

Supplemental digital content is available in the text.
Key Words: placebo, major depressive disorder, trajectories of symptom change, trajectories of change in functioning
Abstract
Purpose/Background
Heterogeneity has been documented in trajectories of symptom change during antidepressant treatment for major depressive disorder (MDD). It is unclear whether distinct trajectories of change exist for functioning during antidepressant treatment.
Methods/Procedures
This analysis explored distinct trajectories of functioning in MDD and tested whether they corresponded to trajectories of symptom change. Data were from 4317 patients and were pooled from 9 randomized placebo-controlled trials. Growth mixture modeling was used to identify trajectories of Hamilton Rating Scale for Depression (HRSD) and Sheehan Disability Scale (SDS) for placebo- and desvenlafaxine-treated patients.
Findings/Results
Three trajectories were identified for symptoms (HRSD) in patients receiving placebo (mean reduction baseline to week 8, −18.4 [most favorable] to −2.6 points [least favorable]). Four HRSD trajectories were identified for patients receiving desvenlafaxine (mean reduction from baseline to week 8, −17.2 [most favorable] to −2.6 points [least favorable]). Four trajectories were identified for functioning (SDS) in patients receiving placebo (mean reduction baseline to week 8, −13.6 [most favorable] to −0.8 points [least favorable]), and 3 for desvenlafaxine (−12.8 to −1.4 points, respectively). Percentages of agreement between most favorable HRSD and SDS trajectories were 75% (placebo) and 85% (desvenlafaxine), and for least favorable trajectories were 88% (placebo) and 80% (desvenlafaxine).
Implications/Conclusions
Distinct trajectories of change based on symptoms and functioning were identified among patients with MDD receiving desvenlafaxine and among patients with MDD receiving placebo. Differentiating subpopulations of patients has the potential to provide a more personalized treatment of patients with MDD.
ClinicalTrials.govIdentifiers: NCT00072774; NCT00277823; NCT00300378; NCT00384033; NCT00798707; NCT00863798; NCT01121484; NCT00824291; NCT01432457.
Antidepressant medications (ADMs) are first-line treatments for major depressive disorder (MDD); however, many patients do not experience sustained remission, even with maximum doses of approved ADMs.1 Distinct trajectories of symptom change in patients receiving ADMs have been identified across a patient's life span.2,3 However, despite these observations, aggregated data may mask differences in patient responses,4 as some patients demonstrate a clear advantage for specific ADMs versus placebo, whereas others do not.5 It is, therefore, essential to describe how patients respond to ADM versus those who respond to placebo.
Although impaired functioning is a critical aspect of MDD and functional recovery is a central, desirable goal,6 the majority of studies focus on identifying trajectories of change in symptoms,2,3,7,8 or clusters of symptoms,9 and there is little knowledge on whether distinct trajectories of change exist for functional improvement. For example, a previous analysis of the same pooled data set of 9 randomized clinical trials (RCTs), as used herein, applied a mixed-effects model for repeated-measures approach and demonstrated that early symptomatic improvement with desvenlafaxine was a significant predictor of later remission.7 The authors suggested that clinicians may be able to use the depression rating score early in treatment as a guide to inform treatment optimization.7 To apply these observations to real-life patient management, it is important to fully understand the diversity of trajectories that exist for symptomatic responses and to understand if all patients who respond to treatment respond in a consistent manner. Furthermore, given that symptoms and functioning are interrelated, and that functional impairments often persists after symptoms resolve,10,11 it is important to determine if similar trajectories can be identified for a functional response. The present analysis explores whether distinct trajectories of change exist for symptoms and for functioning in patients treated with desvenlafaxine or placebo, and investigates the level of agreement between trajectories of functioning and symptoms.
METHODS/PROCEDURES
Sample Population
Data were pooled from 9 double-blind placebo-controlled RCTs for MDD (Table S1, http://links.lww.com/JCP/A773).12–20 Eligible patients had DSM-IV diagnosis of MDD, baseline 17-Item Hamilton Depression Rating Scale total score of 20 or greater and/or Clinical Global Impressions—Severity scale of 4 or greater, and/or Montgomery-Åsberg Depression Rating Scale score of 25 or greater (Table S1, http://links.lww.com/JCP/A773). Patients assigned to receive placebo or desvenlafaxine (50 or 100 mg, only) were included. The Hamilton Rating Scale for Depression (HRSD)21 was used to assess depressive symptoms, and the Sheehan Disability Scale (SDS)22 assessed functioning. For all but 1 study (NCT0112148418), HRSD was measured at least at baseline and weeks 1, 2, 4, 6, and 8, whereas SDS was collected at least at baseline and weeks 2, 4, and 8. Study NCT0143245720 did not collect data on SDS. Studies were conducted according to the principles originating from the Declaration of Helsinki. Full inclusion/exclusion criteria are presented elsewhere, alongside safety and efficacy outcomes.12–20 Written, informed consent was documented before entry into each study, as detailed in the primary publications.12–20
Overview of Statistical Analyses
All analyses were conducted separately for placebo and desvenlafaxine treatment arms, and no comparisons were carried out between treatment arms. To identify distinct trajectories of HRSD and SDS scores over time, a growth mixture model approach was applied,23–25 using the R package lcmm for latent class mixed models26 to estimate the fit of separate models. Models for linear, linear in log time, quadratic, and cubic trends over time with 1 to 4 trajectory classes were considered. Selection of the most appropriate model was based on the Bayesian Information Criterion (BIC), where a lower value indicates a better fit, provided that all trajectory classes included 10% or greater of patients in each treatment arm.
Agreement was expressed as the percentage of patients assigned to the most favorable SDS trajectory from those also assigned to the most favorable HRSD trajectory, and the percentage of patients assigned to the least favorable SDS trajectories from those also assigned to the least favorable HRSD trajectory. This analysis excluded patients without both HRSD and SDS data, and those assigned to the low baseline SDS trajectory class, as these patients had no opportunity to improve an already good functioning score at baseline.
FINDINGS/RESULTS
There were 4317 patients randomized to placebo or desvenlafaxine (50, 100 mg) from 9 RCTs12–20 (Table S1, http://links.lww.com/JCP/A773). Clinical and demographic characteristics are shown in Table S2, http://links.lww.com/JCP/A773.
Trajectories of Change in Symptoms (Based on HRSD Score)
The HRSD scores up to week 8 (N = 4317 patients) were included in the HRSD trajectory analysis. The best-fitting model for the placebo group was a cubic growth mixture model with 3 trajectory classes for HRSD (Table S3, http://links.lww.com/JCP/A773). The trajectory class termed as “fast responders” had 21.8% probability of membership, and a mean reduction of 18.4 points in HRSD from baseline to week 8 (Table 1). “Partial responders” had a 34.4% probability of membership and mean reduction of 11.0 points (baseline to week 8), and “nonresponders” a 43.8% probability of membership and mean reduction of 2.6 points (baseline to week 8) (Table 1).
TABLE 1.
Probability of Membership and Summary Statistics for each HRSD and SDS Trajectory Class
| Placebo | Desvenlafaxine | ||||
|---|---|---|---|---|---|
| Trajectory Class | Probability of Membership, %* | Change From BL, Mean (SD)† | Trajectory Class | Probability of Membership, %* | Change From BL, Mean (SD)† |
| HRSD trajectories | |||||
| Fast responders | 21.8 | −18.4 (3.3) | Fast responders | 12.5 | −17.2 (4.4) |
| Slow responders | 37.6 | −16.2 (4.4) | |||
| Partial responders | 34.4 | −11.0 (3.4) | Partial responders | 33.0 | −7.4 (4.0) |
| Nonresponders | 43.8 | −2.6 (3.7) | Nonresponders | 16.9 | −2.6 (4.3) |
| SDS trajectories | |||||
| Responders | 32.4 | −13.6 (5.2) | Responders | 53.8 | −12.8 (5.8) |
| Nonresponders (moderate BL SDS) | 36.7 | −1.7 (5.6) | Nonresponders | 33.2 | −1.4 (4.7) |
| Nonresponders (severe BL SDS) | 19.6 | −0.8 (4.3) | |||
| Low BL SDS | 11.2 | +0.6 (6.2) | Low BL SDS | 13.0 | 0.0 (5.4) |
*For each group, probabilities of membership were estimated from growth mixture models for trajectories of HRSD and SDS correspondingly.
†Observed mean (SD) change from baseline scores for each trajectory.
BL indicates baseline.
The best-fitting model for desvenlafaxine-treated patients was a cubic growth mixture model with 4 trajectory classes for HRSD (Table S3, http://links.lww.com/JCP/A773). The trajectory class of “fast responders,” with 12.5% probability of membership, had an average reduction of 17.2 points from baseline to week 8 (Table 1). “Slow responders,” with 37.6% probability of membership, had an average reduction of 16.2 points, and “partial responders,” with 33.0% probability of membership, an average reduction of 7.4 points from baseline to week 8. The trajectory class “nonresponders,” with 16.9% probability of membership, had an average reduction of 2.6 points from baseline to week 8 (low baseline SDS) (Table 1). Mean HRSD trajectory classes for placebo- and desvenlafaxine-treated groups are depicted in Figure 1A.
FIGURE 1.
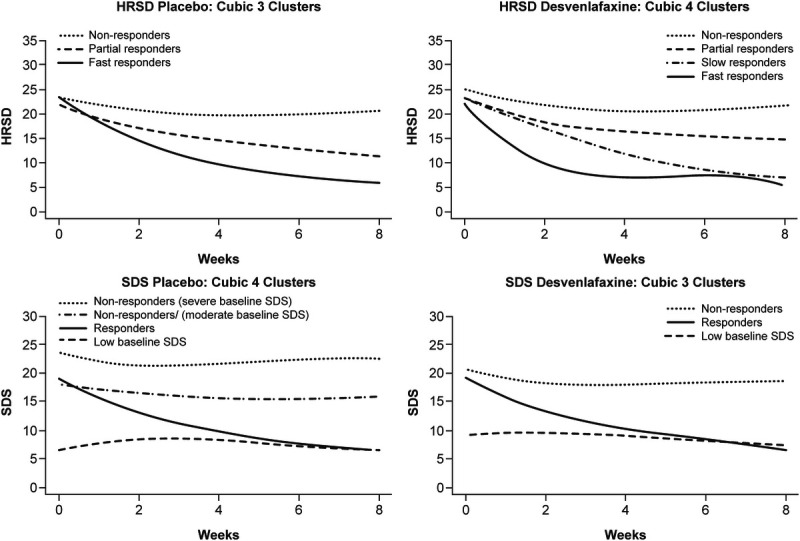
Mean HRSD (top panels) and SDS (bottom panels) scores estimated from cubic trajectories for placebo- and desvenlafaxine-treated patients over 8 weeks of treatment.
Trajectories of Change in Functioning (Based on SDS Score)
The SDS scores up to week 8 (N = 3321 patients) were included in the SDS trajectory analysis. The best-fitting model for the placebo group was a cubic growth mixture model with 4 trajectory classes (Table S3, http://links.lww.com/JCP/A773). The trajectory class termed “responders,” with 32.4% probability of membership, had an average reduction of 13.6 points in SDS from baseline to week 8 (Table 1). Two trajectory classes termed “nonresponders” with 36.7% and 19.6% probability of membership, had average reductions in SDS of 1.7 and 0.8 points from baseline to week 8, respectively. Both of these trajectory classes represent “nonresponders,” but the latter class had more severe baseline SDS scores. The fourth trajectory class (low baseline SDS), with 11.2% probability of membership and an average increase from baseline to week 8 of +0.6 points, representing patients with low baseline SDS scores and thus no opportunity to improve an already good functioning score at baseline (Table 1).
The best-fitting model for desvenlafaxine-treated patients was a cubic growth mixture model with 3 trajectory classes (Table S3, http://links.lww.com/JCP/A773). The trajectory class of “responders,” with 53.8% probability of membership, had an average reduction in SDS score of 12.8 points from baseline to week 8 (Table 1). The trajectory class of “nonresponders,” with 33.2% probability of membership, had an average reduction of 1.4 points from baseline to week 8. The last trajectory class (low baseline SDS) represents those patients with low baseline SDS scores and therefore no functional impairment at baseline, with 13.0% probability of membership, and an average change of 0 points from baseline to week 8 (Table 1). Mean SDS trajectory classes for placebo and desvenlafaxine groups are depicted in Figure 1B.
Agreement Between HRSD and SDS Trajectory Assignment
To assess agreement between HRSD and SDS trajectory assignments, SDS trajectories that started at high functioning were excluded (ie, labeled low baseline SDS; Table 1). For placebo-treated patients, the majority of the 250 patients assigned to the most favorable HRSD trajectory (fast responders) were assigned to the most favorable SDS trajectory (responders [75%]; Table S4, http://links.lww.com/JCP/A773). Among 545 patients assigned to the least favorable HRSD trajectory (nonresponders), 88% were also assigned to 1 of the 2 less favorable nonresponders SDS trajectories (39% and 49%, severe and moderate baseline SDS trajectories, respectively; Table S4A, http://links.lww.com/JCP/A773). Similarly, in desvenlafaxine-treated patients, among the 219 assigned to the most favorable HRSD trajectory (fast responders), 85% were also assigned to the most favorable SDS trajectory (Table S4B, http://links.lww.com/JCP/A773). Among the 302 patients assigned to the least favorable HRSD trajectory (nonresponders), 80% were also assigned to the least favorable SDS trajectory (nonresponders).
IMPLICATIONS/DISCUSSION
Although functional impairment is a main characteristic of MDD,6 little is known about patterns of functional change over the course of ADM treatment, or whether it is possible to infer trajectories of change in functioning from trajectories of change in symptoms. The present analysis identified distinct trajectories of change in symptoms and in functioning, for both placebo- and desvenlafaxine-treated patients, and demonstrated high percentages of agreement between trajectories of symptoms and functioning. Moreover, we identified distinct trajectories of “responders” that suggest not all patients who respond to desvenlafaxine or to placebo respond similarly. Improving our understanding of how MDD patients respond to ADMs may help physicians identify patients likely to respond to treatment, allowing a more “personalized” approach to MDD management.
This overall pool of more than 4300 patients with MDD facilitated a rigorous investigation of trajectories of change in symptoms, alongside changes in function (n = 3321), for patients treated with desvenlafaxine or placebo for up to 8 weeks. We identified 3 distinct trajectories of symptoms (based on HRSD scores) and 4 of functioning (based on SDS scores) for patients treated with placebo, as well as 4 distinct trajectories of symptoms and 3 of functioning, for patients treated with desvenlafaxine. For symptomatic responses, trajectories showed a 15.8-point difference (18.4 to 2.6) on the HRSD between most favorable (fast responders) and least favorable (nonresponders) trajectories with placebo, and for desvenlafaxine, a 14.6-point difference (17.2 to 2.6) between the most (fast responders) and least favorable (nonresponders) trajectories. Previous studies have also investigated trajectories of symptomatic improvement using different statistical approaches.2,3,8 The methodological approach for the present analysis was a growth-mixture model using the Schwartz-BIC for selecting the best model. Gueorguieva et al,3 also used a growth mixture model with selection of the best model based on the BIC, but they also used the Lo-Mendell-Rubin likelihood ratio test to classify patients into their most likely trajectory based on improvement on HAMD17 scores after duloxetine treatment. For this different ADM, Gueorguieva et al,3 identified 2 classes of trajectories of change in symptoms for patients receiving active drug, and 1 for placebo. By identifying 3 trajectories of change in symptoms for placebo-treated patients, our findings differ from observations by Gueorguieva et al,3 where a single trajectory was described for symptomatic change with placebo, and on average these patients demonstrated gradual improvement over time.3
Our larger sample size may have facilitated identification of trajectories that have gone undetected when observations were based on smaller sample sizes in other publications. Historically, the placebo response was thought to be predominantly associated with a rapid, but transient clinical improvement.27 By expanding this understanding to outline distinct trajectories for placebo responders, our observations are consistent with clinical observations and unfolding evidence, which suggest placebo responders are likely to be as heterogeneous as responders to ADMs.28 This view makes intuitive sense given the diverse mechanisms underlying the placebo response, which encompass complex changes in autonomic systems, neuroendocrine responses, and neural circuitry, all of which likely vary across subgroups of MDD patients.28 Taking time to consider subpopulations of patients, such as those described by our trajectories, as well as considering bias around outcome scales,29 and having this broader understanding of the placebo effect,30 may help in the future design of clinical studies for MDD.
The present analysis also revealed trajectories that have no equivalent. First, these are trajectories identified in both placebo and desvenlafaxine groups that showed a certain “floor effect,” that is, where no change in functioning (low baseline SDS trajectories) as patients have a high level of functioning at baseline (scores equal to the level of remission on the SDS). This “floor” highlights individuals who are minimally impacted by symptoms as no reduction in SDS score can be observed because of the already low baseline score. Second, we identified 3 trajectories of change in functioning in desvenlafaxine-treated patients, with the most favorable trajectory (fast responders) showing an 11.4-point difference (12.8 to 1.4) on the SDS compared with the least favorable (nonresponders), excluding the low baseline SDS trajectory as no significant change was possible. Similar patterns and degrees of change were seen for trajectories of functioning with placebo, where a 12.8-point difference (13.6 to 0.8) was seen between most and least favorable SDS trajectories, again excluding the low baseline SDS trajectory. Collectively, these finding shed new light on previous meta-analyses reporting the limited effect that ADMs have on functional improvement, vis-à-vis placebo.31,32 Our observations suggest patients who showed little or no functional improvement with desvenlafaxine belonged to 1 of 2 trajectories: (1) patients for whom desvenlafaxine did not significantly reduce symptoms; and (2) patients with a high level of functioning at treatment onset. Because previous analyses do not distinguish patients who belong to these trajectories (ie, responders, nonresponders, high level of functioning at baseline), it is reasonable to assume that previous results may underestimate the effect of desvenlafaxine on functioning, and furthermore, the degree to which desvenlafaxine produces favorable trajectories in functioning. We, thus, suggest the effect of desvenlafaxine on functioning within 8 weeks is because of the significant improvements in functional impairment, and a lack of change owing to an absence of functional impairment at baseline.
In addition to analyzing trajectories of change, we evaluated agreement between trajectories of symptomatic and functional response to investigate disparate biases that exist for raters and participants. Our findings suggest a high level of agreement between the proportion of patients in the most favorable symptomatic or functioning trajectory assignment, possibly indicating an absence of differential biases between raters and participants. This was true for both placebo- and desvenlafaxine-treated patients, where patients assigned to the most favorable HRSD trajectory (fast responders) had an 85% probability of being assigned to the most favorable SDS trajectory (responders). Similarly, patients assigned to the least favorable HRSD trajectory (nonresponders) had an 80% probability of being assigned to the least favorable SDS trajectory (nonresponders). For placebo, the percentages of agreement were 75% and 88% for patients assigned to the most- and least-favorable trajectories, respectively. The heterogeneity identified in trajectories of symptoms and functioning questions whether individuals who respond in terms of symptomatic changes are more likely to demonstrate a functional response. Although interrelated, they should not however be considered the same. Although symptoms were evaluated by blinded raters, function was self-reported, and different biases may operate on specific measures. For example, findings suggest that rater-administered measures of symptom change demonstrate a greater change in response to ADM treatment than for self-reported.29,30 Baseline score inflation may be introduced because of the motivation of raters to identify eligible patients for recruitment.33 Finally, response biases, according to which participants report more symptom improvement than they actually feel, may contribute toward the placebo effect.30
Some limitations should be noted. Although the RCTs may have some common characteristics, inherent differences may affect trajectory membership based on both symptoms or functioning. In addition, trajectories based on functioning (SDS) were constructed using fewer measurements than trajectories based on symptoms (HRSD), this was because of the study designs, where SDS was a secondary outcome and collected less often than the primary outcome (ie, HRSD). More frequent SDS measurements may have produced more nuanced findings. A number of different assessment scales have been developed for depression, and further study is needed to determine if trajectories can be identified from other assessments of symptoms, function or quality of life. Although some RCTs captured these other assessments, not enough data points were available across studies to carry out additional trajectory analyses. Larsen et al,8 for example, used a growth mixture model to describe fast-responder, slow-responder, and nonresponder trajectories in symptoms after escitalopram treatment, using the Montgomery-Åsberg Depression Rating Scale. We likewise adopted descriptive terms to aid clinical understanding of the trajectory pathway, and these should not be taken to reflect a real “speed” of response. In addition, the term “responder” refers to a specific clinical trajectory and does not indicate that patients achieved a “responder” status defined in the original studies. Desvenlafaxine and placebo arms were analyzed separately in the pooled data, and we cannot assume similar observations should individual studies be analyzed. We did not set out to correlate placebo trajectories with desvenlafaxine trajectories of change and draw no conclusions of the clinical importance of the differences between most and least favorable responders or nonresponders on individual scales. Finally, the RCTs were all conducted by the same sponsor and focused on 1 drug, which may affect generalizability to other ADM-treated patients. Additional studies are needed to determine whether the identified trajectories are specific to desvenlafaxine or can be applied to other ADMs, over the longer term, or in the real-world setting. To our knowledge, this is a first iteration of this statistical approach to analyze response trajectories and needs further validation. With a growing number of publications in this area, we hope to see trajectory assignment guiding clinical management of MDD in the future.
In conclusion, distinct trajectories of symptoms and of functioning were identified among MDD patients treated with desvenlafaxine or with placebo. Differentiating between subpopulations of patients with distinct trajectories has the potential to improve detection of patients showing clinical benefit, and we believe, represents a step toward the ultimate aim of more “personalized” treatment of patients with MDD. The variety of trajectories, including those of placebo-treated patients, may also point to distinct mechanisms of change that could help direct future research and/or clinical study design in this area.
Supplementary Material
AUTHOR DISCLOSURE INFORMATION
S.Z.-M., S.A.F., and B.R.R. declare no conflicts of interest. X.W. was a full-time employee of Syneos Health when the study was conducted. Syneos Health were paid consultants to Pfizer in connection with the development of this manuscript. X.W. also owns Syneos Health stock. D.B.W. is a full-time employee of Pfizer and holds Pfizer stock and/or stock options. M.B. was an employee of Pfizer Canada at the time of the study.
This study was sponsored by Pfizer. Medical writing support was provided by Karen Burrows, MPhil, CMPP, of Engage Scientific Solutions and was funded by Pfizer.
Data Sharing Statement: Upon request, and subject to certain criteria, conditions, and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual de-identified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines, and medical devices (1) for indications that have been approved in the United States and/or Europe or (2) in programs that have been terminated (ie, development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The deidentified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
Footnotes
Supplemental digital content is available for this article. Direct URL citation appears in the printed text and is provided in the HTML and PDF versions of this article on the journal’s Web site (www.psychopharmacology.com).
Contributor Information
Xuemei Wang, Email: xuemei.wang@syneoshealth.com.
Dalia B. Wajsbrot, Email: Dalia.Wajsbrot@pfizer.com.
Matthieu Boucher, Email: matthieu.boucher@me.com.
Stuart A. Fine, Email: s.fine@columbia.edu.
Bret R. Rutherford, Email: brr8@cumc.columbia.edu.
REFERENCES
- 1.Rush AJ Trivedi MH Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163:1905–1917. [DOI] [PubMed] [Google Scholar]
- 2.Zilcha-Mano S Roose SP Brown PJ, et al. Early symptom trajectories as predictors of treatment outcome for citalopram versus placebo. Am J Geriatr Psychiatry. 2017;25:654–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gueorguieva R, Mallinckrodt C, Krystal JH. Trajectories of depression severity in clinical trials of duloxetine: insights into antidepressant and placebo responses. Arch Gen Psychiatry. 2011;68:1227–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeRubeis RJ Cohen ZD Forand NR, et al. The Personalized Advantage Index: translating research on prediction into individualized treatment recommendations. a demonstration. PLoS One. 2014;9:e83875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thase ME, Larsen KG, Kennedy SH. Assessing the ‘true’ effect of active antidepressant therapy v. placebo in major depressive disorder: use of a mixture model. Br J Psychiatry. 2011;199:501–507. [DOI] [PubMed] [Google Scholar]
- 6.Greer TL, Kurian BT, Trivedi MH. Defining and measuring functional recovery from depression. CNS Drugs. 2010;24:267–284. [DOI] [PubMed] [Google Scholar]
- 7.Katzman MA Nierenberg AA Wajsbrot DB, et al. Speed of improvement in symptoms of depression with desvenlafaxine 50 mg and 100 mg compared with placebo in patients with major depressive disorder. J Clin Psychopharmacol. 2017;37:555–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Larsen KG Kennedy SH Reines EH, et al. Patient response trajectories in major depressive disorder. Psychopharmacol Bull. 2020;50:8–28. [PMC free article] [PubMed] [Google Scholar]
- 9.Katzman MA Wang X Wajsbrot DB, et al. Effects of desvenlafaxine versus placebo on MDD symptom clusters: a pooled analysis. J Psychopharmacol. 2020;34:280–292. [DOI] [PubMed] [Google Scholar]
- 10.Novick D Montgomery W Vorstenbosch E, et al. Recovery in patients with major depressive disorder (MDD): results of a 6-month, multinational, observational study. Patient Prefer Adherence. 2017;11:1859–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sheehan DV Harnett-Sheehan K Spann ME, et al. Assessing remission in major depressive disorder and generalized anxiety disorder clinical trials with the discan metric of the Sheehan disability scale. Int Clin Psychopharmacol. 2011;26:75–83. [DOI] [PubMed] [Google Scholar]
- 12.DeMartinis NA Yeung PP Entsuah R, et al. A double-blind, placebo-controlled study of the efficacy and safety of desvenlafaxine succinate in the treatment of major depressive disorder. J Clin Psychiatry. 2007;68:677–688. [DOI] [PubMed] [Google Scholar]
- 13.Liebowitz MR Tourian KA Hwang E, et al. A double-blind, randomized, placebo-controlled study assessing the efficacy and tolerability of desvenlafaxine 10 and 50 mg/day in adult outpatients with major depressive disorder. BMC Psychiatry. 2013;13:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liebowitz MR Manley AL Padmanabhan SK, et al. Efficacy, safety, and tolerability of desvenlafaxine 50 mg/day and 100 mg/day in outpatients with major depressive disorder. Curr Med Res Opin. 2008;24:1877–1890. [DOI] [PubMed] [Google Scholar]
- 15.Boyer P Montgomery S Lepola U, et al. Efficacy, safety, and tolerability of fixed-dose desvenlafaxine 50 and 100 mg/day for major depressive disorder in a placebo-controlled trial. Int Clin Psychopharmacol. 2008;23:243–253. [DOI] [PubMed] [Google Scholar]
- 16.Tourian KA Padmanabhan SK Groark J, et al. Desvenlafaxine 50 and 100 mg/d in the treatment of major depressive disorder: an 8-week, phase III, multicenter, randomized, double-blind, placebo-controlled, parallel-group trial and a post hoc pooled analysis of three studies. Clin Ther. 2009;31(Pt 1):1405–1423. [DOI] [PubMed] [Google Scholar]
- 17.Iwata N Tourian KA Hwang E, et al. Efficacy and safety of desvenlafaxine 25 and 50 mg/day in a randomized, placebo-controlled study of depressed outpatients. J Psychiatr Pract. 2013;19:5–14. [DOI] [PubMed] [Google Scholar]
- 18.Clayton AH Kornstein SG Dunlop BW, et al. Efficacy and safety of desvenlafaxine 50 mg/d in a randomized, placebo-controlled study of perimenopausal and postmenopausal women with major depressive disorder. J Clin Psychiatry. 2013;74:1010–1017. [DOI] [PubMed] [Google Scholar]
- 19.Dunlop BW Reddy S Yang L, et al. Symptomatic and functional improvement in employed depressed patients: a double-blind clinical trial of desvenlafaxine versus placebo. J Clin Psychopharmacol. 2011;31:569–576. [DOI] [PubMed] [Google Scholar]
- 20.Clayton AH Tourian KA Focht K, et al. Desvenlafaxine 50 and 100 mg/d versus placebo for the treatment of major depressive disorder: a phase 4, randomized controlled trial. J Clin Psychiatry. 2015;76:562–569. [DOI] [PubMed] [Google Scholar]
- 21.Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sheehan DV, Harnett-Sheehan K, Raj BA. The measurement of disability. Int Clin Psychopharmacol. 1996;11(suppl 3):89–95. [DOI] [PubMed] [Google Scholar]
- 23.Verbeke G, Lesaffre E. A linear mixed-effects model with heterogeneity in the random-effects population. JSTOR. 1996;91:217–221. [Google Scholar]
- 24.Muthén B, Shedden K. Finite mixture modeling with mixture outcomes using the EM algorithm. Biometrics. 1999;55:463–469. [DOI] [PubMed] [Google Scholar]
- 25.Proust C, Jacqmin-Gadda H. Estimation of linear mixed models with a mixture of distribution for the random effects. Comput Methods Prog Biomed. 2005;78:165–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Proust-Lima C, Philipps V, Liquet B. Estimation of extended mixed models using latent classes and latent processes: the R package lcmm. J Stat Softw. 2017;78:56. [Google Scholar]
- 27.Quitkin FM Rabkin JG Ross D, et al. Identification of true drug response to antidepressants. Use of pattern analysis. Arch Gen Psychiatry. 1984;41:782–786. [DOI] [PubMed] [Google Scholar]
- 28.Wager TD, Atlas LY. The neuroscience of placebo effects: connecting context, learning and health. Nat Rev Neurosci. 2015;16:403–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kobak KA Kane JM Thase ME, et al. Why do clinical trials fail? The problem of measurement error in clinical trials: time to test new paradigms? J Clin Psychopharmacol. 2007;27:1–5. [DOI] [PubMed] [Google Scholar]
- 30.Rutherford BR, Roose SP. A model of placebo response in antidepressant clinical trials. Am J Psychiatry. 2013;170:723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Evans VC Alamian G McLeod J, et al. The effects of newer antidepressants on occupational impairment in major depressive disorder: a systematic review and meta-analysis of randomized controlled trials. CNS Drugs. 2016;30:405–417. [DOI] [PubMed] [Google Scholar]
- 32.Salanti G Chaimani A Furukawa TA, et al. Impact of placebo arms on outcomes in antidepressant trials: systematic review and meta-regression analysis. Int J Epidemiol. 2018;47:1454–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Landin R DeBrota DJ DeVries TA, et al. The impact of restrictive entry criterion during the placebo lead-in period. Biometrics. 2000;56:271–278. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.