

ABSTRACT
The Yersinia pestis pH 6 antigen (PsaA) forms fimbria-like structures and is required for full virulence during bubonic plague. High temperature and low pH regulate PsaA production, and while recent work has uncovered the molecular aspects of temperature control, the mechanisms underlying this unusual regulation by pH are poorly understood. Using defined growth conditions, we recently showed that high levels of PsaE and PsaF (two regulatory proteins required for expression of psaA) are present at mildly acidic pH, but these levels are greatly reduced at neutral pH, resulting in low psaA expression. In prior work, the use of translational reporters suggested that pH had no impact on translation of psaE and psaF, but rather affected protein stability of PsaE and/or PsaF. Here, we investigated the pH-dependent posttranslational mechanisms predicted to regulate PsaE and PsaF stability. Using antibodies that recognize the endogenous proteins, we showed that the amount of PsaE and PsaF is defined by a distinct pH threshold. Analysis of histidine residues in the periplasmic domain of PsaF suggested that it functions as a pH sensor and indicated that the presence of PsaF is important for PsaE stability. At neutral pH, when PsaF is absent, PsaE appears to be targeted for proteolytic degradation by regulated intramembrane proteolysis. Together, our work shows that Y. pestis utilizes PsaF as a pH sensor to control psaA expression by enhancing the stability of PsaE, an essential psaA regulatory protein.
IMPORTANCE Yersinia pestis is a bacterial pathogen that causes bubonic plague in humans. As Y. pestis cycles between fleas and mammals, it senses the environment within each host to appropriately control gene expression. PsaA is a protein that forms fimbria-like structures and is required for virulence. High temperature and low pH together stimulate psaA transcription by increasing the levels of two essential integral membrane regulators, PsaE and PsaF. Histidine residues in the PsaF periplasmic domain enable it to function as a pH sensor. In the absence of PsaF, PsaE (a DNA-binding protein) appears to be targeted for proteolytic degradation, thus preventing expression of psaA. This work offers insight into the mechanisms that bacteria use to sense pH and control virulence gene expression.
KEYWORDS: PsaE, PsaF, PsaA, pH 6 antigen, Y. pestis, pH sensing, ToxR/S, TcpP/H, DegP, Tsp, regulated intramembrane proteolysis, PsaE, Yersinia pestis
INTRODUCTION
Yersinia pestis is a vector-borne bacterial pathogen that cycles between flea and mammalian hosts to cause bubonic plague, one of three forms of the disease plague (1, 2). Bubonic plague is the most common form of the disease in humans and occurs when bacteria are deposited into the dermal layer of skin during the bite of an infected flea. From the skin, bacteria disseminate through lymphatic vessels to a draining lymph node (3, 4), where they proliferate to high numbers, resulting in enlarged lymph nodes known as “buboes,” a hallmark of bubonic plague. At late stages of disease, bacteria enter the bloodstream and can cause a fatal septicemia. Transcriptome analyses have revealed distinct expression profiles within the flea and mammalian host (5–7), suggesting that Y. pestis has regulatory mechanisms to distinguish between these two environments. While the cues that Y. pestis encounters within each host environment are not well defined, temperature is a key distinguishing signal, as many Y. pestis virulence genes necessary for colonizing mammalian hosts are expressed following an upshift in temperature from 26°C to 37°C (7, 8). One such virulence factor upregulated at 37°C is the “pH 6 antigen” (PsaA). PsaA is produced during mammalian infection and is required for full virulence of Y. pestis in multiple murine models of disease (9–13). Despite this impact, the exact role of PsaA during infection is not known. PsaA forms homopolymeric fimbria-like structures on the bacterial cell surface (14), and there is evidence suggesting that PsaA functions to both promote host cell adherence and inhibit phagocytosis (15, 16). Interestingly, high temperature and low pH are both required to activate psaA transcription and PsaA production in vitro (9, 10, 17). Despite this unusual regulation, the underlying molecular mechanisms have not been thoroughly investigated.
Two regulatory proteins encoded upstream of psaA, PsaE and PsaF, are required for psaA transcription (10, 14, 17, 18) and are predicted to play a role in the unusual expression pattern of psaA (18, 19). PsaE and PsaF belong to a family of regulatory protein pairs that localize to the inner membrane and function as transcriptional activators (20–27). The ToxR/ToxS and TcpP/TcpH protein pairs in Vibrio cholerae are the most studied members of this family (21–23, 28–33). ToxR and TcpP are integral membrane proteins that directly coactivate expression of toxT, which encodes the major V. cholerae virulence regulator ToxT (28, 34–36). Like ToxR and TcpP, PsaE contains an N-terminal cytoplasmic OmpR-like DNA-binding domain, a single transmembrane domain, and a C-terminal periplasmic domain (19), and PsaE is predicted to directly activate psaA transcription (37). The periplasmic domain of PsaE constitutes ∼25% of the protein, but the contribution of this domain to the function of PsaE is not known.
The topology of PsaE seems well suited to both sense environmental signals and activate gene expression, yet PsaE alone is not sufficient to activate psaA transcription. PsaF is also required and appears to act at least in part by enhancing PsaE stability (18). The PsaF-like proteins TcpH and ToxS inhibit degradation of TcpP and ToxR, respectively, and are thus thought to play indirect roles in toxT transcription (29, 31, 38, 39). Like TcpH and ToxS, PsaF contains a single transmembrane domain near the N terminus, with the majority of the protein located in the periplasm. PsaF may also enhance the ability of PsaE to activate psaA transcription, as the very low levels of PsaE in the absence of PsaF are unable to activate expression of psaA (18).
We previously reported that temperature and pH influence levels of both PsaE and PsaF in Y. pestis via distinct posttranscriptional mechanisms (18). High temperature promotes the production of both PsaE and PsaF; a predicted RNA thermometer located in the 5′ untranscribed region (UTR) of psaE modulates psaE translation, and psaF translation is controlled through an independent mechanism by sequences upstream of psaF (18). The levels of PsaE and PsaF detected at high temperature are controlled by environmental pH. High levels of both PsaE and PsaF are present at mildly acidic pH (pH 6.3), but at neutral pH (pH 7.3), PsaE is present at very low levels and PsaF is undetectable (18). Since translation of psaE and psaF does not appear to be impacted by pH, we predicted that additional pH-dependent posttranslational mechanisms were involved. To further understand how pH regulates transcription of psaA in Y. pestis, we set out to define the mechanism(s) by which pH influences the levels of PsaE and PsaF. Thus, we analyzed PsaE and PsaF levels following growth in media buffered to different pH levels. Additionally, we identified residues in the periplasmic domains of PsaE and PsaF that influence protein stability. Our data indicate that the stability of PsaE and PsaF is mutually dependent and that both proteins are sensitive to pH, further supporting the idea that Y. pestis utilizes these integral membrane proteins to control the expression of psaA in response to precise environmental cues.
RESULTS
A distinct pH threshold controls the levels of PsaE and PsaF.
We previously reported that high levels of PsaE and PsaF are detected at 37°C and pH 6.3 and that both proteins are greatly reduced at 37°C and pH 7.3 (18). Translation of psaE and psaF is not impacted by pH (18), and thus it is likely that posttranslational mechanisms are involved. To begin to address how PsaE and PsaF are affected by pH, Y. pestis strain CO92 cured of the virulence plasmid pCD1 (YP6; here referred to as the wild type [WT]) was grown at 37°C in brain heart infusion (BHI) buffered to pH 6.3, 6.5, 6.7, 7.0, or 7.3. Consistent with our previous findings (18), high levels of PsaE and PsaF were detected at pH 6.3, and levels were much lower at pH 7.3 (Fig. 1A). While both proteins were readily detected at pH 6.5 and pH 6.7, PsaF was undetectable at a pH of >6.7. PsaE levels were noticeably reduced at pH 6.7 and were very low at pH 7 and 7.3. These data confirm that pH impacts PsaE and PsaF levels and reveal a distinct threshold between pH 6.7 and 7 that defines the levels of these proteins.
FIG 1.

A distinct pH threshold defines PsaE and PsaF levels and impacts PsaF stability. (A) The wild type (WT; YP6) was grown at 37°C in brain heart infusion (BHI) buffered to pH 6.3, 6.5, 6.7, 7.0, and 7.3, and PsaE and PsaF were analyzed via Western blot as described in Materials and Methods. As a control, whole-cell lysates of the ΔpsaEF mutant (YPA18) grown at 37°C in BHI pH 6.3 were also analyzed. (B) WT was grown in BHI pH 6.3 for 8 h, cells were pelleted, washed and suspended in phosphate-buffered saline (PBS) adjusted to pH 6.3 or pH 7.3 as indicated in Materials and Methods, and PsaE and PsaF were analyzed over time. LC, loading control from Ponceau S-stained membrane.
Because pH does not appear to affect translation of psaF (18), we hypothesized that pH affects the stability of PsaF. To test this, the WT strain was first grown at 37°C pH 6.3 to the late exponential phase to allow production of PsaE and PsaF. Bacterial cells were then pelleted and suspended in phosphate-buffered saline (PBS) (a nutrient-limiting condition) at pH 6.3 or pH 7.3, and PsaF levels were monitored over time (Fig. 1B). At pH 7.3 in PBS, PsaF levels were reduced within 2 h, whereas a similar change in PsaF levels was not observed at pH 6.3 in PBS. A comparable number of bacteria were recovered after 2 h from both conditions, and this was comparable to the number of bacteria present when they were initially suspended in PBS (data not shown), indicating that cell viability did not contribute to the changes in PsaF levels at pH 7.3. In contrast, once PsaE was produced during growth at pH 6.3, its stability was minimally affected by the shift to pH 7.3 in PBS (Fig. 1B). These data suggest that PsaF is less stable at pH 7.3 than at pH 6.3, but PsaE is relatively stable at both pH levels under these conditions. The relative stability of PsaE at pH 7.3 could be explained by low levels of PsaF being sufficient to stabilize PsaE, or it could indicate that, once produced, PsaE is stable under these conditions.
DegP and Tsp influence levels of PsaE.
As with PsaF, translation of psaE is not affected by pH (18), and we thus predicted that during bacterial growth, pH may also control the stability of PsaE. Regulated intramembrane proteolysis (RIP) is a two-step degradation of membrane proteins that occurs in most domains of life, and recent work shows that bacteria utilize RIP to control virulence gene expression (40, 41). During RIP, a target protein is sequentially cleaved, first by a periplasmic protease (site 1 protease) and then by an integral membrane protease (site 2 protease), resulting in complete degradation of the target protein. The PsaE-like proteins, TcpP and ToxR, are degraded via RIP (30, 38, 39), and ToxS and TcpH (PsaF-like proteins) inhibit degradation of ToxR and TcpP, respectively (29, 31). Therefore, we wanted to determine if PsaE also could be subjected to proteolytic degradation via RIP. Tsp and YaeL (RseP) are the site 1 and site 2 proteases, respectively, that degrade TcpP in Vibrio cholerae, and since deletion of tsp and/or yaeL restores the ability of TcpP to activate transcription of toxT (38, 39), we speculated that preventing degradation of PsaE at pH 7.3 would restore transcription of psaA. To test this, genes predicted to encode periplasmic (site 1; tsp, degS, degQ, and degP) and membrane (site 2; rseP, ypfJ, ypo0398, and ftsH) proteases were mutated and psaA expression was analyzed after growth at 37°C and pH 6.3 or pH 7.3. The insertion mutations in degS, degQ, ypfJ, ypo0398, and ftsH did not impact psaA expression, suggesting that the proteases encoded by these genes are not required for degradation of PsaE. However, we cannot exclude the possibility that these insertions did not completely disrupt gene function.
Of the eight mutants tested, only the tsp and degP insertion mutants exhibited increased psaA expression at pH 7.3 relative to that of the WT strain (Fig. 2A). Mutations in the tsp and degP genes largely attenuated Y. pestis growth, indicating these proteases play a critical role in Y. pestis physiology. Despite the lower growth rate of these mutants, these data suggest that DegP and Tsp individually contribute to low psaA expression under this noninducing condition. The rseP insertion mutant had reduced psaA expression at pH 6.3 and was severely attenuated for growth on agar plates and in broth medium. We were unable to generate an ΔrseP mutant, which is consistent with previous reports that rseP is an essential gene in Y. pestis (42, 43). Thus, it is difficult to determine if RseP plays a role in PsaE degradation or if the effect on psaA expression of the rseP insertion mutant is a consequence of the growth defect.
FIG 2.

DegP and Tsp contribute to low levels of PsaE and psaA transcription at neutral pH. Strains with mutations in putative protease genes were grown at 37°C for 8 h in buffered BHI, and psaA transcription and PsaE were analyzed as indicated in Materials and Methods. (A) The psaA-gfp reporter (pEW102) was introduced into the indicated strains with insertion disruption mutations in the indicated gene, and expression was measured. ***, P < 0.0001; one-way analysis of variance (ANOVA) and Dunnett’s multiple-comparison test. (B) Whole-cell lysates of WT (YP6), ΔpsaEF (YPA18), ΔdegP (YPA425), and ΔdegP ΔpsaEF (YPA469) strains grown at 37°C in BHI buffered to pH 6.3 and 7.3 were probed for PsaE via Western blot. LC, loading control. (C) Whole-cell lysates of WT, ΔpsaEF, Δtsp (YPA350), and Δtsp ΔpsaEF (YPA379) strains were grown and analyzed as described for panel B. A band that reacted with anti-PsaE serum was used as a loading control (LC). (D) The WT strain, the ΔdegP strain, and the strain with degP strain restored at the native site (degP-C; YPA476) were transformed with psaA-gfp, and expression was measured. Student’s t test was used to compare mean values. ***, P < 0.0001; ns, not significant. (E) The WT strain, the Δtsp strain, and the strain with tsp restored at the native site (tsp-C; YPA438) were transformed with psaA-gfp, and expression was measured. Student’s t test was used to compare mean values. ***, P < 0.0001; ns, not significant.
The increase in psaA expression in the degP and tsp mutants at 37°C and pH 7.3 suggested that PsaE levels and/or activity may be partially restored by loss of these site 1 proteases. As insertion disruption mutations can have polar effects on downstream genes, we constructed individual degP and tsp deletion mutants (ΔdegP and Δtsp). These deletion mutants were then tested to determine if the altered psaA expression at pH 7.3 was associated with changes in PsaE levels. The WT and mutant strains were grown at pH 6.3 and pH 7.3, and PsaE was analyzed via Western blot. The amount of PsaE present at pH 7.3 was elevated in both the ΔdegP and Δtsp mutants relative to that in WT, suggesting that increases in psaA transcription were due to an increase in PsaE levels (Fig. 2B and C). Yet, PsaF remained undetectable in both mutants at pH 7.3, indicating that Tsp and DegP are not individually responsible for low levels of PsaF under this condition (see Fig. S1 in the supplemental material).
To validate the impact of the predicted site 1 proteases on psaA expression, strains with degP or tsp restored were generated by reintroducing degP (YPA476) or tsp (YPA438) at the native site of the ΔdegP and Δtsp mutants, respectively. As seen with the insertion mutants, expression of psaA in both the Δtsp and ΔdegP mutants was higher relative to that in the WT at pH 7.3, suggesting that the absence of these proteases is sufficient to partially restore PsaE activity at pH 7.3 (Fig. 2D and E). Reintroduction of tsp and degP into the Δtsp and ΔdegP mutants resulted in psaA expression comparable to that of the WT at 37°C pH 7.3, indicating that the observed effect of the Δtsp and ΔdegP mutations was due to the loss of Tsp and DegP, respectively, rather than due to secondary site mutations. While expression of psaA increased in the ΔdegP mutant (4-fold) and in the Δtsp mutant (5-fold) relative to that in the WT at pH 7.3, expression levels remained low relative to those at pH 6.3. It is possible that this is due to the potential redundancy in Tsp and DegP activity. Our attempt to generate a ΔdegP Δtsp double mutant to examine this was unsuccessful, likely due to the significant impacts on growth observed with the individual mutants. Under the growth conditions used for these assays, the final optical density at 600 nm (OD600) for the degP and tsp mutants were 17% and 24% that of the WT at pH 7.3, respectively. Together, these data suggest DegP and Tsp individually influence PsaE levels, but it is difficult to determine if these proteases have redundant roles at 37°C and pH 7.3.
Cysteine residues in PsaE are required for PsaE and PsaF stability.
While PsaE is thought to directly activate psaA transcription via its DNA-binding domain (37), the role of the periplasmic domain is unknown. Cysteines in the periplasmic domain affect the stability of TcpP and are predicted to impact the function of ToxR (33, 44–47). Like ToxR and TcpP, PsaE contains two cysteine residues in its periplasmic domain. PsaE variants with individual cysteine-to-serine substitutions (PsaEC206S and PsaEC211S) and a double cysteine-to-serine substitution (PsaEC206S/C211S) were constructed. Expression of psaA was similar to that in the ΔpsaEF mutant in all three PsaE cysteine-to-serine mutants, indicating the importance of these cysteine residues (Fig. 3A).
FIG 3.

Cysteine residues in the PsaE periplasmic domain impact PsaE and PsaF stability. Strains containing alleles encoding wild-type PsaE or PsaE mutants with cysteine-to-serine substitutions were grown at 37°C in BHI buffered to pH 6.3 or 7.3, and psaA transcription, PsaF, and PsaE were analyzed as indicated in Materials and Methods. (A) The psaA-gfp reporter was introduced into the ΔpsaEF mutant (YPA18), or into derivations of the ΔpsaEF mutant expressing wild-type psaEF (YPA260; +psaEF), psaEC206S (YPA275; C206S), psaEC211S (YPA277; C211S), or psaEC206S/C211S (YPA276; C206S/C211S), and psaA expression was measured. ***, P < 0.0001 using one-way ANOVA and Dunnett’s multiple-comparison test. (B) Whole-cell lysates from the same strains shown in panel A (lacking psaA-gfp) were analyzed for PsaF via Western blot. (C) Whole-cell lysates from the strains shown in panel B transformed with pPsaF or vector (pWKS30) were used to analyze PsaE. A band that cross-reacted with anti-PsaE serum was used as a loading control (LC).
In V. cholerae, substitution of either cysteine in the TcpP periplasmic domain greatly reduces the stability of both TcpP and TcpH (33). To determine if the cysteine substitutions in PsaE impacted the stability of PsaF, the PsaE cysteine-to-serine mutants were grown at pH 6.3, and PsaF was analyzed via Western blot (Fig. 3B). Strikingly, PsaF was undetectable in all three PsaE mutants. Because the psaE open reading frame overlaps with that of psaF, it is possible that the nucleotide alterations encoding PsaEC206S (25 nucleotides upstream of psaF) or PsaEC211S (10 nucleotides upstream of psaF) disrupted translation of psaF. As PsaF is required for PsaE stability (18), a disruption of psaF translation could lead to an absence of PsaE in strains encoding PsaE cysteine-to-serine mutations. To test this, psaF was expressed in trans (pPsaF) and introduced in the psaE cysteine mutants. Introduction of pPsaF into a ΔpsaF mutant (psaE+) restores PsaE levels (18); however, the presence of pPsaF was not sufficient to restore detection of PsaEC206S, PsaEC211S, or PsaEC206S/C211S, indicating that reduced translation of psaF in these mutants likely was not responsible for the lack of detectable PsaE (Fig. 3C). Together, these data indicate that both cysteines in the C-terminal periplasmic domain of PsaE impact the stability of both PsaE and PsaF.
PsaF is a pH sensor.
The large periplasmic domain of PsaF seems ideal for sensing environmental cues such as extracellular pH. This region contains nine histidine residues. The pKa of histidine imidazole is ∼6, and histidine protonation at mildly acidic pH levels can impart a “pH-sensing” role shown to modulate the conformation and/or activity of multiple bacterial two-component sensor kinases and viral membrane proteins (48–54). The Yersinia enterocolitica PsaF homologue, MyfF, also contains a histidine-rich periplasmic domain, and six of these histidines are conserved in PsaF (Fig. 4A). While the effect of pH on MyfF has not been reported, the expression of myfA (psaA homologue) is elevated at acidic pH (55, 56). Thus, we wanted to determine if these histidines drive pH-dependent function and/or stability of PsaF. To test this, psaF mutants encoding individual histidine-to-alanine substitutions were expressed with a wild-type psaE allele on the chromosome at the native site. Individual substitutions were made for the six histidines shared between PsaF and MyfF. Western blot analysis of lysates from the six individual PsaF mutants grown at 37°C and pH 6.3 indicated that only PsaFH40A was readily detected at levels similar to those of WT PsaF (Fig. 4B). Corresponding with the presence of PsaF, psaA was highly expressed only in the WT strain and the mutant PsaFH40A strain (Fig. 4C). A triple His-to-Ala substitution was also made for the three histidines unique to PsaF (PsaFH38A, PsaFH116A, and PsaFH159A). Of note, this triple His-to-Ala substitution mutant also produced PsaA (see Fig. S2 in the supplemental material), suggesting that H40 and the three histidine residues unique to PsaF do not significantly impact PsaF function under this condition. The other five PsaF mutants (H54A, H87A, H141A, H153A, and H155A) were undetectable at pH 6.3, suggesting these residues are essential for PsaF stability under this condition. Consistent with the absence of PsaF, very low levels of psaA expression were detected in these mutants (Fig. 4C).
FIG 4.
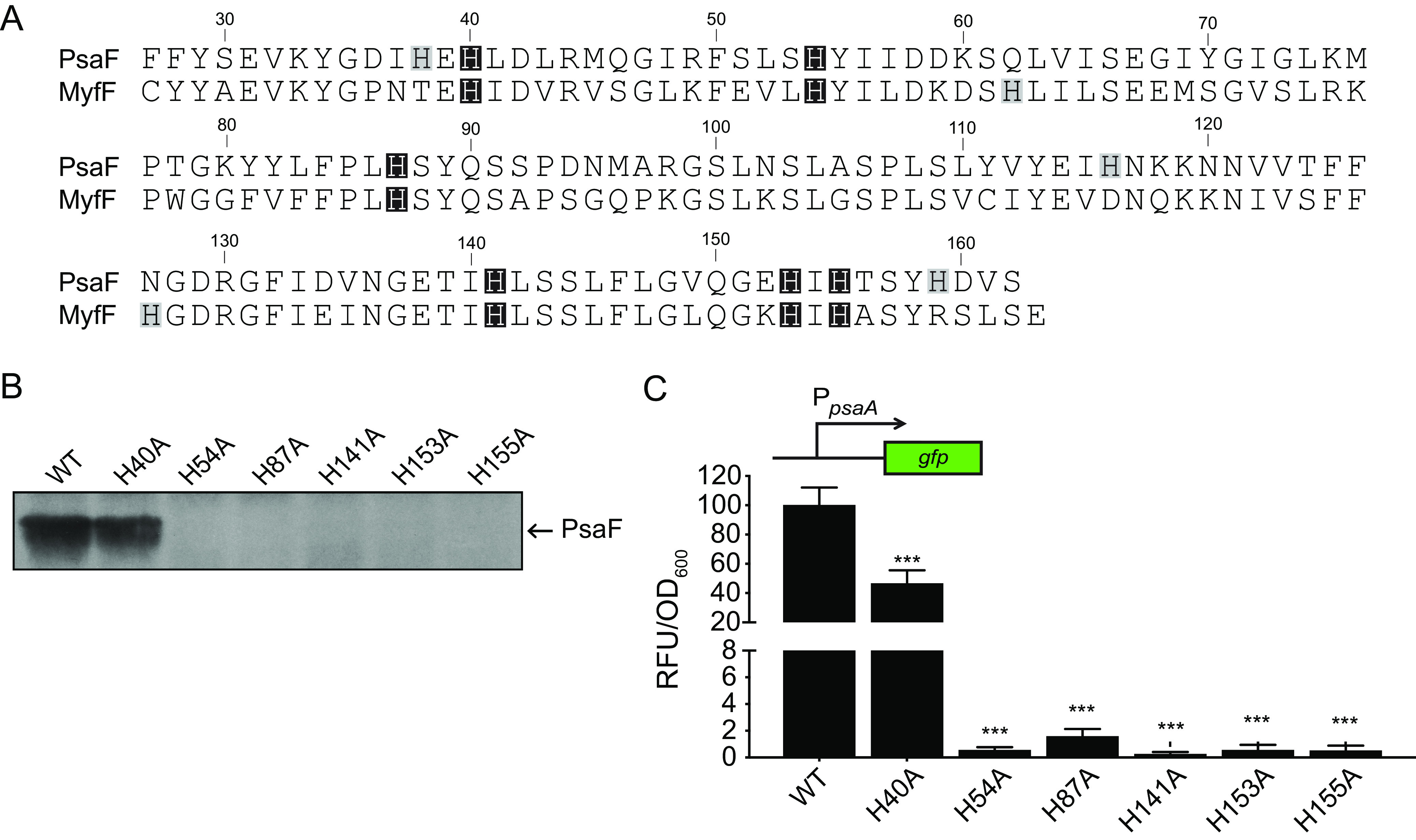
Histidine residues in the PsaF periplasmic domain contribute to PsaF stability and transcription of psaA. (A) Amino acid alignment of the PsaF and MyfF periplasmic domains. DNA sequences were obtained from NCBI and translated using Geneious Prime 2019.2.3 (PsaF, Y. pestis CO92, GenBank accession number NC_003143; MyfF, Y. enterocolitica 8081, accession number NC_008800). Histidine residues conserved between both proteins are highlighted with a black box, and histidine residues unique to each protein are highlighted with a gray box. (B) Wild-type and mutant psaF alleles were introduced at the native site in the ΔpsaEF mutant (YPA18) to generate mutants expressing wild-type psaF (YPA260; WT), psaFH40A (YPA271; H40A), psaFH54A (YPA317; H54A), psaFH87A (YPA274; H87A), psaFH141A (YPA278; H141A), psaFH153A (YPA325; H153A), and psaFH155A (YPA267; H155A). These strains were grown at 37°C in BHI buffered to pH 6.3, and whole-cell lysates were used to analyze PsaF. (C) These same strains were transformed with psaA-gfp, grown at 37°C in BHI buffered to pH 6.3, and psaA expression was determined. ***, P < 0.0001 using one-way ANOVA and Dunnett’s multiple-comparison test.
Since histidine protonation can influence protein conformation (57), we wanted to determine if the stability of the PsaF histidine substitution mutants was impacted by pH. The same strains described above were grown at 37°C and pH 6.5 or 6.0, and PsaE and PsaF were analyzed by Western blotting. The Y. enterocolitica homologue of psaA, myfA, is only expressed at an even lower pH, pH 5 (56); therefore, we also grew these strains at 37°C and pH 5.5 for Western blot analysis of PsaE and PsaF. None of the PsaF mutants, including PsaFH40A, were detected at pH 6.5, a condition under which WT PsaF is abundant (Fig. 5A). However, in samples from three of the six mutants (PsaFH40A, PsaFH87A, and PsaFH153A), PsaF was detected when the pH was lowered to 6.0. Of these mutant proteins, only PsaFH40A was present at pH 6.3, suggesting that both PsaFH87A and PsaFH153A are not stable at pH of >6.0. When the pH was further acidified to 5.5, all six PsaF mutant proteins were detected (PsaFH54A was detected at a low level), indicating that the stability of PsaF histidine mutants can be rescued by decreasing the pH.
FIG 5.
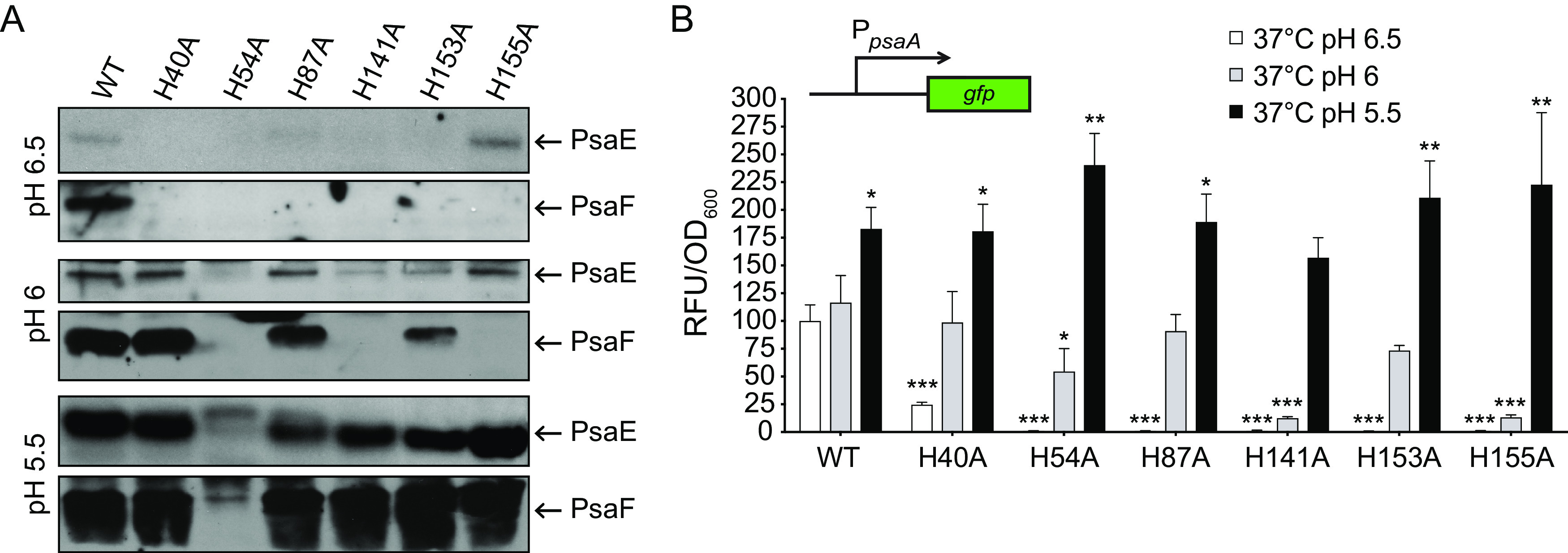
The stability and function of PsaF histidine-to-alanine substitution mutants is restored by low pH. (A) The same strains shown in Fig. 4B were grown at 37°C in BHI buffered to pH 6.5, 6.0, and 5.5, and whole-cell lysates were used to analyze PsaE and PsaF via Western blot. (B) The psaA-gfp plasmid was introduced into these same strains, grown as in panel A, and expression of psaA was determined. ***, P < 0.0001; **, P < 0.001; *, P < 0.01; one-way ANOVA and Dunnett’s multiple-comparison test to compare mean values of each sample to those of the WT at pH 6.5.
The presence of near-WT levels of PsaE correlated with the presence of each PsaF histidine mutant. One exception to this was that at pH 6.0 or 6.5, PsaE appeared to be present at levels comparable to those in the WT in the absence of PsaFH155A. To determine if these PsaF mutant proteins influence psaA expression in response to pH, the WT strain and these psaF histidine mutants were then assayed for psaA expression after growth at 37°C in BHI at pH 6.5, 6.0, and 5.5 (Fig. 5B). Expression of psaA in the WT increased when the pH was more acidic, and this corresponded with an apparent increase in levels of PsaE and PsaF (Fig. 5). Expression of psaA in the PsaF His-to-Ala mutants was roughly comparable to that in the WT when the stability of PsaF was restored by growth at lower pH. Collectively, these data suggest that these histidine residues of PsaF are involved in sensing pH and impact stability of both PsaF and PsaE.
DISCUSSION
PsaA is a bubonic plague virulence factor and is produced when Y. pestis is grown in vitro under high temperature and low pH conditions (9, 10, 17–19). Despite the strong impact of temperature and pH on psaA expression, the underlying mechanisms have remained largely unknown. Using defined growth conditions, we recently demonstrated that high temperature and low pH activate psaA transcription in Y. pestis, and our work suggests that posttranscriptional regulation of two key psaA regulatory proteins, PsaE and PsaF, is primarily responsible for this unusual expression pattern (18). From our previous work, we predicted that posttranslational mechanisms also control the observed pH-dependent effect on PsaE and PsaF levels. Our earlier study examining the absence of PsaE or PsaF in ΔpsaF or ΔpsaE mutants, respectively, indicated that PsaE and PsaF exhibit codependent stability (18). Here, we provide evidence that residues in the periplasmic domains of each protein impact their stability. Building upon our previous work, we propose a model by which PsaF uses its histidine-rich periplasmic domain to sense pH (Fig. 6). As PsaE is thought to directly activate transcription of psaA, we predict that Y. pestis utilizes PsaF to stabilize PsaE and control psaA transcription in response to pH.
FIG 6.

Proposed model for pH control of psaA expression by PsaE and PsaF. PsaF contains a histidine-rich periplasmic domain that impacts the folding and stability of PsaF in response to environmental pH. Intrachain disulfide bond formation in the periplasmic domain of PsaE promotes interaction with PsaF at pH of <6.7, thus blocking proteolytic degradation of PsaE via regulated intramembrane proteolysis (RIP). At pH of >6.7, conformational changes in PsaF prevent effective interaction with PsaE, and PsaE is targeted for proteolytic degradation. Expression of psaA increases as the pH decreases, and thus pH may also impact the activity of PsaE and PsaF through additional mechanisms that have yet to be determined.
Acidic pH is known to influence gene expression in multiple bacteria (58–62). Yet, knowledge of the proteins and mechanisms underlying these pH-sensing pathways is limited. The data presented in this study into how PsaE and PsaF control expression of psaA offer insight into the mechanisms bacteria use to sense pH and activate gene expression. We show that a distinct pH threshold (pH > 6.7) determines whether or not PsaF is present. Because translation of psaF occurs at both pH 6.3 and pH 7.3 (18), it appears that pH controls protein stability rather than production. After production at pH 6.3, PsaF levels decrease following a shift to neutral pH, whereas levels remain high when left at mildly acidic pH; thus, PsaF functions as a membrane-localized pH-sensor, with its stability linked to extracellular pH. As with other proteins that have been implicated as pH sensors (48–50, 54, 63), PsaF has a periplasmic domain that is ideally positioned for such environmental sensing. This domain contains nine histidine residues, and we found that five were essential for PsaF stability at pH 6.3. The stability of PsaF in these five mutants is rescued by further reducing the pH, indicating that these mutants can sense and respond to this signal. While additional studies are needed to determine how histidines enhance PsaF stability, the involvement of pH suggests that their ionization state plays a role such that their protonation promotes stability. Stability of individual PsaF histidine mutants is rescued by growth at lower pH. One possible explanation for this is that individual residues become protonated within the folded protein at different pH levels. While the pKa of histidine imidazole is ∼6.0, protein structure can influence the pKa of individual residues within a protein (64, 65).
We previously reported that the expression of psaF from a plasmid restores PsaE to high levels in a ΔpsaF mutant, suggesting that one key role of PsaF is to enhance PsaE stability (18). The ability of PsaF His-to-Ala mutants to rescue PsaE stability and activate psaA transcription at lower pH levels, when the stability of these PsaF mutants themselves is restored, further supports this role. We predict that low pH alters the structural conformation of PsaF and that this affects its ability to interact with and enhance the stability of PsaE. PsaE is present at very low levels at pH of >6.7, and the absence of PsaF above this pH threshold is likely responsible for the observed loss of PsaE. However, we cannot exclude the possibility that PsaE also senses pH.
Recent work on PsaE-like proteins (ToxR and TcpP) identified proteases involved in targeted protein degradation that indicated these proteins are degraded by RIP (30, 31, 38, 39). Here, we present evidence that DegP and Tsp, two proteases that contribute to ToxR and TcpP degradation, respectively (30, 39, 66), also contribute to the low levels of PsaE observed at pH 7.3. Tsp and DegP are both periplasmic proteases and may directly recognize and target the PsaE periplasmic domain. As PsaF is absent at pH 7.3, it seems likely that PsaF serves to block access of these proteases to PsaE and thus plays a similar role to that of ToxS and TcpH, which inhibit RIP of ToxR and TcpP, respectively (29, 31).
The cytoplasmic N-terminal domain of PsaE contains an OmpR-like winged helix-turn-helix DNA-binding domain that is thought to bind the psaA promoter to directly activate psaA expression (19, 37). While PsaE-like proteins possess a conserved topology, the primary sequences of their periplasmic domains do not resemble each other, and a full understanding of the function of this domain is lacking. As was observed with TcpP (33), PsaE contains two cysteines in the periplasmic domain and our data indicate that both influence protein stability. Conversely, loss of either cysteine does not as readily impact the overall stability of ToxR but may alter its activity (44–47, 66). In both TcpP and ToxR, the cysteines influence protein conformation (33, 44–46, 66, 67). Notably, mutation of the cysteines in PsaE also negatively impacts PsaF stability, supporting our previous work indicating that the stability of PsaE and PsaF is mutually dependent (18). A similar effect of cysteine mutations in TcpP on TcpH stability has been observed (33). A recent study using the purified periplasmic domains of ToxR and ToxS suggested that intradomain disulfide bond formation of ToxR promotes binding to ToxS (68). This finding raises the possibility that intradomain disulfide bond formation in the periplasmic domain of PsaE is necessary for interaction with PsaF. In this scenario, if PsaF does not fold properly due to pH levels above 6.7 and cannot bind PsaE, then PsaE is susceptible to proteolysis via Tsp and DegP. When unable to interact with PsaE, PsaF itself appears to be unstable. Unlike that of PsaE, loss of Tsp or DegP alone is not sufficient to restore stability of PsaF at pH 7.3. These data are consistent with the observation that cysteine-to-serine mutations of PsaE result in loss of stability of both PsaE and PsaF.
While the topology of PsaE seems to be ideal for both environmental sensing and gene activation, it alone cannot activate psaA transcription. In a previous study, the absence of PsaF was associated with low psaA expression even when PsaE was produced at low temperatures (18). At pH levels of >6.7, where only low levels of PsaE and no PsaF are detected, there is negligible psaA transcription and PsaA cannot be detected. In most cases, PsaE levels were significantly reduced when PsaF was undetectable. The analysis of the PsaF histidine mutants, and their rescue by lowering the pH, indicated that in all growth conditions when both PsaE and PsaF were detectable, psaA expression was comparable to that in the WT. Together, these data indicate that PsaF may serve to do more than solely inhibit PsaE degradation and may also influence PsaE activity.
Our data resemble that of a recent report that suggested a direct interaction between the ToxR and ToxS periplasmic domains; the strength of this interaction was negatively impacted at alkaline pH, a growth condition under which ToxR is degraded (32). It is tempting to speculate that an analogous phenomenon occurs with PsaE and PsaF. In a similar manner, pH influences the stability of PsaE. The amount of PsaE is slightly lower at pH 6.7 relative to that at pH 6.3, yet PsaF levels are not altered at this pH range. This may indicate that PsaF and PsaE do not interact as effectively at intermediate pH (e.g., pH 6.7) resulting in reduced amounts of PsaE. In the absence of PsaF at pH of >6.7, levels of PsaE are even further reduced.
Bacteria control protein stability as a means of rapidly modulating gene expression in response to changes in environmental cues via RIP (69), and our data suggest that regulation of PsaE and PsaF stability drives pH-dependent psaA transcription. PsaE and PsaF belong to an unusual family of paired transcriptional regulators that are localized to the inner membrane (19, 21, 23), and most studies have focused on the function and regulation of the “PsaE-like” effector (i.e., ToxR and TcpP), likely because of their predicted role as a direct transcriptional regulator. PsaF clearly plays a vital role in enhancing PsaE stability (18), as has been proposed for the PsaF-like proteins TcpH and ToxS (29, 31). Yet despite this role, little attention has been focused on PsaF-like proteins. This work provides new information on the role of PsaF by defining it as the pH sensor contributing to the pH dependent expression of psaA. As with PsaE and PsaF, TcpP and TcpH exhibit codependent stability; however, it is unknown if TcpP or TcpH levels are regulated in response to specific environmental signals. It is also not known if the PsaF-like proteins TcpH and ToxS directly function as sensors of specific environmental signals. Our work supports the current paradigm while offering new insights into mechanisms that influence the codependent stability of these protein pairs by revealing critical regulation of PsaF in response to an environmental signal, pH. The observation that PsaE is stable following a shift from pH 6.3 to 7.3 but that PsaF is not further suggests that PsaF serves as the direct pH sensor. Thus, we propose the temperature- and pH-dependent regulation of psaA transcription can be explained by temperature-dependent translation of psaE and psaF, an effect of pH on folding of PsaF and its interaction with PsaE, which in turn impacts the susceptibility of PsaE to proteolysis.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Bacterial strains and plasmids used in this study are listed in Table 1. Y. pestis strains were cultivated on brain heart infusion (BHI) agar (BD Biosciences, Bedford, MA) at 26°C for 48 h and in BHI broth with aeration at 26°C or at 37°C. Escherichia coli strains were grown in LB (BD Biosciences) at 37°C. When indicated, bacteria were grown in BHI broth that was buffered to the indicated pH and filter sterilized as described previously (18). When necessary, antibiotics were added at the following concentrations: kanamycin (Kan), 50 μg/ml; carbenicillin (Carb), 100 μg/ml; and Irgasan (Irg), 2 μg/ml.
TABLE 1.
Bacterial strains and plasmids
| Species, strain, or plasmid | Descriptiona | Reference or source |
|---|---|---|
| Escherichia coli | ||
| DH5α | F− ϕ80ΔlacZM15 Δ(lacZYA-argF)U169 deoP recA1 endA1 hsdR17(rK− mK−) | Invitrogen |
| S17-1λpir | Tpr Strr recA thi pro hsdR hsdM+ RP4::2-Tc::Mu::Km Tn7 λpir | 72 |
| Yersinia pestis | ||
| YP6 | CO92, pCD1− | 11 |
| YPA18 | YP6 ΔpsaEF | 18 |
| YPA260 | YP18 with pEW104 at the native site | 18 |
| YPA425 | YP6 ΔdegP | This work |
| YPA469 | YPA18 ΔdegP | This work |
| YPA476 | YPA425 with pJQ054 at the native site | This work |
| YPA350 | YP6 Δtsp | This work |
| YPA379 | YPA18 Δtsp | This work |
| YPA438 | YPA350 with pJQ050 at the native site | This work |
| YPA370 | YP6 tsp::pJQ014 | This work |
| YPA391 | YP6 degS::pJQ032 | This work |
| YPA387 | YP6 degQ::pDF001 | This work |
| YPA395 | YP6 degP::pDF004 | This work |
| YPA382 | YP6 rseP::pJQ031 | This work |
| YPA393 | YP6 ypfJ::pDF002 | This work |
| YPA388 | YP6 ypo0398::pDF003 | This work |
| YPA396 | YP6 ftsH::pDF005 | This work |
| YPA275 | YPA18 with pEW107 at the native site | This work |
| YPA277 | YPA18 with pEW108 at the native site | This work |
| YPA276 | YPA18 with pEW109 at the native site | This work |
| YPA273 | YPA18 with pEW110 at the native site | This work |
| YPA271 | YPA18 with pEW111 at the native site | This work |
| YPA317 | YPA18 with pEW112 at the native site | This work |
| YPA274 | YPA18 with pEW113 at the native site | This work |
| YPA278 | YPA18 with pEW114 at the native site | This work |
| YPA325 | YPA18 with pEW115 at the native site | This work |
| YPA267 | YPA18 with pEW116 at the native site | This work |
| Plasmids | ||
| pSR47S | Kanr, MobRP4 oriR6K sacB suicide vector | 70 |
| pPROBE-AT | Apr, gfp reporter vector | 73 |
| pWKS30 | Apr cloning vector | 74 |
| pEW102 | psaA promoter in pPROBE-AT | 18 |
| pEW104 | psaEF and flanking sequences in pSR47S | 18 |
| pEW105 | psaEF promoter and psaE in pSR47S | 18 |
| pEW106 | psaEF promoter and psaF in pSR47S | 18 |
| pPsaF | psaEF promoter and psaF coding sequence in pWKS30 | 18 |
| pJQ014 | pSR47S with an internal fragment from YP6_1705 (tsp) | This work |
| pJQ032 | pSR47S with an internal fragment from YP6_ 3568 (degS) | This work |
| pDF001 | pSR47S with an internal fragment from YP6_ 3566 (degQ) | This work |
| pDF004 | pSR47S with an internal fragment from YP6_ 3382 (degP) | This work |
| pJQ031 | pSR47S with an internal fragment from YP6_ 1051 (rseP) | This work |
| pDF002 | pSR47S with an internal fragment from YP6_ 3058 (ypfJ) | This work |
| pDF003 | pSR47S with an internal fragment from YP6_ 0398 (peptidase family M48) | This work |
| pDF005 | pSR47S with an internal fragment from YP6_ 3502 (ftsH) | This work |
| pJQ018 | tsp flanking sequences in pSR47S | This work |
| pJQ047 | degP flanking sequences in pSR47S | This work |
| pJQ054 | degP coding and flanking sequences in pSR47S | This work |
| pJQ050 | tsp coding and flanking sequences in pSR47S | This work |
| pEW107 | psaEF and flanking sequences with psaEC206S mutant allele in pSR47S | This work |
| pEW108 | psaEF and flanking sequences with psaEC211S mutant allele in pSR47S | This work |
| pEW109 | psaEF and flanking sequences with psaEC206S/C211S mutant allele in pSR47S | This work |
| pEW110 | psaEF and flanking sequences with psaFH38A/H116A/H159A mutant allele in pSR47S | This work |
| pEW111 | psaEF and flanking sequences with psaFH40A mutant allele in pSR47S | This work |
| pEW112 | psaEF and flanking sequences with psaFH54A mutant allele in pSR47S | This work |
| pEW113 | psaEF and flanking sequences with psaFH87A mutant allele in pSR47S | This work |
| pEW114 | psaEF and flanking sequences with psaFH141A mutant allele in pSR47S | This work |
| pEW115 | psaEF and flanking sequences with psaFH153A mutant allele in pSR47S | This work |
| pEW116 | psaEF and flanking sequences with psaFH155A mutant allele in pSR47S | This work |
Str, streptomycin; Kan, kanamycin; Ap, ampicillin; r, resistance; Tp, trimethoprim.
Plasmid and strain construction.
Primers used in this study are listed in Table 2. All in-frame deletion mutants and mutants in which alleles were introduced at the native site were constructed via allelic exchange using the pSR47S suicide vector (70) as previously described (18). All plasmids were constructed via Gibson assembly (NEB) and were confirmed by sequencing.
TABLE 2.
Primers used in this study
| Primer | Sequencea (5′ to 3′) | Descriptionb |
|---|---|---|
| psaEFcompF | ATCGATCCTCTAGAGTCGACATTAACGGGGGCGCTGTCTATGG | F pEW104 5′ |
| psaEFcompR | GCTCTAGAACTAGTGGATCCATAACTCAGTCGCAGACCTATAG | R pEW104 3′ |
| psaF_3HA_R | CATTCTTAAATCAAGATGCTCAGCGATATCGCCATATTTCACTTC | R pEW110 internal 3′ |
| psaF_3HA_F | GAAGTGAAATATGGCGATATCGCTGAGCATCTTGATTTAAGAATG | F pEW110 internal 5′ |
| psaF_H40A_R | CTTAAATCAAGAGCCTCATGGATATCGCCATATTTC | R pEW111 3′ |
| psaF_H40A_F | GGCGATATCCATGAGGCTCTTGATTTAAGAATGCAAGG | F pEW111 5′ |
| psaF_H54A_R | CAATAATATAAGCTGAGAGGCTAAATCTTATCCC | R pEW112 3′ |
| psaF_H54A_F | GATTTAGCCTCTCAGCTTATATTATTGATGATAAGTCT | F pEW112 5′ |
| psaF_H87A_R | GACTGATATGAAGCAAGAGGGAATAGGTAATACTTCC | R pEW113 3′ |
| psaF_H87A_F | CCTATTCCCTCTTGCTTCATATCAGTCATCCCCTGAT | F pEW113 5′ |
| psaF_H141A_R | GAGAAGATAAGGCGATCGTTTCTCCATTGACATC | R pEW114 3′ |
| psaF_H141A_F | GGAGAAACGATCGCCTTATCTTCTCTGTTTCTCGGG | F pEW114 5′ |
| psaF_H153A_R | CGTATGGATAGCTTCTCCTTGTACCCCGAGAAAC | R pEW115 3′ |
| psaF_H153A_F | GGTACAAGGAGAAGCTATCCATACGTCCTATCATG | F pEW115 5′ |
| psaF_H155A_R | CATGATAGGACGTAGCGATATGTTCTCCTTGTACCCC | R pEW116 3′ |
| psaF_H155A_F | GGAGAACATATCGCTACGTCCTATCATGACGTTAG | F pEW116 5′ |
| JQ076 | AAAAAGGATCGATCCTCTAGAGCGCGGATAAAAAGTATTCGCTGG | F pJQ014 5′ |
| JQ077 | GCCGCTCTAGAACTAGTGGATCCCGCTATCGTTTTACTCTTCGC | R pJQ014 3′ |
| JQ107 | AAAGGATCGATCCTCTAGAGGCCAGCAGCTCCCTAGCCTGG | F pDF004 5′ |
| JQ108 | GCTCCACCGCGGTGGCGGCCGCGCCGCCGGAATTACCACGGT | R pDF004 3′ |
| JQ110 | AAAAAGGATCGATCCTCTAGAGCCCAGCGAATCGAATGGCCG | F pDF005 5′ |
| JQ111 | GCTCCACCGCGGTGGCGGCCGGACACGGGATGCACCGACAC | R pDF005 3′ |
| VM1 | GCGTCGACGATGAGCGTGTAGAGGCTGTTGCGCC | F pJQ031 5′ |
| VM2 | ATAAGAATGCGGCCGCCCCAACATGCTTACCGTCAACCGC | R pJQ031 3′ |
| VM3 | GCGTCGACCAGTACGTCGTGCCGCACCGGCGG | F pJQ032 5′ |
| VM4 | ATAAGAATGCGGCCGCCCGATGCCTTCCGGTGTTTCGCC | R pJQ032 3′ |
| VM5 | GCGTCGACGAGAGCAGCCGGCCATTCGAAGGC | F pDF001 5′ |
| VM6 | ATAAGAATGCGGCCGCGGTTTGATCCCGGCTTTCGCTGCCG | R pDF001 3′ |
| VM7 | GCGTCGACGGTCGCTATCCTGATTATTGTGTTGG | F pDF002 5′ |
| VM8 | ATAAGAATGCGGCCGCCCGATAAGCGATTACCTTCCGCCTGAG | R pDF002 3′ |
| VM9 | GCGTCGACCCACTGAACCTGACATTTCCCGATGG | F pDF003 5′ |
| VM10 | ATAAGAATGCGGCCGCGTCATCCACATAAAGGTCAAGGCGAC | R pDF003 3′ |
| JQ080 | AAAAAGGATCGATCCTCTAGAGGGTCAAAGTCGGTGCTGAGC | F pJQ050 5′ |
| JQ083 | GCCGCTCTAGAACTAGTGGATCCCGCCAGGTTGGTGAGAAGG | R pJQ050 3′ |
| JQ081 | GCGACAGGGCGAGATGCAAGCCTAGGTTGGCCTCCGTATC | R pJQ018 up 3′ |
| JQ082 | TTGCATCTCGCCCTGTCGCTGCTGCTACCGCTGGGGCAACGG | F pJQ018 down 5′ |
| JQ134 | AAAAAGGATCGATCCTCTAGAGGGGCTGCTTGATATTTATAGC | F pJQ054 5′ |
| JQ137 | CCGCTCTAGAACTAGTGGATCGGGCACAGGCTGTCAGGCAAGC | R pJQ054 3′ |
| JQ135 | ACTGTATCAATACCTTACTGCATATTCGTGTTCTCACTATAC | R pJQ047 up 3′ |
| JQ136 | GCAGTAAGGTATTGATACAGTAAGGTATTGATACAGTAAGG | F pJQ047 down 5′ |
| psaEC206S_F | AAACAGCAATGAAAGCAAAATCACTTACTCTCATA | F pEW107 internal 5′ |
| psaEC206S_R | CTTTCATTGCTGTTTGCATTCCGATTGATCAGAGATAATGACTAAACCAAAATC | R pEW107 internal 3′ |
| psaEC211S_F | AAACAGCAATGAAAGCAAAATCACTTACTCTCATA | F pEW108 internal 5′ |
| psaEC211S_R | CTTTCATTGCTGTTTGGATTCCGATTGATCACAGATAATGACTAAACCAAAATC | R pEW108 internal 3′ |
| psaEC206-211S_F | AAACAGCAATGAAAGCAAAATCACTTACTCTCATA | F pEW109 internal 5′ |
| psaEC206-211S_R | CTTTCATTGCTGTTTGGATTCCGATTGATCAGAGATAATGACTAAACCAAAATC | R pEW109 internal 3′ |
Restriction sites are shown in bold. Sequence overlap for Gibson assembly is underlined.
F, forward primer; R, reverse primer.
(i) In-frame deletions.
The plasmids for in-frame deletions of degP and tsp were constructed by amplifying ∼500-bp DNA fragments upstream and downstream of the target gene. These fragments were cloned into pSR47S, and the resulting plasmids, pJQ047 (ΔdegP) and pJQ018 (Δtsp), were introduced into YP6 (WT) or YPA18 (ΔpsaEF) via conjugation as previously described (18). Briefly, transconjugants were selected on BHI plates with Kan50 and Irg2. The second recombination event was selected for by streaking Kanr (Kan-resistant)/Irgr colonies onto BHI agar plates containing 5% sucrose. Deleted genes were confirmed by PCR.
(ii) Reintroduction of degP and tsp in ΔdegP and Δtsp mutants.
The plasmids used to reintroduce degP and tsp at the native site were constructed as follows. The target gene coding sequence, including 500-bp upstream and downstream flanking regions, was amplified and cloned into pSR47S, and the resulting plasmids, pJQ054 and pJQ050, were introduced into YPA425 (ΔdegP) and YPA350 (Δtsp), respectively, via conjugation and integration of the plasmids, and were identified by selection on BHI plates with Kan50 and Irg2.
(iii) Protease insertion mutants.
All plasmids for generating insertion mutations in putative protease genes were constructed by amplifying ∼500-bp DNA fragments of internal coding sequence of the gene. These fragments were cloned into pSR47S, the resulting plasmids (pJQ014, pJQ031, pJQ032, pDF001, pDF002, pDF003, pDF004, and pDF005) were introduced into YP6 via conjugation, and insertion disruption mutants generated by single crossover were identified by selection on BHI plates with Kan50 and Irg2. The resulting strains are YPA370 (tsp), YPA382 (rseP), YPA391 (degS), YPA387 (degQ), YPA393 (ypfJ), YPA388 (ypo0398), YPA395 (degP), and YPA396 (ftsH), respectively.
(iv) psaE and psaF substitution mutants.
The plasmids for generating psaE mutant alleles encoding cysteine-to-serine substitutions were constructed by amplifying the psaEF coding sequence with primers containing the substituted nucleotides. These products were then cloned into pSR47S to generate pEW107 (psaEC206S), pEW108 (psaEC211S), and pEW109 (psaEC206S/C211S). The plasmids for generating psaF mutant alleles encoding histidine-to-alanine substitutions were constructed using a similar procedure as for the psaE cysteine-to-serine mutants, using primers containing the substituted nucleotides. These products were then cloned into pSR47S to generate pEW110 (psaFH38A/H116A/H159A), pEW111 (psaEH40A), pEW112 (psaEH54A), pEW113 (psaEH87A), pEW114 (psaEH141A), pEW115 (psaEH153A), and pEW116 (psaEH155A). These plasmids were conjugated into YPA18 (the ΔpsaEF mutant), and integration of these plasmids into the chromosome was identified by selection on BHI plates with Kan50 and Irg2.
gfp transcriptional reporter assay.
To analyze psaA promoter activity, a gfp transcriptional reporter plasmid (pEW102) was electroporated into the indicated strains and assayed as previously described (18). Saturated cultures grown in unbuffered BHI were subcultured to an OD600 of 0.2 in buffered BHI and grown for 8 h with aeration at 37°C. Relative fluorescent units (RFU) were measured using a Synergy HT microplate reader (BioTek Instruments, Winooski, VT) and normalized to OD600. Data are represented as RFU/OD600 ± standard deviation.
PsaF stability assay.
Saturated cultures of YP6 (WT) were subcultured to an OD600 of 0.2 in BHI buffered to pH 6.3 and grown for 8 h at 37°C. Cells (OD = 10) were pelleted, washed once with PBS adjusted to pH 6.3, split into equal volumes, pelleted, and suspended in PBS adjusted to either pH 6.3 or 7.3, and incubated at 37°C for 4 h. At 0 h, 2 h, and 4 h, cells (OD = 2) from each condition were collected and prepared for Western blot analysis.
Western blot analysis.
Saturated cultures grown in unbuffered BHI were subcultured to an OD600 of 0.2 in BHI buffered to the indicated pH and grown for 8 h at 37°C. PsaE, PsaF, and PsaA were analyzed as previously described (18). Briefly, cell lysates were resuspended in Laemmli buffer, boiled, separated via SDS-PAGE, and transferred to a polyvinylidene difluoride (PVDF) membrane. Loading of protein samples was qualitatively assessed via Ponceau S staining. Anti-PsaE, anti-PsaF, and anti-PsaA sera were used as previously described (18). Anti-IgG horseradish peroxidase (HRP)-conjugated secondary antibodies were used at a titer of 1:20,000.
Statistical analysis and reproducibility.
Analyses were performed using Prism 8 (GraphPad Software, San Diego, CA). All experiments were performed at least three times, with biological triplicates in each assay. Unless otherwise noted, representative assays are shown.
ACKNOWLEDGMENTS
We thank members of the Miller and Tamayo labs for insightful discussions pertaining to the work described in this study and Kimberly A. Walker for assistance with preparation of the manuscript. We thank Jason Cathelyn, Carolina Lin, and Daniel Frederick for technical assistance that involved the construction of plasmids and bacterial strains.
This work was supported by funds from the National Institutes of Health RO1AI119032 (to V.L.M.) and by grant U54AI057157 (Southeast Regional Center for Biodefense and Emerging Infectious Diseases, project 006, to V.L.M.).
Footnotes
Supplemental material is available online only.
Contributor Information
Virginia L. Miller, Email: vlmiller@med.unc.edu.
Yves V. Brun, Université de Montréal
REFERENCES
- 1.Perry RD, Fetherston JD. 1997. Yersinia pestis—etiologic agent of plague. Clin Microbiol Rev 10:35–66. 10.1128/CMR.10.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Demeure CE, Dussurget O, Mas Fiol G, Le Guern A-S, Savin C, Pizarro-Cerdá J. 2019. Yersinia pestis and plague: an updated view on evolution, virulence determinants, immune subversion, vaccination, and diagnostics. Genes Immun 20:357–370. 10.1038/s41435-019-0065-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gonzalez RJ, Miller VL. 2016. A deadly path: bacterial spread during bubonic plague. Trends Microbiol 24:239–241. 10.1016/j.tim.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gonzalez RJ, Lane MC, Wagner NJ, Weening EH, Miller VL. 2015. Dissemination of a highly virulent pathogen: tracking the early events that define infection. PLoS Pathog 11:e1004587. 10.1371/journal.ppat.1004587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lathem WW, Crosby SD, Miller VL, Goldman WE. 2005. Progression of primary pneumonic plague: a mouse model of infection, pathology, and bacterial transcriptional activity. Proc Natl Acad Sci U S A 102:17786–17791. 10.1073/pnas.0506840102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sebbane F, Lemaître N, Sturdevant DE, Rebeil R, Virtaneva K, Porcella SF, Hinnebusch BJ. 2006. Adaptive response of Yersinia pestis to extracellular effectors of innate immunity during bubonic plague. Proc Natl Acad Sci U S A 103:11766–11771. 10.1073/pnas.0601182103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Motin VL, Georgescu AM, Fitch JP, Gu PP, Nelson DO, Mabery SL, Garnham JB, Sokhansanj BA, Ott LL, Coleman MA, Elliott JM, Kegelmeyer LM, Wyrobek AJ, Slezak TR, Brubaker RR, Garcia E. 2004. Temporal global changes in gene expression during temperature transition in Yersinia pestis. JB 186:6298–6305. 10.1128/JB.186.18.6298-6305.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Straley SC, Perry RD. 1995. Environmental modulation of gene expression and pathogenesis in Yersinia. Trends Microbiol 3:310–317. 10.1016/S0966-842X(00)88960-X. [DOI] [PubMed] [Google Scholar]
- 9.Ben-Efraim S, Aronson M, Bichowsky-Slomnicki L. 1961. New antigenic component of Pasteurella pestis formed under specified conditions of pH and temperature. J Bacteriol 81:704–714. 10.1128/JB.81.5.704-714.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lindler LE, Klempner MS, Straley SC. 1990. Yersinia pestis pH 6 antigen: genetic, biochemical, and virulence characterization of a protein involved in the pathogenesis of bubonic plague. Infect Immun 58:2569–2577. 10.1128/IAI.58.8.2569-2577.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cathelyn JS, Crosby SD, Lathem WW, Goldman WE, Miller VL. 2006. RovA, a global regulator of Yersinia pestis, specifically required for bubonic plague. Proc Natl Acad Sci U S A 103:13514–13519. 10.1073/pnas.0603456103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonzalez RJ, Weening EH, Lane MC, Miller VL. 2015. Comparison of models for bubonic plague reveals unique pathogen adaptations to the dermis. Infect Immun 83:2855–2861. 10.1128/IAI.00140-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weening EH, Cathelyn JS, Kaufman G, Lawrenz MB, Price P, Goldman WE, Miller VL. 2011. The dependence of the Yersinia pestis capsule on pathogenesis is influenced by the mouse background. Infect Immun 79:644–652. 10.1128/IAI.00981-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindler LE, Tall BD. 1993. Yersinia pestis pH 6 antigen forms fimbriae and is induced by intracellular association with macrophages. Mol Microbiol 8:311–324. 10.1111/j.1365-2958.1993.tb01575.x. [DOI] [PubMed] [Google Scholar]
- 15.Huang X-Z, Lindler LE. 2004. The pH 6 antigen is an antiphagocytic factor produced by Yersinia pestis independent of Yersinia outer proteins and capsule antigen. Infect Immun 72:7212–7219. 10.1128/IAI.72.12.7212-7219.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Felek S, Tsang TM, Krukonis ES. 2010. Three Yersinia pestis adhesins facilitate Yop delivery to eukaryotic cells and contribute to plague virulence. Infect Immun 78:4134–4150. 10.1128/IAI.00167-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Price SB, Freeman MD, Yeh KS. 1995. Transcriptional analysis of the Yersinia pestis pH 6 antigen gene. J Bacteriol 177:5997–6000. 10.1128/JB.177.20.5997-6000.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quinn JD, Weening EH, Miner TA, Miller VL. 2019. Temperature control of psaA expression by PsaE and PsaF in Y. pestis. J Bacteriol 201:e217-19. 10.1128/JB.00217-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang Y, Isberg RR. 1997. Transcriptional regulation of the Yersinia pseudotuberculosis pH 6 antigen adhesin by two envelope-associated components. Mol Microbiol 24:499–510. 10.1046/j.1365-2958.1997.3511719.x. [DOI] [PubMed] [Google Scholar]
- 20.Watson N, Dunyak DS, Rosey EL, Slonczewski JL, Olson ER. 1992. Identification of elements involved in transcriptional regulation of the Escherichia coli cad operon by external pH. J Bacteriol 174:530–540. 10.1128/JB.174.2.530-540.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller VL, Taylor RK, Mekalanos JJ. 1987. Cholera toxin transcriptional activator ToxR is a transmembrane DNA binding protein. Cell 48:271–279. 10.1016/0092-8674(87)90430-2. [DOI] [PubMed] [Google Scholar]
- 22.Miller VL, DiRita VJ, Mekalanos JJ. 1989. Identification of toxS, a regulatory gene whose product enhances toxR-mediated activation of the cholera toxin promoter. J Bacteriol 171:1288–1293. 10.1128/JB.171.3.1288-1293.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Häse CC, Mekalanos JJ. 1998. TcpP protein is a positive regulator of virulence gene expression in Vibrio cholerae. Proc Natl Acad Sci U S A 95:730–734. 10.1073/pnas.95.2.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alam A, Tam V, Hamilton E, Dziejman M. 2010. vttRA and vttRB encode ToxR family proteins that mediate bile-induced expression of type three secretion system genes in a non-O1/non-O139 Vibrio cholerae strain. Infect Immun 78:2554–2570. 10.1128/IAI.01073-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dalia AB, Lazinski DW, Camilli A. 2014. Identification of a membrane-bound transcriptional regulator that links chitin and natural competence in Vibrio cholerae. mBio 5:e01028-13. 10.1128/mBio.01028-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamamoto S, Mitobe J, Ishikawa T, Wai SN, Ohnishi M, Watanabe H, Izumiya H. 2014. Regulation of natural competence by the orphan two-component system sensor kinase ChiS involves a non-canonical transmembrane regulator in Vibrio cholerae. Mol Microbiol 91:326–347. 10.1111/mmi.12462. [DOI] [PubMed] [Google Scholar]
- 27.Li P, Rivera-Cancel G, Kinch LN, Salomon D, Tomchick DR, Grishin NV, Orth K. 2016. Bile salt receptor complex activates a pathogenic type III secretion system. Elife 5:e15718. 10.7554/eLife.15718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krukonis ES, Yu RR, DiRita VJ. 2000. The Vibrio cholerae ToxR/TcpP/ToxT virulence cascade: distinct roles for two membrane-localized transcriptional activators on a single promoter. Mol Microbiol 38:67–84. 10.1046/j.1365-2958.2000.02111.x. [DOI] [PubMed] [Google Scholar]
- 29.Beck NA, Krukonis ES, DiRita VJ. 2004. TcpH influences virulence gene expression in Vibrio cholerae by inhibiting degradation of the transcription activator TcpP. J Bacteriol 186:8309–8316. 10.1128/JB.186.24.8309-8316.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Almagro-Moreno S, Kim TK, Skorupski K, Taylor RK. 2015. Proteolysis of virulence regulator ToxR is associated with entry of Vibrio cholerae into a dormant state. PLoS Genet 11:e1005145. 10.1371/journal.pgen.1005145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Almagro-Moreno S, Root MZ, Taylor RK. 2015. Role of ToxS in the proteolytic cascade of virulence regulator ToxR in Vibrio cholerae. Mol Microbiol 98:963–976. 10.1111/mmi.13170. [DOI] [PubMed] [Google Scholar]
- 32.Midgett CR, Almagro-Moreno S, Pellegrini M, Taylor RK, Skorupski K, Kull FJ. 2017. Bile salts and alkaline pH reciprocally modulate the interaction between the periplasmic domains of Vibrio cholerae ToxR and ToxS. Mol Microbiol 105:258–272. 10.1111/mmi.13699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morgan SJ, French EL, Thomson JJ, Seaborn CP, Shively CA, Krukonis ES. 2016. Formation of an intramolecular periplasmic disulfide bond in TcpP protects TcpP and TcpH from degradation in Vibrio cholerae. J Bacteriol 198:498–509. 10.1128/JB.00338-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murley YM, Carroll PA, Skorupski K, Taylor RK, Calderwood SB. 1999. Differential transcription of the tcpPH operon confers biotype-specific control of the Vibrio cholerae ToxR virulence regulon. Infect Immun 67:5117–5123. 10.1128/IAI.67.10.5117-5123.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krukonis ES, DiRita VJ. 2003. DNA binding and ToxR responsiveness by the wing domain of TcpP, an activator of virulence gene expression in Vibrio cholerae. Mol Cell 12:157–165. 10.1016/S1097-2765(03)00222-3. [DOI] [PubMed] [Google Scholar]
- 36.Crawford JA, Krukonis ES, DiRita VJ. 2003. Membrane localization of the ToxR winged-helix domain is required for TcpP-mediated virulence gene activation in Vibrio cholerae. Mol Microbiol 47:1459–1473. 10.1046/j.1365-2958.2003.03398.x. [DOI] [PubMed] [Google Scholar]
- 37.Li P, Wang X, Smith C, Shi Y, Wade JT, Sun W. 2019. Dissecting psa locus regulation in Yersinia pestis. bioRxiv 10.1101/782003. [DOI] [PMC free article] [PubMed]
- 38.Matson JS, DiRita VJ. 2005. Degradation of the membrane-localized virulence activator TcpP by the YaeL protease in Vibrio cholerae. Proc Natl Acad Sci U S A 102:16403–16408. 10.1073/pnas.0505818102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Teoh WP, Matson JS, DiRita VJ. 2015. Regulated intramembrane proteolysis of the virulence activator TcpP in Vibrio cholerae is initiated by the tail-specific protease (Tsp). Mol Microbiol 97:822–831. 10.1111/mmi.13069. [DOI] [PubMed] [Google Scholar]
- 40.Brown MS, Ye J, Rawson RB, Goldstein JL. 2000. Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell 100:391–398. 10.1016/S0092-8674(00)80675-3. [DOI] [PubMed] [Google Scholar]
- 41.Urban S. 2009. Making the cut: central roles of intramembrane proteolysis in pathogenic microorganisms. Nat Rev Microbiol 7:411–423. 10.1038/nrmicro2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang ZR, Bullifent HL, Moore K, Paszkiewicz K, Saint RJ, Southern SJ, Champion OL, Senior NJ, Sarkar-Tyson M, Oyston PCF, Atkins TP, Titball RW. 2017. A noise trimming and positional significance of transposon insertion system to identify essential genes in Yersinia pestis. Sci Rep 7:41923. 10.1038/srep41923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Senior NJ, Sasidharan K, Saint RJ, Scott AE, Sarkar-Tyson M, Ireland PM, Bullifent HL, Rong Yang Z, Moore K, Oyston PCF, Atkins TP, Atkins HS, Soyer OS, Titball RW. 2017. An integrated computational-experimental approach reveals Yersinia pestis genes essential across a narrow or a broad range of environmental conditions. BMC Microbiol 17:163. 10.1186/s12866-017-1073-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ottemann KM, Mekalanos JJ. 1996. The ToxR protein of Vibrio cholerae forms homodimers and heterodimers. J Bacteriol 178:156–162. 10.1128/JB.178.1.156-162.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lembke M, Höfler T, Walter AN, Tutz S, Fengler V, Schild S, Reidl J. 2020. Host stimuli and operator binding sites controlling protein interactions between virulence master regulator ToxR and ToxS in Vibrio cholerae. Mol Microbiol 114:262–278. 10.1111/mmi.14510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fengler VHI, Boritsch EC, Tutz S, Seper A, Ebner H, Roier S, Schild S, Reidl J. 2012. Disulfide bond formation and ToxR activity in Vibrio cholerae. PLoS One 7:e47756. 10.1371/journal.pone.0047756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fan F, Liu Z, Jabeen N, Dillon Birdwell L, Zhu J, Kan B. 2014. Enhanced interaction of Vibrio cholerae virulence regulators TcpP and ToxR under oxygen-limiting conditions. Infect Immun 82:1676–1682. 10.1128/IAI.01377-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perez JC, Groisman EA. 2007. Acid pH activation of the PmrA/PmrB two-component regulatory system of Salmonella enterica. Mol Microbiol 63:283–293. 10.1111/j.1365-2958.2006.05512.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mulder DT, Mcphee JB, Reid-Yu SA, Stogios PJ, Savchenko A, Coombes BK. 2015. Multiple histidines in the periplasmic domain of the Salmonella enterica sensor kinase SsrA enhance signaling in response to extracellular acidification. Mol Microbiol 95:678–691. 10.1111/mmi.12895. [DOI] [PubMed] [Google Scholar]
- 50.Müller S, Götz M, Beier D. 2009. Histidine residue 94 is involved in pH sensing by histidine kinase ArsS of Helicobacter pylori. PLoS One 4:e6930. 10.1371/journal.pone.0006930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mueller DS, Kampmann T, Yennamalli R, Young PR, Kobe B, Mark AE. 2008. Histidine protonation and the activation of viral fusion proteins. Biochem Soc Trans 36:43–45. 10.1042/BST0360043. [DOI] [PubMed] [Google Scholar]
- 52.Kampmann T, Mueller DS, Mark AE, Young PR, Kobe B. 2006. The role of histidine residues in low-pH-mediated viral membrane fusion. Structure 14:1481–1487. 10.1016/j.str.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 53.Do H, Makthal N, VanderWal AR, Saavedra MO, Olsen RJ, Musser JM, Kumaraswami M. 2019. Environmental pH and peptide signaling control virulence of Streptococcus pyogenes via a quorum-sensing pathway. Nat Commun 10:1–14. 10.1038/s41467-019-10556-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goers Sweeney E, Henderson JN, Goers J, Wreden C, Hicks KG, Foster JK, Parthasarathy R, Remington SJ, Guillemin K. 2012. Structure and proposed mechanism for the pH-sensing Helicobacter pylori chemoreceptor TlpB. Structure 20:1177–1188. 10.1016/j.str.2012.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Iriarte M, Vanooteghem J-C, Delor I, Diaz R, Knutton S, Cornelis GR. 1993. The Myf fibrillae of Yersinia enterocolitica. Mol Microbiol 9:507–520. 10.1111/j.1365-2958.1993.tb01712.x. [DOI] [PubMed] [Google Scholar]
- 56.Iriarte M, Cornelis GR. 1995. MyfF, an element of the network regulating the synthesis of fibrillae in Yersinia enterocolitica. J Bacteriol 177:738–744. 10.1128/JB.177.3.738-744.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Achilonu I, Fanucchi S, Cross M, Fernandes M, Dirr HW. 2012. Role of individual histidines in the pH-dependent global stability of human chloride intracellular channel 1. Biochemistry 51:995–1004. 10.1021/bi201541w. [DOI] [PubMed] [Google Scholar]
- 58.Bearson S, Bearson B, Foster JW. 1997. Acid stress responses in enterobacteria. FEMS Microbiol Lett 147:173–180. 10.1111/j.1574-6968.1997.tb10238.x. [DOI] [PubMed] [Google Scholar]
- 59.Tucker DL, Tucker N, Conway T. 2002. Gene expression profiling of the pH response in Escherichia coli. J Bacteriol 184:6551–6558. 10.1128/JB.184.23.6551-6558.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McGowan CC, Necheva AS, Forsyth MH, Cover TL, Blaser MJ. 2003. Promoter analysis of Helicobacter pylori genes with enhanced expression at low pH. Mol Microbiol 48:1225–1239. 10.1046/j.1365-2958.2003.03500.x. [DOI] [PubMed] [Google Scholar]
- 61.Weinrick B, Dunman PM, McAleese F, Murphy E, Projan SJ, Fang Y, Novick RP. 2004. Effect of mild acid on gene expression in Staphylococcus aureus. J Bacteriol 186:8407–8423. 10.1128/JB.186.24.8407-8423.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Krulwich TA, Sachs G, Padan E. 2011. Molecular aspects of bacterial pH sensing and homeostasis. Nat Rev Microbiol 9:330–343. 10.1038/nrmicro2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sen H, Aggarwal N, Ishionwu C, Hussain N, Parmar C, Jamshad M, Bavro VN, Lund PA. 2017. Structural and functional analysis of the Escherichia coli acid-sensing histidine kinase EvgS. J Bacteriol 199:e00310-17. 10.1128/JB.00310-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Antosiewicz J, McCammon JA, Gilson MK. 1996. The determinants of pKas in proteins. Biochemistry 35:7819–7833. 10.1021/bi9601565. [DOI] [PubMed] [Google Scholar]
- 65.Geierstanger B, Jamin M, Volkman BF, Baldwin RL. 1998. Protonation behavior of histidine 24 and histidine 119 in forming the pH 4 folding intermediate of apomyoglobin. Biochemistry 37:4254–4265. 10.1021/bi972516+. [DOI] [PubMed] [Google Scholar]
- 66.Lembke M, Pennetzdorfer N, Tutz S, Koller M, Vorkapic D, Zhu J, Schild S, Reidl J. 2018. Proteolysis of ToxR is controlled by cysteine-thiol redox state and bile salts in Vibrio cholerae. Mol Microbiol 110:796–810. 10.1111/mmi.14125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang M, Liu Z, Hughes C, Stern AM, Wang H, Zhong Z, Kan B, Fenical W, Zhu J. 2013. Bile salt-induced intermolecular disulfide bond formation activates Vibrio cholerae virulence. Proc Natl Acad Sci U S A 110:2348–2353. 10.1073/pnas.1218039110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Midgett CR, Swindell RA, Pellegrini M, Jon KF. 2020. A disulfide constrains the ToxR periplasmic domain structure, altering its interactions with ToxS and bile-salts. Sci Rep 10:9002. 10.1038/s41598-020-66050-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pennetzdorfer N, Lembke M, Pressler K, Matson JS, Reidl J, Schild S. 2019. Regulated proteolysis in Vibrio cholerae allowing rapid adaptation to stress conditions. Front Cell Infect Microbiol 9:214. 10.3389/fcimb.2019.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Merriam JJ, Mathur R, Maxfield-Boumil R, Isberg RR. 1997. Analysis of the Legionella pneumophila fliI gene: intracellular growth of a defined mutant defective for flagellum biosynthesis. Infect Immun 65:2497–2501. 10.1128/IAI.65.6.2497-2501.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reference deleted.
- 72.Simon R, Priefer U, Pühler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Biotechnology 1:784–791. [Google Scholar]
- 73.Miller WG, Leveau JHJ, Lindow SE. 2000. Improved gfp and inaZ broad-host-range promoter-probe vectors. MPMI 13:1243–1250. 10.1094/MPMI.2000.13.11.1243. [DOI] [PubMed] [Google Scholar]
- 74.Wang RF, Kushner SR. 1991. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 100:195–199. 10.1016/0378-1119(91)90366-J. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 and S2. Download JB00165-21_Supp_1_seq14.pdf, PDF file, 1.1 MB (1.1MB, pdf)