

Supplemental digital content is available in the text.
Key Words: esketamine, major depressive disorder, suicidal ideation, rapid acting
Abstract
Purpose/Background
Numerous health authority approvals of esketamine nasal spray, combined with oral antidepressant, to treat depressive symptoms in adults with major depressive disorder and acute suicidal ideation or behavior were based on 2 identically designed, double-blind, phase 3 studies.
Methods/Procedures
Across both ASPIRE studies (NCT03039192, NCT03097133), patients (N = 456) were randomized to esketamine 84 mg or placebo nasal spray twice weekly for 4 weeks plus comprehensive standard of care, including hospitalization and newly initiated or optimized antidepressant(s). In post hoc analyses of pooled data, changes from baseline at 24 hours after the first dose in Montgomery-Åsberg Depression Rating Scale total score and Clinical Global Impression–Severity of Suicidality–Revised, in the full cohort and in subgroups, were analyzed using analysis of covariance.
Findings/Results
Esketamine plus standard of care demonstrated significantly greater improvement in Montgomery-Åsberg Depression Rating Scale total score versus placebo plus standard of care at 24 hours (least square mean difference [95% confidence interval], −3.8 [−5.75 to −1.89]) and at earlier (4 hours: −3.4 [−5.05 to −1.71]) and later time points (day 25: −3.4 [−5.36 to −1.36]). The between-group difference (95% confidence interval) for change in Clinical Global Impression–Severity of Suicidality–Revised at 24 hours was −0.20 (−0.43 to 0.04) for all patients and −0.31 (−0.61 to −0.01) for those with a history of suicide attempt. Common adverse events (≥20%) during esketamine treatment were dizziness, dissociation, nausea, somnolence, and headache.
Implications/Conclusions
Esketamine plus comprehensive standard of care rapidly reduces depressive symptoms in patients with major depressive disorder who have acute suicidal ideation or behavior, especially in those with a history of suicide attempt, providing a new treatment option for this particularly ill and vulnerable population.
Major depression is a common, serious, and burdensome psychiatric illness.1 Major depressive disorder (MDD) has a devastating impact on the lives of patients, affecting their relationships, parental functioning, work performance, and various other aspects of role performance, as well as health2 and disability years.3,4 Major depressive disorder is commonly associated with suicide,5–7 posing a major public health concern. Up to 60% of patients with MDD experience suicidal ideation, and up to 20% attempt suicide over their lifetime,6,8 with an estimated lifetime risk of 3.4% for completed suicide in this population.9
Patients with MDD who have active suicidal ideation with intent constitute an extremely ill subpopulation. These patients manifest more severe depressive symptoms, greater psychiatric comorbidities, worse functioning and quality of life, and more prior suicide attempts, compared with patients with MDD without suicidal ideation.10–12 Almost all patients with MDD who attempt or complete suicide experience suicidal ideation beforehand.10,13 In addition, those with more severe suicidal ideation, as evidenced by intent or intent with plan, are at greater risk of attempted or completed suicide than those without suicidal intent.10,14
Patients with MDD who have active suicidal ideation with intent require immediate and comprehensive intervention to avert self-harm. Traditionally, optimal care for these patients in crisis included initiation or optimization of oral antidepressants and hospitalization for many.15,16 However, the utility of standard antidepressants is limited in such situations because of the 4- to 6-week delay in the onset of their full effect.16,17 Although hospitalization is generally helpful in providing a safe environment for evaluation and the initiation of treatment, the risks for attempted and completed suicide remain high in the weeks immediately after discharge.18,19
Some studies have found that patients with MDD who have suicidal ideation are less likely to respond to treatment than patients with MDD without suicidal ideation.20,21 In a study of more than 4000 patients with depression treated with antidepressants, multivariate analyses (odds ratio [OR], 95% confidence interval [CI]) revealed that active suicidal ideation (OR, 1.40; 95% CI, 1.18 to 1.65) and history of suicide attempt (OR, 1.39; 95% CI, 1.16 to 1.66) were the best predictors of non-remission.20 Thus, there was a clear unmet medical need for a novel treatment to rapidly control the symptoms of depression in adult patients with MDD who have active suicidal ideation with intent.
A small study (n = 68) suggested that esketamine nasal spray, a first-in-class glutamatergic N-methyl-d-aspartate receptor antagonist, given in conjunction with an oral antidepressant, may provide rapid relief of depressive symptoms and suicidality in this acutely ill patient population.22 This proof-of-concept study gave rise to 2 identically designed, phase 3 global studies of adults with MDD who had active suicidal ideation with intent (ASPIRE I and ASPIRE II).23,24 In the United States, these studies formed the basis of the approval of esketamine for the treatment of depressive symptoms in adults with MDD with acute suicidal ideation or behavior.25 In the European Union, the product, coadministered with oral antidepressant therapy, is approved in adults with a moderate to severe episode of MDD, as acute short-term treatment, for the rapid reduction of depressive symptoms, which according to clinical judgment constitute a psychiatric emergency.26 We pooled and analyzed efficacy and safety data from these 2 pivotal studies to better characterize the effects of esketamine in treating these acutely suicidal patients with MDD based on a larger study cohort than that available from the individual studies. Specifically, pooling of data increases the sample sizes for subgroup analyses, thereby improving the precision around treatment differences in depressive symptoms and severity of suicidality that were observed among patients with a history of suicide attempts.
MATERIALS AND METHODS
Methods of the ASPIRE I and ASPIRE II trials are described in detail in the primary manuscripts.23,24 In brief, both trials enrolled patients aged between 18 and 64 years with a diagnosis of MDD based on the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-V). Patients were screened for study participation shortly after presenting to an emergency department or inpatient psychiatric unit. At that time, eligible patients provided affirmative answers to Mini-International Neuropsychiatric Interview27 questions B3 (“Think about suicide [killing yourself]?”) and B10 (“Intend to act on thoughts of killing yourself in the past 24 hours?”), warranted acute psychiatric hospitalization due to their imminent suicide risk, and had a Montgomery-Åsberg Depression Rating Scale (MADRS)28 total score greater than 28 before the first dose of study drug on day 1. In addition, patients must have agreed to comprehensive standard-of-care treatment composed of initial hospitalization and initiation or optimization of antidepressant(s) during double-blind treatment. Patients who had a current or prior DSM-V diagnosis of psychotic disorder or certain other psychiatric illnesses (eg, current DSM-V diagnosis of bipolar disorder, obsessive-compulsive disorder, antisocial personality disorder, borderline personality disorder) were excluded from study participation, as were those with moderate to severe DSM-V substance or alcohol use disorder within the most recent 6 months (12 months in some countries).
The ASPIRE I and ASPIRE II were double-blind, randomized, placebo-controlled, multicenter, multicountry trials conducted between June 2017 and April 2019. All patients provided written informed consent after procedures and possible adverse effects were explained to them and before their study participation commenced. The trials were conducted with full approval of an independent review board/ethics committee for each site and are registered at ClinicalTrials.gov (ASPIRE I, NCT03039192; ASPIRE II, NCT03097133).
Both ASPIRE studies comprised a 24- to 48-hour screening phase during which patients' eligibility for study enrollment was assessed, a 4-week (days 1–25) double-blind treatment phase, and a 9-week follow-up phase (days 26–90) during which patients discontinued esketamine or placebo nasal spray, but were continued on standard-of-care oral antidepressant(s).
Within each ASPIRE trial, eligible patients were randomized (1:1) to 84 mg of esketamine nasal spray or matching placebo nasal spray, which they self-administered twice weekly for 4 weeks (days 1, 4, 8, 11, 15, 18, 22, 25) under the supervision of a study site healthcare professional. The first dose of study drug was administered in an emergency department or after patients were admitted to an inpatient psychiatric unit, where they were to remain for a recommended 5 days (14 days recommended in several of the European Union countries). In addition, the choice of standard-of-care oral antidepressant treatment (monotherapy or augmentation therapy [ie, a second antidepressant, atypical antipsychotic, or mood stabilizer]) initiated or optimized at randomization was at the discretion of the investigator; the dose could be adjusted during the first 2 weeks of double-blind treatment, if needed, but was to remain stable thereafter.
Patients were permitted to take benzodiazepines (dosage equivalent to lorazepam ≤6 mg/d) during their participation in the study, except during the 12 hours around the first intranasal study drug dose (ie, restricted from 8 hours before to 4 hours after dosing), the 8 hours preceding all subsequent intranasal study drug doses, and the 8 hours before the day 2 assessments (ie, in the window from 16 to 24 hours after the first dose of intranasal study drug). Patients were also allowed to receive psychotherapy.
Efficacy and Safety Assessments
To prevent functional unblinding, different raters were used to conduct efficacy and safety assessments. Efficacy raters could not be involved in patient care during the study.
Using the Structured Interview Guide for MADRS,28 trained and certified raters assessed the severity of patients' depressive symptoms at baseline (before dosing), 4 and 24 hours after the first dose of intranasal study drug (days 1–2), predose at all ensuing visits during double-blind treatment, and 4 hours after the final dose on day 25. The 10 MADRS items, which evaluate core symptoms of depression, were rated based on a 0 to 6 scale (0 = no abnormality to 6 = severe).
Existing scales (such as the Columbia-Suicide Severity Rating Scale29 and the Scale for Suicide Ideation30) were not designed to be sensitive to rapid changes in suicidality.31 Given this limitation, the Suicide Ideation and Behavior Assessment Tool (SIBAT),32 a computerized instrument, was developed to assess suicidal ideation and behavior in the ASPIRE studies. The main SIBAT outcome is the clinician-reported Clinical Global Impression–Severity of Suicidality–revised (CGI-SS-r, rated on a 0–6 scale [0 = normal, not at all suicidal, to 6 = among the most extremely suicidal patients]). Other SIBAT outcomes are the clinician-reported Clinical Global Impression of Imminent Suicide Risk (CGI-SR-I, rated on a 0–6 scale [0 = no imminent suicidal risk to 6 = extreme imminent suicidal risk]), the clinician-reported Frequency of Suicidal Thinking (FoST, rated on a 0–5 scale [0 = never to 5 = all of the time]), and patient-reported FoST (rated on a 0–4 scale [0 = no suicidal thoughts to 4 = suicidal thoughts all the time]). The SIBAT assessments were conducted on all visit days during the double-blind treatment phase (predose, 4 hours after the first dose).
Adverse events were monitored throughout the study. The details of other safety assessments are presented in the primary manuscripts.23,24
Statistical Methods
Post hoc analyses of efficacy and safety were conducted on pooled data from the ASPIRE I and ASPIRE II trials. Pooling of data from the identically designed ASPIRE studies provides a larger study cohort and larger sample sizes for subgroup analyses, thereby providing more precise estimates for treatment differences.
Point estimates of the treatment differences (either difference in means, proportions, or odds ratios [ORs]) and 95% CIs are reported. Of note, 95% CIs that do not include zero for differences in means and proportions or do not include 1 for ORs correspond to a 2-sided P value of less than 0.05. Because this was a post hoc analysis, no adjustments for multiple comparisons were made.
The safety analysis data set included all randomized patients who received at least 1 dose of study drug. The efficacy analysis data set included all patients in the safety analysis data set who had baseline and at least 1 postbaseline evaluation for the MADRS or CGI-SS-r. The follow-up analysis set included all patients who completed the double-blind treatment phase and either entered the follow-up phase or provided adverse event data after the double-blind treatment phase.
In both ASPIRE studies, the primary efficacy end point was change in the MADRS total score from baseline (day 1, predose) to 24 hours after the first dose (day 2). In the pooled data analysis, the primary end point was analyzed on last observation carried forward (LOCF) data using analysis of covariance (ANCOVA) with study number, treatment, analysis center within study, and standard-of-care antidepressant at time of randomization (monotherapy or antidepressant plus augmentation therapy) as factors, and baseline MADRS total score as a continuous covariate.
Subgroup analyses on change in MADRS total score at 24 hours after the first dose were performed using an ANCOVA model; the subgroups are shown in Figure 1. Because patients were hospitalized at the time of the primary end point, missing data were infrequent. Only 6 patients (3 in each group) had their 4-hour value carried forward to day 2.
FIGURE 1.
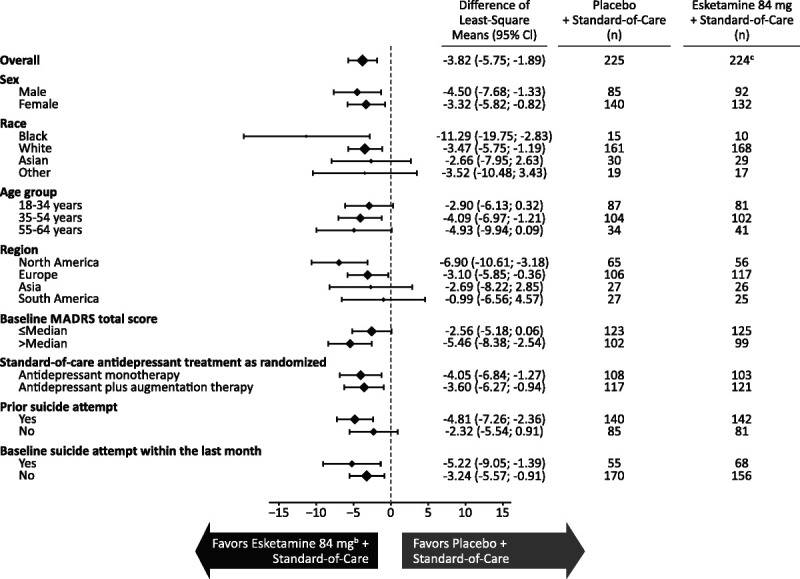
Least squares mean (95% CI) treatment difference of change in the MADRS total score from baseline to 24 hours after the first dose by subgroupa. a Change in the MADRS total score was analyzed using ANCOVA with LOCF. Negative change in score indicates improvement. Patients were hospitalized at the time of the primary end point; therefore, missing data were infrequent. b Includes patients who had their dose reduced because of tolerability issues. c One patient in the esketamine 84 mg + standard-of-care group had missing MADRS data at baseline. One additional patient in esketamine 84 mg + standard-of-care group was missing both the day 1, 4 hour, and day 2 values and therefore was excluded from subgroup analyses.
Treatment effect over time for MADRS total score during the double-blind phase was analyzed using a mixed model for repeated measures based on observed case data. The model included baseline MADRS total score as a continuous covariate; study number, day, treatment, analysis center within study, standard-of-care antidepressant at time of randomization, and day-by-treatment interaction as fixed effects; and a random patient effect. Point estimates and 95% CIs for the treatment differences were provided for each time point.
Remission (defined as MADRS total score ≤12) and response (≥50% reduction in MADRS total score) over time were summarized, and estimates of the treatment difference in proportions and 95% CIs were determined.
Generalized estimation equations of logistic regression models were used to estimate the likelihood (OR, 95% CI) of clinically meaningful improvement on individual MADRS items (defined by the authors as a decrease of ≥2 points).
Change in CGI-SS-r from baseline to 24 hours after the first dose, the key secondary end point of both ASPIRE studies, and to 4 hours after the first dose was analyzed using an ANCOVA model based on LOCF data with the same factors that were used for the primary end point and baseline CGI-SS-r score as a covariate. Subgroup analyses on change in CGI-SS-r at 4 and 24 hours after the first dose were also performed using an ANCOVA model. Resolution of suicidality, defined as CGI-SS-r score of 0 to 1, was also analyzed.
Differences in least squares (LS) means and 95% CIs were calculated for other indices of suicidality (MADRS suicide item, CGI-SR-I, clinician- and patient-rated FoST) based on ANCOVA modeling similar to that described for the primary end point analysis.
Frequency distributions are provided for adverse events.
RESULTS
Altogether, 543 patients (ASPIRE I: 270, ASPIRE II: 273) were screened and 456 patients (ASPIRE I: 226, ASPIRE II: 230; 229 to esketamine plus standard of care and 227 to placebo plus standard of care) were randomized, 4 of whom (2 in each treatment group) were excluded from safety analyses because they did not receive a dose of intranasal study drug. One additional patient (esketamine plus standard of care) was excluded from efficacy analyses because the patient discontinued after the first dose of study agent on day 1 and did not provide any postbaseline efficacy data. Most randomized patients (esketamine: 192 of 229, 83.8%; placebo: 187 of 227, 82.4%) completed the 4-week double-blind treatment phase (Fig. S1, http://links.lww.com/JCP/A780).
The treatment groups were similar based on demographics, baseline clinical characteristics, and baseline psychiatric history (Table 1). The mean (SD) age was 40.1 (13.00) years. The population was composed of more women (60.8%) than men; the most prevalent racial subgroup was White (73.2%). The mean (SD) baseline MADRS total score was 40.4 (5.82), representing severe depression. Most patients (90.0%) were moderately to extremely suicidal, according to the investigator-rated CGI-SS-r. Nearly two thirds (63.1%) of the patients reported a prior lifetime suicide attempt, with approximately one quarter of patients reporting an attempt within the last month (30.1% vs 24.4% in the esketamine and placebo groups, respectively).
TABLE 1.
Demographics, Baseline Clinical Rating, and Psychiatric History (Efficacy Analysis Data Set)
| Parameter | Placebo + Standard of Care (n = 225) | Esketamine 84 mg* + Standard of Care (n = 226) | All Patients (N = 451) |
|---|---|---|---|
| Age, mean (SD), y | 39.6 (13.08) | 40.5 (12.92) | 40.1 (13.00) |
| Sex, n (%) | |||
| Female | 140 (62.2) | 134 (59.3) | 274 (60.8) |
| Male | 85 (37.8) | 92 (40.7) | 177 (39.2) |
| Race, n (%) | |||
| White | 161 (71.6) | 169 (74.8) | 330 (73.2) |
| Asian | 30 (13.3) | 29 (12.8) | 59 (13.1) |
| Black or African American | 15 (6.7) | 11 (4.9) | 26 (5.8) |
| Other/not reported | 19 (8.4) | 17 (7.5) | 36 (8.0) |
| Region, n (%) | |||
| North America | 65 (28.9) | 58 (25.7) | 123 (27.3) |
| Europe | 106 (47.1) | 117 (51.8) | 223 (49.4) |
| Asia | 27 (12.0) | 26 (11.5) | 53 (11.8) |
| South America | 27 (12.0) | 25 (11.1) | 52 (11.5) |
| MADRS total score,† mean (SD) | 40.4 (6.04) | 40.3 (5.60) | 40.4 (5.82) |
| CGI-SS-r,† n (%) | |||
| Normal, not at all suicidal | 0 | 0 | 0 |
| Questionably suicidal | 6 (2.7) | 6 (2.7) | 12 (2.7) |
| Mildly suicidal | 17 (7.6) | 16 (7.1) | 33 (7.3) |
| Moderately suicidal | 61 (27.1) | 64 (28.4) | 125 (27.8) |
| Markedly suicidal | 84 (37.3) | 86 (38.2) | 170 (37.8) |
| Severely suicidal | 55 (24.4) | 46 (20.4) | 101 (22.4) |
| Among the most extremely suicidal patients | 2 (0.9) | 7 (3.1) | 9 (2.0) |
| Suicide attempt, n (%) | |||
| Attempt in the last month | 55 (24.4) | 68 (30.1) | 123 (27.3) |
| Attempt during lifetime† | 140 (62.2) | 144 (64.0) | 284 (63.1) |
| Standard-of-care antidepressant,‡ n (%) | |||
| Antidepressant monotherapy | 108 (48.0) | 104 (46.0) | 212 (47.0) |
| Antidepressant plus augmentation therapy§ | 117 (52.0) | 122 (54.0) | 239 (53.0) |
*Includes patients who had their dose reduced because of tolerability issues.
†Two hundred twenty-five for the esketamine + standard-of-care group.
‡As randomized.
§Augmentation therapy included an agent, such as a second antidepressant, an atypical antipsychotic, or a mood stabilizer.
At randomization, the population was balanced between patients who were to receive antidepressant monotherapy (212 of 451, 47.0%) as standard-of-care treatment and patients who were to receive antidepressant plus augmentation therapy as standard-of-care treatment (239 of 451, 53.0%; Table 1). The most frequently used standard-of-care antidepressant medications (≥10% of patients) during the double-blind phase were venlafaxine (26.5%), quetiapine (21.2%), escitalopram (16.6%), duloxetine (15.0%), and mirtazapine (14.2%). Concomitant use of benzodiazepines (73.6% and 66.7% in the esketamine and placebo groups, respectively) and nonbenzodiazepine hypnotics and anxiolytics (36.0% and 31.7% in the esketamine and placebo groups, respectively) during double-blind treatment was similar between the treatment groups.
Efficacy Results
The MADRS total score decreased from baseline to 24 hours after the first dose in both the esketamine plus standard-of-care group (mean [SD], −16.1 [11.73]) and the placebo plus standard-of-care group (−12.6 [10.56]), with greater improvement of depressive symptoms with esketamine (LS mean difference [SE], −3.8 [0.98]; 95% CI, −5.75 to −1.89). In pooling of data from the ASPIRE I and ASPIRE II studies, clear treatment benefit with esketamine was observed across subgroups (Fig. 1), importantly, among the patients who had more severe depression (ie, baseline MADRS score > median of 41.0; LS mean difference [95% CI], −5.46 [−8.38 to −2.54]) and among the patients who reported prior suicide attempts (LS mean difference [95% CI], −4.81 [−7.26 to −2.36]).
Improvement in depressive symptoms in the full study cohort was documented first at 4 hours after the first dose (LS mean difference, −3.4; 95% CI, −5.05 to −1.71) and at all subsequent visits during the double-blind treatment phase (Fig. 2). The MADRS total scores remained low during the follow-up phase (through follow-up end point/day 90; Fig. S2, http://links.lww.com/JCP/A780).
FIGURE 2.
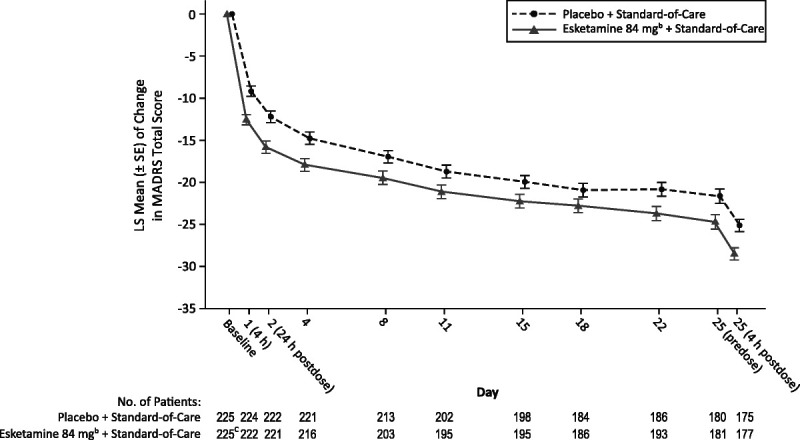
Least squares mean (± SE) change in the MADRS total score from baseline during the double-blind treatment phasea. MMRM, mixed-effects model using repeated measures; SE, standard error. a MMRM analysis with observed cases. Negative change in score indicates improvement. b Includes patients who had their dose reduced because of tolerability issues. c One patient in the esketamine 84 mg + standard-of-care group had missing MADRS data at baseline.
The percentage of patients who achieved remission was greater in the esketamine plus standard-of-care group than the placebo plus standard-of-care group at all time points during double-blind treatment (Fig. S3, http://links.lww.com/JCP/A780). The remission rate was 20.4% in the esketamine plus standard-of-care group and 9.8% in the placebo plus standard-of-care group (difference [95% CI] between treatment groups, 10.6% [4.05 to 17.10]) at 24 hours after the first dose and 50.4% and 37.3%, respectively (difference [95% CI] between treatment groups, 13.1% [4.03 to 22.19]) at 4 hours after dose on day 25.
The response rate was 34.5% in the esketamine plus standard-of-care group and 25.3% in the placebo plus standard-of-care group (difference [95% CI] between treatment groups, 9.2% [0.77 to 17.59]) at 24 hours after first dose and 64.6% and 58.2%, respectively (difference [95% CI] between treatment groups, 6.4% [−2.59 to 15.35]) at 4 hours after dose on day 25 (Fig. S4, http://links.lww.com/JCP/A780).
With respect to the change in individual items of the MADRS, esketamine-treated patients had a greater likelihood of achieving clinically meaningful improvement on all symptoms of depression, as measured by the MADRS items (Fig. 3). Early benefit with esketamine was greatest (ie, lower limit of 95% CI >1.0) on reported sadness (OR = 2.29), apparent sadness (OR = 1.99), inner tension (OR = 1.92), suicidal thoughts (OR = 1.91), inability to feel (OR = 1.75), and pessimistic thoughts (OR = 1.63) at 4 hours after the first dose and on concentration difficulties (OR = 2.47), apparent sadness (OR = 2.13), inner tension (OR = 2.13), inability to feel (OR = 1.95), reported sadness (OR = 1.77), reduced sleep (OR = 1.67), and pessimistic thoughts (OR = 1.62) at 24 hours.
FIGURE 3.
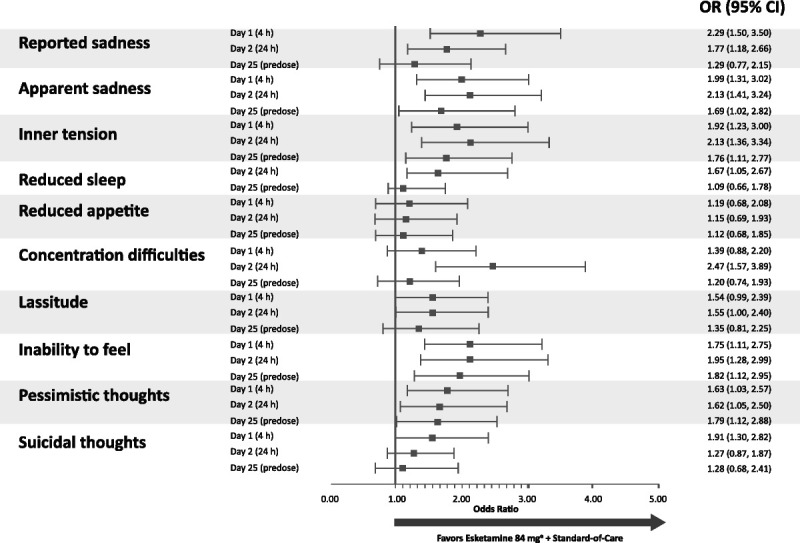
Odds ratios (95% CI) for a ≥2-point improvement in the MADRS item scores during the double-blind treatment phase. a Includes patients who had their dose reduced because of tolerability issues. Notes: MADRS items are rated on a scale of 0 to 6 where 0 indicates no abnormality and 6 indicates severe. The reduced sleep item was not assessed at the 4-hour post–first dose time point.
Patients in both treatment groups experienced rapid reduction in the severity of their suicidality as measured by the CGI-SS-r. Although the difference between groups was not statistically significant (LS mean difference [95% CI], −0.20 [−0.43 to 0.04]), CGI-SS-r decreased from baseline to 24 hours after the first dose in the esketamine plus standard-of-care group (mean [SD], −1.5 [1.55] points) and in the placebo plus standard-of-care group (−1.3 [1.45] points). At 4 hours after the first dose, the mean (SD) change from baseline was −1.4 (1.49) and −0.9 (1.26) for the respective treatment groups (between-group difference, −0.41 [−0.63 to −0.18]). Severity of suicidality was also improved in both treatment groups at the end of double-blind treatment without significant difference between treatment groups (Fig. 4). In the subgroup of patients with a history of prior suicide attempt(s), LS mean treatment difference (95% CI) was −0.31 (−0.61 to −0.01) and −0.40 (−0.68 to −0.11) at 24- and 4-hour post–first dose time points, respectively.
FIGURE 4.
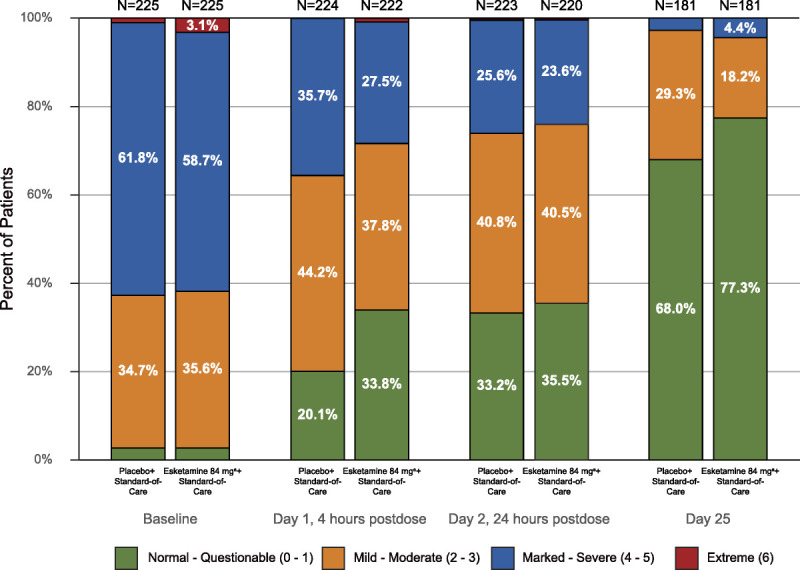
Frequency distribution of CGI-SS-r score at baseline, 4, and 24 hours after the first dose, and day 25 (observed cases). a Includes patients who had their dose reduced because of tolerability issues.
Resolution of suicidality was achieved by 33.2% of the patients in the esketamine plus standard-of-care group and 20.0% of the patients in the placebo plus standard-of-care group at 4 hours after the first dose, 34.5% and 32.9%, respectively, at 24 hours after the first dose, and 61.9% and 54.7%, respectively, at day 25 before dose. The differences (95% CI) between treatment groups were 13.2% (5.12 to 21.25), 1.6% (−7.10 to 10.35), and 7.3% (−1.80 to 16.36) at the respective time points.
Results for other indices of clinician- and patient-rated suicidality are presented in Figure S5, http://links.lww.com/JCP/A780.
Safety Results
The most frequently reported adverse events (≥10% of the patients in either treatment group) during the double-blind treatment phase are listed in Table 2 and during the follow-up phase in Table S1, http://links.lww.com/JCP/A780. The most common (≥20%) adverse events with esketamine were dizziness (38.3% vs 13.8% for placebo), dissociation (33.9% vs 5.8%), nausea (26.9% vs 13.8%), somnolence (20.7% vs 10.2%), and headache (20.3% vs 20.4%). Most events in the esketamine plus standard-of-care group (89.9%) and placebo plus standard-of-care group (68.9%) occurred on intranasal dosing days, and most of these events (94.9% and 85.2%, respectively) resolved on the same day they began. No symptoms or adverse events of psychosis were reported during treatment with intranasal study drug, and no symptoms or adverse events consistent with withdrawal or abuse were reported during the follow-up period. Serious adverse events are presented in Table S2, http://links.lww.com/JCP/A780 and Table S3, http://links.lww.com/JCP/A780.
TABLE 2.
Summary of Most Frequently Reported* Treatment-Emergent Adverse Events During Double-Blind Phase
| No. (%) of Patients | ||
|---|---|---|
| Adverse Event | Placebo + Standard of Care (n = 225) | Esketamine 84 mg† + Standard of Care (n = 227) |
| Dizziness | 31 (13.8) | 87 (38.3) |
| Dissociation | 13 (5.8) | 77 (33.9) |
| Nausea | 31 (13.8) | 61 (26.9) |
| Somnolence | 23 (10.2) | 47 (20.7) |
| Headache | 46 (20.4) | 46 (20.3) |
| Dysgeusia | 29 (12.9) | 45 (19.8) |
| Blurred vision | 11 (4.9) | 27 (11.9) |
| Blood pressure increased | 9 (4.0) | 26 (11.5) |
| Paresthesia | 7 (3.1) | 26 (11.5) |
| Vomiting | 12 (5.3) | 26 (11.5) |
| Anxiety | 17 (7.6) | 23 (10.1) |
| Sedation | 5 (2.2) | 23 (10.1) |
*Most frequently reported is defined as ≥10% of patients in either treatment group during the double-blind phase. Events are presented in descending order in the esketamine group and in alphabetical order for events with the same incidence.
†Includes patients who had their dose reduced because of tolerability issues.
In the esketamine plus standard-of-care group, 15.4% of the subjects experienced 1 or more adverse events leading to dose reduction (to 56 mg) or interruption compared with 1.8% of the patients in the placebo plus standard-of-care group. In the esketamine plus standard-of-care group, the most common (≥1%) adverse events that led to dose reduction or interruption were dissociation (5.7%), nausea (4.8%), dizziness (3.1%), somnolence (2.6%), vomiting (2.2%), and blood pressure increased (1.3%).
Altogether, 14 patients (6.2%) in the esketamine plus standard-of-care group and 8 patients (3.6%) in the placebo plus standard-of-care group experienced adverse events leading to discontinuation of intranasal study medication. Adverse events that led to discontinuation of more than 1 esketamine-treated patient were dissociation (n = 3), depersonalization/derealization disorder, nausea, and increased blood pressure (n = 2 each).
Four patients in each treatment group attempted suicide during the double-blind treatment phase. Seven patients previously treated with esketamine plus standard of care and 3 patients previously treated with placebo plus standard of care attempted suicide in the follow-up phase when patients were only receiving standard-of-care antidepressant treatment. Of these, 1 patient in each treatment group attempted suicide in both phases. The occurrence of suicide attempts during the follow-up phase was dispersed over the 9-week phase without an apparent pattern suggestive of rebound. One patient died by suicide in the follow-up phase, 3 days after the last esketamine dose. Eight of the 10 patients in the esketamine plus standard-of-care group and 3 of the 6 patients in the placebo plus standard-of-care group who attempted suicide had a history of prior suicide attempt(s).
DISCUSSION
The ASPIRE trials were among the first to enroll patients with MDD at imminent risk of suicide, a group that had been excluded from most earlier antidepressant registration studies. The combined data from the ASPIRE I and ASPIRE II studies result in a cohort of 451 adults with MDD with suicidal ideation or behavior, which to our knowledge is the largest sample of acutely suicidal patients to be studied in an antidepressant treatment trial.
Among the patients with MDD who had acute suicidal ideation or behavior, treatment with esketamine plus standard of care resulted in greater improvement of depressive symptoms at 24 hours after the first dose, compared with placebo plus standard of care, based on decrease in the MADRS total score. Results of the pooled analysis on the primary end point are consistent with those of the individual trials, allowing a larger cohort to inform on particular patient subgroups. In this regard, noteworthy are the between-group differences for the patients with more severe depression at baseline as well as the patients with a history of prior suicide attempt, including those having attempted suicide in the month before study enrollment, a group most prone to poor treatment response.20
Rapid improvement in depressive symptoms at 4 hours after the first dose of esketamine is an especially important finding for patients with severe depression who are acutely suicidal. Rapid improvement was also observed across all symptoms of depression, reflected in the individual MADRS items. Furthermore, treatment benefit with esketamine continued throughout 4-week double-blind treatment, at the end of which half of esketamine-treated patients had achieved remission.
Results of the pooled analyses are also consistent with the findings of the individual trials on the key secondary end point, in that patients in both treatment groups experienced a rapid, clinically meaningful reduction (ie, ≥1-point reduction in CGI scale, at the individual patient level33,34) in the severity of their suicidality, as measured by the CGI-SS-r at 24 hours after the first dose without statistically significant treatment differences.23,24 This may be due in part to the benefit of hospitalization, the intensive clinical contact patients received as study participants,35 and/or the method used to assess suicidality. Methodological challenges of measuring rapid changes in suicidality are well described.31 Ecological momentary assessment studies have identified fluctuations in suicidal ideation, frequency, and intensity, as markers of increased suicide risk.36 These findings highlight that suicidal ideation is highly variable over short periods, and traditional 1-time assessment methods or once-daily measurement may not adequately capture the impact of interventions on this symptom.
It is important to note that esketamine nasal spray is approved in conjunction with an oral antidepressant to treat depressive symptoms, but not for reducing suicidal ideation or behavior, in adults with MDD and acute suicidal ideation or behavior.25,26 Nonetheless, rapid reduction of depressive symptoms may serve as an important approach in this highly vulnerable population, bridging the gap until additional comprehensive therapeutic measures (including the onset of full oral antidepressant effect) may be set in place.
The adverse events observed in the ASPIRE studies are consistent with the established safety profile of esketamine nasal spray.37–39 In general, adverse events occurred on intranasal dosing days and resolved the same day.
Even in an enriched population, the event of suicidality is relatively rare, underscoring the value of a large database in a vulnerable population. Suicide-related events were reported among the extremely vulnerable patients enrolled in the ASPIRE studies, not unexpectedly given their high risk. Ten patients in the esketamine plus standard-of-care group and 6 patients in the placebo plus standard-of-care group attempted suicide during the double-blind treatment and/or follow-up phases. This difference in numbers of attempted suicides between groups may be related to the 5% greater rate of recent suicide attempts reported by patients assigned to esketamine at randomization, as recent suicidal behavior is a known risk factor for subsequent suicide attempt.40 One patient treated with esketamine during the double-blind phase died by suicide during the follow-up phase. This patient had a history of multiple prior suicide attempts, including one attempt within the month before randomization. Although tragic, this was the only suicide across the clinical development program that included more than 500 patients with MDD who had active suicidal ideation with intent, a high rate of prior suicide attempts, and moderate to severe depressive symptoms at baseline, all significant risk factors or predictors for suicide.41–43
Limitations
To ensure ethical treatment, patients in the control arm of the ASPIRE studies received comprehensive standard of care, including initial psychiatric hospitalization and optimized oral antidepressant therapy. The well-known placebo response in antidepressant clinical trials,44,45 coupled with the frequent and intensive clinical visits (contacts) in the ASPIRE studies, should be considered when interpreting the results. Standard-of-care interventions provided in these global studies may differ across regions, also potentially limiting generalizations based on the findings.
CONCLUSIONS
The pooled results from the ASPIRE I and ASPIRE II trials affirm and strengthen the results of the individual studies: esketamine nasal spray, given in the context of comprehensive standard of care, provides rapid relief of depressive symptoms in patients with MDD and acute suicidal ideation or behavior, especially so in those with a history of prior suicide attempt, a severely ill, vulnerable population in need of urgent symptom control.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Sandra Norris, PharmD (Norris Communications Group, LLC) for medical writing assistance, and Ellen Baum, PhD (Janssen Global Services, LLC) for additional editorial support.
AUTHOR DISCLOSURE INFORMATION
C.M.C., D.F.I., X.L., X.Q., I.T., D.-J.F., and R.L. are employees of Janssen Research & Development, LLC, and A.I.N. and T.J.L. are employees of Janssen Scientific Affairs, LLC; all authors hold company equity.
The ASPIRE I and ASPIRE II studies were funded by Janssen Research & Development, LLC, Titusville, NJ. Employees of the sponsor, as noted in author contributions, were involved in trial design; data analysis or interpretation; and/or other aspects pertinent to the study. The authors had full access to all of the data in the study, were involved in writing and/or revising the manuscript, and had final responsibility for the decision to submit for publication. C.M.C., D.-J.F., and D.F.I. participated in study design and data collection. X.L., X.Q., I.T., and R.L. participated in the statistical design and data analysis. All authors substantially contributed in interpretation of data, writing and critical revision of manuscript, and consented to the final version of the manuscript. All authors had full access to all the data in the study and take responsibility for integrity of the data and the accuracy of the data analysis. All authors meet the International Committee of Medical Journal Editors criteria, and all those who fulfilled those criteria are listed as authors.
Footnotes
A podcast discussing this article is available online at the journal website.
Pooled data from the ASPIRE I and ASPIRE II studies were presented in an oral presentation at the 32nd ECNP Congress, Copenhagen, Denmark, September 7–10, 2019, and in poster sessions at the 58th Annual Meeting of American College of Neuropsychopharmacology, Orlando, FL, December 8–11, 2019, and the 2020 Virtual Annual Meeting of American Society of Psychopharmacology (ASCP), May 29–30, 2020.
Supplemental digital content is available for this article. Direct URL citation appears in the printed text and is provided in the HTML and PDF versions of this article on the journal’s Web site (www.psychopharmacology.com).
Contributor Information
Dawn F. Ionescu, Email: dionesc@its.jnj.com.
Xiang Li, Email: xli256@its.jnj.com.
Xin Qiu, Email: xqiu24@its.jnj.com.
Rosanne Lane, Email: RLANE@its.jnj.com.
Ibrahim Turkoz, Email: ITurkoz@its.jnj.com.
Abigail I. Nash, Email: anash2@its.jnj.com.
Tricia J. Lopena, Email: tlopena@its.jnj.com.
Dong-Jing Fu, Email: dfu@its.jnj.com.
REFERENCES
- 1.World Health Organization . Depression. 30 January 2020. Available at: https://www.who.int/news-room/fact-sheets/detail/depression. Accessed February 26, 2021.
- 2.Kessler RC. The costs of depression. Psychiatr Clin North Am. 2012;35:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Global Burden of Disease Study 2013 Collaborators . Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;386:743–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greenberg PE Fournier AA Sisitsky T, et al. The economic burden of adults with major depressive disorder in the United States (2005 and 2010). J Clin Psychiatry. 2015;76:155–162. [DOI] [PubMed] [Google Scholar]
- 5.Harris EC, Barraclough B. Suicide as an outcome for mental disorders. A meta-analysis. Br J Psychiatry. 1997;170:205–228. [DOI] [PubMed] [Google Scholar]
- 6.Hasin DS Sarvet AL Meyers JL, et al. Epidemiology of adult DSM-5 major depressive disorder and its specifiers in the United States. JAMA Psychiatry. 2018;75:336–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Angst J, Angst F, Stassen HH. Suicide risk in patients with major depressive disorder. J Clin Psychiatry. 1999;60:57–62. [PubMed] [Google Scholar]
- 8.Holma KM Melartin TK Haukka J, et al. Incidence and predictors of suicide attempts in DSM-IV major depressive disorder: a five-year prospective study. Am J Psychiatry. 2010;167:801–808. [DOI] [PubMed] [Google Scholar]
- 9.Blair-West GW Cantor CH Mellsop GW, et al. Lifetime suicide risk in major depression: sex and age determinants. J Affect Disord. 1999;55:171–178. [DOI] [PubMed] [Google Scholar]
- 10.Sokero TP Melartin TK Rytsala HJ, et al. Suicidal ideation and attempts among psychiatric patients with major depressive disorder. J Clin Psychiatry. 2003;64:1094–1100. [DOI] [PubMed] [Google Scholar]
- 11.van Ballegooijen W Eikelenboom M Fokkema M, et al. Comparing factor structures of depressed patients with and without suicidal ideation, a measurement invariance analysis. J Affect Disord. 2019;245:180–187. [DOI] [PubMed] [Google Scholar]
- 12.Wolfersdorf M. Depression and suicidal behaviour: psychopathological differences between suicidal and non-suicidal depressive patients. Arch Suicide Res. 1995;1:273–288. [Google Scholar]
- 13.McGirr A Renaud J Seguin M, et al. An examination of DSM-IV depressive symptoms and risk for suicide completion in major depressive disorder: a psychological autopsy study. J Affect Disord. 2007;97:203–209. [DOI] [PubMed] [Google Scholar]
- 14.Brown GK Beck AT Steer RA, et al. Risk factors for suicide in psychiatric outpatients: a 20-year prospective study. J Consult Clin Psychol. 2000;68:371–377. [PubMed] [Google Scholar]
- 15.Practice guideline for the assessment and treatment of patients with suicidal behaviors. Am J Psychiatry. 2003;160:1–60. [PubMed] [Google Scholar]
- 16.Wasserman D Rihmer Z Rujescu D, et al. European Psychiatric Association . The European Psychiatric Association (EPA) guidance on suicide treatment and prevention. Eur Psychiatry. 2012;27:129–141. [DOI] [PubMed] [Google Scholar]
- 17.Simon GE, Savarino J. Suicide attempts among patients starting depression treatment with medications or psychotherapy. Am J Psychiatry. 2007;164:1029–1034. [DOI] [PubMed] [Google Scholar]
- 18.Chung D Hadzi-Pavlovic D Wang M, et al. Meta-analysis of suicide rates in the first week and the first month after psychiatric hospitalisation. BMJ Open. 2019;9:e023883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacobs DG Baldessarini RJ Conwell Y, et al. Practice guideline for the assessment and treatment of patients with suicidal behaviors. Available at: https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/suicide.pdf. Accessed February 26, 2021. [PubMed]
- 20.Lopez-Castroman J Jaussent I Gorwood P, et al. Suicidal depressed patients respond less well to antidepressants in the short term. Depress Anxiety. 2016;33:483–494. [DOI] [PubMed] [Google Scholar]
- 21.Pompili M Baldessarini RJ Tondo L, et al. Response to intravenous antidepressant treatment by suicidal vs. nonsuicidal depressed patients. J Affect Disord. 2010;122:154–158. [DOI] [PubMed] [Google Scholar]
- 22.Canuso CM Singh JB Fedgchin M, et al. Efficacy and safety of intranasal esketamine for the rapid reduction of symptoms of depression and suicidality in patients at imminent risk for suicide: results of a double-blind, randomized, placebo-controlled study. Am J Psychiatry. 2018;175:620–630. [DOI] [PubMed] [Google Scholar]
- 23.Fu DJ Ionescu DF Li X, et al. Esketamine nasal spray for rapid reduction of major depressive disorder symptoms in patients who have active suicidal ideation with intent: double-blind, randomized study (ASPIRE I). J Clin Psychiatry. 2020;81:19m13191. [DOI] [PubMed] [Google Scholar]
- 24.Ionescu DF Fu D-J Qiu X, et al. Esketamine nasal spray for rapid reduction of depressive symptoms in patients with major depressive disorder who have active suicide ideation with intent: results of a phase 3, double-blind, randomized study (ASPIRE II). Int J Neuropsychopharmacol. 2021;24:22–31. doi: 10.1093/ijnp/pyaa068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spravato® (Esketamine) Nasal Spray. Prescribing Information. Titusville, NJ: Janssen Pharmaceutical Companies; 2020. [Google Scholar]
- 26.European Medicines Agency . Esketamine nasal spray. Summary of product characteristics. Available at: https://www.ema.europa.eu/en/documents/product-information/spravato-epar-product-information_en.pdf. Accessed April 30, 2021.
- 27.Sheehan DV Lecrubier Y Sheehan KH, et al. The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry. 1998;59:22–33. [PubMed] [Google Scholar]
- 28.Williams JB, Kobak KA. Development and reliability of a structured interview guide for the Montgomery Asberg Depression Rating Scale (SIGMA). Br J Psychiatry. 2008;192:52–58. [DOI] [PubMed] [Google Scholar]
- 29.Posner K Brown GK Stanley B, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168:1266–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beck AT, Kovacs M, Weissman A. Assessment of suicidal intention: the Scale for Suicide Ideation. J Consult Clin Psychol. 1979;47:343–352. [DOI] [PubMed] [Google Scholar]
- 31.Ballard ED Gilbert JR Wusinich C, et al. New methods for assessing rapid changes in suicide risk. Front Psychiatry. 2021;12:598434. doi: 10.3389/fpsyt.2021.598434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alphs L Fu D-J Williamson D, et al. Suicide Ideation and Behavior Assessment Tool (SIBAT): evaluation of intra- and inter-rater reliability, validity, and mapping to Columbia Classification Algorithm of Suicide Assessment. Psychiatry Res. 2020;294:113495. doi: 10.1016/j.psychres.2020.113495. [DOI] [PubMed] [Google Scholar]
- 33.Busner J, Targum SD. The Clinical Global Impressions Scale: applying a research tool in clinical practice. Psychiatry (Edgmont). 2007;4:28–37. [PMC free article] [PubMed] [Google Scholar]
- 34.Wyrwich K, Reeve B, Coon CD. What's the score? Moving from items to scores -methods, considerations, and case examples. Presented at: Eighth Annual Patient-Reported Outcome Consortium Workshop. 2017. Bethesda, MD. Available at: https://c-path.org/eighth-annual-patient-reported-outcome-consortium-workshop-2/. Accessed April 30, 2021. [Google Scholar]
- 35.Pompili M. Intranasal esketamine and current suicidal ideation with intent in major depression disorder: beat the clock, save a life, start a strategy. Front Psychiatry. 2020;11:325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kleiman EM Turner BJ Fedor S, et al. Examination of real-time fluctuations in suicidal ideation and its risk factors: results from two ecological momentary assessment studies. J Abnorm Psychol. 2017;126:726–738. [DOI] [PubMed] [Google Scholar]
- 37.Daly EJ Trivedi MH Janik A, et al. Efficacy of esketamine nasal spray plus oral antidepressant treatment for relapse prevention in patients with treatment-resistant depression: a randomized clinical trial. JAMA Psychiatry. 2019;76:893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fedgchin M Trivedi M Daly EJ, et al. Efficacy and safety of fixed-dose esketamine nasal spray combined with a new oral antidepressant in treatment-resistant depression: results of a randomized, double-blind, active-controlled study (TRANSFORM-1). Int J Neuropsychopharmacol. 2019;22:616–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Popova V Daly EJ Trivedi M, et al. Efficacy and safety of flexibly dosed esketamine nasal spray combined with a newly initiated oral antidepressant in treatment-resistant depression: a randomized double-blind active-controlled study. Am J Psychiatry. 2019;176:428–438. [DOI] [PubMed] [Google Scholar]
- 40.Gramaglia C Feggi A Bergamasco P, et al. Clinical characteristics associated with suicide attempts in clinical settings: a comparison of suicidal and non-suicidal depressed inpatients. Front Psychiatry. 2016;7:109. doi: 10.3389/fpsyt.2016.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Coryell W, Young EA. Clinical predictors of suicide in primary major depressive disorder. J Clin Psychiatry. 2005;66:412–417. [DOI] [PubMed] [Google Scholar]
- 42.Hawton K Casañas I Comabella C, et al. Risk factors for suicide in individuals with depression: a systematic review. J Affect Disord. 2013;147:17–28. [DOI] [PubMed] [Google Scholar]
- 43.Oquendo MA, Currier D, Mann JJ. Prospective studies of suicidal behavior in major depressive and bipolar disorders: what is the evidence for predictive risk factors? Acta Psychiatr Scand. 2006;114:151–158. [DOI] [PubMed] [Google Scholar]
- 44.Leuchter AF Hunter AM Tartter M, et al. Role of pill-taking, expectation and therapeutic alliance in the placebo response in clinical trials for major depression. Br J Psychiatry. 2014;205:443–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rutherford BR, Roose SP. A model of placebo response in antidepressant clinical trials. Am J Psychiatry. 2013;170:723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.