40; studies = 1);at week 16 (RR 1.00, 95% CI 0.29 to 3.45; participants = 40; studies = 1); and week 20 (RR 1.00, 95% CI 0.29 to 3.45; participants = 40; studies = 1). The responder rate, by physician assessment, was higher with onabotulinumtoxinA 60 U than onabotulinumtoxinA 40 U at week 8 (RR 1.90, 95% CI 1.21 to 2.98; participants = 40; studies = 1) (Analysis 24.1).
24.1. Analysis.

Comparison 24: OnabotulinumtoxinA 60 units versus 40 units one cycle of treatment glabellar lines, Outcome 1: Physician assessment of success by analysing scores and scales
Any adverse event
No difference between groups was found, but the confidence interval was wide (RR 0.40, 95% CI 0.09 to 1.83 participants = 40; studies = 1) (Analysis 24.2).
24.2. Analysis.

Comparison 24: OnabotulinumtoxinA 60 units versus 40 units one cycle of treatment glabellar lines, Outcome 2: Total adverse events
Duration of treatment effect
The duration of treatment effect was 21.7 weeks in onabotulinumtoxinA 40U and 22.8 weeks in onabotulinumtoxinA 60 U.
COMPARISON 25. OnabotulinumtoxinA 80 units versus 40 units one cycle of treatment, glabellar lines
Only one RCT (n = 40 participants) assessed this comparison (Carruthers 2005a). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
This RCT did not assess this outcome.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by physician assessment were similar with onabotulinumtoxinA 80 U and onabotulinumtoxinA 40 U at week 4 (RR 1.17, 95% CI 0.96 to 1.43; participants = 40; studies = 1); at week 8 (RR 1.60, 95% CI 0.98 to 2.61; participants = 40; studies = 1), at week 12 (RR 1.75, 95% CI 0.95 to 3.22; participants = 40; studies = 1); and at week 16 (RR 1.00, 95% CI 0.29 to 3.45; participants = 40; studies = 1). At week 20 (RR 0.50, 95% CI 0.10 to 2.43; participants = 40; studies = 1) and at week 24 (RR 5.00, 95% CI 0.26 to 98.00; participants = 40; studies = 1) the effect size is less certain due to wide confidence intervals and/or the inclusion of 1 in the confidence interval (Analysis 25.1).
25.1. Analysis.

Comparison 25: OnabotulinumtoxinA 80 units versus 40 units one cycle of treatment glabellar lines, Outcome 1: Physician assessment of success by analysing scores and scales
Total adverse event
No difference between groups was found, but the confidence interval was wide (RR 0.80, 95% CI 0.25 to 2.55; participants = 40; studies = 1) (Analysis 25.2).
25.2. Analysis.

Comparison 25: OnabotulinumtoxinA 80 units versus 40 units one cycle of treatment glabellar lines, Outcome 2: Total adverse events
Duration of treatment effect
The duration of treatment effect was 21.7 weeks in onabotulinumtoxinA 40 U, and 24.2 weeks in onabotulinumtoxinA 80U.
COMPARISON 26. OnabotulinumtoxinA 80 units versus 60 units one cycle of treatment, glabellar lines
One RCT (n = 40 participants) assessed this comparison (Carruthers 2005a).
For this reason (one study), we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
This RCT did not assess this outcome.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major event.
Physician assessment of success by analysing scores and scales
The responder rate, by physician assessment, were similar with onabotulinumtoxinA 80 U and onabotulinumtoxinA 60 U at week 4 (RR 1.05, 95% CI 0.92 to 1.20; participants = 40; studies = 1) and at week 16 (RR 1.00, 95% CI 0.29 to 3.45; participants = 40; studies = 1), but was higher in the onabotulinumtoxin 60 U group at week 8 (RR 0.53, 95% CI 0.34 to 0.83; participants = 40; studies = 1). At week 12 it was higher in the onabotulinumtoxinA 80 U group, but the result is less certain due to the confidence interval including 1 (RR 1.75, 95% CI 0.95 to 3.22; participants = 40; studies = 1). At week 20 (RR 0.50, 95% CI 0.10 to 2.43; participants = 40; studies = 1) and at week 24 (RR 5.00, 95% CI 0.26 to 98.00; participants = 40; studies = 1) the effect size is uncertain due to the wide confidence intervals (Analysis 26.1).
26.1. Analysis.

Comparison 26: OnabotulinumtoxinA 80units versus 60units one cycle of treatment glabellar lines, Outcome 1: Physician assessment of success by analysing scores and scales
Total adverse events
No difference between groups was found, but the confidence interval was wide (RR 2.00, 95% CI 0.41 to 9.71; participants = 40; studies = 1) (Analysis 26.2).
26.2. Analysis.

Comparison 26: OnabotulinumtoxinA 80units versus 60units one cycle of treatment glabellar lines, Outcome 2: Total adverse events
Duration of treatment effect
The duration of treatment effect was 22.8 weeks in onabotulinumA 60 U, and 24.2 weeks in onabotulinumtoxinA 80 U.
COMPARISON 27. OnabotulinumtoxinA 12 units versus 7.5 units one cycle of treatment, perioral lines
Only one RCT (n = 60 participants) assessed this comparison (Cohen 2012). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
This RCT did not assess this outcome.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by physician assessment, were similar with onabotulinumtoxinA 12 U and onabotulinumtoxinA 7.5 U at week 4 (RR 1.27, 95% CI 0.96 to 1.69; participants = 60; studies = 1), at week 8 (RR 0.90, 95% CI 0.68 to 1.19; participants = 60; studies = 1), at week 16 (RR 1.32, 95% CI 0.78 to 2.23; participants = 60; studies = 1), and at week 20 (RR 1.23, 95% CI 0.72 to 2.12; participants = 60; studies = 1). The responder rate, by physician assessment, were higher with onabotulinumtoxinA 12U than onabotulinumtoxinA 7.5 U at week 12 (RR 2.14, 95% CI 1.27 to 3.59; participants = 60; studies = 1) (Analysis 27.1).
27.1. Analysis.

Comparison 27: OnabotulinumtoxinA 12 units versus 7.5 units one cycle of treatment perioral lines, Outcome 1: Physician assessment of success by analysing scores and scales
Total adverse events
Total adverse events were higher with onabotulinumtoxinA 12 U than onabotulinumtoxinA 7.5 U (RR 1.55, 95% CI 1.00 to 2.42 participants = 60; studies = 1) (Analysis 27.2).
27.2. Analysis.

Comparison 27: OnabotulinumtoxinA 12 units versus 7.5 units one cycle of treatment perioral lines, Outcome 2: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 28. OnabotulinumtoxinA 20 units versus placebo three cycles of treatment, glabellar lines
Only one RCT (n = 537 participants) assessed this comparison (Carruthers 2004). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
For the participants who received two or more onabotulinumtoxinA injections, the responder rate were significantly higher after the third cycle of treatment than after the first and second cycle of treatment (P < 0.005). No numeric data were provided.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
The number of major adverse events in the first cycle was higher in the onabotulinumtoxinA 20 U group compared with placebo, but the result is very imprecise due to the wide confidence intervals (RR 8.84, 95% CI 0.53 to 147.78; participants = 537; studies = 1) (Analysis 28.1).
28.1. Analysis.

Comparison 28: OnabotulinumtoxinA 20units versus placebo in glabellar lines three cycles of treatment, Outcome 1: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
For the participants who received two or more onabotulinumtoxinA injections the responder rate were higher in the third cycle of treatment than in the first and second cycles at day 30, 60, 90, and 120 days (P < 0.007). No numeric data were provided.
Total adverse events
No difference between groups was found (RR 0.76, 95% CI 0.58 to 0.99; participants = 537; studies = 1) (Analysis 28.2).
28.2. Analysis.

Comparison 28: OnabotulinumtoxinA 20units versus placebo in glabellar lines three cycles of treatment, Outcome 2: Total adverse events
The number of adverse events across first, second and third cycle were 106/501 (21.2%) in the first cycle of BontA treatment, 17/362 (4.7%) in the second cycle of treatment, and 5/258 (2%) in the third cycle of treatment. But no numerical data related to placebo group were provided.
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 29. OnabotulinumtoxinA 24 units versus placebo, crow's feet lines
Four RCTs (n = 1675 participants) assessed this comparison (Carruthers 2014; Harii 2017; Moers‐Carpi 2015; Wu 2019).
Harii 2017 was a two‐phase study, so we considered only the first phase (randomised, double‐blind).
Primary outcomes
Participant assessment of success by analysing scores and scales
The meta‐analysis showed a difference in favour of onabotulinumtoxinA 24;U group at week 4 (RR 12.71, 95% CI 8.67 to 18.63; participants = 1474; studies = 3; I2 = 0%), week 8 (RR 10.25, 95% CI 7.02 to 14.98; participants = 1474; studies = 3; I2 = 0%); and week 12 (RR 7.70, 95% CI 4.81 to 12.33; participants = 1474; studies = 3; I2 = 28%). Only Wu 2019 showed results of onaboltulinuntoxinA versus placebo at week 20 (RR 10.33, 95% CI 3.35 to 31.89; participants = 417; studies = 1) (Analysis 29.1).
29.1. Analysis.

Comparison 29: OnabotulinumtoxinA24 units versus placebo one treatment in crow's feet lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Two studies (Harii 2017; Wu 2019) assessed this outcome. There was no difference between onabotulinumtoxinA 24 U group and placebo group (Peto OR 1.28, 95% CI 0.12 to 13.10; participants = 665; studies = 2; I2 = 0%) (Analysis 29.2).
29.2. Analysis.

Comparison 29: OnabotulinumtoxinA24 units versus placebo one treatment in crow's feet lines, Outcome 2: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
The meta‐analysis showed a difference in favour of OnabotulinumtoxinA 24U group at week 4 (RR 12.38, 95% CI 8.93 to 17.16; participants = 1675; studies = 4; I2 = 0%); at week 8 (RR 10.13, 95% CI 5.34 to 19.23; participants = 1258; studies = 3; I2 = 63%); at week 12 (RR 9.29, 95% CI 5.95 to 14.50; participants = 1258; studies = 3; I2 = 0%); and week 16 (RR 5.46, 95% CI 3.19 to 9.32; participants = 1057; studies = 2; I² = 0%) (Analysis 29.3).
29.3. Analysis.

Comparison 29: OnabotulinumtoxinA24 units versus placebo one treatment in crow's feet lines, Outcome 3: Physician assessment of success by analysing scores and scales
Total adverse events
Moers‐Carpi 2015 showed the number of adverse events per group of intervention not per cycle of treatment. Therefore, only three RCTs (Carruthers 2014; Harii 2017; Wu 2019) were included in the meta‐analysis. Total adverse events were similar in both groups (RR 1.17, 95% CI 0.94 to 1.45; participants = 692; studies = 2; I2 = 0%) (Analysis 29.4).
29.4. Analysis.

Comparison 29: OnabotulinumtoxinA24 units versus placebo one treatment in crow's feet lines, Outcome 4: Total adverse events
Duration of treatment effect
Three RCTs assessed this outcome (Carruthers 2014; Harii 2017; Wu 2019), but both studies described the results narratively. The duration of treatment effect for responders (none‐ mild at maximum smile) was 16.8 weeks (118 days) (physician assessment), and 16.5 (116 days) (participant assessment). The median duration of effect of treatment at day 30, responders with an improvement from baseline of at least 1 grade on the facial wrinkle scale (FWS) ‐ maximum smile, was 17.8 weeks (125 days) (physician assessment) and 20.5 weeks (144 days) (participant assessment) (Carruthers 2014). Harii 2017 showed a duration of treatment effect around 95 days. And Wu 2019 assessed the duration of onabotulinumtoxinA 24 U effect around 150‐157 by investigator assessment and 150‐157 by participant assessment.
COMPARISON 30. OnabotulinumtoxinA 12 units versus placebo, crow's feet lines
Two RCTs (n = 657 participants) assessed this comparison (Harii 2017, Wu 2019). Harii 2017 was a two‐phase study, so we considered only the first phase (randomised, double‐blind).
Primary outcome
Participant assessment of success by analysing scores and scales
Neither RCT assessed this outcome.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Harii 2017 assessed this outcome and there was no difference between groups was found, but the confidence interval was wide (RR 1.36, 95% CI 0.12 to 14.76; participants = 240; studies = 1) (Analysis 30.1).
30.1. Analysis.

Comparison 30: OnabotulinumtoxinA 12 units versus placebo one treatment in crow's feet lines, Outcome 1: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
Harii 2017 assessed this outcome and the results favoured onabotulinumtoxinA 12 U group at week 4 (RR 6.86, 95% CI 3.45 to 13.62; participants = 196; studies = 1); at week 8 (RR 4.29, 95% CI 2.10 to 8.76; participants = 196; studies = 1); and at week 12 (RR 5.55, 95% CI 1.68 to 18.34; participants = 196; studies = 1) (Analysis 30.2).
30.2. Analysis.

Comparison 30: OnabotulinumtoxinA 12 units versus placebo one treatment in crow's feet lines, Outcome 2: Physician assessment of success by analysing scores and scales
Total adverse events
Both studies reported the total adverse events and they were similar across groups (RR 1.33. 95% CI 0.87 to 2.02; participants= 657, studies=2, I2 = 0%) (Analysis 30.3).
30.3. Analysis.

Comparison 30: OnabotulinumtoxinA 12 units versus placebo one treatment in crow's feet lines, Outcome 3: Total adverse events
Duration of treatment effect
Harii 2017 described the results narratively. The duration of treatment effect for responders (none‐ mild at maximum smile) was 85 days in onabotulinumtoxinA 12 U group.
COMPARISON 31. OnabotulinumtoxinA 24 units versus onabotulinumtoxinA 12 units, crow's feet lines
One RCT (n = 294 participants) assessed this comparison (Harii 2017).
Harii 2017 was a two‐phase study, we considered only the first phase (randomised, double‐blind).
Primary outcomes
Participant assessment of success by analysing scores and scales
This RCT did not assess this outcome.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Harii 2017 assessed this outcome and there was no difference between onabotulinumtoxinA 24 U group and onabotulinumtoxinA 12 U group (RR 0.95, 95% CI 0.14 to 6.63; participants = 294; studies = 1) (Analysis 31.1).
31.1. Analysis.

Comparison 31: OnabotulinumtoxinA 24 units versus OnabotulinumtoxinA 12 units one treatment in crow's feet lines, Outcome 1: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
The analysis showed a difference in favour of onabotulinumtoxinA 24 U group than onabotulinumtoxinA 12 U group at week 12 (RR 1.90, 95% CI 1.14 to 3.18; participants = 203; studies = 1); but there was a similar response between groups at week 4 (RR 1.21, 95% CI 0.97 to 1.50; participants = 203; studies = 1); and at week 8 (RR 1.36, 95% CI 0.97 to 1.90; participants = 203; studies = 1) (Analysis 31.2).
31.2. Analysis.

Comparison 31: OnabotulinumtoxinA 24 units versus OnabotulinumtoxinA 12 units one treatment in crow's feet lines, Outcome 2: Physician assessment of success by analysing scores and scales
Total adverse events
In Harii 2017, the total adverse events were similar in both groups (RR 0.86, 95% CI 0.59 to 1.25; participants = 294; studies = 1) (Analysis 31.3).
31.3. Analysis.

Comparison 31: OnabotulinumtoxinA 24 units versus OnabotulinumtoxinA 12 units one treatment in crow's feet lines, Outcome 3: Total adverse events
Duration of treatment effect
Harii 2017 described the results narratively. The duration of treatment effect for responders (none‐ mild at maximum smile) was 85 days in onabotulinumtoxinA 12 U group and 95 days in onabotulinumtoxinA 24 U group.
COMPARISON 32. OnabotulinumtoxinA 44 units versus placebo one cycle of treatment, glabellar lines and crow's feet lines
Three RCTs (n = 808 participants) assessed this comparison (Carruthers 2015; Moers‐Carpi 2015; Rivers 2015).
Primary outcomes
Participant assessment of success by analysing scores and scales
The meta‐analysis showed a benefit with onabotulinumtoxinA 44 U at week 4 (RR 16.33, 95% CI 9.27 to 28.76; participants = 808; studies = 2; I² = 0%) (Analysis 32.1).
32.1. Analysis.

Comparison 32: OnabotulinumtoxinA 44 units versus placebo one treatments in glabellar lines and crow's feet lines, Outcome 1: Participant assessment of success by analysing scores and scales
Only one RCT assessed success in the following weeks (Moers‐Carpi 2015). The responder rate by participant assessment was higher with onabotulinumtxinA 44 U than placebo at week 8 (RR 8.95, 95% CI 5.03 to 15.90; participants = 611; studies = 1); at week 12 (RR 5.10, 95% CI 2.80 to 9.28; participants = 611; studies = 1); and at week 16 (RR 2.51, 95% CI 1.31 to 4.81; participants = 611; studies = 1) (Analysis 32.1).
Rivers 2015 did not report this outcome.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
These RCTs did not report any major event (Moers‐Carpi 2015; Rivers 2015). One RCT did not assess this outcome (Carruthers 2015).
Secondary outcomes
Physician assessment of success by analysing scores and scales
Two RCTs (Carruthers 2015; Moers‐Carpi 2015) studied this outcome. The meta‐analysis showed benefit with onabotulinumtoxinA 44 U at week 4 (RR 11.09, 95% CI 4.12 to 29.83; n = 808; studies = 2; I² = 80%); and week 8 (RR 9.94, 95% CI 1.78 to 55.44; n = 808; studies = 2; I² = 91%). However, a less certain difference was observed at week 12 due to the wide confidence interval (RR 5.96, 95% CI 0.60 to 58.98; n = 808) (Analysis 32.2).
32.2. Analysis.

Comparison 32: OnabotulinumtoxinA 44 units versus placebo one treatments in glabellar lines and crow's feet lines, Outcome 2: Physician assessment of success by analysing scores and scales
One study (Rivers 2015), assessed this outcome separately. At week 4, the responders rate (score of none or mild on the glabellar lines (G)L facial wrinkle scale (FWS) compared with the placebo group‐FWS) was 50/60 (83.3%) of participants in the onabotulinumtoxinA group, and 1/57(1.8%) in placebo group. The improvement of at least one point was 52/60 (86.7%) in the onabotulinumtoxinA group and 5/57 (8.8%) in placebo group. At week 12, 40/60 (66%) of participants in onabotulinumtoxinA group were responders.
Total adverse events
Carruthers 2015 and Moers‐Carpi 2015 reported total adverse events; however, the authors showed the number of adverse events per group of intervention not per cycle of treatment.
For this reason (one study), we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plot.
One study assessed this outcome (Rivers 2015), total adverse events were similar in both groups (RR 1.16, 95% CI 0.56 to 2.40; participants = 125; studies = 1) (Analysis 32.3).
32.3. Analysis.

Comparison 32: OnabotulinumtoxinA 44 units versus placebo one treatments in glabellar lines and crow's feet lines, Outcome 3: Total adverse events
Duration of treatment effect
These RCTs did not assess this outcome.
COMPARISON 33. OnabotulinumtoxinA 44 units versus placebo two cycles of treatment, glabellar lines and crow's feet lines
Two RCTs (n = 808 participants) assessed this comparison (Carruthers 2015; Moers‐Carpi 2015).
Primary outcomes
Participant assessment of success by analysing scores and scales
The meta‐analysis showed a benefit with onabotulinumtoxinA 44 U than placebo at week 4 (RR 10.12, 95% CI 6.36 to 16.09; participants = 808; studies = 2; I² = 0%) (Analysis 33.1).
33.1. Analysis.

Comparison 33: OnabotulinumtoxinA 44 units versus placebo two cycles of treatments in crow's feet lines, Outcome 1: Participant assessment of success by analysing scores and scales
One RCT (Moers‐Carpi 2015) assessed responder rate at week 8 (RR 8.95, 95% CI 5.03 to 15.90; participants = 611; studies = 1), and at week 12 (RR 4.85, 95% CI 2.66 to 8.84; participants = 611; studies = 1), finding a benefit favouring onabotulinumtoxinA 44 U (Analysis 33.1).
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Two RCTs assessed this outcome (Moers‐Carpi 2015; Carruthers 2015). These RCTs did not report any major event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The meta‐analysis showed a benefit with onabotulinumtoxinA 44 U at week 4 (RR 17.63, 95% CI 9.50 to 32.69; participants = 808; studies = 2; I² = 19%), and week 8 (RR 19.86, 95% CI 9.90 to 39.87; participants = 808; studies = 2; I² = 0%) (Analysis 33.2).
33.2. Analysis.

Comparison 33: OnabotulinumtoxinA 44 units versus placebo two cycles of treatments in crow's feet lines, Outcome 2: Physician assessment of success by analysing scores and scales
Total adverse events
The meta‐analysis showed no difference between OnabotulinumtoxinA 44 U and placebo (RR 1.13, 95% CI 0.98 to 1.30; n = 808; studies = 2; I² = 0%) (Analysis 33.3).
33.3. Analysis.

Comparison 33: OnabotulinumtoxinA 44 units versus placebo two cycles of treatments in crow's feet lines, Outcome 3: Total adverse events
Duration of treatment effect
These RCTs did not assess this outcome.
COMPARISON 34. OnabotulinumtoxinA 24 units versus 12 units five cycles of treatment, crow's feet lines
Only one RCT (n = 300 participants) assessed this comparison (Harii 2017). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
The authors considered only one point of improvement as a cut‐off value. For this reason, we did not analyse this outcome.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not show any major adverse events.
Secondary outcomes
Physician assessment of success by analysing scores and scales
This RCT did not assess this outcome.
Total adverse events
The total adverse events rate was higher with onabotulinumtoxinA 24 U than onabotulinumtoxinA 12 U (RR 1.42, 95% CI 0.41 to 4.93; participants = 294; studies = 1) (Analysis 34.1).
34.1. Analysis.

Comparison 34: OnabotulinumtoxinA 24 units versus 12 units five cycles of treatment in crow's feet lines, Outcome 1: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 35. AbobotulinumtoxinA 25 units versus placebo one cycle of treatment, glabellar lines
Only one RCT (n = 45 participants) assessed this comparison (Ascher 2004). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
This RCT did not assess this outcome.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus
This RCT did not show any major adverse event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responders' rates, by physician assessment, were higher with abobotulinumtoxinA 25 U than placebo at week 4 (RR 7.50, 95% CI 1.09 to 51.52; participants = 45; studies = 1); at week 8(RR 17.03, 95% CI 1.09 to 265.91; participants = 45; studies = 1), at week 12 (RR 10.84, 95% CI 0.68 to 173.34; participants = 45; studies = 1); and at week 24 (RR 4.65, 95% CI 0.27 to 81.01; participants = 45; studies = 1) (Analysis 35.1).
35.1. Analysis.

Comparison 35: AbobotulinumtoxinA 25 units versus placebo one cycle of treatment, glabellar lines, Outcome 1: Physician assessment of success by analysing scores and scales
Total adverse events
This RCT showed more adverse events in abobotulinumtoxinA 25U than placebo (Peto OR 5.21, 95% CI 0.74 to 36.60; participants = 45; studies = 1) (Analysis 35.2).
35.2. Analysis.

Comparison 35: AbobotulinumtoxinA 25 units versus placebo one cycle of treatment, glabellar lines, Outcome 2: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 36. AbobotulinumtoxinA 30 units versus placebo one cycle of treatment, glabellar lines
One RCT (n = 99 participants) assessed this comparison (Rzany 2006). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plot.
Primary outcomes
Participant assessment of success by analysing scores and scales
This RCT did not assess this outcome.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder's rates, by physician assessment, were higher with abobotulinumtoxinA 30U than placebo at week 4: (RR 4.55, 95% CI 2.32 to 8.93; participants = 109; studies = 1) (Analysis 36.1).
36.1. Analysis.

Comparison 36: AbobotulinumtoxinA 30 units versus placebo one cycle of treatment in glabellar lines, Outcome 1: Physician assessment of success by analysing scores and scales
Total adverse events
This RCT did not report any adverse events.
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 37. AbobotulinumtoxinA 50 units versus placebo one cycle of treatment, glabellar lines
Nine RCTs (n = 1333 participants) assessed this comparison (Ascher 2004; Ascher 2005; Ascher 2018; Brandt 2009; Kane 2009; Monheit 2007; Monheit 2019; Rzany 2006; NCT02450526), and the results are included in Table 2.
Primary outcomes
Participant assessment of success by analysing scores and scales
The meta‐analysis (Ascher 2018; Brandt 2009; Kane 2009; Monheit 2019; NCT02450526) showed a benefit with abobotulinumtoxinA when compared with placebo at week 4 (RR 21.22, 95% CI 7.43 to 60.56, participants = 915; studies = 5; I2 = 54%, high‐certainty evidence); at week 8 (RR 39.74, 95% CI 14.04 to 112.44; participants = 714, studies = 3; I2 = 0%); and week 12 (RR 28.71, 95% CI 10.12 to 81.49; participants = 724; studies = 3; I2 = 0%, low‐certainty evidence) (Analysis 37.1).
37.1. Analysis.

Comparison 37: AbobotulinumtoxinA 50 units versus placebo one treatment glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Only Monheit 2019 assessed the following weeks: 16 week (RR 10.67, 95% CI 3.44 to 33.11; participants = 300; studies = 1); and at 20 weeks (RR 5.33, 95% CI 1.67 to 16.99; participants = 300; studies = 1) (Analysis 37.1).
Ascher 2004; Ascher 2005 did not assess this comparison, and Monheit 2007 and Rzany 2006 did not provide data.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
A meta‐analysis (Ascher 2004; Ascher 2005; Brandt 2009; Rzany 2006; Monheit 2007; Monheit 2019; NCT02450526) showed more adverse events in abobotulinum group when compared to placebo group (RR 3.36, 95% CI 0.88 to 12.87; participants = 1294; studies = 7; I2 = 0%, moderate‐certainty evidence) (Analysis 37.2).
37.2. Analysis.

Comparison 37: AbobotulinumtoxinA 50 units versus placebo one treatment glabellar lines, Outcome 2: Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Kane 2009 and Ascher 2018 did not report this outcome.
Secondary outcomes
Physician assessment of success by analysing scores and scales
Ascher 2004; Ascher 2005; Ascher 2018; Brandt 2009; Kane 2009; Monheit 2019; NCT02450526 assessed this comparison. The meta‐analysis showed a benefit with abobotulinumtoxinA 50u than placebo at week 4 RR 15.78, 95% CI 8.75 to 28.45; participants = 1060; studies = 7; I2 = 7%; moderate‐certainty evidence); at week 8 (RR 30.84, 95% CI 11.58 to 82.12; participants = 802; studies = 5; I2 = 2%); at week 12 (RR 17.79, 95% CI 6.70 to 45.28; participants = 900; studies = 6; I2 = 29%); and at week 16 (RR 29.88, 95% CI 6.01 to 148.52; participants = 371; studies = 2; I2 = 0%) (Analysis 37.3).
37.3. Analysis.

Comparison 37: AbobotulinumtoxinA 50 units versus placebo one treatment glabellar lines, Outcome 3: Physician assessment of success by analysing scores and scales
Only Monheit 2019 assessed this outcome at 20 weeks (RR 17.00, 95% CI 2.36 to 122.39; participants = 300; studies = 1) (Analysis 37.3).
At 24 weeks in Ascher 2004, the responder rates were 13.8% in abobotulinumtoxinA group and 0% in placebo group. We did not perform a meta‐analysis because only one RCT showed data in this period (Ascher 2004).
Total adverse events
Ascher 2004; Ascher 2005; Ascher 2018; Brandt 2009; Monheit 2007; Rzany 2006; Monheit 2019; NCT02450526 showed data for this analysis. The total adverse events were higher with abobotulinumtoxinA than the placebo group (RR 1.25, 95% CI 1.05 to 1.49; participants = 1471; studies = 7; I2 = 0%, low‐certainty evidence) (Analysis 37.4).
37.4. Analysis.

Comparison 37: AbobotulinumtoxinA 50 units versus placebo one treatment glabellar lines, Outcome 4: Total adverse events
Kane 2009 did not report this outcome.
Duration of treatment effect
Ascher 2005 showed abobotulinumtoxinA reporting a longer duration of effect compared to placebo (MD 17.30, 95% CI 15.82 to 18.78; participants = 100; studies = 1; moderate certainty evidence) (Analysis 37.5).
37.5. Analysis.

Comparison 37: AbobotulinumtoxinA 50 units versus placebo one treatment glabellar lines, Outcome 5: Duration of treatment (days)
Brandt 2009 showed median duration of effect of BontA of 12 weeks (85 days).
Kane 2009 showed a duration of effect of 15.3 weeks (107 days) in BontA group.
Monheit 2019 showed median duration of effect of 117 days.
Ascher 2004; Ascher 2018; Monheit 2007; Rzany 2006 and NCT02450526 did not study this outcome.
COMPARISON 38. AbobotulinumtoxinA 50 units versus 25 units one cycle of treatment, glabellar lines
Only one RCT (n = 59 participants) assessed this comparison (Ascher 2004). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
This RCT did not assess this outcome.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus
This RCT did not show any major adverse events.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responders' rates, by physician assessment, were higher with abobotulinumtoxinA 50 U than abobotulinumtoxinA 25 U at week 4 (RR 1.52, 95% CI 1.00 to 2.29; participants = 59; studies = 1); at week 12 (RR 1.45, 95% CI 0.77 to 2.72; participants = 59; studies = 1), but there was no difference between abobotulinumtoxinA 50 U and abobotulinumtoxinA 25 U at week 8 (RR 1.03, 95% CI 0.65 to 1.65; participants = 59; studies = 1); and at week 24 (RR 1.03, 95% CI 0.29 to 3.75; participants = 59; studies = 1) (Analysis 38.1).
38.1. Analysis.

Comparison 38: AbobotulinumtoxinA 50 units versus 25 units one cycle of treatment, glabellar lines, Outcome 1: Physician assessment of success by analysing scores and scales
Total adverse events
No difference between groups was found, but the confidence interval was wide (Peto OR 0.59, 95% CI 0.13 to 2.58; participants = 59; studies = 1) (Analysis 38.2).
38.2. Analysis.

Comparison 38: AbobotulinumtoxinA 50 units versus 25 units one cycle of treatment, glabellar lines, Outcome 2: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 39. AbobotulinumtoxinA 60 units versus placebo one cycle of treatment, glabellar lines
Only one RCT (n = 423 participants) assessed this comparison (Kane 2009). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
The responders' rates, by participant assessment, were higher with abobotulinumtoxinA 60 U than placebo at week 4 (RR 11.48, 95% CI 6.50 to 20.29; participants = 423; studies = 1) (Analysis 39.1).
39.1. Analysis.

Comparison 39: AbobotulinumtoxinA 60 units versus placebo one cycle of treatment in glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major event by units.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder's rates, by physician assessment, were higher with abobotulinumtoxinA 60 U than placebo at week 4 (RR 15.98, 95% CI 8.14 to 31.36; participants = 423; studies = 1) (Analysis 39.2).
39.2. Analysis.

Comparison 39: AbobotulinumtoxinA 60 units versus placebo one cycle of treatment in glabellar lines, Outcome 2: Physician assessment of success by analysing scores and scales
Total adverse events
This RCT did not report any adverse events by units.
Duration of treatment effect
This RCT did not assess this outcome by units.
COMPARISON 40. AbobotulinumtoxinA 70 units versus placebo one cycle of treatment, glabellar lines
Only one RCT(n = 291 participants) assessed this comparison (Kane 2009). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
The responders' rates, by participant assessment, were higher with abobotulinumtoxinA 70 U than placebo however the confidence interval was very wide showing we are very uncertain with this result; at week 4 (RR 151.39, 95% CI 9.54 to 2402.95; participants = 291; studies = 1) (Analysis 40.1).
40.1. Analysis.

Comparison 40: AbobotulinumtoxinA 70 units versus placebo one cycle of treatment in glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major adverse events by units.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responders' rates, by physician assessment, were higher with abobotulinumtoxinA 70 U than placebo at week 4; however, the confidence interval is very wide showing uncertainty in the effect size (RR 71.36, 95% CI 10.15 to 501.56; participants = 289; studies = 1) (Analysis 40.2).
40.2. Analysis.

Comparison 40: AbobotulinumtoxinA 70 units versus placebo one cycle of treatment in glabellar lines, Outcome 2: Physician assessment of success by analysing scores and scales
Total adverse events
This RCT did not report any adverse events by units.
Duration of treatment effect
This RCT did not assess this outcome by units.
COMPARISON 41. AbobotulinumtoxinA 75 units versus placebo one cycle of treatment, glabellar lines
Only one RCT (n = 45 participants) assessed this comparison (Ascher 2004). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
This RCT did not assess this outcome.
Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus
This RCT did not show any major adverse events.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responders' rates, by physician assessment, were higher with abobotulinumtoxinA 75 U than placebo at week 4 (RR 11.50, 95% CI 1.71 to 77.18; participants = 45; studies = 1); at week 8 (RR 9.50, 95% CI 1.40 to 64.35; participants = 45; studies = 1); at week 12 (RR 16.00, 95% CI 1.02 to 250.48; participants = 45; studies = 1); and at week 24 (RR 3.61, 95% CI 0.20 to 65.73; participants = 45; studies = 1) (Analysis 41.1).
41.1. Analysis.

Comparison 41: AbobotulinumtoxinA 75 units versus placebo one cycle of treatment in glabellar lines, Outcome 1: Physician assessment of success by analysing scores and scales
Total adverse events
This RCT did not show any adverse events.
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 42. AbobotulinumtoxinA 75 units versus 25 units one cycle of treatment, glabellar lines
Only one RCT (n = 60 participants) assessed this comparison (Ascher 2004). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
This RCT did not assess this outcome.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus
This RCT did not show any major adverse event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responders' rates, by physician assessment, were higher with AaobotulinumtoxinA 75 U than abobotulinumtoxinA 25 U at week 4 (RR 11.50, 95% CI 2.97 to 44.51; participants = 60; studies = 1).
Although the responders' rates, by physician assessment, were higher with abobotulinumtoxinA 75 U than abobotulinumtoxinA 25 U, the results were very uncertain because of the wide confidence intervals: at week 8 (RR 39.00, 95% CI 2.46 to 617.81; participants = 60; studies = 1); at week 12 (RR 31.00, 95% CI 1.94 to 495.61; participants = 60; studies = 1); and at week 24 (RR 7.00, 95% CI 0.38 to 129.93; participants = 60; studies = 1) (Analysis 42.1).
42.1. Analysis.

Comparison 42: AbobotulinumtoxinA 75 units versus 25 units one cycle of treatment in glabellar lines, Outcome 1: Physician assessment of success by analysing scores and scales
Total adverse events
This RCT did not assess this outcome.
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 43. AbobotulinumtoxinA 75 units versus 50 units one cycle of treatment, glabellar lines
Only one RCT (n = 60 participants) assessed this comparison (Ascher 2004). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
This RCT did not show any major adverse event.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus
This RCT did not assess this outcome.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responders' rates, by physician assessment, did not show any difference with abobotulinumtoxinA 75 U or abobotulinumtoxinA 50 U at week 4 (RR 1.01, 95% CI 0.76 to 1.34; participants = 59; studies = 1); at week 8 (RR 1.15, 95% CI 0.75 to 1.76; participants = 59; studies = 1); at week 12 (RR 1.04, 95% CI 0.62 to 1.74; participants = 59; studies = 1); at week 24 (RR 0.72, 95% CI 0.18 to 2.96; participants = 59; studies = 1) (Analysis 43.1).
43.1. Analysis.

Comparison 43: AbobotulinumtoxinA 75 units versus 50 units one cycle of treatment in glabellar lines, Outcome 1: Physician assessment of success by analysing scores and scales
Total adverse events
This RCT did not show any difference between AbobotulinumtoxinA 75U and AbobotulinumtoxinA 50U (Peto OR 0.12, 95% CI 0.01 to 1.22; participants = 59; studies = 1) (Analysis 43.2).
43.2. Analysis.

Comparison 43: AbobotulinumtoxinA 75 units versus 50 units one cycle of treatment in glabellar lines, Outcome 2: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 44. AbobotulinumtoxinA 80 units versus placebo one cycle of treatment, glabellar lines
Only one RCT (n = 59 participants) assessed this comparison (Kane 2009). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
The responders' rates, by participant assessment, were higher with abobotulinumtoxinA 80 U than placebo at week 4; however, because of the confidence interval the results is very uncertain (RR 40.50, 95% CI 2.58 to 635.35; participants = 59; studies = 1) (Analysis 44.1).
44.1. Analysis.

Comparison 44: AbobotulinumtoxinA 80 units versus placebo one cycle of treatment in glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report major adverse events by units.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responders' rates, by physician assessment, were higher with abobotulinumtoxinA 8 0U than placebo at week 4; however, because of the confidence interval the results is very uncertain (RR 32.56, 95% CI 2.06 to 514.19; participants = 59; studies = 1) (Analysis 44.2).
44.2. Analysis.

Comparison 44: AbobotulinumtoxinA 80 units versus placebo one cycle of treatment in glabellar lines, Outcome 2: Physician assessment of success by analysing scores and scales
Total adverse events
This RCT did not report any adverse events by units.
Duration of treatment effect
This RCT did not assess this outcome by units.
COMPARISON 45. AbobotulinumtoxinA 15units versus placebo one cycle of treatment, crow's feet lines
Only one RCT(n = 109 participants) assessed this comparison (Ascher 2009). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
This RCT did not assess this outcome.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responders' rates, by physician assessment, were higher with abobotulinumtoxinA 15U versus placebo at week 4 (RR 4.52, 95% CI 1.85 to 11.01; participants = 109; studies = 1) (Analysis 45.1).
45.1. Analysis.

Comparison 45: AbobotulinumtoxinA 15 units versus placebo one cycle of treatment crow's feet lines, Outcome 1: Physician assessment of success by analysing scores and scales
Total adverse events
One RCT assessed this outcome (Ascher 2009). Adverse events frequency was 5/42 (13%) in abobotulinumtoxinA 15 U group, but no information was given for the placebo group.
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 46. AbobotulinumtoxinA 30 units versus placebo one cycle of treatment, crow's feet lines
Only one RCT (n = 108 participants) assessed this comparison (Ascher 2009). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
This RCT did not assess this outcome.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responders' rates, by physician assessment, were higher with abobotulinumtoxinA 30 U versus placebo week 4 (RR 6.40, 95% CI 2.70 to 15.18; participants = 108; studies = 1) (Analysis 46.1).
46.1. Analysis.

Comparison 46: AbobotulinumtoxinA 30 units versus placebo one cycle of treatment crow's feet lines, Outcome 1: Physician assessment of success by analysing scores and scales
Total adverse events
The adverse events frequency was 4/37(11%) in abobotulinumtoxinA 30U group, but no information was given for placebo.
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 47. AbobotulinumtoxinA 45units versus placebo one cycle of treatment, crow's feet lines
Only one RCT (n = 108 participants) assessed this comparison (Ascher 2009). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
This RCT did not assess this outcome.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
One case of eyelid ptosis in AbobotulinumtoxinA 45U was reported.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responders' rates, by physician assessment, were higher with abobotulinumtoxinA 4 5U versus placebo week 4 (RR 6.20, 95% CI 2.61 to 14.74; participants = 108; studies = 1) (Analysis 47.1).
47.1. Analysis.

Comparison 47: AbobotulinumtoxinA 45 units versus placebo one cycle of treatment crow's feet lines, Outcome 1: Physician assessment of success by analysing scores and scales
Total adverse events
The adverse events frequency was 5/40 (13%) in abobotulinumtoxinA 45 U, but no information was given for the placebo group.
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 48. AbobotulinumtoxinA 50 units versus placebo three cycles of treatment, glabellar lines
Only one RCT (n = 142 participants) assessed this comparison (Rubin 2009). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
The responders'rates, by participant assessment, were higher with abobotulinumtoxinA 50 U versus placebo week 4 in the third cycle (RR 8.00, 95% CI 3.92 to 16.33; participants = 142; studies = 1) (Analysis 48.1).
48.1. Analysis.

Comparison 48: AbobotulinumtoxinA 50 units versus placebo, three cycles of treatment in glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responders' rates, by physician assessment, were higher with AbobotulinumtoxinA 50U versus placebo week 4 in the third cycle (RR 20.00, 95% CI 6.58 to 60.80; participants = 142; studies = 1) (Analysis 48.2).
48.2. Analysis.

Comparison 48: AbobotulinumtoxinA 50 units versus placebo, three cycles of treatment in glabellar lines, Outcome 2: Physician assessment of success by analysing scores and scales
Total adverse events
No difference between groups was found, but the confidence interval was wide at week 4 in the third cycle (RR 1.29; 95% CI 0.81 to 2.05; participants = 142; studies = 1) (Analysis 48.3).
48.3. Analysis.

Comparison 48: AbobotulinumtoxinA 50 units versus placebo, three cycles of treatment in glabellar lines, Outcome 3: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 49. IncobotulinumtoxinA 20 units versus placebo one cycle of treatment, glabellar lines
Two RCTs (n = 547 participants) assessed this comparison (Carruthers 2013; Hanke 2013), and the results are included in Table 3.
Primary outcomes
Participant assessment of success by analysing scores and scales.
The meta analysis showed a benefit favours incobotulinumtoxinA at week 4 (RR 66.57, 95% CI 13.50 to 328.28; participants = 547; studies = 2; I² = 0%); week 8 (RR 7.35, 95% CI 4.79 to 11.29; participants = 547; studies = 2; I² = 0%); week 12 (RR 7.29, 95% CI 4.38 to 12.13; participants = 547; studies = 2; I² = 0%); and week 16 (RR 4.40, 95% CI 2.61 to 7.41; participants = 547; studies = 2; I² = 0%) (moderate‐certainty evidence) (Analysis 49.1).
49.1. Analysis.

Comparison 49: IncobotulinumtoxinA 20 units versus placebo one treatment glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
These RCTs did not report any major event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The meta‐analysis showed a benefit favouring incobotulinumtoxinA at week 4 (RR 134.62, 95% CI 19.05 to 951.45; participants = 547; studies = 2; I² = 0%), but the confidence interval was very wide showing uncertainty (Analysis 49.2). No studies assessed this outcome at other time points (moderate‐certainty evidence).
49.2. Analysis.

Comparison 49: IncobotulinumtoxinA 20 units versus placebo one treatment glabellar lines, Outcome 2: Physician assessment of success by analysing scores and scales
Total adverse events
The meta‐analysis did not show any clear difference between intervention groups (RR 1.17, 95% CI 0.90 to 1.53; participants = 547; studies = 2; I² = 0%, low‐certainty evidence) (Analysis 49.3).
49.3. Analysis.

Comparison 49: IncobotulinumtoxinA 20 units versus placebo one treatment glabellar lines, Outcome 3: Total adverse events
Duration of treatment effect
These RCTs did not assess this outcome.
COMPARISON 50. IncobotulinumtoxinA 54 to 64 units versus placebo one cycles of treatment, glabellar lines, forehead lines, crow's feet lines
Only one RCT (n = 156 participants) assessed this comparison (Kerscher 2015). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
This RCT did not assess this outcome.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
The major adverse event were higher with incotulinumtoxinA 54 U to 64 U (one unilateral blepharoptosis and the other one bilateral blepharoptosis) and placebo (dry eyes) ,but the RR is uncertain (RR 0.49; 95% CI 0.07 to 3.35; n = 156) (Analysis 50.1).
50.1. Analysis.

Comparison 50: IncobotulinumtoxinA 54 to 64units versus placebo one cycles of treatment glabellar lines, forehead liens, crow's feet lines, Outcome 1: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by physician assessment, was higher with IncobotulinumtoxinA than placebo at week 4 in glabellar lines ((GL) RR 87.81, 95% CI 5.56 to 1386.93; participants = 156; studies = 1); in forehead lines (RR 35.94, 95% CI 5.14 to 251.27; participants = 156; studies = 1); and in crow's feet lines (RR 32.54, 95% CI 4.65 to 227.82; participants = 156; studies = 1) (Analysis 50.2). However, because of the wide confidence intervals the results are very uncertain.
50.2. Analysis.

Comparison 50: IncobotulinumtoxinA 54 to 64units versus placebo one cycles of treatment glabellar lines, forehead liens, crow's feet lines, Outcome 2: Physician assessment of success by analysing scores and scales
Total adverse events
The frequency of total adverse events were similar with IncobotulinumtoxinA and placebo (RR 1.13, 95% CI 0.84 to 1.51; participants = 156; studies = 1) (Analysis 50.3).
50.3. Analysis.

Comparison 50: IncobotulinumtoxinA 54 to 64units versus placebo one cycles of treatment glabellar lines, forehead liens, crow's feet lines, Outcome 3: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 51. HBTX‐A 10units versus placebo one cycle of treatment, glabellar lines
Only one RCT (n = 305 participants) assessed this comparison (Feng 2015). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales.
The responder rate, by participant assessment, was higher with HBTX‐A 10 U than placebo at week 4 (RR 14.78, 95% CI 6.74 to 32.41; participants = 305; studies = 1; at week 8 (RR 13.78, 95% CI 6.27 to 30.25; participants = 305; studies = 1); and at week 16 (RR 9.44, 95% CI 4.26 to 20.93; participants = 305; studies = 1) (Analysis 51.1).
51.1. Analysis.

Comparison 51: HBTX‐A 10 units versus placebo one cycle of treatment in glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major adverse event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
This RCT did not study this outcome.
Total adverse events
One RCT assessed this outcome (Feng 2015), the total adverse events were higher with HBTX‐A 10 U than placebo (RR 4.95, 95% CI 2.33 to 10.54; participants = 305; studies = 1) (Analysis 51.2).
51.2. Analysis.

Comparison 51: HBTX‐A 10 units versus placebo one cycle of treatment in glabellar lines, Outcome 2: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 52. HBTX‐A 20 units versus placebo one cycle of treatment, glabellar lines
One RCT (n = 305 participants) assessed this comparison (Feng 2015).
For this reason (one study), we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcome
Participant assessment of success by analysing scores and scales.
The responder's rate, by participant assessment, was higher with HBTX‐A 20 U than placebo at week 4 (RR 18.33, 95% CI 8.39 to 40.06; participants = 305; studies = 1); at week 8 (RR 17.00, 95% CI 7.77 to 37.19; participants = 305; studies = 1); and at week 16 (RR 15.22, 95% CI 6.95 to 33.36; participants = 305; studies = 1) (Analysis 52.1).
52.1. Analysis.

Comparison 52: HBTX‐A 20 units versus placebo one cycle of treatment in glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major adverse event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
This RCT did not assess this outcome.
Total adverse events
The total adverse events were higher with HBTX‐A 20 U than placebo (RR 4.67; 95% CI 2.19 to 9.96; participants = 305; studies = 1) (Analysis 52.2).
52.2. Analysis.

Comparison 52: HBTX‐A 20 units versus placebo one cycle of treatment in glabellar lines, Outcome 2: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 53. HBTX‐A 50 units versus placebo one cycle of treatment, glabellar lines
One RCT (n = 190 participants) assessed this comparison (NCT02493946).
For this reason (one study), we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate, by participant assessment, was higher with HBTX‐A 50 U than placebo at week 4 (RR 42.68, 95% CI 6.08 to 299.41; participants = 187; studies = 1); at week 8 (RR 43.28, 95% CI 6.18 to 303.19; participants = 183; studies = 1); and at week 12 (RR 21.67, 95% CI 3.06 to 153.72; participants = 185; studies = 1) (Analysis 53.1).
53.1. Analysis.

Comparison 53: HBTX‐A 50 units versus placebo one cycle of treatment, glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major adverse event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by physician assessment, was higher with HBTX‐A 50 U than placebo at week 4 (RR 49.69; 95% CI 7.10 to 347.71, participants = 185, studies = 1); at week 8 (RR 53.52; 95% CI 7.65 to 376.34, participants = 167, studies =1); at week 12 (RR 33.17; 95% CI 4.72 to 233.16, participants = 182, studies = 1 (Analysis 53.2).
53.2. Analysis.

Comparison 53: HBTX‐A 50 units versus placebo one cycle of treatment, glabellar lines, Outcome 2: Physician assessment of success by analysing scores and scales
Total adverse events
The total adverse event was similar with HBTX‐A 50 U and placebo (Peto OR 0.22, 95% CI 0.02 to 2.50; participants = 190, studies = 10) (Analysis 53.3).
53.3. Analysis.

Comparison 53: HBTX‐A 50 units versus placebo one cycle of treatment, glabellar lines, Outcome 3: Total adverse events
Duration of treatment effect
This RCT reported only HBTX‐A 50 U mean duration, which was 113 days.
COMPARISON 54. HBTX‐A 20 units versus 10 units one cycle of treatment, glabellar lines
One RCT (n = 366 participants) assessed this comparison (Feng 2015).
For this reason (one study), we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales.
The responder rate, by participant assessment, was higher with HBTX‐A 20 U than HBTX‐A 10 U at week 4 (RR 1.24, 95% CI 1.12 to 1.37; participants = 366; studies = 1); at week 8 (RR 1.23, 95% CI 1.10 to 1.39; participants = 366; studies = 1); and at week 16 (RR 1.61, 95% CI 1.35 to 1.92; participants = 366; studies = 1) (Analysis 54.1).
54.1. Analysis.

Comparison 54: HBTX‐A 20 units versus 10 units one cycle of treatment in glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major adverse event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
This RCT did not assess this outcome.
Total adverse events
The total adverse event was similar with HBTX‐A 20 U and HBTX‐A 10 U (RR 0.94, 95% CI 0.68 to 1.31; participants = 366; studies = 1) (Analysis 54.2).
54.2. Analysis.

Comparison 54: HBTX‐A 20 units versus 10 units one cycle of treatment in glabellar lines, Outcome 2: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 55. AbobotulinumtoxinA 50 units versus OnabotulinumtoxinA 20 units one cycle of treatment, glabellar lines
Two RCTs (n = 479 participants) assessed this comparison (Lowe 2006; NCT02450526). The results are included in Table 4.
Primary outcomes
Participant assessment of success by analysing scores and scales
Only NCT02450526 assessed this outcome, n = 388.
The responder rate, by participant's assessment, was similar with abobotulinumtoxinA 50 U and onabotulinumtoxinA 20 U at week 4 (RR 1.00, 95% CI 0.92 to 1.08; participants 388, study = 1, high‐certainty evidence); at week 8 (RR 0.96, 95% CI 0.88 to 1.05; participants 388, study =1 ; at week 12 (RR 0.94, 95% CI 0.81 to 1.09; participants 388, study = 1).
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Only one study assessed this outcome (NCT02450526).
The frequency of major adverse events was higher in abobotulinumtoxinA 50 U when compared to onabotulinumtoxinA 20 U (Peto OR 2.65, 95% CI 0.77 to 9.09; participants = 433, study = 1, moderate‐certainty evidence) (Analysis 55.2).
55.2. Analysis.

Comparison 55: AbobotulinumtoxinA 50 units versus OnabotulinumtoxinA 20 units one cycle of treatment in glabellar lines, Outcome 2: Major adverse events
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by physician's assessment, was similar with abobotulinumtoxinA 50 U and onabotulinumtoxinA 20 U at week 4 (RR 1.01, 95% CI 0.95 to 1.06; participants = 388, studies = 1, high‐certainty evidence); at week 8 (RR 0.95, 95% CI 0.89 to 1.02; participants = 449, studies = 2, I2 = 0% moderate‐certainty evidence); at week 12 (RR 0.92, 95% CI 0.60 to 1.40; participants = 488; studies = 2, I2 = 55%); and at week 16 (RR 0.44, 95% CI 0.13 to 1.55; participants = 59; studies = 1) the RR favours the onabotulinumtoxinA 20 U, but the results are uncertain due to the confidence intervals including 1 (Analysis 55.3).
55.3. Analysis.

Comparison 55: AbobotulinumtoxinA 50 units versus OnabotulinumtoxinA 20 units one cycle of treatment in glabellar lines, Outcome 3: Physician assessment of success by analysing scores and scales
Total adverse events
The frequency of total adverse events was similar with abobotulinumtoxinA 50 U and onabotulinumtoxinA 20 U (RR 1.02, 95% CI 0.67 to 1.54; participants = 492; studies = 1, moderate‐certainty evidence) (Analysis 55.4).
55.4. Analysis.

Comparison 55: AbobotulinumtoxinA 50 units versus OnabotulinumtoxinA 20 units one cycle of treatment in glabellar lines, Outcome 4: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 56. IncobotulinumtoxinA 30 units versus OnabotulinumtoxinA 20 units one cycle of treatment, glabellar lines
Only one RCT (n = 224 participants) assessed this comparison (Moers‐Carpi 2012). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate, by participant assessment, was similar with IncobotulinumtoxinA 30 U and OnabotulinumtoxinA 20 U at week 4 (RR 0.90, 95% CI 0.78 to 1.04; participants = 224; studies = 1); at week 12 (RR 0.84, 95% CI 0.65 to 1.09; participants = 224; studies = 1); at week 14 (RR 0.93, 95% CI 0.68 to 1.29; participants = 224; studies = 1); and at week 16 (RR 0.94, 95% CI 0.63 to 1.40; participants = 224; studies = 1) (Analysis 56.1).
56.1. Analysis.

Comparison 56: IncobotulinumtoxinA 30 units versus OnabotulinumtoxinA 20 units one cycle of treatment in glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by physician assessment, was similar with IncobotulinumtoxinA 30 U and onabotulinumtoxinA 20 U at week 4 (RR 0.98, 95% CI 0.93 to 1.04; participants = 224; studies = 1); at week 12 (RR 0.90, 95% CI 0.77 to 1.04; participants = 224; studies = 1); at week 14 (RR 0.96, 95% CI 0.79 to 1.16; participants = 224; studies = 1); and at week 16 (RR 0.90, 95% CI 0.70 to 1.16; participants = 224; studies = 1) (Analysis 56.2).
56.2. Analysis.

Comparison 56: IncobotulinumtoxinA 30 units versus OnabotulinumtoxinA 20 units one cycle of treatment in glabellar lines, Outcome 2: Physician assessment of success by analysing scores and scales
Total adverse events
One RCT assessed this outcome (Moers‐Carpi 2012); the total adverse events were higher with incobotulinumtoxinA 30 U compared with onabotulinumtoxinA 20 U, but the result is uncertain due to the low number of events (two events versus one event) (RR 1.96, 95% CI 0.20 to 19.03, participants = 224; studies = 1 (Analysis 56.3).
56.3. Analysis.

Comparison 56: IncobotulinumtoxinA 30 units versus OnabotulinumtoxinA 20 units one cycle of treatment in glabellar lines, Outcome 3: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 57. IncobotulinumtoxinA 24 units versus onabotulinumtoxinA 24 units one cycle of treatment, glabellar lines
Only one RCT (n = 381 participants) assessed this comparison (Satler 2010). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots. Results are also shown in Table 5.
Primary outcomes
Participant assessment of success by analysing scores and scales
This RCT did not assess this outcome.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Ptosis was reported in one participant in onabotulinumtoxinA group (OR 0.02 95% CI 0.00 to 1.77; participants = 381; studies = 1, very low‐certainty evidence) (Analysis 57.1).
57.1. Analysis.

Comparison 57: IncobotulinumtoxinA 24 units versus OnabotulinumtoxinA 24 units one treatment in glabellar lines, Outcome 1: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by physician assessment, was similar with incobotulinumtoxinA 24 U and onabotulinumtoxinA 24 U at week 4 (RR 1.01, 95% CI 0.96 to 1.05; participants = 381; studies = 1); and at week 12 (RR 1.02, 95% CI 0.91 to 1.15; participants = 381; studies = 1) (moderate‐certainty evidence) (Analysis 57.2).
57.2. Analysis.

Comparison 57: IncobotulinumtoxinA 24 units versus OnabotulinumtoxinA 24 units one treatment in glabellar lines, Outcome 2: Physician assessment of success by analysing scores and scales
Total adverse events
There was only one adverse event in the onabotulinumtoxinA 24 U group and zero events in the IncobotulinumtoxinA 24 U group (OR 0.02, 95% CI 0.00 to 1.77; participants = 381; studies = 1) (very low‐certainty evidence) (Analysis 57.3).
57.3. Analysis.

Comparison 57: IncobotulinumtoxinA 24 units versus OnabotulinumtoxinA 24 units one treatment in glabellar lines, Outcome 3: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 58. IncobotulinumtoxinA 20 units versus OnabotulinumtoxinA 20 units one cycle of treatment, glabellar lines
Two RCTs (n = 250 participants) assessed this comparison (Kane 2015; Rappl 2013).
Primary outcomes
Participant assessment of success by analysing scores and scales
These RCTs did not assess this outcome.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Rappl 2013 did not report any major adverse event. One RCT studied this outcome (Kane 2015). No difference between groups (Peto OR 0.14, 95% CI 0.01 to 2.26; participants = 250; studies = 1) (Analysis 58.1).
58.1. Analysis.

Comparison 58: IncobotulinumtoxinA 20 units versus OnabotulinumtoxinA 20 units one treatment in glabellar lines, Outcome 1: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
Rappl 2013 did not assess this outcome.
Only one RCT studied this outcome (Kane 2015). The responder rate, by independent observer assessment, was similar with incobotulinumtoxinA 20 U and onabotulinumtoxinA 20 U at week 4 (RR 0.98, 95% CI 0.92 to 1.04; participants = 250; studies = 1); at week 8 (RR 0.97, 95% CI 0.88 to 1.06; participants = 250; studies = 1); at week 12 (RR 0.97, 95% CI 0.85 to 1.12; participants = 250; studies = 1); and at week 16 (RR 0.98, 95% CI 0.80 to 1.20; participants = 250; studies = 1) (Analysis 58.2).
58.2. Analysis.

Comparison 58: IncobotulinumtoxinA 20 units versus OnabotulinumtoxinA 20 units one treatment in glabellar lines, Outcome 2: Physician assessment of success by analysing scores and scales ‐ injector
The responder rate, by physician assessment, was similar with incobotulinumtoxinA 20 U and onabotulinumtoxinA 20 U at week 4 (RR 0.97, 95% CI 0.93 to 1.01; participants = 250; studies = 1); at week 8 (RR 0.96, 95% CI 0.90 to 1.03; participants = 250; studies = 1); at week 12 (RR 1.00, 95% CI 0.88 to 1.13; participants = 250; studies = 1); and at week 16 (RR 0.93, 95% CI 0.77 to 1.11; participants = 250; studies = 1) (Analysis 58.3).
58.3. Analysis.

Comparison 58: IncobotulinumtoxinA 20 units versus OnabotulinumtoxinA 20 units one treatment in glabellar lines, Outcome 3: Physician assessment of success by analysing scores and scales ‐ independent observer
Total adverse events
Rappl 2013 did not report total adverse events. One RCT studied this outcome (Kane 2015). The frequency of total adverse events was similar with incobotulinumtoxinA 20 U and onabotulinumtoxinA 20 U (RR 1.05, 95% CI 0.41 to 2.71; participants = 250; studies = 1) (Analysis 58.4).
58.4. Analysis.

Comparison 58: IncobotulinumtoxinA 20 units versus OnabotulinumtoxinA 20 units one treatment in glabellar lines, Outcome 4: Total adverse events
Duration of treatment effect
One RCT assessed this outcome by gender (Rappl 2013). The duration of treatment effect was 20.8 weeks (146.12 days) for female and 17.3 weeks (121.14 days) for male in incobotulinumtoxinA group.The duration of treatment effect was 20 weeks (140.65 days for female and 16.6 weeks (116.61 days) for male) in onabotulinumtoxinA group. After 180 days, four in the incobotulinumtoxinA group, and two in onabotulinumtoxinA, still showed an effect. All of these participants were botulinum toxin treatment naïve, and all had mild glabellar frown lines at baseline.
Kane 2015 did not study this outcome.
COMPARISON 59. NewBontA [Medytox®] 20 units versus OnabotulinumtoxinA 20 units one cycle of treatment, glabellar lines
Only one RCT (n = 291 participants) assessed this comparison (Lee 2013). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
This RCT did not assess this outcome.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by physician assessment, was similar with NewBontA 20U and onabotulinumtoxinA 20 U (RR 0.99, 95% CI 0.94 to 1.05; participants = 291; studies = 1) (Analysis 59.1).
59.1. Analysis.

Comparison 59: NewBontA [Medytox®] 20 units versus OnabotulinumtoxinA 20 units one treatment in glabellar lines, Outcome 1: Physician assessment, maximum contraction (responder rate)
Total adverse events
This RCT did not report any adverse event.
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 60. NewBontA [Neuronox®] 20 units versus OnabotulinumtoxinA 20 units one cycle of treatment, glabellar lines
Only one RCT (n = 314 participants) assessed this comparison (Won 2013). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
One RCT assessed this outcome (Won 2013) where the responder rate, by participant assessment, was similar with NewBontA 20 U and onabotulinumtoxinA 20 U at week 4 (RR 0.94, 95% CI 0.87 to 1.02; participants = 314; studies = 1); at week 8 (RR 0.96, 95% CI 0.88 to 1.04; participants = 314; studies = 1); at week 12 (RR 1.02, 95% CI 0.90 to 1.14; participants = 314; studies = 1); and at week 16 (RR 1.07, 95% CI 0.92 to 1.24; participants = 314; studies = 1) (Analysis 60.1).
60.1. Analysis.

Comparison 60: NewBontA [Neuronox®] 20 units versus OnabotulinumtoxinA 20 units one treatment in glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
The frequency of eyelid was similar with NewBontA 20U and onabotulinumtoxinA 20 U (RR 0.86, 95% CI 0.30 to 2.51; participants = 313; studies = 1) (Analysis 60.2).
60.2. Analysis.

Comparison 60: NewBontA [Neuronox®] 20 units versus OnabotulinumtoxinA 20 units one treatment in glabellar lines, Outcome 2: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by physician assessment, was similar with NewBontA 20 U and onabotulinumtoxinA 20 U at week 4 (RR 0.99, 95% CI 0.94 to 1.05; participants = 314; studies = 1); at week 8 (RR 1.01, 95% CI 0.92 to 1.10; participants = 314; studies = 1); at week 12 (RR 1.06, 95% CI 0.93 to 1.22; participants = 314; studies = 1); and at week 16 (RR 0.95, 95% CI 0.75 to 1.20; participants = 314; studies = 1) (Analysis 60.3).
60.3. Analysis.

Comparison 60: NewBontA [Neuronox®] 20 units versus OnabotulinumtoxinA 20 units one treatment in glabellar lines, Outcome 3: Physician assessment of success by analysing scores and scales
Total adverse events
The frequency of adverse events was similar with NewBontA 20 U and onabotulinumtoxinA 20 U (RR 1.21, 95% CI 0.82 to 1.78; participants = 313; studies = 1) (Analysis 60.4).
60.4. Analysis.

Comparison 60: NewBontA [Neuronox®] 20 units versus OnabotulinumtoxinA 20 units one treatment in glabellar lines, Outcome 4: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 61. NewBontA [Neuronox®] 24 units versus OnabotulinumtoxinA 24 units one cycle of treatment, crow's feet lines
Only one RCT (n = 220 participants) assessed this comparison (Cheon 2019). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
One RCT assessed this outcome (Cheon 2019). The responder rate, by participant assessment, was similar with NewBontA 24 U and onabotulinumtoxinA 24 U at week 4 (RR 1.01, 95% CI 0.89 to 1.15; participants = 220; studies = 1); at week 8 (RR 0.93, 95% CI 0.83 to 1.03; participants = 220; studies = 1); at week 12 (RR 1.02, 95% CI 0.90 to 1.16; participants = 220; studies = 1); at week 16 (RR 0.84, 95% CI 0.70 to 1.01; participants = 220; studies = 1) (Analysis 61.1).
61.1. Analysis.

Comparison 61: NewBontA [Neuronox®] 20 units versus OnabotulinumtoxinA 20 units one treatment in crow’s feet lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
No difference between groups (Peto OR 7.39, 95% CI 0.15 to 372.38); participants = 220; studies = 1) (Analysis 61.2).
61.2. Analysis.

Comparison 61: NewBontA [Neuronox®] 20 units versus OnabotulinumtoxinA 20 units one treatment in crow’s feet lines, Outcome 2: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by physician assessment, was similar with NewBontA 24U 24 U and onabotulinumtoxinA 24 U at week 4 (RR 1.00, 95% CI 0.89 to 1.13; participants = 220; studies = 1); at week 8 (RR 0.98, 95% CI 0.86 to 1.11; participants = 220; studies = 1); at week 12 (RR 1.08, 95% CI 0.88 to 1.32; participants = 220; studies = 1); at week 16 (RR 1.14, 95% CI 0.83 to 1.56; participants = 220; studies = 1) (Analysis 61.3).
61.3. Analysis.

Comparison 61: NewBontA [Neuronox®] 20 units versus OnabotulinumtoxinA 20 units one treatment in crow’s feet lines, Outcome 3: Physician assessment of success by analysing scores and scales
Total adverse events
The frequency of adverse events was similar with NewBontA 24Us and onabotulinumtoxinA 24 U (RR 0.97, 95% CI 0.65 to 1.45; participants = 220; studies = 1) (Analysis 61.4).
61.4. Analysis.

Comparison 61: NewBontA [Neuronox®] 20 units versus OnabotulinumtoxinA 20 units one treatment in crow’s feet lines, Outcome 4: Total adverse events
Duration of treatment effect
The median duration of the treatment effect was similar in both groups (112 days after week 4).
COMPARISON 62. Liquid BontA (MT10109L) 20 units versus OnabotulinumtoxinA 20 units one cycle of treatment, glabellar lines
Only one RCT (n = 159 participants) assessed this comparison (Kim 2015). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate, by participant assessment, was similar with NewBontA 20 U and onabotulinumtoxinA 20 U at week 4 (RR 0.97, 95% CI 0.88 to 1.07; participants = 159; studies = 1); at week 10 (RR 0.90, 95% CI 0.79 to 1.02; participants = 159; studies = 1); and at week 16 (RR 1.04, 95% CI 0.84 to 1.28; participants = 159; studies = 1) (Analysis 62.1).
62.1. Analysis.

Comparison 62: Liquid BontA 20 units versus OnabotulinumtoxinA 20 units one treatment in glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate, by physician assessment, was similar with NewBontA 20 U and onabotulinumtoxinA 20 U at week 4 (RR 0.99, 95% CI 0.88 to 1.12; participants = 159; studies = 1). The responder rate was higher with NewBontA 20 U than onabotulinumtoxinA 20 U at week 16 (RR 1.54, 95% CI 1.12 to 2.12; participants = 156; studies = 1) (Analysis 62.2).
62.2. Analysis.

Comparison 62: Liquid BontA 20 units versus OnabotulinumtoxinA 20 units one treatment in glabellar lines, Outcome 2: Physician assessment of success by analysing scores and scales
Total adverse events
The total adverse events were similar with NewBontA 20U and onabotulinumtoxinA 20 U (RR 1.27, 95% CI 0.69 to 2.32; participants = 156; studies = 1) (Analysis 62.3).
62.3. Analysis.

Comparison 62: Liquid BontA 20 units versus OnabotulinumtoxinA 20 units one treatment in glabellar lines, Outcome 3: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 63. NewBontA [Prosigne®] 20 units versus OnabotulinumtoxinA 20 units one cycle of treatment, glabellar lines
Only one RCT (n = 157 participants) assessed this comparison (Costa 2016). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
This RCT did not assess this outcome.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate by physician assessment was higher in the onabotulinumtoxinA 20 U group at week 12 (RR 0.49, 95% CI 0.21 to 1.19; participants = 157; studies = 1) (Analysis 63.1).
63.1. Analysis.

Comparison 63: NewBontA (Prosigne®) 20 units versus OnabotulinumtoxinA 20 units one treatment in glabellar lines, Outcome 1: Physician assessment of success by analysing scores and scales
Total adverse events
This RCT did not assess this outcome.
Duration of treatment effect
The duration of treatment effect was 12 weeks (84.5 ± 38.8 days) for NewBontA 20U (Prosigne®), and 12.8 weeks (89.9 ± 41.1 days) for OnabotulinumtoxinA 20 U assessed by three independent observers. The duration of treatment effect by physician assessment was 10.9 weeks: 76.8 ± 46.6 days for NewBontA 20 U (Prosigne®), and 12.5 weeks (88.1 ± 43.6 days) for onabotulinumtoxinA 20 U.
COMPARISON 64. CBFC26 20 units versus OnabotulinumtoxinA 20 units one cycle of treatment, glabellar lines
Only one RCT (n = 249 participants) assessed this comparison (Kim 2014). Hence, we were unable to undertake a meta‐analysis and only present results in a forest plot for visual representation.
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate by participant assessment was higher with NewBontA 20 U than onabotulinumtoxinA 20 U at week 4 (RR 1.22, 95% CI 1.10 to 1.35; participants = 249; studies = 1); and at week 8 (RR 1.14, 95% CI 1.01 to 1.29; participants = 249; studies = 1). The responder rate was similar with NewBontA 20 U and onabotulinumtoxinA 20 U at week 12 (RR 1.08, 95% CI 0.91 to 1.28; participants = 249; studies = 1); and at week 16 (RR 1.15, 95% CI 0.92 to 1.44; participants = 249; studies = 1) (Analysis 64.1).
64.1. Analysis.

Comparison 64: CBFC26 20 units versus OnabotulinumtoxinA 20 units one treatment in glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
The major adverse events were higher with NewBontA 20 U than onabotulinumtoxinA 20 U, but the result is uncertain due to the wide confidence interval (RR 2.04, 95% CI 0.52 to 8.01; participants = 271; studies = 1) (Analysis 64.2).
64.2. Analysis.

Comparison 64: CBFC26 20 units versus OnabotulinumtoxinA 20 units one treatment in glabellar lines, Outcome 2: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate by physician assessment was similar with NewBontA 20 U and onabotulinumtoxinA 20 U at week 4 (RR 1.09, 95% CI 0.99 to 1.21; participants = 249; studies = 1); at week 8 (RR 1.09, 95% CI 0.92 to 1.29; participants = 249; studies = 1); at week 12 (RR 1.21, 95% CI 0.96 to 1.51; participants = 249; studies = 1); and at week 16 (RR 1.26, 95% CI 0.94 to 1.68; participants = 249; studies = 1) (Analysis 64.3).
64.3. Analysis.

Comparison 64: CBFC26 20 units versus OnabotulinumtoxinA 20 units one treatment in glabellar lines, Outcome 3: Physician assessment of success by analysing scores and scales
Total adverse events
The total adverse events were similar with NewBontA 20 U andoonabotulinumtoxinA 20 U (RR 0.86, 95% CI 0.60 to 1.24; participants = 249; studies = 1) (Analysis 64.4).
64.4. Analysis.

Comparison 64: CBFC26 20 units versus OnabotulinumtoxinA 20 units one treatment in glabellar lines, Outcome 4: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 65. Liquid AbobotulinumtoxinA 20 units versus placebo one cycle of treatment, glabellar lines
Only one RCT (n = 71 participants) assessed this comparison (Ascher 2018). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate by participant assessment was similar with liquid abobotulinumtoxinA 20 U and placebo at week 4 (RR 66.81, 95% CI 4.25 to 1050.36, participants = 71; studies = 1); at week 8 (RR 42.14, 95% CI 2.65 to 670.89), participants = 71; studies = 1); at week 12 (RR 44.19, 95% CI 2.78 to 702.51 participants = 71; studies = 1); at week 16 (RR 27.75, 95% CI 1.71 to 449.60, participants = 71; studies = 1) (Analysis 65.1).
65.1. Analysis.

Comparison 65: Liquid AbobotulinumtoxinA 20 units versus placebo one treatment in glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
There was no difference between groups (Peto OR 7.60, 95% CI 0.15 to 383.33, participants = 71; studies = 1) (Analysis 65.2).
65.2. Analysis.

Comparison 65: Liquid AbobotulinumtoxinA 20 units versus placebo one treatment in glabellar lines, Outcome 2: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate by physician assessment was similar with liquid abobotulinumtoxinA 20 U and placebo at week 4 (RR 66.81, 95% CI 4.25 to 1050.36; participants = 71; studies = 1); at week 8 (RR 58.58, 95% CI 3.71 to 923.86, participants = 71, studies = 1); at week 12 (RR 40.08, 95% CI 2.51 to 639.27, participants = 71, studies = 1); at week 16 (RR 19.53, 95% CI 1.18 to 323.24, participants = 71, studies = 1) (Analysis 65.3).
65.3. Analysis.

Comparison 65: Liquid AbobotulinumtoxinA 20 units versus placebo one treatment in glabellar lines, Outcome 3: Physician assessment of success by analysing scores and scales
Total adverse events
There was no difference between groups (RR 1.23, 95% CI 0.41 to 3.68, participants = 71; studies = 1) (Analysis 65.4).
65.4. Analysis.

Comparison 65: Liquid AbobotulinumtoxinA 20 units versus placebo one treatment in glabellar lines, Outcome 4: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 66. Liquid AbobotulinumtoxinA 50 units versus placebo one cycle of treatment, glabellar lines
Two RCTs (n = 255 participants) assessed this comparison (Ascher 2018; Ascher 2020).
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate by participant's assessment was higher than placebo at week 4 (RR 46.95, 95% CI 9.57 to 230.36, participants = 255, studies = 2); at week 8 (RR 41.87, 95% CI 8.52 to 205.88, participants = 253, studies = 2), at week 12 (RR 24.61, 95% CI 4.97 to 121.91, participants= 253, studies =2); at week 16 (RR 6.54, 95% CI 1.70 to 25.15, participants= 254, studies =2), at week 20 (RR 3.35, 95% CI 1.71 to 6.58, participants= 183, studies =1) (see Analysis 66.1).
66.1. Analysis.

Comparison 66: Liquid AbobotulinumtoxinA 50 units versus placebo one treatment in glabellar lines, Outcome 1: Participant assessment success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
No difference between groups (Peto OR 4.39, 95% CI 0.07 to 289.31, participants = 256, studies = 2, Analysis 66.2).
66.2. Analysis.

Comparison 66: Liquid AbobotulinumtoxinA 50 units versus placebo one treatment in glabellar lines, Outcome 2: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate by physician assessment was higher than placebo at week 4 (RR 16.73, 95% CI 2.84 to 98.58, participants = 255, studies = 2); at week 8 (RR 48.98, 95% CI 9.99 to 240.21, participants = 255, studies = 2); at week 12 (RR 35.93, 95% CI 7.30 to 176.90, participants = 255, studies = 2); at week 16 (RR 21.25, 95% CI 2.95 to 152.88, participants = 255, studies = 2); and at week 20 (RR 25.86, 95% CI 1.60 to 417.34, participants = 184, studies = 1) (see Analysis 66.3).
66.3. Analysis.

Comparison 66: Liquid AbobotulinumtoxinA 50 units versus placebo one treatment in glabellar lines, Outcome 3: Physician assessment of success by analysing scores and scales
Total adverse events
There was no difference between the groups (RR 1.11, 95% CI 0.72 to1.71, participants = 255, studies = 2).
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 67. Liquid AbobotulinumtoxinA 75 units versus placebo one cycle of treatment, glabellar lines
One RCT (n = 71 participants) assessed this comparison (Ascher 2018).
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate by participant's assessment was higher with liquid abobotulinumtoxinA 7 5U than placebo at week 4 (RR 60.64, 95% CI 3.85 to 955.48, participants = 71, studies = 1); at week 8 (RR 62.69, 95% CI 3.98 to 987.11, participants = 71, studies = 1); at week 12 (RR 44.19, 95% CI 2.78 to 702.51, participants = 71, studies = 1); at week 16 (RR 35.97, 95% CI 2.25 to 576.04, participants = 71, studies = 1) (Analysis 67.1).
67.1. Analysis.

Comparison 67: Liquid AbobotulinumtoxinA 75 units versus placebo one treatment in glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
There was no difference between groups (Peto 7.83, 95% CI 0.48 to 127.75, participants = 71, studies = 1) (Analysis 67.2).
67.2. Analysis.

Comparison 67: Liquid AbobotulinumtoxinA 75 units versus placebo one treatment in glabellar lines, Outcome 2: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate by physician assessment was higher with liquid abobotulinumtoxinA 20 U and placebo at week (RR 64.75, 95% CI 4.12 to 1018.73; participants = 71; studies = 1); at week 8 (RR 60.64, 95% CI 3.85 to 955.48, participants = 71, studies = 1); at week 12 (RR 56.53 95% CI 3.58 to 892.24, participants = 71, studies = 1); at week 16 (RR 42.14 95% CI2.65 to 670.89, participants = 71, studies = 1) (Analysis 67.3).
67.3. Analysis.

Comparison 67: Liquid AbobotulinumtoxinA 75 units versus placebo one treatment in glabellar lines, Outcome 3: Physician assessment of success by analysing scores and scales
Total adverse events
There was no difference between groups (RR 0.82 95% CI 0.24 to 2.81, participants = 71, studies = 1) (Analysis 67.4).
67.4. Analysis.

Comparison 67: Liquid AbobotulinumtoxinA 75 units versus placebo one treatment in glabellar lines, Outcome 4: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 68. Liquid AbobotulinumtoxinA 20 units versus Liquid AbobotulinumtoxinA 50 units one cycle of treatment, glabellar lines
One RCT (n = 70 participants) assessed this comparison (Ascher 2018).
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate by participant's assessment was similar with Liquid Abobotulinumtoxin 20 U and Liquid abobotulinumtoxin 50 U at week 4 (RR 1.07, 95% CI 0.90 to 1.26, participants = 70,studies = 1); at week 8 (RR 1.05, 95% CI 0.69 to 1.60, participant s = 70, studies = 1); at week 12 (RR 1.31, 95% CI 0.84 to 2.06, participants = 70, studies = 1); at week 16 (RR 1.08, 95% CI 0.58 to 2.03, participants = 70, studies = 1) (Analysis 68.1).
68.1. Analysis.

Comparison 68: Liquid AbobotulinumtoxinA 20 units versus Liquid AbobotulinumtoxinA 50 units one cycle of treatment, glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
There was no difference between groups (Peto OR 7.39, 95% CI 0.15 to 372.38, participants = 70, studies = 1) (Analysis 68.2).
68.2. Analysis.

Comparison 68: Liquid AbobotulinumtoxinA 20 units versus Liquid AbobotulinumtoxinA 50 units one cycle of treatment, glabellar lines, Outcome 2: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate by participant's assessment was similar with Liquid abobotulinumtoxin 20 U and Liquid abobotulinumtoxin 50 U at week 4 (RR 1.07, 95% CI 0.90 to 1.26; participants = 70; studies = 1); at week 12 (RR 1.19, 95% CI 0.74 to 1.90, participants = 70, studies = 1); at week 16 (RR 0.75, 95% CI 0.36 to 1.55, participants = 70, studies = 1).
The responder rate by participant's assessment was higher with Liquid abobotulinumtoxin 50 U and Liquid abobotulinumtoxin 20 U at week 8 (R 1.47, 95% CI 1.04 to 2.08, participants =70, studies = 1) (Analysis 68.3).
68.3. Analysis.

Comparison 68: Liquid AbobotulinumtoxinA 20 units versus Liquid AbobotulinumtoxinA 50 units one cycle of treatment, glabellar lines, Outcome 3: Physician assessment of success by analysing scores and scales
Total adverse events
There was no difference between groups (RR 1.50, 95% CI 0.46 to 4.86, participants = 70, studies = 1) (Analysis 68.4).
68.4. Analysis.

Comparison 68: Liquid AbobotulinumtoxinA 20 units versus Liquid AbobotulinumtoxinA 50 units one cycle of treatment, glabellar lines, Outcome 4: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 69. Liquid AbobotulinumtoxinA 20 units versus Liquid AbobotulinumtoxinA 75 units one cycle of treatment, glabellar lines
One RCT (n = 70 participants) assessed this comparison (Ascher 2018).
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate by participant's assessment was similar with Liquid abobotulinumtoxin 20 U and Liquid abobotulinumtoxin 75 U at week 4 (RR 1.10, 95% CI 0.92 to 1.32, participants = 70, studies = 1); at week 8 (RR 0.67, 95% CI 0.49 to 0.92, participants = 70, studies = 1); at week 12 (RR 0.95, 95% CI 0.66 to 1.38, participants = 70, studies = 1); at week 16 (RR 0.76, 95% CI 0.44 to 1.32, participants = 70, studies = 1) (Analysis 69.1).
69.1. Analysis.

Comparison 69: Liquid AbobotulinumtoxinA 20 units versus Liquid AbobotulinumtoxinA 75 units one cycle of treatment, glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
There was no difference between groups (Peto OR 0.50, 95% CI 0.05 to 5.00, participants = 70, studies = 1) (Analysis 69.2).
69.2. Analysis.

Comparison 69: Liquid AbobotulinumtoxinA 20 units versus Liquid AbobotulinumtoxinA 75 units one cycle of treatment, glabellar lines, Outcome 2: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate by physician assessment was similar with Liquid abobotulinumtoxin 20 U and Liquid abobotulinumtoxin 75 U at week 4 (RR 1.03, 95% CI 0.88 to 1.21; participants = 70; studies = 1); at week 8 (RR 0.97, 95% CI 0.77 to 1.21, participants = 70, studies = 1); at week 12 (RR 0.70, 95% CI 0.49 to 1.00, participants = 70, studies = 1); at week 16 (RR 0.55, 95% CI 0.29 to 1.06, participants = 70, studies = 1) (Analysis 69.3).
69.3. Analysis.

Comparison 69: Liquid AbobotulinumtoxinA 20 units versus Liquid AbobotulinumtoxinA 75 units one cycle of treatment, glabellar lines, Outcome 3: Physician assessment of success by analysing scores and scales
Total adverse events
There was no difference between groups (RR 2.50, 95% CI 0.87 to 7.22, participants = 70, studies = 1) (Analysis 69.4).
69.4. Analysis.

Comparison 69: Liquid AbobotulinumtoxinA 20 units versus Liquid AbobotulinumtoxinA 75 units one cycle of treatment, glabellar lines, Outcome 4: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 70. Liquid AbobotulinumtoxinA 50 units versus Liquid AbobotulinumtoxinA 75 units one cycle of treatment, glabellar lines
One RCT (n = 70 participants) assessed this comparison (Ascher 2018).
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate by participant's assessment was similar with liquid abobotulinumtoxin 50 U and Liquid abobotulinumtoxin 75 U at week 4 (RR 1.00, 95% CI 0.83 to 1.21 participants = 70, studies = 1; at week 8 (RR 0.63, 0.88 95% CI 0.45 to 0.88, participants = 70, studies = 1); at week 12 (RR 0.76, 95% CI 0.49 to 1.20, participants = 70, studies = 1); at week 16 (RR 0.71, 95% CI 0.40 to 1.25, participants = 70, studies = 1) (Analysis 70.1).
70.1. Analysis.

Comparison 70: Liquid AbobotulinumtoxinA 50 units versus Liquid AbobotulinumtoxinA 75 units one cycle of treatment, glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
There was no difference between groups (Peto 0.13, 95% CI 0.01 to 2.14, participants = 70, studies = 1) (Analysis 70.2).
70.2. Analysis.

Comparison 70: Liquid AbobotulinumtoxinA 50 units versus Liquid AbobotulinumtoxinA 75 units one cycle of treatment, glabellar lines, Outcome 2: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate by physician assessment was similar with Liquid abobotulinumtoxin 50 U and Liquid abobotulinumtoxin 75 U at week 4 (RR 0.97, 95% CI 0.81 to 1.16; participants = 70; studies = 1), at week 8 (RR 0.66, 95% CI 0.47 to 0.92, participants = 70, studies = 1); at week 12 (RR 0.59, 95% CI 0.40 to 0.89, participants = 70, studies = 1); at week 16 (RR 0.60, 95% CI 0.35 to 1.03, participants = 70, studies = 1) (Analysis 70.3).
70.3. Analysis.

Comparison 70: Liquid AbobotulinumtoxinA 50 units versus Liquid AbobotulinumtoxinA 75 units one cycle of treatment, glabellar lines, Outcome 3: Physician assessment of success by analysing scores and scales
Total adverse events
There was no difference between groups (RR 1.00, 95% CI 0.27 to 3.69, participants = 70, studies = 1) (Analysis 70.4).
70.4. Analysis.

Comparison 70: Liquid AbobotulinumtoxinA 50 units versus Liquid AbobotulinumtoxinA 75 units one cycle of treatment, glabellar lines, Outcome 4: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 71. Liquid AbobotulinumtoxinA 20 units versus AbobotulinumtoxinA 50 units one cycle of treatment, glabellar lines
One RCT (n = 70 participants) assessed this comparison (Ascher 2018).
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate by participant's assessment was similar with Liquid abobotulinumtoxin 20 U and Liquid abobotulinumtoxin 50 U at week 4 (RR 1.10, 95% CI 0.92 to 1.32, participants = 70, studies = 1); at week 8 (RR 0.91, 95% CI 0.62 to 1.33 participants = 70, studies = 1); at week 12 (RR 1.50, 95% CI 0.92 to 2.44, participants = 70, studies = 1); at week 16 (RR 0.93, 95% CI 0.51 to 1.68, participants = 70, studies = 1) (Analysis 71.1).
71.1. Analysis.

Comparison 71: Liquid AbobotulinumtoxinA 20 units versus AbobotulinumtoxinA 50 units one cycle of treatment, glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
There was no difference between groups (Peto OR 7.39, 95% CI 0.15 to 372.38, participants = 70, studies = 1) (Analysis 71.2).
71.2. Analysis.

Comparison 71: Liquid AbobotulinumtoxinA 20 units versus AbobotulinumtoxinA 50 units one cycle of treatment, glabellar lines, Outcome 2: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate by physician assessment was similar with Liquid abobotulinumtoxin 20 U and Liquid abobotulinumtoxin 50 U at week (RR 1.1, 95% CI 0.96 to 1.46; participants = 70; studies = 1); at week 8 (RR 1.00, 95% CI 0.79 to 1.26, participants = 70, studies = 1); at week 12 (RR 1.00, 95% CI 0.65 to 1.54, participants = 70, studies = 1); at week 16 (RR 0.69, 95% CI 0.34 to 1.41, participants = 70, studies = 1) (Analysis 71.3).
71.3. Analysis.

Comparison 71: Liquid AbobotulinumtoxinA 20 units versus AbobotulinumtoxinA 50 units one cycle of treatment, glabellar lines, Outcome 3: Physician assessment of success by analysing scores and scales
Total adverse events
There was no difference between groups (RR 3.00, 95% CI 0.65 to 13.86), participants = 70, studies = 1) (Analysis 71.4).
71.4. Analysis.

Comparison 71: Liquid AbobotulinumtoxinA 20 units versus AbobotulinumtoxinA 50 units one cycle of treatment, glabellar lines, Outcome 4: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 72. Liquid AbobotulinumtoxinA 75 units versus AbobotulinumtoxinA 50 units one cycle of treatment, glabellar lines
One RCT (n = 70 participants) assessed this comparison (Ascher 2018).
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate by participant's assessment was similar with Liquid abobotulinumtoxin 75 U and abobotulinumtoxin 50 U at week 4 (RR 1.00, 95% CI 0.81 to 1.24, participants = 70, studies = 1); at week 12 (RR 1.57, 95% CI 0.97 to 2.54, participants = 70, studies = 1); at week 16 (RR 1.70, 95% CI 0.91 to 3.18, participants = 70, studies = 1).
The responder rate by participant's assessment was higher with Liquid abobotulinumtoxin 75 U than abobotulinumtoxin 50 U at week 8 (RR 1.36, 95% CI 1.02 to 1.82, participants = 70, studies = 1) (Analysis 72.1).
72.1. Analysis.

Comparison 72: Liquid AbobotulinumtoxinA 75 units versus AbobotulinumtoxinA 50 units one cycle of treatment, glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
There was no difference between groups (Peto OR 7.61, 95% CI 0.47 to 124.15, participants = 70, studies = 1) (Analysis 72.2).
72.2. Analysis.

Comparison 72: Liquid AbobotulinumtoxinA 75 units versus AbobotulinumtoxinA 50 units one cycle of treatment, glabellar lines, Outcome 2: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate by physician assessment was similar with Liquid abobotulinumtoxin 75 U and abobotulinumtoxin 50 U at week 4 (RR 1.15, 95% CI 0.93 to 1.43 participants = 70; studies = 1); at week 8 (RR 1.04, 95% CI 0.83 to 1.30, participants = 70, studies = 1); at week 12 (RR 1.42, 95% CI 1.00 to 2.02), participants = 70, studies = 1); at week 16 (RR 1.54, 95% CI 0.92 to 2.58, participants = 70, studies = 1) (Analysis 72.3).
72.3. Analysis.

Comparison 72: Liquid AbobotulinumtoxinA 75 units versus AbobotulinumtoxinA 50 units one cycle of treatment, glabellar lines, Outcome 3: Physician assessment of success by analysing scores and scales
Total adverse events
There was no difference between groups (RR 2.00, 95% CI 0.39 to 10.22, participants = 70, studies = 1) (Analysis 72.4).
72.4. Analysis.

Comparison 72: Liquid AbobotulinumtoxinA 75 units versus AbobotulinumtoxinA 50 units one cycle of treatment, glabellar lines, Outcome 4: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
Comparison 73. DaxibotulinumtoxinA 60 units versus placebo one cycle of treatment, glabellar lines
One RCT (n = 76 participants) assessed this comparison (Carruthers 2017).
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate by participant assessment was higher with daxibotulinumtoxinA 60 U and placebo at week 4 (RR 52.29, 95% CI 3.31 to 825.07; participants = 76; studies = 1); at week 16 (RR 18.00, 95% CI 1.09 to 296.56; participants = 76; studies = 1); and at week 24 (RR 6.00, 95% CI 0.32 to 112.32; participants = 76; studies = 1) (Analysis 73.1).
73.1. Analysis.

Comparison 73: DaxibotulinumtoxinA 60 units versus placebo one cycle of treatment in glabellar lines, Outcome 1: Participants assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
No difference between groups (Peto 7.71, 95% CI 0.43 to 138.48; participants = 76; studies = 1) (Analysis 73.2).
73.2. Analysis.

Comparison 73: DaxibotulinumtoxinA 60 units versus placebo one cycle of treatment in glabellar lines, Outcome 2: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate by physician assessment was higher with daxibotulinumtoxinA 60 U and placebo at week 4 (RR 69.43, 95% CI 4.42 to 1089.43; participants = 76; studies = 1); at week 8 (RR 55.71, 95% CI 3.54 to 877.94; participants = 76; studies = 1); at week 12 (RR 31.71, 95% CI 1.98 to 507.90; participants = 76; studies = 1); at week 16 (RR 14.57, 95% CI 0.87 to 243.78; participants = 76; studies = 1); at week 20 (RR 14.57, 95% CI 0.87 to 243.78; participants = 76; studies = 1); and at week 24 (RR 4.29, 95% CI 0.21 to 86.39; participants = 76; studies = 1). Although, the effect sizes are uncertain due to the very large confidence interval, which includes 1 at week 16, week 20 and week 24 (Analysis 73.3).
73.3. Analysis.

Comparison 73: DaxibotulinumtoxinA 60 units versus placebo one cycle of treatment in glabellar lines, Outcome 3: Physician assessment of success by analysing scores and scales
Total adverse events
The total adverse events was higher in daxibotulinumtoxinA 60 U group than in placebo group (RR 2.89, 95% CI 1.23 to 6.75; participants = 107; studies = 1) (Analysis 73.4)
73.4. Analysis.

Comparison 73: DaxibotulinumtoxinA 60 units versus placebo one cycle of treatment in glabellar lines, Outcome 4: Total adverse events
Duration of treatment effect
The duration of effect of daxibotulinumtoxinA 60 U was 22.5 weeks and 0.4 weeks for placebo group (MD 22.10, 95% CI 20.24 to 23.96; participants = 76; studies = 1) (Analysis 73.5).
73.5. Analysis.

Comparison 73: DaxibotulinumtoxinA 60 units versus placebo one cycle of treatment in glabellar lines, Outcome 5: Duration of treatment
COMPARISON 74. DaxibotulinumtoxinA 40 units versus placebo one cycle of treatment, glabellar lines
Two RCTs (n = 683 participants) assessed this comparison (Bertucci 2020; Carruthers 2017). Results are also shown in Table 6.
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate by participant assessment was higher with daxibotulinumtoxinA 40 U and placebo at week 4 (RR 21.10, 95% CI 11.31 to 39.34; participants = 683; studies = 2; I2 = 0%, moderate‐certainty evidence); at week 8 (RR 15.75, 95% CI 8.85 to 28.03; participants = 609; studies = 1); at week 12 (RR 24.51, 95% CI 11.12 to 54.05; participants = 609; studies = 1); at week 16 (RR 12.74, 95% CI 6.80 to 23.89; participants = 683; studies = 2; I2 = 0%); at week 20 (RR 13.60, 95% CI 6.13 to 30.18; participants = 609; studies = 1); and at week 24 (RR 3.09, 95% CI 0.13 to 73.21; participants = 683; studies = 2; I2 = 0%). (Analysis 74.1)
74.1. Analysis.

Comparison 74: DaxibotulinumtoxinA 40 units versus placebo one cycle of treatment in glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
These RCTs did not show any major event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate by physician assessment was higher with daxibotulinumtoxinA 40 U and placebo at week 4 (RR 23.40, 95% CI 12.56 to 43.61; participants = 683; studies = 2; I2 = 0%, moderate‐certainty evidence); at week 8 (RR 18.09, 95% CI 10.30 to 31.78; participants = 683; studies = 2; I2 = 0%); at week 12 (RR 29.46, 95% CI 13.79 to 62.94; participants = 683; studies = 2; I2 = 0%); at week 16 (RR 16.84, 95% CI 9.01 to 31.47; participants = 683; studies = 2; I2 = 0%); at week 20 (RR 18.06, 95% CI 8.42 to 38.76; participants = 683; studies = 2; I2 = 0%); and at week 24 (RR 15.33, 95% CI 6.06 to 38.78; participants = 683; studies = 2; I2 = 0%) (Analysis 74.2).
74.2. Analysis.

Comparison 74: DaxibotulinumtoxinA 40 units versus placebo one cycle of treatment in glabellar lines, Outcome 2: Physician assessment of success by analysing scores and scales
Total adverse events
The risk of any adverse events was higher in daxibotulinumtoxinA group (RR 2.23, 95% CI 1.46 to 3.40; participants = 716; studies = 2; I2 = 0%, moderate‐certainty evidence) (Analysis 74.3).
74.3. Analysis.

Comparison 74: DaxibotulinumtoxinA 40 units versus placebo one cycle of treatment in glabellar lines, Outcome 3: Total adverse events
Duration of treatment effect
One RCT assesses this outcome (Carruthers 2017).The duration of treatment effect was 23.2 weeks for daxibotulinumtoxinA 60 U and 0.4 weeks for placebo group (MD 22.80, 95% CI 20.74 to 24.86; participants = 74; studies = 1) (Analysis 74.4).
74.4. Analysis.

Comparison 74: DaxibotulinumtoxinA 40 units versus placebo one cycle of treatment in glabellar lines, Outcome 4: Duration of treatment effect, weeks
Bertucci 2020 pre‐specified 24 weeks as duration of treatment effect.
COMPARISON 75. DaxibotulinumtoxinA 20 units versus placebo one cycle of treatment, glabellar lines
One RCT (n = 69 participants) assessed this comparison (Carruthers 2017).
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate by participant assessment was higher with daxibotulinumtoxinA 20 U and placebo at week 4 (RR 50.40, 95% CI 3.19 to 797.13; participants = 69; studies = 1); at week 16 (RR 11.31, 95% CI 0.65 to 197.06; participants = 69; studies = 1); at week 24 (RR 3.09, 95% CI 0.13 to 73.21; participants = 69; studies = 1), although the effect size is uncertain due to the very large confidence interval (Analysis 75.1).
75.1. Analysis.

Comparison 75: DaxibotulinumtoxinA 20units versus placebo one cycle of treatment in glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate by physician assessment was higher with daxibotulinumtoxinA 20 U and placebo at week 4 (RR 60.69, 95% CI 3.86 to 955.19; participants = 69; studies = 1); at week 8 (RR 33.94, 95% CI 2.12 to 544.26; participants = 69; studies = 1); at week 12 (RR 21.60, 95% CI 1.32 to 354.72; participants = 69; studies = 1); at week 16 (RR 15.43, 95% CI 0.92 to 260.05; participants = 69; studies = 1); at week 20 (RR 9.26, 95% CI 0.52 to 165.65; participants = 83; studies = 1); at week 24 (RR 7.20, 95% CI 0.39 to 134.36; participants = 69; studies = 1), although the effect size is uncertain due to the very large confidence interval (Analysis 75.2).
75.2. Analysis.

Comparison 75: DaxibotulinumtoxinA 20units versus placebo one cycle of treatment in glabellar lines, Outcome 2: Physician assessment of success by analysing scores and scales
Total adverse events
The total adverse events was higher in daxibotulinumtoxinA 40 U group than in placebo group (RR 2.17, 95% CI 0.89 to 5.28; participants = 108; studies = 1) (Analysis 75.3).
75.3. Analysis.

Comparison 75: DaxibotulinumtoxinA 20units versus placebo one cycle of treatment in glabellar lines, Outcome 3: Total adverse events
Duration of treatment effect
The duration of treatment effect of daxibotulinumtoxinA 60 U was 20.8 weeks and 0.4 weeks for placebo group (MD 20.40, 95% CI 18.56 to 22.24; participants = 69; studies = 1) (Analysis 75.4).
75.4. Analysis.

Comparison 75: DaxibotulinumtoxinA 20units versus placebo one cycle of treatment in glabellar lines, Outcome 4: Duration of treatment
COMPARISON 76. DaxibotulinumtoxinA 60 units versus OnabotulimtoxinA 20units one cycle of treatment, glabellar lines
One RCT (n = 83 participants) assessed this comparison (Carruthers 2017).
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate by participant assessment was higher with daxibotulinumtoxinA 60 U and onabotulinumtoxinA 20 U at week 4(RR 1.14, 95% CI 0.85 to 1.52; participants = 83; ; studies = 1); and at week 16 (RR 2.05, 95% CI 0.77 to 5.48; participants = 83; studies = 1); but similar at week 24 (RR 2.05, 95% CI 0.77 to 5.48; participants = 83; studies = 1) (Analysis 76.1).
76.1. Analysis.

Comparison 76: DaxibotulinumtoxinA 60 units versus OnabotulinumtoxinA 20 units one cycle of treatment in glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
No difference between groups (Peto OR 3.62, 95% CI 0.60 to 21.86; participants = 83; studies = 1) (Analysis 76.2).
76.2. Analysis.

Comparison 76: DaxibotulinumtoxinA 60 units versus OnabotulinumtoxinA 20 units one cycle of treatment in glabellar lines, Outcome 2: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate by physician assessment was higher with daxibotulinumtoxinA 60 U and onabotulinumtoxinA 20 U at week 8 (RR 1.49, 95% CI 1.07 to 2.07; participants = 83; studies = 1); but a similar response at week 4 (RR 1.24, 95% CI 1.05 to 1.46); participants = 83; studies = 1); at week 12 (RR 0.97, 95% CI 0.60 to 1.57; participants = 83; studies = 1); at week 16 (RR 2.05, 95% CI 0.67 to 6.28; participants = 83; studies = 1); at week 20 (RR 1.02, 95% CI 0.15 to 6.9; participants = 83; studies = 1); and at week 24 (RR 1.02, 95% CI 0.15 to 6.93; participants = 83; studies = 1) (Analysis 76.3).
76.3. Analysis.

Comparison 76: DaxibotulinumtoxinA 60 units versus OnabotulinumtoxinA 20 units one cycle of treatment in glabellar lines, Outcome 3: Physician assessment of success by analysing scores and scales
Total adverse events
There was no difference between total adverse events between groups (RR 1.15, 95% CI 0.65 to 2.07; participants = 107; studies = 1) (Analysis 76.4).
76.4. Analysis.

Comparison 76: DaxibotulinumtoxinA 60 units versus OnabotulinumtoxinA 20 units one cycle of treatment in glabellar lines, Outcome 4: Total adverse events
Duration of treatment effect
There was no difference of duration of treatment between groups (MD 3.70, 95% CI 0.91 to 6.49; participants = 83; studies = 1) (Analysis 76.5).
76.5. Analysis.

Comparison 76: DaxibotulinumtoxinA 60 units versus OnabotulinumtoxinA 20 units one cycle of treatment in glabellar lines, Outcome 5: Duration of treatment
COMPARISON 77. DaxibotulinumtoxinA 40 units versus OnabotulimtoxinA 20 units one cycle of treatment, glabellar lines
One RCT (n = 250 participants) assessed this comparison (Carruthers 2017).
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate by participant assessment was similar between daxibotulinumtoxinA 40 U and onabotulinumtoxinA 20 U at week 4(RR 0.88, 95% CI 0.61 to 1.25; participants = 81; studies = 1); at week 16 (RR 2.15, 95% CI 0.81 to 5.74; participants = 81; studies = 1); and at week 24 (RR 1.08, 95% CI 0.16 to 7.28; participants = 81; studies = 1) (Analysis 77.1).
77.1. Analysis.

Comparison 77: DaxibotulinumtoxinA 40 units versus OnabotulinumtoxinA 20 units one cycle of treatment in glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
There was no difference between groups (Peto OR 0.15, 95% CI 0.00 to 7.35; participants = 81; studies = 1) (Analysis 77.2).
77.2. Analysis.

Comparison 77: DaxibotulinumtoxinA 40 units versus OnabotulinumtoxinA 20 units one cycle of treatment in glabellar lines, Outcome 2: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate by physician assessment was higher with daxibotulinumtoxinA 40 U and onabotulinumtoxinA 20 U at week 4(RR 1.21, 95% CI 1.01 to 1.44; participants = 81; studies = 1); at week 8 (RR 1.42, 95% CI 1.01 to 2.00; participants = 81; studies = 1); at week 16 (RR 3.77, 95% CI 1.36 to 10.48; participants = 81; studies = 1); but similar at week 12 (RR 0.96, 95% CI 0.59 to 1.57; participants = 81; studies = 1); at week 20 (RR 2.87, 95% CI 0.82 to 10.06; participants = 81; studies = 1); and at week 24 (RR 2.15, 95% CI 0.42 to 11.11; participants = 81; studies = 1) (Analysis 77.3).
77.3. Analysis.

Comparison 77: DaxibotulinumtoxinA 40 units versus OnabotulinumtoxinA 20 units one cycle of treatment in glabellar lines, Outcome 3: Physician assessment of success by analysing scores and scales
Total adverse events
There was no difference between total adverse events between groups (RR 0.95, 95% CI 0.51 to 1.77; participants = 107; studies = 1) (Analysis 77.4).
77.4. Analysis.

Comparison 77: DaxibotulinumtoxinA 40 units versus OnabotulinumtoxinA 20 units one cycle of treatment in glabellar lines, Outcome 4: Total adverse events
Duration of treatment effect
The duration of treatment effect was similar in the daxibotulinumtoxinA 40 U group and OnabotulinumtoxinA 20 U group (MD 4.40, 95% CI 1.47 to 7.33; participants = 81; studies = 1) ( Analysis 77.5).
77.5. Analysis.

Comparison 77: DaxibotulinumtoxinA 40 units versus OnabotulinumtoxinA 20 units one cycle of treatment in glabellar lines, Outcome 5: Duration of treatment
COMPARISON 78. DaxibotulinumtoxinA 20 units versus onabotulimtoxinA 20units one cycle of treatment, glabellar lines
One RCT (n = 76 participants) assessed this comparison (Carruthers 2017).
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate by participant assessment showed no difference between daxibotulinumtoxinA 20 U and onabotulinumtoxinA 20 U at week 4 (RR 1.10, 95% CI 0.80 to 1.50; participants = 76; studies = 1); at week 16 (RR 1.24, 95% CI 0.39 to 3.92; participants = 76; studies = 1); and at week 24(RR 0.25, 95% CI 0.01 to 4.95; participants = 76; studies = 1) (Analysis 78.1).
78.1. Analysis.

Comparison 78: DaxibotulinumtoxinA 20 units versus OnabotulinumtoxinA 20 units one cycle of treatment in glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
There was no difference between groups (Peto OR 0.16, 95% CI 0.00 to 8.43; participants = 76; studies = 1) (Analysis 78.2).
78.2. Analysis.

Comparison 78: DaxibotulinumtoxinA 20 units versus OnabotulinumtoxinA 20 units one cycle of treatment in glabellar lines, Outcome 2: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate by physician assessment was similar with daxibotulinumtoxinA 20 U and onabotulinumtoxinA 20 U at week 4 (RR 1.09, 95% CI 0.88 to 1.34; participants = 76; studies = 1); at week 8 (RR 0.90, 95% CI 0.57 to 1.42; participants = 76; studies = 1); at week 12 (RR 1.24, 95% CI 0.58 to 2.62; participants = 76; studies = 1); at week 16 (RR 2.16, 95% CI 0.69 to 6.77; participants = 76; studies = 1); at week 20 (RR 1.65, 95% CI 0.40 to 6.86; participants = 76; studies = 1); and at week 24 (RR 1.85, 95% CI 0.33 to 10.46; participants = 76; studies = 1) (Analysis 78.3).
78.3. Analysis.

Comparison 78: DaxibotulinumtoxinA 20 units versus OnabotulinumtoxinA 20 units one cycle of treatment in glabellar lines, Outcome 3: Physician assessment of success by analysing scores and scales
Total adverse events
There was no difference between daxibotulinumtoxinA 20U and onabotulinumtoxinA 20 U (RR 0.87, 95% CI 0.46 to 1.64; participants = 108; studies = 1) (Analysis 78.4).
78.4. Analysis.

Comparison 78: DaxibotulinumtoxinA 20 units versus OnabotulinumtoxinA 20 units one cycle of treatment in glabellar lines, Outcome 4: Total adverse events
Duration of treatment effect
The duration of treatment effect was similar between of daxibotulinumtoxinA 20U and onabotulinumtoxinA 20 U (MD 2.00, 95% CI ‐0.78 to 4.78; participants = 76; studies = 1) (Analysis 78.5).
78.5. Analysis.

Comparison 78: DaxibotulinumtoxinA 20 units versus OnabotulinumtoxinA 20 units one cycle of treatment in glabellar lines, Outcome 5: Duration of treatment
COMPARISON 79. PrabotulinumtoxinA 20 units versus placebo one cycle of treatment, glabellar lines
Three RCTs (n = 948 participants) assessed this comparison (Beer 2019a; Beer 2019b; Rzany 2019).
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate by participant assessment was higher withdaxibotulinumtoxinA 20 U than placebo at week 4 (RR 18.34, 95% CI 9.68 to 34.76; participants = 930; studies = 3; I2 = 0%). (Analysis 79.1).
79.1. Analysis.

Comparison 79: PrabotulinumtoxinA 20 units versus placebo one cycle of treatment, glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Two RCTs (Beer 2019a; Beer 2019b) showed this outcome at week 8, 12 and 16, but the authors did not separate participant and physician assessment scores.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
There was no difference between prabotulinumtoxinA 20U and placebo (RR 0.60, 95% CI 0.06 to 5.65; participants = 939; studies = 3; I2 = 0%) (Analysis 79.2).
79.2. Analysis.

Comparison 79: PrabotulinumtoxinA 20 units versus placebo one cycle of treatment, glabellar lines, Outcome 2: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate by physician assessment was similar with prabotulinumtoxinA 20 U and placebo at week 4 (RR 23.96, 95% CI 9.35 to 61.40; participants = 929; studies = 3; I2 = 26%) (Analysis 79.3).
79.3. Analysis.

Comparison 79: PrabotulinumtoxinA 20 units versus placebo one cycle of treatment, glabellar lines, Outcome 3: Physician assessment of success by analysing scores and scales
Two RCTs (Beer 2019a; Beer 2019b) showed this outcome at weeks 8, 12, and 16, but the authors did not separate participant and physician assessment scores.
Total adverse events
There was no difference between prabotulinumtoxinA 20 U and placebo (RR 1.14, 95% CI 0.91 to 1.43; participants = 948; studies = 3; I2 = 0%) (Analysis 79.4).
79.4. Analysis.

Comparison 79: PrabotulinumtoxinA 20 units versus placebo one cycle of treatment, glabellar lines, Outcome 4: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 80. PrabotulinumtoxinA 20 units versus OnabotulimtoxinA 20 units one cycle of treatment, glabellar lines
Two RCTs (n = 759 participants) assessed this comparison (Rzany 2019; Won 2015).
Primary outcomes
Participant assessment of success by analysing scores and scales
The responder rate by participant assessment was higher with daxibotulinumtoxinA 20 U than onabotulinumtoxinA 20 U at week 4 (RR 1.04, 95% CI 1.00 to 1.09; participants = 749; studies = 2; I2 = 0%) (Analysis 80.1).
80.1. Analysis.

Comparison 80: PrabotulinumtoxinA 20 units versus OnabotulimtoxinA 20units one cycle of treatment, glabellar lines, Outcome 1: Participant assessment of success by analysing scores and scales
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
There was no difference between prabotulinumtoxinA 20 U and onabotulinumtoxinA (Peto OR 2.75, 95% CI 0.38 to 19.61; participants = 491; studies = 1) (Analysis 80.2).
80.2. Analysis.

Comparison 80: PrabotulinumtoxinA 20 units versus OnabotulimtoxinA 20units one cycle of treatment, glabellar lines, Outcome 2: Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate by physician assessment was similar with prabotulinumtoxinA 20 U and onabotulinumtoxinA at week 4 (RR 1.04, 95% CI 0.94 to 1.14; participants = 483; studies = 1) (Analysis 80.3).
80.3. Analysis.

Comparison 80: PrabotulinumtoxinA 20 units versus OnabotulimtoxinA 20units one cycle of treatment, glabellar lines, Outcome 3: Physician assessment of success by analysing scores and scales
Total adverse events
There was no difference between prabotulinumtoxinA 20U and onabotulinumtoxinA (RR 0.91, 95% CI 0.74 to 1.13; participants = 759; studies = 2; I2 = 0%) (Analysis 80.4).
80.4. Analysis.

Comparison 80: PrabotulinumtoxinA 20 units versus OnabotulimtoxinA 20units one cycle of treatment, glabellar lines, Outcome 4: Total adverse events
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISON 81. OnabotulinumtoxinA 9 units versus hyaluronic acid [JUVEDERM ULTRA ® and/or JUVEDERM ULTRA PLUS®] one cycle of treatment, lips and perioral lines
Only one RCT (n = 60 participants) assessed this comparison (Carruthers 2010). Therefore, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
This did not assess this outcome.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The authors showed a significant difference among the groups (data not shown). No numeric data were provided.
Total adverse events
The total adverse events were higher with onabotulinumtoxinA than hyaluronic acid, but the result is uncertain due to the wide confidence interval (RR 2.00, 95% CI 0.40 to 10.11; participants = 60; studies = 1) (Analysis 81.1).
81.1. Analysis.

Comparison 81: OnabotulinumtoxinA 9 units versus hyaluronic acid [JUVEDERM ULTRA ® and/or JUVEDERM ULTRA PLUS®] one treatment in lips and perioral lines, Outcome 1: Total adverse events
Duration of treatment effect
The mean duration of treatment effect by physician assessment at maximum contraction of each treatment were: 9.9 weeks (69.3 ± 11.0 days) for onabotulinumtoxinA group; 15.5 weeks (108.9 ± 12 days) 24 g/mL cohesive gel group.
COMPARISON 82. OnabotulinumtoxinA 9 units associated with hyaluronic acid [JUVEDERM ULTRA ® and/or JUVEDERM ULTRA PLUS®] versus onabotulinumtoxinA 9 units one cycle of treatment, lips and perioral lines
Only one RCT (n = 60 participants) assessed this comparison (Carruthers 2010). Hence, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
This RCT did not assess this outcome.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The authors showed a significant difference among the groups (data not shown). No numerical data were provided.
Total adverse events
The frequency of adverse events was similar with onabotulinumtoxinA and onabotulinumtoxinA associated with filler (RR 0.75, 95% CI 0.18 to 3.07; participants = 60; studies = 1) (Analysis 82.1).
82.1. Analysis.

Comparison 82: OnabotulinumtoxinA 9 units associated with hyaluronic acid [JUVEDERM ULTRA ® and/or JUVEDERM ULTRA PLUS®] versus OnabotulinumtoxinA one treatment in lips and perioral lines, Outcome 1: Total adverse events
Duration of treatment effect
The mean duration of treatment effect by investigator assessment at maximum contraction were 15.2 weeks (106.6 ± 10.6 days) in the BontA plus 24‐mg/mL cohesive gel and 9.9 weeks (69.3 ± 11.0 days) for onabotulinumtoxinA.
COMPARISON 83. Hyaluronic acid versus OnabotulinumtoxinA 9 units associated with hyaluronic acid [JUVEDERM ULTRA ® and/or JUVEDERM ULTRA PLUS®] one cycle of treatment, lips and perioral lines
Only one RCT (n = 60 participants) assessed this comparison (Carruthers 2010). For this reason, we were not able to undertake a meta‐analysis, and only a visual representation of the results is shown in the forest plots.
Primary outcomes
Participant assessment of success by analysing scores and scales
This RCT did not assess this outcome.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
This RCT did not report any major event.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The authors showed a significant difference among the groups. No numeric data were provided.
Total adverse events
no difference between groups (RR 0.67; 95% CI 0.12 to 3.71, participants = 60; studies = 1) (Analysis 83.1).
83.1. Analysis.

Comparison 83: Hyaluronic acid [JUVEDERM ULTRA ® and/or JUVEDERM ULTRA PLUS®]versus OnabotulinumtoxinA 9 units associated with hyaluronic acid one treatment in lips and perioral lines, Outcome 1: Total adverse events
Duration of treatment effect
The mean duration of treatment effect by investigator assessment at maximum contraction were 15.2 (106.6 ± 10.6 days) in the onabotulinumtoxinA plus 24‐mg/mL cohesive gel group, and 15.5 weeks (108.9 ± 12 days) in 24‐mg/mL cohesive gel.
COMPARISON 84. IncobotulinumtoxinA 21 units versus AbobotulinumtoxinA 63 units, one cycle of treatment, glabellar lines
Only one RCT (n = 120 participants) assessed this comparison (Rappl 2013).
Primary outcomes
Participant assessment of success by analysing scores and scales
This RCT did not assess this outcome.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
No serious adverse events were reported.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The authors showed a significant difference among the groups. No numerical data were provided.
Total adverse events
Two participants reported mild bruising, which resolved within 2 to 3 days (the authors did not provide this results by intervention group) (very low‐certainty evidence).
Duration of treatment
For females, the mean duration of treatment effect was 146.12 days for incobotulinumtoxinA versus 139.69 days for abobotulinumtoxinA. For males, these results were 121.14 and 115.81, respectively (no SD or P value were provided).
COMPARISON 85. AbobotulinumtoxinA 50 units versus OnabotulinumtoxinA 18 units one cycle of treatment, glabellar and crow's feet lines
One RCT (n = 85 participants) assessed this comparison (Kassir 2013). This was a split‐face study design, so the results are described narratively.
Primary outcomes
Participant assessment of success by analysing scores and scales
This RCT did not assess this outcome.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
One participant in the abobotulinumtoxinA group reported ptosis.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The responder rate for glabellar lines, by physician assessment, was similar with abobotulinumtoxinA 30 U compared to onabotulinumtoxinA 8 U from week 4 to week 16.
The responder rate for crow's feet lines, by physician assessment, was similar with AbobotulinumtoxinA 30U and onabotulinumtoxinA 10 U from week 4 to week 16.
Total adverse events
This RCT did not assess this outcome.
Duration of treatment
This RCT did not assess this outcome.
COMPARISON 86. AbobotulinumtoxinA 30 units versus OnabotulinumtoxinA 10 units one cycle of treatment, crow's feet lines
One RCT (n = 90 participants) assessed this comparison (Nettar 2011).
Primary outcomes
Participant assessment of success by analysing scores and scales
The score (Merz scale) rate by participant assessment was higher with abobotulinumtoxinA treatment (2.34) than onabotulinumtoxinA (2.13), P = 0.03 at week 4.
Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus)
No major adverse events were reported.
Secondary outcomes
Physician assessment of success by analysing scores and scales
The score (Merz scale) rate by participant assessment was higher with abobotulinumtoxinA treatment (2.60) than onabotulinumtoxinA (2.33), P = 0.01 at week 4.
Total adverse events
One patient from onabotulinumtoxinA group showed a bruise.
Duration of treatment effect
This RCT did not assess this outcome.
COMPARISONS WITHOUT USABLE DATA
We were unable to collect usable data on the following comparisons, because the studies did not collect or report relevant data:
AbobotulinumtoxinA different doses comparison (full face treatment): one RCT (n = 90, Hexsel 2013). The doses compared were too similar and did not allow any validate comparisons (no clinical relevance). Moreover, there was dose overlap among the dose range (Hexsel 2013).
OnabotulinumtoxinA versus facial cream (Strivectin®) one cycle of treatment (glabellar lines): one RCT (n = 32, Beer 2006).
OnabotulinumtoxinA versus facial cream (Wrinklerelax™) one cycle of treatment (glabellar lines): one RCT (n = 31, Beer 2006).
OnabotulinumtoxinA versus facial cream (Hydroderm™) one cycle of treatment (glabellar lines): one RCT (n = 31, Beer 2006).
OnabotulinumtoxinA 25 units versus abobotulinumtoxinA 62.5 units one cycle of treatment (glabellar lines, crow's feet, and forehead lines (split‐face)): One RCT (n = 53, Michaels 2012).
OnabotulinumtoxinA 20 units versus abobotulinumtoxinA 50 units one cycle of treatment (crow's feet lines): one RCT (n = 90, Nettar 2011).
OnabotulinumtoxinA 7.5 units versus incobotulinumtoxinA 7.5 units one cycle of treatment (crow's feet lines): one RCT (n = 56, Park 2014).
AbobotulinumtoxinA 63 units versus onabotulinumtoxinA 32 units one cycle of treatment (glabellar lines): one RCT (n = 119, Rappl 2013).
OnabotulinumtoxinA 25 units versus onabotulinumtoxinA 25 units associated to collagen one cycle of treatment (glabellar lines): One RCT (n = 45, Patel 2004).
OnabotulinumtoxinA 25 units versus collagen one cycle of treatment (glabellar lines): one RCT (n = 42, Patel 2004).
OnabotulinumtoxinA 25 units associated with collagen versus collagen one cycle of treatment (glabellar lines): one RCT (n = 45, Patel 2004).
OnabotulinumtoxinA units not shown versus abobotulinumtoxin units not shown one cycle of treatment of glabellar lines and frontal lines, split‐face study: one study (Firoz 2012).
OnabotulinumtoxinA 25 units versus abobotulinumtoxin 62.5 units one cycle of treatment of glabellar lines, periorbital lines and frontal lines (Michaels 2012).
Discussion
Summary of main results
We included 65 randomised controlled trials (RCTs), involving 14,919 randomised participants. Here we summarise the results for the main comparisons that assessed the treatment of the facial region glabellar lines.
Table 1: based on moderate‐certainty evidence, onabotulinumtoxinA‐20 units (U) probably has a higher success rate than placebo when measured at four weeks by participants or physicians. OnabotulinumtoxinA‐20 U probably increased the risk of major adverse events compared to placebo (moderate‐certainty evidence), but there may be no difference between groups in any adverse events (low‐certainty evidence). Adverse events were collected over the duration of these studies, which ranged from four weeks to 24 weeks.
Table 2: abobotulinumtoxinA‐50 U showed a higher success rate than placebo at week four by participant assessment (high‐certainty evidence), and abobotulinumtoxinA‐50 U probably has a higher success rate by physician assessment (moderate‐certainty evidence). Compared to placebo, abobotulinumtoxinA‐50 U probably increases the occurrence of major adverse effects(moderate‐certainty evidence; collected in studies of four to 12 weeks duration) and may increase the occurrence of any adverse events (low‐certainty evidence; collected in studies of four to 16 weeks duration).
Table 3: there is probably a higher success rate with incobotulinumtoxinA‐20 U than placebo at week four by both participant assessment (moderate‐certainty evidence) and physician assessment (moderate‐certainty evidence). Major adverse events were not observed (moderate‐certainty evidence), and there may be no difference between groups in any adverse events (low‐certainty evidence; collected in studies of four to 16 weeks duration).
Table 4: there is no difference in the participant‐assessed or physician‐assessed success rate between abobotulinumtoxinA‐50 U and onabotulinumtoxinA‐20 U at four weeks (high‐certainty evidence). AbobotulinumtoxinA‐50 U probably increases the occurrence of major adverse events compared to onabotulinumtoxinA‐20 U (moderate‐certainty evidence), but there is probably no difference in any adverse events (moderate‐certainty evidence; collected in studies of four to 12 weeks duration).
Table 5: there may be no difference in the success rate between incobotulinumtoxinA‐24 U and onabotulinumtoxinA‐24 U at four weeks (by physician assessment; low‐certainty evidence). Participant assessment was not measured. Ptosis was reported in one participant in the onabotulinumtoxinA group, but the certainty of this evidence is very low, so we are uncertain of the risk of adverse events (collected in studies of four to 12 weeks duration).
Table 6: daxibotulinumtoxinA 40 U probably has a higher success rate than placebo when assessed at four weeks by either participants or physicians (moderate‐certainty evidence). Major adverse events were not observed. There may be an increase in any adverse events with daxibotulinumtoxinA than with placebo (moderate‐certainty evidence; collected in studies of four to 24 weeks duration).
Adverse events: ptosis was the main major adverse event. Botulinum toxin type A (BontA) are associated with a risk of strabismus or eyelid sensory disorders.
Overall completeness and applicability of evidence
Analysing the data, some ethnicities (Middle East, Latin America) and males are underrepresented. More than 80% of the total number of study participants were female. Ethnicity and anatomic characteristics such as skin width, oily skin, the skeletal structure can potentially impact on outcomes for BontA treatment. In general, men have stronger muscles than women, and this fact could interfere in the relationship between the units of BontA needed to treat and duration of effect.
Our review aimed to assess treatment of any type of facial wrinkle, but almost two‐thirdsof studies assessed treatment of the glabella region (43/65 studies). The other types of wrinkles assessed in this review: crow's feet lines: seven RCTs; forehead lines: two RCTs; forehead lines and crow's feet lines: one RCT; upper lines (glabellar lines, crow's feet lines, and forehead lines): three RCTs; forehead lines and glabellar lines: three RCTs; crow's feet lines and glabellar lines: three RCTs; full face: one RCT; perioral area: two RCTs.
Most of the studies that treated glabellar lines included moderate‐to‐severe glabellar lines according to Facial Wrinkle Scale score (FWS)or Glabellar Lines Severity Scale (GLSS). The studies that assessed others regions did not show details about wrinkle severity. In clinical practice, physicians treat mild‐to‐moderate glabellar lines.
The main commercial BontA treatments were addressed in the included studies. These included onabotulinumtoxinA, abobotulinumtoxinA, incobotulinumtoxinA, HBTX‐A, NewBontA (Medytox®, Neuronox®, Prosigne®), liquid BontA (MT101109L), daxibotulinumtoxinA (DWP450), praxibotulinumtoxinA, liquid BontA (Ipsen), and CBFC26. However, these treatments were not assessed by equal numbers of studies. Half of the studies assessed onabotulinumtoxinA, with 71% of the rest of the studies assessing abobotulinumtoxinA or incobotulinumtoxinA. Some types of BontA (e.g. liquid BontA (MT101109L)) were only assessed by single studies, or two studies (HBTXA).
The mean study duration was 20.75 weeks ± 11.7 (range: 1 to 52 weeks).
There was huge variation in doses, number of cycles, and duration of follow‐up. These factors precluded data pooling and compromised the robustness of evidence. We assessed the following comparisons: BontA versus placebo, at least one cycle of treatment (36 studies); BontA at different doses, one cycle of treatment (21 studies); BontA versus placebo, at least two cycles of treatment (11 studies); BontA versus facial cream (one study); BontA associated to fillers (two studies). Eighty‐three per cent of the studies assessed a single cycle of treatment, and as BontA has a temporary effect on wrinkles, the single cycle does not reflect the need for repeated treatments. Some studies administered much higher doses than those recommended by two different consensus groups (Carruthers 2008a; Sundaram 2016).
The most common comparator was placebo (36 studies). An active control was used in 19 studies. There were eight dose‐ranging studies of onabotulinumtoxinA, and a small number of studies compared against fillers. Direct comparisons of different BontA treatments were lacking. OnabotulinumtoxinA and abobotulinumtoxinA were the most common BontA comparators (seven studies each), followed by incobotulinumtoxinA (five studies). OnabotulinumtoxinA was compared with abobotulinumtoxinA (five studies), onabotulinumtoxinA was compared with incobotulinumtoxinA (four studies), and the following were compared in single studies against onabotulinumtoxinA: liquid BontA (MT101109L), New BontA (Medytox®), and Neuronox.
Duration of treatment was the least‐reported outcome of interest (21 studies). Physician assessment of success was more often reported (49 studies) than participant assessment of success (35 studies). Seventy‐one per cent of studies assessed major adverse events, and 78% evaluated the occurrence of any adverse events. However, long‐term adverse effects were not measured.
Unfortunately, there was disparity in the definition of outcome tools, inhibiting pooling.
Most of the studies had pharmaceutical support with no description about the role of the research development, planning, conduct, statistical analysis and reporting.
Quality of the evidence
As presented in the summary of findings tables, the certainty of the body of evidence obtained for each outcome was usually rated as very low, low or moderate certainty. Only three effect sizes were rated as high certainty. Most outcomes were downgraded for study limitations. Such limitations included an unclear/high risk of bias of selection bias from random sequence generation and allocation concealment, and unclear/high risk of performance or detection bias from blinding of participants, study personnel and outcome assessors. Many outcomes were also downgraded for imprecision as the estimates had wide confidence intervals which crossed the null effect.
Moreover, the studies were poorly reported, and patient double count was difficult to detect. Design study complexity was another obstacle to data collection.
Potential biases in the review process
This review followed strictly all the recommendations of the Cochrane Handbook (Higgins 2020) on searching, study selection, data collection, and data analysis to avoid bias.
Strengths of this review include a wide and recently updated literature search. The limitations of this review include no assessment of publication bias through funnel plot analysis because there were less than 10 studies included in each meta‐analysis.
We changed some items from protocol to the review as a tentative measure to decrease potential bias: minimal number of participants for included studies from 20 to 50; we assessed the responder rates only during 'muscle contraction', rather than 'at rest', as: for clinical practice this last approach was less relevant, and at label, BontA was indicated for hyperdynamic facial wrinkles; and we did meta‐analysis only in parallel group studies; we did not include split face studies in this analysis.
The 24 studies in 'Studies awaiting classification' may alter the conclusions of the review once assessed.
Agreements and disagreements with other studies or reviews
In general, all systematic reviews (SRs) showed similar results.
The majority of the studies injected onabotulinumtoxinA, followed by abbobotulinumtoxinA and incobotulinumtoxinA.
The most frequent facial region was glabellar lines, crow's feet lines and forehead lines.
The frequency of adverse effects is similar between BontA and placebo, except for the occurrence of blepharoptosis. Blepharoptosis is higher in the BontA groups than the placebo groups.
Ghadia and colleagues (Ghadia 2009) analysed 11 articles (1063 participants). The primary endpoint was the efficacy of BontA in facial wrinkles, and the secondary endpoint was safety. This systematic review studied the effect of treatment of onabotulinumtoxinA and abobotulinumtoxinA. The age ranged from 31 to 59 years, whereas in our systematic review, age range was broader (18 to 65 years). Regarding efficacy, we found similar results to Ghadia and colleagues: BontA in facial wrinkles was more effective than the placebo group, but because of a high level of heterogeneity this conclusion was uncertain. In Ghadia 2009, the overall quality of evidence was considered good (Jadad scale). In our systematic review, we largely showed a range from very low‐ to moderate‐certainty evidence according to GRADE pro GDT (GRADEpro GDT).
Cavallini and colleagues studied 35 articles, 8787 participants (Cavallini 2014). In this systematic review, the authors only considered onabotulinumtoxinA, abobotulinumtoxinA, and incobotulinumtoxinA, and analysed only safety outcomes(Cavallini 2014). Because of limited search on the most common BontA brands, this review also limited their results (Cavallini 2014).
Jia and colleagues reported another systematic review of adverse events. The authors limited their search strategy to articles in the English language, some BontA (onabotulinumtoxinA, abobotulinumtoxinA, incobotulinumtoxinA, and HBTX‐A), and some regions (glabellar and crow’s feet lines). This systematic review studied 34 articles, 42,405 participants (Jia 2016). The meta‐analysis was different, because they used a fixed‐effect analysis, whereas our systematic review used random‐effects analysis, In general, both systematic reviews agreed that BontA was safe for glabellar and crow’s feet lines (Jia 2016).
Authors' conclusions
Implications for practice.
This systematic review found that BontA is effective for reducing facial wrinkles within four weeks of treatment, but carries a risk of ptosis. There was a lack of long‐term data, and data on adverse effects were limited. The formulations of BontA tested appeared to be similarly effective in short‐term assessment. Longer‐term effects of treatment are unclear, and this applies to both efficacy and adverse effects.
Analysing the data, some ethnicities (Middle East, Latin America) and gender (male) are underrepresented. Depending on the ethnicity, anatomic characteristics such as skin width, oiliness of the skin, and the skeletal structure, can interfere in the BontA treatment.
In general, males have stronger muscles than females; this fact could interfere in the relationship between the units of BontA needed to treat and duration of effect. Most of the studies analysed moderate‐to‐severe glabellar lines, but in clinical practice, physicians treat mild‐to‐moderate glabellar lines.
Regarding major adverse events, only ptosis was reported in the trials; however, BontA is known to carry a risk of strabismus and eyelid sensory disorders. The trials reported ptosis in less than 5% of BontA‐treated participants, and no episodes of ptosis were reported in placebo‐treated trial participants.
Implications for research.
In recent years, new BontA formulations have become available. However, despite all the safety and clinical indications, some crucial questions remain that future BontA research should aim to address.
Therefore, according to the PICOT acronym, further research could focus on the following.
Population: different ethnicities should be assessed.
Intervention: studies should assess multiples cycles of treatment and seek to find the most effective dilution. There should be a standard conversion ratio among all available BontA brands.
Comparator: it would help to compare all available BontA brands regarding the duration of treatment effect (time between wrinkles treatment session) with assessment of different facial regions (glabella, canthal lines, frontal lines), liquid BontA versus lyophylised BontA, what is the most effective treatment association (hyaluronic acid, peeling, laser, radiofrequency, surgery), and what are the consequences of therapy of several facial regions in the same session.
Outcome: helpful outcomes to measure in future studies include duration of treatment in multiples sessions, the time between BontA dilution and clinical application, the effectiveness and safety of multiple cycles of treatment, and the longer‐term effectiveness of multiple cycles of treatment.
Future studies should also aim to improve study methodology. The main issue identified in the existing literature was failure to describe adequate methods of randomisation or allocation concealment. There is a need for better powered studies and more homogeneity of study design. There is a need for new studies that answer clinical questions such as the duration of the effect of BontA, one cycle versus multiple cycles, as well as active comparator studies, so we can more confidently compare different BontA treatment options.
What's new
| Date | Event | Description |
|---|---|---|
| 19 January 2022 | Amended | There was a mistake in one comparison: we included the wrong study. We have now rectified this. |
History
Protocol first published: Issue 9, 2014 Review first published: Issue 7, 2021
Acknowledgements
The authors would like to thank the Brazilian Cochrane Centre Handbook Study Group for their methodological support and the Cochrane Skin editorial base.
The Cochrane Skin editorial base wishes to thank Michael Bigby, Cochrane Dermatology Editor; Ben Carter, Statistical Editor; Laurence Le Cleach, Methods Editor; the clinical referee, Berthold Rzany; Shunjie (Sean) Chua, the consumer referee; Heather Maxwell who copy‐edited the review; and Carolyn Hughes who wrote the plain language summary.
Appendices
Appendix 1. Glossary
| Term | Definition |
| Adverse event | Any undesirable harmful effect caused by medication, surgery, or other medical procedures Major adverse events will be defined when will be defined when there is partial or total loss of function of a given organ (E.g., ptosis‐the patient can open her/his eyes). Total adverse events‐ when there is no dysfunction(pain, bruise, facial asymmetry). |
| Asthenotopia | Problem of vision, with pain in the eyes, back of the head and the neck |
| Cosmeceutical | A cosmetic pharmaceutical |
| Dynamic wrinkles | Wrinkles that appear only during muscle contraction, e.g. crow's feet in young people when smiling |
| Glabellar | Adjective for anatomical region between the eyebrows |
| Off‐label | Term given to the use of medicine to treat a condition for which it is not licensed |
| Observational study | A study that describes a condition according to the presence or absence of a factor, e.g. smoking habit and lung cancer |
| Quasi‐randomised clinical trial | Very similar to a randomised clinical trial, but the participants are not randomised to the study |
| Periorbicular wrinkles | Crow's feet |
| Randomisation | Process of selecting participants for a clinical trial; this process assures an equal chance of treatment assignment for each participant |
| Split‐face | Some studies apply treatment X to the right side of the face and treatment Y to the left side of the face. It works like 2 separate faces |
| Randomised clinical trial | A type of comparative scientific research. It is the best evidence to prove causation between an intervention (e.g. drug, surgery, or devices) and outcome |
| Muscle tonus | The passive and continuous muscle tension at rest |
| Static wrinkles | Visible wrinkles not related to muscle activity (at rest or contraction) but that develop with age |
| Strabismus | Misalignment of the eyes. If a physician injects botulinum toxin to treat crow's feet around the eyes, the toxin can cause strabismus, because it relaxes intrinsic orbital muscles |
Appendix 2. Cochrane Skin Specialised Register (CRSW)
(onabotulinum* or abobotulinum* or incobotulinum* or Botulinum* or botox or vistabel or vistabex or dysport or reloxin or azzalure or bocoture or xeomin or xeomeen or prosigne or cbtx‐a or nt201 or dps refinex or pur tox) and (wrinkl* or rhytid* or glabellar or forehead or frown or marionette or crow* or ((aging or age or aged) near skin))
Appendix 3. CENTRAL (Cochrane Library) search strategy
#1 MeSH descriptor: [Botulinum Toxins] explode all trees #2 MeSH descriptor: [Botulinum Toxins, Type A] explode all trees #3 (onabotulinum* or abobotulinum* or incobotulinum*):ti,ab,kw #4 (botox or vistabel or vistabex or dysport or reloxin or azzalure or bocoture or xeomin or xeomeen or prosigne or cbtx‐a or nt201 or dps refinex or pur tox):ti,ab,kw #5 {or #1‐#4} #6 MeSH descriptor: [Skin Aging] explode all trees #7 (wrinkl* or rhytid*):ti,ab,kw #8 ((aging or age or aged) near skin):ti,ab,kw #9 (glabellar near line*):ti,ab,kw #10 ((forehead or frown* or marionette*) and line*):ti,ab,kw #11 (crow* feet):ti,ab,kw #12 {or #6‐#11} #13 #5 and #12
Appendix 4. MEDLINE (Ovid) search strategy
1. exp Botulinum Toxins/ or exp Botulinum Toxins, Type A/ 2. onabotulinum$.mp. 3. abobotulinum$.mp. 4. incobotulinum$.mp. 5. (botox or vistabel or vistabex or dysport or reloxin or azzalure or bocoture or xeomin or xeomeen or prosigne or cbtx‐a or nt201 or dps refinex or pur tox).mp. 6. or/1‐5 7. exp Skin Aging/ 8. wrinkl$3.ti,ab. 9. rhytid$.ti,ab. 10. (glabellar adj2 line$).mp. 11. ((aging or age or aged) adj3 skin).mp. 12. forehead line$.mp. 13. frown line$.mp. 14. crow's feet.mp. 15. marionette line$.mp. 16. or/7‐15 17. randomized controlled trial.pt. 18. controlled clinical trial.pt. 19. randomized.ab. 20. placebo.ab. 21. clinical trials as topic.sh. 22. randomly.ab. 23. trial.ti. 24. 17 or 18 or 19 or 20 or 21 or 22 or 23 25. exp animals/ not humans.sh. 26. 24 not 25 27. 6 and 16 and 26
[Lines 17‐26: Cochrane Highly Sensitive Search Strategy for identifying randomized trials in MEDLINE: sensitivity‐ and precision‐maximizing version (2008 revision); Ovid format, from section 3.6.1 in Lefebvre C, Glanville J, Briscoe S, Littlewood A, Marshall C, Metzendorf M‐I, et al. Technical Supplement to Chapter 4: Searching for and selecting studies. In: Higgins JPT, Thomas J, Chandler J, Cumpston MS, Li T, Page MJ, Welch VA (eds). Cochrane Handbook for Systematic Reviews of Interventions Version 6. Cochrane, 2019. Available from: www.training.cochrane.org/handbook]
Appendix 5. Embase (Ovid) search strategy
1. botulinum toxin/ or botulinum toxin a/ 2. onabotulinum$.mp. 3. abobotulinum$.mp. 4. incobotulinum$.mp. 5. (botox or vistabel or vistabex or dysport or reloxin or azzalure or bocoture or xeomin or xeomeen or prosigne or cbtx‐a or nt201 or dps refinex or pur tox).mp. 6. or/1‐5 7. wrinkl$3.ti,ab. 8. rhytid$.ti,ab. 9. (glabellar adj2 line$).mp. 10. ((aging or age or aged) adj3 skin).mp. 11. forehead line$.mp. 12. frown line$.mp. 13. crow's feet.mp. 14. marionette line$.mp. 15. cutaneous parameters/ 16. wrinkle/ 17. or/7‐16 18. crossover procedure.sh. 19. double‐blind procedure.sh. 20. single‐blind procedure.sh. 21. (crossover$ or cross over$).tw. 22. placebo$.tw. 23. (doubl$ adj blind$).tw. 24. allocat$.tw. 25. trial.ti. 26. randomized controlled trial.sh. 27. random$.tw. 28. or/18‐27 29. exp animal/ or exp invertebrate/ or animal experiment/ or animal model/ or animal tissue/ or animal cell/ or nonhuman/ 30. human/ or normal human/ 31. 29 and 30 32. 29 not 31 33. 28 not 32 34. 6 and 17 and 33
[Lines 18‐28: Based on terms suggested for identifying RCTs in Embase (section 3.6.2) in Lefebvre C, Glanville J, Briscoe S, Littlewood A, Marshall C, Metzendorf M‐I, et al. Technical Supplement to Chapter 4: Searching for and selecting studies. In: Higgins JPT, Thomas J, Chandler J, Cumpston MS, Li T, Page MJ, Welch VA (eds). Cochrane Handbook for Systematic Reviews of Interventions Version 6. Cochrane, 2019. Available from: www.training.cochrane.org/handbook]
Appendix 6. LILACS search strategy
((onabotulinum$ or abobotulinum$ or incobotulinum$ or botulinum$ or botox) and (wrinkl$ or rhytid$ or skin or piel or arruga$))
In LILACS we searched using the above terms and the Controlled clinical trials topic‐specific query filter.
Appendix 7. Trials register search strategies
These search terms were used for all the trials register searches:
1. botulinumtoxin A 2. facial wrinkles 3. skin ageing 4. skin aging 5. 1 and 2 6. 1 and 3 7. 1 and 4 8. 2 and 3 9. 2 and 4
Data and analyses
Comparison 1. OnabotulinumtoxinA 10units versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.2 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.2.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.2.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.2.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.2.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.3 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 2. OnabotulinumtoxinA 20 units versus placebo one cycle of treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 Participant assessment of success by analysing scores and scales | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1.1 4 weeks | 4 | 575 | Risk Ratio (M‐H, Random, 95% CI) | 19.45 [8.60, 43.99] |
| 2.1.2 8 weeks | 2 | 204 | Risk Ratio (M‐H, Random, 95% CI) | 28.45 [5.92, 136.74] |
| 2.1.3 12 weeks | 2 | 203 | Risk Ratio (M‐H, Random, 95% CI) | 12.77 [2.78, 58.72] |
| 2.1.4 16 weeks | 2 | 167 | Risk Ratio (M‐H, Random, 95% CI) | 20.71 [2.82, 151.91] |
| 2.1.5 24 weeks | 1 | 77 | Risk Ratio (M‐H, Random, 95% CI) | 4.19 [0.21, 84.41] |
| 2.2 Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 8 | 1390 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 3.62 [1.50, 8.74] |
| 2.3 Physician assessment of success by analysing scores and scales | 7 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.3.1 4 weeks | 7 | 1339 | Risk Ratio (M‐H, Random, 95% CI) | 17.10 [10.07, 29.05] |
| 2.3.2 8 weeks | 6 | 1046 | Risk Ratio (M‐H, Random, 95% CI) | 21.50 [9.68, 47.75] |
| 2.3.3 12 weeks | 6 | 1046 | Risk Ratio (M‐H, Random, 95% CI) | 10.81 [5.79, 20.16] |
| 2.3.4 16 weeks | 5 | 933 | Risk Ratio (M‐H, Random, 95% CI) | 15.13 [5.98, 38.27] |
| 2.3.5 20 weeks | 1 | 77 | Risk Ratio (M‐H, Random, 95% CI) | 5.86 [0.31, 109.74] |
| 2.3.6 24 weeks | 1 | 77 | Risk Ratio (M‐H, Random, 95% CI) | 4.19 [0.21, 84.41] |
| 2.4 Total adverse events | 8 | 1388 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.89, 1.45] |
| 2.5 Duration of treatment (weeks) | 1 | 77 | Mean Difference (IV, Random, 95% CI) | 18.40 [16.17, 20.63] |
Comparison 3. OnabotulinumtoxinA 20units versus 10 units one treatment glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 Participant assessment of success by analysing scores and scales | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1.1 4 weeks | 2 | 131 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.96, 1.29] |
| 3.1.2 8 weeks | 2 | 131 | Risk Ratio (M‐H, Random, 95% CI) | 1.24 [1.01, 1.52] |
| 3.1.3 16 weeks | 2 | 131 | Risk Ratio (M‐H, Random, 95% CI) | 1.23 [0.75, 2.01] |
| 3.2 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 2 | 131 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.93 [0.20, 18.70] |
| 3.3 Physician assessment of success by analysing scores and scales | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.3.1 4 weeks | 2 | 131 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.91, 1.20] |
| 3.3.2 8 weeks | 2 | 131 | Risk Ratio (M‐H, Random, 95% CI) | 1.53 [0.76, 3.10] |
| 3.3.3 16 weeks | 2 | 131 | Risk Ratio (M‐H, Random, 95% CI) | 1.43 [0.76, 2.69] |
| 3.4 Total adverse events | 2 | 131 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.49, 2.37] |
Comparison 4. OnabotulinumtoxinA 64units versus placebo one cycle of treatment, upper wrinkles.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 4.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1.5 20 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1.6 24 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.2 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.3 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.3.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.3.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.3.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.3.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.3.5 20 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.3.6 24 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.4 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 5. OnabotulinumtoxinA 40units versus placebo one cycle of treatment, upper wrinkles.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 5.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1.1 4 weeks | 1 | 474 | Risk Ratio (M‐H, Random, 95% CI) | 7.41 [4.02, 13.64] |
| 5.1.2 8 weeks | 1 | 474 | Risk Ratio (M‐H, Random, 95% CI) | 86.13 [5.38, 1378.82] |
| 5.1.3 12 weeks | 1 | 474 | Risk Ratio (M‐H, Random, 95% CI) | 39.87 [2.47, 644.08] |
| 5.1.4 16 weeks | 1 | 474 | Risk Ratio (M‐H, Random, 95% CI) | 11.32 [0.67, 190.86] |
| 5.1.5 20 weeks | 1 | 474 | Risk Ratio (M‐H, Random, 95% CI) | 2.46 [0.12, 50.95] |
| 5.1.6 24 weeks | 1 | 474 | Risk Ratio (M‐H, Random, 95% CI) | 1.48 [0.06, 36.04] |
| 5.2 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5.3 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.3.1 4 weeks | 1 | 474 | Risk Ratio (M‐H, Random, 95% CI) | 7.41 [4.02, 13.64] |
| 5.3.2 8 weeks | 1 | 474 | Risk Ratio (M‐H, Random, 95% CI) | 86.13 [5.38, 1378.82] |
| 5.3.3 12 weeks | 1 | 474 | Risk Ratio (M‐H, Random, 95% CI) | 39.87 [2.47, 644.08] |
| 5.3.4 16 weeks | 1 | 474 | Risk Ratio (M‐H, Random, 95% CI) | 11.32 [0.67, 190.86] |
| 5.3.5 20 weeks | 1 | 474 | Risk Ratio (M‐H, Random, 95% CI) | 2.46 [0.12, 50.95] |
| 5.3.6 24 weeks | 1 | 474 | Risk Ratio (M‐H, Random, 95% CI) | 1.48 [0.06, 36.04] |
| 5.4 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 6. OnabotulinumtoxinA 64units versus OnabotulinumoxinA 40U one cycle of treatment, upper wrinkles.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 6.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.1.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.1.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.1.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.1.5 20 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.1.6 24 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.2 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.3 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.3.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.3.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.3.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.3.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.3.5 20 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.3.6 24 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.4 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 7. OnabotulinumtoxinA 64units versus 32 units one cycle of treatment upper wrinkles.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 7.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.1.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.1.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.1.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.1.5 20 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.1.6 24 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.2 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.2.1 4 and 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.2.2 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.2.3 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.2.4 20 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.3 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only |
Comparison 8. OnabotulinumtoxinA 96 units versus 32 units one cycle of treatment upper wrinkles.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 8.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.1.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.1.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.1.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.1.5 20 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.1.6 24 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.1.7 28 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.1.8 32 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.2 Physician assessment of success by analysing scores and scales | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.2.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.2.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.2.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.2.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.2.5 20 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.2.6 24 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.2.7 28 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.2.8 32 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.3 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 9. OnabotulinumtoxinA 96 units versus 64 units one cycle of treatment upper wrinkles.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 9.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.1.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.1.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.1.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.1.5 20 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.1.6 24 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.1.7 28 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.1.8 32 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.2 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.2.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.2.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.2.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.2.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.2.5 20 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.2.6 24 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.2.7 28 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.2.8 32 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.3 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 10. OnabotulinumtoxinA 32 units versus 16 units one cycle of treatment forehead lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 10.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.1.1 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.2 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.3 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.3.1 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 11. OnabotulinumtoxinA 60 units versus 20 units one cycle of treatment glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 11.1 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.1.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.1.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.1.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.1.5 20 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.2 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 12. OnabotulinumtoxinA 48 units versus 32 units one cycle of treatment forehead lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 12.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12.1.1 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12.2 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12.3 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12.3.1 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 13. OnabotulinumtoxinA 40 units versus placebo one cycle of treatment for forehead lines and glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 13.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.1.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.1.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.1.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.1.5 20 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.1.6 24 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.2 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.2.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.2.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.2.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.2.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.2.5 20 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.2.6 24 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.3 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 14. OnabotulinumtoxinA 30 units versus placebo one cycle of treatment for forehead lines and glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 14.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.1.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.1.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.1.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.1.5 20 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.1.6 24 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.2 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.3 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 14.3.1 4 weeks | 1 | 118 | Risk Ratio (M‐H, Random, 95% CI) | 24.00 [6.11, 94.23] |
| 14.3.2 8 weeks | 1 | 118 | Risk Ratio (M‐H, Random, 95% CI) | 43.00 [6.12, 302.08] |
| 14.3.3 12 weeks | 1 | 118 | Risk Ratio (M‐H, Random, 95% CI) | 8.67 [2.77, 27.08] |
| 14.3.4 16 weeks | 1 | 118 | Risk Ratio (M‐H, Random, 95% CI) | 3.33 [1.44, 7.71] |
| 14.3.5 20 weeks | 1 | 118 | Risk Ratio (M‐H, Random, 95% CI) | 15.00 [2.05, 109.93] |
| 14.3.6 24 weeks | 1 | 118 | Risk Ratio (M‐H, Random, 95% CI) | 8.00 [1.03, 61.98] |
| 14.4 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 15. OnabotulinumtoxinA 40 units versus OnabotulinumonabotulinumtoxinA 30 units one cycle of treatment for forehead lines and glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 15.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15.1.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15.1.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15.1.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15.1.5 20 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15.1.6 24 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15.2 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15.3 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15.3.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15.3.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15.3.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15.3.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15.3.5 20 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15.3.6 24 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15.4 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 16. OnabotulinumtoxinA 40 units versus 20 units one treatment glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 16.1 Participant assessment of success by analysing scores and scales | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 16.1.1 4 weeks | 2 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 1.63 [1.13, 2.35] |
| 16.1.2 8 weeks | 2 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 1.52 [0.77, 2.97] |
| 16.1.3 16 weeks | 2 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 2.51 [0.88, 7.16] |
| 16.2 Physician assessment of success by analysing scores and scales | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 16.2.1 4 weeks | 2 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 1.48 [0.93, 2.36] |
| 16.2.2 8 weeks | 2 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 1.35 [0.82, 2.23] |
| 16.2.3 16 weeks | 2 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 1.34 [0.51, 3.53] |
| 16.3 Total adverse events | 2 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 1.93 [0.53, 7.05] |
Comparison 17. OnabotulinumtoxinA 48 units versus 16 units one cycle of treatment forehead lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 17.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 17.1.1 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 17.2 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 17.3 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 17.3.1 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 18. OnabotulinumtoxinA 80 units versus 20 units one cycle of treatment glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 18.1 Participant assessment, maximum contraction (responder rate) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18.2 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18.2.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18.2.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18.2.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18.2.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18.2.5 20 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18.2.6 24 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18.3 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 19. OnabotulinumtoxinA 80 units versus 60 units one cycle of treatment glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 19.1 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 19.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 19.1.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 19.1.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 19.1.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 19.1.5 20 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 19.1.6 24 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 19.2 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 20. OnabotulinumtoxinA 30 units versus 10 units one cycle of treatment glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 20.1 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 20.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 20.1.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 20.1.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 20.1.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 20.2 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 21. OnabotulinumtoxinA 40 units versus 10 units one cycle of treatment glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 21.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 21.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 21.1.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 21.1.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 21.1.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 21.2 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 21.2.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 21.2.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 21.2.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 21.2.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 21.3 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 22. OnabotulinumtoxinA 40 units versus 30 units one cycle of treatment glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 22.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.1.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.1.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.1.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.2 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.2.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.2.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.2.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.2.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22.3 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 23. OnabotulinumtoxinA 30 units versus 20 units one cycle of treatment glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 23.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 23.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 23.1.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 23.1.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 23.1.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 23.2 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 23.2.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 23.2.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 23.2.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 23.2.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 23.3 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 24. OnabotulinumtoxinA 60 units versus 40 units one cycle of treatment glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 24.1 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 24.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 24.1.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 24.1.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 24.1.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 24.1.5 20 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 24.2 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 25. OnabotulinumtoxinA 80 units versus 40 units one cycle of treatment glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 25.1 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 25.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 25.1.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 25.1.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 25.1.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 25.1.5 20 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 25.1.6 24 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 25.2 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 26. OnabotulinumtoxinA 80units versus 60units one cycle of treatment glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 26.1 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 26.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 26.1.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 26.1.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 26.1.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 26.1.5 20 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 26.1.6 24 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 26.2 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 27. OnabotulinumtoxinA 12 units versus 7.5 units one cycle of treatment perioral lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 27.1 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 27.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 27.1.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 27.1.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 27.1.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 27.1.5 20 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 27.2 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 28. OnabotulinumtoxinA 20units versus placebo in glabellar lines three cycles of treatment.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 28.1 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 28.2 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 29. OnabotulinumtoxinA24 units versus placebo one treatment in crow's feet lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 29.1 Participant assessment of success by analysing scores and scales | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 29.1.1 4 weeks | 3 | 1474 | Risk Ratio (M‐H, Random, 95% CI) | 12.71 [8.67, 18.63] |
| 29.1.2 8 weeks | 3 | 1474 | Risk Ratio (M‐H, Random, 95% CI) | 10.25 [7.02, 14.98] |
| 29.1.3 12 weeks | 3 | 1474 | Risk Ratio (M‐H, Random, 95% CI) | 7.70 [4.81, 12.33] |
| 29.1.4 16 weeks | 1 | 417 | Risk Ratio (M‐H, Random, 95% CI) | 12.31 [4.68, 32.36] |
| 29.1.5 20 weeks | 1 | 417 | Risk Ratio (M‐H, Random, 95% CI) | 10.33 [3.35, 31.89] |
| 29.2 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 2 | 665 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.28 [0.12, 13.10] |
| 29.3 Physician assessment of success by analysing scores and scales | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 29.3.1 4 weeks | 4 | 1675 | Risk Ratio (M‐H, Random, 95% CI) | 12.38 [8.93, 17.16] |
| 29.3.2 8 weeks | 3 | 1258 | Risk Ratio (M‐H, Random, 95% CI) | 10.13 [5.34, 19.23] |
| 29.3.3 12 weeks | 3 | 1258 | Risk Ratio (M‐H, Random, 95% CI) | 9.29 [5.95, 14.50] |
| 29.3.4 16 weeks | 2 | 1057 | Risk Ratio (M‐H, Random, 95% CI) | 5.46 [3.19, 9.32] |
| 29.4 Total adverse events | 2 | 692 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.94, 1.45] |
Comparison 30. OnabotulinumtoxinA 12 units versus placebo one treatment in crow's feet lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 30.1 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 30.2 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 30.2.1 4 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 30.2.2 8 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 30.2.3 12 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 30.3 Total adverse events | 2 | 657 | Risk Ratio (IV, Random, 95% CI) | 1.33 [0.87, 2.02] |
Comparison 31. OnabotulinumtoxinA 24 units versus OnabotulinumtoxinA 12 units one treatment in crow's feet lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 31.1 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 31.2 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 31.2.1 4 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 31.2.2 8 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 31.2.3 12 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 31.3 Total adverse events | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected |
Comparison 32. OnabotulinumtoxinA 44 units versus placebo one treatments in glabellar lines and crow's feet lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 32.1 Participant assessment of success by analysing scores and scales | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 32.1.1 4 weeks | 2 | 808 | Risk Ratio (M‐H, Random, 95% CI) | 16.33 [9.27, 28.76] |
| 32.1.2 8 weeks | 1 | 611 | Risk Ratio (M‐H, Random, 95% CI) | 8.95 [5.03, 15.90] |
| 32.1.3 12 weeks | 1 | 611 | Risk Ratio (M‐H, Random, 95% CI) | 5.10 [2.80, 9.28] |
| 32.1.4 16 weeks | 1 | 611 | Risk Ratio (M‐H, Random, 95% CI) | 2.51 [1.31, 4.81] |
| 32.2 Physician assessment of success by analysing scores and scales | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 32.2.1 4 weeks | 2 | 808 | Risk Ratio (M‐H, Random, 95% CI) | 11.09 [4.12, 29.83] |
| 32.2.2 8 weeks | 2 | 808 | Risk Ratio (M‐H, Random, 95% CI) | 9.94 [1.78, 55.44] |
| 32.2.3 12 weeks | 2 | 808 | Risk Ratio (M‐H, Random, 95% CI) | 5.96 [0.60, 58.98] |
| 32.3 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 33. OnabotulinumtoxinA 44 units versus placebo two cycles of treatments in crow's feet lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 33.1 Participant assessment of success by analysing scores and scales | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 33.1.1 4 weeks | 2 | 808 | Risk Ratio (M‐H, Random, 95% CI) | 10.12 [6.36, 16.09] |
| 33.1.2 8 weeks | 1 | 611 | Risk Ratio (M‐H, Random, 95% CI) | 8.95 [5.03, 15.90] |
| 33.1.3 12 weeks | 1 | 611 | Risk Ratio (M‐H, Random, 95% CI) | 4.85 [2.66, 8.84] |
| 33.2 Physician assessment of success by analysing scores and scales | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 33.2.1 4 weeks | 2 | 808 | Risk Ratio (M‐H, Random, 95% CI) | 17.63 [9.50, 32.69] |
| 33.2.2 8 weeks | 2 | 808 | Risk Ratio (M‐H, Random, 95% CI) | 19.86 [9.90, 39.87] |
| 33.3 Total adverse events | 2 | 808 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [0.98, 1.30] |
Comparison 34. OnabotulinumtoxinA 24 units versus 12 units five cycles of treatment in crow's feet lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 34.1 Total adverse events | 1 | 294 | Risk Ratio (M‐H, Random, 95% CI) | 1.42 [0.41, 4.93] |
Comparison 35. AbobotulinumtoxinA 25 units versus placebo one cycle of treatment, glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 35.1 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 35.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 35.1.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 35.1.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 35.1.4 24 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 35.2 Total adverse events | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Totals not selected |
Comparison 36. AbobotulinumtoxinA 30 units versus placebo one cycle of treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 36.1 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 37. AbobotulinumtoxinA 50 units versus placebo one treatment glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 37.1 Participant assessment of success by analysing scores and scales | 5 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 37.1.1 4 weeks | 5 | 915 | Risk Ratio (M‐H, Random, 95% CI) | 21.22 [7.43, 60.56] |
| 37.1.2 8 weeks | 3 | 725 | Risk Ratio (M‐H, Random, 95% CI) | 39.89 [14.10, 112.88] |
| 37.1.3 12 weeks | 3 | 725 | Risk Ratio (M‐H, Random, 95% CI) | 28.83 [10.16, 81.81] |
| 37.1.4 16 weeks | 2 | 371 | Risk Ratio (M‐H, Random, 95% CI) | 11.78 [4.12, 33.66] |
| 37.1.5 20 weeks | 1 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 5.33 [1.67, 16.99] |
| 37.2 Major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 7 | 1294 | Risk Ratio (M‐H, Random, 95% CI) | 3.36 [0.88, 12.87] |
| 37.3 Physician assessment of success by analysing scores and scales | 7 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 37.3.1 4 weeks | 7 | 1060 | Risk Ratio (M‐H, Random, 95% CI) | 15.78 [8.75, 28.45] |
| 37.3.2 8 weeks | 5 | 802 | Risk Ratio (M‐H, Random, 95% CI) | 30.84 [11.58, 82.12] |
| 37.3.3 12 weeks | 6 | 900 | Risk Ratio (M‐H, Random, 95% CI) | 17.79 [6.70, 47.28] |
| 37.3.4 16 weeks | 2 | 371 | Risk Ratio (M‐H, Random, 95% CI) | 29.88 [6.01, 148.52] |
| 37.3.5 20 weeks | 1 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 17.00 [2.36, 122.39] |
| 37.4 Total adverse events | 8 | 1471 | Risk Ratio (M‐H, Random, 95% CI) | 1.25 [1.05, 1.49] |
| 37.5 Duration of treatment (days) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
Comparison 38. AbobotulinumtoxinA 50 units versus 25 units one cycle of treatment, glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 38.1 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 38.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 38.1.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 38.1.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 38.1.4 24 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 38.2 Total adverse events | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Totals not selected |
Comparison 39. AbobotulinumtoxinA 60 units versus placebo one cycle of treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 39.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 39.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 39.2 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 39.2.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 40. AbobotulinumtoxinA 70 units versus placebo one cycle of treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 40.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 40.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 40.2 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 40.2.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 41. AbobotulinumtoxinA 75 units versus placebo one cycle of treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 41.1 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 41.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 41.1.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 41.1.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 41.1.4 24 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 42. AbobotulinumtoxinA 75 units versus 25 units one cycle of treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 42.1 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 42.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 42.1.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 42.1.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 42.1.4 24 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 43. AbobotulinumtoxinA 75 units versus 50 units one cycle of treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 43.1 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 43.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 43.1.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 43.1.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 43.1.4 24 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 43.2 Total adverse events | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Totals not selected |
Comparison 44. AbobotulinumtoxinA 80 units versus placebo one cycle of treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 44.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 44.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 44.2 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 44.2.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 45. AbobotulinumtoxinA 15 units versus placebo one cycle of treatment crow's feet lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 45.1 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 45.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 46. AbobotulinumtoxinA 30 units versus placebo one cycle of treatment crow's feet lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 46.1 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 46.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 47. AbobotulinumtoxinA 45 units versus placebo one cycle of treatment crow's feet lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 47.1 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 47.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 48. AbobotulinumtoxinA 50 units versus placebo, three cycles of treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 48.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 48.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 48.2 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 48.2.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 48.3 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 49. IncobotulinumtoxinA 20 units versus placebo one treatment glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 49.1 Participant assessment of success by analysing scores and scales | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 49.1.1 4 weeks | 2 | 547 | Risk Ratio (M‐H, Random, 95% CI) | 66.57 [13.50, 328.28] |
| 49.1.2 8 weeks | 2 | 547 | Risk Ratio (M‐H, Random, 95% CI) | 7.35 [4.79, 11.29] |
| 49.1.3 12 weeks | 2 | 547 | Risk Ratio (M‐H, Random, 95% CI) | 7.29 [4.38, 12.13] |
| 49.1.4 16 weeks | 2 | 547 | Risk Ratio (M‐H, Random, 95% CI) | 4.40 [2.61, 7.41] |
| 49.2 Physician assessment of success by analysing scores and scales | 2 | 547 | Risk Ratio (M‐H, Random, 95% CI) | 134.62 [19.05, 951.45] |
| 49.3 Total adverse events | 2 | 547 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.90, 1.53] |
Comparison 50. IncobotulinumtoxinA 54 to 64units versus placebo one cycles of treatment glabellar lines, forehead liens, crow's feet lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 50.1 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 50.2 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 50.2.1 4 weeks‐ Glabella | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 50.2.2 4 weeks ‐ forehead | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 50.2.3 4 weeks crow's feet line | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 50.3 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 51. HBTX‐A 10 units versus placebo one cycle of treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 51.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 51.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 51.1.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 51.1.3 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 51.2 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 52. HBTX‐A 20 units versus placebo one cycle of treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 52.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 52.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 52.1.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 52.1.3 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 52.2 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 53. HBTX‐A 50 units versus placebo one cycle of treatment, glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 53.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 53.1.1 4 weeks | 1 | 187 | Risk Ratio (M‐H, Random, 95% CI) | 42.68 [6.08, 299.41] |
| 53.1.2 8 weeks | 1 | 182 | Risk Ratio (M‐H, Random, 95% CI) | 43.28 [6.18, 303.19] |
| 53.1.3 12 weeks | 1 | 185 | Risk Ratio (M‐H, Random, 95% CI) | 21.67 [3.06, 153.72] |
| 53.2 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 53.2.1 4 weeks | 1 | 185 | Risk Ratio (M‐H, Random, 95% CI) | 49.69 [7.10, 347.71] |
| 53.2.2 8 weeks | 1 | 167 | Risk Ratio (M‐H, Random, 95% CI) | 53.52 [7.65, 374.34] |
| 53.2.3 12 weeks | 1 | 183 | Risk Ratio (M‐H, Random, 95% CI) | 33.17 [4.72, 233.16] |
| 53.3 Total adverse events | 1 | 190 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.22 [0.02, 2.50] |
| 53.3.1 Total adverse events | 1 | 190 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.22 [0.02, 2.50] |
Comparison 54. HBTX‐A 20 units versus 10 units one cycle of treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 54.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 54.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 54.1.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 54.1.3 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 54.2 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 55. AbobotulinumtoxinA 50 units versus OnabotulinumtoxinA 20 units one cycle of treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 55.1 Participant assessment success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 55.1.1 4 weeks | 1 | 388 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.92, 1.08] |
| 55.1.2 8 weeks | 1 | 388 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.88, 1.05] |
| 55.1.3 12 weeks | 1 | 388 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.81, 1.09] |
| 55.2 Major adverse events | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Subtotals only | |
| 55.2.1 Any major adverse events (eyelid ptosis, eyelid sensory disorder, s | 1 | 433 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 2.65 [0.77, 9.09] |
| 55.3 Physician assessment of success by analysing scores and scales | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 55.3.1 4 weeks | 1 | 388 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.95, 1.06] |
| 55.3.2 8 weeks | 2 | 449 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.89, 1.02] |
| 55.3.3 12 weeks | 2 | 448 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.60, 1.40] |
| 55.3.4 16 weeks | 1 | 59 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.13, 1.55] |
| 55.4 Total adverse events | 2 | 492 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.67, 1.54] |
55.1. Analysis.

Comparison 55: AbobotulinumtoxinA 50 units versus OnabotulinumtoxinA 20 units one cycle of treatment in glabellar lines, Outcome 1: Participant assessment success by analysing scores and scales
Comparison 56. IncobotulinumtoxinA 30 units versus OnabotulinumtoxinA 20 units one cycle of treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 56.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 56.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 56.1.2 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 56.1.3 14 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 56.1.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 56.2 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 56.2.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 56.2.2 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 56.2.3 14 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 56.2.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 56.3 Total adverse events | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Totals not selected |
Comparison 57. IncobotulinumtoxinA 24 units versus OnabotulinumtoxinA 24 units one treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 57.1 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Totals not selected | |
| 57.2 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 57.2.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 57.2.2 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 57.3 Total adverse events | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Totals not selected |
Comparison 58. IncobotulinumtoxinA 20 units versus OnabotulinumtoxinA 20 units one treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 58.1 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Totals not selected | |
| 58.2 Physician assessment of success by analysing scores and scales ‐ injector | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 58.2.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 58.2.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 58.2.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 58.2.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 58.3 Physician assessment of success by analysing scores and scales ‐ independent observer | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 58.3.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 58.3.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 58.3.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 58.3.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 58.4 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 59. NewBontA [Medytox®] 20 units versus OnabotulinumtoxinA 20 units one treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 59.1 Physician assessment, maximum contraction (responder rate) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 59.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 60. NewBontA [Neuronox®] 20 units versus OnabotulinumtoxinA 20 units one treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 60.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 60.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 60.1.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 60.1.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 60.1.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 60.2 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 60.3 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 60.3.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 60.3.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 60.3.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 60.3.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 60.4 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 61. NewBontA [Neuronox®] 20 units versus OnabotulinumtoxinA 20 units one treatment in crow’s feet lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 61.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 61.1.1 4 weeks | 1 | 220 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.89, 1.15] |
| 61.1.2 8 weeks | 1 | 220 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.83, 1.03] |
| 61.1.3 12 weeks | 1 | 220 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.90, 1.16] |
| 61.1.4 16 weeks | 1 | 220 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.70, 1.01] |
| 61.2 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Subtotals only | |
| 61.3 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 61.3.1 4 weeks | 1 | 220 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.89, 1.13] |
| 61.3.2 8 weeks | 1 | 220 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.86, 1.11] |
| 61.3.3 12 weeks | 1 | 220 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.88, 1.32] |
| 61.3.4 16 weeks | 1 | 220 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.83, 1.56] |
| 61.4 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 61.4.1 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 62. Liquid BontA 20 units versus OnabotulinumtoxinA 20 units one treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 62.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 62.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 62.1.2 10 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 62.1.3 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 62.2 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 62.2.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 62.2.2 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 62.3 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 63. NewBontA (Prosigne®) 20 units versus OnabotulinumtoxinA 20 units one treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 63.1 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 63.1.1 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 64. CBFC26 20 units versus OnabotulinumtoxinA 20 units one treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 64.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 64.1.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 64.1.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 64.1.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 64.1.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 64.2 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 64.3 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 64.3.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 64.3.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 64.3.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 64.3.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 64.4 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 65. Liquid AbobotulinumtoxinA 20 units versus placebo one treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 65.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 65.1.1 4 weeks | 1 | 71 | Risk Ratio (M‐H, Random, 95% CI) | 66.81 [4.25, 1050.36] |
| 65.1.2 8 weeks | 1 | 71 | Risk Ratio (M‐H, Random, 95% CI) | 42.14 [2.65, 670.89] |
| 65.1.3 12 weeks | 1 | 71 | Risk Ratio (M‐H, Random, 95% CI) | 44.19 [2.78, 702.51] |
| 65.1.4 16 weeks | 1 | 71 | Risk Ratio (M‐H, Random, 95% CI) | 27.75 [1.71, 449.60] |
| 65.2 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Subtotals only | |
| 65.2.1 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | 71 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 7.60 [0.15, 383.33] |
| 65.3 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 65.3.1 4 weeks | 1 | 71 | Risk Ratio (M‐H, Random, 95% CI) | 66.81 [4.25, 1050.36] |
| 65.3.2 8 weeks | 1 | 71 | Risk Ratio (M‐H, Random, 95% CI) | 58.58 [3.71, 923.86] |
| 65.3.3 12 weeks | 1 | 71 | Risk Ratio (M‐H, Random, 95% CI) | 40.08 [2.51, 639.27] |
| 65.3.4 16 weeks | 1 | 71 | Risk Ratio (M‐H, Random, 95% CI) | 19.53 [1.18, 323.24] |
| 65.4 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 65.4.1 Totl adverse events | 1 | 71 | Risk Ratio (M‐H, Random, 95% CI) | 1.23 [0.41, 3.68] |
Comparison 66. Liquid AbobotulinumtoxinA 50 units versus placebo one treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 66.1 Participant assessment success by analysing scores and scales | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 66.1.1 4 weeks | 2 | 255 | Risk Ratio (M‐H, Random, 95% CI) | 46.95 [9.57, 230.36] |
| 66.1.2 8 weeks | 2 | 253 | Risk Ratio (M‐H, Random, 95% CI) | 41.87 [8.52, 205.88] |
| 66.1.3 12 weeks | 2 | 254 | Risk Ratio (M‐H, Random, 95% CI) | 24.61 [4.97, 121.91] |
| 66.1.4 16 weeks | 2 | 254 | Risk Ratio (M‐H, Random, 95% CI) | 6.54 [1.70, 25.15] |
| 66.1.5 20 weeks | 1 | 183 | Risk Ratio (M‐H, Random, 95% CI) | 3.35 [1.71, 6.58] |
| 66.2 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 2 | 256 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 4.39 [0.07, 289.13] |
| 66.3 Physician assessment of success by analysing scores and scales | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 66.3.1 4 weeks | 2 | 255 | Risk Ratio (M‐H, Random, 95% CI) | 16.73 [2.84, 98.58] |
| 66.3.2 8 weeks | 2 | 255 | Risk Ratio (M‐H, Random, 95% CI) | 48.98 [9.99, 240.21] |
| 66.3.3 12 weeks | 2 | 255 | Risk Ratio (M‐H, Random, 95% CI) | 35.93 [7.30, 176.90] |
| 66.3.4 16 weeks | 2 | 255 | Risk Ratio (M‐H, Random, 95% CI) | 21.25 [2.95, 152.88] |
| 66.3.5 20 weeks | 1 | 184 | Risk Ratio (M‐H, Random, 95% CI) | 25.86 [1.60, 417.34] |
| 66.4 Total adverse events | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 66.4.1 Total adverse events | 2 | 255 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.72, 1.71] |
66.4. Analysis.

Comparison 66: Liquid AbobotulinumtoxinA 50 units versus placebo one treatment in glabellar lines, Outcome 4: Total adverse events
Comparison 67. Liquid AbobotulinumtoxinA 75 units versus placebo one treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 67.1 Participant assessment of success by analysing scores and scales | 1 | 284 | Risk Ratio (M‐H, Random, 95% CI) | 49.64 [12.47, 197.64] |
| 67.1.1 4 weeks | 1 | 71 | Risk Ratio (M‐H, Random, 95% CI) | 60.64 [3.85, 955.48] |
| 67.1.2 8 weeks | 1 | 71 | Risk Ratio (M‐H, Random, 95% CI) | 62.69 [3.98, 987.11] |
| 67.1.3 12 weeks | 1 | 71 | Risk Ratio (M‐H, Random, 95% CI) | 44.19 [2.78, 702.51] |
| 67.1.4 16 weeks | 1 | 71 | Risk Ratio (M‐H, Random, 95% CI) | 35.97 [2.25, 576.04] |
| 67.2 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | 71 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 7.83 [0.48, 127.75] |
| 67.3 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 67.3.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 67.3.2 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 67.3.3 12 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 67.3.4 16 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 67.4 Total adverse events | 1 | 71 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.24, 2.81] |
Comparison 68. Liquid AbobotulinumtoxinA 20 units versus Liquid AbobotulinumtoxinA 50 units one cycle of treatment, glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 68.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 68.1.1 4 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.90, 1.26] |
| 68.1.2 8 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.69, 1.60] |
| 68.1.3 12 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.31 [0.84, 2.06] |
| 68.1.4 16 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.58, 2.03] |
| 68.2 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Subtotals only | |
| 68.2.1 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | 70 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 7.39 [0.15, 372.38] |
| 68.3 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 68.3.1 4 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.90, 1.26] |
| 68.3.2 8 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.47 [1.04, 2.08] |
| 68.3.3 12 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.19 [0.74, 1.90] |
| 68.3.4 16 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.36, 1.55] |
| 68.4 Total adverse events | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.50 [0.46, 4.86] |
| 68.4.1 Total adverse events | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.50 [0.46, 4.86] |
Comparison 69. Liquid AbobotulinumtoxinA 20 units versus Liquid AbobotulinumtoxinA 75 units one cycle of treatment, glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 69.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 69.1.1 4 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.10 [0.92, 1.32] |
| 69.1.2 8 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.49, 0.92] |
| 69.1.3 12 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.66, 1.38] |
| 69.1.4 16 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 0.76 [0.44, 1.32] |
| 69.2 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Subtotals only | |
| 69.2.1 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | 70 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.50 [0.05, 5.00] |
| 69.3 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 69.3.1 4 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.88, 1.21] |
| 69.3.2 8 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.77, 1.21] |
| 69.3.3 12 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 0.70 [0.49, 1.00] |
| 69.3.4 16 weeks | 1 | 78 | Risk Ratio (M‐H, Random, 95% CI) | 0.55 [0.29, 1.06] |
| 69.4 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 69.4.1 Total adverse events | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 2.50 [0.87, 7.22] |
Comparison 70. Liquid AbobotulinumtoxinA 50 units versus Liquid AbobotulinumtoxinA 75 units one cycle of treatment, glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 70.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 70.1.1 4 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.83, 1.21] |
| 70.1.2 8 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 0.63 [0.45, 0.88] |
| 70.1.3 12 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 0.76 [0.49, 1.20] |
| 70.1.4 16 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.40, 1.25] |
| 70.2 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Subtotals only | |
| 70.2.1 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | 70 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.13 [0.01, 2.14] |
| 70.3 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 70.3.1 4 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.81, 1.16] |
| 70.3.2 8 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 0.66 [0.47, 0.92] |
| 70.3.3 12 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 0.59 [0.40, 0.89] |
| 70.3.4 16 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 0.60 [0.35, 1.03] |
| 70.4 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 70.4.1 Total adverse events | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.27, 3.69] |
Comparison 71. Liquid AbobotulinumtoxinA 20 units versus AbobotulinumtoxinA 50 units one cycle of treatment, glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 71.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 71.1.1 4 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.10 [0.92, 1.32] |
| 71.1.2 8 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.62, 1.33] |
| 71.1.3 12 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.50 [0.92, 2.44] |
| 71.1.4 16 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.51, 1.68] |
| 71.2 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Subtotals only | |
| 71.2.1 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | 70 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 7.39 [0.15, 372.38] |
| 71.3 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 71.3.1 4 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.19 [0.96, 1.46] |
| 71.3.2 8 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.79, 1.26] |
| 71.3.3 12 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.65, 1.54] |
| 71.3.4 16 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 0.69 [0.34, 1.41] |
| 71.4 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 71.4.1 Total adverse events | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 3.00 [0.65, 13.86] |
Comparison 72. Liquid AbobotulinumtoxinA 75 units versus AbobotulinumtoxinA 50 units one cycle of treatment, glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 72.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 72.1.1 4 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.81, 1.24] |
| 72.1.2 8 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.36 [1.02, 1.82] |
| 72.1.3 12 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.57 [0.97, 2.54] |
| 72.1.4 16 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.70 [0.91, 3.18] |
| 72.2 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Subtotals only | |
| 72.2.1 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | 70 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 7.61 [0.47, 124.15] |
| 72.3 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 72.3.1 4 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.15 [0.93, 1.43] |
| 72.3.2 8 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.83, 1.30] |
| 72.3.3 12 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.42 [1.00, 2.02] |
| 72.3.4 16 weeks | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 1.54 [0.92, 2.58] |
| 72.4 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 72.4.1 Total adverse events | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 2.00 [0.39, 10.22] |
Comparison 73. DaxibotulinumtoxinA 60 units versus placebo one cycle of treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 73.1 Participants assessment of success by analysing scores and scales | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 73.1.1 4 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 73.1.2 16 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 73.1.3 24 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 73.2 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 73.3 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 73.3.1 4 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 73.3.2 8 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 73.3.3 12 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 73.3.4 16 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 73.3.5 20 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 73.3.6 24 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 73.4 Total adverse events | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 73.5 Duration of treatment | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
Comparison 74. DaxibotulinumtoxinA 40 units versus placebo one cycle of treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 74.1 Participant assessment of success by analysing scores and scales | 2 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 74.1.1 4 weeks | 2 | 683 | Risk Ratio (IV, Random, 95% CI) | 21.10 [11.31, 39.34] |
| 74.1.2 8 weeks | 1 | 609 | Risk Ratio (IV, Random, 95% CI) | 15.75 [8.85, 28.03] |
| 74.1.3 12 weeks | 1 | 609 | Risk Ratio (IV, Random, 95% CI) | 24.51 [11.12, 54.05] |
| 74.1.4 16 weeks | 2 | 683 | Risk Ratio (IV, Random, 95% CI) | 12.74 [6.80, 23.89] |
| 74.1.5 20 weeks | 1 | 609 | Risk Ratio (IV, Random, 95% CI) | 13.60 [6.13, 30.18] |
| 74.1.6 24 weeks | 2 | 683 | Risk Ratio (IV, Random, 95% CI) | 10.25 [4.01, 26.20] |
| 74.2 Physician assessment of success by analysing scores and scales | 2 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 74.2.1 4 weeks | 2 | 683 | Risk Ratio (IV, Random, 95% CI) | 23.40 [12.56, 43.61] |
| 74.2.2 8 weeks | 2 | 683 | Risk Ratio (IV, Random, 95% CI) | 18.09 [10.30, 31.78] |
| 74.2.3 12 weeks | 2 | 683 | Risk Ratio (IV, Random, 95% CI) | 29.46 [13.79, 62.94] |
| 74.2.4 16 weeks | 2 | 683 | Risk Ratio (IV, Random, 95% CI) | 16.84 [9.01, 31.47] |
| 74.2.5 20 weeks | 2 | 683 | Risk Ratio (IV, Random, 95% CI) | 18.06 [8.42, 38.76] |
| 74.2.6 24 weeks | 2 | 683 | Risk Ratio (IV, Random, 95% CI) | 15.33 [6.06, 38.78] |
| 74.3 Total adverse events | 2 | 716 | Risk Ratio (IV, Random, 95% CI) | 2.23 [1.46, 3.40] |
| 74.4 Duration of treatment effect, weeks | 1 | 74 | Mean Difference (IV, Random, 95% CI) | 22.80 [20.74, 24.86] |
Comparison 75. DaxibotulinumtoxinA 20units versus placebo one cycle of treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 75.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 75.1.1 4 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 75.1.2 16weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 75.1.3 24 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 75.2 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 75.2.1 4 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 75.2.2 8 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 75.2.3 12 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 75.2.4 16 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 75.2.5 20 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 75.2.6 24 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 75.3 Total adverse events | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 75.4 Duration of treatment | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
Comparison 76. DaxibotulinumtoxinA 60 units versus OnabotulinumtoxinA 20 units one cycle of treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 76.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 76.1.1 4 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 76.1.2 16 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 76.1.3 24 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 76.2 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Totals not selected | |
| 76.3 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 76.3.1 4 weeks | 1 | 83 | Risk Ratio (IV, Random, 95% CI) | 1.24 [1.05, 1.46] |
| 76.3.2 8 weeks | 1 | 83 | Risk Ratio (IV, Random, 95% CI) | 1.49 [1.07, 2.07] |
| 76.3.3 12 weeks | 1 | 83 | Risk Ratio (IV, Random, 95% CI) | 0.97 [0.60, 1.57] |
| 76.3.4 16 weeks | 1 | 83 | Risk Ratio (IV, Random, 95% CI) | 2.05 [0.67, 6.28] |
| 76.3.5 20 weeks | 1 | 83 | Risk Ratio (IV, Random, 95% CI) | 2.73 [0.78, 9.58] |
| 76.3.6 24 weeks | 1 | 83 | Risk Ratio (IV, Random, 95% CI) | 1.02 [0.15, 6.93] |
| 76.4 Total adverse events | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 76.5 Duration of treatment | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
Comparison 77. DaxibotulinumtoxinA 40 units versus OnabotulinumtoxinA 20 units one cycle of treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 77.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 77.1.1 4 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 77.1.2 16 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 77.1.3 24 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 77.2 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Totals not selected | |
| 77.3 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 77.3.1 4 weeks | 1 | 81 | Risk Ratio (IV, Random, 95% CI) | 1.21 [1.01, 1.44] |
| 77.3.2 8 weeks | 1 | 81 | Risk Ratio (IV, Random, 95% CI) | 1.42 [1.01, 2.00] |
| 77.3.3 12 weeks | 1 | 81 | Risk Ratio (IV, Random, 95% CI) | 0.96 [0.59, 1.57] |
| 77.3.4 16 weeks | 1 | 81 | Risk Ratio (IV, Random, 95% CI) | 3.77 [1.36, 10.48] |
| 77.3.5 20 weeks | 1 | 81 | Risk Ratio (IV, Random, 95% CI) | 2.87 [0.82, 10.06] |
| 77.3.6 24 weeks | 1 | 81 | Risk Ratio (IV, Random, 95% CI) | 2.15 [0.42, 11.11] |
| 77.4 Total adverse events | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 77.5 Duration of treatment | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
Comparison 78. DaxibotulinumtoxinA 20 units versus OnabotulinumtoxinA 20 units one cycle of treatment in glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 78.1 Participant assessment of success by analysing scores and scales | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 78.1.1 4 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 78.1.2 16 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 78.1.3 24 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 78.2 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Totals not selected | |
| 78.3 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 78.3.1 4 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 78.3.2 8 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 78.3.3 12 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 78.3.4 16 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 78.3.5 20 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 78.3.6 24 weeks | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 78.4 Total adverse events | 1 | 108 | Risk Ratio (IV, Random, 95% CI) | 0.87 [0.46, 1.64] |
| 78.5 Duration of treatment | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
Comparison 79. PrabotulinumtoxinA 20 units versus placebo one cycle of treatment, glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 79.1 Participant assessment of success by analysing scores and scales | 3 | 930 | Risk Ratio (M‐H, Random, 95% CI) | 18.34 [9.68, 34.76] |
| 79.1.1 4 weeks | 3 | 930 | Risk Ratio (M‐H, Random, 95% CI) | 18.34 [9.68, 34.76] |
| 79.2 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 3 | 939 | Risk Ratio (M‐H, Random, 95% CI) | 0.60 [0.06, 5.65] |
| 79.3 Physician assessment of success by analysing scores and scales | 3 | 929 | Risk Ratio (M‐H, Random, 95% CI) | 23.96 [9.35, 61.40] |
| 79.3.1 4 weeks | 3 | 929 | Risk Ratio (M‐H, Random, 95% CI) | 23.96 [9.35, 61.40] |
| 79.4 Total adverse events | 3 | 948 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.91, 1.43] |
Comparison 80. PrabotulinumtoxinA 20 units versus OnabotulimtoxinA 20units one cycle of treatment, glabellar lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 80.1 Participant assessment of success by analysing scores and scales | 2 | 1538 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.99, 1.06] |
| 80.1.1 4 weeks | 2 | 749 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [1.00, 1.09] |
| 80.1.2 8weeks | 1 | 263 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.92, 1.05] |
| 80.1.3 12 weeks | 1 | 263 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.97, 1.14] |
| 80.1.4 16 weeks | 1 | 263 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.91, 1.13] |
| 80.2 Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus) | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Subtotals only | |
| 80.3 Physician assessment of success by analysing scores and scales | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 80.3.1 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 80.4 Total adverse events | 2 | 759 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.74, 1.13] |
Comparison 81. OnabotulinumtoxinA 9 units versus hyaluronic acid [JUVEDERM ULTRA ® and/or JUVEDERM ULTRA PLUS®] one treatment in lips and perioral lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 81.1 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 82. OnabotulinumtoxinA 9 units associated with hyaluronic acid [JUVEDERM ULTRA ® and/or JUVEDERM ULTRA PLUS®] versus OnabotulinumtoxinA one treatment in lips and perioral lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 82.1 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 83. Hyaluronic acid [JUVEDERM ULTRA ® and/or JUVEDERM ULTRA PLUS®]versus OnabotulinumtoxinA 9 units associated with hyaluronic acid one treatment in lips and perioral lines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 83.1 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Ascher 2004.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised, double‐blind, parallel‐design in glabellar lines Study date‐ start date March and June 2002” no information about end date Study centre‐ outpatients from three centres |
|
| Participants |
Randomised 119 participants with mean age of 48. 6 ±7.9 years in BontA 25 U group; 50.9 ± 7.5 years in BontA 50 U group; 48.8 ± 7.7 years in BontA75u group; 48.3 ± 6.4 years in placebo group; 49. 3 ±7.5 years total population. Gender: 114/119 (95.8%) female and 5/119 (4.2%) male in total population, 33/34 (97.1%) female and 1/34 (2.9%) male in BontA 25 U group; 32/34 (94.1%) female and 2/34 (5.9%) male in BontA 50 U group; 32/34 (94.1%) female and 2/34 (5.9%) male in BontA75 U group; 17/17 (100% female) and 0/17 (0%) male in placebo group Inclusion criteria
Exclusion criteria
Severity of disease‐ moderate to severe glabellar lines at rest Ethnicity‐ no information |
|
| Interventions |
Duration of study‐ 24 weeks Intervention
Comparator
|
|
| Outcomes |
Primary outcome
Secondaries outcomes
|
|
| Notes | “Supported by Beaufour Ipsen Pharma SAS. Disclosure: Dr Zakine is an employee of Beaufour Ipsen Pharma” |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "This multicenter, randomised, double‐blind study compared..." page 224 "Patients were randomly assigned to receive BTX‐A, according to a computer‐generated randomisation schedule" page 225 Comment: we considered low risk of bias |
| Allocation concealment (selection bias) | Unclear risk | No information available to allow a judgement Comment: we consider this unclear risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "To maintain the study blind, treatments were reconstituted and syringes for injection prepared by a third party not involved with the patient treatment or assessment" page 225 Comment: we considered this low risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "double‐blind... study" To maintain the study blind, treatments were reconstituted and syringes for injection prepared by a third party not involved with the patient treatment or assessment" page 224/225 Comment: we considered this low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote:"two patients withdrew before the first month" page 227 Comment: we considered a unclear risk of bias because the authors did not explain the reason of drop outs |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported. Comment: we considered this low risk of bias |
| Other bias | High risk | Quote: "the two series of glabellar severity scores were not fully superimposable: taking the objective double‐blind, digital photographic baseline results as a reference, it appears that clinical scoring overestimated the occurrence of medium scores (scores of 2) by including patients with a mild score (score of 1). Conversely, severe scores were slightly underestimated by the investigators." page 227 Comment: to clarify this information an e‐mail was sent on 23 May 2015, we did not receive any answer. |
Ascher 2005.
| Study characteristics | ||
| Methods |
Study design ‐multicentre, parallel‐design. First phase randomised, double‐blind, month to month evaluation until 6 months. Second phase, open‐label, pragmatic study, month to month evaluation between month 3 and month 6 in glabellar lines Study date‐ no information Study setting‐ outpatients from five centres |
|
| Participants |
Randomised 100 participants, mean age of 50.4 ± 7.7 years in BontA group, 49.1 ±8.6 years in placebo group, 49.8 ± 8.2 years total population. Gender: 94/100 (94%) female and 6/100 (6.0%) male total population; 48/50 (96%) female and 2/50 (4.0%) male in BontA group; 46/50 (92%) female and 4/50 (8.0%) male in placebo group Inclusion criteria
Exclusion criteria
Severity of disease‐ moderate to severe glabellar rhytides at rest Ethnicity‐ no information |
|
| Interventions |
Duration‐ two phases (12 weeks and 24 weeks) Intervention
Comparator
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | "This study was funded by Beaufour Ipsen SAS, Paris, France. Dr. Zakine is an employee of Beaufour Ipsen SAS. Drs. Ascher, Kestemont, Baspeyras, Niforos, Malet, and Santini received payment for conducting the study." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A multicenter, randomised, placebo‐controlled study" page 366 "Patients were randomly assigned to receive 50 U BoNT‐A or placebo according to a computer‐generated randomisation schedule prepared prior to the start of the study" page 367 Comment: we consider this low risk of bias |
| Allocation concealment (selection bias) | Unclear risk | No information available to allow a judgement Comment: we consider this unclear risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "The first phase was conducted according to a double‐blind...The second injection, which started the second phase of the study, was pragmatically" page 366 Comment: we consider this unclear risk of bias (first phase), because the authors did not mention the method used for blinding the participants and personnel |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "The first phase was conducted according to a double‐blind...The second injection, which started the second phase of the study, was pragmatically" page 366 Comment: we consider this unclear risk of bias (first phase), because the authors did not mention how they blinded the outcome assessor |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "One patient of BontA group did not receive the second injection.The patient was "completely satisfied" from Month 1 to Month 6 and then refused the second injection. Also, 2 patients in each group withdrew from the study after the second injection, none of them for AEs. Protocol deviation was responsible for the withdrawal of 2 patients, and a professional problem and an administrative reason for the other 2 cases" page 370 Comment: we consider this low risk of bias (first phase) due to number of withdraw was low and there was a balance between groups regarding this number and the reasons of withdraws |
| Selective reporting (reporting bias) | Unclear risk | The percentages of responders (i.e. patients with clinical scores of 0 or 1) at rest and at maximum frown ‐ only P value was provided (page 372) Comment: we consider this unclear risk of bias. An e‐mail was sent for the authors on 23 May 2015. No reply until the date |
| Other bias | Unclear risk | Quote: "Dr. Zakine is an employee of Beaufour Ipsen SAS." page 375 Comment: We considered this unclear risk of bias |
Ascher 2009.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised, double‐blind, parallel‐design, dose‐ranging in crow's feet lines Study date‐ no information Study setting‐ outpatients from nine centres |
|
| Participants |
Randomised‐ 220 participants, with mean age of 47.6 ± 7.6 years in BontA 15 U group; 47.8 ± 9.5 years in BontA 30 U group; 48.3 ± 10 years in BontA 45 U group; 47.6± 8.2 years in placebo group. Gender: 48/55 (87%) female, 7/55(13%) male in BontA 15 U group; 48/54 (89%) female, 6/54 (11%) male in BontA 30 U group; 46/55 (84%) female, 9/55 (16%) male in BontA 45 U group; 50/53 (93%) female, 4/54 (7%) male in placebo group Inclusion criteria
Exclusion criteria
Severity of disease‐ “moderate to severe crow’s feet at maximum smile” Ethnicity‐ BontA(15u) 54/55 (98%) Caucasian; 1/55 (2%) non‐Caucasian; BontA (30u) 54/54 (100%) Caucasian; BontA (45u) 55/55 (100%) Caucasian; placebo 54/54 (100%) Caucasian |
|
| Interventions |
Duration of study‐ 16 weeks Interventions
Comparator
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | "Ipsen, Ltd. provided the Dysport and the funding for this study." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "a multicenter, randomised, double‐blind" page 1479 "Subjects were randomly assigned to one of four treatment arms (BoNT‐A 15, 30, or 45 U per eye or placebo) according to a computer‐generated" page 1479 Comment: we consider this low risk of bias |
| Allocation concealment (selection bias) | Unclear risk | No information available to allow a judgement Comment: we consider this unclear risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "An independent reconstitutor reconstituted the study medication (active treatment or placebo) to maintain the study blind" page 1479 Comment: we consider this low risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "assessment by an independent panel of four expert clinicians blinded to treatment group and study time point." page 1480 Comment: we consider this low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "200 patients completed the study. "Because of a high incidence of protocol violations at one centre, efficacy data for all subjects from that site were excluded, resulting in an ITT population of 193 subjects. The mITT (maximum smile) population consisted of 162 subjects; 31 were excluded because severity ratings were missing or were rated none or mild at baseline by the independent panel. The mITT (rest) population consisted of 188 subjects; five subjects were excluded because severity ratings were missing or were rated none at baseline by the independent panel" page 1481 Coment: we consider a high risk of bias, protocol violation |
| Selective reporting (reporting bias) | Unclear risk | Subject evaluation, only graphic no data showed Comment: we consider this unclear risk of bias, because the authors did not show data. We sent an e‐mail to authors on 23 May 2015. No reply until the date |
| Other bias | Low risk | We considered this study at low risk of other bias |
Ascher 2018.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised, phase II, double‐blind, parallel‐design, placebo‐controlled in glabellar lines Study date‐ start in March 2011 ; end in DecembeR 2011 Study centre‐ outpatients eight centres |
|
| Participants |
Randomised 176 female participants with mean age of 47.0 ± 6.6 years in AbobotulinumtoxinA 50 group; 47.9 ± 6.0 years in Liquid AbobotulinumtoxinA 75 U group; 48.1± 6.9 years in Liquid AbobotulinumtoxinA 50 U group; 46.7 ± 8.4 years in Liquid AbobotulinumtoxinA 20 U group; 46.8 ± 6.4 years in placebo group.100% female. Inclusion criteria
Exclusion criteria
Severity of disease‐ 17/35 (48.6%) severe and 18/35 (51.4%) moderate in AbobotulinumtoxinA 50 group; 15/35 (42.8%) severe and 20/35(57.1%) moderate in Liquid AbobotulinumtoxinA 75 U group; 15/35 (42.8%) severe and 20/35(57.1%) moderate in Liquid AbobotulinumtoxinA 50 U group; 14/36(38.9%) severe and 22/36(61.1%) moderate in Liquid AbobotulinumtoxinA 20 U group; 14/35 (60%) severe and 14/35 (61.1%0 moderate in placebo group). Ethnicity‐ 5/176 (2.8%) Hispanic/Latina, 171/176 (97.2%) not Hispanic/Latina |
|
| Interventions |
Duration of study‐ 26 weeks Interventions
Comparators
|
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes | Philippe Picaut was Ipsen employee | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Computer‐generated randomization lists were cre‐ ated by a sponsor statistician independent from the study using a validated in‐house system developed with SAS software (SAS Institute, Inc., Cary, NC). The randomization schedule is generated using the SAS procedure PLAN "...page2 Comment: we consider this low risk of bias |
| Allocation concealment (selection bias) | Low risk | No information available to allow a judgement Comment: we consider this unclear risk of bi |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | No information available to allow a judgement Comment: we consider this unclear risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information available to allow a judgement Comment: we consider this unclear risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information available to allow a judgement Comment: we consider this unclear risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported. Comment: we considered this a low risk of bias |
| Other bias | Unclear risk | Group liquid formulation 20u showed an imbalance (fewer participants with severe GL at maximum frown). Comment: we consider this unclear risk of bias |
Ascher 2020.
| Study characteristics | ||
| Methods | Randomised, double‐blind (first cycle), placebo‐controlled and open‐label Phase (2‐5 cycles) multicentre study, phase III trial | |
| Participants | 185 participants Inclusion criteria
Exclusion Criteria
|
|
| Interventions |
Intervention Clostridium Botulinum Toxin Type A (BTX A HAC NG), total treatment volume 0.25 mL will be divided into 5 injections (0.05 mL per injections) injected in 5 pre‐defined sites across the glabellar region. A total of 50 U of BTX‐A‐HAC NG will be injected/cycle. Comparator Placebo volume 0.25 mL will be divided into 5 injections (0.05 mL per injections) injected in 5 pre‐defined sites across the glabellar region. Administered in Cycle 1 of the double‐blind phase only. |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | Sponsor Ipsen Other study ID Y‐52‐52120‐214 We sent an email on April,28 2019. “The Ipsen company answered:These trials have not been published unfortunately.” on June, 27, 2019. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "computer‐generated randomization lists that were created by "...page95 Comment: we considered it low risk of bias |
| Allocation concealment (selection bias) | Low risk | Quote: "computer‐generated randomization lists that were created by "...page95 Comment: we considered it low risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: No information Comment: we considered it unclear risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: No information Comment: we considered it unclear risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote:Patient disposition is shown in Figure 1...page 95 and 97 Comment: we considered it low risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered this low risk of bias |
| Other bias | Low risk | no other risk of bias was identified |
Beer 2006.
| Study characteristics | ||
| Methods |
Study design‐ single‐centre, randomised, investigator‐blinded, placebo controlled, parallel‐design. Two phases in glabellar lines Study date‐ no information Study setting‐ outpatients from one centre (USA) |
|
| Participants |
Randomised 77 women, age range from 31.5 to 67.5years (age range, by treatment group, from 46.8 to 55.8 years). Gender 100% female Inclusion criteria
Exclusion criteria
Severity of disease‐ moderate‐to‐severe glabellar lines at maximum frown Ethnicity‐ 86% to 100% white |
|
| Interventions |
Duration of study‐ 12 weeks (phase 1) and 4 weeks (phase 2) Intervention
Comparator‐ Facial creams in glabellar lines
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | “This study was supported by an unrestricted educational grant from Allergan, Inc.” | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Subjects randomised to receive either botulinum toxin type A or placebo injection" page 186 Comment: we considered this unclear risk of bias because the authors did not explain how they randomised the participants |
| Allocation concealment (selection bias) | Unclear risk | No information available to allow a judgment. Comment: we consider this unclear risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "In order to maintain the blind, the injection syringes for botulinum toxin type A treatment and for placebo injection were prepared by a qualified staff member rather than the principal investigator. In addition, injections were given by another physician who had not prepared the syringes and who was not involved in the clinical evaluation of the subjects." page 186 Comment: we consider this low risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "In order to maintain the blind, the injection syringes for botulinum toxin type A treatment and for placebo injection were prepared by a qualified staff member rather than the principal investigator. In addition, injections were given by another physician who had not prepared the syringes and who was not involved in the clinical evaluation of the subjects." page 186 Comment: we considered this low risk of bias to OnabotulinumtoxinA versus placebo, but a high risk of bias for the other groups (creams) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "More than 80% of the subjects in each treatment group completed the study as planned and no differences between groups in subject disposition were observed" page 188 Comment: we considered this unclear risk of bias because the percentage of drop outs were almost 20% and we are not sure in which extension this fact could affect the results |
| Selective reporting (reporting bias) | High risk | For patient satisfaction, only P value was provided and no numeric data was showed. Comment: we considered a high risk of bias. We sent an e‐mail on 21 November 2015. No reply until the date |
| Other bias | Low risk | We considered this study at low risk of other bias |
Beer 2019a.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised (3:1) , phase II, double‐blind, parallel‐design, placebo‐controlled, single‐dose in glabellar lines (EV‐001) Study date‐ start in March 2015 ; end in September 2015 Study centre outpatient, 10 centres (USA) |
|
| Participants |
Randomised 330 participants, with a mean age of 50.2 ±11.76 years in PrabotulinumtoxinA group and 50.4 ±11.95 years in placebo group. Gender: 220/246 (92.3%) female in PrabotulinumtoxinA group, 79/84 (94%) female in placebo group. Previous botulinum toxin treatment: 103/246 (41.9%) in PrabotulinumtoxinA group and 31/84(36.9%) in placebo group Inclusion criteria
Exclusion criteria
Severity of the disease‐ by investigator assessment moderate 78/246 (31.7%) and severe 168/246(68.3%) in PrabotulinumtoxinA group, and moderate 28/84 (33.3%) and severe 56/84 (66.7%) in placebo group ; by participant assessment moderate 56/246 (22.8%) and severe 190/84(77.2%) in PrabotulinumtoxinA group, and moderate 18/84 (21.4%) and severe 66/84 (78.6%) in placebo group. Ethnicity‐ 205/246 (83.3%) white, 18/246 (7.3%) African‐American, 2/246 (0.8%) Asian, 21/246 (8.5%) other in PrabotulinumtoxinA group; 63/84 (75%) white, 7/84 (8.3%) African‐American, 4/84 (4.8%) Asian, 10/84(11.9%) other in placebo group. |
|
| Interventions |
Duration of study‐ 20weeks Intervention PrabotulinumtoxinA (20 U) (N = 246), 0.1 mL injected into each of 5 sites) of either(administered as 4 U/0.1 mL) Comparator Placebo (N = 84), 0.1 mL injected into each of 5 sites) |
|
| Outcomes |
Primary endpoint
Secondary outcome
|
|
| Notes | "As sponsor of the EV‐001 and EV‐002 studies, Evolus, Inc., of Newport Beach, CA, was involved in the design of these studies and provided funding, study materials, equipment, and medications to all investigational sites. Evolus also provided funding to contract organizations involved in data collection, analysis, and reporting of the results. R. L. Avelar is the Head of R&D and Chief Medical Officer for Evolus, Inc., and receives compensation in salary, stock, and stock options. Before and during the time of these studies and manuscript preparation, J. E. Gross was the Chief Scientific Officer at Evolus, Inc.; he will receive royalty and milestone payments should the product be approved. Anneke Jonker of Medical Writing Associates, West Vancouver, BC, Canada, provided technical assistance with manuscript preparation and submission; she holds stock in Evolus, Inc." This study was a phase III trial identical to NCT02334436. Both studies had the same reference than the NCT02334436 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Random numbers had been generated using SAS PROC PLAN (SAS Institute, Inc.,
Cary NC); a block randomization scheme with no stratification was used, with each block containing assignments for 3 prabotulinumtoxinA subjects and 1 placebo subject"... page 1382‐3 Comment: we considered this low risk of bias |
| Allocation concealment (selection bias) | Unclear risk | No information Comment: we considered this unclear risk of bias because the authors did not explain the methods used to maintain the allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote:"At each study site, designated protocoltrained study personnel selected a study vial, which contained either 100 U of prabotulinumtoxinA or placebo according to the randomization schedule, reconstituted the vial with 2.5 mL of 0.9% sterile saline, filled the injection syringe, and provided it to the investigator in a blinded manner."...page 1383 Comment: we considered this low risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote:"At each study site, designated protocoltrained study personnel selected a study vial, which contained either 100 U of prabotulinumtoxinA or placebo according to the randomization schedule, reconstituted the vial with 2.5 mL of 0.9% sterile saline, filled the injection syringe, and provided it to the investigator in a blinded manner."...page 1383 Comment: we considered this low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: " figur2 3...page 1385 Comment: we considered this low risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered this low risk of bias |
| Other bias | Low risk | We considered this study at low risk of other bias |
Beer 2019b.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised (3:1), phase II, double‐blind, parallel‐design, placebo controlled, single‐dose in glabellar lines (EV‐002) Study date‐ start in March 2015 ; end in September 2015 Study centre outpatient, 10 centres (USA) |
|
| Participants |
Randomised 324 participants, with a mean age of 51.5 ± 11.54 years in PrabotulinumtoxinA group and 50.4 ± 10.14 years in placebo group. Gender: 219/246 (89%) female in PrabotulinumtoxinA group, 72/78 (92.3%) female in placebo group. Previous botulinum toxin treatment: 91/246 (37%) in PrabotulinumtoxinA group and 26/78(33.3%) in placebo group Inclusion criteria
Exclusion criteria
Severity of the disease‐ by investigator assessment moderate 42/246 (17.1%) and severe 204/246(82.9%) in PrabotulinumtoxinA group, and moderate 12/78 (15.4%) and severe 66/78 (84.6%) in placebo group ; by participant assessment moderate 46/246 (18.7%) and severe 200/84(81.3%) in PrabotulinumtoxinA group, and moderate 13/78 (16.7%) and severe 65/78 (83.3%) in placebo group. Ethnicity‐ 215/246 (87.4%) white, 19/246 (7.7%) African‐American, 5/246 (2%) Asian, 7/246 (2.8%) other in PrabotulinumtoxinA group; 69/78 (88.5%) white, 6/78 (7.7%) African‐American, 2/78 (2.6%) Asian, 1/78(1.3%) other in placebo group |
|
| Interventions |
Duration of study‐ 20 weeks Intervention PrabotulinumtoxinA (20 U) (N = 246), 0.1 mL injected into each of 5 sites of either(administered as 4 U/0.1 mL) Comparator Placebo (N=78), 0.1 mL injected into each of 5 sites |
|
| Outcomes |
Primary endpoint
Secondary outcome
|
|
| Notes | "As sponsor of the EV‐001 and EV‐002 studies, Evolus, Inc., of Newport Beach, CA, was involved in the design of these studies and provided funding, study materials, equipment, and medications to all investigational sites. Evolus also provided funding to contract organizations involved in data collection, analysis, and reporting of the results. R. L. Avelar is the Head of R&D and Chief Medical Officer for Evolus, Inc., and receives compensation in salary, stock, and stock options. Before and during the time of these studies and manuscript preparation, J. E. Gross was the Chief Scientific Officer at Evolus, Inc.; he will receive royalty and milestone payments should the product be approved. Anneke Jonker of Medical Writing Associates, West Vancouver, BC, Canada, provided technical assistance with manuscript preparation and submission; she holds stock in Evolus, Inc." This study was a phase III trial identical to NCT02334423. Both studies had the same reference than the NCT02334423 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Random numbers had been generated using SAS PROC PLAN (SAS Institute, Inc.,
Cary NC); a block randomization scheme with no stratification was used, with each block containing assignments for 3 prabotulinumtoxinA subjects and 1 placebo subject"... page 1382‐3 Comment: we considered this low risk of bias |
| Allocation concealment (selection bias) | Unclear risk | No information Comment: we considered this unclear risk of bias because the authors did not explain the methods used to maintain the allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote:"At each study site, designated protocoltrained study personnel selected a study vial, which contained either 100 U of prabotulinumtoxinA or placebo according to the randomization schedule, reconstituted the vial with 2.5 mL of 0.9% sterile saline, filled the injection syringe, and provided it to the investigator in a blinded manner."...page 1383 Comment: we considered this low risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote:"At each study site, designated protocoltrained study personnel selected a study vial, which contained either 100 U of prabotulinumtoxinA or placebo according to the randomization schedule, reconstituted the vial with 2.5 mL of 0.9% sterile saline, filled the injection syringe, and provided it to the investigator in a blinded manner."...page 1383 Comment: we considered this low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: " figur2 3...page 1385 Comment: we considered this low risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered this low risk of bias |
| Other bias | Low risk | We considered this study at low risk of other bias |
Bertucci 2020.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised, double‐blinded, placebo‐controlled, parallel‐design. Phase III in glabellar lines (SAKURA 1 and SAKURA 2). NCT03014622 and NCT03014635 Study date‐started December 5, 2016, and ended on 14 November 2017 Study setting‐ outpatients, 30 centres (24 in the USA and 6 in Canada |
|
| Participants |
Randomised 609 participants with age range 50.2 ± 10.56 years in DaxibotulinumtoxinA group, and 49.8 ±10.58 years in placebo group. Gender 335/405(88.1%) females in DaxibotulinumtoxinA group, and 175/204 (85.8%) in placebo group. Prior treatment‐ 213/405(52.6%) in DaxibotulinumtoxinA group and 105/204(51.5%) in placebo group. Inclusion criteria
Exclusion criteria
Severity of the disease‐ moderate 252/405(62.2%) in DaxibotulinumtoxinA group, and 133/204(65.2%) in placebo group. Ethinicity‐ caucasian 353/405 (87.2%),19/405(4.7%) African American, 18/405 (4.4%) Asian, 15/405 (3.7%) other in DaxibotulinumtoxinA group; Caucasian 173/204 (87.4.8%),11/204(5.4%) African American, 7/204 (3.4%) Asian, 13/204 (6.4%) other in |
|
| Interventions |
Duration: 36 weeks Intervention DaxibotulinumtoxinA 40 U, (N = 405), two 0.1‐mL injections into each corrugator muscle and one 0.1‐mL injection into the procerus muscle. Intramuscular injection in glabella region Comparator Placebo (N = 204), two 0.1‐mL injections into each corrugator muscle and one 0.1‐mL injection into the procerus muscle.Iintramuscular injection in glabella region. |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes | "We thank Revance Therapeutics, Inc, for sponsoring the studies and funding the development of the manuscript." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote:"An independent statistician produced a computer‐generated randomization
code (using SAS PROC PLAN [SAS Institute, Inc, Cary, NC])...page 840 Comment: we considered this low risk of bias |
| Allocation concealment (selection bias) | Low risk | Quote:" Study treatments were provided in sequentially numbered clinical trial kits containing single use 50‐U vials,"... page 840 |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote:"All vials looked identical to each other before and after reconstitution"...page 840 Comment: we considered this low risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote:"All vials looked identical to each other before and after reconstitution"...page 840 Comment: we considered this low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote:"Discontinuations were attributable predominantly to withdrawal of consent and loss to follow‐up, with none being due to adverse events (Supplemental Fig 1)...page 841 Comment: we considered this low risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we consider low risk of bias |
| Other bias | Low risk | Pharmaceutical sponsored |
Brandt 2009.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised, double‐blind, placebo‐controlled, parallel‐design, phase III trial in glabellar lines Study date‐ start (18 Nov 2005), end (28 July 2006) Study setting‐ outpatients from three centres |
|
| Participants |
Randomised‐ 158 participants, with mean age of 43.1 ± 10.3 years in BontA group; 42.7 ± 9.1 years in placebo group; 42.9 ±9.9 years total population. Gender: 90/105 (86%) female, 15/105 (14%) male in BontA group; 45/53 (85%) female, 8/53 (15%) male in placebo group; 135/158 (85%) female, 23/158 (15%) male total population Inclusion criteria
Exclusion criteria
Severity of disease‐ moderate to severe glabellar lines Ethnicity‐ BontA 52/105 (50%) white, 50/105 (48%) Hispanic, 2/105 (2%) African American, 1/105 (1%) Asian; placebo 25/53 (47%) Caucasian, 25/53 (47%) Hispanic, 2/53 (4%) Asian, 1/53 (2%) other; Total population 77/158 (49%) Caucasian, 75/158 (47%) Hispanic, 2/158 (1%) African American, 3/158 (2%) Asian, 1/158 (1%) other |
|
| Interventions |
Duration of study‐ 24 weeks Intervention
Comparator
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | "Medicis Aesthetics, Inc. provided funding and the material Dysport for this study. Dr. Baumann is a consultant and advisory board member for the both Medicis and Allergan." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Subjects were randomised using a unique randomisation code generated by a Beaufour Ipsen Industrie SAS randomisation manager (SAS Institute, Inc., Cary, NC)." page 1895 Comment: we considered this low risk of bias |
| Allocation concealment (selection bias) | Unclear risk | No information available to allow a judgment Comment: we considered this unclear risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Treatment vials (BoNT‐A or placebo) were not identifiable apart from a unique sequential number, and the investigator (or trained designee) prepared the individual’s study dose for injection" page 1895 Comment: we considered this low risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Investigators did not evaluate subjects for safety at Day 14 or until after the efficacy assessment at Day 30 to maintain blinding, and the blind was not broken during the study," page 1895 Comment: we considered this low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Of the 158 subjects enrolled, 143 (91%) completed the 180‐day study: 92% in the active group and 87%in the placebo group" page 1896‐7 Comment: we considered this low risk of bias |
| Selective reporting (reporting bias) | High risk | Quote: "the co‐primary endpoint‐ investigator assessment: 89.5%, vs 7.5% placebo", but in the graphic figure 2,placebo = 3.9% page 1897 Data was different from table to the graphic about placebo Comment: we considered this high risk of bias. We sent an e‐mail to Dr Brandt on 27 October 2015. Dr Brandt passed away. Another two messages by e‐mail were sent to Dr Baumann on 14 November 2015 and on 21 November 2015. No reply until the date. |
| Other bias | Low risk | We considered this study at low risk of other bias |
Carruthers 2002.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, double‐blind, randomised, placebo‐controlled parallel design study in glabellar lines Study date‐ no information Study setting‐ outpatients of 14 centres |
|
| Participants |
Randomised‐ 264 participants, with a mean age of 44.7 ± 11 years in BontA group; 44.3 ± 11.3 years in placebo group. Gender: 173/203 (85.2%) female, 30/203 (14.8%) male in BontA group; female 47/61 (77%), 14/61 (23%) male in placebo group Inclusion criteria
Exclusion criteria
Severity of disease‐ moderate to severe glabellar lines at maximum frown Ethnicity‐ 174/203 (85.7%) white, Hispanic 16/203 (7.9%), black 7/203 (3.4%), Asian 3/203 (1.5%), other 3/203(1.5%) in BontA; 49/61 (80.3%) white, Hispanic 6/61 (9.8%), black 1/61 (6.6%), Asian 4/61 (6.6%), other 1/61(1.6%) in placebo |
|
| Interventions |
Duration of study‐ 4 weeks Intervention
Comparator
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | "Funding sources: Allergan, Inc. Disclosure: Drs Carruthers and Lowe are paid consultants of Allergan" |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "At each study centre, randomisation to treatment was stratified by age group (≤50 years and ≥ 51 years). Within each age group, patients were randomly assigned in blocks of 8, with a ratio of 3:1 (BTX‐A/placebo)." page 842 Comment: we considered this unclear risk of bias because the authors did not explain how they randomised the participants |
| Allocation concealment (selection bias) | Unclear risk | Quote: "At each study centre, randomisation to treatment was stratified by age group (≤50 years and ≥ 51 years). Within each age group, patients were randomly assigned in blocks of 8, with a ratio of 3:1 (BTX‐A/placebo)." page 842 Comment: we considered this unclear risk of bias because the authors did not explain the methods used for maintaining the allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Randomization block size was not divulged to the physician investigators to help maintain blinding. In addition, two investigators co‐evaluated each patient on day 0, one of whom was to do the day 7 assessment, while the other was to do the day 30 assessment. This prevented the evaluation on day 30 from being influenced by the patient’s appearance on day 7." page 842 Comment: we considered this unclear risk of bias due to the visual aspect of BontA and placebo was not described. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Randomization block size was not divulged to the physician investigators to help maintain blinding. In addition, two investigators co‐evaluated each patient on day 0, one of whom was to do the day 7 assessment, while the other was to do the day 30 assessment. This prevented the evaluation on day 30 from being influenced by the patient’s appearance on day 7." page 842 Comment: we considered this low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "two patients (both in the BTX‐A group) did not complete the study. One moved out of state, and the other was unable to keep to scheduled appointments for personal reasons. A third patient was randomly assigned (placebo group) but declined treatment because of the visit schedule requirements." page 841 Comment: we considered this low risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered this low risk of bias |
| Other bias | Low risk | We considered this study at low risk of other bias |
Carruthers 2003a.
| Study characteristics | ||
| Methods |
Study design‐ randomised, double‐blind, dose‐ranging, parallel‐design in forehead lines in female Study date‐ no information Study setting‐ no information |
|
| Participants |
Randomised‐ 59 women, with age between 18‐50 years old. Gender 100% female Inclusion criteria
Exclusion criteria
Severity of disease‐ moderate to severe horizontal forehead rhytides at maximum brow elevation Ethnicity‐ no information |
|
| Interventions |
Duration of study‐ 48 weeks Intervention/Comparator
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | "Drs. Carruthers are consultants to Allergan, and supplies for this study were provided by Allergan." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomised to receive 16, 32, or 48 U of" page 462 Comment: we considered this unclear risk of bias because the authors did not detailed how they randomised the participants |
| Allocation concealment (selection bias) | Unclear risk | No information available to allow a judgment Comment: we considered this unclear risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "A Prospective, Double‐Blind, Randomized" page 461 Comment: we considered this unclear risk of bias because the authors did not explain how they blinded the participants and personnel |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "A Prospective, Double‐Blind, Randomized" page 461 Comment: we considered this unclear risk of bias because the authors did not explain how they blinded the outcome assessor |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "Only two patients were lost to follow‐up and did not complete treatment" page 461 Comment: we considered this unclear risk of bias because the authors did not mention the reasons for lost to follow up or which group these patients came from |
| Selective reporting (reporting bias) | High risk | Quote: "No difference between the groups (observer evaluation), although by self‐assessment, patients in the 32‐U botulinum toxin type A group had a significantly higher FWS at contraction than either the 16‐ or 48‐U botulinum toxin type A treatment groups" page 461 ‐There was missing data, and figure seven showed a divergent data from text. page 461 Comment: we considered high risk of bias because group imbalance and missing data, we sent an e‐mail on June 24 2015, answered on June 25 2015 "The study was done almost 10 years ago. The data will be buried in our basement. Getting out the raw data would be very labor intensive. Alastair." |
| Other bias | Low risk | We considered this study at low risk of other bias |
Carruthers 2003b.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, double‐blind, randomised, placebo‐controlled, parallel‐design, in glabellar lines Study date‐ start (April 1999), end (November 1999) Study setting‐ outpatients |
|
| Participants |
Randomised‐ 273 participants, with a mean age of 47.7 ± 11.4 years in BontA group, 46.4 ± 12 years in placebo group. Gender: 161/202(79.7%) female, 41/202 (20.3%) male in BontA group; 59/71(83.1%) female, 12/71(16.9%) male in placebo group Inclusion criteria
Exclusion criteria
Severity of disease‐ glabellar lines of at least moderate severity at maximal frown Ethnicity‐ 167/202 (82.7%) Caucasian, 14/202 (6.9%) black, 6/202 (3%) Asian, 14/202 (6.9%) Hispanic, 1/202 (0.5%) other in BontA; 60/71 (84.5%) Caucasian, 6/71 (8.5%) black, 0 (0%) Asian, 5/71 (7%) Hispanic, 0 (0%) other in placebo |
|
| Interventions |
Duration of study‐ 4 weeks Intervention
Comparator
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | "This study was sponsored by Allergan, Inc. (Irvine, Calif.). Dr. Carruthers owns stock in Allergan, Inc. Dr. Lowe has received research grants and consultant payments from and owns stock in Allergan, Inc." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomly assigned to receive either botulinum toxin or placebo, in a 3:1 ratio, by using a block‐of‐eight design," page 1090 Comment: we considered this unclear risk of bias because the authors did not explain how they randomised the participants |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Patients were randomly assigned to receive either botulinum toxin or placebo, in a 3:1 ratio, by using a block‐of‐eight design" page 1090 Comment: we considered this unclear risk of bias because the authors did not explain the methods used for maintaining the allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "vials of Botox and placebo had identical investigational labels, which prevented identification of the contents." page 1090 Comment: we considered this low risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "vials of Botox and placebo had identical investigational labels, which prevented identification of the contents." page 1090 Comment: we considered this low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Four subjects (two in the botulinum toxin group and two in the placebo group) were lost to follow‐up monitoring during the study (Table I). One additional patient was enrolled but not treated. The remaining 268 patients completed the study" page 1092 Comment: we considered this low risk of bias |
| Selective reporting (reporting bias) | High risk | Subgroup analysis, better results in subpopulation younger than 50 years old. No data showed Comment: we considered this high risk of bias |
| Other bias | Low risk | We considered this study at low risk of other bias |
Carruthers 2004.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised, placebo‐ controlled, parallel ‐design in glabellar lines. Phase one‐ randomised clinical trial, double‐blind, duration 4 months two arms (BontA versus placebo), and phase two‐ open‐label, duration 8 months Study date‐ start (Feb 1999), end (June 2000) Study setting‐ outpatients from 30 centres |
|
| Participants |
Randomised‐ 537 participants, with a mean age of 46.2 years in BontA group; 45.5 years in placebo group; and 46 years total population. Gender 334/405 (82.5%) female, 71/405 (17.5%) male in BontA group; 106/132 (80.3%) female, 26/132 (19.7%) in placebo group; 440/537 (81.9%) female, 97/537 (18.1%) total population Inclusion criteria
Exclusion criteria
Severity of the disease‐ moderate to severe glabellar frown lines at maximum frown Ethnicity‐ BontA: 341/405 (84.2%) Caucasian, 21/405 (5.2%) black, 9/405 (2.2%) Asian, 30/405 (7.4%) Hispanic, 4/405 (1%) other; placebo: 109/132 (82.6%) Caucasian, 28/132 (5.3%) black, 4/132 (3%) Asian, 11/132 (8.3%) Hispanic,1/132 (0.8%) other; total 450/537 (83.8%) Caucasian, 28/537(5.2%) black, 13/537(2.4%) Asian, 41/537 (7.6%) Hispanic, 5/537 (0.9%) other |
|
| Interventions |
Duration of study‐ Phase one‐ 16 weeks, phase two 32 weeks Intervention
Comparator
|
|
| Outcomes |
Primary outcome
Secondary outcome
|
|
| Notes | The first phase of this study was previous published as Carruthers 2002; Carruthers 2003b so this first phase was not reconsidered double count of patients | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "4‐month, randomised, double‐blind" page2 Comment: we considered this unclear risk of bias because the authors did not show how they randomised the participants (phase 1). Phase 2 was open‐label |
| Allocation concealment (selection bias) | Unclear risk | No information available to allow a judgment Comment: we considered this an unclear risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "vials of botulinum toxin patients and placebo were identical,identified only by patient number and study number, and required identical dilution and injection procedure" page 3‐4 Comment: we considered this low risk of bias (Phase 1). Phase 2 was open‐label. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "vials of botulinum toxin patients and placebo were identical,identified only by patient number and study number, and required identical dilution and injection procedures.To help maintain blinding randomisation block size was not divulged to the physician investigator" page2 Comment: we considered this low risk of bias (Phase 1). Phase 2 was open‐label |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | At phase 1, there were 4 withdraws at BontA group due to the following reasons: lost of follow‐up (2), personal reasons (1) and other (1). For placebo group, the chart showed 4 withdraws but in table 2, there were 5 patients (inconsistence) due to the following reasons: lost of follow‐up (1), personal reasons (2), other (2). Comment:we considered an unclear risk of bias. Despite a low number of withdraw and a balance between interventions group regarding number and reason of withdraw, we are not sure about in which extension the inconsistence stated below could affect the results An e‐mail sent on 24 June 2015, answer on 25 June 2015 "The study was done almost 10 years ago. The data will be buried in our basement. Getting out the raw data would be very labor intensive. Alastair." |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported. Comment: we considered this a low risk of bias |
| Other bias | Low risk | We considered this study at low risk of other bias |
Carruthers 2005a.
| Study characteristics | ||
| Methods |
Study design‐ single‐centre, double‐blind, randomised, parallel‐design, dose‐ranging in glabellar lines in men Study date‐ no information Study setting‐ outpatients, one centre (Canada) |
|
| Participants |
Randomised 80 men, with a mean age of 44.2 ± 14.6 years in BontA 20u group; 38.6 ± 8.2 years in BontA 40u group; 44.0 ± 12.8 years in BontA 60u group; 39.6 ± 13.2 years in BontA 80u group. Gender 100% male Inclusion criteria
Exclusion criteria
Severity of disease‐ moderate to severe glabellar lines at maximum frown Ethnicity‐ BontA 20 U‐ 100% white; 40 U‐ 18/20 white, 2/20 other; 60 U‐ 19/20 white and 1/20 other; 80 U 16/20 white, 1/20 Hispanic and 3/20 other |
|
| Interventions |
Duration of study‐ 52 weeks Intervention/Comparator
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | Drs Carruthers are consultants of Allergan | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "prospective, double‐blind, randomised" page 1297 "participants were randomly assigned into one of four possible treatment groups using a block‐of‐eight design" page 1298 Comment: we considered this unclear risk of bias because the authors did not explain how they randomised the participants |
| Allocation concealment (selection bias) | Unclear risk | No information available to allow a judgment Comment: we considered this unclear risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "to maintain blind, vials were prepared by a registered nurse who took no further part in the study" page 1298 Comment: we considered this unclear risk of bias due to the visual aspect of intervention and placebo was not detailed |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "to maintain blind, vials were prepared by a registered nurse who took no further part in the study" page 1298 Comment: we considered this unclear risk of bias due to the visual aspect of intervention and placebo was not detailed. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "Two participants withdrew consent, and one discontinued without providing information" page 1299 Comment: we considered this unclear risk of bias, because the authors did not mention the reason of drop out neither which group these patients came from |
| Selective reporting (reporting bias) | High risk | Patient self assessment no data, only P value. Comment: e‐mail sent on 24 June 2015, answer on 25 June 2015 "The study was done almost 10 years ago. The data will be buried in our basement. Getting out the raw data would be very labor intensive. Alastair." |
| Other bias | Low risk | We considered this study at low risk of other bias |
Carruthers 2005b.
| Study characteristics | ||
| Methods |
Study design‐ single centre, double‐blind, randomised, dose‐ranging trial followed by a 1‐year open‐label extension period. Study date‐ no information Study setting‐ outpatients from a private clinic, one centre (Canada) |
|
| Participants |
Randomised 80 women, with a mean age of 49 ± 8.9 years in BontA 10u group; 49.9 ± 9.3 years in BontA 20u group; 46.2 ± 9.1 years in BontA 30u group; and 45.3 ± 8.2years in BontA 40u group. Gender 100% female Inclusion criteria
Exclusion criteria
Reported by self‐evaluation questionnaire: severe at rest: 3/20(15%) BontA 10 U, 3/20 (16%) BontA 20 U, 9/20 (45%) BontA 30 U, 4/20 (20%) BontA 40 U maximum frown 13/20 (65%) BontA 10 U, 15/20 (79%) BontA 20 U, 19/20 (95%) BontA 30 U, 14/20 (70%) BontA 40 U. Ethnicity‐ BontA 10 U, 20U, 30 U‐ 95% Caucasian, 40 U‐ 100% Caucasian. |
|
| Interventions |
Duration of study‐ phase one‐ one year, phase two‐ one year Comparator/Comparator‐ (phase one)
Phase two
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | "This study was funded by a grant from Allergan, INC. Alastair Carruthers and Jean Carruthers are Allergan consultants." |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "This study was a 1‐year, double‐blind, randomised, dose‐ ranging trial" page 414 Comment: we considered this a unclear risk of bias because the authors did not explain how they randomised the participants |
| Allocation concealment (selection bias) | Unclear risk | Quote: "For the double‐blind trial, subjects were randomised into one of four possible treatment groups." page 415 Comment: we considered an unclear risk of bias, the authors did not mentioned methods to maintain allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "One physician (S.S.) prepared all doses in identical syringes marked only with the subject number. This physician did not see the subjects or administer any of the injections" page 415 Comment: we considered this low risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "One physician (S.S.) prepared all doses in identical syringes marked only with the subject number. This physician did not see the subjects or administer any of the injections" page 415 Comment: we considered this a low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "80 subjects participated, 74 completed the study. Four patients withdrew the consent, and two were lost to follow‐up." page 416 Comment: we considered this a low risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported. Comment: we considered this a low risk of bias |
| Other bias | High risk | Quote: "Group 40u and 30u of BontA were younger than the others. A post hoc pair‐wise comparison of trained observer assessments, however, demonstrated that baseline Facial Wrinkle Scale scores at maximum frown were significantly higher in the 20 U group than in both the 10 and 40 U groups (P = 0.047)." page 416 Quote: "percentage of subjects rated as severe in the 20 U group at baseline was significantly higher (by 25%) than both the 10 and 40 U groups " page 421 Comment: we considered this a high risk of bias |
Carruthers 2009.
| Study characteristics | ||
| Methods |
Study design‐ single‐centre, randomised, double‐blind, dose‐ranging, parallel‐design in upper facial wrinkles Study date‐ no information Study setting‐ outpatients from one centre (Canada) |
|
| Participants |
Randomised‐ 60 women, with a mean age of 42.6 ±8.3 years in BontA 32 U group, 42.5 ± 9.0 years in BontA 64 U group, and 39.5 ± 8.8 years in BontA 96u group. Gender 100% female Inclusion criteria
Exclusion criteria
Severity of disease‐ upper rhydites, glabella, crow's feet Ethnicity‐ BontA20 U group 19/20 white, 1/20 Asian; BontA 64 U group 19/20 white, 1/20 Asian; BontA 96 U group 19/20 white, 1/20 other |
|
| Interventions |
Duration of study‐ 52 weeks Intervention/Comparator
|
|
| Outcomes |
Primary outcome
Secondary outcome
|
|
| Notes | "Supported by a grant from Allergan Inc, Irvine, California. Disclosure: Drs Carruthers and Carruthers are consultants and investigators for and receive honoraria from Allergan Inc, which sponsored the study and the preparation of the article. They are also consultants and investigators for Merz GmbH and Solstice Neurosciences Inc." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "a single‐center, prospective, double‐ blind, randomised, dose‐comparisons" page 973 Comment: we considered this unclear risk of bias because the authors did not show how they randomised the participants |
| Allocation concealment (selection bias) | Unclear risk | Quote: "female patients randomly assigned to one of 3 treatment groups of 20 patients per group: 32, 64, and 96 U" page 973 Comment: we considered this an unclear risk of bias because the authors did not describe the methods used to maintain the allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "All injection volumes were 0.06 mL. To maintain the study blind, vials were not prepared by the same person who performed the injections." page 973 Comment: we considered this as low risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "All injection volumes were 0.06 mL. To maintain the study blind, vials were not prepared by the same person who performed the injections." page 973 Comment: we considered this as low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "Discontinued before 52 weeks: group 32u = 0, 64u = 2/20 (10%), 96u = 2/20 (10%)" table 1, page 974 Comment: we considered this as unclear risk of bias, because the authors did not mention the reason of drop outs. |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported. Comment: we considered this a low risk of bias |
| Other bias | Low risk | We considered this study at low risk of other bias |
Carruthers 2010.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, single‐blind, randomised, parallel‐design Study date‐ no information Study setting‐ outpatients from three centres |
|
| Participants |
Randomised‐ 90 women, with mean age of 48.4 ± 5.5 years, median 49.9 years in 24‐mg/mLCohesive Gel group; 48.6 ± 4.4 years, median 49.1 years in BontA 20 U group; 47.3 ± 5.3 years, median 48 years in BontA Plus 24‐mg/mL Cohesive gel group; 48.1 ± 5.1 years, median 49.1 years total population. Gender 100% female Inclusion criteria
Exclusion criteria
Severity of disease‐ investigator's assessment scale: perioral (upper lip at maximum contraction): 2.43 ± 0.6 total population, 2.63 ± 0.49 24‐mg/mL Cohesive gel, 2.43 ±0.63 BontA, 2.23 ± 0.63 (BontA Plus 24‐mg/mL Cohesive gel. Lip fulness: 1.52 ± 0.5 total population, 1.53 ± 0.51 24‐mg/mL Cohesive gel, 1.53 ± 0.51 BontA, 1.50 ± 0.51 (BontA Plus 24‐mg/mL Cohesive gel. Oral commissure: 1.90 ± 0.3 total population, 1.90 ± 0.31 24‐mg/mL Cohesive gel, 1.90 ± 0.31 BontA, 1.90 ± 0.3 (BontA Plus 24‐mg/mL Cohesive gel). Assessing investigator's assessment: perioral 1.53±0.64 total population, 1.50 ± 0.57 24‐mg/mL Cohesive gel, 1.60 ±0.77 BontA, 1.50 ± 0.57 (BontA Plus 24‐mg/mL Cohesive gel. Lip fullness: 1.63±0.57 total population, 1.63 ± 0.61 24‐mg/mL Cohesive gel, 1.67 ±0.55 BontA, 1.60 ± 0.56 (BontA Plus 24‐mg/mL Cohesive gel. Oral commissure: 1.86±0.57 total population, 1.83 ± 0.53 24‐mg/mL Cohesive gel, 1.93 ±0.58 BontA, 1.80 ± 0.61 (BontA Plus 24‐mg/mL Cohesive gel Ethnicity‐ 29/30 (96.7%) Caucasian, 1/30 (3.3%) Hispanic, 0% Asian, 0% other in 24‐mg/mL Cohesive Gel group; 25/30 (83.3%) Caucasian, 2/30 (6.7%) Hispanic, 2/30 (6.7%) Asian, 1/30 (3.3%) in other BontA; 28/30 (93.3%) Caucasian, 1/30 (3.3%) Hispanic,1/30 (3.3%) Asian, 0% other in BontA Plus 24‐mg/mL Cohesive gel |
|
| Interventions |
Duration of study‐ 26 weeks Intervention/Comparator
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | "The authors received research grant support from Allergan, Inc. for this study and for manuscript preparation." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "multicenter (3 site), prospective, single‐blind, randomised, parallel‐group study comprising three treatment groups" page 2123 Comment: we considered this unclear risk of bias because the authors did not show how they randomised the participants |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Subjects were randomly assigned to one of the three treatment groups on a 1:1:1 basis." page 2123 Comment: we considered unclear risk, because the authors did not explain the methods used to maintain the allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "maintain the single blind, a treating investigator performed injections" page2123 Comment: we considered this high risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "maintain the single blind, a treating investigator performed injections, and an assessing investigator who was masked to the treatment that the subject received conducted effectiveness evaluations." page 2123 Comment: we considered this low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Ninety subjects were enrolled and had at least one postbaseline visit. Seventeen (18.9%) discontinued before week 24. Significantly more subjects (n = 9) in the onabotulinumtoxinA‐alone group discontinued than those in the combination group (n = 1). A total of two subjects withdrew because of an AE, 11 were lost to follow‐up, and four withdrew consent." page2126 Comment: we considered this low risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered this low risk of bias |
| Other bias | Low risk | We considered this study at low risk of other bias |
Carruthers 2013.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised, double‐blind, placebo‐controlled, parallel design, phase III study in glabellar lines Study date‐ no information Study setting‐ outpatients from eight centres |
|
| Participants |
Randomised‐ 276 participants, with a mean age of 46.6 ± 9.87 years, median 46 years in BontA group; 46.4 ± 10.56 years, median 48 years in placebo group. Gender: 162/184 (88%) female, 22/184 (12.0%) male in BontA group; 76/92 (82.6%) female, 16/92 (17.4%) male in placebo group Inclusion criteria
Exclusion criteria
Severity of disease‐ moderate to severe glabellar frown lines at maximum frown (FWS) Ethnicity‐ 163/184 (88.6%) Caucasian, 3/184 (1.6%) black, 10/184 (5.4%) Asia, 7/184 (3.8%) Hispanic, 1/184 (0.5%) other in BontA group ; 83/92 (90.2%) Caucasian, 3/92 (3.3%) black, 1/92 (1.1%) Asian, 3/92 (3.3%) Hispanic, 2/92 (2.2%) other in placebo group |
|
| Interventions |
Duration of study‐ 28 weeks Intervention
Comparator
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | "This study was funded by Merz Pharmaceuticals GmbH. Editorial assistance was provided by Ogilvy 4D, Oxford, UK. Alastair Carruthers, Jean Carruthers, William P. Coleman III, Lisa Donofrio, Timothy Flynn, Michael Gold, Moritz Heinz, Derek Jones, David McDaniel, Thomas Rohrer, Nowell Solish, and Robert Weiss are paid investigators in this Phase III trial. Andrea Schlo€be and Moritz Heinz are employees of Merz Pharmaceuticals GmbH, and Laura Harrington is an employee of Ogilvy 4D." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were randomised 2:1 to receive 20 U of incobotulinumtoxinA or placebo" page 552 Comment: we considered this unclear risk of bias because the authors did not show how they randomised the participants |
| Allocation concealment (selection bias) | Unclear risk | no information Comment: we considered this unclear risk of bias because the authors did not presented the methods used to maintain the allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double‐blind, placebo‐controlled," page 552 Comment: we considered this unclear risk of bias because the authors did not explain how they blinded the participants |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double‐blind, placebo‐controlled," page 552 Comment: we considered this unclear risk of bias because the authors did not explain how they blinded the participants |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Nine patients discontinued the study; three from the incobotulinumtoxinA group withdrew consent (with one case of an adverse event), and three were lost
to follow‐up, and three in the placebo group withdrew consent.One hundred seventy‐eight completed the study in the incobotulinumtoxinA group and 89 in the placebo group." page 554 Comment: we considered this low risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered this low risk of bias |
| Other bias | Unclear risk | Pharmaceutical support and some authors were Allergan employees. Comment: we considered unclear risk of bias |
Carruthers 2014.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised, double‐blind, placebo‐controlled parallel design study, in crow's feet lines Study date‐ no information Study setting‐ outpatients of 23 centres |
|
| Participants |
Randomised 445 participants, with a mean age of 46.7 years in BontA group; 46.2 years in placebo group; and 46.4 years total population. Gender: female 86.9%, in BontA group; female 85.7% in placebo group; female 86.3% total population Inclusion criteria
Exclusion criteria
Severity of disease‐ moderate‐to‐severe CFL (maximum smile) Ethnicity‐ white 88.7%, black 3.2%, Asian 1.8%, Hispanic 5.4,other 0.9% in BontA group; white 88.8%, black 3.1%, Asian 2.2 %, Hispanic 4.9%, other 0.9% in placebo group; white 88.8%, black 3.1%, Asian 2.0%, Hispanic 5.2%, other 0.9% in total population |
|
| Interventions |
Duration of study‐ 20 weeks Intervention
Comparator
|
|
| Outcomes |
Primary outcome‐
Secondary outcome‐
|
|
| Notes | "Dr Carruthers is a consultant and investigator for Allergan, Inc., Merz Pharmaceuticals, Merz Aesthetics USA, and Solstice Neurosciences" | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Eligible subjects were randomised in a 1:1 ratio"...page 1182 Comment: we considered this unclear risk of bias because the authors did not show how they randomised the participants |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Eligible subjects were randomised in a 1:1 ratio."page 1182 Comment: we considered this unclear risk of bias due to the authors did not clarify the methods used to maintain the allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Reconstitution and preparation of syringes were undertaken by individuals with no study responsibilities involving interactions with subjects" page 1182 Comment:we considered this unclear risk of bias due to the visual aspect of BontA and placebo was not described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "Reconstitution and preparation of syringes were undertaken by individuals with no study responsibilities involving interactions with subjects" page 1182 Comment:we considered this unclear risk of bias due to the visual aspect of BontA and placebo was not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Botox, 210/222 (94.5%) completed the study, 12 withdrew (lost of follow up = 8, personal reasons = 2, protocol violation = 1, not treated = 1). Placebo group 205/223 (91.9%), 10 withdrew (lost of follow up = 12, personal reasons = 4, protocol violation = 1, not treated = 1)" page 1185 (figure 2) Comment: we considered low risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered low risk of bias |
| Other bias | Low risk | We considered this study at low risk of other bias |
Carruthers 2015.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, double‐blind, randomised, parallel‐design, placebo‐controlled extension study of participants who completed the 7‐ month phase 3 study (Study 191622‐099; www. clinicaltrials.gov identifier: NCT01224015) in crow's feet and/or glabellar lines Study date‐ no information Study setting‐ outpatients |
|
| Participants |
Randomised‐ 684 participants, with a mean age of 49.7 ± 9.48 years in BontA 44 U/44U group; 49.4 ± 9.35 years in BontA 24 U/24 U group; 49.4 ± 9.23 in Placebo/BontA 0/44 U group; 49.1 ± 9.32 in Placebo/placebo group; 49.4 ± 9.36 years total population. Gender: 226/260 (86.9%) female, 34/260 (13.1%) male BontA 44 U/44 group; 203/227 (89.4%) female, 24/227 (11.9%) male in BontA 24 U/24 U group; 89/101 (88.1%) female, 12/101 (11.9%) male in Placebo/BontA 0/44 U group; 80/96 (83.3%) female, 16/96 (16.7%) male in Placebo/placebo group; 598/684 (87.4%) female, 86/684 (12.6%) male total population Inclusion criteria
Exclusion criteria
Severity of disease‐ moderate‐to‐severe bilaterally symmetrical CFL at maximum smile and moderate‐to‐severe GL at maximum frown Ethnicity‐ white 232/260 (89.2%), non‐white 28/260 (10.8%) in Botox 44/44 U, white 200/227 (88.1%), non‐white 27/227 (11.9%) in BontA 24/24 U group; white 89/101 (88.1%, non‐white 12/101 (11.9%) in placebo group; BontA44 U group; white 84/96 (87.5%), non‐white 12/96 (12.5%) in placebo/placebo group; white 605/684 (88.5%), non‐white 79/684 (11.5%) in total population |
|
| Interventions |
Duration of study‐ 20 weeks Intervention/ Comparator
Total (N = 684)
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | "J. Carruthers and A. Rivkin are consultants and investigators for Allergan, Inc. L. Donofrio is an investigator for Allergan, Inc. V. Bertucci is a speaker, consultant, and investigator for Allergan, Inc. X. Lei is an employee of Allergan, Inc., and receives compensation in salary, as well as stock or stock options (or both). C. Somogyi and F. C. Beddingfield were employees of Allergan, Inc. at the time of this study and received compensation in salary, as well as stock or stock options (or both). The other authors have indicated no significant interest with commercial supporters." This RCT was a continuation of previous RCT published (Moers‐Carpi 2015), so we will consider only the patients that received placebo (Moers‐Carpi 2015) and were re randomised for BotoxR 44u and placebo in this study to avoid double count of participants. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Randomization took place on Day 1 of this study, corresponding to the last day of Study 191622‐099. Subjects who had received onabotulinumtoxinA in Study 191622‐099 continued to receive the same dose (44 U for CFL + GL, 24 U for CFL alone) in this study. Subjects who had received placebo in Study 191622‐ 099 were re randomised in a double‐blind fashion to either 44 U onabotulinumtoxinA (CFL + GL) or to placebo in a 1:1 ratio" page 703 Comment: we considered this unclear risk of bias because the authors did not explain how they randomised the participants |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Randomization took place on Day 1 of this study, corresponding to the last day of Study 191622‐099. Subjects who had received onabotulinumtoxinA in Study 191622‐099 continued to receive the same dose (44 U for CFL + GL, 24 U for CFL alone) in this study. Subjects who had received placebo in Study 191622‐ 099 were re‐randomized in a double‐blind fashion to either 44 U onabotulinumtoxinA (CFL + GL) or to placebo in a 1:1 ratio" page 703 Comments: we considered this unclear risk of bias because the authors did not explain the methods used for maintaining the allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "To maintain the blind, all medications were reconstituted and prepared by individuals who had no interactions with subjects." page 104 (from Moers‐Carpi 2015) Comment: we considered this unclear risk of bias due to the visual aspect of BontA and placebo was not described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "To maintain the blind, all medications were reconstituted and prepared by individuals who had no interactions with subjects." (from Moers‐Carpi 2015) Comment: we considered this unclear risk of bias due to the author did not provide information about blinding of outcome assessor |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Of 684 subjects enrolled, 641 (93.7%) completed this study A total of 667 subjects (97.5%) received the third treatment. Most subjects who received a third dose (80.2%; 535/667) received their dose at Day 1 visit of Study 191622‐104. A total of 414 subjects (60.5%) received 2 treatments (treatment cycles 3 and 4): 149 onabotulinumtoxinA 24 U/24 U, 123 onabotulinumtoxinA 44 U/44 U, 69 placebo/ onabotulinumtoxinA 44 U, and 73 placebo/placebo. In this study, 253 subjects (37.0%) received only 1 treatment (treatment cycle 3): 74 onabotulinumtoxinA 24 U/24 U, 126 onabotulinumtoxinA 44 U/44 U, 31 placebo/onabotulinumtoxinA 44 U, and 22 placebo/placebo. Seventeen subjects failed to meet re‐treatment criteria after they received treatment 2 in Study 191622‐099 and therefore did not receive any treatment in this study: 4 onabotulinumtoxinA 24 U/ 24 U, 11 onabotulinumtoxinA 44 U/44 U, 1 placebo/ onabotulinumtoxinA 44 U, and 1 placebo/placebo" page 705 Comment: we considered this low risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered this low risk of bias |
| Other bias | High risk | Comment: we considered a high risk of bias because C. Somogyi and F. C. Beddingfield were employees of Allergan and conducted this trial |
Carruthers 2017.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, double‐blind, randomised, parallel‐design,dose‐‐ranging, placebo‐controlled (www. clinicaltrials.gov identifier: NCT0020303002) in glabellar lines Study date‐ start December 2014; end December 2015 Study setting‐ outpatients, 9 private practice settings |
|
| Participants |
Randomised‐ 268 participants with a mean age of 50 years in OnabotulinumtoxinA 20U group; 47 years in DaxibotulinumtoxinA 60 U groups; 50 years in DaxibotulinumtoxinA 40 U groups; 49 years in DaxibotulinumtoxinA 20U groups and 50 years in placebo group. Gender: 38/42(90%) female and 4/42(10%) male onabotulinumtoxinA 20U group; 37/41(76%) female and 4/41(24%) male in DaxibotulinumtoxinA 60 U groups; 36/39(92%) female and 3/39(8%) male in DaxibotulinumtoxinA 40 U groups; 36/39(91%) female and 3/34(9%) male in DaxibotulinumtoxinA 20 U groups and 31/35(89%) female and 4/35(11%) male in placebo group. Inclusion criteria
Exclusion criteria
Severity of disease‐ IGA‐FWSrating at maximum frown 28/42(67%) moderate and 14/42 (33%) severe in OnabotulinumtoxinA 2 0U group; 24/41(59%) moderate and 14/41 (41%) severe in DaxibotulinumtoxinA 60U groups; 27/39(69%) moderate and 12/39 (31%) severe in DaxibotulinumtoxinA 40 U groups; 22/34(65%) moderate and 12/34 (35%) severe in DaxibotulinumtoxinA 20 U groups and 24/35(69%) moderate and 11/35 (31%) severe in placebo group Ethinicity‐Caucasian 38/42 (90%) in OnabotulimtoxinA 20 U group; Caucasian 38/41 (93%) in DaxibotulinumtoxinA 60 U groups; Caucasian 37/39(95%) in DaxibotulinumtoxinA 40 U groups; Caucasian 31/34 (91%) in DaxibotulinumtoxinA 20 U groups and 31/35 (89%) in placebo group. |
|
| Interventions |
Duration of study‐ 36 weeks Intervention/ Comparator
|
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes | "Supported by Revance Therapeutics, Inc. J.D. Carruthers is a consultant and researcher for Revance, Allergan, Merz, and Alphaeon. N. Solish received a grant from Revance for participating in this study and is a consultant to Revance, Allergan, and Galderma. S. Humphrey has received research grants from Revance Therapeutics. V. Bertucci is a consultant to, and receives payment for lectures, including service on speaker bureaux, from Allergan, Galderma, and Merz. He is also an investigator for Allergan, Galderma, Alphaeon, Merz, and Revance. A. Swift received an investigator fee from Revance Therapeutics, Inc. A. Metelitsa has been a consultant for Galderma and Merz. R.G. Rubio is an employee of, and holder of stock/stock options in, Revance Therapeutics, Inc. J. Waugh was an employee of, and held stock/stock options in, Revance Therapeutics, Inc. J. Quiring is an employee of QST Consultations, Ltd., which has received fees from Revance Therapeutics, Inc. for performing statistical analyses. G. Shears is an employee of Write on Target Ltd., which has received fees from Revance Therapeutics, Inc. for medical writing services. A. Carruthers is a consultant and researcher for Revance, Allergan, Merz, and Alphaeon. The remaining authors have indicated no significant interest with commercial supporters. DaxibotulinumtoxinA is an investigational agent." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "An independent statistician provided a randomization scheme of treatment assignments for each study site and subjects eligible for randomization were given the next available subject number"...page1322 Comment: we considered this unclear risk of bias because the authors did not explain how they randomised |
| Allocation concealment (selection bias) | Unclear risk | Quote:"An independent statistician provided a randomization scheme of treatment assignments for each study site and subjects eligible for randomization were given the next available subject number"...page1322 Comment: we considered this unclear risk of bias because the authors did not explain how they randomised |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote:"assigned product was reconstituted by an unblinded preparer and the masked product was provided to the investigator in a syringe. The subjects, investigators, and other site staff remained blinded to the treatment assignments."...page 1323 Comment: we considered this low risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote:"assigned product was reconstituted by an unblinded preparer and the masked product was provided to the investigator in a syringe. The subjects, investigators, and other site staff remained blinded to the treatment assignments."...page 1323 Comment: we considered this low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Overall, 98% of subjects completed the study (3 discontinued from the placebo group and 2 from the 20U daxibotulinumtoxinA group due to subject withdrawals or loss to follow‐up). Per protocol analyses required the exclusion of 77 subjects, most of these (57/77) being attributable to the Week 24 visit being more than 5 days off schedule (Table 2)."...page 1324 Comment: we considered this low risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered this low risk of bias |
| Other bias | Unclear risk | Comment: we considered a unclear risk of bias because J. Waugh was an employee of, and held stock/stock options in, Revance Therapeutics, Inc. J. Quiring is an employee of QST Consultations, Ltd. and G. Shears is an employee of Write on Target Ltd., which has received fees from Revance Therapeutics, Inc. for medical writing services. |
Cheon 2019.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, double‐blind, randomised, active‐drug controlled, phase I/III study designed to determine the non‐inferiority of Neuronox® compared to Onabotulinumtoxin A in the treatment of moderate to severe lateral canthal lines. NCT03317574 Study date‐ no information Study setting‐ outpatients, 5 centres (South Korea) |
|
| Participants |
Randomised 220 participants with a mean age of 47.14 ±7.87yearsinn Nuronox® group and 49.03± 8.28 years in onabotulinumtoxinA group. Gender 88/110 (80%) female and 22/110 (20) males in Neuronox® group, and 92/110 (83.64%) female and 18/110(16.36%) male in onabotulinumtoxinA group. Previous BontA treatment 14/110(12.73%) in Neuronox® group and 17/110 (15.45%) in OnabotulinumtoxinA group. Inclusion criteria
severity scale Exclusion criteria
Severity of the disease‐ moderate 29/110(26%), severe 81/110 (74%) in Neuronox® group; moderate 35/110 (32%), severe 75/110 (68%) in onabotulinumtoxinA group. |
|
| Interventions |
Duration of study‐ 16 weeks Intervention Neuronox® 24u (N=110)‐ 4 U (0.1 mL), 3 sites each side Comparator OnabotulinumtoxinA 24 U (N=110)‐ 4 U (0.1 mL), 3 sites each side |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | "This study was sponsored by Medytox Inc., Korea. W.S. Lee is an employee of Medytox Inc." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote:"Dynamic Allocation was used to randomize eligible subjects"...page no number Comment: we considered this an unclear risk of bias because the authors did not explain how thy randomise the participants |
| Allocation concealment (selection bias) | Unclear risk | Quote:"Dynamic Allocation was used to randomize eligible subjects"...page no number Comment: we considered this an unclear risk of bias because the authors did not explain how thy randomise the participants |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The pharmacist or designee
responsible for the reconstitution was kept unblinded, and performed the dilution and
preparation of the syringe in a separate room. All other individuals, including investigators and subjects were kept blinded throughout the study."... page no information. Comment: we considered this low risk of bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The pharmacist or designee
responsible for the reconstitution was kept unblinded, and performed the dilution and
preparation of the syringe in a separate room. All other individuals, including investigators and subjects were kept blinded throughout the study."... page no information. Comment: we considered this low risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote:" due to major protocol deviation by seven subjects (two from Neuronox® and five from ONA (Figure 2)."...pgae no information Comment; we considered this a low risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered this low risk of bias |
| Other bias | Low risk | Quote:"This study was sponsored by Medytox Inc., Korea. W.S. Lee is an employee of Medytox Inc."..page no information Comment: we considered this a low risk of bias |
Cohen 2012.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised, double‐blind, parallel‐design, in perioral lines Study date‐ no information Study setting‐ no information |
|
| Participants |
Randomised‐ 60 women, with a mean age of 41. 9 ± 8 years total population; 40.8 ± 7.1 years in BontA 7.5 U group; 43.2 ± 8.7 years in BontA 12 U group. Gender 100% female Inclusion criteria
Exclusion criteria
Severity of disease‐ POL score of 2 or 3 (moderate or severe) at maximal attempted muscle contraction based on a 4‐point scale that considered the upper and lower lips Ethnicity‐ 54/60 (90%) Caucasian, 3/60 (5%) black, 3/60 (5%) Hispanic total population; 30/31 (96.8%) Caucasian, 1/31 (3.2%) black, 0% Hispanic in BontA 7.5u group; 24/29 (82.8%) Caucasian, 2/29 (6.9%) black, 3/29 (10.3%), Hispanic in BontA 12u group |
|
| Interventions |
Duration of study‐ 20 weeks. Intervention
Comparator
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | The authors received research grant support from Allergan Inc. for this study and for manuscript preparation. Dr Cohen and two other authors are consultants and investigators for Allergan Inc. This study finished in 2016 ‐ results added to clinical trial register May 28, 2019. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "this study was a multicenter, randomised... parallel‐design, in which subjects were randomised in a 1:1 ratio" page 1498. Comment: we considered this unclear risk of bias because the authors did not show how they randomised the participants |
| Allocation concealment (selection bias) | Unclear risk | No information Comment: we considered this unclear risk of bias because the authors did not show the methods used for maintaining the allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "this study was...double‐blind.Onabotulinumtoxin A was reconstituted in preserved saline. The volume of reconstituted toxin in each syringe was 0.30mL to maintain the blind." page 1498 Comment: we considered this low risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "this study was...double‐blind..Onabotulinumtoxin A was reconstituted in preserved saline. The volume of reconstituted toxin in each syringe was 0.30mL to maintain the blind." page 1498 Comment: we considered this low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "Fifty‐one subjects (85%) completed the study, of whom 26 and 25 subjects were from the 12.0‐U and 7.5‐U arms, respectively." page 1499 Comment: we consider unclear risk of bias, the authors did not mention the reason of drop outs An e‐mail was sent to the authors on 21 November 2015. Answer on 22 November 2015: "I'm not sure I actually remember this. And the study was done so long ago, that I'm not sure we have these records on site any longer. You might want to inquire with Allergan, as they have the documents. I would imagine several patients were simply lost to follow up between the 3 sites" |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered this low risk of bias |
| Other bias | Low risk | We considered this study at low risk of other bias |
Costa 2016.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, double‐blind, randomised, parallel‐design in glabellar lines Study date‐ started 2012, ended 2014 Study setting‐ outpatients three centres |
|
| Participants |
Randomised‐ 157 female participants with mean age of 43.9 years in BontA1 (Prosigne®) group and 43.7 years in OnabotulinumtoxinA group Inclusion criteria
Exclusion criteria
Severity of disease‐ moderate to severe glabellar lines at maximum contraction and mild to moderate glabellar lines at rest Ethnicity‐ no information |
|
| Interventions |
Duration of study‐ 12 weeks Intervention
Comparator
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | Pharmaceutical support | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Realizou‐se a aleatorização em blocos de quatro, utilizando‐se o Random Allocation Software 1.0 para alocar os pacientes nos grupos" page 35 Comment: we consider low risk of bias (translation of quote: the authors wrote they use a software to randomise the patients) |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Realizou‐se a aleatorização em blocos de quatro, utilizando‐se o Random Allocation Software 1.0 para alocar os pacientes nos grupos." page 35 Comment: we considered unclear risk of bias because the authors did not explain how the methods used for maintaining the allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Um investigador reconstituiu os frascos da Toxina 1 e da Toxina 2, aspirou 20 unidades de cada produto com seringa BD com capacidade para 1ml, agulha curta, e entregou para o segundo investigador que injetou a toxina já diluída sem saber qual produto havia na seringa."... page 35 (translation‐ one of the investigator reconstituted toxin 1 and toxin 2, he used 20u for each toxin, seringe BD, 1mL, short needle, and delivered to the second blinded investigator that injected the diluted toxin )Comment: we considered unclear risk of bias due to the authors did not reported methods for blinding visual aspect of interventions |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "Três avaliadores independentes analisaram todas as fotografias realizadas durante o estudo e classificaram a gravidade das rugas glabelares".. page 35 (translation‐ Three independents investigators evaluated all the study pictures and they classified all of them) Comment: we consider unclear risk of bias due to the due to the authors did not reported methods for blinding visual aspect of interventions |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Houve seis perdas de seguimento na visita 2 (V2) (uma da Toxina 1, e cinco da Toxina 2), pela dificuldade de as pacientes seguirem as datas de retorno do protocolo. Da visita V2 (15 dias) até a V6 (120 dias), 16 sujeitos de pesquisa perderam o seguimento por faltar às visitas, mesmo após inúmeras tentativas de contato pelo centro do estudo (também por dificuldade de seguir o calendário do estudo). Completaram o estudo 119 pacientes (63 e 56 nos braços Toxina 1 e Toxina 2, respectivamente."... page 36 (translation‐ There was six drop outs in visit 2 (one in toxin 1 group and five in toxin 2 group), because the patients lost their follow‐up visit. From visit 2(15 days) to visit 6 (120 days), 16 patients did not show up, even though several previous contact. 119 subjects (63 and 56 in toxin1 and toxin 2 groups, respectively) completed the study Comment: we consider low risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we consider low risk of bias |
| Other bias | High risk | High number of protocol violation (17 in BontA (Prosigne®) group and 4 in OnabotulinumtoxinA group). Comment: we consider this high risk of bias |
Dayan 2010.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, double‐blind, randomised, placebo‐controlled, parallel‐design in glabellar lines, forehead lines, and crow’s feet lines Study date‐ no information Study setting‐ outpatients |
|
| Participants |
Randomised 100 participants, with a mean age of 48.3 ± 9.3 years (range 25‐73). Gender 96% female, 4% male Inclusion criteria
Exclusion criteria
Severity of disease‐ glabellar lines, forehead and crow's feet lines Ethnicity‐ no information |
|
| Interventions |
Duration of study‐ 12 weeks Intervention
Comparator
|
|
| Outcomes |
Primary outcome
Secondary outcome
|
|
| Notes | "The authors have indicated no significant interest with commercial supporters." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "double‐blind, randomised, placebo‐controlled" page 2089 Comment: we consider unclear risk of bias because the authors did not explain how they randomised the participants |
| Allocation concealment (selection bias) | Unclear risk | No information Comment: we considered unclear risk of bias because the authors did not explain how the methods used for maintaining the allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "An unblinded nurse prepared the injections, keeping the physician and participant blinded to treatment for the duration of the 3‐month survey." page 2089 Comment: we considered unclear risk of bias due to the authors did not reported methods for blinding participants, including visual aspect of interventions |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "An unblinded nurse prepared the injections, keeping the physician and participant blinded to treatment for the duration of the 3‐month survey." page 2089 Comment: we considered unclear risk of bias due to the authors did not reported methods for blinding assessors, including visual aspect of interventions |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "Follow‐up data at the time of the telephone call were available for 97% of the entire cohort (BoNTA, 96%; placebo, 98%) and for 80% of the entire cohort at 3 months (BoNTA, 74%; placebo, 86%)." page 2090 Comment: we considered unclear risk of bias because the authors did not explain drop‐out reason |
| Selective reporting (reporting bias) | High risk | Only P value was shown. Comment: we considered this high risk of bias, we sent an e‐mail on 22 November 2015. We received no answer. |
| Other bias | Low risk | We considered this study at low risk of other bias |
De Boulle 2018.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, double‐blind, randomised, multicentre, placebo‐controlled, parallel‐design in upper facial lines, forehead lines, and crow’s feet lines. Period 1 (Days 1–180) comprised a double‐blind, placebo‐controlled, single‐treatment parallel‐group study design comparing onabotulinumtoxinA and placebo. Period 2 (Days 180–360), participants could receive up to 2 additional open‐label treatments (Cycles 2 and 3, >= 84 days apart) with onabotulinumtoxinA 64 U, administered using the same 16‐point pattern Study date‐ started October 2014 and finished April 2016 Study setting‐ outpatient 24 sites (10 in the USA, 14 in Europe) |
|
| Participants | Randomised 787 participants. Age ranging: Onabot40 U group‐47.6 years old (range 21‐75), Onabot 64 U group‐45.5 years old (range 21‐76), placebo group‐ 48.1 (range 22‐73). Gender: Onabot40 U group 87.4% female, Onabot64 U group 90.7% female, placebo group 88.7% female. Inclusion criteria
Exclusion criteria
Severity of disease (according investigator rating of FHL severity eyebrow elevation in baseline): Onabot40U group 54.1% (moderate), 45.9%(severe); Onabot64U group 51.8% (moderate), 48.2%(severe); placebo group 51.9% moderate, 48.1% severe Ethnicity‐ 90.3% Caucasian in Onabot40U group; 91.1% Caucasian in Onabot64U group; 92.9% Caucasian in placebo group |
|
| Interventions |
Duration of study: Period 1 (Days 1–180). Period 2 (Days 180–360)
Intervention
Comparator
were administered as 0.1 mL injections distributed over 16 sites: 5 in FHL, 5 in GL, and 3 in CFLon each side Ratio: period 1 (2:2:1 Onabot44 U:Onabot64 U: placebo) Period 2 no information |
|
| Outcomes |
Primary outcomes
subject FWS ratings of FHL severity at maximum eyebrow elevation on Day 30 of the double‐blind period
Secondary outcomes
|
|
| Notes | "This study was sponsored by Allergan plc. Writing and editorial assistance was provided by K.E. Larsen and J. Street of PelotonAdvantage, Parsippany, NJ, and was funded by Allergan plc Dublin, Ireland. K. De Boulle has served as a consultant on an advisory board and speakers’ bureau, and has received honoraria from Allergan plc. W.P. Werschler has served on an advisory board, as a speaker, and as a consultant and/or received research funding from Allergan plc.M.H. Gold serves as a consultant and has received research funding from Allergan plc. S. Bruce has served on an advisory board and on a speakers’ bureau, has received research grants, and serves as an investigator trainer forAllergan plc. G. Sattler has received a research grant for participation in this study. P. Ogilvie received research grants from Allergan plc and serves as a consultant, advisory board member, and trainer for Allergan plc. D. Vitarella was an employee of Allergan plc at the time of this study. J. Street and K.E. Larsen serve as medical writers forPeloton Advantage, which received funding for editorial services from Allergan plc. E. Lee, X. Lei, C. Mao, and I.Yushmanova are employees of Allergan plc and may own stock or options in the company." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "On Day 1, after randomization (2:2:1), subjects"...page 1438 Comment: we considered unclear risk of bias because the authors did not mention how they randomise the participants |
| Allocation concealment (selection bias) | Unclear risk | Quote:"On Day 1, after randomization (2:2:1), subjects"...page 1438 Comment: we considered unclear risk of bias because the authors did not mention how they randomise the participants |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote:"Period 1 (Days 1–180) comprised a double‐blind, placebo‐controlled"...page 1438 Comment: we considered unclear risk of bias because the authors did not mention how they blind the participants |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote:"Period 1 (Days 1–180) comprised a double‐blind, placebo‐controlled"...page 1438 Commet: we considered unclear risk of bias because the authors did not mention how they keep the participants blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: Figure 2 ...page1440 Coment: we considered low risk of bias |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes were reported Comment: e consider a low risk of bias |
| Other bias | Unclear risk | Quote:"E. Lee, X. Lei, C. Mao, and I.Yushmanova are employees of Allergan plc and may own stock or options in the company"....page1437 Comment: we consider unclear risk of bias because professional aspects of some authors |
Fagien 2007a.
| Study characteristics | ||
| Methods |
Study design‐ double‐blind, randomised, placebo‐controlled, parallel‐design, in glabellar lines Study date‐ no information Study setting‐ no information |
|
| Participants |
Randomised‐ 70 participants, with mean age of 44 years (30‐54 years). Gender‐ no information Inclusion criteria
Exclusion criteria
Severity of disease‐ moderate to severe glabellar lines at contraction Ethnicity‐ 97% Caucasian |
|
| Interventions |
Duration of study‐ 12 weeks Intervention
Comparator
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | “This research was funded by Allergan, Inc. All authors received compensation for their work in the study. Jonathan Kowalski is a paid employee/researcher for Allergan. Drs. Kowalski, Cox, and Finn are stockholders in Allergan.” | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "A Double‐Blind, Randomized, Placebo‐Controlled Study" page S3 " The patients were assigned to one of the treatments groups (in a 1:1 ratio) in accordance with a randomisation schedule generated by the independent clinical research organization." page S3 Comment: we considered this unclear risk of bias because the authors did not explain how they randomised the participants |
| Allocation concealment (selection bias) | Unclear risk | Quote: "The patients were assigned to one of the treatments groups (in a 1:1 ratio) in accordance with a randomisation schedule generated by the independent clinical research organization." page S3 Comment: we considered this unclear risk of bias because the authors did not explain the methods used for maintaining the allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "To maintain the study blinding, the syringes for treatment were prepared with botulinum toxin type A or placebo by an assistant and the injector was unaware of which treatment was in each syringe." page S4 Comment: we considered low risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "To maintain the study blinding, the syringes for treatment were prepared with botulinum toxin type A or placebo by an assistant and the injector was unaware of which treatment was in each syringe." page S4 Comment: we consider low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "A total of 70 patients were enrolled, of whom 65 (93%) completed" page S5 Comment: we considered unclear risk of bias because the authors did not explain the drop‐out reason |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered low risk of bias |
| Other bias | Unclear risk | One of the authors (V) was Allergan employee. Pharmaceutical support Comment: we considered this a unclear risk of bias because one of the authors was Allergan employee |
Feng 2015.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised, dose‐ranging placebo‐controlled, parallel‐design in glabellar lines Study date‐ start (25 November 2009), end (27 November 2010) Study setting‐ outpatients from seven centres |
|
| Participants |
Randomised‐ 448 participants, with mean age of 44.34 years in placebo group; 44.2 years in low‐dose BontA (10 u) group; 42.79 years in high‐dose BontA (20 u). Gender: male 17/122 (13.93%), female 105 /122 (86.07%) in placebo group; male 29/183 (15.85%), female 154/183 (84.15%) in BontA low‐dose group; male 27/183(14.75%), female 156/183 (85.25%) in BontA hig‐ dose group Inclusion criteria
Exclusion criteria
Severity of disease‐ Participants with moderate‐to‐severe glabellar lines Ethnicity‐ no information |
|
| Interventions |
Duration of study‐ 16 weeks Intervention
Comparator
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | “The authors have indicated no significant interest with commercial supporters.” | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "double‐blind trial and randomly divided into" page S56 Comment: we consider unclear risk of bias because the authors did not explain how they randomised the participants |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Participants were assigned to low‐dose (10 units, n = 183), high‐dose (20 units, n = 183), or saline" page S57 Commen: we consider unclear risk of bias because the authors did not explain the methods used for maintaining the allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Participants who met the inclusion and exclusion criteria were enrolled in a double‐blind, placebo‐controlled" page S57 Comment: we consider unclear risk of bias because the authors did not explain how they blinded the participants |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "Participants who met the inclusion and exclusion criteria were enrolled ina double‐blind, placebo‐controlled" page S57 Comment: we consider unclear risk of bias because the authors did not explain how they blinded the outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Four participants were lost to follow‐up (one in the placebo group, 2 in the low dose, and 1 in the high‐dose group), and excluded from primary end point data. Another 13 participants statistically 'out of the time window' were excluded from analysis, including 5 participants in the placebo group, 5 in the low‐dose group, and 3 in the high‐ dose groups, respectively. One participant in the high‐dose group receiving combined therapy was not included in the per protocol set (PPS). The final PPS consisted of 449 (92.01%) participants, including 111, 167, and 171 in the placebo, low‐dose and high‐dose arms, respectively." page S58 Comment: we consider low risk of bias |
| Selective reporting (reporting bias) | High risk | The authors did not mention if the investigator assessment was done at rest or at contraction Comment: we consider this high risk of bias, we sent an e‐mail on 21 November 2015. We received no answer |
| Other bias | Low risk | We considered this study at low risk of other bias |
Firoz 2012.
| Study characteristics | ||
| Methods |
Study design‐ randomised, double‐blind, split‐face design, active controlled, parallel 2‐arm, onabotulinumtoxinA versus AbobotulinumtoxinA in glabellar and forehead lines (Poster) Study date‐ no information Study setting‐ no information |
|
| Participants |
Randomised 74 participants, with mean age of 50 years (32‐65 years). Gender‐ no information Inclusion criteria‐ no information Exclusion criteria‐ no information Severity of disease‐ moderate to severe glabellar lines and raising forehead‐ split face Ethnicity‐ 43.2% Caucasian, 2.8% black, 51.4% Hispanic, 1.4% Asian and 1.4% other |
|
| Interventions |
Duration of study‐ 24 weeks Intervention
Comparator
Ratio‐ no information |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | “Commercial support: None identified” | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Using a random number generator, patients received" page AB210 Comment: We considered low risk of bias |
| Allocation concealment (selection bias) | Unclear risk | No information Comment: we considered unclear risk of bias due to the authors did not provide the methods used for maintain the allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "patients received identical volumes and injection patterns of one product in the right corrugator and frontalis, and the other product in the left." (page AB21) Comment: We considered low risk of bias since the authors adopted proper actions to assure blinding of personnel and patients. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information about outcome assessors blinding Comment: we considered unclear risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information about losses Comment: we considered unclear risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered low risk of bias |
| Other bias | Low risk | We considered this study at low risk of other bias |
Hanke 2013.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised, double‐blind, placebo‐controlled, parallel ‐design, phase III in glabellar lines Study date‐ no information Study setting‐ outpatients from eight centres |
|
| Participants |
Randomised‐ 271 participants, with mean age of 46.9 ± 9.3 years (median = 46.5 years) BontA group; 45.7 ± 11.4 years (median 46 years) placebo group. Gender: 170/182 (93.4%) female, 12/182 (6.6%) male in BontA group; placebo‐ 84/89 (94.4%) female, 5/89 (5.%) male in placebo group Inclusion criteria
Exclusion criteria
8 months
Severity of disease‐ moderate to severe glabellar frown lines at maximum frown (FWS) Ethnicity‐ 119/182 (65.4%) Caucasian, 8/182 (4.4%) black, 51/182 (28%) Hispanic, 2/182 (1.1%) Asian, and 2/182 (1.0%) other in BontA group; 58/89 (65.2%) Caucasian, 3/89 (3.4%) black, 28/89 (31.5%) Hispanic, 0% Asian, and 0% other in placebo group |
|
| Interventions |
Duration of study‐ 16 weeks Intervention
Comparator
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | "This study was supported by Merz Pharmaceuticals GmbH. The sponsor was involved in the design and conduct of the study; in the collection, analysis, and interpretation of data; and in the preparation, review, and approval of this manuscript. C. William Hanke, Rhoda S. Narins, Fredric S. Brandt, Joel L. Cohen, Lisa M. Donofrio, Jeanine Downie, David H. McDaniel, Mark Nestor, Joel Schlessinger, Amy Taub, and Robert Weiss are paid consultants and researchers for Merz. Moritz Heinz and Andrea Schlo€ be are employees of Merz Pharmaceuticals GmbH." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "At Visit 1 (Day 0), subjects were randomised 2:1 to receive a total dose of 20 U of incobotulinumtoxinA" page 893 Comment: we considered unclear risk of bias because the authors did not mention how they randomised the participants |
| Allocation concealment (selection bias) | Unclear risk | Quote: "At Visit 1 (Day 0), subjects were randomised 2:1 to receive a total dose of 20 U of incobotulinumtoxinA" page 893 Comment: we considered unclear risk of bias because the authors did not mention due to the authors did not provide the methods used for maintain the allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The study investigators, centre staff, and subjects were blinded to the assigned medication" page 893 Comment: we considered low risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The study investigators, centre staff, and subjects were blinded to the assigned medication" page 893 Comment: we considered low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Three subjects (1.1%) discontinued the study prematurely; one (0.5%) in the incobotulinumtoxinA group withdrew consent, and two (2.2%) in the placebo group were lost to follow‐up. Twenty‐one subjects were major protocol deviators and were not included in the PPS; 6.6% (n = 12) and 10.1% (n = 9) in the incobotulinumtoxinA and placebo groups, respectively." page 894 Comment: we considered low risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered low risk of bias |
| Other bias | Unclear risk | Quote: "This study was supported by Merz Pharmaceuticals GmbH. The sponsor was involved in the design and conduct of the study; in the collection, analysis, and interpretation of data; and in the preparation, review, and approval of this manuscript." Comment: we considered this a unclear risk of bias |
Harii 2008.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, double‐blind, randomised, placebo‐controlled, two‐ dose, parallel design in glabellar lines in Japanese participants Study date‐ no information Study setting‐ outpatients |
|
| Participants |
Randomised 142 participants, with mean age of 45.7± 9.1 years. Gender: 90% female, 10% male Inclusion criteria
Exclusion criteria
Severity of disease‐Quote: “All subjects had either moderate (50.7%) or severe (49.3%) glabellar lines at maximal contraction.” Ethnicity‐ 100% Japanese |
|
| Interventions |
Duration of study‐ 16 weeks Intervention
Comparator
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | “This study was funded by Allergan, Inc., Irvine, California.” | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Using the random‐number‐generation function of SAS (Statistical Institute, Inc., Cary, NC), subjects were allocated to one of three treatment groups: 10‐U BoNTA, 20‐U " page 725 Comment: we considered this low risk |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Using the random‐number‐generation function of SAS (Statistical Institute, Inc., Cary, NC), subjects were allocated to one of three treatment groups: 10‐U BoNTA, 20‐U" page 725 Comment: we considered unclear risk of bias due to the authors did not provide the methods used for maintain the allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Vials used for treatment administration were coded to maintain the blind." page 725 Comment: we considered unclear risk of bias due to no information about visual aspect of interventions was provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "Vials used for treatment administration were coded to maintain the blind" page 725 Comment: we considered unclear risk of bias due to no information about visual aspect of interventions was provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "Six subjects in the full analysis data set discontinued: two in the 20‐U group discontinued before treatment, two in the 10‐U group moved away, one in the 20‐U group retracted consent, and one in the placebo group became pregnant." page 726 At the beginning the authors mentioned 140 participants, but they treated 139 participants and there was no explanation about the missing one. Comment: We considered unclear risk of bias because data discrepancy. We sent an e‐mail on 23 November 2015, but the electronic address was wrong and we could not find a valid e‐mail. |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered low risk of bias |
| Other bias | Low risk | We considered this study at low risk of other bias |
Harii 2017.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, phase III. First period (6 months) was a double‐blind, randomised, placebo‐controlled, two‐ dose, parallel design in crow's feet lines in Japanese participants. Second period was an open‐label study (7 months) Study date‐ no information Study setting‐ outpatients |
|
| Participants |
Randomised 300 participants, with mean age of 50.2± 6.05 years in OnabotulinumtoxinA 24U (first period)/OnabotulinumtoxinA 24U (second period) group; 50.6 ± 6.11 years in OnabotulinumtoxinA 12 U (first period)/OnabotulinumtoxinA 12 U (second period) group; 49.3 ± 7.24 years in Placebo(first period)/OnabotulinumtoxinA 24 U (second period); 48.3 ± 8.10 years in placebo(first period)/OnabotulinumtoxinA 12 U (second period); 49.7 ± 6.64 years in total populatio. Gender: 84/104(80.8%) female and 20/104(9.2%) male in OnabotulinumtoxinA 24 U (first period)/OnabotulinumtoxinA 24 U (second period) group; 70/99(70.7%) female and 29/99(29.3%) male in OnabotulinumtoxinA 12 U (first period)/OnabotulinumtoxinA 12 U (second period) group; 36/48(75%) female and 12/48(25%) male in placebo(first period)/OnabotulinumtoxinA 24 U (second period); 34/49(69.4%) female and 15/39(29.6%) male in Placebo(first period)/OnabotulinumtoxinA 12 U (second period); 224/300(74.7%) female and 76/300 (25.33%) in total population. Inclusion criteria
Exclusion criteria
Severity of disease‐ according to investigator assessment using FWS‐A at maximum smile: 51/104(49%) moderate and 53/104(51%) severe in OnabotulinumtoxinA 24 U (first period)/OnabotulinumtoxinA 24 U (second period) group; 49/99(49.5%) moderate and 50/99(50.5%) in OnabotulinumtoxinA 12 U (first period)/OnabotulinumtoxinA 12 U (second period) group; 23/48(47.9%) moderate and 25/48(52.1%) severe in Placebo(first period)/OnabotulinumtoxinA 24U (second period); 24/49(49%) moderate and 25/49(51%) severe in Placebo(first period)/OnabotulinumtoxinA 12 U (second period); 147/300(49%) moderate and 153/300(51%) severe in total population. Ethnicity‐ 100% Japanese |
|
| Interventions |
Duration of study‐ first period 24 weeks and second period 35 weeks Interventions
Comparator
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | "This study was sponsored by Allergan plc, Dublin, Ire‐ land. Writing and editorial assistance was provided to the authors by Emily H. Seidman, MSc, of Peloton Advantage, Parsippany, NJ, and was funded by Allergan plc." Elisabeth Lee was Alergan employee. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote:"Subjects were assigned a randomization number (not disclosed to the study center),"...page 1187 Comment: we considered unclear risk of bias because the authors did not explain how they randomised the participants |
| Allocation concealment (selection bias) | Low risk | Quote:"An interactive voice or web response system designed by Allergan Data Management provided a specific kit number for each subject, and the study center administered treatment. ,"...page 1188 Comment: we considered low risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: no information Comment: we considered an unclear risk of bias, because the authors did not explain how and who prepared the medication. We sent an e‐mail on November 12, 2017 |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: no information Comment: we considered an unclear risk of bias, because the authors did not explain how and who prepared the medication. We sent an e‐mail on November 12, 2017 |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote:" The majority of subjects completed the study (89.3%)"...page1189 Coment: we considered an unclear risk of bias, because the author did not explain the drop outs. We sent an e‐mail on November 12, 2017 |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered low risk of bias |
| Other bias | Unclear risk | Comment: we considered unclear risk of bias because one of the authors (Elisabeth Lee) was Allergan employee |
Hexsel 2013.
| Study characteristics | ||
| Methods |
Study design‐ single‐centre, randomised, open‐label study of full face, parallel‐ design, phase IV Study date‐ start October 2009, end December 2010 Study setting‐ outpatients from one private clinic (Brazil) |
|
| Participants |
Randomised‐ 90 participants, with mean age of 48.3 ± 7.2 years (30‐60 years). Gender: 82/90 (96.5%) female, 8/90 (3.5%) male. Other demographic data: 62.4% nonsmokers, 60% BontA naive, non fillers‐63.5% Inclusion criteria
Exclusion criteria
Severity of disease‐ wrinkles in full face Ethnicity‐ 79/90 (92.9%) Caucasian |
|
| Interventions |
Duration of study‐ 16 weeks Intervention/Comparator‐ AbobotulinumtoxinA group 1 (166 U ± 4), group 2 (194 U ± 12), and group 3 (214 U ± 11) |
|
| Outcomes |
Primary outcome
Secondary outcome
|
|
| Notes | "Dr Hexsel has conducted clinical trials for Ipsen, Allergan, Galderma, and Medicis. The other authors have no relevant conflicts of interest to declare. Scientific grant was received from Galderma Inc., France" | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The randomisation list was generated by a statistician and subjects were sequentially allocated into 3 groups" page 1356 Comment: we considered this low risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Quote: "The randomisation list was generated by a statistician and subjects were sequentially allocated into 3 in a 1:1:1 proportion" page 1356 Comment: we consider this unclear risk of bias, because the authors did not explain the methods used for maintain the allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "open‐label study" page 1356 Comment: we considered this high risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "open‐label study" page 1356 Comment: we considered this high risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "85 completed the study: 5 discontinued due to loss of follow‐up" page 1357 Comment: we considered this low risk of bias |
| Selective reporting (reporting bias) | Unclear risk | Only P value was showed. Comment: we considered this unclear risk of bias. We e‐mailed the author on 6 March 2015. Answer on 10 March 10 2015: the authors sent a SPSS file |
| Other bias | Low risk | We considered this study at low risk of other bias |
Kane 2009.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised, double‐blind, placebo‐controlled study, parallel‐design, phase III Study date‐ no information Study setting‐ outpatients from 27 centres |
|
| Participants |
Randomised‐ 816 participants, with mean age of 49.2 ± 10.31 years in placebo group, 48.7 ± 10.33 years in AbobotulinumtoxinA group. Gender: 238/272 (88%) female, 34/272 (13%) male in placebo group; 481/544 (88%) female, 63/544 (12%) male in BontA group. Patients stratified by race/ethnicity. Severity of disease‐ moderate to severe glabellar lines Other demographic data‐ Fitzpatrick skin type: Placebo‐ 1 (extremely fair, always burns, never tans) 7/272 (3%), 2 (white, always burns, sometimes tans) 91/272 (33%), 3 (white, sometimes burns, always tans) 79/271 (29%), 4 (olive brown, rarely burns, always tans) 47/272 (17%), 5 (brown, never burns) 30/272 (11%), 6 (heavily pigmented or black, never burns) 18/272 (7%). Dysport‐ 1 (extremely fair, always burns, never tans) 19/544 (3%), 2 (white, always burns, sometimes tans) 161/544(30%), 3 (white, sometimes burns, always tans) 185/544 (34%), 4 (olive brown, rarely burns, always tans) 86/544 (67%), 5 (brown, never burns) 53/544 (10%), 6 (heavily pigmented or black, never burns) 140/544 (7%). BontA naive 221/272 (81%) in placebo group; 51/272 (10%) not naive in placebo group. BontA naive in AbobotulinumtoxinA group 437/544 (80%) BontA naive; 107/544(20%) not BontA naive Inclusion criteria
Exclusion criteria
Severity of disease‐ patients with grade 2 or 3, corresponding to moderate to severe wrinkles at maximum contraction Ethnicity‐ 191/271 (70%) Caucasian, 54/272 (20%) African American, 19/271 (7%) Hispanic, 2/272 (<1%) Asian, 2/272(<1%) Native American, 3/272 (<1%) other in placebo group; 364/544 (67%) Caucasian, 106/544 (19%) African American, 57/544 (10%) Hispanic, 8/544 (1%) Asian, 1/544 (<1%) Native American, 8/544 (1%) other in BontA group |
|
| Interventions |
Duration of study‐ 20 weeks Intervention
Comparator‐
|
|
| Outcomes |
Primary outcome‐
Secondary outcome‐
|
|
| Notes | "Medicis Aesthetics, Inc. (Scottsdale, Ariz.) provided Dysport and study funding to the authors. Michael A. C. Kane is a consultant, speaker, stockholder, and investigator for Medicis; a consultant, speaker, and stockholder for Allergan; a consultant and stockholder for Mentor; a consultant and speaker for QMed; and a consultant, investigator, and stock option holder for Revan" | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomised in a 2:1 ratio to one treatment of variably dosed" page 1620 Comment: we considered unclear risk of bias because the authors did not explain how they randomised the participants |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Patients were randomised in a 2:1 ratio to one treatment of variably dosed" page 1620 Comment: we considered unclear risk of bias because the authors did not explain the methods used for maintain the allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Double‐Blind, Placebo‐Controlled" page 1620 Comment: we considered unclear risk of bias because the authors did not explain how they blinded the participants |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Double‐Blind, Placebo‐Controlled" page 1620. "Duration of Response Assessed by Blinded Evaluator at Maximum Frown"page 1623 Comment: we considered low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Patient disposition can be seen in Table 3. No patient discontinued because of an adverse event or lack of product efficacy". Placebo completed 265/272 (97%). Withdraw reasons: lost of follow‐up 1/272(<1%), patient decision 6/272 (2%), patient not compliant study requirements 0%. Dysport completed 534/544 (98%). Withdraw reasons: lost of follow‐up 7/544 (1%), patient decision 2/544 (<1%), patient not compliant study requirement 1/544 (<1%) page 1622 Comment: we considered this low risk of bias |
| Selective reporting (reporting bias) | Low risk | Duration of Response Assessed by Blinded Evaluator at Maximum Frown (table 4). In the second row: No. (%) of patients who became non responders during the study observation period, the authors reported 30/272 = 97%, but the correct percentage was 30/272 = 11% Comment: we considered this low risk of bias. We sent an e‐mail on 1st November 2015. No answer to date |
| Other bias | High risk | One of the authors was stockholder of pharmaceutical company Comment: we considered this high risk of bias |
Kane 2015.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised, double‐blind, parallel‐design in glabellar lines Study date‐ start February 2014, end no information Study setting‐ outpatients from ten centres |
|
| Participants |
Randomised‐ 250 females participants, with mean age of 39.3 ± 7.4 years in incobotulinumtoxinA group, 39.4 ± 7.8 years in onabotulinumtoxinA group, 39.3 ± 7.6 years total population Inclusion criteria
Exclusion criteria
Severity of disease‐ moderate‐to‐severe glabellar lines at maximum frown Ethnicity‐ IncobotulinumtoxinA 23/122 (18.9%) Hispanic, 99/122 (81.1%) non‐Hispanic. OnabptulinumtoxinA 35/128 (27.3%) Hispanic, 93/128 (72.7%) non‐Hispanic, total population 58/250(23.2%) Hispanic, 192/250(76.8%) non‐Hispanic. Race‐ IncobotulinumtoxinA 104/122 (85.2%) white, 14/122 (11.5%) black or African American, 4/122 (3.3%) Asian, 0/122 (0%) American Indian or Alaska native, 0/122(0%) other; OnabotulinumtoxinA 107/128 (83.6%) white, 13/128 (10.2%) black or African American, 4/128 (3.1%) Asian, 1/128 (0.8%) American Indian or Alaska native,3/128 (2.3%) other; total population 211/250 (84.4%) white, 27/250(10.8%) black or African American, 8/250 (3.2%) Asian, 1/250 (0.4%) American Indian or Alaska native,3/250 (1.2%) other |
|
| Interventions |
Duration of study‐ 16 weeks Intervention
Comparator
|
|
| Outcomes |
Responder rate to treatment, defined as a ≥1‐point improvement from base‐ line on the FWS at maximum frown Secondary outcomes
|
|
| Notes | "The study was sponsored by Merz North America, Inc. All authors except E. Finn have been consultants and/or investigators for Merz North America, Inc. E. Finn (on behalf of Complete Medical Communications, which provided editorial support funded by Merz North provides services to the biopharmaceutical industry) America, Inc." |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "An independent biostatistician created the randomization schedule to obtain a balanced 1:1 randomization. As a result, blocks of appropriate" page 1311 Comment: we considered this unclear risk of bias because the authors did not detail how they randomised the participants |
| Allocation concealment (selection bias) | Unclear risk | No information available to allow a judgment Comment: we considered this unclear risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "IncobotulinumtoxinA and onabotulinumtoxinA were reconstituted out of view of the treating physician and the subject by designated unblinded site personnel. Site personnel were monitored to ensure that exactly the same reconstitution volume was added to each vial." page 1312 Comment: we considered this low risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Both an independent masked panel of physicians and the treating physician who was also masked evaluated subject photographs in a blinded fashion" page 1312 Commet: we consider low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: figure 2 Comment: we consider low risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we consider low risk of bias |
| Other bias | Low risk | We considered this study at low risk of other bias |
Kassir 2013.
| Study characteristics | ||
| Methods |
Study design‐ single‐centre, randomised, triple‐blind, active‐controlled, split‐face in glabellar lines and crow's feet lines Study date‐ no information Study setting‐ outpatients, one‐ centre (USA) |
|
| Participants |
Randomised‐ 85 participants, with mean age of 47 years. Gender: 87 women, 6 men Inclusion criteria
Exclusion criteria
Severity of disease‐ moderate to severe wrinkles in either the glabellar or crow’s feet area, or both Ethnicity‐ most of the patients were Caucasian |
|
| Interventions |
Duration of study‐ 20 weeks Intervention
Comparator
Ratio‐ glabella 2.5:1 (AbobotulinumtoxinA: OnabotulinumtoxinA) crow's feet 3:1 (AbobotulinumtoxinA: OnabotulinumtoxinA ) |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | "Dr Ramtin Kassir is a national trainer for Medicis. Dr Aparanjita Kolluru and Dr Martin Kassir declare no conflicts of interest." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomised into one of two groups using a computer‐generated randomisation list" page 181 Comment: we considered low risk of bias |
| Allocation concealment (selection bias) | Low risk | Quote: "A medical assistant kept the randomisation list and was the only person who knew which side" page 181 Comment: we considered low risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "A medical assistant kept the randomisation list and was the only person who knew which side" page 181 Comment: we considered unclear risk of bias due to the authors did not reported methods for blinding participants, including visual aspect of interventions |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "A medical assistant kept the randomisation list and was the only person who knew which side" page 181 Comment: we considered unclear risk of bias due to the authors did not reported methods for blinding participants, including visual aspect of interventions |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "No patients discontinued visits due to adverse effects from the treatment." page183 Comment: we considered this low risk of bias |
| Selective reporting (reporting bias) | High risk | The number of participants did not match Quote: "A total of 85 patients with moderate to severe wrinkle" page 179 Quote: "Ninety‐three patients were treated with ABO and ONA over a period of 2 months (87 women and 6 men)" page 183 Patient satisfaction was not showed. Comment: We considered high risk of bias. We sent an e‐mail on June, 21 2015 and November 23 2015. He sent the full paper but no additional information was provided about the missing data |
| Other bias | Low risk | We considered this study at low risk of other bias |
Kerscher 2015.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised, double‐blind, parallel‐design in forehead lines, glabellar lines, and crow's feet lines Study date‐ start July 2012, end October 2013 Study setting‐ outpatients from ten centres |
|
| Participants |
Randomised 156 participants, with a mean age of 47.4 ± 10.1 years in IncobotulinumtoxinA group, and 47.5 ± 8.4 years in placebo group. Gender: 94/105 (89.5%) females and 11/105 (10.5%) males in the incobotulinumtoxinA group, and 41/51 (80.4%) females and 10/51 (19.6%) males in the placebo group. Inclusion criteria
Exclusion criteria
Severity of disease‐ glabellar lines: moderate 32/105(30.5%), severe 73/105 (69.5%) in IncobotulinumtoxinA group; 13/51 (25.5%), severe 38/51 (74.5%) in placebo group. Forehead lines: moderate 18/105 (17.1%), severe 87/105 (82.9%) in IncobotulinumtoxinA group; 13/51 (25.5%), severe 38/51 (74.5%) in placebo group. Crow's feet lines: moderate 28/105 (26.7%), severe 76/105 (72.4%) in IncobotulinumtoxinA group; 14/51 (27.5%), severe 37/51 (72.5%) in placebo group Ethnicity‐ no information |
|
| Interventions |
Duration of study‐ 16 weeks Intervention
Comparator
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | Pharmaceutical support | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "using the computerized randomisation program RANCODE (Version 3.6; IDV Datenanalyse und Versuchsplanung, Gauting, Germany). Randomization in blocks of appropriate size and the blockwise distribution" page 1150 Comment: we consider low risk of bias |
| Allocation concealment (selection bias) | Low risk | Quote: "The randomisation schedule was sealed and locked in the total quality management depart‐ ment of the study sponsor and was not accessible before database close." page 1150 Comment: we consider low risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Placebo vials had the same appearance as the test product vials to ensure that the identity of the individual study materials remained unknown to the investigator, medical staff, and all subjects. All other individuals involved in the study also remained blinded, with the exception of one individual who was responsible for reporting adverse events (AEs) to the relevant authorities" page 1150 Comment: we consider low risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "All other individuals involved in the study also remained blinded, with the exception of one individual who was responsible for reporting adverse events (AEs) to the relevant authorities" page 1150 Comment: we consider low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "In total, 240 subjects were screened and 156 randomised as detailed in Figure 3" page 1152 Comment: we consider low risk of bias |
| Selective reporting (reporting bias) | High risk | The response rate by participant assessment, the response rate at rest by investigator assessment, the proportion of 1‐point responders based on the investigator’s rating of GFL and HFL at rest, investigator‐assessed and subject‐assessed ratings only P value Comment: we consider this high risk of bias. We sent an e‐mail to the authors (22 May 2016), no answer to date |
| Other bias | Low risk | We considered this study at low risk of other bias |
Kim 2014.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised, double‐blind, parallel‐design in glabellar lines Study date‐ start July 2011, end December 2012 Study setting‐ outpatients from six centres |
|
| Participants |
Randomised‐ 271 participants with mean age of 48 ± 9.8 years in NTC group; 49 ± 9.4 years in onbotulinumtoxinA group; total population 48.5 ± 9.6 years. Gender: 113/134 (84.3%) female and 21/134 (15.7%) male in NTC group; 111/134 (82.8%) female and 23/134 (17.2%) male in OnabotulinumtoxinA group; total population 224/268 (83.6%) female and 38/268 (14.2%) male. Previous botulinum toxin treatment 20/134 (14.9%) in NTC group, 18/134 (13.4%) onabotulinumtoxinA group; total population 38/268 (14.2%) Inclusion criteria
Exclusion criteria
Severity of disease‐ moderate‐to‐severe glabellar lines Ethnicity‐ no information |
|
| Interventions |
Duration of study‐ 16 weeks Intervention
Comparator
Ratio‐ 1:1 |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | Pharmaceutical support (This study was supported by SNUH research fund (H 1106‐010‐364).) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A blocked random allocation sequence was created by computer‐generated random numbers," page 1762 Comment: we consider low risk of bias |
| Allocation concealment (selection bias) | Low risk | Quote: "allocation to the either one of the two was performed by a third party. All dermatologists, managing nurses and patients were unaware of group assignments. Randomization codes were secured until all data entry was complete." page 1762 Comment: we consider low risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information available to allow a judgment Comment: we consider this unclear risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "All dermatologists, managing nurses and patients were unaware of group assignments. Randomization codes were secured until all data entry was complete." page1762 Comment: we consider low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "In summary, 262 subjects (n = 130, NTC; n = 132, onabotulinumtoxin A) completed the study (Fig. 2)" page 1763 Comment: we consider low risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered this low risk of bias |
| Other bias | Low risk | We considered this study at low risk of other bias |
Kim 2015.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised, double‐blind, active–controlled, parallel‐design, phase III trial Study date‐ no information Study setting‐ outpatients from three centres |
|
| Participants |
Randomised‐ 168 participants, with a mean age of 48.94 ± 9.13 years in M10109L group; 49.86 ± 9.13 years in BontA group years. Gender: 59/78 (75.64%) female, 19/78 (24.36%) male in M10109L group; 66/81 (81.48%) female, 15/81 (18.52%) male in BontA group Inclusion criteria
Exclusion criteria
Severity of disease‐ moderate to severe (severity score 2 to 3) glabellar frown lines according to the Facial Wrinkle Scale Ethnicity‐ 100% Korean |
|
| Interventions |
Duration of study‐ 16 weeks Intervention
Comparator
Ratio‐ 1:1 (MT10109L:OnabotulinumtoxinA) |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | "This study was supported by Medytox, Inc., Republic of Korea, and the province of Chungcheongbuk‐do, Republic of Korea. Medytox, Inc., is the manufacturer of MT10109L." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "All eligible subjects were randomised into two groups at a 1:1 ratio" page 733 Comment: we considered this unclear risk of bias because the authors did not explain how they allocated the participants |
| Allocation concealment (selection bias) | Unclear risk | Quote: "All eligible subjects were randomised into two groups at a 1:1 ratio" page 733 Comment: we considered this unclear risk of bias because the authors did not explain the methods used for maintain the allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "in a double‐blind manner." page 733 Coment: we consider this unclear risk of bias because the authors did not explain how they blinded the participants |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Three blinded raters assessed the photographs at maximum frown" page 733 Comment: we considered this low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Of 168 subjects who were enrolled, 159 completed the study and therefore constituted the per‐protocol set, of which 78 subjects were in the MT10109L group and 81 subjects were in the Botox group." page 735 Comment: we considered this low risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered this low risk of bias |
| Other bias | Low risk | We considered this study at low risk of other bias |
Lee 2013.
| Study characteristics | ||
| Methods |
Study design‐ double‐blinded, randomised, active‐controlled, parallel‐design, phase III study in glabellar lines (Poster) Study date‐ no information Study setting‐ no information |
|
| Participants |
Randomised 314 participants. Age‐ no information. Gender ‐ no information Inclusion criteria‐ no information Exclusion criteria‐ no information Severity of disease‐ moderate to severe glabellar lines at maximal contraction Ethnicity‐ no information |
|
| Interventions |
Duration of study‐ 16 weeks Intervention
Comparator
Ratio‐ 1:1 (New BontA[Medytox®]: OnabotulinumtoxinA) |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | WS Lee worked in the Medical Department of Medytox Inc., Ochang, South Korea | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients was randomised at a 1:1 ratio to receive 20U of toxin" page 116 Comment: we considered unclear risk of bias because the authors did not explain how they randomised the participants |
| Allocation concealment (selection bias) | Unclear risk | Quote: "patients was randomised at a 1:1 ratio to receive 20U of toxin" page 116 Comment: we considered unclear risk of bias because the authors did not explain the methods used for maintain the allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double‐blinded" page 116 Comment: we considered this unclear risk of bias because the authors did not explain how they blinded the participants |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double‐blinded" page 116 Comment: we considered this unclear risk of bias because the authors did not explain how they blinded the outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information about losses Comment: we considered unclear risk of bias |
| Selective reporting (reporting bias) | High risk | Incomplete data. The authors reported only the outcomes assessed at week 4 Comment: we considered this high risk of bias We e‐mailed authors on June 21, 2018, but the electronic address was wrong and we could not find a valid e‐mail. |
| Other bias | Unclear risk | The author worked for Medy‐tox Comment: we considered this a unclear risk of bias |
Lowe 2006.
| Study characteristics | ||
| Methods |
Study design‐ single centre, double‐blind, randomised study, active‐controlled, parallel‐design Study date‐ start (March 2003), end (March 2005) Study setting‐ outpatients from one private clinic |
|
| Participants |
Randomised 62 participants, with mean age of 41 years (range 27‐60 years) total population; 44 ± 7.3 years in BontA1 group; 39 ± 6.6 years in BontA2 group. Gender‐ 90% female, 10% male Inclusion criteria
Exclusion criteria
Severity of disease‐ moderate or severe glabellar lines at maximum contraction. BontA group comprised 15 of 31 patients (48%) with moderate glabellar lines and 16 patients (52%) with severe glabellar lines. In the BontA2 group, 17 of 31 patients (55%) had moderate glabellar lines and 14 (45%) had severe glabellar lines at baseline. Ethnicity‐ 97% Caucasian |
|
| Interventions |
Duration of study‐ 16 weeks Intervention
Comparator
Ratio‐ 1:2.5 (OnabotulinumtoxinA: AbobotulinumtoxinA) |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | "Drs P. Lowe and R. Patnaik have received research grants from Allergan, Inc. Dr N. Lowe owns stock in Allergan, Inc, and has received research grants, consulting payments, and educational grants from Allergan, Inc. He has also received research grants and consulting payments from Medicis. Gill Shears, PhD (of Gill Shears, Inc), provided assistance with the writing of the manuscript." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "a computer‐generated randomisation code, in block sizes of 6, that determined treatment assignments for each individual. "Randomization cards" were prepared" page 976 Comment: we considered this low risk of bias |
| Allocation concealment (selection bias) | Low risk | Quote: "randomisation number and contained the treatment assignment. These were kept in a secure location and neither the investigator nor the patients had access to them or their contents. The treatment assigned to each patient was determined at the baseline visit by a pharmacist who opened the card with the lowest available randomisation number in order to discover the treatment assignment and then prepared the appropriate syringe" page 976 Comment: we considered this low risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The investigator and the patients were masked as to which product was being used—the syringes were identical in appearance and the volume to be injected was the same regardless of the product" page 976 Comment: we considered this low risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The investigator and the patients were masked as to which product was being used—the syringes were identical in appearance and the volume to be injected was the same regardless of the product." page 976 Comment: we considered this low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "A total of 62 patients were enrolled, of whom 59 (95%) completed the study (Fig 1). No patient discontinued because of lack of efficacy or adverse effects and one each discontinued for personal reasons, withdrawal of consent, and need for surgery." page 977 Comment: we considered this low risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported. Comment: we considered this low risk of bias |
| Other bias | Unclear risk | Dr N. Lowe owns stock in Allergan, Inc. Comment: we considered this unclear risk of bias |
Michaels 2012.
| Study characteristics | ||
| Methods |
Study design‐ single centre, randomised, double‐ blind, active ‐controlled, split‐face design in glabellar lines, forehead lines, and crow's feet lines Study date‐ no information Study setting‐ outpatients from one private clinic (USA) |
|
| Participants |
Randomised‐ 53 participants, with mean of age of 50 years (range 34‐65 years). Gender: 52 female, one male. Other demographic data: eight were smokers, and 26 had undergone treatment with BoNT‐ONA in the past Inclusion criteria
Exclusion criteria
Severity of disease‐ glabella, crow's feet, forehead lines. Ethnicity‐ no information |
|
| Interventions |
Duration of study‐ 20 weeks Intervention
Comparator
Ratio‐ 1:2.5 (OnabotulinumtoxinAOna:AbobotulinumtoxinA) |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | "The authors declared no potential conflicts of interest with respect to the research, authorship, and publication of this article." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A coin toss was used to randomly assign the side of the face to receive BoNT‐ONA as follows: “heads” meant that BoNT‐ONA was injected on the right side, whereas “tails” meant that BoNT‐ONA was injected on the left" page 98 Comment: we considered this low risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Quote: "A coin toss was used to randomly assign the side of the face to receive BoNT‐ONA as follows: “heads” meant that BoNT‐ONA was injected on the right side, whereas “tails” meant that BoNT‐ONA was injected on the left" page 98 Comment: we considered this unclear risk of bias due to the authors did not reported methods of allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Patients were blinded to the laterality of treatments" page 97 Comment: we considered this unclear risk of bias because the authors did not explain how they blinded the participants |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double‐blinded" page 97 Comment: we considered this unclear risk of bias because the authors did not explain how they blinded the investigators |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information about losses Comment: we considered this unclear risk of bias |
| Selective reporting (reporting bias) | High risk | Only P value, no data were showed Comment: we considered this a high risk of bias. We sent an e‐mail to authors on 23 November 2015. No answer to date |
| Other bias | Low risk | We considered this study at low risk of other bias |
Moers‐Carpi 2012.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, double‐blind, randomised, active controlled, parallel‐ design in glabellar lines, one cycle Study date‐ start November 2010, end April 2011 Study setting‐ outpatients from seven centres |
|
| Participants |
Randomised‐ 224 participants, with mean age of 45.0 ±10.8 years in OnabotulinumtoxinA group; 45.4 ± 9.8 years in IncobotulinumtoxinA group. Gender: 102/112 (91.1%) female, 10/112 (8.9%) male in OnabotulinumtoxinA group; 98/112 (87.5%) female, 14/112 (12.5%) male in IncobotulinumtoxinA group Inclusion criteria
Exclusion criteria
Severity of disease‐ Moderate glabellar lines 47.3% BontA1 group and 45.5% BontA2 group; Severe glabellar lines 52.7% BontA1 group and 54.5% BontA2 group Ethnicity‐ 110/112 (98.2%) white, 2/112 (1.8%) other in BontA1 group; 109/112(97.3%) white, 3 (2.7%) other in BontA2 group |
|
| Interventions |
Duration of study‐ 14.9 weeks Intervention
Comparator
Ratio‐ Quote: "higher dose of incobotulinumtoxinA would produce results comparable to the established 20 units of onabotulinumtoxinA in the treatment of glabellar lines. An incobotulinumtoxinA dose of 30 units was selected for comparison based on the product’s label, the disparity in biological activity testing" |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | "This study was sponsored by Allergan, Inc. Dr. Moers‐Carpi has acted as a lecturer for Allergan. Dr. Phillip‐Dormston has acted as a lecturer, consultant and has received educational grants from Allergan, Ipsen and Merz. Dr. Hoffman has acted as a consultant for Allergan. Prof Dirschka has acted as a consultant for Allergan. Drs. Fulford‐ Smith and Tan are employed by Allergan, Marlow, UK and Mary Ann Chapman, PhD, is a paid writer/ consultant to Allergan. Dr. Rütter, Dr. Feller‐Heppt and Dr. Hilton report no conflicts of interest." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Subjects were assigned to a treatment group based on a computer generated paper randomisation list at each centre" page 297 Comment: we considered this low risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Comment: we considered unclear risk of bias because the authors did not explain the methods used for maintain the allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "In preparation for injection, clinic staff member(s) with no other study responsibilities reconstituted vials containing,..The injectors, who were blinded to study treatment..The products were diluted to different concentrations so that they could be injected in equal volumes (0.5 mL) in order to maintain study blinding" page 297 Comment: we considered this low risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "In preparation for injection, clinic staff member(s) with no other study responsibilities reconstituted vials containing ,...The injectors, who were blinded to study treatment...The products were diluted to different concentrations so that they could be injected in equal volumes (0.5 mL) in order to maintain study blinding," page 297 Commment: we considered this low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Of the 224 subjects in the intent to treat analysis set, 16 subjects violated protocol criteria: 4 randomisation failures, 6 unauthorised concomitant therapy, 5 missing primary endpoint data or visit outside of allowable window on day 28, and 1 study site staff" page 299 Comment: we considered this low risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered this low risk of bias |
| Other bias | Unclear risk | Drs. Fulford‐Smith and Tan are employed by Allergan Quote: "Limitations+Possible limitations with this study include the following. The study incorporated a 1‐point change on the FWS as the primary endpoint, whereas placebo‐controlled trials have generally used ratings of none or mild on the FWS as the primary outcome measure" Comment: we considered this unclear risk of bias because two authors were Allergan employees |
Moers‐Carpi 2015.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised, double‐blind, placebo‐controlled, parallel‐design, Phase III study with 2 treatment cycles in crow's feet lines and/or glabellar lines Study date‐ no information Study setting‐ outpatients from 34 centres |
|
| Participants |
Randomised 917 participants, with mean age of 50 ±9.7 years in OnabotulinumtoxinA 44 U group; 49.6 ± 9.5 years in OnabotulinumtoxinA 24 U group; 49 ± 9.3 years in placebo group; 49.5±9.5 years total population. Gender: female = 87.5% in OnabotulinumtoxinA 44 U group; female = 89.2% in OnabotulinumtoxinA 24 U group; female = 86.9% in placebo group; female = 87.6% total population Inclusion criteria
Exclusion criteria
Severity of disease‐ Moderate wrinkles (investigator rating crow's feet lines at maximum smile) 35.7% in OnabotulinumtoxinA44 U group, 37.6% in OnabotulinumtoxinA 24 U group, 36.9% in placebo group, 36.8% total population Severe (investigator rating crow's feet lines at maximum smile) 64.3% in OnabotulinumtoxinA 44 U group, 62.4% in BontA 24 U group, 63.1% in placebo group, 63.2% total population Moderate participant assessment (maximum smile at baseline)) 36.1% in OnabotulinumtoxinA 44 U group, 36.9% in OnabotulinumtoxinA24 U group, 36.6% in placebo group, 36.5% total population. Severe (participant assessment (maximum smile at baseline)) 63.9% in OnabotulinumtoxinA 44 U, 63.1% in OnabotulinumtoxinA 24 U, 63.4% in placebo group, 63.5% total population Ethnicity ‐ 88.25% white in OnabotulinumtoxinA 44 U group; 88.2% white in OnabotulinumtoxinA24 U group; 86.6% white in placebo group; 87.7% white in total population |
|
| Interventions |
Duration of study‐ 28 weeks (7 months), two treatment cycles Intervention
Comparator
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | "M. Moers‐Carpi, J. Carruthers, S. Fagien, M. Lupo, H. Delmar, and D. Jones are consultants and/or investigators for Allergan, Inc. C. Somogyi, E. Lee, X. Lei, S. MacKinnon, and F. C. Beddingfield are employees of Allergan, Inc., and receive compensation in salary, as well as stock or stock options (or both), at the time the study was conducted. The remaining authors have indicated no significant interest with commercial supporters." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Eligible subjects were randomised 1:1:1 to 1 of 3 groups on Day 1" page 104 Comment: we considered this unclear risk of bias because the authors did not explain how they randomised the patients |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Eligible subjects were randomised 1:1:1 to 1 of 3 groups on Day 1" page 104 Comment: we considered this unclear risk of bias because the authors did not explain the methods used to maintain the allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "To maintain the blind, all medications were reconstituted and prepared by individuals who had no interactions with subjects." page 104 Comment: we considered this unclear risk of bias due to, for instance, the visual aspect of interventions were not described; and it is not possible to know if the participants were aware about the allocation. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "To maintain the blind, all medications were reconstituted and prepared by individuals who had no interactions with subjects." page 104 Comment: we considered this low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Of 684 subjects enrolled, 641 (93.7%) completed this study A total of 667 subjects (97.5%) received the third treatment. Most subjects who received a third dose (80.2%; 535/667) received their dose at Day 1 visit of Study 191622‐104. A total of 414 subjects (60.5%) received 2 treatments (treatment cycles 3 and 4): 149 onabotulinumtoxinA 24 U/24 U, 123 onabotulinumtoxinA 44 U/44 U, 69 placebo/ onabotulinumtoxinA 44 U, and 73 placebo/placebo. In this study, 253 subjects (37.0%) received only 1 treatment (treatment cycle 3): 74 onabotulinumtoxinA 24 U/24 U, 126 onabotulinumtoxinA 44 U/44 U, 31 placebo/onabotulinumtoxinA 44 U, and 22 placebo/placebo. Seventeen subjects failed to meet retreatment criteria after they received treatment 2 in Study 191622‐099 and therefore did not receive any treatment in this study: 4 onabotulinumtoxinA 24 U/ 24 U, 11 onabotulinumtoxinA 44 U/44 U, 1 placebo/ onabotulinumtoxinA 44 U, and 1 placebo/placebo" page 106 (figure 2) Comment: we considered this low risk of bias |
| Selective reporting (reporting bias) | High risk | Investigator‐Assessed Responder Rates on Crow's Feet Lines–Facial Wrinkle Scale, Subject's Global Assessment of Change in Crow's Feet Lines, Patient‐Reported Outcomes, no data shown Comment: we considered this high risk of bias We sent an e‐mail on 23 October 2015. No answer to date |
| Other bias | Unclear risk | C. Somogyi, E. Lee, X. Lei, S. MacKinnon, and F. C. Beddingfield are employees of Allergan, Inc Comment: we considered this unclear risk of bias because some authors were Allergan employees |
Monheit 2007.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised, double‐blind,dose‐ranging, placebo‐controlled, parallel design in glabellar lines Study date‐ no information Study setting‐ outpatients |
|
| Participants |
Randomised‐ 373 participants, with a mean age of 41.5 ± 9.7 years in AbobotulinumtoxinA 20 U group; 41.9 ±10.1 years in AbobotulinumtoxinA 50 U group; 42.1 ±10.3 years in AbobotulinumtoxinA 75 U group; 42.5 ± 9.9 years in placebo group; 42.0 ±10.0 total population. Gender: 79/91 (86.8%) female, 12/91 (13.2%) male in AbobotulinumtoxinA 20 U group; 72/93 (77.4%) female, 21/93 (22.6%) male in AbobotulinumtoxinA 50 U group; 78/95 (82.1%) female, 17/95 (17.9%) male in AbobotulinumtoxinA 75 U group; 84/94 (89.4%) female, 10/94(10.6%) male in placebo group; 313/373 (83.9%) female, 60/373 (16.1%) male in total population. Subgroup ≤ 50 years 77/91 (84.6%), > 50 years 14/91 (15.4%) AbobotulinumtoxinA 20 U group; ≤ 50 years 74/93 (79.6%), > 50 years 19/93 (20.4%) in AbobotulinumtoxinA 50 U group; ≤ 50 years 43/95 (77.9%), > 50 years 21/95 (22.1%) in AbobotulinumtoxinA 75 U group; placebo ≤ 50 years 73/94 (77.7%), > 50 years 21/94 (22.3%); ≤ 50 years 298/373 (80%), > 50 years 75/373 (20%) total population Inclusion criteria
Exclusion criteria‐ no information Severity of disease‐ investigator's assessment (glabellar lines at maximum frown), moderate 43/91 (47.3%) in AbobotulinumtoxinA20 U group, 35/93 (37.6%) in AbobotulinumtoxinA 50 U group, 48/95 (50.5%) in AbobotulinumtoxinA 75 U group, 42/94 (44.7%) in placebo group. Severe 48/91 (52.7%) in AbobotulinumtoxinA 2 Uu group, 58/93 (62.4%) in AbobotulinumtoxinA50 U group, 47/95 (49.5%) in AbobotulinumtoxinA 75 U group, 52/94 (55.3%) in placebo group. Investigators' assessment (glabellar lines at rest), none 5/91 (5.5%) in AbobotulinumtoxinA 20 U group, 4/93 (4.3%) in AbobotulinumtoxinA 50 U group, 5/95 (5.3%) in AbobotulinumtoxinA 75 U group, 3/94 (3.2%) in placebo group. Mild 35/91 (38.5%) in AbobotulinumtoxinA 20 U group, 42/93 (45.2%) in AbobotulinumtoxinA 50 U group, 40/95 (42.1%) in AbobotulinumtoxinA 75 U group, 42/94 (44.7%) in placebo group. Moderate 47/91 (51.6%) in AbobotulinumtoxinA 20 U group, 41/93 (44.1%) in AbobotulinumtoxinA 50u group, 46/95 (48.4%) in AbobotulinumtoxinA 75u group, 44/94 (46.8%) in placebo group. Severe 4/91 (4.4%) in AbobotulinumtoxinA 20u group, 6/93 (6.5%) in AbobotulinumtoxinA 50 U group, 4/95 (4.2%) in AbobotulinumtoxinA 75 U group, 5/94 (5.3%) in placebo group Ethnicity‐ 70/91 (76.9%) white, 21/91 (25.5%) non‐Caucasian in AbobotulinumtoxinA 20 U group; 64/93 (68.8%) Caucasian, 29/93 (31.2%) non‐Caucasian in AbobotulinumtoxinA 50 U group; 74/95 (77.9%) Caucasian, 21/95 (22.1%) non‐Caucasian in AbobotulinumtoxinA 75 U group; 70/94 (74.5%) Caucasian, 24/94 (25.5%) non‐Caucasian in placebo group; 278/373 (74.5%) Caucasian, 95/373 (25.5%) non‐Caucasian total population |
|
| Interventions |
Duration of study‐ 16 weeks Intervention
Comparator
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | "This study was a Phase II FDA investigation and was supported by funds from Ipsen Biopharm Limited and Inamed Corporation. Each of the authors was a paid investigator for the study by the sponsoring companies" | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "A Randomized, Double‐Blind" page S52 Comment: we considered this unclear risk of bias because the authors did not explain how they randomised the participants |
| Allocation concealment (selection bias) | Unclear risk | No information Comment: we considered this unclear risk of bias because the authors did not explain the methods used to maintain the allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Both participants and investigators were blinded to the treatment" page S52 Comment: we considered this low risk os bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Both participants and investigators were blinded to the treatment" page S52 Comment: we considered this low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "The demographic data relating to the ITT population are shown " page S55. "No deaths were reported and no adverse event led to withdrawal of a participant from the study." page S57 Comment: we considered this low risk of bias |
| Selective reporting (reporting bias) | Low risk | The authors showed participant assessment, but they did not specify if the outcome was at maximum frown or at rest Comment: we considered this low risk of bias We sent an e‐mail on November 1st, 2015. The author answered on 1st November 2015: "The assessment for primary response was at maximal contraction" |
| Other bias | Low risk | We considered this study at low risk of other bias |
Monheit 2019.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised (2:1), double‐blind,dose‐ranging, placebo‐controlled, parallel design in glabellar lines Study date‐ no information Study setting‐ outpatients, 5 centres (USA) |
|
| Participants |
Randomised‐ 300 participants, with a mean age of 44.7 (21‐71) years in AbobotulinumtoxinA group; 43.2 (24 to 66) years in placebo group. Gender‐ 28/200(14%) male and 172/200(86%) female in AbobotulinumtoxinA group, 12/100 (12%)male and 88/100(88%) male in placebo group. Inclusion criteria
Exclusion criteria
Ethnicity‐ Caucasian 149/200(75%), Native American 1/200(0.5%), Hispanic 37/200(19%), Asian 5/200(3%), 4/200(2%) other in Abobotulinumtoxin group.Caucasian 76/100(76%), Native American 0, Hispanic 18/100(18%), Asian 0, 1/100(1%) other in placebo group. |
|
| Interventions |
Duration of study‐ 20 weeks Intervention AbobotulinumtoxinA (50 U) (N = 200),,0.05mL/site, 5 points in procerus muscle corrugator muscle and orbicularis muscle, Comparator
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | Quote: "Medicis Pharmaceutical corp., funded the study and provided the study products. G.D. Monheit, R. Rand, C. Maas, and L. Baumann serve as consultants for Galderma and Ipsen. R. Down of Zenith Healthcare Communications Ltd., provided medical writing assistance, funded by Galderma. The authors have indicated no significant interest with commercial supporters." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote:"prospective, single‐dose, multicenter, randomized, parallel‐group,
placebo‐controlled, double‐blind "...page 61 Comment: we consider unclear risk of bias because the authors did not explain how they randomise the participants |
| Allocation concealment (selection bias) | Unclear risk | Quote:"prospective, single‐dose, multicenter, randomized, parallel‐group,
placebo‐controlled, double‐blind "...page 61 Comment: we consider unclear risk of bias because the authors did not explain how they randomise the participants |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote:"prospective, single‐dose, multicenter, randomized, parallel‐group,
placebo‐controlled, double‐blind "...page 61 Comment: we consider unclear risk of bias because the authors did not explain how they blind the participants |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote:"prospective, single‐dose, multicenter, randomized, parallel‐group,
placebo‐controlled, double‐blind "...page 61 Comment: we consider unclear risk of bias because the authors did not explain how they blind the participants |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote‐ the authors did not mention any drop out during the study Comment: we consiider a low risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered this low risk of bias |
| Other bias | Low risk | We considered this study at low risk of other bias |
NCT02450526.
| Study characteristics | ||
| Methods |
Study design‐ randomised, first phase double‐blind, second phase open‐label, multicentre, active andpPlacebo‐controlled study. Phase III Study date‐ start December 2009, end August 2010 Study setting |
|
| Participants | 520 participants, with a mean age raging from 18‐65 years. Gender: 86.8% (282/325) femal,13.2%, (9/325) male in Abobotulinumtoxin 50 U group, 86.4% (57/66) female, 13.6%, (9/66) male in Abobotulin placebo group, 87.9% (94/107) female, 12.1% (13/107) male in Onabotulinumtoxin 20u group, 86.4% (19/22) female, 13.6% (3/22) male in Onabotulintoxin ‐placebo group. Inclusion criteria
Exclusion criteria
Ethnicity:100% Asian (Chinese) Country: China |
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | Sponsor Ipsen Other study ID Y‐52‐52120‐158. We sent an email on April,28,2019.The Ipsen company answered:"These trials have not been published unfortunately." on June, 27, 2019. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: No information Comment: we considered it unclear risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Quote:No information Comment: we considered it unclear risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote:No information Comment: we considered it unclear risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote:No information Comment: we considered it unclear risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote:The authors described in the table reasons not complete the study Comment: we considered it low risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered this low risk of bias |
| Other bias | Unclear risk | This study was in clinical trial register site. We have contacted Ipsen pharmaceutical several times (last one January, 24, 2020) asking for publishing information. Answer‐ They did not have published it yet |
NCT02493946.
| Study characteristics | ||
| Methods | Randomised clinical trial, placebo‐controlled, double ‐blinded, 2 phases.Phase 1‐ Cycle 1 double‐blind. Phase 2‐ 2‐5 cycles. Start April 2015 Finish December 2016 Multicentre‐ Europe (France, UK Germany) |
|
| Participants |
Participants: 190 participants randomised to the 1st phase of the study. Participants age varied from 18‐65 years: 123/126 (97.6%) in HBTX‐A groups and 64/64 (100%) placebo group; > 65 years 3/126(2.42%) in HBTX‐A group, and 0 in placebo group. Gender: female 115/126 (913%)in HBTX‐A group and 56/64(90.6%) in placebo group. Ethnicity: Caucasian 125/126 (99.2%) in HBTX‐A group and 100% in placebo group. Black 1/126(0.8%) in HBTX‐A group and 0% in placebo group Inclusion Criteria
Exclusion Criteria
|
|
| Interventions |
Intervention Clostridium Botulinum Toxin Type A (BTX A HAC NG), total treatment volume 0.25 mL will be divided into 5 injections (0.05 mL per injections) injected in 5 pre‐defined sites across the glabellar region. A total of 50 U of BTX‐A‐HAC NG will be injected/ cycle. Comparator The total placebo volume 0.25 mL will be divided into 5 injections (0.05 mL per injections) injected in 5 pre‐defined sites across the glabellar region. Administered in Cycle 1 of the double‐blind phase only. |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | We used only phase 1 data | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: no information provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Subjects will first be enrolled to enter the double blind (DB) period Cycle 1" Comment: not clear who was blinded to treatment allocation. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "Subjects will first be enrolled to enter the double blind (DB) period Cycle 1" Comment: not clear who was blinded to treatment allocation. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: 8/126 and 5/64 did not complete the 1st phase of the study ‐ but no reasons given for withdrawal.. |
| Selective reporting (reporting bias) | Low risk | Comment: All prespecified outcomes were reported |
| Other bias | Unclear risk | Comment: limited information about the study provided on the clinical trial website. Therefore, unable to judge if any other biases were present. |
Nettar 2011.
| Study characteristics | ||
| Methods |
Study design‐ randomised, double‐blind, split‐face design in crow's feet lines Study date‐ start December 2009, end August 2010 Study setting‐ no information |
|
| Participants |
Randomised‐ 90 participants, with a mean age of 54.5 years (range 31‐78 years). Gender: no information Inclusion criteria
Exclusion criteria
Severity of disease‐ moderate to severe lateral orbital rhytids at maximal contraction Ethnicity‐ no information |
|
| Interventions |
Duration of study‐ 4 weeks Intervention
Comparator
Ratio‐ 1:3 (OnabotulinumtoxinA:AbobotulinumtoxinA) |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | "Dr Maas is a consultant and owns stock in both Medicis Aesthetics Inc (makers of abobotulinumtoxinA) and Allergan Inc (makers of onabotulinumtoxinA)." "Funding for this study was solicited from both Medicis Aesthetics Inc and Allergan Inc. Medicis Aesthetics Inc funded this study" |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Treatment sides of the face were randomised with computer‐ aided software" page 381 Comment: we considered this low risk of bias |
| Allocation concealment (selection bias) | Unclear risk | No information Comment: we considered this unclear risk of bias due to the authors did not explain the methods used to maintain the allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Preparation of product was performed by an unblinded registered nurse who was responsible for maintaining the blind" page 381 ..."identical volumes (0.2 mL) of each were drawn into tuberculin syringes to ensure the blindness" Comment: we considered this unclear risk of bias due to no information was provided about blinding of participants |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Preparation of product was performed by an unblinded registered nurse who was responsible for maintaining the blind"......"identical volumes (0.2 mL) of each were drawn into tuberculin syringes to ensure the blindness of the injector" page 381 Comment: we considered this low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No losses Comment: we considered this low risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered this low risk of bias |
| Other bias | High risk | Dr Maas was stockholder of Medicia Inc. Comment: we considered this a high risk of bias |
Ogilvie 2019.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised, double‐blind, placebo‐controlled, parallel ‐design,phase III in glabellar lines (Period 1) and an open‐label extension (period 2) Study date‐ Start October 2014. Finished April 2016 Study setting‐ outpatients from 16 centres (9‐USA, 5‐Canada, 2‐Europe) |
|
| Participants |
Randomised 391 participants. Age 44.5 ±11.2 Onabotulinum group and 42.4 ±10.6 placebo group. Gender 85.9% female in OnabotulinumtoxinA group and 86.1% in placebo group. Inclusion criteria
Exclusion criteria
Ethnicity: Caucasian 89.7% in OnabotulinumtoxinA group and 86.1% in placebo group. Asian 3.1 in OnabotulinumtoxinA group and 5% in placebo. Others 7.2% in OnabotulinumtoxinA group and 8.9% in placebo group. Severity of the disease:(maximum frown):Moderate 47.6% in OnabotulinumtoxinA group and 47.5% in Placebo group. Severe 52.4 % in OnabotulinumtoxinA group and 52.4% in Placebo group. |
|
| Interventions |
Duration:Period 1‐ 6 months. Period 2‐6 months Intervention OnabotulinumtoxinA 40 U (20 U in FHL and 20 U in GL) administered at 10 injection sites Comparator Placebo same volume in FHL and in GL) administered at 10 injection sites Ratio: 3:1 (OnabotulinumtoxinA: placebo) |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | This study was funded by Allergan plc, Dublin, Ireland. Editorial support for this article was provided by Peloton Advantage, Parsippany, New Jersey, and was funded by Allergan plc. P. Ogilvie has received research support or speaking/consultant fees from Allergan plc, Evolus, Inc., Galderma, Merz Aesthetics, and Revance. A.Z. Rivkin serves as a consultant and investigator for Allergan plc and Merz Aesthetics. S. Dayan has received research support or speaking/consultant fees from Allergan plc, Galderma, Merz Aesthetics, and Valeant. S.G. Yoelin serves as a consultant and investigator for Allergan plc. B.M. Weichman was employed by Peloton Advantage, which received funding from Allergan plc for medical editing and editorial support. J.K. Garcia is an employee of Allergan plc and owns stock/options in the company. The opinions expressed in this article are those of the authors. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote:"The randomization assignment was obtained from an interactive voice/web response system,
which was based on a randomization scheme prepared by Allergan Biostatistics"...Page3 Comment: we consider a low risk of bias |
| Allocation concealment (selection bias) | Low risk | Quote:"The randomization assignment was obtained from an interactive voice/web response system,
which was based on a randomization scheme prepared by Allergan Biostatistics"...Page3 Comment: we consider a low risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote:The study comprised a 6‐month, double‐blind,placebo‐controlled"...Page 3 Comment: we consider unclear bias because the authors did not mention how they blinded the participants and staff |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote:The study comprised a 6‐month, double‐blind,placebo‐controlled"...Page 3 Comment: we consider unclear bias because the authors did not mention how they blinded the participants and staff |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote:"early discontinuations were mostly attributable to personal reasons or being lost to follow‐up (Figure 2...Page4 Comment: we consider unclear risk of bias because the authors did not mention the reasons of the drop‐out |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comments: we consider low risk of bias |
| Other bias | Unclear risk | One of the authors was an employee of the sponsor |
Park 2014.
| Study characteristics | ||
| Methods |
Study design‐ randomised, double‐blind, split‐face design in crow's feet lines and masseter Study date‐ no information Study setting‐ no information |
|
| Participants |
Randomised 56 participants, with mean age of 43.4 years, range 23‐69 years. Gender: 94.6% female, 5.4% male Inclusion criteria
Exclusion criteria
Severity of disease‐ crow's feet lines, masseter Ethnicity‐ no information |
|
| Interventions |
Duration of study‐ 16 weeks Intervention
Comparator
Ratio‐ 1:1 (IncobotulinumtoxinA:OnabotulinumtoxinA) |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | Quote: "Authors do not have any kind of conflict of interest regarding this study" | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "a randomised,double‐blind" page 326 Comment: we considered this unclear risk of bias because the authors did not explain how they randomised the participants |
| Allocation concealment (selection bias) | Unclear risk | No information Comment: we considered this unclear risk of bias due to the authors did not explain the methods used to maintain the allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double‐blind, split‐face" page 326 Comment: we considered this unclear risk of bias because the authors did not explain how they blinded the participants |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double‐blind,split‐face" page 326 Comment: we considered this unclear risk of bias because the authors did not explain how they blinded the participants |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "in the periocular rhytides group 56 subjects completed the study" page 328 Comment: we considered this unclear risk of bias because the authors did not explain the reason of drop outs |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered this low risk of bias |
| Other bias | Low risk | We considered this study at low risk of other bias |
Patel 2004.
| Study characteristics | ||
| Methods |
Study design‐ randomised, double‐blind, parallel‐design in glabellar lines Study date‐ no information Study setting‐ no information |
|
| Participants |
Randomised‐ 65 participants. No age or gender information Inclusion criteria
Exclusion criteria
Severity of disease‐ moderate or severe vertical glabellar lines Ethnicity‐ no information |
|
| Interventions |
Duration of study‐ 12 weeks Intervention/Comparator
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | No information about conflict of interest | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "prospectively randomised to receive" page 442 Comment: we considered this unclear risk of bias because the authors did not explain how they randomised the participants |
| Allocation concealment (selection bias) | Unclear risk | No information Comment: we considered this unclear risk of bias due to the authors did not explain the methods used to maintain the allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Subsequently, these study participants were randomised and blinded to 1 of 3 treatment arms." page 442 Comment: we considered this unclear risk of bias because the authors did not explain how they blinded the participant |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Patients then returned for 3 follow‐up visits at 1 week, 1 month, and 3 months for an independent physician evaluation" page 443 Comment: we considered this low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No withdrawals Comment: we considered this low risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered this a low risk of bias |
| Other bias | Low risk | No other source of bias was found Comment: we considered this low risk of bias |
Rappl 2013.
| Study characteristics | ||
| Methods |
Study design‐ single‐centre, randomised, double‐blind, placebo‐controlled, parallel‐ design in glabellar lines Study date‐ start 2008 end 2011 Study setting‐ outpatient from one centre (Austria) |
|
| Participants |
Randomised‐ 180 participants, with mean age of 40.3± 6.8 years in IncobotulinumtoxinA group; 39.7 ± 6.1 years in OnabotulinumtoxinA group; 40.7± 6.5 years in AbobotulinumtoxinA group; 40.2 ± 6.4 years total population. Gender‐ 51/60 (85%) female, 9/60 (15%) male in IncobotulinumtoxinA group; 50/59 (85%) female, 9/59 (15%), male in OnabotulinumtoxinA group; 51/60 (85%) female, 9/60 (15%) male in AbobotulinumtoxinA group; 152/179 (85%) female, 27/179 (15%) male total population Inclusion criteria
Exclusion criteria
Severity of disease‐ Quote: "26 subjects had a score of 1 on the validated Merz 5‐point scale, 136 subjects had a score of 2, and 18 subjects had a score of 3" Ethnicity‐ 100% Caucasian |
|
| Interventions |
Duration of study‐ 24 weeks Intervention
Comparator‐ OnabotulinumtoxinA (21 U), 0.21 mL (N = 60)
Ratio‐ 1:1:3 (IncobotulinumtoxinA : OnabotulinumtoxinA: AbobotulinumtoxinA) |
|
| Outcomes |
Primary outcomes
Secondary outcome
|
|
| Notes | "Thomas Rappl has conducted speaker activities for Merz, Croma Pharma, MD‐Skin Solutions, Johnson & Johnson, and Smith & Nephew. The authors report no other conflicts of interest in this work." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Eligible subjects were assigned to groups of 60 and then randomised, using the randomizer program available at the Medical University of Graz (http:// www.randomizer.at)" page 213 Comment: we considered this low risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Eligible subjects were assigned to groups of 60 and then randomised, using the randomizer program available at the Medical University of Graz (http:// www.randomizer.at)" page 213 Comment: we considered this unclear risk of bias due to the authors did not reported methods for allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "In order for the injecting physician to remain blinded to the product used, an assistant prepared the products for injection using special syringes" page 213 Comment: we considered this low risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "In order for the injecting physician to remain blinded to the product used, an assistant prepared the products for injection using special syringes" page 213 Comment: we considered this low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "One female subject in the onabotulinumtoxinA group was excluded from the analysis due to failure to respond (in order to establish and compare the onset and duration of effect of the different products, it was essential that an effect was demonstrated" page 214 Comment: we considered this low risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered this low risk of bias |
| Other bias | Unclear risk | In figure 3 of the publication, the total dose of BontA1 and BontA2 was 23u but in the material and methods the authors described 21u Comment: we considered this unclear risk of bias, we sent an e‐mail to the authors to clarify this discrepancy on November 28 2015. The author answer that 21u was the correct dose |
Rivers 2015.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised, double‐blind, parallel‐design in glabellar lines and crow's feet lines Study date‐ start July 2013, end no information Study setting‐ outpatients from eight centres |
|
| Participants |
Randomised‐ 117 participants with mean age of 45.9 ± 9.66 years in onabotulinumtoxinA group, 47.1 ± 9.76 years in placebo group. Gender 48/60 (80%) females and 12/60 (20%) males in onabotulinumtoxinA group; 50/57 (87.7%) female and 7/57 (12.3%) male in placebo group Inclusion criteria
Exclusion criteria
Severity of disease‐ glabellar lines at maximum frown severe 24/60 (40%), moderate 36/60 (60%) in onabotulinumtocxinA; severe 30/57 (52.6%), moderate 27/57 (47.4%) in placebo group. Crow's feet lines at maximum frown severe 23/60 (38.3%), moderate 34/60 (56.7%), mild 3/60 (5%) in onabotulinumtocxinA; severe 18/57 (31.6%), moderate 38/57 (66.7%), mild 1/57 (1.8%) in placebo group Ethnicity‐ white 59/60 (98.3%) in onabotulinumtoxinA group; 56/57 (98.2%) white in placebo group |
|
| Interventions |
Duration of study‐ 16 weeks Intervention
Comparator Placebo 0.5 mL, 5 injections to the glabellar area (1 in the procerus muscle, 2 in each corrugator muscle) and 6 injections to the crow’s feet region (N = 57) |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes | Pharmaceutical support | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available to allow a judgment Comment: we considered this unclear risk of bias |
| Allocation concealment (selection bias) | Unclear risk | No information available to allow a judgment Comment: we considered this unclear risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information available to allow a judgment Comment: we considered this unclear risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information available to allow a judgment Comment: we considered this unclear risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "The study enrolled 125 subjects (Figure 2)." and there were no losses page 953 Comment: we consider this low risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we consider this ow risk of bias |
| Other bias | High risk | Imbalance in baseline glabellar lines (more severe lines in placebo group) Comment: we consider high risk of bias |
Rubin 2009.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised, double‐blind, placebo‐controlled, parallel‐design, phase III, two‐three cycles (Cycle A1, A2, B, C). Study date‐ start 20 June 2005, end 11 April 2007 Study setting‐ outpatients from six centres |
|
| Participants |
Randomised‐ 311 participants with a mean age of: In Cycle A1 (N = 311) mean 46.6 ± 9.72 years; Cycle A2 (N = 190) mean 46.4±9.51 years; Cycle B placebo (N = 84) 49.3 ± 9.8 years; Cycle B BontA (N = 171) 44.5 ± 8.92 years; Cycle C placebo (N = 71) mean 44.7 ± 8.73 years; Cycle C BontA (N = 71) BontA mean 44.7 ± 9.36 years. Gender Cycle A1 overall (N = 311) 269/311 (86%) female, 42/ 311 (14%) male; Cycle A2 (N = 190) 160/190 (84%) female, 30/190 (16%) male; Cycle B placebo (N = 84) 78/84 (93%) female, 6/84 (7%); Cycle B BontA (N = 171) 144/171 (84%) female, 27/171 (16%) male; Cycle C placebo (N = 71) 62/71 (87%) female, 9/71 (13%); Cycle C BontA (N = 71) BontA 60/71 (85%) female, 11/71 (15%) male Inclusion criteria
Exclusion criteria
Severity of disease‐ moderate to severe glabellar rhytids (GLSS scale), at maximum frown Ethnicity‐ Total population (N = 311) 249/311 (80%) Caucasian, 4/311 (1%) native American, 32/311 (10%) Hispanic, 9/311 (3%) African American, 13/ 311 (4%) Asian, 4/311 (1%) other |
|
| Interventions |
Duration of study‐ 52 weeks Intervention‐ AbobotulinumtoxinA (50 U), 0.05 mL, 4 points(10 U/point)
Comparator‐ placebo, 0.05 mL, 4 points
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | "Medicis Aesthetics Inc. (Scottsdale, AZ) provided Reloxin® and study funding to all of the authors" | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: "following one or two open‐label treatments and one randomised treatment" page440 "The protocol was modified due to a randomisation error in cycle B" page 439 Comment: we considered this unclear risk of bias because the authors did not explain if participants in cycle A1 and A2 were randomised |
| Allocation concealment (selection bias) | Unclear risk | Quote: "in Cycle C were randomly assigns to receive either Bont‐A (50) units or placebo" page 440 Comment: we considered this unclear risk of bias because the authors did not explain the methods for allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Patients were treated once during the double‐blind portion of the study" page 441 Comment: we considered this unclear risk of bias due to the authors did not describe methods for blinding the participants |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "Patients were treated once during the double‐blind portion of the study" page 441 Comment: we considered this unclear risk of bias because the authors did not explain how they blinded the outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information about losses Comment: we considered this unclear risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we consider this low risk of bias |
| Other bias | Low risk | We considered this study at low risk of other bias |
Rzany 2006.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised, double‐blind, placebo‐controlled, parallel‐design in forehead lines Study date‐ no information Study setting‐ outpatients from 23 centres |
|
| Participants |
Randomised‐ 221 participants, with mean age of 6.6 ± 9.2 years in arm 1; 46.4 ± 8.1 years in arm 2. Gender: 98/ 109(89.9%) female in arm 1, 100/111 (90.1%) female in arm 2 Inclusion criteria
Exclusion criteria‐ no information Severity of disease‐ moderate or severe vertical or diagonal glabellar wrinkles (scores of 2 or 3 on a standardised 4‐point clinical scale ranging from 0 [no wrinkles] to 3 [severe wrinkles]) at maximum frown; and had mild, moderate, or severe (scores of 1, 2, or 3) vertical or diagonal glabellar wrinkles at rest. Ethnicity‐ Quote:Quote: "Only 1 patient, in study arm 2, was not white" |
|
| Interventions |
Duration of study‐ 16 weeks Arm 1‐ 3 injections (N = 110) Intervention
Comparator
Arm 2‐ 5 injections (N = 111) Intervention
Comparator
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | "Financial Disclosure: Dr Rzany has received grants from Ipsen Pharma, Ettlingen, as well as from Pharma Allergan, Ettlingen, for other clinical trials not related to this study. Dr Rzany also has received honoraria from Ipsen Pharma for consulting and for conducting educational workshops. Funding/Support: This study was supported by a grant from Ipsen Pharma." |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "This was a double‐blind, placebo‐controlled, randomised" page 321 Comment: we considered this unclear risk of bias because the authors did not explain how they randomised the participants |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Within each centre, patients were randomised 2:1 to receive botulinum toxin A or placebo" page 321 Comment: we considered this unclear risk of bias because the authors did not explain the methods for allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "This was a double‐blind, placebo‐controlled,"page 321 Comment: we considered this unclear risk of bias because the authors did not explain how they blinded the participants (patients) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "This was a double‐blind, placebo‐controlled" page 321 Comment: we considered this unclear risk of bias because the authors did not explain how they blinded the participants (patients) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "All but 1 patient (in study arm 1) were included in the intention‐to‐treat analysis." page 322 Comment: we considered this unclear risk of bias, because the authors did not explain the reason of drop outs |
| Selective reporting (reporting bias) | High risk | Quote: "the scores at maximum frown and at rest (evaluated by the investigator) at weeks 0, 2, 4, 12, and 16 (data not shown); the subjective assessment of improvement since the first visit (evaluated by the patient) at weeks 2, 4, 12, and 16 (data not shown)" page 322 Comment: we considered a high risk of bias. We sent an e‐mail on 28 November 2015. He answer on 14 December 2015: "Concerning the data. This was an IPSEN initiated trial. All analysis was done through IPSEN. I would suggest that you contact IPSEN directly. I cced Dr. Caird in who was at that time responsible for the study." |
| Other bias | High risk | Pharmaceutical support: "Concerning the data. This was an IPSEN initiated trial. All analysis was done through IPSEN. I would suggest that you contact IPSEN directly. I cced Dr. Caird in who was at that time responsible for the study." Comment: we considered this high risk of bias |
Rzany 2019.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised, double‐blind, placebo‐controlled, parallel‐design, single dose for the treatment in glabellarlines Study date‐ started June 2015. Finished April 2016 Study setting‐ outpatients from 19 centres (7‐ USA, 5‐ France, 7‐ Germany, 2‐ Sweden, 1‐UK, 4‐Canada |
|
| Participants | Randomised 540 participants. Age: 48.8 ± 10.73 in PRA group; 49.7 ± 10.41 ONA group; 48.4 ± 10.84 in placebo group. Gender 89.8%females in PRA group; 92.3% in ONA group and 91.8% in placebo group. Inclusion criteria
Exclusion criteria
Ethnicity: Caucasian ‐ 67.3% PRA group, 74.4% ONA group, 73.5% placebo group. Afro‐America‐1.2% PRA group; 0.4% ONA group and 2.0% in placebo group. Asian‐ 2.4% PRA group, 2.0% ONA group, 2.0% placebo group. Multiple 0.4% PRA group, 0.4% ONA group, 0% placebo group. Other 3.3% PRA group, 0.8% ONA group, 2.0% placebo group. Missing 25.3% PRA group, 22.0% ONA group, 20.4% placebo group. Severity of disease: Investigator assessment maximum frown Moderate 25.3% PRA group, 28.5% ONA group, 26.5% placebo group. Severe‐ 74.7% PRA group, 71.5% ONA group, 73.5% placebo group |
|
| Interventions |
Duration: 5 months (150 days) Intervention
Comparators
Ratio: 5:5:1 (PRA: ONA: Placebo) |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | It is a non ‐inferiority trial between PrabotulinumtoxinA and OnabotulinumtoxinA Sponsor Evolus, Inc Dr Avelar is Chief Medical Officer and Head of Research and Development, Evolus, Inc., Newport Beach, CA. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote:"Random numbers were generated utilizing SAS PROC PLAN (SAS Institute, Inc., Cary NC); a block randomization scheme
with no stratification was employed where each block contained assignments for 5 prabotulinumtoxinA patients, 5 onabotulinumtoxinA patients, and 1 placebo patient".....Page3 Comment: we consider a low risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Quote:"Random numbers were generated utilizing SAS PROC PLAN (SAS Institute, Inc., Cary NC); a block randomization scheme
with no stratification was employed where each block contained assignments for 5 prabotulinumtoxinA patients, 5 onabotulinumtoxinA patients, and 1 placebo patient".....Page3 Comment: we consider a low risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote:"Trained individuals were responsible for reconstituting the vial with 2.5 mL of saline
and filling the injection syringe. The loaded syringe was then provided to the investigator while maintaining
appropriate spatial separation to ensure blinding" ...Page 3 Comment: we consider a low risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote:"Trained individuals were responsible for reconstituting the vial with 2.5 mL of saline
and filling the injection syringe. The loaded syringe was then provided to the investigator while maintaining
appropriate spatial separation to ensure blinding" ...Page 3 Comment: we consider a low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: Figure 1 ...Page 6 Comment: we consider a low risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we consider a low risk of bias |
| Other bias | Unclear risk | An author worked for sponsor |
Satler 2010.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised, non‐inferiority, active‐controlled, parallel‐design in glabellar lines Study date‐ no information Study setting‐ outpatients from 20 centres |
|
| Participants |
Randomised 381 participants, with a mean age of 41.7± 5.7 years, median 42 years, (range 22 to 50) in IncobotulinumtoxinA group; 42 ± 6.0 years, median 42 years, (range 24 to 51) in OnabotulinumtoxinAgroup. Gender‐ 100% female Inclusion criteria
Exclusion criteria
Severity of disease‐ Facial wrinkle score at rest, none 11/277 (4%) in IncobotulinumtoxinA group; 5/93 (5.4%) in OnabotulinumtoxinAgroup. Mild 92/277 (33.2%) in IncobotulinumtoxinA group, 32/93 (34.4%) in OnabotulinumtoxinA group. Moderate 174/277 (62.8%) in IncobotulinumtoxinA group; 56/93 (60.2%) in OnabotulinumtoxinA group. Severe 0/277 (0%) in IncobotulinumtoxinA group; 0/93 (0%) in OnabotulinumtoxinA group. Facial wrinkle scale at maximum contraction, none 0/277 (0%) in IncobotulinumtoxinA group; 0/93 (0%) in OnabotulinumtoxinA group. Mild 0/277 (0%) in IncobotulinumtoxinA group; 0/93 (0%) in OnabotulinumtoxinA group. Moderate 90/277 (32.5%) in IncobotulinumtoxinA group; 27/93 (29%) in OnabotulinumtoxinA group. Severe 187/277 (67.5%) in BIncobotulinumtoxinA group; 66/93 (71%) in OnabotulinumtoxinA group. Ethnicity‐ BontA1275/277 (99.3%) Caucasian, other 2/277 (0.7%); BontA292/93 (98.9%) Caucasian, 1/93 (1.1%) other Received at least one previous BontA treatment IncobotulinumtoxinA 84/277 (30.3%), OnabotulinumtoxinA 28/93 (30.1%) |
|
| Interventions |
Duration of study‐ 12 weeks Intervention
Comparator
Ratio‐ 1:1 (IncobotulinumtoxinA: OnabotulinumtoxinA) |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | "This study was funded by Merz Pharmaceuticals GmbH. Editorial assistance was provided by Ogilvy 4D, Oxford, UK." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomised into two groups in a 3:1" page 2147 Comment: we considered this unclear risk of bias because the authors did not explain how they randomised the participants |
| Allocation concealment (selection bias) | Unclear risk | no information Comment: we considered this unclear risk of bias because the authors did not explain the methods for allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "The blinding referred to the patients and the independent rates" page 2147 Comment: we considered this unclear risk of bias due to the authors did not report the methods used for blinding the participants, including the visual aspect of interventions |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "Three independent raters individually performed the assessment of the photographs" page 2147 Comment: we considered this unclear risk of bias due to the authors did not report the methods used for blinding the outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "These 381 patients had at least an observed baseline value of the primary efficacy variable and were therefore included in the FAS. Eleven patients showed major deviations from the study protocol, so the PPS comprised 370 patients (n = 277, incobotulinumtoxinA; n= 93, on‐ abotulinumtoxinA). Major deviations were missing efficacy measurements, time schedule deviations such as premature study termination and visits not done or done outside the visit window, taking medi‐ cation excluded from the study, and deviation of exclusion criteria" page 2148‐9 Comment: we considered this low risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered this low risk of bias |
| Other bias | Low risk | We considered this study at low risk of other bias |
Solish 2016.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised, dose‐ranging placebo‐controlled, parallel‐design in glabellar lines and in forehead lines Study date‐ no information Study setting‐ outpatients from seven centres |
|
| Participants |
Randomised‐ 175 participants, with mean age of 45.5 ± 9.8 years in OnabotulinumtoxinA 40u group; 47.6 ± 11 years in OnabotulinumtoxinA 30u group; 47.2 ± 8.6 years in placebo group; total population 46.8 ± 9.8 years. Gender 50/57 (87.7%) female, 7/57 (12.3%) male in OnabotulinumtoxinA 40u group; 49/59 (83.3%) female, 10/59 (16.7%) male in OnabotulinumtoxinA 30u group; 53/59 (89.7%) female, 6/57 (10.3%) male in placebo group; total population 152/175 (86.9%) female, 23/175 (13.2%)male Inclusion criteria
Exclusion criteria
Severity of disease‐ glabellar line severity (maximum frown): 25/57 (43.9%) moderate, 32/57 (56.1%) severe in OnabotulinumtoxinA 40 U group; 30/59 (50.8%) moderate, 27/59 (45.8%) severe in OnabotulinumtoxinA 30 U group; 34/59 (57.6%) moderate, 24/59 (40.7%) severe in placebo group; total population 89/175 (50.9%) moderate and 83/175 (47.4%) severe Ethnicity‐ white 55/57 (96.5%) in OnabotulinumtoxinA 40 U group; white 53/59 (89.8%) in OnabotulinumtoxinA 40 U group;52/59 (88.1%) in placebo group; total population 160/175 (91.4%) |
|
| Interventions |
Duration of study‐ 24 weeks Intervention
Comparator
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | Quote: "Dr. X. Lei, Ms. M. Bhogal, and Dr. C. Caulkins are employees of Allergan plc and receive compensation in salary, as well as stock and/or stock options. Ms. C. Somogyi, currently an employee of Kythera Pharmaceuticals, was an employee of Allergan plc at the time of this study and during manuscript preparation. The authors received research grant support from Allergan plc, Dublin, Ireland, for this study and for manuscript preparation. Funding for editorial support was provided by Allergan plc. Writing and editorial assistance was provided by SCI Scientific Communications and Information (SCI), Parsippany, NJ. Editorial assistance was provided by Beta Bowen of Allergan plc" FLO‐116 and SPA5 are validated instruments developed in accordance with the US Food and Drug Administration Guidance for Industry– Patient‐Reported Outcome Measures: Use in Medical Product Development to Support Labeling Claims" |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Subjects were randomised in a 1:1:1 ratio" page 411 Comment: we considered this unclear risk of bias because the authors did not explain how they randomised the participants |
| Allocation concealment (selection bias) | Unclear risk | no information Comment: we considered this unclear risk of bias because the authors did not explain the methods for allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "An independent drug reconstitutor/injector was used in this study to ensure maintenance of blinding of study drug and placebo" Page 411 Comment: we considered this unclear risk of bias due to the authors did not report the methods used for blinding the participants, including the visual aspect of interventions |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "An independent drug reconstitutor/injector was used in this study to ensure maintenance of blinding of study drug and placebo. A separate blinded evaluator conducted all efficacy and safety assessments." page 411 Comment: we considered this unclear risk of bias due to the authors did not report the methods used for blinding the outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | no information Comment: we consider unclear risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered a low risk of bias |
| Other bias | Unclear risk | Quote: "However, comparison between the 30 U and placebo groups suggested a possible difference between the 2 groups in subject assessment of GL appearance at baseline" page 414 Quote: "Dr. X. Lei, Ms. M. Bhogal, and Dr. C. Caulkins are employees of Allergan plc and receive compensation in salary, as well as stock and/or stock options. Ms. C. Somogyi, currently an employee of Kythera Pharmaceuticals, was an employee of Allergan plc at the time of this study and during manuscript preparation. The authors received research grant support from Allergan plc, Dublin, Ireland, for this study and for manuscript preparation. Funding for editorial support was provided by Allergan plc. Writing and editorial assistance was provided by SCI Scientific Communications and Information (SCI), Parsippany, NJ. Editorial assistance was provided by Beta Bowen of Allergan plc" Comment: we consider unclear risk of bias because wrinkles baseline severity imbalance, and some authors were employees and stockholders of Allergan. |
Solish 2018.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised, dose‐ranging placebo‐controlled, parallel‐design in glabellar lines (BELMONT STUDY). Phase II Study date‐ no information Study setting‐ outpatients |
|
| Participants |
Randomised 268 participants aged 30‐65 years of age. Gender: 80% of participants were female Incluion criteria: no information Exclusion criteria : no information Ethinicity‐ 85% were "white" Severity of the disease: moderate‐to‐severe glabellar lines |
|
| Interventions |
Duration: from 24 weeks to 36 weeks Intervention DaxibotulinumtoxinA 20U, 40 U, 60 U in the glabellar lines Comparator OnabotulinumtoxinA 20 U in the glabellar lines Placebo in the glabellar lines Ratio: (1:1:1) |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | Poster | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "The BELMONT study was a randomized, "....page S106 Comment: we considered this unclear risk of bias because the authors did not explain how they randomised the participants |
| Allocation concealment (selection bias) | Unclear risk | Quote: "The BELMONT study was a randomized, "....page S106 Comment: we considered this unclear risk of bias because the authors did not explain how they randomised the participants |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "The BELMONT study was a randomized, "....page S10 Comment: we considered this unclear risk of bias because the authors did not explain how they blinded the participants |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "The BELMONT study was a randomized, "....page S10 we considered this unclear risk of bias because the authors did not explain how theey keep the participants blinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: no information Comment: we consider an unclear risk of bias |
| Selective reporting (reporting bias) | Unclear risk | The authors did not show all the endpoints Comment: we consider an unclear risk of bias |
| Other bias | Unclear risk | This study is a poster |
Won 2013.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised, double‐blind, parallel‐design, active‐controlled, phase III clinical trial Study date‐ no information Study setting‐ outpatient from six centres |
|
| Participants |
Randomised‐ 314 participants, with mean age of 48 ± 8.8 years in NewBontA group, median 49 years (range 25‐64); 47 ± 8.8 years, median 82 years, (range 27 to 64) in OnabotulinumtoxinA group. Younger than 50 years: 79/157 (50.3%) in New BontA group; 85/157 (54.1%) OnabotulinumtoxinA. Equal or older than 50 years: 78/157 (49.7%) in New BontA group; 72/157 (45.9%) in OnabotulinumtoxinA group. Gender: 135/157 (86%) female, 22/157 (14%) male in New BontA group; 124/157 (79%) female, 33/157 (21%) male in OnabotulinumtoxinAgroup. Botulinum treatment naive: New BontA 93%, OnabotulinumtoxinA 90.4% Inclusion criteria
Exclusion criteria
Severity of disease‐ moderate to severe glabellar frown lines at maximum frown (severity score of 2 or 3 on the Facial Wrinkle Scale (FWS) (FWS at rest) 8/142 (5.5%) none, 58/142 (40.3%) mild,38/142 (26.4%) moderate, 40/142 (27.8%) severe in New BontA group; (FWS at rest) 16/146 (10.9%) none, 48/146 (32.6%) mild, 46/146 (31.3%), 37/146 (25.2%) severe in OnabotulinumtoxinA group. FWS at maximum contraction 0 none,0 mild, 66/142 (47.5%) moderate, 76/142 (53.5%) severe in New BontA group; OnabotulinumtoxinA (FWS at maximum contraction) 0 none, 0 mild, 66/146 (47.5%) moderate, 80/146 (53.5%) severe in OnabotulinumtoxinA group. Ethnicity‐ no information |
|
| Interventions |
Duration of study‐ 16 weeks Intervention
Comparator
Ratio‐ 1:1 (New BontA [Neurono®]:OnabotulinumtoxinA) |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | "This study was sponsored by Medytox Inc. Dr. Woo Shun Lee was an employee of Medytox Inc., Korea." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "After confirmation of eligibility, patients were randomised into two groups at a 1:1 ratio" page 173 Comment: we considered this unclear risk of bias because the authors did not explain how they randomised the participants |
| Allocation concealment (selection bias) | Unclear risk | Quote: "After confirmation of eligibility, patients were randomised into two groups at a 1:1 ratio" page 173 Comment: we considered this unclear risk of bias because the authors did not explain the methods for allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Each patient received a total dose of 20 U (4 U/0.1 mL) of NBoNT or OBoNT in a double‐blind manner." page 173 Comment: we considered this unclear risk of bias because the authors did not explain how they blinded the participants |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Three blinded raters assessed the photographs according to the FWS." page 173 Comment: we considered this a low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "Two hundred ninety‐one of 314 patients enrolled completed the study without major deviation and therefore constituted the PP set: 142 in the NBoNT group and 146 in the OBoNT group" page 174 Comment: we considered unclear risk because the authors did not explain the drop‐out reason |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered a low risk of bias |
| Other bias | Unclear risk | Dr. Woo Shun Lee was an employee of Medytox Inc., Korea." Comment: we considered this a unclearrisk of bias |
Won 2015.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, double‐blinded, randomised, active‐controlled parallel design Study date‐ no information Study setting‐ outpatients from three centres |
|
| Participants |
Randomised‐ 268 participants, with mean age of 47.82 ± 9.15 years in DWP450 group; 47.31 ± 8.57 years in OnabotulinumtoxinA group; 47.57 ± 8.85 years total population. Gender 106/133 (79.70%) female, 27/133(20.30%) male in DWP450 group; 111/132 (84.09%) female, 21/132 (15.91%) male inOnabotulinumtoxinA group; 217/265 (81.89%) female, 48/265 (18.11%) male total population Inclusion criteria
Exclusion criteria
Severity of disease‐ baseline severity glabellar lines at maximum frown moderate 51/133 (38.35%) in BontA1 group, 52/132 (39.9%) in BontA2 group, 103/265 (38.87%) total population. Severe 82/133 (61.65%) in BontA1 group, 80/132 (60.61%%) in BontA2 group, 162/265 (61.13%) total population. Severity of glabellar lines at rest‐ mild 51/133 (38.35%) in BontA1 group, 47/132 (35.61%) in BontA2 group, 98/265 (36.98%) total population. Moderate‐ 26/133 (19.55%) in BontA1 group, 28/132 (21.21%) in BontA2 group, 54/265 (20.38%) total population. Severe ‐56/133 (42.11%) in BontA1 group, 57/132 (43.18%) in BontA2 group, 113/265 (42.64%) total population Ethnicity‐ no information |
|
| Interventions |
Duration of study‐ 16 weeks Intervention
Comparator
Ratio‐ 1:1 (DWP450: OnabotulinumtoxinA) |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | "Su‐Young Lee and Chung‐Sei Kim are employees of Daewoong Pharmaceutical. This study was sponsored by Daewoong Pharmaceutical" | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "At each centre, eligible patients were randomly assigned to either the DWP450 or OBoNT group in a 1:1 ratio using a computer‐generated randomisation schedule" page 228 Comment: we considered this low risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Quote: "At each centre, eligible patients were randomly assigned to either the DWP450 or OBoNT group in a 1:1 ratio using a computer‐generated randomisation schedule" page 228 Comment: we considered this unclear risk of bias |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Investigators were blinded to medication type throughout the study." page228 Comment: we considered this unclear because the authors did not explain how they blinded the participants (patients) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "This prospective, double‐blinded, randomised, active‐controlled"...page 228"Three blinded independent investigators conducted photographic assessment, and all the blinded raters additionally received" page 229 Comment: we considered this low risk of bias |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Of the 281 subjects screened, 268 were randomised, so that 135 were assigned to the DWP450 group and 133 to the OBoNT group. Of these, 263 completed the study, so that 265 of 268 randomised subjects composed the FAS population. The PPS1 population consisted of 263 subjects, excluding two patients who violated the visit window period and had committed concomitant medication violations. PPS2 population consisted of 245 patients, 18 of whom were excluded for the following reasons: eight for visit window violations, six for concomitant medication violations, and four for omissions of secondary efficacy endpoint assessment." page 229‐30 Comment: we considered this low risk of bias |
| Selective reporting (reporting bias) | High risk | Patient satisfaction rate data not shown Comment: we considered this high risk of bias, we sent an e‐mail on 22 November 2015. No answer |
| Other bias | Unclear risk | Su‐Young Lee and Chung‐Sei Kim are employees of Daewoong Pharmaceutical Comment: we considered this unclear risk of bias |
Wu 2010.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised double‐blind, placebo‐controlled, parallel‐design in glabellar lines Study date‐ no information Study setting‐ outpatient from four centres |
|
| Participants |
Randomised 227 participants, with median age of 41.7 years in BontA group, and 44.1years in placebo group. Gender: 142/170 (83.5%) female, 28/170 (16.5%) male in BontA group; 53/57 (93%) female, 4/57 (7%) male in placebo group Inclusion criteria
Exclusion criteria
Severity of disease‐ moderate severity at maximum frown (graded on a 4‐point facial wrinkle scale: 0 = none, 1 = mild, 2 = moderate to 3 = severe) BontA (FWS at maximum frown) 86/170 (50.6%) moderate, 84/170 (49.4%) severe Placebo (FWS at maximum frown) 27/57 (47.4%) moderate, 30/57 (52.6%) severe Ethnicity‐ 100% Chinese |
|
| Interventions |
Duration‐ 16 weeks Intervention
Comparator
|
|
| Outcomes |
Primary outcome
Secondary outcome
|
|
| Notes | "GlaxoSmithKline provided funding and study material." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "A total of 227 patients were randomised to receive a single treatment of 20 U of BontA or identical placebo in a ratio of 3:1" page 102‐3 Comment: we considered unclear risk of bias because the authors did not explain how they allocated the participants |
| Allocation concealment (selection bias) | Unclear risk | Quote: "A total of 227 patients were randomised to receive a single treatment of 20U of BontA or identical placebo in a ratio of 3:1" page 102‐3 Comment: we considered unclear risk of bias because the authors did not explain the methods used for allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Vials of BoNTA and placebo had identical investigational labels, which prevented identification of the contents." page 103 Comment: we considered low risk of bias |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | no information Comment: we considered unclear risk of bias because the authors did not explain how they maintained the blindness of the assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Of 258 subjects screened, 227 were randomised, with 170 in the BoNTA group and 57 in the placebo group; 222 completed the study. Reasons for discontinuation included SAEs (one breast cancer and one gastric cancer) that the investigator considered to be unrelated to the treatment, withdrawal from the study (N = 2), and loss to follow‐up (n = 1)" page 104 Comment: we considered this low risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered this low risk of bias |
| Other bias | Low risk | We considered this study at low risk of other bias |
Wu 2019.
| Study characteristics | ||
| Methods |
Study design‐ multicentre, randomised, double‐blind, placebo‐controlled, parallel design in crow's feet lines, phase III Study date‐ Started ‐September 2014.Finished ‐June 2015 Study setting‐ outpatients (nine centers in China) |
|
| Participants |
Randomised‐ 417 participants were randomised in a 3:1 ratio to receive a single treatment of OnabotulinumtoxinA 24 U or placebo, with a mean age of 46.3 ± 9.64 years in OnabotulinumtoxinA group, 46.6 ± 9.39 years in placebo group. Gender, female 273/316 (86.4%) in OnabotulinumtoxinA group, 87/101 (86.1%) in placebo group. Inclusion criteria
Exclusion criteria
Severity of the disease FWS‐at maximum smile(investigator rated), moderate 153/319 (48.4%) in OnabotulinumtoxinA group, 49/101 (48.5%) in placebo group. Severe 163/319 (51.6%) in OnabotulinumtoxinA group, 52/101 (51.5%) in placebo group. FWS‐at maximum smile(subject rated), moderate 154/319 (48.7%) in OnabotulinumtoxinA group, 49/101 (48.5%) in placebo group. Severe 162/319 (51.3%) in OnabotulinumtoxinA group, 52/101 (51.5%) in placebo group. Ethnicity 100% Asian Country China |
|
| Interventions |
Duration of study‐ 20 weeks Intervention
Comparator‐
|
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes | Sponsor Allergan. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "At study entry, subjects were assigned a randomization number, and an interactive voice response system was used to manage the randomization and treatment assignment based on a randomization scheme prepared by Allergan
Biostatistics"...page 2 Comments: we consider low risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Quote:"This 5‐month, multicenter, double‐blind, randomized, parallel‐group,"..page 2 Coment" we consider a unclear risk of bias, because the authors did not explain how they do concealed allocation |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Investigators and subjects were blinded to the treatment administered...page 2 Comment: we consider unclear risk of bias because the authors did not explain how they blinded the participants |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "Investigators and subjects were blinded to the treatment administered...page 2 Comment: we consider unclear risk of bias because the authors did not explain how they blinded the participants |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote:"Reasons for discontinuation in both groups were limited to lost to follow‐up (n = 4, onabotulinumtoxinA; n = 1, placebo) and personal reasons (n = 1, onabotulinumtoxinA; n = 1, placebo)."...page 3 Commenst: we consider a low risk of bias |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported Comment: we considered this low risk of bi |
| Other bias | Low risk | We considered this study at low risk of other bias |
ABO: AbobotulinumtoxinA;BTX: Botulinum toxin; CETS: composite endpoint treatment success; CFL: crow's feet lines ; FWS: facial wrinkle scale;GL: : glabellar lines; GFL: glabellar frown lines;HADS: Hospital Anxiety and Depression Scale; (HFL: horizontal forehead lines;ILA: Investigator's Live Assessment; ITT: intention‐to‐treat; LPL: lateral periorbital lines;MAS: Merz Aesthetics Scales; ONA: nabotulinumtoxinA; SGA: Subject's Global Assessment; SPA: Self‐Perception of Age; U: unit
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| 2014‐003770‐16 | Not randomised clinical trial |
| Cartier 2020 | Other intervention |
| Hexsel 2018 | Non randomised clinical trial |
| Mahmoud 2016 | Non randomised clinical trial |
| NCT00752050 | J&J ended plans to produce BontA |
| NCT00752297 | J&J ended plans to produce BontA |
| NCT02297516 | other intervention |
| Punga 2016 | Other intervention (same units in different dilution) |
| Rzany 2013 | Non randomised clinical trial |
| Wilson 2016 | Other intervention‐ the authors assessed the BontA treatment by speckle tracking with digital image correlation. This measurement was different from all the available tools. |
| Zhang 2018 | Other intervention |
Characteristics of studies awaiting classification [ordered by study ID]
NCT01180348.
| Methods | Randomised, non‐inferiority trial, split‐face,active‐controlled, facial wrinkles, phase III trial |
| Participants | 192 randomised female participants, age ranging from 18 years old to 65 years old Inclusion Criteria
Exclusion Criteria
Country: Brazil |
| Interventions |
Intervention:3 applications on each side of the face in 15 predetermined sites in three regions of the face (front, glabellar, periocular). Botulift® 90 U Comparator: 3 applications on each side of the face in 15 predetermined sites in three regions of the face (front, glabellar, periocular).OnabotulinumtoxinA |
| Outcomes |
Primary Outcomes
Secondary Outcomes
|
| Notes | Sponsor Azidus Brasil We sent an email to this company (http://laboratoriobergamo.com.br/contato/) on June, 23rd, 2018. No answer. |
NCT01358695.
| Methods | Randomised, multicentre, factorial design, placebo‐controlled, masking quadruple (participant, care provider, investigator, outcomes assessor). phase II trial |
| Participants | 111 participants, age ranging from 30 to 70 years old, both gender Inclusion criteria
Exclusion Criteria
Country: USA |
| Interventions |
Intervention ANT‐1207 five different doses (no information about the units) Comparator Placebo (no information about volume) |
| Outcomes |
Primary outcome
Secondary outcomes
|
| Notes | Supported by Anterios InC. Allergan bought Anteris in 2014 Other study ID: ANT‐1207‐201‐LCL |
NCT01485601.
| Methods | Randomised, double‐blind, multi‐centre, Active‐control, optimal dose‐finding of MT10109, phase II trial |
| Participants | 121 participants, age raging from 18 to 75 years old, both gender Inclusion criteria
Exclusion Criteria
Country: Australia |
| Interventions |
Intervention
Comparator
|
| Outcomes |
Primary outcome
No secondary outcomes. |
| Notes | other register MT‐GPRT‐GL01 support Medy‐tox® The title was double‐blind study, but in the study descriptions it was a Quadruple (Participant, Care Provider, Investigator, Outcomes Assessor) |
NCT01583478.
| Methods | Open‐label, dose ranging, active‐controlled trial |
| Participants | 60 participants, older than 18 years old, both gender Inclusion criteria
Exclusion criteria
Country: USA |
| Interventions | Treatment/comparator: IncobotulinumtoxinA doses of 20‐, 40‐, 60‐, 80‐ or 100‐units divided among 5 injection points (0.25 mL total) in the glabellar region |
| Outcomes |
Primary Outcome
Secondary outcomes
|
| Notes | Other ID study number: ITGR‐2012.This trials ended on September 30, 2015. We sent an email to Merz |
NCT01791920.
| Methods | Double‐blinded, randomised, active control comparative, multicentre‐designed, phase III trial |
| Participants | 262 participants Inclusion Criteria
Exclusion Criteria
Country: Republic of Korea |
| Interventions |
Intervention
Single administration, Day 0, 20 units Comparator
Single administration, Day 0, 20 units |
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Notes | Sponsor Hugel We sent an email in April 2019 in the Hugel site. We did not receive any answer. |
NCT01951742.
| Methods | Randomised, parallel‐design, double‐blind, dose‐ranging, phase II |
| Participants | 145 participants, age ranging from 30 to 60 years old, both gender Inclusion Criteria
Exclusion Criteria
Country : USA |
| Interventions |
Intervention: five different dose of ANT‐1401. The units were not specified in crow's feet lines Comparator: placebo in crow's feet lines |
| Outcomes |
Primary Outcome
Secondary Outcomes
|
| Notes | ANT‐1401 is a BontA From Anterios Inc. I sent an email for this company on June 23, 2018 |
NCT02236312.
| Methods | Randomised, parallel‐design, double‐blind, phase II trial |
| Participants | 350 participants, older than 18 years old, both gender. Inclusion criteria
Exclusion Criteria
Country: USA |
| Interventions |
Intervention: AbobotulinumtoxinA 30 U, 45 U, and 60 U in glabellar region Comparator: placebo in glabellar region |
| Outcomes |
Primary outcome
Secondary outcome
|
| Notes | Other study ID number: 43QM1313 Q‐med. This trial finished on June 2, 2016. We resent an email on July 25, 2019 |
NCT02677298.
| Methods | Randomised, double‐blind trial to assess the efficacy and safety of BoNT/A‐DP in the treatment of glabellar lines in comparison with placebo, followed by an open‐label extension study, phase III |
| Participants | 700 participants, male and female. Inclusion Criteria
Exclusion criteria
|
| Interventions |
Intervention BoNT/A‐DP‐ Intramuscular injection, 20 Units divided in five 0.1 mL i.m. injections into the glabellar area Comparator
|
| Outcomes |
Primary outcome
Secondary outcomes
|
| Notes | Sponsor Croma‐Pharma GmbH |
NCT02677805.
| Methods | Randomised, double‐blind, parallel design, placebo‐controlled, phase 3 trial |
| Participants | 200 participants, both genders Inclusion criteria
Exclusion criteria
|
| Interventions |
Intervention Botulinum toxin A will be administered in double‐blind fashion in cycle 1. 20 units will be administered (divided in five 0.1 mL i.m. injections) into glabellar area Comparator Placebo will be administered in double‐blind fashion in cycle 1 divided in five 0.1 mL injections into the glabellar area |
| Outcomes |
Primary outcome
Secondary outcomes
Country: no information |
| Notes | Sponsor Croma‐Pharma GmbH Other study id CPH‐302‐201030 |
NCT02882893.
| Methods | Randomised, active controlled, double‐blind, multi‐centre, phase II/III trial |
| Participants | 238 participants Inclusion criteria
Exclusion criteria: no criteria Country: Republic of Korea |
| Interventions |
Intervention
Comparator
|
| Outcomes |
Primary outcome
Secondary outcome: no secondary outcome |
| Notes | Sponsor Daewoong |
NCT02961673.
| Methods | Randomised, double‐blind (participants and investigator), active‐control, phase I/II trial A phase I/Ⅱ clinical trial to compare the safety and efficacy of HU‐014 versus Botox® in subject with moderate to severe glabellar lines |
| Participants | 57 participants, older than 19 years old, both gender Inclusion Criteria
Exclusion Criteria
|
| Interventions |
Intervention
Comparator:
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Notes | Sponsor Huons Co., Ltd. This trial finished on April 2018 |
NCT03317574.
| Methods | Randomised, double‐blind, active drug‐controlled, multicentre phase Ⅰ/Ⅲ trial |
| Participants | 250 participants Inclusion criteria
Exclusion criteria
Country: Republic of Korea |
| Interventions |
Intervention
Comparator
|
| Outcomes |
Primary outcome
Secondary outcomes: none. |
| Notes | Sponsor Medy‐tox Other study id MT01‐KR17CFL903 This trial finished on March 27, 2019 |
NCT03440671.
| Methods | Randomised, multicentre, active control, double‐blind. phase III trial |
| Participants | 260 participant, older than 18 years old, both gender Inclusion Criteria
Exclusion Criteria
Country: Republic of Korea |
| Interventions |
Intervention
Comparator
|
| Outcomes |
Primary outcome
Change from baseline of glabellar lines improvement rate(frown) |
| Notes | Sponsor Huons Co., Ltd. |
NCT03721016.
| Methods | Randomised, multicentre, double‐blind, placebo‐controlled, parallel‐group study to evaluate thesSafety and efficacy of MT10109L (NivobotulinumtoxinA) for the treatment of glabellar lines With or without concurrent treatment of lateral Canthal lines, phase III trial |
| Participants | 375participants, older than 18 years old, both genders Inclusion Criteria
Exclusion Criteria
Countries: USA and Canada |
| Interventions |
Intervention
Comparator
|
| Outcomes |
Primary outcome
Secondary outcomes
|
| Notes | Sponsor Allergan |
NCT03736928.
| Methods | Randomised, dose‐ranging, double‐blind, placebo‐controlled, multicentre Study, phase II trial |
| Participants | 401 participants, age ranging from 18 to 65 years old, both genders. Inclusion criteria
Exclusion Criteria
Country : USA |
| Interventions |
Intervention
Comparator
|
| Outcomes |
Primary outcome
|
| Notes | Sponsor Q‐Med AB Other study id 43USD1801 |
NCT03806933.
| Methods | Randomised, double‐blind, multicentre study to investigate the safety and duration of effect of different NT 201 Dose groups following the treatment of glabellar frown lines, phase II trial |
| Participants | 240 participants, older than 18 years old, both genders. Inclusion criteria
Exclusion criteria
Countries: USA and Germany |
| Interventions | Intervention ‐dose‐rangingIncobotulinumtoxinA, intramuscular injection into the glabellar area Open‐label extension |
| Outcomes |
Primary outcome
Secondary outcomes
|
| Notes | Supported by Merz Pharmaceuticals GmbH |
NCT03837561.
| Methods | Randomised, double‐blind, active‐controlled, multicentre, phase III trial |
| Participants | 136 participants Inclusion criteria
Exclusion Criteria
Country: Russia |
| Interventions |
Intervention
Comparator
|
| Outcomes |
Primary outcome
Secondary outcome: no secondary outcome |
| Notes | Sponsor Medy‐tox Other study id MT01‐RU18GBL301 |
NCT03970876.
| Methods | Phase I clinical trial, participants with moderate to severe glabellar lines at maximum frown are enrolled and safety is assessed after 12 weeks of administration of 20 U of ATGC‐100. In phase II clinical trial, participants with moderate to severe glabellar lines at maximum frown are enrolled, and efficacy and safety are assessed by comparing with Botox (Allergan). Phase II‐ randomised, double‐blind, active‐controlled, multicentre trial |
| Participants | 60 participants. Inclusion Criteria
Exclusion Criteria
|
| Interventions |
Intervention
Comparator
|
| Outcomes | Primary outcomes
Secondary outcomes
Country: Republic of Korea |
| Notes | EuBiologics Co.,Ltd |
NCT03985982.
| Methods | Randomised, double‐blind, placebo‐controlled trial |
| Participants | 353 participants Inclusion criteria
Exclusion criteria
Country: USA |
| Interventions |
Intervention
Comparator
|
| Outcomes |
Primary outcome
Secondary outcomes
|
| Notes | Croma‐Pharma GmbH |
NCT04081402.
| Methods | Randomised, quadruple‐blind, active control, parallel arm, phase I/III trial |
| Participants | 290 participants Inclusion Criteria
Exclusion Criteria
Country Republic of Korea |
| Interventions |
Intervention
Comparator
|
| Outcomes |
Primary outcome
Secondary outcome No information |
| Notes | Huons Co., Ltd. |
NCT04143815.
| Methods | Randomised, double‐blind, placebo‐controlled, multicentre, dose‐ranging and open‐lLabel extension study |
| Participants | 200 participants Inclusion criteria
Exclusion Criteria
Country Australia |
| Interventions |
Intervention
Comparator
|
| Outcomes |
Primary outcome
Secondary outcome No information |
| Notes | Medy‐Tox |
NCT04247074.
| Methods | Randomised, double‐blind, placebo‐controlled, multicentre trial |
| Participants | 350 participants Inclusion Criteria
Exclusion Criteria
Country‐ USA |
| Interventions |
Intervention
Comparator
|
| Outcomes |
Primary outcome
Secondary outcomes
|
| Notes | Q‐Med AB |
NCT04249583.
| Methods | Randomised, double‐blind, placebo‐controlled, multicentre trial |
| Participants | 300 participants Inclusion criteria
Exclusion Criteria
Country USA |
| Interventions |
Intervention
Comparator
|
| Outcomes |
Primary outcome
Secondary outcome
|
| Notes | Q‐Med AB |
NCT04281095.
| Methods | Randomised, double‐blind, active‐controlled, single‐centre clinical trial |
| Participants | 60 participants Inclusion criteria
Exclusion criteria
Country Republic of Korea |
| Interventions |
Intervention
Comparator
|
| Outcomes |
Primary outcome
Secondary outcomes
|
| Notes | ATGC Co., Ltd. |
AEs: adverse events; CFL: Crow's feet lines; FWS: facial wrinkle scale;IGA: Investigator's Global Assessment;I.M.: intramuscular; NSAIDs: non‐steroidal anti‐inflammatory drugs; PP: per protocol;SAEs: serious adverse events; IC: informed consent; U: unit.
Differences between protocol and review
In the review version we made the following changes:
We expanded the search for all relevant plastic and dermatologic conference proceedings, because some RCTs were published only in medical conference proceedings.
We included an additional secondary outcome “duration of the effect of treatment”, since it was a relevant clinical question to guide decision‐making.
We changed the “Minor adverse effects (headache, haematoma, pain in the site of injection)” to “Total adverse events”. This was because most of the studies adopted major and total adverse events' categorisation.
We changed the minimal number of participants for include studies from 20 to 50 (actually it was a typing error type of protocol version that was amended at review).
We assessed the responder rates only during “muscle contraction”, rather than “at rest”, due to (a) for clinical practice this last approach was less relevant and (b) at label, BontA was indicated for hyperdynamic facial wrinkles.
We performed meta‐analysis only in parallel group studies, we did not include split face studies in this analysis.
We changed the following terms: 1) “Responder rate by participant assessment” for “Participant assessment of success by analysing scores and scales”; 2) “major events” to “Any major adverse events (eyelid ptosis, eyelid sensory disorder, strabismus”; 3) “Responder rate by physician assessment” for “An assessment of the many physician scores or scales”; 4) “Any total adverse event” for “Total adverse event”; 5) “Duration of treatment effect” for “Duration of treatment”; according to editorial board suggestion.
We were unable to assess reporting bias and perform subgroup and sensitivity analyses due to a limited number of studies.
We planned to perform analyses using fixed‐effect model by default. However, due to clinical and/or methodological heterogeneity among included studies, we used a random‐effects model instead.
We added that we would use Peto odds ratios for rare event meta‐analyses.
In our protocol we did not specify which comparisons we would create 'summary of findings tables' for. We therefore made the decision post‐hoc to include six 'summary of findings tables' for the most clinically import comparisons of this review. We also specified the time points we would include (four weeks for effectiveness outcomes, and study duration for safety outcomes).
Contributions of authors
CPC and JX was the contact person with the editorial base. CPC and RR co‐ordinated contributions from the co‐authors and wrote the final draft of the review. CPC and RG screened papers against eligibility criteria. CPC and CSC obtained data on ongoing and unpublished studies. CPC, RG appraised the quality of papers. CPC, RR and JX extracted data for the review and sought additional information about papers. CPC and RR entered data into RevMan. CPC and RR analysed and interpreted data. CPC and MB undertook statistical analysis. CPC and RR worked on the methods sections. CPC, JX, and RR drafted the clinical sections of the Background and responded to the clinical comments of the referees. CPC, RR responded to the methodology and statistics comments of the referees. MCT was the consumer co‐author and checked the review for readability and clarity, as well as ensuring outcomes are relevant to consumers. CPC is the guarantor of the update.
Disclaimer
This project was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Skin Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
Sources of support
Internal sources
No sources of support provided
External sources
-
The National Institute for Health Research (NIHR), UK
The NIHR, UK, is the largest single funder of the Cochrane Skin Group.
Declarations of interest
Cristina Pires Camargo: nothing to declare. Jun Xia: nothing to declare. Caroline S Costa: nothing to declare. Rolf Gemperli: nothing to declare. Maria DC Tatini: nothing to declare. Max K Bulsara: nothing to declare. Rachel Riera: nothing to declare.
Professor Berthold Rzany, clinical referee, declared the following: "I am, or have been in the past, a speaker and advisor for IPSEN, Q‐Med/Galderma and Merz." BR reports using botulinum toxin in his practice for aesthetic indications for over 20 years, and he is an author of the following included studies: Ascher 2009, Kerscher 2015, Rzany 2006, Rzany 2019, Satler 2010; and excluded study Rzany 2013.
Edited (no change to conclusions)
References
References to studies included in this review
Ascher 2004 {published data only}
- Ascher B, Zakine B, Kestemont P, Baspeyras M, Bougara A, Santini J.A multicenter, randomized, double-blind, placebo-controlled study of efficacy and safety of 3 doses of botulinum toxin A in the treatment of glabellar lines. Journal of the American Academy of Dermatology 2004;51(2):223-33. [PMID: ] [DOI] [PubMed] [Google Scholar]
Ascher 2005 {published data only}
- Ascher B, Zakine B, Kestemont P, Baspeyras M, Bougara A, Niforos F, et al.Botulinum toxin A in the treatment of glabellar lines: scheduling the next injection. Aesthetic Surgery Journal 2005;25(4):365-75. [PMID: ] [DOI] [PubMed] [Google Scholar]
Ascher 2009 {published data only}
- Ascher B, Rzany BJ, Grover R.Efficacy and safety of botulinum toxin type A in the treatment of lateral crow's feet: double-blind, placebo-controlled, dose-ranging study. Dermatologic Surgery 2009;35(10):1478-86. [DOI: 10.1111/j.1524-4725.2009.01261.x] [PMID: ] [DOI] [PubMed] [Google Scholar]
Ascher 2018 {published data only}
- Ascher B, Kestemont P, Boineau D, Bodokh I, Stein A, Heckmann M, et al.Liquid formulation of abobotulinumtoxinA exhibits a favorable efficacy and safety profile in moderate to severe glabellar lines: a randomized, double-blind, placebo- and active comparator-controlled trial. Aesthetic Surgery Journal 2018;38(2):183-91. [PMID: ] [DOI] [PubMed] [Google Scholar]
Ascher 2020 {published data only}
- Ascher B, Rzany B, Kestemont P, Hilton S, Heckmann M, Bodokh I, et al.Liquid formulation of abobotulinumtoxinA: A 6-month, phase 3, double-blind,randomized, placebo-controlled study of a single treatment, ready-to-use toxin for moderate-to-severe glabellar lines. Aesthetic Surgery Journal 2020;40(1):93-104. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT02353871.Efficacy and safety of clostridium botulinum toxin type A to improve appearance of moderate to severe glabellar lines (BTX-A-HAC NG). clinicaltrials.gov/ct2/show/NCT02353871 (first received 3 February 2015).
Beer 2006 {published data only}
- Beer KR.Comparative evaluation of the safety and efficacy of botulinum toxin type A and topical creams for treating moderate-to-severe glabellar rhytids. Dermatologic Surgery 2006;32(2):184-97. [DOI: 10.1111/j.1524-4725.2006.32036.x] [PMID: ] [DOI] [PubMed] [Google Scholar]
Beer 2019a {published data only}
- Beer KR, Shamban AT, Avelar RL, Gross JE, Jonker A.Efficacy and safety of prabotulinumtoxinA for the treatment of glabellar lines in adult subjects: results from 2 identical phase III studies. Dermatologic Surgery 2019;45(11):1381-93. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Beer 2019b {published data only}
- Beer KR, Shamban AT, Avelar RL, Gross JE, Jonker A.Efficacy and safety of prabotulinumtoxinA for the treatment of glabellar lines in adult subjects: results From 2 identical phase III studies. Dermatologic Surgery 2019;45(11):1381-93. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bertucci 2020 {published data only}
- Bertucci V, Solish N, Kaufman-Janette J, Yoelin S, Shamban A, Schlessinger J, et al.DaxibotulinumtoxinA for Injection has a prolonged duration of response in the treatment of glabellar lines: pooled data from two multicenter, randomized, double-blind, placebo-controlled, phase 3 studies (SAKURA 1 and SAKURA 2). Journal of the American Academy of Dermatology 2020;82(4):838-45. [PMID: ] [DOI] [PubMed] [Google Scholar]
- Carruthers JD, Fagien S, Joseph JH, Humphrey SD, Biesman BS, Gallagher CJ, et al.DaxibotulinumtoxinA for injection for the treatment of glabellar lines: results from each of two multicenter, randomized, double-blind, placebo-controlled, phase 3 studies (SAKURA 1 and SAKURA 2). Plastic and Reconstructive Surgery 2020;145(1):45-58. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Brandt 2009 {published data only}
- Brandt F, Swanson N, Baumann L, Huber B.Randomized, placebo-controlled study of a new botulinum toxin type a for treatment of glabellar lines: efficacy and safety. Dermatologic Surgery 2009;35(12):1893-901. [CENTRAL: CN-00734159] [DOI] [PubMed] [Google Scholar]
Carruthers 2002 {published data only}
- Carruthers JA, Lowe NJ, Menter MA, Gibson J, Nordquist M, Mordaunt J, et al.A multicenter, double-blind, randomized, placebo-controlled study of the efficacy and safety of botulinum toxin type A in the treatment of glabellar lines. Journal of the American Academy of Dermatology. 2002;46(6):840-9. [CENTRAL: CN-00389568] [PMID: ] [DOI] [PubMed] [Google Scholar]
Carruthers 2003a {published data only}
- Carruthers A, Carruthers J, Cohen J.A prospective, double-blind, randomized, parallel- group, dose- ranging study of botulinum toxin type a in female subjects with horizontal forehead rhytides. Dermatologic Surgery 2003;29(5):461-7. [CENTRAL: CN-00437536] [DOI] [PubMed] [Google Scholar]
Carruthers 2003b {published data only}
- Carruthers JD, Lowe NJ, Menter MA, Gibson J, Eadie N, Botox Glabellar Lines II Study Group.Double-blind, placebo-controlled study of the safety and efficacy of botulinum toxin type A for patients with glabellar lines. Plastic and Reconstructive Surgery 2003;112(4):1089-98. [CENTRAL: CN-00450428] [PMID: ] [DOI] [PubMed] [Google Scholar]
Carruthers 2004 {published data only}
- Carruthers A, Carruthers J, Lowe NJ, Menter A, Gibson J, Nordquist M, et al.One-year, randomised, multicenter, two-period study of the safety and efficacy of repeated treatments with botulinum toxin type A in patients with glabellar lines. Journal of Clinical Research 2004;7:1-20. [CENTRAL: CN-00516138] [Google Scholar]
Carruthers 2005a {published data only}
- Carruthers A, Carruthers J.Prospective, double-blind, randomized, parallel- group, dose-ranging study of botulinum toxin type A in men with glabellar rhytids. Dermatologic Surgery 2005;31(10):1297-303. [PMID: ] [DOI] [PubMed] [Google Scholar]
Carruthers 2005b {published data only}
- Carruthers A, Carruthers J, Said S.Dose-ranging study of botulinum toxin type A in the treatment of glabellar rhytids in females. Dermatologic Surgery 2005;31(4):414-22. [PMID: ] [DOI] [PubMed] [Google Scholar]
Carruthers 2009 {published data only}
- Carruthers A, Carruthers J.A single-center, dose-comparison, pilot study of botulinum neurotoxin type A in female patients with upper facial rhytids: safety and efficacy. Journal of the American Academy of Dermatology 2009;60(6):972-9. [CENTRAL: CN-00699077] [DOI] [PubMed] [Google Scholar]
- Carruthers A, Carruthers J.A single-center dose-comparison study of botulinum neurotoxin type A in females with upper facial rhytids: assessing patients' perception of treatment outcomes. Journal of Drugs in Dermatology. 2009;8(10):924-9. [CENTRAL: CN-00722569] [PubMed] [Google Scholar]
Carruthers 2010 {published data only}
- Carruthers A, Carruthers J, Monheit GD, Davis PG, Tardie G.Multicenter, randomized, parallel-group study of the safety and effectiveness of onabotulinumtoxinA and hyaluronic acid dermal fillers (24-mg/ml smooth, cohesive gel) alone and in combination for lower facial rejuvenation. Dermatologic Surgery 2010;36(Suppl 4):2121-34. [PMID: ] [DOI] [PubMed] [Google Scholar]
Carruthers 2013 {published data only}
- Carruthers A, Carruthers J, Coleman WP, Donofrio L, Flynn T, Gold M, et al.Multicenter, randomized, phase III study of a single dose of incobotulinumtoxinA, free from complexing proteins, in the treatment of glabellar frown lines. Dermatologic Surgery 2013;39(4):551-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Carruthers 2014 {published data only}
- Carruthers A, Bruce S, Coninck A, Connoly S, Cox SE, Davis PG, et al.Efficacy and safety of onabotulinumtoxinA for the treatment of crow’s feet lines: a multicenter, randomized, controlled trial. Dermatologic Surgery 2014;40(11):1181-90. [PMID: ] [DOI] [PubMed] [Google Scholar]
Carruthers 2015 {published data only}
- Carruthers J, Rivkin A, Donofrio L, Bertucci V, Somogyi C, Lei X, et al.A multicenter, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of repeated onabotulinumtoxinA treatments in subjects with crow's feet lines and glabellar lines. Dermatologic Surgery 2015;41(6):702-11. [CENTRAL: CN-01083939] [DOI] [PubMed] [Google Scholar]
- NCT01224015.Safety and efficacy study of botulinum toxin type A for the treatment of crow's feet lines and frown lines. clinicaltrials.gov/ct2/show/NCT01224015 (first received 19 October 2010).
Carruthers 2017 {published data only}
- Carruthers J, Solish N, Humphrey S, Rosen N, Muhn C, Bertucci V, et al.Injectable daxibotulinumtoxinA for the treatment of glabellar lines: a phase 2, randomized, dose-ranging, double-blind, multicenter comparison with onabotulinumtoxinA and placebo. Dermatologic Surgery 2017;43(11):1321-31. [PMID: ] [DOI] [PubMed] [Google Scholar]
- NCT02303002.Efficacy and safety of botulinum toxin type A for injection to treat glabellar lines. clinicaltrials.gov/ct2/show/NCT02303002 (first received 27 November 2014).
Cheon 2019 {published data only}
- Cheon HI, Jung N, Won CH, Kim BJ, Lee YW.Efficacy and safety of prabotulinumtoxin A and onabotulinumtoxin A for crow's feet A phase 3, multicenter, randomized, double-blind, split-face study. Dermatologic Surgery 2019;45(12):1610-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Cohen 2012 {published data only}
- Cohen JL, Dayan SH, Cox SE, Yalamanchili R, Tardie G.OnabotulinumtoxinA dose-ranging study for hyperdynamic perioral lines. Dermatologic Surgery 2012;38(9):1497-505. [CENTRAL: CN-00969333] [DOI] [PubMed] [Google Scholar]
Costa 2016 {published data only}
- Costa A, Talarico-Filho S, Arruda LH, Pecora CS, Ortolan DG, Monteiro EO, et al.Multicenter, prospective, comparative, randomized, double-blind clinical study comparing two botulinum toxin type A formulations registered in Brazil for the treatment of glabellar wrinkles [Estudo clínico multicêntrico, prospectivo, comparativo, randomizado e duplo cego, entre duas formulações de toxina botulínica tipo A registradas no Brasil para o tratamento das rugas da glabela]. Surgical and Cosmetic Dermatology 2016;8(1):33-40. [Google Scholar]
Dayan 2010 {published data only}
- Dayan SH, Arkins JP, Patel AB, Gal TJ.A double-blind, randomized, placebo-controlled health- outcomes survey of the effect of botulinum toxin type a injections on quality of life and self-esteem. Dermatologic Surgery 2010;36(Suppl 4):2088-97. [CENTRAL: CN-00779861] [DOI] [PubMed] [Google Scholar]
De Boulle 2018 {published data only}
- De Boulle K, Werschler WP, Gold MH, Bruce S, Satler G, Ogilvie P, et al.Phase 3 study of onabotulinumtoxinA distributed between frontalis, glabellar complex, and lateral canthal areas for treatment of upper facial lines. Dermatologic Surgery 2018;44(11):1437-48. [CENTRAL: CN-01650294] [DOI] [PubMed] [Google Scholar]
Fagien 2007a {published data only}
- Fagien S, Cox SE, Finn JC, Werschler WP, Kowalski JW.Patient-reported outcomes with botulinum toxin type A treatment of glabellar rhytids: a double-blind, randomized, placebo-controlled study. Dermatologic Surgery 2007;33(1 Spec No):S2-9. [CENTRAL: CN-00575067] [PMID: ] [DOI] [PubMed] [Google Scholar]
Feng 2015 {published data only}
- Feng Z, Sun Q, He L, Wu Y, Xie H, Zhao G, et al.Optimal dosage of botulinum toxin type A for treatment of glabellar lines: efficacy and safety in a clinical trial. Dermatologic Surgery 2015;41(Suppl 1):S56-63. [CENTRAL: CN-01111346] [DOI] [PubMed] [Google Scholar]
Firoz 2012 {published data only}
- Firoz B, Pilato T, Zhou W.A randomized, double blind, split-face comparison of Botox verses dysport for glabellar and forehead lines. Journal of the American Academy of Dermatology 2012;66(4 Suppl 1):AB21. [EMBASE: 70703936] [Google Scholar]
Hanke 2013 {published data only}
- Hanke CW, Narins RS, Brandt F, Cohen JL, Donofrio LM, Downie J, et al.A randomized, placebo-controlled, double-blind phase III trial investigating the efficacy and safety of incobotulinumtoxinA in the treatment of glabellar frown lines using a stringent composite endpoint. Dermatologic Surgery 2013;39(6):891-9. [CENTRAL: CN-00964813] [DOI] [PubMed] [Google Scholar]
Harii 2008 {published data only}
- Harii K, Kawashima M.A double-blind, randomized, placebo-controlled, two- dose comparative study of botulinum toxin type A for treating glabellar lines in Japanese subjects. Aesthetic Plastic Surgery 2008;32(5):724-30. [CENTRAL: CN-00669238] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima M, Harii K.An open-label, randomized, 64-week study repeating 10- and 20-U doses of botulinum toxin type A for treatment of glabellar lines in Japanese subjects. International Journal of Dermatology 2009;48(7):768-76. [CENTRAL: CN-00720265] [DOI] [PubMed] [Google Scholar]
Harii 2017 {published data only}
- Harii K, Kawashima M, Furuyama N, Lei X, Hopfinger R, Lee E.Onabotulinumtoxina (botox) in the treatment of crow's feet lines in Japanese subjects. Aesthetic Plastic Surgery 2017;41(5):1186-97. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT01797094.BOTOX® in the treatment of upper facial lines in Japan. clinicaltrials.gov/ct2/show/NCT01797094 (first received 22 February 2013).
Hexsel 2013 {published data only}
- Hexsel D, Brum C, Porto MD, Soirefmann M, Siega C, Schilling-Souza J, et al.Full-face injections of variable total doses of abobotulinum toxin type A: A randomized, phase IV clinical trial of safety and efficacy. Journal of Drugs in Dermatology 2013;12(12):1356-62. [CENTRAL: CN-00961073] [PubMed] [Google Scholar]
Kane 2009 {published data only}
- Kane MA, Brandt F, Rohrich RJ, Narins RS, Monheit GD, Huber MB, et al.Evaluation of variable-dose treatment with a new U.S. botulinum toxin type A (Dysport) for correction of moderate to severe glabellar lines: results from a phase III, randomized, double-blind, placebo-controlled study. Plastic and Reconstructive Surgery 2009;124(5):1619-29. [CENTRAL: CN-00728882] [DOI] [PubMed] [Google Scholar]
Kane 2015 {published data only}
- Kane MA, Gold MH, Coleman WP, Jones DH, Tanghetti EA, Alster TS, et al.A randomized, double-blind trial to investigate the equivalence of IncobotulinumtoxinA and OnabotulinumtoxinA for glabellar frown lines. Dermatologic Surgery 2015;41(11):1310-9. [CENTRAL: CN-01105599] [DOI] [PubMed] [Google Scholar]
Kassir 2013 {published data only}
- Kassir R, Kolluru A, Kassir M.Triple-blind, prospective, internally controlled comparative study between AbobotulinumtoxinA and OnabotulinumtoxinA for the treatment of facial rhytids. Dermatology and Therapy 2013;3(2):179-89. [CENTRAL: CN-00993785] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kerscher 2015 {published data only}
- Kerscher M, Rzany B, Prager W, Turnbull C, Trevidic P, Inglefield C.Efficacy and safety of incobotulinumtoxina in the treatment of upper facial lines: results from a randomized, double-blind, placebo-controlled, phase iii study. Dermatologic Surgery 2015;41(10):1149-57. [CENTRAL: CN-01108580] [DOI] [PubMed] [Google Scholar]
Kim 2014 {published data only}
- Kim BJ, Kwon HH, Park SY, Min SU, Yoon JY, Park YM, et al.Double-blind, randomized non-inferiority trial of a novel botulinum toxin a processed from the strain cbfc26, compared with onabotulinumtoxin a in the treatment of glabellar lines. Journal of the European Academy of Dermatology and Venereology : JEADV 2014;28(12):1761-7. [CENTRAL: CN-01118064] [DOI] [PubMed] [Google Scholar]
Kim 2015 {published data only}
- Kim JE, Song EJ, Choi GS, Lew BL, Sim WY, Kang H.The efficacy and safety of liquid-type botulinum toxin type A for the management of moderate to severe glabellar frown lines. Plastic and Reconstructive Surgery 2015;135(3):732-41. [CENTRAL: CN-01076800] [DOI] [PubMed] [Google Scholar]
Lee 2013 {published data only}
- Lee WS, Won CH, Huh CH, Kang H, Kim BJ, Lee JH, et al.Efficacy and safety of a novel botulinum toxin type A product for the treatment of moderate to severe glabellar lines: a randomized, double-blinded, active controlled multicenter study. Toxicon 2013;68:116. [EMBASE: 71313875] [DOI] [PubMed] [Google Scholar]
Lowe 2006 {published data only}
- Lowe P, Patnaik R, Lowe N.Comparison of two formulations of botulinum toxin type A for the treatment of glabellar lines: a double-blind, randomized study. Journal of the American Academy of Dermatology 2006;55(6):975-80. [CENTRAL: CN-00573598] [DOI] [PubMed] [Google Scholar]
Michaels 2012 {published data only}
- Michaels BM, Csank GA, Ryb GE, Eko FN, Rubin A.Prospective randomized comparison of onabotulinumtoxinA (Botox) and abobotulinumtoxinA (Dysport) in the treatment of forehead, glabellar, and periorbital wrinkles. Aesthetic Surgery Journal 2012;32(1):96-102. [CENTRAL: CN-00832322] [DOI] [PubMed] [Google Scholar]
Moers‐Carpi 2012 {published data only}
- Moers-Capri M, Dirschka T, Feller-Heppt G, Hiltons S, Hoffmann K, Philipp-Dormston WG, et al.A randomised, double-blind comparison of 20 units of onabotulinumtoxinA with 30 units of incobotulinumtoxinA for glabellar lines. Journal of Cosmetic and Laser Therapy 2012;14(6):296-303. [CENTRAL: CN-00967309] [DOI] [PubMed] [Google Scholar]
Moers‐Carpi 2015 {published data only}
- Moers-Capri M, Carruthers J, Fagien S, Lupo M, Delmar H, Jones D, et al.Efficacy and safety of onabotulinumtoxinA for treating crow's feet lines alone or in combination with glabellar lines: a multicenter, randomized, controlled trial. Dermatologic Surgery 2015;41(1):102-12. [CENTRAL: CN-01049373] [DOI] [PubMed] [Google Scholar]
Monheit 2007 {published data only}
- Monheit G, Carruthers A, Brandt F, Rand R.A randomized, double-blind, placebo-controlled study of botulinum toxin type A for the treatment of glabellar lines: determination of optimal dose. Dermatologic Surgery 2007;33(1 Spec No):S51-9. [CENTRAL: CN-00575071] [DOI] [PubMed] [Google Scholar]
Monheit 2019 {published data only}
- Monheit GD, Baumann L, Maas C, Rand R, Down R.Efficacy, safety, and subject satisfaction after abobotulinumtoxinA treatment for moderate to severe glabellar lines. Dermatologic Surgery 2020;46(1):61-9. [DOI] [PubMed] [Google Scholar]
NCT02450526 {published data only}
- NCT02450526.Dysport in the treatment of glabellar lines in Chinese subjects. clinicaltrials.gov/ct2/show/NCT02450526 (first received 21 May 2015).
NCT02493946 {published data only}
- NCT02493946.Efficacy and safety of botulinum toxin type A haemagglutinin complex next generation (BTX-A-HAC NG) in glabellar lines. clinicaltrials.gov/ct2/show/NCT02493946 (first received 10 July 2015).
Nettar 2011 {published data only}
- Nettar KD, Yu KC, Bapna S, Boscardin J, Maas CS.An internally controlled, double-blind comparison of the efficacy of onabotulinumtoxinA and abobotulinumtoxinA. Archives of Facial Plastic Surgery 2011;13(6):380-6. [CENTRAL: CN-00814326] [DOI] [PubMed] [Google Scholar]
- Yu KC, Nettar KD, Bapna S, Boscardin WJ, Maas CS.Split-face double-blind study comparing the onset of action of onabotulinumtoxinA and abobotulinumtoxinA. Archives of Facial Plastic Surgery 2012;14(3):198-204. [CENTRAL: CN-00840586] [DOI] [PubMed] [Google Scholar]
Ogilvie 2019 {published data only}
- Ogilvie P, Rivkin AZ, Dayan S, Yoelin SG, Weichman BM, Garcia JK.OnabotulinumtoxinA for treatment of forehead and glabellar lines: subject-reported satisfaction and impact from phase 3 double-blind study. Dermatologic Surgery 2019;45(5):689-99. [CENTRAL: CN-01941030] [DOI] [PubMed] [Google Scholar]
- Rivkin AZ, Ogilvie P, Dayan S, Yoelin SG, Weichman BM, Garcia JK.OnabotulinumtoxinA for simultaneous treatment of upper facial lines: subject-reported satisfaction and impact from a phase 3 study. Dermatologic Surgery 2020;46(1):50-60. [PMID: ] [DOI] [PubMed] [Google Scholar]
Park 2014 {published data only}
- Lee JH, Park JH, Lee SK, Han KH, Kim SD, Yoon CS, et al.Efficacy and safety of incobotulinum toxin A in periocular rhytides and masseteric hypertrophy: side-by-side comparison with onabotulinum toxin A. Journal of Dermatological Treatment 2014;25(4):326-30. [CENTRAL: CN-00986202] [DOI] [PubMed] [Google Scholar]
Patel 2004 {published data only}
- Patel MP, Talmor M, Nolan WB.Botox and collagen for glabellar furrows: advantages of combination therapy. Annals of Plastic Surgery 2004;52(5):442-7. [CENTRAL: CN-00489363] [DOI] [PubMed] [Google Scholar]
Rappl 2013 {published data only}
- Rappl T, Parvizi D, Friedl H, Wiedner M, May S, Kranzelbinder B, et al.Onset and duration of effect of incobotulinumtoxinA, onabotulinumtoxinA, and abobotulinumtoxinA in the treatment of glabellar frown lines: a randomized, double-blind study. Clinical, Cosmetic and Investigational Dermatology 2013;6:211-9. [CENTRAL: CN-00914061] [DOI] [PMC free article] [PubMed] [Google Scholar]
Rivers 2015 {published data only}
- Rivers JK, Bertucci V, McGillivray W, Muhn C, Rosen N, Solish N, et al.Subject satisfaction with onabotulinumtoxina treatment of glabellar and lateral canthal lines using a new patient-reported outcome measure. Dermatologic Surgery 2015;41(8):950-9. [CENTRAL: CN-01091229] [DOI] [PubMed] [Google Scholar]
Rubin 2009 {published data only}
- Rubin MG, Dover J, Glogau RG, Goldberg DJ, Goldman MP, Schlessinger J.The efficacy and safety of a new U.S. Botulinum toxin type A in the retreatment of glabellar lines following open-label treatment. Journal of Drugs in Dermatology 2009;8(5):439-44. [CENTRAL: CN-00698408] [PubMed] [Google Scholar]
Rzany 2006 {published data only}
- Rzany B, Ascher B, Fratila A, Monheit GD, Talarico S, Sterry W.Efficacy and safety of 3- and 5-injection patterns (30 and 50 U) of botulinum toxin A (Dysport) for the treatment of wrinkles in the glabella and the central forehead region. Archives of Dermatology 2006;142(3):320-6. [CENTRAL: CN-00555812] [DOI] [PubMed] [Google Scholar]
Rzany 2019 {published data only}
- Rzany BJ, Ascher B, Avelar RL, Bergdahl J, Bertucci V, Bodokh I, et al.A multicenter, randomized, double-blind, placebo-controlled, single-dose, phase III, non-inferiority study comparing prabotulinumtoxinA and onabotulinumtoxinA for the treatment of moderate to severe glabellar lines in adult subjects. Aesthetic Surgery Journal 2019 Apr 5 [Epub ahead of print]. [CENTRAL: CN-01941776] [DOI: 10.1093/asj/sjz110] [DOI] [PubMed]
Satler 2010 {published data only}
- Sattler G, Callander MJ, Grablowitz D, Walker T, Bee EK, Rzany B, et al.Noninferiority of incobotulinumtoxinA, free from complexing proteins, compared with another botulinum toxin type A in the treatment of glabellar frown lines. Dermatologic Surgery 2010;36(Suppl 4):2146-54. [CENTRAL: CN-00778835] [DOI] [PubMed] [Google Scholar]
Solish 2016 {published data only}
- Solish N, Rivers JK, Humphrey S, Muhn C, Somogyi C, Lei X, et al.Efficacy and safety of onabotulinumtoxina treatment of forehead lines: a multicenter, randomized, dose-ranging controlled trial. Dermatologic Surgery 2016;42(3):410-9. [CENTRAL: CN-01141742] [DOI] [PubMed] [Google Scholar]
Solish 2018 {published data only}
- Solish N, Liu Y, Rubio RG, Gallagher CJ.A head-to-head dose-ranging study of daxibotulinumtoxinA for injection in the treatment of glabellar lines: results of the BELMONT study. Toxicon 2018;156(Suppl 1):S106. [CENTRAL: CN-01794190] [DOI: 10.1016/j.toxicon.2018.11.255] [DOI] [Google Scholar]
Won 2013 {published data only}
- Won CH, Lee HM, Lee WS, Kang H, Kim BJ, Kim WS, et al.Efficacy and safety of a novel botulinum toxin type A product for the treatment of moderate to severe glabellar lines: a randomized, double-blind, active- controlled multicenter study. Dermatologic Surgery 2013;39(1 Pt 2):171-8. [CENTRAL: CN-00841439] [DOI] [PubMed] [Google Scholar]
Won 2015 {published data only}
- Won CH, Kim HK, Kim BJ, Kang H, Hong JP, Lee SY, et al.Comparative trial of a novel botulinum neurotoxin type A versus onabotulinumtoxinA in the treatment of glabellar lines: a multicenter, randomized, double-blind, active-controlled study. International Journal of Dermatology 2015;54(2):227-34. [CENTRAL: CN-01048870] [DOI] [PubMed] [Google Scholar]
Wu 2010 {published data only}
- Wu Y, Zhao G, Li H, Zheng Z, Zhong S, Yang Z, et al.Botulinum toxin type A for the treatment of glabellar lines in Chinese: a double-blind, randomized, placebo-controlled study. Dermatologic Surgery 2010;36(1):102-8. [CENTRAL: CN-00743925] [DOI] [PubMed] [Google Scholar]
- Wu Y, Zhao G, Li H, Zheng Z.Botulinum toxin type A for the treatment of glabellar lines in Chinese subjects: a multicenter, double-blind, placebo-controlled trial. Journal of the American Academy of Dermatology. 2009;60(3 Suppl 1):AB80. [EMBASE: 70142039] [Google Scholar]
Wu 2019 {published data only}
- Wu Y, Wang G, Li C, Mao C, Lei X, Lee E.Efficacy of onabotulinumtoxinA for treatment of crow's feet lines in Chinese subjects. Plastic and Reconstructive Surgery - Global Open 2019;7(1):e2079. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
2014‐003770‐16 {published data only}
- 2014-003770-16.A prospective, open-label, multicenter, repeat-dose trial to investigate the safety and efficacy of NT 201 (incobotulinumtoxinA) in the combined treatment of upper facial lines (horizontal forehead lines, glabellar frown lines, and lateral periorbital lines). www.clinicaltrialsregister.eu/ctr-search/search?query=2014-003770-16 (first received 22 January 2015).
Cartier 2020 {published data only}
- Cartier H, Hedén P, Delmar H, Bergentz P, Skoglund C, Edwartz C, et al.Repeated full-face aesthetic combination treatment with abobotulinumtoxinA, hyaluronic acid filler, and skin-boosting hyaluronic acid after monotherapy with abobotulinumtoxinA or hyaluronic acid filler. Dermatologic Surgery 2020;46(4):475-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hexsel 2018 {published data only}
- Hexsel D, Cartier H, Hedén P, Delmar H, Bergentz P, Camozzato F, et al.Efficacy, safety, and subject satisfaction after abobotulinumtoxinA treatment of upper facial lines. Dermatologic Surgery 2018;44(12):1555-64. [DOI: 10.1097/DSS.0000000000001679] [DOI] [PubMed] [Google Scholar]
Mahmoud 2016 {published data only}
- Mahmoud BH, Ozog D, Burnett C, Cohen JL.Prospective randomized split-face comparative study between topical botulinum toxin A surface application and local injection for crow's feet. Dermatologic Surgery 2016;42(4):554-6. [CENTRAL: CN-01154179] [DOI] [PubMed] [Google Scholar]
NCT00752050 {unpublished data only}
- NCT00752050.Safety and efficacy study of repeat treatment with PurTox® botulinum toxin type A for frown lines between the eyebrows (PT-03b). clinicaltrials.gov/ct2/show/NCT00752050 (first received 15 September 2008).
NCT00752297 {published data only}
- NCT00752297.Safety and efficacy study of PurTox® botulinum toxin type A to treat frown lines between the eyebrows (PT-03a). clinicaltrials.gov/ct2/show/NCT00752297 (first received 15 September 2008).
NCT02297516 {published data only}
- NCT02297516.Safety and efficacy of azzalure/dysport, restylane/emervel filler and restylane skinbooster treatment. clinicaltrials.gov/ct2/show/NCT02297516 (first received 21 November 2014).
Punga 2016 {published data only}
- Punga AR, Alimohammadi M, Fagrell D, Nyberg F, Rees D, Wong C.A randomized, comparative study to evaluate the efficacy and safety of two injection volumes of abobotulinumtoxinA in treatment of glabellar lines. Dermatologic Surgery 2016;42(8):967-76. [CENTRAL: CN-01342684] [DOI] [PMC free article] [PubMed] [Google Scholar]
Rzany 2013 {published data only}
- Rzany B, Flynn TC, Schlobe A, Heinz M, Harrington L.Long-term results for incobotulinumtoxinA in the treatment of glabellar frown lines. Dermatologic Surgery 2013;39(1 Pt 1):95-103. [CENTRAL: CN-00841632] [DOI] [PubMed] [Google Scholar]
Wilson 2016 {published data only}
- Wilson AJ, Chang B, Taglienti AJ, Chin BC, Chang CS, Folsom N, et al.Quantitative analysis of onabotulinumtoxinA, abobotulinumtoxinA, and incobotulinumtoxinA: a randomized, double-blind, prospective clinical trial of comparative dynamic strain reduction. Plastic and Reconstructive Surgery 2016;137(5):1423-33. [CENTRAL: CN-01260494] [DOI] [PubMed] [Google Scholar]
Zhang 2018 {published data only}
- Zhang W, Xie Y, Liu W, Lei J, Liu Y.Combination therapy of botulinum toxin type A and hyaluronic acid filler for facial rejuvenation. International Journal of Clinical and Experimental Medicine 2018;11(5):5033-8. [CENTRAL: CN-01920192] [Google Scholar]
References to studies awaiting assessment
NCT01180348 {published data only}
- NCT01180348.Efficacy and safety of a new botulinum toxin type A for treatment of facial expression lines. clinicaltrials.gov/ct2/show/NCT01180348 (first received 12 August 2010).
NCT01358695 {published data only}
- NCT01358695.Clinical trial to evaluate ANT-1207 in subjects with crow's feet. clinicaltrials.gov/ct2/show/NCT01358695 (first received 24 May 2011).
NCT01485601 {published data only}
- NCT01485601.Safety and efficacy study of botulinum toxin type A to treat glabellar lines. clinicaltrials.gov/ct2/show/study/NCT01485601 (first received 5 December 2011).
NCT01583478 {published data only}
- NCT01583478.Comparison of escalating doses of incobotulinumtoxin A (Xeomin®) in the treatment of glabellar rhytids. clinicaltrials.gov/ct2/show/NCT01583478 (first received 20 March 2012).
NCT01791920 {published data only}
- NCT01791920.To evaluate the safety and efficacy of Botulax® are not inferior to those of Botox® in the treatment of glabellar lines. clinicaltrials.gov/ct2/show/NCT01791920 (first received 15 February 2013).
NCT01951742 {published data only}
- NCT01951742.Dose finding study in subjects with crow's feet. clinicaltrials.gov/ct2/show/NCT01951742 (first received 27 September 2013).
NCT02236312 {published data only}
- NCT02236312.Safety and efficacy study of botulinum toxin for the treatment of glabellar frown lines. clinicaltrials.gov/ct2/show/NCT02236312 (first received 10 September 2014).
NCT02677298 {published data only}
- NCT02677298.Botulinum toxin treatment of glabellar lines: efficacy and safety study I (BLESS I). clinicaltrials.gov/ct2/show/NCT02677298 (first received 9 February 2016).
NCT02677805 {published data only}
- NCT02677805.Botulinum toxin treatment of glabellar lines: efficacy and safety study II (BLESS II). clinicaltrials.gov/ct2/show/NCT02677805 (first received 9 February 2016).
NCT02882893 {published data only}
- NCT02882893.Efficacy and safety of DWP450 for the treatment of crows feet lines. clinicaltrials.gov/ct2/show/NCT02882893 (first received 30 August 2016).
NCT02961673 {published data only}
- NCT02961673.A phase I/II clinical trial to compare the safety and efficacy of HU-014 versus Botox® in subject with moderate to severe glabellar lines. clinicaltrials.gov/ct2/show/nct02961673 (first received 11 November 2016).
NCT03317574 {published data only}
- NCT03317574.MEDITOXIN® in treatment of crow's feet line. clinicaltrials.gov/ct2/show/NCT03317574 (first received 23 October 2017).
NCT03440671 {published data only}
- NCT03440671.The safety and efficacy study of Hutox versus Botox® in subject with moderate to severe glabellar lines. clinicaltrials.gov/ct2/show/nct03440671 (first received 21 February 2018).
NCT03721016 {published data only}
- NCT03721016.MT10109L in the treatment of glabellar lines with or without concurrent treatment of lateral canthal lines. clinicaltrials.gov/ct2/show/NCT03721016 (first received 26 October 2018).
NCT03736928 {published data only}
- NCT03736928.Safety and efficacy of abobotulinumtoxinA for the treatment of glabellar lines. clinicaltrials.gov/ct2/show/NCT03736928 (first received 9 November 2018).
NCT03806933 {published data only}
- NCT03806933.Study to investigate the safety and duration of effect of different Botulinum Toxin A (NT 201) dose groups following the treatment of glabellar frown lines. clinicaltrials.gov/ct2/show/NCT03806933 (first received 16 January 2019).
NCT03837561 {published data only}
- NCT03837561.The efficacy and safety of CUNOX® in patients with moderate to severe glabellar lines. clinicaltrials.gov/ct2/show/NCT03837561 (first received 12 February 2019).
NCT03970876 {published data only}
- NCT03970876.Safety and efficacy study of ATGC-100 for the treatment of moderate to severe glabellar lines in adult subjects. clinicaltrials.gov/ct2/show/NCT03970876 (first received 3 June 2019).
NCT03985982 {published data only}
- NCT03985982.Botulinum toxin treatment of glabellar lines: Efficacy and safety study III (BLESS III) (BLESS III). clinicaltrials.gov/ct2/show/NCT03985982 (first received 14 June 2019).
NCT04081402 {published data only}
- NCT04081402.A phase 1/3 clinical trial to evaluate the safety and efficacy of HUTOX compared to Botox® in subjects with moderate to severe crow's feet lines. clinicaltrials.gov/ct2/show/NCT04081402 (first received 9 September 2019).
NCT04143815 {published data only}
- NCT04143815.Clinical trial to evaluate the efficacy and safety of MBA-P01 in treatment of glabellar lines. clinicaltrials.gov/ct2/show/NCT04143815 (first received 29 October 2019).
NCT04247074 {published data only}
- NCT04247074.Treatment of moderate to severe lateral canthal lines and glabellar lines alone or in combination (READY-3). clinicaltrials.gov/ct2/show/NCT04247074 (first received 29 January 2020).
NCT04249583 {published data only}
- NCT04249583.Treatment of moderate to severe glabellar lines (READY-1). clinicaltrials.gov/ct2/show/NCT04249583 (first received 31 January 2020).
NCT04281095 {published data only}
- NCT04281095.A phase I/II randomized, double blind, active-controlled, single center clinical trial for evaluation of safety and efficacy of ATGC-110, an intramuscularly administered clostridium botulinum neurotoxin type A, in adult patients with moderate to severe glabellar frown lines. clinicaltrials.gov/ct2/show/NCT04281095 (first received 24 February 2020).
Additional references
ASAPS 2016
ASAPS 2017
- The American Society for Aesthetic Plastic Surgery.Botulinum Toxin. www.plasticsurgery.org/cosmetic-procedures/botulinum-toxin/cost (accessed 9 November 2017).
Ascher 2010
- Ascher B, Talarico S, Cassuto D, Escobar S, Hexsel D, Jaen P, et al.International consensus recommendations on the aesthetic usage of botulinum toxin type A (Speywood Unit) – Part 1: upper facial wrinkles. Journal of the European Academy of Dermatology & Venereology 2010;24(11):1278-84. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Berry 2012
- Berry MG, Stanek JJ.Botulinum neurotoxin A: a review. Journal of Plastic, Reconstructive & Aesthetic Surgery 2012;65(10):1283-91. [MEDLINE: 22552262 ] [DOI] [PubMed] [Google Scholar]
Carruthers 2003
- Carruthers J, Carruthers A.A prospective, randomized, parallel group study analyzing the effect of BTX-A (Botox) and nonanimal sourced hyaluronic acid (NASHA, Restylane) in combination compared with NASHA (Restylane) alone in severe glabellar rhytides in adult female subjects: treatment of severe glabellar rhytides with a hyaluronic acid derivative compared with the derivative and BTX-A. Dermatologic Surgery 2003;29(8):802-9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Carruthers 2008a
- Carruthers JD, Glogau RG, Blitzer AS, Facial Aesthetics Consensus Group Faculty.Advances in facial rejuvenation: botulinum toxin type A, hyaluronic acid dermal fillers, and combination therapies-consensus recommendations. Plastic & Reconstructive Surgery 2008;121(5 Suppl):5S-30S. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Carruthers 2008b
- Carruthers A, Carruthers J, Hardas B, Kaur M, Goertelmeyer R, Jones D, et al.A validated grading scale for forehead lines. Dermatologic Surgery 2008;34(Suppl 2):S155-60. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Carruthers 2008c
- Carruthers A, Carruthers J, Hardas B, Kaur M, Goertelmeyer R, Jones D, et al.A validated brow positioning grading scale. Dermatologic Surgery 2008;34(Suppl 2):S150-4. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Carruthers 2008d
- Carruthers A, Carruthers J, Hardas B, Kaur M, Goertelmeyer R, Jones D, et al.A validated grading scale for crow’s feet. Dermatologic Surgery 2008;34(Suppl 2):S173-8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Carruthers 2012
- Carruthers J, Flynn T, Geister T, Gortelmeyer T, Hardas B, Himmrich S, et al.Validated assessment scales for the mid face. Dermatologic Surgery 2012;38(2 Spec no.):320-32. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Cavallini 2014
- Cavallini M, Cirillo P, Fundaro SP, Quartucci S, Sito C, Sito G, et al.Safety of botulinum toxin A in aesthetic treatments: a systematic review of clinical studies. Dermatologic Surgery 2014;40(5):525-36. [PMID: ] [DOI] [PubMed] [Google Scholar]
Cox 2003
- Cox SE, Finn JC, Stetler L, Mackowiak J, Kowalski JW.Development of the facial lines treatment satisfaction questionnaire and initial results for botulinum toxin type A–treated patients. Dermatologic Surgery 2003;29(5):444-9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Fagien 2003
- Fagien S.Botox for the treatment of dynamic and hyperkinetic facial lines and furrows: adjunctive use in facial aesthetic surgery. Plastic and Reconstructive Surgery 2003;112(Suppl 5):40S-52S. [DOI] [PubMed] [Google Scholar]
Fagien 2007b
- Fagien S, Cox SE, Finn JC, Werschler WP, Kowalski JW.Patient-reported outcomes with botulinum toxin type A treatment of glabellar rhytids: a double-blind, randomized, placebo-controlled study. Dermatologic Surgery 2007;33(1 Spec No):S2-S9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Fagien 2008
- Fagien S, Carruthers JD.A comprehensive review of patient-reposted satisfaction with botulinum toxin type A for aesthetic procedures. Plastic & Reconstructive Surgery 2008;122(6):1915-25. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Flynn 2012
- Flynn T, Carruthers A, Carruthers J, Geister TL, Gortelmeyer R, Hardas B, et al.Validated assessment scales for upper face. Dermatologic Surgery 2012;38(2 Spec no):309-19. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Garnham 2013
- Garnham B.Designing ‘older’ rather than denying ageing: Problematizing anti-ageing discourse in relation to cosmetic surgery undertaken by older people. Journal of Aging Studies 2013;27(1):38-46. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Ghadia 2009
- Gadhia K, Walmsley AD.Facial aesthetics: is botulinum toxin treatment effective and safe? A systematic review of randomised controlled trials. British Dental Journal 2009;207(5):E-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Glogau 2012
- Glogau R, Kane M, Beddingfield F, Somogyi C, Lei X, Caulkins C, et al.OnabotulinumtoxinA: a meta-analysis of duration of effect in the treatment of glabellar lines. Dermatologic Surgery 2012;38(11):1794-803. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
GRADEpro GDT [Computer program]
- McMaster University (developed by Evidence Prime) GRADEpro GDT.Hamilton (ON): McMaster University (developed by Evidence Prime), 2021. Available at gradepro.org.
Higgins 2017
- Higgins JPT, Altman DG, Sterne JAC (editors).Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Churchill R, Chandler J, Cumpston MS (editors), Cochrane Handbook for Systematic Reviews of Interventions version 5.2.0 (updated June 2017). Cochrane, 2017. Available from training.cochrane.org/handbook.
Higgins 2020
- Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s).Cochrane Handbook for Systematic Review of Interventions Version 6.1 (updated September 2020). Cochrane, 2020. Available from training.cochrane.org/handbook.
Honeck 2003
- Honeck P, Weiss C, Sterry W, Rzany B, Gladys StudyGgroup.Reproducibility of a four-point clinical severity score for glabellar frown lines. British Journal of Dermatology 2003;149(2):306-10. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Hund 2006
- Hund T, Ascher B, Rzany B, Smile Study Group.Reproducibility of two four-point clinical severity scores for lateral canthal lines (crow's feet). Dermatologic Surgery 2006;32(10):1256-60. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Jia 2016
- Jia Z, Lu H, Yang X, Jin X, Wu R, Zhao J, et al.Adverse events of botulinum toxin type A in facial rejuvenation: a systematic review and meta-analysis. Aesthetic Plastic Surgery 2016;40(5):769-77. [PMID: ] [DOI] [PubMed] [Google Scholar]
Kane 2012
- Kane MA, Blitzer A, Barndt FS, Glogu G, Monheit GD, Narins RS, et al.Development and validation of a new clinically-meaningful rating scale for measuring lateral canthal line severity. Aesthetic Surgery Journal 2012;32(3):275-85. [MEDLINE: 22395318 ] [DOI] [PubMed] [Google Scholar]
Le Louarn 2007
- Le Louarn C.Botulinum toxin and the Face Recurve concept: decreasing resting tone and muscular regeneration [Toxine botulique et Face Recurve®: actionsur le tonus de repos et la régénération musculaire]. Annales de Chirurgie Plastique et Esthetique 2007;52(3):165-76. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Narins 2012
- Narins RS, Carruthers J, Flynn TC, Geister TL, Gortelmeyer R, Hardas B, et al.Validated assessment scales for the lower face. Dermatologic Surgery 2012;38(2 Spec no.):333-42. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
RevMan [Computer program]
- Nordic Cochrane Centre, The Cochrane Collaboration Review Manager (RevMan 5.4).Version 5.4. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2020.
Rzany 2012
- Rzany B, Carruthers A, Carruthers J, Flynn TC, Geister TL, Gortelmeyer R, et al.Validated composite assessment scales for the global face. Dermatologic Surgery 2012;38(2 Spec no.):294-308. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Samuel 2005
- Samuel M, Brooke R, Hollis S, Griffiths CEM.Interventions for photodamaged skin. Cochrane Database of Systematic Reviews 2005, Issue 1. Art. No: CD001782. [DOI: 10.1002/14651858.CD001782.pub2] [DOI] [PubMed] [Google Scholar]
Sattler 2012
- Sattler G, Carruthers A, Carruthers J, Flynn TC, Geister TL, Gortelmeyer R, et al.Validated assessment scale for neck volume. Dermatologic Surgery 2012;38(2 Spec no):343-50. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Sundaram 2016
- Sundaram H, Signorini M, Liewn S, Trindade de Almeida AR, Wu Y, Braz AV, et al.Global aesthetics consensus: botulinum toxin type A—evidence-based review emerging concepts, and consensus recommendations for aesthetic use, including updates on complications. Plastic and Reconstructive Surgery 2016;137(3):518e-29e. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sveikata 2011
- Sveikata K, Balciuniene I, Tutkuviene J.Factors influencing face aging. Literature review. Stomatologija 2011;13(4):113-6. [MEDLINE: ] [PubMed] [Google Scholar]
References to other published versions of this review
Camargo 2014
- Camargo CP, Costa CS, Gemperli R, Tatini MD, Bulsara MK, Riera R.Botulinum toxin for facial wrinkles. Cochrane Database of Systematic Reviews 2014, Issue 9. Art. No: CD011301. [DOI: 10.1002/14651858.CD011301] [DOI] [PMC free article] [PubMed] [Google Scholar]