

ABSTRACT
Methicillin-resistant Staphylococcus sciuri (MRSS) strain C2865 from a stranded dog in Nigeria was trimethoprim (TMP) resistant but lacked formerly described staphylococcal TMP-resistant dihydrofolate reductase genes (dfr). Whole-genome sequencing, comparative genomics, and pan-genome analyses were pursued to unveil the molecular bases for TMP resistance via resistome and mobilome profiling. MRSS C2865 comprised a species subcluster and positioned just above the intraspecies boundary. Lack of species host tropism was observed. S. sciuri exhibited an open pan-genome, while MRSS C2865 harbored the highest number of unique genes (75% associated with mobilome). Within this fraction, we discovered a transferable TMP resistance gene, named dfrE, which confers high-level TMP resistance in Staphylococcus aureus and Escherichia coli. dfrE was located in a novel multidrug resistance mosaic plasmid (pUR2865-34) encompassing adaptive, mobilization, and segregational stability traits. dfrE was formerly denoted as dfr_like in Exiguobacterium spp. from fish farm sediment in China but escaped identification in one macrococcal and diverse staphylococcal genomes in different Asian countries. dfrE shares the highest identity with dfr of soil-related Paenibacillus anaericanus (68%). Data analysis discloses that dfrE has emerged from a single ancestor and places S. sciuri as a plausible donor. C2865 unique fraction additionally enclosed novel chromosomal mobile islands, including a multidrug-resistant pseudo-SCCmec cassette, three apparently functional prophages (Siphoviridae), and an SaPI4-related staphylococcal pathogenicity island. Since dfrE seems not yet common in staphylococcal clinical specimens, our data promote early surveillance and enable molecular diagnosis. We evidence the genome plasticity of S. sciuri and highlight its role as a resourceful reservoir for adaptive traits.
IMPORTANCE The discovery and surveillance of antimicrobial resistance genes (AMRG) and their mobilization platforms are critical to understand the evolution of bacterial resistance and to restrain further expansion. Limited genomic data are available on Staphylococcus sciuri; regardless, it is considered a reservoir for critical AMRG and mobile elements. We uncover a transferable staphylococcal TMP resistance gene, named dfrE, in a novel mosaic plasmid harboring additional resistance, adaptive, and self-stabilization features. dfrE is present but evaded detection in diverse species from varied sources geographically distant. Our analyses evidence that the dfrE-carrying element has emerged from a single ancestor and position S. sciuri as the donor species for dfrE spread. We also identify novel mobilizable chromosomal islands encompassing AMRG and three unrelated prophages. We prove high intraspecies heterogenicity and genome plasticity for S. sciuri. This work highlights the importance of genome-wide ecological studies to facilitate identification, characterization, and evolution routes of bacteria adaptive features.
KEYWORDS: Staphylococcus sciuri, adaptation, comparative genomics, dfrE, dihydrofolate reductase, methicillin-resistant coagulase-negative staphylococci, mobile genetic elements, multidrug resistance, plasmid, trimethoprim, SCCmec, prophage, PICI, S. sciuri subspecies, intraspecies diversity, evolution, reservoir
INTRODUCTION
Staphylococcus spp. are ubiquitous bacteria present in diverse ecological niches. They are opportunistic pathogens responsible for mild to life-threatening infections (1). Coagulase-negative staphylococci (CoNS), the major group within the genus, now represent one of the major nosocomial pathogens (2). Within this cluster, the Staphylococcus sciuri species group includes five species that are most often present as commensal animal-associated bacteria (3). The ubiquitous presence of S. sciuri represents a continuous source for contamination, colonization, and infection in animals and humans from different niches, including dust and hospital surfaces (4–12). S. sciuri is a natural reservoir of the ancestral β-lactam resistance mecA gene, and it is considered a source for dissemination of mecA via horizontal gene transfer (HGT) by the staphylococcal cassette chromosome (SCCmec) element to other staphylococcal species (13–15). S. sciuri is frequently multidrug resistant (MDR), and novel clinically relevant antimicrobial resistance (AMR) genes have been first detected in this species. This includes the multidrug resistance (PhLOPS phenotype) cfr gene (16), the macrolide/lincosamide/streptogramin B (MLSB) resistance erm(33) (17), the lincosamide/streptogramin A resistance sal(A) (18), the oxazolidinone/phenicol resistance optrA (19), or the coexistence of plasmid-located cfr-optrA (19) and the β-lactam resistance mecA-mecC in hybrid SCCmec elements (20, 21). Subsequently, early discovery of novel AMR genes in this species appears critical to constrain their further expansion into pathogens.
Mobile genetic elements (MGEs) play a key role in intra- and interspecies HGT of AMR and virulence determinants. In S. sciuri, their “mobilome” (pool of genes within MGEs) and diversity remain largely unknown. Particularly, staphylococcal phages are considered ubiquitous in this genus and constitute major contributors to genome modulation and plasticity (22). Yet, phages of S. sciuri have been reported only twice (23, 24). Former reports indicate that staphylococcal phages also contribute to the spread of AMR elements (25–29), including mobilization of the SCCmec (24, 27, 30, 31).
In staphylococci, resistance to trimethoprim (TMP) is mediated by any of the following acquired dihydrofolate reductases (Dfrs): DfrA (DfrS1), DfrD, DfrG, DfrK, and DfrF (32–37). These enzymes are TMP-insensitive variants of the intrinsic dihydrofolate reductase(s) (Dhfr), which converts dihydrofolate into tetrahydrofolate, essential in the synthesis of nucleic acids precursors. In this study, we identified and characterized the TMP resistance dfrE gene in canine MDR S. sciuri C2865 and determined its location in a novel MDR-mobilizable plasmid. We noticed that dfrE is already present in several species from varied sources geographically distant. Our analyses evidence that the dfrE-carrying element has emerged from a single ancestor and position S. sciuri as the donor species. We further identified C2865 complete “resistome” (pool of AMR determinants) and mobilome, which included novel chromosomal and extrachromosomal elements. Finally, comparative genomics of S. sciuri species revealed high intraspecies heterogenicity and high genome plasticity for methicillin-resistant S. sciuri (MRSS) C2865.
RESULTS
Sequencing approach reasoning and general characteristics of S. sciuri C2865 genome.
MRSS C2865 was chosen for whole-genome sequencing (WGS) as it harbored the highest number of detected AMR genes. A summary of MRSS C2865 sequencing and assembly data is shown in Table 1. Illumina assembly retrieved 341 contigs with a contig sum of 2,937,715 bp. Plasmids pUR2865-1 (2,559 bp) and pUR2865-2 (3,830 bp) were identified as single circularized contigs based on contig boundary redundancy. Several fragmented mobile elements were identified; however, multiple attempts to determine their entire structure failed. PacBio sequencing retrieved one single circular chromosomal contig of 2,913,767 bp plus one circularized 40,108-bp plasmid (pUR2865-34), making a sum of 2,953,875 bp. Dot plot analysis of Illumina versus PacBio assemblies is included in the supplemental material and Fig. S1. PacBio data were used for all annotations and downstream analyses, except for both small Illumina-sequenced plasmids.
TABLE 1.
Sequencing and assembly data comparison of the S. sciuri C2865 genome processed with Illumina Miseq and with PacBio RSII
| Data type | Parameter | Illumina MiSeq | PacBio RSII |
|---|---|---|---|
| Sequencing | No. of bp | 662,993,478 | 2,162,733,205 |
| No. of reads | 2,774,428 | 103,918 | |
| Mean read length | 239 | 20,811 | |
| Avg genome coverage | 225× | 730× | |
| Assembly | Total assembled sequence (bp) | 2,937,715 | 2,953,875 |
| Total assembled contigs | 341 | 2 | |
| Mean contig size | 8,615 | 1,488,849 | |
| Maximum contig length | 125,700 | 2,913,767 | |
| Length of chromosome sequence (bp) | 2,893,295 | 2,913,767 | |
| G+C content (%) | 32.7 | 32.5 | |
| N50 contig length | 37,104 | 2,913,767 | |
| ORFs | 3,270 | 3,097 | |
| Gene density (no genes/kb) | 1.13 | 1.06 | |
| Coding (%) | 91% | 89% | |
| Median intergenic spacer (bp) | 51 | 51 | |
| Protein-coding genes | 3,193 | 3,020 | |
| Ribosomal RNAs: 16S, 23S, 5S | 19 | 19 | |
| Transfer RNAs | 58 | 58 | |
| Mobile elements | Preidentified insertion sequences | 22 (15 different) | 78 (18 different) |
| Prophage | ∼2–3 | 3 | |
| SCCmec | 1 | 1 | |
| Staphylococcal pathogenicity island | 1 | 1 | |
| Plasmida | ∼4 | 1 |
Consensus (Illumina + PacBio data) = 3 plasmids. All other definitive values in text were taken from PacBio data.
Dot-plot graphs of MRSS C2865 genome assembled of PacBio and of Illumina reads using D-Genies. The program searches all the query sequences aligned on the diagonal and calculates the target sequence coverage per identity bin (79). Left graph shows the genome-genome alignments using PacBio assembly as reference (chromosomal contig and plasmid contig) (x axes) with Illumina query (y axes), while right graph depicts the genome-genome alignments using Illumina-assembled contigs as reference (x axes) with PacBio query (y axes). The summary identity calculation (>75% identity, <75%, <50%, and <25% and no matches) is made on the target sequence (reference) and represents the percentage of the target genome in base pairs. Note: node 96 (5,513 bp) denotes plausible contamination after contig manual checking. Download FIG S1, EPS file, 0.5 MB (495.4KB, eps) .
Copyright © 2021 Gómez-Sanz et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
S. sciuri exhibits an open pan-genome while MRSS C2865 shows the highest unique genome content and constitutes a subgroup within the species.
Pan-genome analysis of 21 S. sciuri species genomes, which originated from human, animal, food, or environmental samples, including clinical isolates, revealed a pan-genome of 5,721 genes, comprising a core genome of 1.3 Mb with 1,547 shared proteins at 95% of identity (27% of genes) and a flexible genome of 4,174 (73%). The pan-genome curve did not level off, as the addition of each new genome increased the total gene pool. Instead, the core genome appeared to have reached a plateau (<1,600 genes) (Fig. 1A). This ability to acquire exogenous DNA indicates an open pan-genome for S. sciuri (Fig. 1B). This is further supported by a prevalence of “cloud genes” (genes found in up to 15% of the strains), which corresponds with approximately 43.2% of the pan-genome (Fig. 1A). S. sciuri C2865 harbored the highest number of genes (3,063) and the highest number of unique genes (521), whereas the average gene numbers were 2,707 and 95, respectively, considering the 21 strains (Fig. 1C). This was not correlated with the genome size or overall genome coding density (89%, median 89); however, MRSS C2865 encompassed the shortest median intergenic distance (Fig. 1C), mainly included within the flexible unique gene set (Table 2). Several novel chromosomal and extrachromosomal elements were detected (see below), which represented 75% of C2865 unique genes (390/521). Three plasmids were identified within the unique fraction: two small single resistance plasmids (pUR2865-1 and pUR2865-2) (Fig. S4, plus Text S1 for details) and a novel MDR plasmid, pUR2865-34 (see section below). Table 2 summarizes the most relevant characteristics of the MGEs identified.
FIG 1.

Pan-genome analyses for S. sciuri genomes at the species level (n = 21, 95% identity). (A) Pie chart showing the proportions of coding DNA sequence (CDS) in the core, soft core, shell, and cloud genomes. The parameters were defined as follows. Core genes: ≥99% of analyzed genomes, accessory genes: 1 to 99% (soft core 95 to 99%; shell 15 to 95%; cloud ≤15%). (B) Number of core genes (green) and total number of genes (pan genome) (red) curve for 21 S. sciuri strains. The upper and lower edges of the boxes indicate the first quartile (25th percentile of the data) and third quartile (75th percentile), respectively, of 1,000 random different input orders of the genomes. The central horizontal line indicates the sample median (50th percentile). (C) Bar chart of the total number of genes per genome indicating the number of non-unique and unique genes per genome (gray and dark blue, respectively) ordered by the genome size. Secondary axis displays the median intergenic distance per genome in base pairs.
TABLE 2.
General characteristics of most relevant S. sciuri C2865 mobile genetic elements detected as consensus of sequencing data
| Parameter | Value for: |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| pUR2865-1 | pUR2865-2 | pUR2865-34 | SCCmecC2865 | C2865-pp1 | C2865-pp2 | C2865-pp3 | SscPIC2865 | ψTn554 | |
| Contig coverage (compared to chromosome) | 1.52 | 5.66 | 3.48 | NAb | NA | NA | NA | NA | NA |
| Length of sequence (bp) | 2,559 | 3,830 | 40,108 | 55,137 | 41,284 | 45,020 | 126,192 | 9,645 | 7,306 |
| G+C content (%) | 31.3 | 29.0 | 30.1 | 31.5 | 34.6 | 33.8 | 30.8 | 30.1 | 35.3 |
| Protein-coding genes | 2 | 3 | 41 | 61 | 62 | 64 | 166 | 21 | 6 |
| Gene density (no. genes/kb) | 0.78 | 0.78 | 1.02 | 1.11 | 1.50 | 1.42 | 1.32 | 2.18 | 0.82 |
| Coding density (%) | 67 | 89 | 78 | 85 | 95 | 94 | 89 | 87 | 96 |
| Median intergenic spacer (size, bp) | 851 | 203 | 85 | 53 | 9.5 | 11 | 25 | 55 | 6 |
| Replication | RepC | RepC | RepA_N | Primase | |||||
| Recombinasea (no.) | Res/Rec | Res/Rec (3) | LSR, Res/Rec | LSR | LSR | LSR, Y-Int/Rec (2) | Y-Int/Rec | Y-Int/Rec (2) | |
| Resistance gene(s) | lnu(A) | cat pC221 | erm(B), aacA-aphD, dfr_like, tet(K) | mecA, tet(S), aadE, arsC, arsB, copB, arsAD | cadC, cadA, cadD | ||||
| Other genes of interest | mob | ica-locus variant | abiF | hicA-hicB, ardA | mazF | ||||
| Insertion sequences (family) | 5 (IS6) | 4 (IS6) | 1 (IS110) | ||||||
| Transfer RNAs (aa) | 1 (Pro, TGG) | ||||||||
Res/Rec, serine recombinase (S-rec) of the resolvase family; LSR, large serine recombinase; Y-Int/Rec, tyrosine recombinase; aa, amino acids.
NA, not applied.
This file includes a detailed description of methods, further methods not indicated in the main text, additional results, and an extended discussion. Download Text S1, PDF file, 0.5 MB (479KB, pdf) .
Copyright © 2021 Gómez-Sanz et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Graphical representation of the RCR plasmids detected in MRSS C2865 and related plasmids. Arrows denote the genes, length, and orientation. Gene colors other than gray represent the following functions (color): replication and/or mobilization (green), antimicrobial resistance (pink, yellow). Areas of nucleotide similarity plasmids are indicated in gray. (A) Nucleotide sequence comparative analysis of the three RCR plasmids detected in MRSS C2865: pUR2865-1, pUR2865-2, and pUR2865-int, which is integrated in the larger pUR2865-34 plasmid. (B) Nucleotide sequence comparative analysis of pC194-related pUR2865-1 and S. aureus pC194 (GenBank accession no. NC_002013). Estimated double-strand origin (dso) and single-strand origin (sso) of replication are indicated. (C) Nucleotide sequence comparative analysis of the pT181-related pUR2865-2, the integrated pUR2865-int, and the RCR family prototype S. aureus pT181 (GenBank accession no. J01764.1). Putative dso nick and sso sites for pUR2865-1 and pUR2865-2 are denoted (100). Origin-of-transfer (oriT) for pT181-related plasmids is also indicated. (D) Sequence comparison of closest plasmids to pUR2865-1 (> 80% ID, > 70% coverage). (E) Sequence comparison of plasmids closest to pUR2865-2 (> 80% ID, > 70% coverage). Download FIG S4, SVG file, 0.5 MB (387.7KB, svg) .
{kind=link}
Copyright © 2021 Gómez-Sanz et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
A refined S. sciuri phylogenomic tree revealed that MRSS C2865 formed a separate subgroup, together with MDR S. sciuri Z8, isolated from a human skin wound infection in China (38), MDR S. sciuri SNUDS-18, from a duckling with tremor in South Korea (39), and S. sciuri CCUG39509, from a sliced veal leg in Sweden (Fig. 2A). To obtain the most accurate phylogeny possible, we removed from the alignment all regions affected by recombination (see Materials and Methods). Average nucleotide identity (ANI) analysis evidenced that these four genomes shared ≥98% identity, while they displayed an ANI of 96% with respect to the major species cluster, positioning just above the species demarcation threshold (>95%) (Fig. S2). Genomic comparison of MRSS C2865 chromosome against its three closest relatives (Fig. 2C) evidenced the MRSS C2865 unique chromosomal islands. They corresponded to novel site-specific recombinase carrying MGEs (SCCmec, three prophages, staphylococcal pathogenicity island, transposon ψTn554) described in the following sections or in the supplemental material (for ψTn554, see also Fig. S3).
FIG 2.
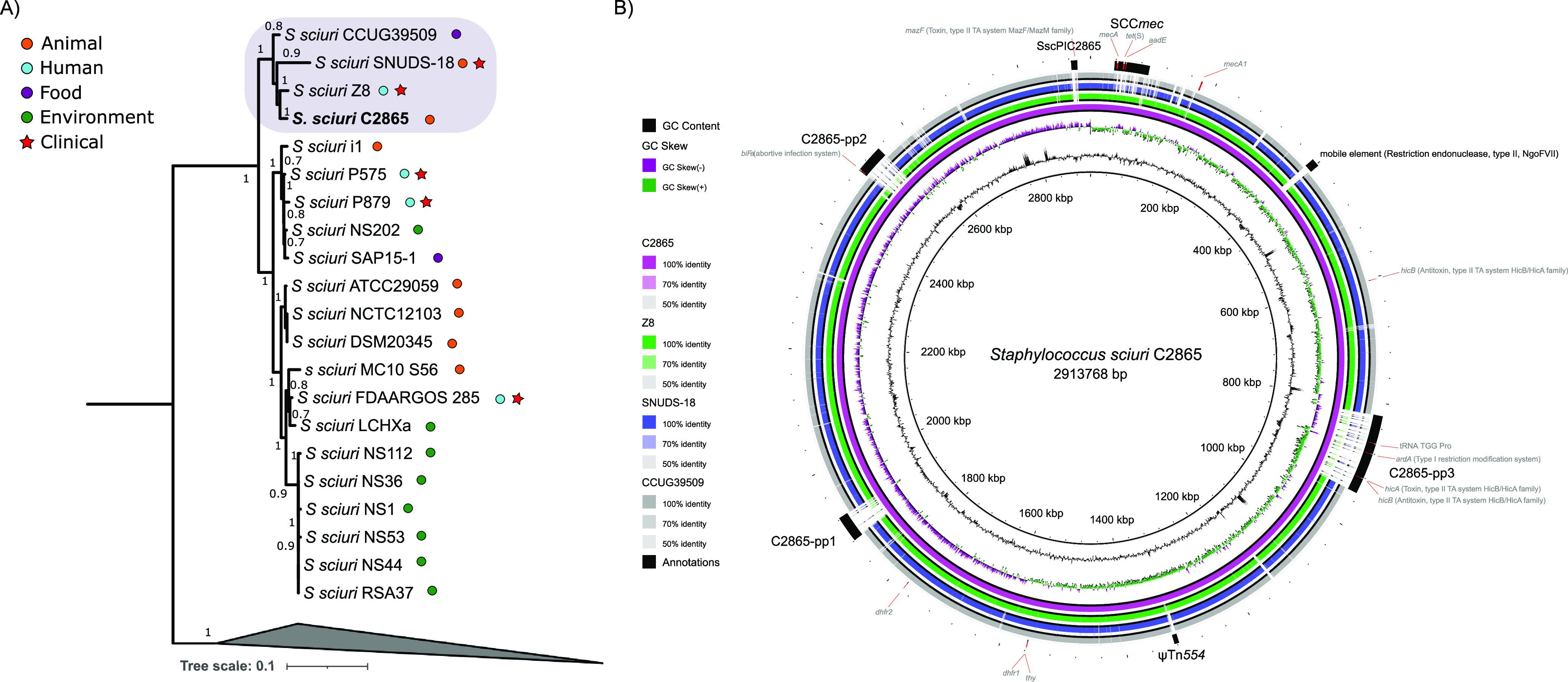
Phylogenomic analysis of S. sciuri and comparative diagram of MRSS C2865 closest relatives. (A) Maximum likelihood phylogenomic tree of S. sciuri C2865 and all 29 Staphylococcus sciuri group genomes deposited in the NCBI database (accessed until April 2018). Phylogenomic view of S. sciuri group species other than S. sciuri is collapsed as an outgroup to facilitate relatedness analysis of S. sciuri strains. Sample source is indicated as human, animal, food, and environment as well as whether the sample was isolated from a clinical infection. Note: strains S. sciuri DSM 20345 and S. sciuri NCTC12103 correspond to the same type strain (also known as ATCC 29062). (B) Comparative diagram of S. sciuri C2865 (pink ring) (reference genome) against its three closest genomes (S. sciuri Z8 [green ring], S. sciuri SNUD18 [blue], and S. sciuri CCUG39509 [gray]) as a set of concentric rings, where color indicates a BLAST match using BLAST Ring Image Generator (BRIG) (83). GC content and GC skew of reference genome is also displayed. Several features of interest are depicted, highlighting the unique presence of remarkable mobile genetic elements (SCCmec, C2865-pp3, ψTn554, C2865-pp1, C2865-pp2, SscPIC2865) in S. sciuri C2865. In addition, the genome location of the methicillin-susceptible mecA1 gene, two intrinsic dhfr genes (dhfr1 and dhfr2, the former next to a thy gene), a bacterial chromosomal hicA-antitoxin hicB (type II TA system), and the most relevant adaptive genes from the novel chromosomally located MGEs are displayed.
Phylogenomic tree and heat map resultant from the average nucleotide identity (ANI) of 30 Staphylococcus sciuri group genomes, plus one Staphylococcus aureus and one Staphylococcus epidermidis reference genome used as outgroup for the genus level. Download FIG S2, EPS file, 0.3 MB (261.9KB, eps) .
Copyright © 2021 Gómez-Sanz et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Graphical overview of MRSS C2865 ψTn554 chromosomal region and comparative elements. The diagram depicts (i) MRSS C2865 ψTn554 chromosomal region including the interrupted radC gene, (ii) its corresponding closest relative S. aureus 6850 (GenBank accession no. CP006706), and (iii) the chromosomal radC region and remnant 3′-end radC of phenicol and oxazolidinone resistant S. sciuri wo22_7 (KX982170) and S. sciuri MS11-3 (KX447571), respectively. Arrows denote the genes, length, and orientation. Gene colors other than gray represent the following functions (color): truncated or remnant chromosomal integration radC in Tn554 family transposons (black), antimicrobial resistance (pink), metal resistance or transport (bright blue), transposition or recombination (yellow), ABC transporter ATP-binding proteins (faint brown). The 6-bp nucleotides resultant from Tn554-like integration are also depicted. Areas of nucleotide similarity (nblastn, >100 bp match, >85% identity) between strains/structures are indicated in gray. Download FIG S3, EPS file, 0.2 MB (196.3KB, eps) .
Copyright © 2021 Gómez-Sanz et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Mosaic mobile adaptive elements within the novel plasmid pUR2865-34.
Plasmid pUR2865-34 was 40,108 bp in size and harbored 41 coding sequences (CDSs) (Fig. 3A). It showed a mosaic IS6-like-delimited modular organization (Fig. 3A), encompassing novel adaptive and backbone modules. pUR2865-34 cargo region enclosed a copy of the tetracycline efflux major facilitator superfamily (MFS) transporter gene tet(K), located within the small rolling-circle replication (RCR)-integrated plasmid, designated pUR2865-int. Sequence identity of RepC (83.1%) and composition of the double- (nick site: 5′-AAAACCGGaTACTCT/AATAGCCGGTT-3′, where capital letters denote conserved bases with respect to pT181, and slash the site of cleavage) and single-strand origin of replication (dso and sso, respectively), typical of RCR plasmids, classified this plasmid as a member of the pT181 family (Fig. 3A; Fig. S4). In addition, pUR2865-34 harbored a macrolide/lincosamide/streptogramin B resistance gene erm(B), coding for a 23S rRNA adenine N-6-methyltransferase and an aminoglycoside modifying aacA-aphD gene, encoding the bifunctional enzyme 6′-aminoglycoside N-acetyltransferase AAC(6′)-Ie aminoglycoside O-phosphotransferase APH(2″)-Ia, located immediately downstream of an IS257/IS1216E copy in the same orientation.
FIG 3.

Comparative analysis of novel pUR2865-34 with the closest sequences in NCBI. Arrows denote the genes, length, and orientation. Gene colors other than gray represent the following genes of interest (color): antimicrobial resistance genes (pink), genes involve in metal resistance or transport (bright blue), intercellular adhesion gene cluster (ica) genes (navy blue), genes involved in transcription regulation (bright green), plasmid backbone genes (dark green), genes involved in transposition or recombination (yellow), and insertion sequences with defined imperfect inverted repeats (boxed and black). Areas of nucleotide similarity (nblastn, >100 bp match, >80% identity) between strains/structures are indicated in gray. For plasmids pSA-01, pAFS11, and S. sciuri FDAARGOS plasmid unnamed, only the area of interest is represented. (A) S. sciuri C2865 pUR2865-34 underlining its modular organization and indicating strain/plasmid sequences detected in NCBI with ≥50% coverage and ≥95% identity carrying those modules. The novel predicted secondary structure of the origin of conjugative transfer mimic (oriT mimic) is depicted, indicating the free energy of the DNA hairpins formation (ΔG). (B) Truncated dfrE-carrying transposon and immediate up- and downstream regions of S. sciuri C2865 pUR2865-34 and comparison with different dfrE-enclosing regions of Exiguobacterium sp. S3-2 pMC1 and pMC2, as well as S. sciuri GN5-1 pSS-04. Black boxes above the graphical display represent the Tn3-like characteristic 38-bp inverted repeats detected, involved in excision and integration of Tn3 related elements. Region conserved in all dfrE-carrying elements except for those graphically represented is underlined in pUR2865-34 segment. (C) ica-locus variant-carrying region in S. sciuri C2865 pUR2865-34 and closest relatives deposited in NCBI database: S. aureus strain 11 pAFS11 and S. sciuri FDAARGOS plasmid. (D) Graphical comparison of pUR2865-34 and closest plasmids in the NCBI. Represented samples correspond to S. arlettae strain SA-01 plasmid pSA-01 and S. sciuri strain wo19-3e plasmid.
Three serine recombinase (S-rec) genes of the resolvase/invertase subfamily (Text S1), named res/rec1, res/rec2, and res/rec3, were identified. Res/Rec2 and Res/Rec3 shared the highest amino acid identity to few staphylococcal isolates (four S. aureus, two S. sciuri, and one Staphylococcus arlettae strains) (>93% identity), while most entries corresponded to resolvases found in other Gram-positive bacteria (Macrococcus caseolyticus, Exiguobacterium sp., Bacillus sp., Enterococcus sp., Lactococcus sp., Streptococcus sp., Lysinibacillus sp., Paenibacillus sp., Clostridium sp.). A dfr gene, designated dfrE, and a thymidylate synthase gene (thy) were detected immediately downstream of res/rec3. DfrE shared 31.5 and 15.5% identity to two additional intrinsic Dhfr detected in S. sciuri C2865 genome, both located in conserved chromosomal regions lacking insertion sequences (ISs) or other mobile elements. DfrE was present in a limited number of strains at 100% sequence identity (Table 3). The dfrE-carrying strains corresponded to all the staphylococcal isolates harboring the res/rec3 plus M. caseolyticus strain JCSC5402 and Exiguobacterium sp. strain S3-2, the latter harboring the dfrE gene in two different coexisting plasmids (Table 3). All dfrE-carrying staphylococcal genomes corresponded to WGS projects with direct submission to NCBI. In all these cases, the dfrE gene was unnoticed and included within MDR plasmids or plasmid-associated elements hosting additional AMR genes (Table 3). Of note, all strains harboring the overlooked dfrE were reported in Asian countries from animal-related or clinical human samples (Table 3). Only Yang et al. (40) denoted an identical gene, designated “dfr_like,” in Exiguobacterium sp. S3-2 from a fish farm sediment in China and proved its ability to confer TMP resistance in Gram-negative Escherichia coli DH5α. Hence, the role of this gene, here renamed dfrE (for Exiguobacterium spp), in TMP resistance in staphylococci remained open. DfrE revealed phylogenetically distant from the intrinsic Dhfr (Fig. S5) and was closest to the Dhfr of soil-related Paenibacillus anaericanus (68% identity) (Fig. 4). Within the staphylococcal TMP resistance genes, DfrE shared closer phylogenetic identity with TMP resistance DfrF, typical of enterococci and streptococci (Fig. S5).
TABLE 3.
General features of dfrE-carrying strains deposited in the NCBI database and genetic platforms containing dfrE
| Bacterial species | Location (plasmid ID) | Plasmid size (bp) | Tn3 family element | Coverage/ID dfrE regionb | Additional resistance pattern in dfrE-carrying element | Source | Country | TMP phenotype (μg/ml) | Reference | NCBI acc. no. |
|---|---|---|---|---|---|---|---|---|---|---|
| Staphylococcus sciuri C2865 | Plasmid (pUR2865-34) | 41,108 | 3′-end region | Reference | erm(B), aacA-aphD, tet(K) | Dog | Nigeria | MIC ≥ 4,096 | This study | SAMN16182282 (PRJNA663854) |
| Exiguobacterium sp. S3-2 | Plasmid (pMC2) | 19,981 | Complete + IRsa | 47/99.95 | Fish farm sediment | China | MIC > 1,024 in E. coli | 40 | KF648875 | |
| Plasmid (pMC1) | 71,276 | 3′-end region | 46/99.85 | aadE, mefA, fexA, mph_like, mph(B), tet(L) | Fish farm sediment | China | MIC > 1,024 in E. coli | 40 | KF648874 | |
| Macrococcus caseolyticus JCSC5402 | Plasmid (pMCCL2) | 80,545 | 3′-end region | 100/100 | erm(B), aacA-aphD, mec(B) | Chicken | Japan | Not tested | 65 | AP009486 |
| Staphylococcus sciuri GN5-1 | Plasmid (pSS-04) | 18,496 (partial) | 3′-end region | 73/99.96 | erm(B), aacA-aphD, fexA, cfr | Swine | China | Not indicated | Direct submission (2016) | KF129410 |
| Staphylococcus sciuri wo19-3e | Genomic sequence (plasmid) | 38,241 | 3′-end region | 100/99.98 | aacA-aphD, tetK, aadD, ble, cfr, copB-mco, arsB | Swine | China | Not indicated | Direct submission (2017) | KX982172 |
| Staphylococcus aureus NTUH_3874 | Plasmid (pNTUH_3874) | 14,566 | 3′-end region | 99/99.98 | erm(B), aacA-aphD | Human blood | Taiwan | Not indicated | Direct submission (2016) | LC102479 |
| Staphylococcus aureus GD1677 | Chromosome (integrated plasmid) | 3′-end region | 99/100 | erm(B), aacA-aphD, aadE, cad | Human | China | Not indicated | Direct submission (2017) | CP019595 | |
| Staphylococcus aureus FORC_039 | Plasmid | 35,415 | 3′-end region | 99/99.98 | aacA-aphD, blaZ, cadAC operon | Food | South Korea | Not indicated | Direct submission (2017) | CP015818 |
| Staphylococcus aureus FORC59 | Plasmid (pFORC59) | 35,269 | 3′-end region | 99/99.11 | erm(B), aacA-aphD, blaZ operon, cadX | Human blood | South Korea | Not indicated | Direct submission (2017) | CP020355 |
| Staphylococcus arlettae SA-01 | Plasmid (pSA-01) | 63,558 | 3′-end region | 99/99.96 | erm(B), aacA-aphD, cfr, erm(C), tet(L), erm(T), aadD, fosD, fexB, ars | Chicken | China | Not indicated | Direct submission (2017) | KX274135 |
Exiguobacterium sp. S3-2 pMC2 harbors a primordial complete dfrE-carrying Tn3 family element, flanked by two 38-bp imperfect inverted repeats (IRs) characteristic of transposases of the Tn3 family, involved in excision and integration of the element.
Coverage/ID in percentage of the remnant Tn-like dfrE-carrying element, which was defined as the region covering the IS1216E-res/rec2-res/rec3-dfrE-thy genes plus the 3′-end 38-bp IR characteristic of Tn3 family elements (4,345 bp), using MRSS C2865 as reference.
FIG 4.

Phylogenetic network of aligned amino acid sequences of the dihydrofolate reductases (Dfr) closest to DfrE. The diagram illustrates all Dfr proteins derived from NCBI NR hits with a percentage of identity of >50% to DfrE of S. sciuri strain C2865 (bold), plus all trimethoprim resistance Dfrs described so far in staphylococci (DfrA, DfrD, DfrG, DfrK, DfrF) (dark red) (32–36). Identical Dfr proteins present in different bacterial classes were labeled with the identity of one representative per class (i.e., Firmicutes: Streptococcus suis BM407 and Coprobacillus sp. AF18-40). The amino acid branch clustering the trimethoprim resistance DfrE is highlighted with a faint green background.
Neighbor-joining tree of aligned amino acid sequences of S. sciuri group dihydrofolate reductases. The alignment was performed with (i) the entire dihydrofolate reductase (Dhfr) protein(s) present in all S. sciuri species group genomes deposited in the NCBI database (accessed until April 2018), (ii) both Dhfrs present in S. sciuri strain C2865, and (iii) the amino acid sequence of all trimethoprim resistance dihydrofolate reductases (Dfr) described so far in staphylococci (DfrA, DfrD, DfrG, DfrK, DfrF) (32–36), as well as (iv) the Dhfr of trimethoprim-susceptible S. epidermidis ATCC 12228 (NC_004461) and S. aureus ATCC 25923 (Z16422) as reference, in blue color. Clusters enclosing the different TMP resistance Dfrs are colored differently. Download FIG S5, EPS file, 0.2 MB (165.3KB, eps) .
Copyright © 2021 Gómez-Sanz et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The dfrE-carrying region suggested a transposon-like structure encompassing an IS1216E copy, the res/rec2 and res/rec3 resolvase genes, and the dfrE and thy genes (4,345 bp) (Fig. 3B). This element seems to be a truncated version of the original dfr_like-carrying transposon (Tnp) of the Tn3 family detected in plasmid pMC2 of Exiguobacterium sp. S3-2 (40), as only the 38-bp imperfect inverted repeats (IRs) downstream of thy were present (Fig. 3B). Here, the pMC2-carrying Tn3-like tnpA gene, including the 38-bp upstream flanking IR, has been replaced by res/rec2. This Tn3-3′-end truncated region (IS1216E-res/rec2-res/rec3-dfrE-thy-IR) was conserved in all dfrE-enclosing strains except Staphylococcus sciuri GN5-1, which lacks the res/rec2 plus immediate downstream coding sequences (CDSs) before the IS1216E (Table 3; Fig. 3B).
Plasmid pUR2865-34 harbored an additional IS257-flanked module, which included a variant of the intercellular adhesion gene cluster (icaADBC) (Fig. 3C). This operon comprised the icaA, icaD, icaB, and icaC genes and the adjacent ica-locus repressor icaR gene (41). The ica-locus is involved in the early steps of biofilm formation (intercellular adhesion and cell agglutination) in staphylococci and appeared functional according to the CRAmod assay (Text S1; Fig. S6). This gene cluster shared the highest identity (97.9%) to the ica-locus variant of S. aureus pAFS11, an apramycin resistance apmA-carrying plasmid (GenBank accession no. FN806789.3) (Fig. 3C), followed by the icaADBC cluster present in S. sciuri FDAARGOS_285 chromosome and respective plasmid (CP022046.2 and CP022047.2, respectively).
Biofilm formation ability of ica-locus variant-carrying S. sciuri strains and transformants. (A) Biofilm formation capacity based on visual analysis of colony colors by a modified Congo red agar (CRAmod) assay. Spots were plated in duplicates. From left to right, (1) the positive controls (strong biofilm formers) S. aureus SA113 (DSM 4910) and S. aureus ATCC 25923 (DSM 1104), (2) negative-control (pUR2865-34 recipient) strain S. aureus RN4220, (3 and 4) the original S. sciuri ica-locus variant-carrying strains C2865, C2853, C2854, and C2855, (5) two selected S. aureus RN4220/pUR2865-34 transformants (S319 and S320). (B) Quantification of biofilm formation by crystal violet (CV) staining of adherent cells. The bar chart displays the results of six independent experiments in triplicates. Original S. sciuri ica-locus variant-carrying strains C2865, C2853, C2854, and C2855, two selected S. aureus RN4220/pUR2865-34 transformants (S319 and S320) as well as an ica-negative non-biofilm-producing methicillin-resistant Staphylococcus lentus strain (C3030) (101) are included. The three bars on the left show the control strains used: the strong biofilm formers S. aureus SA113 (DSM 4910) and S. aureus ATCC 25923 (DSM 1104), as well as S. aureus RN4220 strain. Significant differences by t test or analysis of variance (ANOVA; P < 0.01) are indicated with two stars on the compared cluster, i.e., ica-carrying strains (C2865, C2853, C2854, C2855, S319, S320; ns, not significant differences between values) and C3030, and ica-carrying strains and RN4220. Download FIG S6, EPS file, 0.7 MB (747.2KB, eps) .
Copyright © 2021 Gómez-Sanz et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The pUR2865-34 backbone region contains a type Ib partitioning system and a repA_N replication initiation gene (repA), characteristic of theta-replicating plasmids (42). It also harbors a single relaxase gene (mob) with an origin-of-transfer sequence (oriT) located immediately upstream as part of the integrated pUR2865-int (Fig. 3). In addition, an oriT mimic sequence (positions 6,077 to 6,132) similar to one recently described by Bukowski et al. (43) was observed proximal to the replication and partitioning genes (Fig. 3A). It is important to note that although pUR2865-34 does not carry any conjugative genes, these oriT regions can mediate the transfer of plasmid-carried phenotypes, such as resistance to trimethoprim, in the presence of conjugative elements.
MDR plasmids pSA-01 from S. arlettae strain SA-01 and pW019-3e from S. sciuri strain W019-3e resulted in the closest relatives to mosaic pUR2865-34 (Fig. 3D). MRSS C2853, C2854, and C2855 harbored a similar pUR2865-34 plasmid and enclosed the dfrE and ica-gene cluster (see Text S1 for details).
The novel dfrE gene confers high-level resistance to trimethoprim.
All S. aureus RN4220 transformants carrying the different types of dfrE-carrying constructs exhibited a 2,048-fold increase in TMP resistance with respect to the empty S. aureus RN4220, irrespective of the presence of the natural or constitutive promoter located upstream of dfrE (Table 4). These results report the activity of the dfrE in S. aureus for the first time in literature. Likewise, all E. coli dfrE-carrying DH5aα transformants exhibited a 2,046- or 4,096-fold increase in TMP resistance with respect to the control (Table 4). Lack of synergistic activity was observed when dfrE and thy were cloned together into E. coli DH5α and S. aureus RN4220. Two S. aureus RN4220 transformants carrying entire pUR2865-34, named S319 and S320, exhibited a 2,048-fold increase in TMP resistance with respect to control strains and displayed additional resistance to tetracycline, erythromycin, clindamycin, gentamicin, kanamycin, and tobramycin, as confirmed by disc-diffusion agar tests.
TABLE 4.
MIC values to trimethoprim (TMP) of original strains and respective DH5a and RN4220 constructs
| Species | Strain | Characteristics, origin, or description | Reference or source | Antimicrobial resistance gene(s) | MIC (μg/ml) to TMP |
|---|---|---|---|---|---|
| Escherichia coli | DH5α | Recipient strain for electroporation, plasmid free | Promega | 0.5 | |
| DH5α/pBUS1-HC | DH5α with S. aureus-E. coli shuttle vector pBUS1-HC | 96 | tet(L) | 0.5 | |
| DH5α/pBUS1-Pcap-HC | DH5α with S. aureus-E. coli shuttle vector pBUS1-Pcap-HC | 96 | tet(L) | 0.5 | |
| DH5α/1B-1 | DH5α/pBUS1-Pcap-HC/dfrE alone | This study | tet(L), dfrE | 2,048 | |
| DH5α/2A-1 | DH5α/pBUS1-HC/+dfrE+a | This study | tet(L), dfrE | 2,048 | |
| DH5α/4A-1 | DH5α/pBUS1-HC/+dfrE+thy + | This study | tet(L), dfrE | 1,024 | |
| Staphylococcus aureus | DSM 2569 | Reference strain for MIC (agar dilution method) | DSMZb | 1 | |
| RN4220 | Recipient strain for electroporation, plasmid free | DSMZ | 1 | ||
| RN4220/pBUS1-HC | RN4220 with S. aureus-E. coli shuttle vector pBUS1-HC | This study | tet(L) | 0.5 | |
| RN4220/pBUS1-Pcap-HC | RN4220 with S. aureus-E. coli shuttle vector pBUS1-Pcap-HC | This study | tet(L) | 0.5 | |
| RN4220/1B-1 | RN4220/pBUS1-Pcap-HC/dfrE alone | This study | tet(L), dfrE | 2,048 | |
| RN4220/2A-1 | RN4220/pBUS1-HC/+dfrE+ | This study | tet(L), dfrE | 2,048 | |
| RN4220/4A-1 | RN4220/pBUS1-HC/+dfrE+thy + | This study | tet(L), dfrE | 2,048 | |
| RN4220/pUR2865-34 | RN4220 with natural pUR2865-34 from S. sciuri C2865 | This study | tet(K), erm(B), aacA-aphD, dfrE | 2,048 | |
| Staphylococcus sciuri | C2865 | Groin of dog in Nigeria | 12 | mecA, tet(K), tet(M), tet(S)c, erm(B), lnu(A), aacA-aphD, ant6’, catpC221, dfrE | ≥4,096 |
| C2853 | Groin of dog in Nigeria | 12 | mecA, tet(K), erm(B), aacA-aphD, dfrE | ≥4,096 | |
| C2854 | Groin of dog in Nigeria | 12 | mecA, tet(K), tet(M), erm(B), aacA-aphD, catpC223, dfrE | ≥4,096 | |
| C2855 | Groin of dog in Nigeria | 12 | mecA, tet(K), tet(M), erm(B), lnu(A), aacA-aphD, catpC221, dfrE | ≥4,096 |
“+” represents dfrE immediate upstream or downstream region.
Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures.
The tetracycline resistance tet(S) was only detected after WGS in this study.
Novel SCCmecC2865 element lacking formerly described chromosomal cassette recombinases.
A novel MDR SCCmec cassette, denominated SCCmecC2865, was identified at the 3′-end region of 23S rRNA [pseudouridine(1915)-N(3)]-methyltransferase RlmH gene (rmlH). SCCmecC2865 was 55,137 bp in size and contained a class A mec gene complex (IS431-mecA-mecR1-mecI) (Fig. 5A). None of the so-far-described staphylococcal or macrococcal chromosome cassette recombinase genes (ccr), the resultant large serine recombinases (LSR) of which are responsible for excision and integration of the cassette, were detected. SCCmecC2865 was delimited at both ends by characteristic SCCmec-flanking direct repeats (DRs) with typical insertion site sequences (ISS). Characteristic imperfect inverted repeats (IRs), required for CcrAB or CcrC recognition of the att sites, were located at the internal boundaries of the complete cassette (Fig. 5A; Text. S1) (44). The SCCmecC2865 rlmH-proximal region (attR-attL1) carried a class A mec gene complex and one transposon-like structure consisting of one tetracycline resistance gene tet(S), coding for a translation elongation factor G (EF-G) involved in tetracycline ribosomal protection, flanked by two IS1216E copies in the same orientation. No DRs were present, which could evidence its mobilization as a composite transposon (Tn) or translocatable unit (Fig. 5A). A streptomycin resistance gene aadE, coding for a 6-aminoglycoside adenylyltransferase, was detected immediately downstream of the 3′-end IS1216E copy. In addition, an arsenic resistance operon (arsCBR) and a copper resistance gene (copB), coding for a copper-translocating P-type ATPase, were located (Fig. 5A). SCCmecTXG24 from S. sciuri TXG24 and SCCmecGVGS2 from S. sciuri GVGS2 were identified as closest relatives to SCCmecC2865, sharing 57% and 40% coverage, respectively (Fig. 5A) (20, 45).
FIG 5.

Graphical comparative analysis of the novel SCCmec and related large serine recombinases (LSR) detected in MRSS C2865. (A) Novel SCCmecC2865 (55,137 bp) and its closest elements in NCBI (SCCmecGVGS2-mecC of S. sciuri strain GVGS2 and SCCmecTXG24 of S. sciuri strain TXG24) (20, 45). Arrows denote the genes, length, and orientation. Gene colors other than gray represent the following genes of interest (color): antimicrobial resistance genes (pink), genes involved in metal resistance or transport (bright blue), genes involved in transcription regulation (bright green), genes involved in transposition or recombination (yellow), insertion sequences with defined imperfect inverted repeats (boxed and black), phage-related genes (red), and the SCCmec integration gene (dark red), which strictly does not belong to the SCCmec cassette, except for the rlmH 3′-end terminal 18-bp attachment site (ISS1 or attR 5′-GAAGCATATCATAAATGA-3′). The two perfect direct repeats found at both extremities of the cassette (attR 5′-GAAGCATATCATAAATGA-3′ and attL3 5′-GAAGCATATCATAAATGA-3′) are depicted. Two additional imperfect att sites (attL1 5′-GAAGCGTATCACAAATAA-3′ and attL2 5′-GAGCCATATAATAAATAA-3′) within SCCmecC2865 at base-pair positions 29,884 and 41,929 of the cassette, respectively, are also represented. Attachment sites detected in SCCmecGVGS2-mecC and SCCmecTXG24 are indicated. Unique bases with respect to attR, per SCCmec cassette, are represented in lower case. Areas of nucleotide similarity (nblastn, >100 bp match, >80% identity) between SCCmec cassettes are indicated in grayscale. (B) Phylogenetic network of aligned amino acid sequences of one representative staphylococcal chromosomal cassette recombinase (Ccr) per allotype described in Staphylococcus spp. and in Macrococcus spp. as well as the three LSRs present in S. sciuri C2865 genome comprising the Ccr consensus motif Y-[LIVAC]-R-[VA]-S-[ST]-x(2)-Q or Y-[LIVAC]-R-[VA]-S-[ST]-x(4)-Q. LSRs from S. sciuri C2865 originate from SCCmecC2865 (LSRSCCmec), prophage C2865-pp1 (LSRC2865-pp1) and prophage C2865-pp2 (LSRC2865-pp2).
Protein domain analysis revealed a phage-related LSR, designated LSRSCCmec, which encompassed the typical PF00239, PF07508, and PF13408 domains, also present in Ccrs. As Ccrs can still transpose SCCmec elements when located elsewhere in the genome (46), the possible presence of ccr gene variants outside SCCmecC2865 was determined by a search of the translated genome of MRSS C2865 for the site-specific Ccr S-rec motif Y-[LIVAC]-R-[VA]-S-[ST]-x(2)-Q present in LSR. Two hits were identified: (i) LSR from prophage C2865-pp1, designated LSRC2865-pp1, and (ii) LSR from prophage C2865-pp2, designated LSRC2865-pp2 (see below). LSRSCCmec was identified only when we searched for consensus motif Y-[LIVAC]-R-[VA]-S-[ST]-x(4)-Q. Phylogenetic analyses evidenced these recombinases remarkably distant from known Ccrs. Yet, they revealed phylogenetically closer to CcrCs (Fig. 5B).
Unique Siphoviridae prophages vB_SsS-C2865-pp1, vB_SsS-C2865-pp2, and vB_SsS-C2865-pp3 enclose adaptive features and excise the bacterial chromosome.
We identified three novel prophages designated vB_SsS-C2865-pp1, vB_SsS-C2865-pp2, and vB_SsS-C2865-pp3, according to recommended guidelines (47, 48). ANI revealed that they differed from all available staphylococcal phages (Text S1; Fig. S7). Figures 6 and 7 show a comparative analysis of prophages C2865-pp1 and C2865-pp2, and C2865-pp3, respectively, with their closest relatives. Phylogenetic analysis of the closest integrases (LSR, tyrosine recombinases [Y-Int/Rec]) (>70% coverage, >80% identity), all recovered from phage-related elements, is displayed (Fig. 6B; Fig. 7B). This indicates the distribution of (pro)phages harboring this conserved feature. Full lysogenic modules were detected in all three phages (see Text S1 for details).
FIG 6.

Graphical comparative analysis of prophages vB_SscS-C2865-pp1 (C2865-pp1) and vB_SscS-C2865-pp2 (C2865-pp2) and closest relatives. (A) Comparative analysis of the novel staphylococcal prophages C2865-pp1 and C2865-pp2 integrated in the chromosome of S. sciuri strain C2865 and its closest relatives based on (i) all staphylococcal phages deposited in the Viral RefSeq database and (ii) genetic elements enclosing the closest integrases in the NCBI NR database. Arrows denote the genes, length, and orientation. Gene colors other than gray (hypothetical proteins) and gray-turquoise (others) represent genes involved in the following processes (color): chromosomal integration (yellow), regulation (green), DNA metabolism (orange), DNA packaging (dark green), phage morphogenesis (bright blue), cell lysis (purple), host adaptation (navy blue). Areas of identity (tblastx, >50 aa match, >50% identity) between (pro)phages are indicated in the color scale. (B) Maximum likelihood phylogenetic tree of C2865-pp1 and C2865-pp2 integrases (Int), as well as all integrases sharing >80% amino acid identity with respect to either C2865-pp1 and C2865-pp2 integrases and integrase of Enterobacteria lambda phage, used as outgroup (colored in blue). C2865-pp1 and C2865-pp2 integrases are highlighted in faint yellow. Percentage of identity is indicated, with that sharing highest identity to respective integrase highlighted in faint green.
FIG 7.

Graphical comparative analysis of prophage vB_SscS-C2865-pp3 (C2865-pp3) and closest relatives. (A) Comparative analysis of the novel staphylococcal prophage C2865-pp3 integrated in the chromosome of S. sciuri strain C2865 and its closest relatives based on (i) all staphylococcal phages deposited in the Viral RefSeq database as well as on (ii) genetic elements enclosing the closest integrases in the NCBI NR database. Arrows denote the genes, length, and orientation. Gene colors other than gray (hypothetical proteins) and gray-turquoise (others) represent genes involved in the following processes (color): chromosomal integration (yellow), regulation (green), DNA metabolism (orange), DNA packaging (dark green), phage morphogenesis (bright blue), cell lysis (purple), host adaptation (navy blue), antimicrobial resistance (pink). Areas of identity (tblastx, >50 aa match, >50% identity) between (pro)phages are indicated in the color scale. Staphylococcus phage SPbeta-like was linearized at position of interest. Area with green background denotes the region enclosing an independent mobile genetic region (flanked by two IS257 copies in the same orientation), which encompasses the transposon Tn4001 (IS256-aacA/aphD-IS256) as well as the trimethoprim resistance gene dfrA. (B) Maximum likelihood phylogenetic tree of the three integrases (Int) detected in C2865-pp3, as well as those present in its two closest (pro)phages and closest integrases (>80% amino acid identity) and integrase of Enterobacteria lambda phage, used as outgroup (colored in blue). Integrases from C2865-pp3 are highlighted in faint yellow. Percentage of identity is indicated, with that sharing highest identity to respective integrase highlighted in faint green.
Phylogenomic tree and heat map resultant from the average nucleotide identity (ANI) of a subset of 43 staphylococcal phages deposited in the Viral RefSeq NCBI database positioned within the same subbranch as C2865-pp1 to -pp3, plus the three prophages from S. sciuri strain C2865 colored in green. A phylogenomic tree including all 187 staphylococcal phages included in the analysis is represented in the upper left corner. Download FIG S7, PDF file, 2.5 MB (2.5MB, pdf) .
Copyright © 2021 Gómez-Sanz et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
C2865-pp1 was 41,283 bp long and consisted of 62 CDSs, of which 26 (41.9%) had predicted functions. C2865-pp1 had a GC content of 34.6% and was integrated truncating a VOC family-protein gene, 1,880,677 bp downstream of dnaA. The structural gene distribution of C2865-pp1 shared a typical phage modular organization. Characteristic phage gene groups involved in lysogeny (integrase, repressors, antirepressor; see Text S1 for details), DNA metabolism (single-strand DNA binding, methyltransferase), packaging (HNH endonuclease, terminases TerS, TerL), morphogenesis (major capsid, tail fiber, tape measure), and cell lysis (peptidase, holin, endolysin) were detected from left to right arm (Fig. 6A). Chromosomal integration of C2865-pp1 generated a 6-bp perfect DR (attL and attR) (5′-AATGGT-3′) (Fig. 6A) at its boundaries.
C2865-pp2 was 45,020 bp and consisted of 64 CDSs, of which 27 (42.2%) had predicted functions. C2865-pp2 had a GC content of 33.8% and was integrated truncating a nucleoside hydroxylase gene, 2,501,758 bp downstream of dnaA. C2865-pp2 CDSs were arranged in functional modules in synteny with those detected in C2865-pp1. Unusually, an abortive infection bacteriophage resistance gene, designated abiF (Abi_2 family, PF07751), was detected at its accessory right-arm region. The Abi group of proteins are involved in bacteriophage resistance mediated by abortive infection in Lactococcus species. Two 15-bp imperfect DRs (attL and attR) with consensus sequence 5′-[A/C]GG[A/T]GGAACGTTTGG-3′ were identified at the extremities of the phage integration core site (Fig. 6A).
C2865-pp3 was 126,192 bp and consisted of 166 CDSs, of which only 34 (20.5%) had predicted functions. C2865-pp3 had a GC content of 30.8% and was integrated truncating a yeeE/yedE gene, coding for an inner membrane protein, positioned 819,965 bp downstream of dnaA. C2865-pp3 genome was organized into three proposed modules delimited by three different integrases, one LSR and two Y-Int/Rec. The combined modules contained genes involved in lysogeny, DNA replication and metabolism, virion packaging, phage morphogenesis, cell lysis, and adaptation (Fig. 7A; see Text S1 for details). Two putative elements involved in phage adaption to the host were detected at its left and central regions, hicA-hicB and ardA, respectively. The putative HicA and HicB belong to type II toxin-antitoxin systems, where the toxin (HicA) acts as mRNA interferase and the antitoxin (HicB) as neutralizer. The ardA gene coded for a putative antirestriction protein ArdA with an N-terminal domain (PF07275) involved in evasion of the bacterial type I restriction-modification system. Moreover, C2865-pp3 harbored a tRNA, which shared 86.7% nucleotide identity with the corresponding tRNA of several Listeria phages (see supplemental material for analysis of tRNA codon and amino acid usage; Fig. S9). The integration of C2865-pp3 generated two 8-bp perfect DRs (5′-GTACTTGG-3′) at its boundaries (Fig. 7A). C2865-pp3 shared the closest identity to the prophage-like element of mouse Staphylococcus lentus HT5 and to Staphylococcus phage SPbeta-like, obtained from a clinical Staphylococcus epidermidis 36-1, both carrying three different integrases (Fig. 7B; Text S1). Importantly, the left-arm region of SPbeta-like phage enclosed a composite transposon-like element (IS257-flanked) carrying the TMP resistance gene dfrA and transposon Tn4001 (IS256-aacA/aphD-IS256), which harbors the aminoglycoside resistance gene aacA-aphD (Fig. 7A).
Comparison of codon usage (A) and amino acid (B) percentage between the three prophages (C2865-pp1, C2865-pp2, C2865-pp3) and C2865 host genome. The scale (in percentage) in the left plots is represented at the upper center of the graph. (A) Left, radar plot of the 64 different amino acid codons (at DNA level) highlighting the tRNA gene present in prophage C2865-pp3 genome. Right, specific abundance (%) of TGG codon (UGG). (B) Left, radar plot of the 20 amino acids highlighting proline (P), as resultant amino acid from tRNA present in C2865-pp3. Right, specific abundance (%) of proline (P). Download FIG S9, EPS file, 0.2 MB (168.3KB, eps) .
Copyright © 2021 Gómez-Sanz et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Sequencing analyses of potential circular intermediates (CIs), as well as restorage of respective phage chromosomal integration genes, revealed that the three prophages could excise the bacterial genome. Phylogenetic analysis of terminase TerL plus the presence of an HNH endonuclease gene in front of terminase terS, involved in DNA packaging of cos phages (49), predicted a cohesive end packaging strategy (cos) for C2865-pp1 and C2865-pp2 (Text S1; Fig. S8).
Circular phylogenetic tree of all available staphylococcal terminase large subunit (TerL). The tree built with the amino acid sequences of all identifiable terminase large subunit (TerL) present in 187 staphylococcal phages deposited in the Viral RefSeq database, TerL of C2865-pp1, C2865-pp2, and C2865-pp3, and TerL of Enterobacteria phage lambda, used as outlier. Phage nomenclature: S-phage, which refers to staphylococcal phage, is followed by the phage name, faconcant (refering to concatenated fasta), plus the CDSs number of respective TerL in the phage genome. Branch enclosing the C2865-pp1, C2865-pp2, and C2865-pp3 integrases is bold. C2865-pp1, C2865-pp2, and C2865-pp3 integrases are boxed in green, while phage lambda is boxed in gray. Phages with known packaging mechanism (95), carrying either cos-sites (cos packaging strategy) or pac-sites (headful packaging strategy), are indicated in black and green, respectively. Download FIG S8, PDF file, 1.6 MB (1.6MB, pdf) .
Copyright © 2021 Gómez-Sanz et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The novel S. sciuri pathogenicity island (SscPIC2865) belongs to the SaPI4 family, and related elements are disclosed to be common among S. sciuri genomes.
A novel phage-inducible chromosomal island (PICI) of 13,101 bp, named SscPIC2865, was detected 20,705 bp upstream of dnaA gene. SscPIC2865 was classified as an S. sciuri pathogenicity island (SscPI), as it contains homologues of the entire core set of genes characteristic of S. aureus pathogenicity islands (SaPIs) and displays synteny with previously characterized SaPIs (Text S1). SscPIC2865 consisted of 21 CDSs, of which 9 (42.9%) lacked any predicted function (Fig. 8A). SscPIC2865 enclosed a tyrosine recombinase (Y-Int/Rec) directing chromosomal integration at the end of the 3′-end of the 30S ribosomal protein S18 gene (rpsR). Two SscPIC2865-flanking 15-bp DRs (5′-AAAGAAGAACAATAA-3′) constituted its integrase attachment core sites (Fig. 8A). None of the genes involved in interference of the helper phage reproduction in favor of SscPIC2865 packaging were identified. However, this 3′ region carried a putative transcriptional regulator, an RNA polymerase sigma factor, and a putative helicase, which might be involved in the replication of the element (Fig. 8A). Interestingly, a putative mazF toxin gene, coding for an endoribonuclease (mRNA interferase) of a type II toxin-antitoxin system (MazF/MazE), was identified in the accessory region of SscPIc2865.
FIG 8.

Graphical analysis of the novel staphylococcal pathogenicity island SscPIC2865. (A) Schematic representation of scPIC2865, including its chromosomal integration flanking region, downstream the 30S ribosomal S18 protein (rpsR gene), and comparison of the integration region with that of S. sciuri SNUDS-18. Arrows denote the genes, length, and orientation. The core integration site sequence detected (5′-AAAGAAGAACAATAA-3′) as well as position for integration (attC for putative integration site in S. sciuri SNUDS-18 chromosome; attL and attR at both extremities of SsPIC2865) are indicated. Areas of nucleotide similarity (nblastn, >100 bp match, >99% identity) between both structures are indicated in gray. (B) Maximum likelihood phylogenetic tree of (left) all site-specific integrases (Int) deposited in the protein NCBI database and (right) all terminases small subunit (TerS) sharing >80% amino acid identity with respect to those present in SscPIC2865, in addition to those of prototype SaPI4 from S. aureus MRSA252 (69) (displayed in blue). Strains with Int but either lack of or divergent (<50% ID) TerS, and vice versa, are indicated in yellow or green color, respectively. Percentages of amino acid identity with respect to Int or TerS of SscPIC2865 are displayed on the right of the respective tree. TerS-carrying strains corresponded to those harboring the SscPIC2865-conserved integrase in all cases but one (S. sciuri SNUC70), which was integrated at a different position of the bacterial chromosome (at tRNA, ssrA gene). (C) Sequence comparison analysis of the phage-related chromosomal island att sequence core site region (attL and attR) of SscPIC2865 and corresponding region of all strains carrying a similar integrase (>80% identity), in addition to that of S. aureus MRSA252 (harboring prototype SaPI4), which also integrates at the 3′-end of the S18 ribosomal gene rpsR. Underlined area represents the proposed integration core sequence of SscPIC2865 and related elements. Bases differing from the att region of S. sciuri C2865 SscPIC2865 are indicated in red. Original att core site length of SaPI4 is indicated in dark blue (69), and the additional direct repeat is shown in blue.
Several integrases sharing >80% identity to SscPIc2865 integrase were identified, all truncating rpsR (Fig. 8B). Of these, nine belong to S. sciuri and the rest to Staphylococcus schleiferi, S. lentus, and Staphylococcus fleurettii. Their integrase-surrounding regions share the characteristic structure of SaPIs (data not shown). We denoted that these elements belong to the SaPI4 family (integrase group I), according to the integrase sequence homology, integration site, and conserved core site 5′-AAAGAAGAACAATAA-3′ (Text S1; Fig. 8C). Sequencing analyses of potential circular intermediates (CIs) and restorage of SscPIC2865 chromosomal integration site revealed that SscPIC2865 could excise the bacterial chromosome.
DISCUSSION
The combination of Illumina followed by deep PacBio sequencing using large DNA fragmentation and size selection allowed the resolution of the complete genome of IS-rich MDR S. sciuri C2865 at a minimum error rate. This enabled the discovery of transferable TMP resistance dfrE within a novel mosaic plasmid and additional novel MGEs. MRSS C2865 revealed high genome plasticity with respect to the S. sciuri genomes analyzed, as it was enriched in novel unique chromosomal and extrachromosomal elements.
We display for the first time that S. sciuri has an open pan-genome, with an ever-increasing flexible genome as new strains are added. Similar patterns were observed in other clinically relevant staphylococcal species formerly analyzed, such as S. aureus (50), S. epidermidis (51–53), Staphylococcus haemolyticus (54), Staphylococcus capitis, and Staphylococcus caprae (52). This result could be anticipated, as an open pan-genome is characteristic of species living in multiple environments and/or mixed bacterial communities. This feature facilitates multiple ways of exchanging genetic material, enabling them to unceasingly expand their total gene repertoire. Our data set revealed MRSS C2865 as the major acceptor of adaptive mobile traits, as it exhibited the highest number of unique genes, most of them (75%) corresponding to MGEs. However, this was not correlated with phylogeny, as C2865 closest relatives (SNUDS-18 and Z8) did not disclose this sharp profile. Based on a range of phenotypic, biochemical, physiological, and genetic analyses, Svec et al. (55) recently showed high S. sciuri intraspecies heterogeneity with no clear differentiation into different subspecies. While agreeing with those statements, our whole-genome-based phylogenomic analysis revealed a clear distinction of an intraspecies subcluster, which included MRSS C2865 (96% ANI with the rest of the S. sciuri genomes). This ANI value falls just above the threshold to be considered a different species (≈95%) (56, 57). Hence, we suggest that there indeed may be subspecies discrimination among the S. sciuri species, but such distinction needs to be addressed by WGS comparisons and might not correspond to the outcomes retrieved by the above-mentioned traditional methods. Alternatively, the available metadata of compared genomes evidence the absence of clear phylogeny delineation depending on the origin, source, or host. This corroborates the low host tropism suggested for this species (3).
We reveal that the novel dfrE confers high-level resistance to TMP in both staphylococci and E. coli. DfrE is phylogenetically distant from all staphylococcal TMP resistance genes and displays a common ancestor with Dfrs from soil-associated P. anaericanus. Scarce data are available on P. anaericanus, but bacteria belonging to this genus are ubiquitous in nature, and closest species have been reported in different environmental sources, such as soils and rhizosphere from different crops (58–60). Importantly, dfrE evaded discovery in all available dfrE-carrying genomes except Exiguobacterium sp. (dfr_like), where it proved to confer TMP resistance in E. coli. Exiguobacterium spp. are extremophiles adapted to a wide range of habitats, including cold environments (40, 61–64). We also identify here that dfrE is already distributed in Asia, as it is present in at least three different staphylococcal species (S. sciuri, S. aureus, and S. arlettae) of human and animal origin (including clinical samples) as well as in animal-associated M. caseolyticus strain JCSC5402 (65) from several countries. Importantly, all dfrE-carrying genomes harbored it in MDR plasmids or plasmid-associated elements. This colocalization is of concern, because it enables the transfer of diverse adaptive traits via a single HGT event. Our data reflect the transferability of TMP resistance via dfrE across diverse bacteria from different taxonomic families (order Bacillales), diverse environments, and geographically distant regions. Of note, in Exiguobacterium sp. strain S3-2, dfrE was enclosed within a conserved Tn3-like transposon (40). MRSS C2865 (pUR2865-34) and all additional dfrE-carrying strains harbored a truncated version of this element. In particular, the dfrE-carrying region of S. sciuri GN5-1 (pSS-04) seems to have evolved afterwards by losing an additional recombinase-carrying segment from the Tn3-like remnant. This indicates that Exiguobacterium sp. harbored an ancestral dfrE transposable element that has recombined and jumped to different species from a single ancestor. The fact that the dfrE element is already detected in several unrelated animal S. sciuri strains from China and Nigeria, and that S. sciuri GN5-1 has an evolved dfrE segment, points toward the assumption that S. sciuri is the ancestral species within the genus, as several recombination events have already occurred within this region. Unfortunately, no flanking regions of the dfr gene of P. anaericanus are available, so potential homology with its surrounding area could not be addressed. The backbone of mosaic pUR2865-34 denoted a theta replication mechanism, characteristic of larger staphylococcal plasmids. The putative type Ib partitioning system detected is well studied in Gram-negative bacteria, but little to nothing is known about its functions in Gram-positive cocci. These systems contribute to the prevalence and spread of these plasmids, ensuring stable inheritance and effectively maintaining resistance in the absence of selection. pUR2865-34 could only move by conjugation via the detected oriT mimic sequence in the presence of a pWBG749-like plasmid or via the integrated mob gene and associated oriT of pUR2865-int in the presence of a conjugative plasmid (42). However, MRSS C2865 did not harbor any member of the three known staphylococcal conjugative plasmid families (pSK41, pWBG4, and pWBG749) or any recognizable transfer gene cluster that could act in trans.
We identified a novel ccr-lacking SCCmecC2865 encompassing additional AMR genes, including an IS1216E-flanked region carrying the tetracycline resistance gene tet(S). This gene has been found among Firmicutes and Gammaproteobacteria from diverse ecological sources since the 1950s (66). However, the tet(S) has been detected only twice before in staphylococci: (i) among methicillin-resistant Staphylococcus aureus (MRSA) isolates from animal carcasses (67) and (ii) within the S. sciuri SCCmecC2865-related SCCmecTG24 cassette, from ready-to-eat meat (45). The additional closest cassette corresponded to mecA-mecC hybrid SCCmec-mecC in S. sciuri GVGS2 from a bovine infection (20). These strain sources highlight that S. sciuri isolates from animals behave as reservoirs for mosaic MGEs carrying AMR genes.
MRSS C2865 resulted in a polylysogen with three novel unrelated prophages, two of them harboring several putative adaptive features that may promote bacteria and/or phage survival. Only these prophages constituted over 7% of MRSS C2865 genome, evidencing their role as drivers of bacterial evolution and genome modulation. As recently observed by Oliveira et al. (68), a low rate of staphylococcal phages may exhibit more than one site-specific recombinase, as was the case for phage C2865-pp3. Phage integrases are required for the establishment of the lysogeny, but their maintenance is also dependent on the regulatory proteins next to them (68). At least two of its three integrases were surrounded by transcriptional regulators and/or antirepressor proteins. This may enable the integration at additional chromosomal sites depending on the integrase used. In fact, the closest putative prophage, integrated into S. lentus HT5, was incorporated at a different location. Importantly, C2865-pp3-related S. epidermidis SPbeta-like phage harbored an AMR gene cluster enclosed within a composite transposon-like element, harboring resistance determinants for aminoglycosides and TMP. This is a central feature demonstrating that C2865-pp3-related functional phages can indeed harbor mobilizable AMR genes.
PICIs are characterized by a specific set of phage-related functions that enable them to hijack the phage lytic reproduction cycle of helper phages for their own highly efficient transduction. These elements often carry critical staphylococcal virulence genes (69). Several staphylococcal strains harbored SscPIC2865-related PICIs at the same integration site, disclosing that these elements are common among S. sciuri and contribute to genome plasticity and potential toxigenicity. Hence, the ribosomal protein S18 (rpsR gene) appears as a hub for the integration of mobile islands and should be considered when addressing staphylococcal chromosomal-integrated elements.
The conservation of the integrase-generated DRs flanking the three prophage genomes, together with their ability to excise the bacterial chromosomal DNA and circularize, as well as the high genome synteny observed among functional phages, strongly suggest that the novel prophages are functional. If this is the case, along with progeny generation, also plasmids or chromosomal DNA of the bacterial host may be mistakenly encapsidated (26). Hence, it is conceivable that any of these prophages may package and mobilize the MGEs discovered here, highlighting any of the three nonconjugative plasmids, the novel ccr-lacking SCCmec, and/or SscPIC2865 of the SaPI4 family. In fact, PICIs of the SaPI4 family are only induced by endogenous prophages (69). Upcoming studies are warranted to explore the transduction ability of these prophages.
We unveil a transferable TMP resistance gene that has so far evaded identification; although dfrE is already present in diverse species from varied sources geographically distant, dfrE seems to have emerged from a single ancestor. Molecular data analysis suggests S. sciuri as the donor for transfer within staphylococci. Since dfrE seems not yet common in staphylococcal clinical specimens, the data presented here enable early surveillance and facilitate molecular diagnosis, which could promote spread mitigation. Additional experimental analyses may elucidate the spread vehicle and routes for this evolving element. We highlight S. sciuri as a resourceful hub for diverse mobilizable adaptive traits.
MATERIALS AND METHODS
Bacterial strain selection and characteristics.
MRSS strains C2865, C2853, C2854, and C2855 were obtained in a former study on the occurrence of methicillin-resistant coagulase negative staphylococci from canine samples and were recovered from the groin area of unrelated stranded dogs in Nsukka, Nigeria (12). These strains exhibited TMP resistance, while none of the known staphylococcal TMP resistance genes (dfrS1, dfrD, dfrG, dfrK) (32, 33, 35, 36) nor the typical streptococcal/enterococcal dfrF gene (34, 37) was detected. All strains were MDR and exhibited related AMR patterns. Strain C2865 was selected for WGS to unveil the genetic basis for TMP resistance. MRSS C2865 was chosen for WGS as it harbored the highest number of detected AMR genes.
DNA extraction, whole-genome sequencing, assembly, and annotation.
Detailed DNA extraction procedures are described in the supplemental material. Briefly, genomic DNA was extracted by two different methods: for Illumina sequencing, the Wizard Genomic DNA purification kit was used including both lysozyme and lysostaphin (10 mg/ml each) (A1120, Promega Corporation, Spain), and for PacBio sequencing, DNA was isolated using a phenol-chloroform method with some modifications for improved cell lysis. Plasmid DNA was obtained using the GenElute Plasmid Miniprep kit (PLN350, Sigma) also including a lysis step with lysozyme (2.5 mg/ml) and lysostaphin (0.25 mg/ml) for 20 min at 37°C after the resuspension solution step.
High-throughput WGS of S. sciuri C2865 DNA was performed with Illumina Miseq (2 × 300 bp), with NEBNext Ultra kit. For long-read sequencing, PacBio RSII was used after DNA fragmentation of 15 kb followed by a mild size selection. Illumina read quality was checked by Fastqc (103), and reads were trimmed using Trimmomatic v0.36 (70). Good-quality Illumina reads were de novo assembled using SPAdes (71). PacBio RSII raw reads were assembled using Canu (72). Protein-coding genes, tRNAs, and rRNA operons were predicted using Prodigal (73), tRNAscan-SE, and RNAmmer on both data sets (74). Predicted protein sequences were compared against the NCBI NR database (NCBI nonredundant database) using DIAMOND (75) and against COG (76) and TIGFRAM (77) using HMMscan (78) for taxonomic and functional annotation. Genomic alignment dot plots between Illumina- and PacBio-resultant contigs were generated with D-GENIES software to evaluate consistency and reliability of both sequencing and assembly approaches (79). Amino acid and codon usages were determined for C2865 chromosome and prophages using the compareM package (https://github.com/dparks1134/CompareM).
Pan-genome examination, S. sciuri phylogenomic analysis, and comparative genomics.
All genomes available belonging to the S. sciuri group species were downloaded from the NCBI database (accessed in April 2018). In total, 30 strains (29 from NCBI, MRSS C2865) were included, which belonged to the following species (no. of strains): S. sciuri (21), S. lentus (5), Staphylococcus vitulinus (2), S. fleuretti (1), and Staphylococcus stepanovicii (1) (see the supplemental material for strain characteristics). In order to measure the probability of two genomes belonging to the same species, an ANI of the 30 S. sciuri group genomes was calculated using JSpecies as indicated before (56). A heat map was generated using the ANI matrix output table with R (80). In parallel, a maximum likelihood tree for all the S. sciuri group genomes was generated using RAxML (version 7.2.6) (81) using core alignment obtained with Parsnp software within Harvest Suite package (82). Before phylogenomic analysis, all the genomic regions where recombination was detected were removed from the alignment. These regions were determined using the Gingr software, also included in the Harvest Suite package (82), which uses as input the output obtained directly from the Pasnp software. The results were visualized using iTOL v6 (https://itol.embl.de/). All S. sciuri group species other than S. sciuri were considered an outgroup for graphical representation. BLAST Ring Image Generator (BRIG) was used to evaluate and visualize comparisons between MRSS strain C2865 and its closest genomes (>98% ANI), using C2865 as reference (83). Pan-genome analysis (core plus accessory genome) for the 21 S. sciuri genomes was carried out using Roary with a 95% identity cutoff value (84).
Detection and analysis of resistome and mobilome from S. sciuri C2865.
Antimicrobial resistance (AMR) genes. AMR genes formerly detected in MRSS C2865 (see Table S1 in reference 85) were blasted against the WGS of strain C2865. Contig(s) were manually checked for redundant dhfr genes and for additional resistance genes of interest.
Plasmids. Plasmid contig identification and plasmid reconstruction were achieved by contig coverage, sequence similarity with plasmid backbone genes, gene composition and organization, and contig boundaries redundancy (circularity). Putative oriT and oriT mimics on the mobilizable elements were searched using the core sequence of those from conjugative plasmids (86). For RCR plasmids, dso and sso regions, involved in the initiation of replication of the leading and lagging strand, respectively, were searched using core sequences of an RCR representative per plasmid family (87). The secondary structures of dso, sso, oriT, and oriT mimic were generated using Mfold web server for single-stranded linear DNA at default parameters (88).
Prophages. Detailed analyses are described in the supplemental material. Manual inspection of phage-associated genes (morphology/structure, lysogeny, cell lysis, DNA metabolism) and characteristic functional modular organization was implemented for phage confirmation and integrity. Integrase motif and domain analysis of the translated candidate CDSs was performed against ScanProsite database (89), Pfam database (90), and NCBI conserved domain database (CDD) (91). Integrase-directed generation of DRs as a result of genome integration was investigated manually by sequence comparison of bacterial chromosome-prophage boundaries using a S. sciuri strain prototype (SNUDS-18, GenBank accession no. CP020377) lacking those prophages. ANI pairwise comparison between C2865-enclosed prophages and all staphylococcal phage genomes available in the Viral RefSeq database (accessed until November 2018, n = 187) were calculated using the JSpecies with default parameters (56). A heat map was generated using the ANI matrix output table with R (80).
Staphylococcal chromosomal cassette mecA (SCCmec). ORFs found downstream of the integration gene 23S rRNA [pseudouridine(1915)-N(3)]-methyltransferase RlmH, initially known as OrfX, as well as regions containing characteristic SCCmec genes (mecA, mecR, mecI, ccr) were analyzed. To detect ccr gene(s), whose resultant proteins belong to the large S-rec (LSR) family, consensus motif Y-[LIVAC]-R-[VA]-S-[ST]-x(2)-Q derived from Prosite entry PS00397 (http://prosite.expasy.org) was used (supplemental material). Additional S-rec encompassing the Y-[LIVAC]-R-[VA]-S-[ST]-x(4)-Q motif were likewise screened along the entire MRSS C2865 genome. Putative ISS for SCCmec or att core sites recognized by the typical staphylococcal Ccr were manually identified by searching for the consensus sequence 5′-GAAGC[AG]TATCA[TC]AAAT[AG]A-3′ (supplemental material).
Others. Additional recombinases associated with MGEs were investigated as described above for motif and domain analysis (supplemental material). SscPI chromosomal integration site was compared with similar integrase-carrying genomic islands (supplemental material). Integrase-directed generation of DRs as a result of SscPI integration was analyzed by sequence comparison of bacterial chromosome-SscPI boundaries with corresponding regions of S. sciuri strain SNUDS-18, which lacks any insertion in that region. In addition, ISsaga2 web tool was used for IS identification and quantification (92).
Excision ability of chromosomally located MGEs. Potential excision and circularization of selected chromosomally located mobile elements, in addition to detection of the resultant chromosomal region after excision, were tested by specific inverse and conventional PCR, respectively (supplemental material; see Table S1 in reference 85).
Phylogenetic analyses of proteins of interest.
Phylogenetic analyses for Dhfr/Dfr and Ccr of interest were investigated by the construction of unrooted phylogenetic networks with SplitsTree v4 (93) using a neighbor-net with default parameters. Evidence for phylogenetic heterogeneity due to recombination was conducted with SplitsTree v4 using the Phi test for recombination option.
A maximum likelihood phylogenetic tree for MRSS C2865 prophage integrases and all related integrases was built upon amino acid sequence alignment as indicated above (using CLC Genomics Workbench 11) (supplemental material). To estimate the packaging mechanism of identified prophages, a circular phylogenetic tree of the terminase large subunit (TerL) from MRSS C2865 prophages and those identified in the 187 staphylococcal phages deposited in the Viral RefSeq database was likewise created upon amino acid sequence alignment using CLUSTALW (CLC Genomics Workbench 11). TerL of staphylococcal phages with known packaging mechanism was indicated (95).
Phylogenetic analyses for all site-specific tyrosine recombinases and terminases small subunit (TerS) related to those of SscPI in C2865 were performed by the construction of a maximum likelihood tree using the MEGA 7.0.21 program (supplemental material) (94).
Construction of recombinant plasmids to analyze DfrE functionality, transfer assays, and analysis of native plasmid integrity.
To address the functionality of the candidate TMP resistance gene and for potential synergistic activity by its immediate 3′-end thymidylate synthase gene (thy), three different dfrE-containing regions were amplified and cloned into pBUS-Pcap-HC (constitutive promoter) or pBUS-HC vectors using the Gibson assembly workflow: (i) dfrE gene alone, (ii) dfrE gene plus flanking regions, and (iii) dfrE gene, thy gene, and flanking regions (Table S2; supplemental material) (96). Constructs of interest were transformed into Escherichia coli DH5α and subsequently into S. aureus RN4220 (supplemental material). In addition, native dfrE-containing plasmid was transformed into RN4220.
Purified DNA of two independent transformants containing the native dfrE-carrying plasmid (RN4220/pUR2865-34) were sent for Illumina WGS (HiSeq, 10 million reads, 2 × 150 bp) for confirmation of the integrity of transformed plasmid. In parallel, one microgram of dfrE-containing plasmid DNA from selected transformants (RN4220/pUR2865-34) and from original strains (C2865, C2853, C2854, C2855) was digested with 1 unit of S1 enzyme (1 min at 37°C) (Thermo Fisher Scientific) for plasmid linearization. Plasmid length and integrity were then assayed by gel electrophoresis.
Antimicrobial susceptibility testing.
Minimum inhibitory concentration of TMP was determined in strains of interest by the agar dilution method on Mueller-Hinton plates (MH, Becton, Dickinson) with a concentration range of 0.5 to 4,096 μg/ml (supplemental material) (97). The agar disk-diffusion method was also used to test for AMR profile of transformants (97). Antimicrobials tested were as follows (μg/disk): erythromycin (15), clindamycin (2), gentamicin (10), kanamycin (30), streptomycin (10 U), tobramycin (10), tetracycline (30), trimethoprim (5), sulfonamide (300), and TMP-sulfamethoxazole (1.25 + 23.75).
Phenotypic characterization of biofilm formation.
Biofilm formation ability of the four S. sciuri strains as well as S. aureus RN4220/pUR2865-34 transformants was tested by a modified Congo red agar (CRAmod) assay and by crystal violet microtiter plate assay (see supplemental material for detailed procedures) (98, 99). S. aureus SA113 and S. aureus ATCC 25923 were used as positive controls for their strong biofilm-forming potential, while S. aureus RN4220 (DSM 26309) was selected as negative control for biofilm production.
PCR detection of the TMP resistance gene dfrE and the ica-locus variant.
Primers were designed for the detection of the TMP resistance dfrE gene, the biofilm formation ica-locus (icaADBC) variant genes, and the ica-locus repressor (icaR) (see Table S1 in reference 85). For the dfrE gene, primers designed enclosed the Dhfr superfamily domain region (Cd-Search PF00186). All dhfr genes with ≥45% nucleotide similarity were included to search for specificity of dfrE gene primer set. Original strains (C2853, C2854, C2855), RN4220/pUR2865-34 transformants, and several control strains were tested for sensitivity and specificity (supplemental material).
Data availability.
The whole-genome shotgun sequence data supporting this work are available via NCBI SRA database under BioProject and Biosample accession numbers PRJNA663854 and SAMN16182282, respectively.
ACKNOWLEDGMENTS
We thank Carmen Torres from the University of La Rioja (Spain) and Kennedy Chah from the University of Nigeria Nsukka for kindly providing us with S. sciuri strains C2865, C2853, C2854, and C2855 and Brion Duffy and Theo Smits from the Zurich University of Applied Sciences (ZHAW), Wädenswil (Switzerland) for providing the Illumina sequencing raw data of S. sciuri C2865. We thank the Functional Genomics Center Zurich (FGCZ), particularly Andrea Patrignani, for the PacBio RSII sequencing of C2865. We also thank Vincent Perreten and Sybille Schwendener from the Institute of Veterinary Bacteriology, Bern (Switzerland) for the vectors pBUS-HC and pBUS-Pcap-HC, as well as Francisco Rodriguez-Valera and Martin Loessner for accessibility to computing and/or wet lab resources. We thank Dominique Lorgé for excellent technical assistance and Pauline Göller for facilitating the TerS sequences from the staphylococcal phages.
We also thank Gloria del Solar and Jose R. Penadés for expert discussion on plasmid and PICI biology, respectively, Alexandro Varani for bioinformatic analysis support related to ISs, and Maria de Toro for assistance using BRIG software.
This work was supported by the European Union’s Framework Program for Research and Innovation Horizon 2020 (2014 to 2020) under the Marie Sklodowska-Curie Grant Agreement no. 659314 to E.G.S., by the Swiss National Science Foundation NFP72 “Antimicrobial Resistance” project no. 167090 to E.G.S.; and by the ETH Career Seed Grant Project SEED-01 18-1 to E.G.S. Part of the funding was also provided by ETHZ. J.M.H.M. was supported with a PhD fellowship from the Spanish Ministerio de Economía y Competitividad (BES-2014-067828).
Conceptualization, E.G.S.; data curation, E.G.S., J.M.H.M., M.L.P., S.O.J.; formal analysis, E.G.S., M.L.P., J.M.H.M.; funding acquisition, E.G.S.; investigation, E.G.S.; methodology, E.G.S., J.M.H.M., M.L.P.; project administration, E.G.S.; resources, E.G.S. plus see acknowledgments above; software, J.M.H.M., M.L.P.; supervision, E.G.S.; validation, E.G.S., M.L.P., J.M.H.M., J.J.R.G., S.O.J.; visualization, E.G.S., M.L.P., J.M.H.M.; writing—original draft, E.G.S.; writing—review and editing, E.G.S., M.L.P., J.M.H.M., S.O.J.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors have declared that no competing interests exist.
ADDENDUM IN PROOF
During the time this paper was prepared for publication, the taxonomy of the Staphylococcaceae family has been revisited. Following phylogenomic analyses, Madhaiyan et al. (104) propose species belonging to the S. sciuri species group (S. sciuri, S. fleurettii, S. lentus, S. stepanovicii, and S. vitulinus) be reassigned to the novel genus Mammaliicoccus, with Mammaliicoccus sciuri as the type species.
Contributor Information
Elena Gómez-Sanz, Email: elena.gomez@hest.ethz.ch.
Charles R. Langelier, UCSF
REFERENCES
- 1.Grundmann H, Aires-de-Sousa M, Boyce J, Tiemersma E. 2006. Emergence and resurgence of meticillin-resistant Staphylococcus aureus as a public-health threat. Lancet 368:874–885. doi: 10.1016/S0140-6736(06)68853-3. [DOI] [PubMed] [Google Scholar]
- 2.Becker K, Heilmann C, Peters G. 2014. Coagulase-negative staphylococci. Clin Microbiol Rev 27:870–926. doi: 10.1128/CMR.00109-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nemeghaire S, Argudín MA, Feßler AT, Hauschild T, Schwarz S, Butaye P. 2014. The ecological importance of the Staphylococcus sciuri species group as a reservoir for resistance and virulence genes. Vet Microbiol 171:342–356. doi: 10.1016/j.vetmic.2014.02.005. [DOI] [PubMed] [Google Scholar]
- 4.Hauschild T, Schwarz S. 2003. Differentiation of Staphylococcus sciuri strains isolated from free-living rodents and insectivores. J Vet Med B Infect Dis Vet Public Health 50:241–246. doi: 10.1046/j.1439-0450.2003.00662.x. [DOI] [PubMed] [Google Scholar]
- 5.Dakic I, Morrison D, Vukovic D, Savic B, Shittu A, Jezek P, Hauschild T, Stepanovic S. 2005. Isolation and molecular characterization of Staphylococcus sciuri in the hospital environment. J Clin Microbiol 43:2782–2785. doi: 10.1128/JCM.43.6.2782-2785.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aslantas O, Turkyilmaz S, Yilmaz MA, Erdem Z, Demir C. 2012. Isolation and molecular characterization of methicillin-resistant staphylococci from horses, personnel and environmental sites at an equine hospital in Turkey. J Vet Med Sci 74:1583–1588. doi: 10.1292/jvms.12-0124. [DOI] [PubMed] [Google Scholar]
- 7.Kumar D, Pornsukarom S, Sivaraman GK, Thakur S. 2018. Environmental dissemination of multidrug methicillin-resistant Staphylococcus sciuri after application of manure from commercial swine production systems. Foodborne Pathog Dis 15:210–217. doi: 10.1089/fpd.2017.2354. [DOI] [PubMed] [Google Scholar]
- 8.Ortega-Pena S, Franco-Cendejas R, Salazar-Saenz B, Rodriguez-Martinez S, Cancino-Diaz ME, Cancino-Diaz JC. 2019. Prevalence and virulence factors of coagulase negative Staphylococcus causative of prosthetic joint infections in an orthopedic hospital of Mexico. Cir Cir 87:428–435. doi: 10.24875/CIRU.19000690. [DOI] [PubMed] [Google Scholar]
- 9.Sousa M, Silva V, Silva A, Silva N, Ribeiro J, Tejedor-Junco MT, Capita R, Chenouf NS, Alonso-Calleja C, Rodrigues TM, LeitAo M, GonCalves D, CaniCa M, Torres C, Igrejas G, Poeta P. 2020. Staphylococci among wild European rabbits from the azores: a potential zoonotic issue? J Food Prot 83:1110–1114. doi: 10.4315/0362-028X.JFP-19-423. [DOI] [PubMed] [Google Scholar]
- 10.Lu Y, Lu Q, Cheng Y, Wen G, Luo Q, Shao H, Zhang T. 2020. High concentration of coagulase-negative staphylococci carriage among bioaerosols of henhouses in Central China. BMC Microbiol 20:21. doi: 10.1186/s12866-020-1709-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meservey A, Sullivan A, Wu C, Lantos PM. 2020. Staphylococcus sciuri peritonitis in a patient on peritoneal dialysis. Zoonoses Public Health 67:93–95. doi: 10.1111/zph.12664. [DOI] [PubMed] [Google Scholar]
- 12.Chah KF, Gomez-Sanz E, Nwanta JA, Asadu B, Agbo IC, Lozano C, Zarazaga M, Torres C. 2014. Methicillin-resistant coagulase-negative staphylococci from healthy dogs in Nsukka, Nigeria. Braz J Microbiol 45:215–220. doi: 10.1590/S1517-83822014005000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu S, Piscitelli C, de Lencastre H, Tomasz A. 1996. Tracking the evolutionary origin of the methicillin resistance gene: cloning and sequencing of a homologue of mecA from a methicillin susceptible strain of Staphylococcus sciuri. Microb Drug Resist 2:435–441. doi: 10.1089/mdr.1996.2.435. [DOI] [PubMed] [Google Scholar]
- 14.Rolo J, Worning P, Nielsen JB, Bowden R, Bouchami O, Damborg P, Guardabassi L, Perreten V, Tomasz A, Westh H, de Lencastre H, Miragaia M. 2017. Evolutionary origin of the staphylococcal cassette chromosome mec (SCCmec). Antimicrob Agents Chemother 61. doi: 10.1128/AAC.02302-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miragaia M. 2018. Factors contributing to the evolution of mecA-mediated beta-lactam resistance in staphylococci: update and new insights from whole genome sequencing (WGS). Front Microbiol 9:2723. doi: 10.3389/fmicb.2018.02723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwarz S, Werckenthin C, Kehrenberg C. 2000. Identification of a plasmid-borne chloramphenicol-florfenicol resistance gene in Staphylococcus sciuri. Antimicrob Agents Chemother 44:2530–2533. doi: 10.1128/AAC.44.9.2530-2533.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwarz S, Kehrenberg C, Ojo KK. 2002. Staphylococcus sciuri gene erm(33), encoding inducible resistance to macrolides, lincosamides, and streptogramin B antibiotics, is a product of recombination between erm(C) and erm(A). Antimicrob Agents Chemother 46:3621–3623. doi: 10.1128/AAC.46.11.3621-3623.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hot C, Berthet N, Chesneau O. 2014. Characterization of sal(A), a novel gene responsible for lincosamide and streptogramin A resistance in Staphylococcus sciuri. Antimicrob Agents Chemother 58:3335–3341. doi: 10.1128/AAC.02797-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li D, Wang Y, Schwarz S, Cai J, Fan R, Li J, Feßler AT, Zhang R, Wu C, Shen J. 2016. Co-location of the oxazolidinone resistance genes optrA and cfr on a multiresistance plasmid from Staphylococcus sciuri. J Antimicrob Chemother 71:1474–1478. doi: 10.1093/jac/dkw040. [DOI] [PubMed] [Google Scholar]
- 20.Harrison EM, Paterson GK, Holden MT, Ba X, Rolo J, Morgan FJ, Pichon B, Kearns A, Zadoks RN, Peacock SJ, Parkhill J, Holmes MA. 2014. A novel hybrid SCCmec-mecC region in Staphylococcus sciuri. J Antimicrob Chemother 69:911–918. doi: 10.1093/jac/dkt452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paterson GK. 2020. Genomic epidemiology of methicillin-resistant Staphylococcus sciuri carrying a SCCmec-mecC hybrid element. Infect Genet Evol 79:104148. doi: 10.1016/j.meegid.2019.104148. [DOI] [PubMed] [Google Scholar]
- 22.Xia G, Wolz C. 2014. Phages of Staphylococcus aureus and their impact on host evolution. Infect Genet Evol 21:593–601. doi: 10.1016/j.meegid.2013.04.022. [DOI] [PubMed] [Google Scholar]
- 23.Jurczak-Kurek A, Gąsior T, Nejman-Faleńczyk B, Bloch S, Dydecka A, Topka G, Necel A, Jakubowska-Deredas M, Narajczyk M, Richert M, Mieszkowska A, Wróbel B, Węgrzyn G, Węgrzyn A. 2016. Biodiversity of bacteriophages: morphological and biological properties of a large group of phages isolated from urban sewage. Sci Rep 6:34338. doi: 10.1038/srep34338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeman M, Mašlaňová I, Indráková A, Šiborová M, Mikulášek K, Bendíčková K, Plevka P, Vrbovská V, Zdráhal Z, Doškař J, Pantůček R. 2017. Staphylococcus sciuri bacteriophages double-convert for staphylokinase and phospholipase, mediate interspecies plasmid transduction, and package mecA gene. Sci Rep 7:46319. doi: 10.1038/srep46319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Varga M, Kuntová L, Pantůček R, Mašlaňová I, Růžičková V, Doškař J. 2012. Efficient transfer of antibiotic resistance plasmids by transduction within methicillin-resistant Staphylococcus aureus USA300 clone. FEMS Microbiol Lett 332:146–152. doi: 10.1111/j.1574-6968.2012.02589.x. [DOI] [PubMed] [Google Scholar]
- 26.Haaber J, Leisner JJ, Cohn MT, Catalan-Moreno A, Nielsen JB, Westh H, Penades JR, Ingmer H. 2016. Bacterial viruses enable their host to acquire antibiotic resistance genes from neighbouring cells. Nat Commun 7:13333. doi: 10.1038/ncomms13333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mašlaňová I, Doškař J, Varga M, Kuntová L, Mužík J, Malúšková D, Růžičková V, Pantůček R. 2013. Bacteriophages of Staphylococcus aureus efficiently package various bacterial genes and mobile genetic elements including SCCmec with different frequencies. Environ Microbiol Rep 5:66–73. doi: 10.1111/j.1758-2229.2012.00378.x. [DOI] [PubMed] [Google Scholar]
- 28.Maslanova I, Stribna S, Doskar J, Pantucek R. 2016. Efficient plasmid transduction to Staphylococcus aureus strains insensitive to the lytic action of transducing phage. FEMS Microbiol Lett 363. doi: 10.1093/femsle/fnw211. [DOI] [PubMed] [Google Scholar]
- 29.Stanczak-Mrozek KI, Laing KG, Lindsay JA. 2017. Resistance gene transfer: induction of transducing phage by sub-inhibitory concentrations of antimicrobials is not correlated to induction of lytic phage. J Antimicrob Chemother 72:1624–1631. doi: 10.1093/jac/dkx056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chlebowicz MA, Mašlaňová I, Kuntová L, Grundmann H, Pantůček R, Doškař J, van Dijl JM, Buist G. 2014. The staphylococcal cassette chromosome mec type V from Staphylococcus aureus ST398 is packaged into bacteriophage capsids. Int J Med Microbiol 304:764–774. doi: 10.1016/j.ijmm.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 31.Scharn CR, Tenover FC, Goering RV. 2013. Transduction of staphylococcal cassette chromosome mec elements between strains of Staphylococcus aureus. Antimicrob Agents Chemother 57:5233–5238. doi: 10.1128/AAC.01058-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rouch DA, Messerotti LJ, Loo LS, Jackson CA, Skurray RA. 1989. Trimethoprim resistance transposon Tn4003 from Staphylococcus aureus encodes genes for a dihydrofolate reductase and thymidylate synthetase flanked by three copies of IS257. Mol Microbiol 3:161–175. doi: 10.1111/j.1365-2958.1989.tb01805.x. [DOI] [PubMed] [Google Scholar]
- 33.Dale GE, Langen H, Page MG, Then RL, Stuber D. 1995. Cloning and characterization of a novel, plasmid-encoded trimethoprim-resistant dihydrofolate reductase from Staphylococcus haemolyticus MUR313. Antimicrob Agents Chemother 39:1920–1924. doi: 10.1128/AAC.39.9.1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coque TM, Singh KV, Weinstock GM, Murray BE. 1999. Characterization of dihydrofolate reductase genes from trimethoprim-susceptible and trimethoprim-resistant strains of Enterococcus faecalis. Antimicrob Agents Chemother 43:141–147. doi: 10.1128/AAC.43.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sekiguchi J, Tharavichitkul P, Miyoshi-Akiyama T, Chupia V, Fujino T, Araake M, Irie A, Morita K, Kuratsuji T, Kirikae T. 2005. Cloning and characterization of a novel trimethoprim-resistant dihydrofolate reductase from a nosocomial isolate of Staphylococcus aureus CM.S2 (IMCJ1454). Antimicrob Agents Chemother 49:3948–3951. doi: 10.1128/AAC.49.9.3948-3951.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kadlec K, Schwarz S. 2009. Identification of a novel trimethoprim resistance gene, dfrK, in a methicillin-resistant Staphylococcus aureus ST398 strain and its physical linkage to the tetracycline resistance gene tet(L). Antimicrob Agents Chemother 53:776–778. doi: 10.1128/AAC.01128-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bergmann R, van der Linden M, Chhatwal GS, Nitsche-Schmitz DP. 2014. Factors that cause trimethoprim resistance in Streptococcus pyogenes. Antimicrob Agents Chemother 58:2281–2288. doi: 10.1128/AAC.02282-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu X, Zheng B, Jiang H, Kang Y, Cao Q, Ning H, Shang J. 2015. Draft genome sequence of Staphylococcus sciuri subsp. sciuri Strain Z8, isolated from human skin. Genome Announc 3. doi: 10.1128/genomeA.00714-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Han JE, Hwang SY, Kim JH, Shin SP, Jun JW, Chai JY, Park YH, Park SC. 2013. CPRMethicillin resistant coagulase-negative staphylococci isolated from South Korean ducks exhibiting tremor. Acta Vet Scand 55:88. doi: 10.1186/1751-0147-55-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang J, Wang C, Wu J, Liu L, Zhang G, Feng J. 2014. Characterization of a multiresistant mosaic plasmid from a fish farm sediment Exiguobacterium sp. isolate reveals aggregation of functional clinic-associated antibiotic resistance genes. Appl Environ Microbiol 80:1482–1488. doi: 10.1128/AEM.03257-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ziebuhr W, Krimmer V, Rachid S, Lossner I, Gotz F, Hacker J. 1999. A novel mechanism of phase variation of virulence in Staphylococcus epidermidis: evidence for control of the polysaccharide intercellular adhesin synthesis by alternating insertion and excision of the insertion sequence element IS256. Mol Microbiol 32:345–356. doi: 10.1046/j.1365-2958.1999.01353.x. [DOI] [PubMed] [Google Scholar]
- 42.Firth N, Jensen SO, Kwong SM, Skurray RA, Ramsay JP. 2018. Staphylococcal plasmids, transposable and integrative elements. Microbiol Spectr 6. doi: 10.1128/microbiolspec.GPP3-0030-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bukowski M, Piwowarczyk R, Madry A, Zagorski-Przybylo R, Hydzik M, Wladyka B. 2019. Prevalence of antibiotic and heavy metal resistance determinants and virulence-related genetic elements in plasmids of Staphylococcus aureus. Front Microbiol 10:805. doi: 10.3389/fmicb.2019.00805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jansen WT, Beitsma MM, Koeman CJ, van Wamel WJ, Verhoef J, Fluit AC. 2006. Novel mobile variants of staphylococcal cassette chromosome mec in Staphylococcus aureus. Antimicrob Agents Chemother 50:2072–2078. doi: 10.1128/AAC.01539-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang TY, Hung WW, Lin L, Hung WC, Tseng SP. 2017. mecA-related structure in methicillin-resistant coagulase-negative staphylococci from street food in Taiwan. Sci Rep 7:42205. doi: 10.1038/srep42205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen L, Mediavilla JR, Smyth DS, Chavda KD, Ionescu R, Roberts RB, Robinson DA, Kreiswirth BN. 2010. Identification of a novel transposon (Tn6072) and a truncated staphylococcal cassette chromosome mec element in methicillin-resistant Staphylococcus aureus ST239. Antimicrob Agents Chemother 54:3347–3354. doi: 10.1128/AAC.00001-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adriaenssens E, Brister JR. 2017. How to name and classify your phage: an informal guide. Viruses 9:70. doi: 10.3390/v9040070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kropinski AM, Prangishvili D, Lavigne R. 2009. Position paper: the creation of a rational scheme for the nomenclature of viruses of bacteria and archaea. Environ Microbiol 11:2775–2777. doi: 10.1111/j.1462-2920.2009.01970.x. [DOI] [PubMed] [Google Scholar]
- 49.Quiles-Puchalt N, Carpena N, Alonso JC, Novick RP, Marina A, Penades JR. 2014. Staphylococcal pathogenicity island DNA packaging system involving cos-site packaging and phage-encoded HNH endonucleases. Proc Natl Acad Sci U S A 111:6016–6021. doi: 10.1073/pnas.1320538111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bosi E, Monk JM, Aziz RK, Fondi M, Nizet V, Palsson BO. 2016. Comparative genome-scale modelling of Staphylococcus aureus strains identifies strain-specific metabolic capabilities linked to pathogenicity. Proc Natl Acad Sci US A 113:E3801–E3809. doi: 10.1073/pnas.1523199113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Argemi X, Matelska D, Ginalski K, Riegel P, Hansmann Y, Bloom J, Pestel-Caron M, Dahyot S, Lebeurre J, Prevost G. 2018. Comparative genomic analysis of Staphylococcus lugdunensis shows a closed pan-genome and multiple barriers to horizontal gene transfer. BMC Genomics 19:621. doi: 10.1186/s12864-018-4978-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sun Z, Zhou D, Zhang X, Li Q, Lin H, Lu W, Liu H, Lu J, Lin X, Li K, Xu T, Bao Q, Zhang H. 2020. Determining the genetic characteristics of resistance and virulence of the “epidermidis cluster group” through pan-genome analysis. Front Cell Infect Microbiol 10:274. doi: 10.3389/fcimb.2020.00274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Conlan S, Mijares LA, Program NCS, Becker J, Blakesley RW, Bouffard GG, Brooks S, Coleman H, Gupta J, Gurson N, Park M, Schmidt B, Thomas PJ, Otto M, Kong HH, Murray PR, Segre JA, NISC Comparative Sequencing Program . 2012. Staphylococcus epidermidis pan-genome sequence analysis reveals diversity of skin commensal and hospital infection-associated isolates. Genome Biol 13:R64. doi: 10.1186/gb-2012-13-7-r64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pain M, Hjerde E, Klingenberg C, Cavanagh JP. 2019. Comparative genomic analysis of Staphylococcus haemolyticus reveals key to hospital adaptation and pathogenicity. Front Microbiol 10:2096. doi: 10.3389/fmicb.2019.02096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Svec P, Petras P, Pantucek R, Doskar J, Sedlacek I. 2016. High intraspecies heterogeneity within Staphylococcus sciuri and rejection of its classification into S. sciuri subsp. sciuri, S. sciuri subsp. carnaticus and S. sciuri subsp. rodentium. Int J Syst Evol Microbiol 66:5181–5186. doi: 10.1099/ijsem.0.001493. [DOI] [PubMed] [Google Scholar]
- 56.Richter M, Rossello-Mora R. 2009. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A 106:19126–19131. doi: 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jain C, Rodriguez RL, Phillippy AM, Konstantinidis KT, Aluru S. 2018. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun 9:5114. doi: 10.1038/s41467-018-07641-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xiang W, Wang G, Wang Y, Yao R, Zhang F, Wang R, Wang D, Zheng S. 2014. Paenibacillus selenii sp. nov., isolated from selenium mineral soil. Int J Syst Evol Microbiol 64:2662–2667. doi: 10.1099/ijs.0.063701-0. [DOI] [PubMed] [Google Scholar]
- 59.Huang H, Feng F, Liu M, Zhang F, Sun Q, Qin S, Bao S. 2016. Paenibacillus segetis sp. nov., isolated from soil of a tropical rainforest. Int J Syst Evol Microbiol 66:3703–3707. doi: 10.1099/ijsem.0.001258. [DOI] [PubMed] [Google Scholar]
- 60.Chen Z, Ouyang W, Chen Y, Tian W, Sun L. 2019. Paenibacillus zeisoli sp. nov., isolated from maize-cultivated soil artificially contaminated with cadmium. Int J Syst Evol Microbiol 69:1149–1154. doi: 10.1099/ijsem.0.003288. [DOI] [PubMed] [Google Scholar]
- 61.Noor Uddin GM, Larsen MH, Guardabassi L, Dalsgaard A. 2013. Bacterial flora and antimicrobial resistance in raw frozen cultured seafood imported to Denmark. J Food Prot 76:490–499. doi: 10.4315/0362-028X.JFP-12-402. [DOI] [PubMed] [Google Scholar]
- 62.Tahrani L, Soufi L, Mehri I, Najjari A, Hassan A, Van Loco J, Reyns T, Cherif A, Ben Mansour H. 2015. Isolation and characterization of antibiotic-resistant bacteria from pharmaceutical industrial wastewaters. Microb Pathog 89:54–61. doi: 10.1016/j.micpath.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 63.Castro-Severyn J, Remonsellez F, Valenzuela SL, Salinas C, Fortt J, Aguilar P, Pardo-Este C, Dorador C, Quatrini R, Molina F, Aguayo D, Castro-Nallar E, Saavedra CP. 2017. Comparative genomics analysis of a new Exiguobacterium strain from Salar de Huasco reveals a repertoire of stress-related genes and arsenic resistance. Front Microbiol 8:456. doi: 10.3389/fmicb.2017.00456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lei J, Zheng M, Wang L, Yin G, Lou Y, Shi L. 2020. Complete genome sequence of Exiguobacterium mexicanum A-EM, isolated from seafloor hydrothermal vents in Atlantic Ocean. Mar Genomics doi: 10.1016/j.margen.2020.100801. [DOI] [PubMed] [Google Scholar]
- 65.Baba T, Kuwahara-Arai K, Uchiyama I, Takeuchi F, Ito T, Hiramatsu K. 2009. Complete genome sequence of Macrococcus caseolyticus strain JCSCS5402, [corrected] reflecting the ancestral genome of the human-pathogenic staphylococci. J Bacteriol 191:1180–1190. doi: 10.1128/JB.01058-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Roberts MC. 2005. Update on acquired tetracycline resistance genes. FEMS Microbiol Lett 245:195–203. doi: 10.1016/j.femsle.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 67.Moon DC, Tamang MD, Nam HM, Jeong JH, Jang GC, Jung SC, Park YH, Lim SK. 2015. Identification of livestock-associated methicillin-resistant Staphylococcus aureus isolates in Korea and molecular comparison between isolates from animal carcasses and slaughterhouse workers. Foodborne Pathog Dis 12:327–334. doi: 10.1089/fpd.2014.1868. [DOI] [PubMed] [Google Scholar]
- 68.Oliveira H, Sampaio M, Melo LDR, Dias O, Pope WH, Hatfull GF, Azeredo J. 2019. Staphylococci phages display vast genomic diversity and evolutionary relationships. BMC Genomics 20:357. doi: 10.1186/s12864-019-5647-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Novick RP, Christie GE, Penades JR. 2010. The phage-related chromosomal islands of Gram-positive bacteria. Nat Rev Microbiol 8:541–551. doi: 10.1038/nrmicro2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. 2017. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27:722–736. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Buchfink B, Xie C, Huson DH. 2015. Fast and sensitive protein alignment using DIAMOND. Nat Methods 12:59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 76.Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV. 2001. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res 29:22–28. doi: 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Haft DH, Loftus BJ, Richardson DL, Yang F, Eisen JA, Paulsen IT, White O. 2001. TIGRFAMs: a protein family resource for the functional identification of proteins. Nucleic Acids Res 29:41–43. doi: 10.1093/nar/29.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Eddy SR. 2011. Accelerated profile HMM searches. PLoS Comput Biol 7:e1002195. doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cabanettes F, Klopp C. 2018. D-GENIES: dot plot large genomes in an interactive, efficient and simple way. PeerJ 6:e4958. doi: 10.7717/peerj.4958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.R Development Core Team. 2018. R: a language and environment for statistical computing, R Foundation for Statistical Computing.
- 81.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Treangen TJ, Ondov BD, Koren S, Phillippy AM. 2014. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol 15:524. doi: 10.1186/s13059-014-0524-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. 2011. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MT, Fookes M, Falush D, Keane JA, Parkhill J. 2015. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31:3691–3693. doi: 10.1093/bioinformatics/btv421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gómez-Sanz E, Haro-Moreno JM, Jensen SO, Roda-García JJ, López-Pérez M. 2020. Staphylococcus sciuri C2865 from a distinct subspecies cluster as reservoir of the novel transferable trimethoprim resistance gene, dfrE, and adaptation driving mobile elements. bioRxiv doi: 10.1101/2020.09.30.320143. [DOI]
- 86.O'Brien FG, Yui Eto K, Murphy RJ, Fairhurst HM, Coombs GW, Grubb WB, Ramsay JP. 2015. Origin-of-transfer sequences facilitate mobilisation of non-conjugative antimicrobial-resistance plasmids in Staphylococcus aureus. Nucleic Acids Res 43:7971–7983. doi: 10.1093/nar/gkv755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.del Solar GH, Puyet A, Espinosa M. 1987. Initiation signals for the conversion of single stranded to double stranded DNA forms in the streptococcal plasmid pLS1. Nucleic Acids Res 15:5561–5580. doi: 10.1093/nar/15.14.5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.de Castro E, Sigrist CJ, Gattiker A, Bulliard V, Langendijk-Genevaux PS, Gasteiger E, Bairoch A, Hulo N. 2006. ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res 34:W362–W365. doi: 10.1093/nar/gkl124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, Qureshi M, Richardson LJ, Salazar GA, Smart A, Sonnhammer ELL, Hirsh L, Paladin L, Piovesan D, Tosatto SCE, Finn RD. 2019. The Pfam protein families database in 2019. Nucleic Acids Res 47:D427–D432. doi: 10.1093/nar/gky995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Marchler-Bauer A, Bo Y, Han L, He J, Lanczycki CJ, Lu S, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Lu F, Marchler GH, Song JS, Thanki N, Wang Z, Yamashita RA, Zhang D, Zheng C, Geer LY, Bryant SH. 2017. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res 45:D200–D203. doi: 10.1093/nar/gkw1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Varani AM, Siguier P, Gourbeyre E, Charneau V, Chandler M. 2011. ISsaga is an ensemble of web-based methods for high throughput identification and semi-automatic annotation of insertion sequences in prokaryotic genomes. Genome Biol 12:R30. doi: 10.1186/gb-2011-12-3-r30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 94.Kumar S, Stecher G, Tamura K. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gutiérrez D, Adriaenssens EM, Martínez B, Rodríguez A, Lavigne R, Kropinski AM, García P. 2014. Three proposed new bacteriophage genera of staphylococcal phages: “3alikevirus”, “77likevirus” and “Phietalikevirus”. Arch Virol 159:389–398. doi: 10.1007/s00705-013-1833-1. [DOI] [PubMed] [Google Scholar]
- 96.Schwendener S, Perreten V. 2015. New shuttle vector-based expression system to generate polyhistidine-tagged fusion proteins in Staphylococcus aureus and Escherichia coli. Appl Environ Microbiol 81:3243–3254. doi: 10.1128/AEM.03803-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.CLSI. 2018. Performance standards for antimicrobial susceptibility testing; twenty-eighth informational supplement (M100-S28). Clinical and Laboratory Standards Institute. [Google Scholar]
- 98.Kaiser TD, Pereira EM, Dos Santos KR, Maciel EL, Schuenck RP, Nunes AP. 2013. Modification of the Congo red agar method to detect biofilm production by Staphylococcus epidermidis. Diagn Microbiol Infect Dis 75:235–239. doi: 10.1016/j.diagmicrobio.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 99.Schmelcher M, Shen Y, Nelson DC, Eugster MR, Eichenseher F, Hanke DC, Loessner MJ, Dong S, Pritchard DG, Lee JC, Becker SC, Foster-Frey J, Donovan DM. 2015. Evolutionarily distinct bacteriophage endolysins featuring conserved peptidoglycan cleavage sites protect mice from MRSA infection. J Antimicrob Chemother 70:1453–1465. doi: 10.1093/jac/dku552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.del Solar G, Giraldo R, Ruiz-Echevarria MJ, Espinosa M, Diaz-Orejas R. 1998. Replication and control of circular bacterial plasmids. Microbiol Mol Biol Rev 62:434–464. doi: 10.1128/MMBR.62.2.434-464.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gomez-Sanz E, Ceballos S, Ruiz-Ripa L, Zarazaga M, Torres C. 2019. Clonally diverse methicillin and multidrug resistant coagulase negative staphylococci are ubiquitous and pose transfer ability between pets and their owners. Front Microbiol 10:485. doi: 10.3389/fmicb.2019.00485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhang Q, Xing S, Sun Q, Pei G, Cheng S, Liu Y, An X, Zhang X, Qu Y, Tong Y. 2017. Characterization and complete genome sequence analysis of a novel virulent Siphoviridae phage against Staphylococcus aureus isolated from bovine mastitis in Xinjiang, China. Virus Genes 53:464–476. doi: 10.1007/s11262-017-1445-z. [DOI] [PubMed] [Google Scholar]
- 103.Andrews S. 2010. FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. [Google Scholar]
- 104.Madhaiyan M, Wirth JS, Saravanan VS. 2020. Phylogenomic analyses of the Staphylococcaceae family suggest the reclassification of five species within the genus Staphylococcus as heterotypic synonyms, the promotion of five subspecies to novel species, the taxonomic reassignment of five Staphylococcus species to Mammaliicoccus gen. nov., and the formal assignment of Nosocomiicoccus to the family Staphylococcaceae. Int J Sys Evol Microbiol 70:5926–5936. doi: 10.1099/ijsem.0.004498. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Dot-plot graphs of MRSS C2865 genome assembled of PacBio and of Illumina reads using D-Genies. The program searches all the query sequences aligned on the diagonal and calculates the target sequence coverage per identity bin (79). Left graph shows the genome-genome alignments using PacBio assembly as reference (chromosomal contig and plasmid contig) (x axes) with Illumina query (y axes), while right graph depicts the genome-genome alignments using Illumina-assembled contigs as reference (x axes) with PacBio query (y axes). The summary identity calculation (>75% identity, <75%, <50%, and <25% and no matches) is made on the target sequence (reference) and represents the percentage of the target genome in base pairs. Note: node 96 (5,513 bp) denotes plausible contamination after contig manual checking. Download FIG S1, EPS file, 0.5 MB (495.4KB, eps) .
Copyright © 2021 Gómez-Sanz et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
This file includes a detailed description of methods, further methods not indicated in the main text, additional results, and an extended discussion. Download Text S1, PDF file, 0.5 MB (479KB, pdf) .
Copyright © 2021 Gómez-Sanz et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Graphical representation of the RCR plasmids detected in MRSS C2865 and related plasmids. Arrows denote the genes, length, and orientation. Gene colors other than gray represent the following functions (color): replication and/or mobilization (green), antimicrobial resistance (pink, yellow). Areas of nucleotide similarity plasmids are indicated in gray. (A) Nucleotide sequence comparative analysis of the three RCR plasmids detected in MRSS C2865: pUR2865-1, pUR2865-2, and pUR2865-int, which is integrated in the larger pUR2865-34 plasmid. (B) Nucleotide sequence comparative analysis of pC194-related pUR2865-1 and S. aureus pC194 (GenBank accession no. NC_002013). Estimated double-strand origin (dso) and single-strand origin (sso) of replication are indicated. (C) Nucleotide sequence comparative analysis of the pT181-related pUR2865-2, the integrated pUR2865-int, and the RCR family prototype S. aureus pT181 (GenBank accession no. J01764.1). Putative dso nick and sso sites for pUR2865-1 and pUR2865-2 are denoted (100). Origin-of-transfer (oriT) for pT181-related plasmids is also indicated. (D) Sequence comparison of closest plasmids to pUR2865-1 (> 80% ID, > 70% coverage). (E) Sequence comparison of plasmids closest to pUR2865-2 (> 80% ID, > 70% coverage). Download FIG S4, SVG file, 0.5 MB (387.7KB, svg) .
Copyright © 2021 Gómez-Sanz et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Phylogenomic tree and heat map resultant from the average nucleotide identity (ANI) of 30 Staphylococcus sciuri group genomes, plus one Staphylococcus aureus and one Staphylococcus epidermidis reference genome used as outgroup for the genus level. Download FIG S2, EPS file, 0.3 MB (261.9KB, eps) .
Copyright © 2021 Gómez-Sanz et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Graphical overview of MRSS C2865 ψTn554 chromosomal region and comparative elements. The diagram depicts (i) MRSS C2865 ψTn554 chromosomal region including the interrupted radC gene, (ii) its corresponding closest relative S. aureus 6850 (GenBank accession no. CP006706), and (iii) the chromosomal radC region and remnant 3′-end radC of phenicol and oxazolidinone resistant S. sciuri wo22_7 (KX982170) and S. sciuri MS11-3 (KX447571), respectively. Arrows denote the genes, length, and orientation. Gene colors other than gray represent the following functions (color): truncated or remnant chromosomal integration radC in Tn554 family transposons (black), antimicrobial resistance (pink), metal resistance or transport (bright blue), transposition or recombination (yellow), ABC transporter ATP-binding proteins (faint brown). The 6-bp nucleotides resultant from Tn554-like integration are also depicted. Areas of nucleotide similarity (nblastn, >100 bp match, >85% identity) between strains/structures are indicated in gray. Download FIG S3, EPS file, 0.2 MB (196.3KB, eps) .
Copyright © 2021 Gómez-Sanz et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Neighbor-joining tree of aligned amino acid sequences of S. sciuri group dihydrofolate reductases. The alignment was performed with (i) the entire dihydrofolate reductase (Dhfr) protein(s) present in all S. sciuri species group genomes deposited in the NCBI database (accessed until April 2018), (ii) both Dhfrs present in S. sciuri strain C2865, and (iii) the amino acid sequence of all trimethoprim resistance dihydrofolate reductases (Dfr) described so far in staphylococci (DfrA, DfrD, DfrG, DfrK, DfrF) (32–36), as well as (iv) the Dhfr of trimethoprim-susceptible S. epidermidis ATCC 12228 (NC_004461) and S. aureus ATCC 25923 (Z16422) as reference, in blue color. Clusters enclosing the different TMP resistance Dfrs are colored differently. Download FIG S5, EPS file, 0.2 MB (165.3KB, eps) .
Copyright © 2021 Gómez-Sanz et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Biofilm formation ability of ica-locus variant-carrying S. sciuri strains and transformants. (A) Biofilm formation capacity based on visual analysis of colony colors by a modified Congo red agar (CRAmod) assay. Spots were plated in duplicates. From left to right, (1) the positive controls (strong biofilm formers) S. aureus SA113 (DSM 4910) and S. aureus ATCC 25923 (DSM 1104), (2) negative-control (pUR2865-34 recipient) strain S. aureus RN4220, (3 and 4) the original S. sciuri ica-locus variant-carrying strains C2865, C2853, C2854, and C2855, (5) two selected S. aureus RN4220/pUR2865-34 transformants (S319 and S320). (B) Quantification of biofilm formation by crystal violet (CV) staining of adherent cells. The bar chart displays the results of six independent experiments in triplicates. Original S. sciuri ica-locus variant-carrying strains C2865, C2853, C2854, and C2855, two selected S. aureus RN4220/pUR2865-34 transformants (S319 and S320) as well as an ica-negative non-biofilm-producing methicillin-resistant Staphylococcus lentus strain (C3030) (101) are included. The three bars on the left show the control strains used: the strong biofilm formers S. aureus SA113 (DSM 4910) and S. aureus ATCC 25923 (DSM 1104), as well as S. aureus RN4220 strain. Significant differences by t test or analysis of variance (ANOVA; P < 0.01) are indicated with two stars on the compared cluster, i.e., ica-carrying strains (C2865, C2853, C2854, C2855, S319, S320; ns, not significant differences between values) and C3030, and ica-carrying strains and RN4220. Download FIG S6, EPS file, 0.7 MB (747.2KB, eps) .
Copyright © 2021 Gómez-Sanz et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Phylogenomic tree and heat map resultant from the average nucleotide identity (ANI) of a subset of 43 staphylococcal phages deposited in the Viral RefSeq NCBI database positioned within the same subbranch as C2865-pp1 to -pp3, plus the three prophages from S. sciuri strain C2865 colored in green. A phylogenomic tree including all 187 staphylococcal phages included in the analysis is represented in the upper left corner. Download FIG S7, PDF file, 2.5 MB (2.5MB, pdf) .
Copyright © 2021 Gómez-Sanz et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Comparison of codon usage (A) and amino acid (B) percentage between the three prophages (C2865-pp1, C2865-pp2, C2865-pp3) and C2865 host genome. The scale (in percentage) in the left plots is represented at the upper center of the graph. (A) Left, radar plot of the 64 different amino acid codons (at DNA level) highlighting the tRNA gene present in prophage C2865-pp3 genome. Right, specific abundance (%) of TGG codon (UGG). (B) Left, radar plot of the 20 amino acids highlighting proline (P), as resultant amino acid from tRNA present in C2865-pp3. Right, specific abundance (%) of proline (P). Download FIG S9, EPS file, 0.2 MB (168.3KB, eps) .
Copyright © 2021 Gómez-Sanz et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Circular phylogenetic tree of all available staphylococcal terminase large subunit (TerL). The tree built with the amino acid sequences of all identifiable terminase large subunit (TerL) present in 187 staphylococcal phages deposited in the Viral RefSeq database, TerL of C2865-pp1, C2865-pp2, and C2865-pp3, and TerL of Enterobacteria phage lambda, used as outlier. Phage nomenclature: S-phage, which refers to staphylococcal phage, is followed by the phage name, faconcant (refering to concatenated fasta), plus the CDSs number of respective TerL in the phage genome. Branch enclosing the C2865-pp1, C2865-pp2, and C2865-pp3 integrases is bold. C2865-pp1, C2865-pp2, and C2865-pp3 integrases are boxed in green, while phage lambda is boxed in gray. Phages with known packaging mechanism (95), carrying either cos-sites (cos packaging strategy) or pac-sites (headful packaging strategy), are indicated in black and green, respectively. Download FIG S8, PDF file, 1.6 MB (1.6MB, pdf) .
Copyright © 2021 Gómez-Sanz et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
The whole-genome shotgun sequence data supporting this work are available via NCBI SRA database under BioProject and Biosample accession numbers PRJNA663854 and SAMN16182282, respectively.