

Abstract
Reactivation of fetal hemoglobin (HbF) is being pursued as a treatment strategy for hemoglobinopathies. Here, we evaluated the therapeutic potential of hematopoietic stem and progenitor cells (HSPCs) edited with the CRISPR/Cas9 nuclease platform to recapitulate naturally occurring mutations identified in individuals who express increased amounts of HbF, a condition known as hereditary persistence of HbF. CRISPR/Cas9 treatment and transplantation of HSPCs purified on the basis of surface expression of the CD34 receptor in a nonhuman primate (NHP) autologous transplantation model resulted in up to 30% engraftment of gene-edited cells for >1 year. Edited cells effectively and stably reactivated HbF, as evidenced by up to 18% HbF-expressing erythrocytes in peripheral blood. Similar results were obtained by editing highly enriched stem cells, defined by the markers CD34+CD90+CD45RA–, allowing for a 10-fold reduction in the number of transplanted target cells, thus circumventing issues associated with scale-up and considerably reducing the need for editing reagents. The frequency of engrafted, gene-edited cells persisting in vivo using this approach may be sufficient to ameliorate the phenotype for a number of genetic diseases.
One Sentence Summary:
CRISPR/Cas9-edited hematopoietic stem cells produce long-term engraftment and fetal hemoglobin reactivation in nonhuman primates.
Introduction
Beta-hemoglobinopathies such as sickle cell disease (SCD) and β-thalassemia are the most common monogenic disorders, representing a substantial public health burden caused by mutations in the β-globin gene (1). Currently, the only curative treatment for hemoglobinopathies is an allogeneic stem cell transplant which carries a marked risk of complications, and which is additionally limited by donor availability (2). Recent advances in stem cell gene therapy have raised promise of additional curative methods, some of which are currently under clinical investigation (reviewed in Esrick et al. (3)).
Beyond the benefit brought by gene addition strategies, the pathological manifestations of these disorders are naturally improved in patients who continue to express γ-globin postnatally. γ-globin associates with α-globin to form fetal hemoglobin (HbF, α2γ2) and serves as a substitute for defective or insufficient adult hemoglobin (HbA, α2β2). This benign genetic condition, known as hereditary persistence of fetal hemoglobin (HPFH), is often linked to single nucleotide (nt) variants or large deletions spanning the β-globin locus and is accompanied by varying degrees of HbF reactivation (reviewed in Forget et al. (4)).
We were particularly interested in a potent and naturally occurring 13-nt HPFH deletion in the HBG1 promoter (−102 to −114) identified in SCD patients (5). The therapeutic potential of this target was recently validated with genome engineering tools that included the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 system (6) and Transcription activator-like effector nucleases (TALENs) (7). This chromosomal region contains a CCAAT box that recruits transcription factors such as NF-Y (8), as well as the TGACCA motif recently identified as a binding site for the HbF repressor BCL11A (9, 10).
Here, we used a nonhuman primate (NHP) autologous transplantation model to assess the curative potential of this approach for hemoglobinopathies using gene editing of hematopoietic stem and progenitor cells (HSPCs). This pre-clinical model closely reproduces parameters from human stem cell transplants (kinetics of hematopoietic recovery, immunophenotypic markers, cross reactivity between cytokines, etc.) and offers an opportunity to monitor both long-term engraftment and peripheral blood (PB) hemoglobin production. Using this model, we previously identified an hematopoietic stem cell (HSC)-enriched target cell population (CD34+CD90+CD45RA–) found to be required for both rapid short-term and durable multilineage hematopoietic reconstitution (11).
In this report, we demonstrate 1) reproducible and stable engraftment of CRISPR/Cas9-edited NHP HSPCs over 1 year after transplant, 2) persistent HbF reactivation that correlates with the extent of in vivo editing; and 3) reduction in the number of target cells by over 10-fold by targeting a HSC-enriched CD34+CD90+CD45RA– population. These results demonstrate stable engraftment of CRISPR/Cas9-edited HSPCs, with over 25% editing frequency detected in PB at >1 year after transplant in a large animal model. These findings should help to facilitate the clinical translation of HSPC-based editing approaches for hemoglobinopathies as well as other genetic diseases.
Results
Recapitulation of HPFH mutations in NHP HSPCs
We aimed to generate an array of potentially therapeutic insertions or deletions (indels) that would target the recently characterized BCL11A binding site found in the promoter region of the two γ-globin genes (HBG) (9, 10), including edits that recapitulated the naturally occurring 13-nt HPFH deletion. To achieve this goal, we used a CRISPR binding site conserved between human and rhesus macaque, which overlaps with the 5’-TGACCA-3’ BCL11A binding site (Fig. 1A). Conditions were first optimized for efficient editing of NHP CD34+ cells using CRISPR/Cas9 ribonucleoprotein (RNP) electroporation. Our results indicated that greater molar ratios of Cas9 protein to chemically-modified guide RNA (gRNA) give the best editing efficiencies in NHP cells (fig. S1A), in comparison to lower molar ratios that are optimal in human CD34+ cells (12) (fig. S1B).
Fig. 1. Recapitulation of HPFH genotype in NHP HSPCs.

(A) Schematic of β-globin locus with CCAAT repressor motifs (underlined), putative BCL11a binding sequence TGACCA (green box), and CRISPR/Cas9 target site (red arrow). Sequences highlighted in orange show HPFH sites for 13-nt deletion [−114/−102] and −117 G/A substitution. (B) Deletion profile in NHP CD34+ edited cells (animal A17117) at 4 days post-editing. Colored boxes show identified distinct deletions relative to the total sequencing pool, and the white portion shows all combined deletions contributing less than 1%. The 13-nt deletion is on top in the dark blue box. (C) Genomic sequences of the most common deletions from (B) with length of deletions on the left in nucleotides (nt). Boxes highlight 8-nt microhomology repeats. (D) Immunophenotypic separation of HSPC subsets after CD34+ enrichment (A17117). (E) Flow cytometric validation of the indicated sorted HSPCs subsets from (D). (F) HBG editing efficiency measured at 24 hours post-treatment in sorted subsets from (E). (G) Contribution of 13-nt HPFH deletion relative to all other deletions in edited subsets from (C). (H) CFC assay of CD34+ and HSPC subsets taken at 24 hours after mock electroporation (left) or CRISPR/Cas9 RNP (right) treatment (n=1). CFU=Colony-forming unit, CFU-M=macrophages, CFU-G=granulocytes, CFU-GM=granulocyte/macrophage, BFU-E=erythroid. In panels F and G, results are means and standard deviations from A17117 and A17114. *denotes statistical significance (two-tailed unpaired t-test, P<0.05) of the difference in 13-nt deletion in CD90+ subset as compared to CD34+ cells.
All subsequent NHP experiments were conducted with a 1:10 molar ratio of Cas9 protein to gRNA, which was the most cost-effective in rhesus CD34+ HSPCs. Editing efficiency in these cells averaged 75%, with up to 39% consisting of the natural HPFH 13-nt deletion and the remainder comprised of small deletions ranging from 1 to 6 nts in length (Fig. 1B–C, fig. S1C, data file S1). The prevalence of the 13-nt HPFH deletion is consistent with results from editing of human HSPCs (6) and is likely mediated by the microhomology-mediated end joining (MMEJ) repair pathway using 8-nt tandem repeats (Fig. 1C). In vitro erythroid differentiation of gene-edited CD34+ cells confirmed reactivation of HbF by CRISPR/Cas9 treatment and correlated with the extent of editing (fig. S1D). To determine whether long-term engrafting stem cells were efficiently edited, phenotypically defined CD34+ subsets enriched for HSCs (CD90+CD45RA–), multipotent progenitor cells (MPPs: CD90–CD45RA–), and lympho-myeloid progenitors (LMPs: CD45RA+) were sorted by fluorescence-activated cell sorting (FACS) for analysis (Fig. 1D,E, fig. S1E) (11). All subsets, including the HSC-enriched phenotype, showed comparable degrees of HBG editing (Fig. 1F and fig. S1F). Notably, the frequency of the 13-nt HPFH deletion was reduced in HSC-enriched CD90+CD45RA– as compared to bulk CD34+ cells (24.60% vs. 35.05%, P<0.05, Fig. 1G), suggesting that the MMEJ pathway is less active in this more primitive cell population. We further confirmed that the colony-forming cell (CFC) potential was not impacted by our editing approach. We observed equivalent number and composition of CFCs in mock-electroporated versus CRISPR-treated cells for all HSPC subsets (Fig. 1H).
Recent studies have demonstrated induction of the p53-dependent DNA damage response in human CD34+ HSPCs treated with CRISPR/Cas9 (13, 14). Consistent with activation of the p53 pathway, we observed a transient upregulation in p53 protein expression that peaked at 24 to 48 hours after electroporation with CRISPR/Cas9 and that was prolonged in NHP as compared to human CD34+ HSPCs (fig. S2A). Similarly, mRNA transcripts of the downstream effector gene CDKN1A (p21) were transiently induced by treatment in NHP cells (fig. S2B). Cytotoxicity of CRISPR/Cas9-electroporation was also assessed by Annexin V/7-AAD staining, which revealed a transient increase in apoptosis and cell death in both human and NHP cells, peaking at 24 and 48 hour after treatment, respectively (fig. S3 and data file S2).
Persistence of HBG-edited bulk CD34+ HSPCs and HSC-enriched CD90+CD45RA– cells in transplanted rhesus macaques
To assess long-term in vivo persistence of gene-edited cells and corresponding HbF reactivation, we transplanted a first cohort of three rhesus macaques with CRISPR/Cas9-treated bone marrow (BM)-enriched CD34+ HSPCs. Autologous CD34+ cells were electroporated with CRISPR/Cas9 RNPs as described above. Editing efficiency in the infusion product was comparable among animals and ranged from 70% to 75% as measured by next-generation sequencing (Fig. 2A,B). The phenotype (fig. S4A) and CFC potential (fig. S4B) of these cells were not affected by the editing procedure. Ultimately, a total of 83 to 204 million gene-edited CD34+ cells were infused into each recipient animal after total body irradiation (TBI). Additional transplantation and quality control parameters are summarized in Table 1.
Fig. 2. Tracking of HBG editing in all transplanted animals.

(A) Editing efficiency was measured in PB white blood cells from transplanted animals. Inset shows magnification for the early time points. (B) Editing efficiency measured in the infusion product of transplanted animals at 3–5 days after treatment (n=3). (C) Flow cytometric validation of CD90+ and CD90–-sorted populations after CD34+ enrichment (A17116). (D) CFC assay of sorted populations from (C) before editing (left) and 24 hours after editing (right). CFU-M=macrophages, CFU-G=granulocytes, CFU-GM=granulocyte/macrophage, BFU-E=erythroid. (E) Normalized frequency of the 13-nt HPFH deletion in reactions from (B), n=3. * denotes statistical significance (two-tailed unpaired t-test, P<0.05) of the difference in 13-nt deletion in CD90+ subset as compared to CD34+. Deletion profile of CD34+/CRISPR animal A17114 (F) or CD90+/CRISPR animal A17116 (G) after transplant. In panels F and G, colored boxes show identified distinct deletions relative to the total sequencing pool, and the white portion shows all combined deletions contributing less than 1%. The 13-nt deletion is on top in the dark blue box. (H) Contribution of the 13-nt HPFH deletion in the same animals as in (A) after normalization of all deletion frequencies to 100%.
Table 1.
Transplant summary.
| CD34+ cohort | CD34+CD90+CD45RA− cohort | |||||
|---|---|---|---|---|---|---|
| Animal ID | A17117 | A17114 | A17112 | A17116 | A17115 | A17113 |
| Date of infusion | 7/14/17 | 8/4/17 | 2/8/18 | 9/28/17 | 11/17/17 | 3/23/18 |
| Animal weight (kg) | 5 | 4.9 | 7.3 | 5.25 | 4.5 | 4.55 |
|
| ||||||
| Infusion producta: | ||||||
| CD34+ fraction | 125E+06 | 83.5E+06 | 204E+06 | — | — | — |
| CD90+ fraction | — | — | — | 15.0E+06 | 8.65E+06 | 5.15E+06 |
| CD90– fraction | — | — | — | 266E+06 | 213E+06 | 50.0E+06 |
| Total cells infused | 125E+06 | 83.5E+06 | 204E+06 | 281E+06 | 221E+06 | 55.2E+06 |
|
| ||||||
| Calculated number of cells infusedb: | ||||||
| CD34+ cells | 53.0E+06 | 33.5E+06 | 148E+06 | 64.6E+06 | 77.4E+06 | 15.7E+06 |
| CD90+ cells | 3,537,500 | 3,582,150 | 4,998,000 | 5,223,540 | 3,053,730 | 1,639,000 |
| CD34+ cells/kg | 10.6E+06 | 6.84E+06 | 20.3E+06 | 12.3E+06 | 17.2E+06 | 3.45E+06 |
| CD90+ cells/kg | 707,500 | 731,051 | 684,657 | 994,960 | 678,606 | 360,219 |
Based on the number of cells harvested on the day of infusion
Adjusted based on flow cytometric analysis of cells on the day of infusion
One major challenge for the clinical translation of gene therapy and gene editing approaches lies in the upscaling of conditions and associated cost of agents required for the modification of CD34+ HSPCs. Addressing these concerns, our laboratory previously identified an HSC-enriched target cell population (CD34+CD90+CD45RA–) capable of both rapid short-term and durable multilineage hematopoietic reconstitution (11). Thus, we investigated whether editing and transplantation of this refined phenotypic subset, comprising less than 10% of the total CD34+ cell number, would result in comparable engraftment and HbF reactivation as compared to the bulk CD34+ transplant approach. We first verified that the FACS-sorted CD34+CD90+ fraction could be efficiently edited without compromising viability and differentiation potential of these cells. As shown in fig. S5, editing in sorted CD34+CD90+ was comparable to bulk CD34+ and to sorted CD34+CD90– cells, and did not affect the CFC potential. Based on these findings, we transplanted a second cohort of three rhesus macaques using a modified protocol in which BM-enriched CD34+ HSPCs were FACS-sorted into two separate fractions: 1) a CD90+CD45RA– subset that was treated by CRISPR/Cas9 RNP electroporation of 5 to 15 million cells per animal (Table 1); and 2) a CD90+CD45RA–-depleted subset consisting of non-edited cells that were combined with the edited CD90+CD45RA– cells at the time of infusion (Fig. 2C, fig. S6A). Consistent with previous results and despite upfront FACS-sorting, editing did not affect CFC potential (Fig. 2D, fig. S6B), and the editing efficiency in the infused CD90+CD45RA– subset was comparable to that obtained in bulk CD34+ cells (Fig. 2B). In addition, the frequency of the 13-nt HPFH deletion was significantly reduced in the CD90+CD45RA– cells as compared to bulk CD34+ cells (Fig. 2E, 28.26% for CD90+CD45RA– vs. 38.47% for CD34+, P<0.05).
All six animals recovered promptly without complications, and blood counts including neutrophils, lymphocytes, platelets, as well as monocytes rapidly stabilized and remained within a normal range during post-transplant monitoring (fig. S7). A delay in platelet recovery was noted in 2 of 3 animals in the CD90+ cohort (fig. S7C,D), likely caused by myelosuppression-associated reactivation of cytomegalovirus (CMV) at the time of transplantation (table S1). Gene-editing in PB nucleated cells stabilized after approximately one month and remained at frequencies ranging from 8% to 27% for the entire follow-up period (Fig. 2A). An initial drop in the percentage of in vivo editing was observed in the CD90+ transplanted animals (Fig. 2A inset) because these cells constitute less than 10% of the CD34+ bulk cell number and were combined with non-edited, CD90-CD45RA–-depleted cells at the time of infusion. However, editing efficiency rapidly rebounded to near infusion percentage after just 7 days, confirming that the edited CD90+CD45RA– population is required for hematopoietic reconstitution beyond 2 weeks after transplant. Longitudinal analysis of CRISPR/Cas9-induced mutation profiles in PB showed persistence of all types of deletions (Fig. 2F,G, fig. S8, data file S1), with 8% to 14% of them consisting of the 13-nt HPFH genotype, which was maintained during the entire follow-up (Fig. 2H). In summary, our results demonstrated rapid recovery and robust multilineage engraftment after transplantation of CRISPR/Cas9 HBG-edited CD34+ or of HSC-enriched CD90+CD45RA– cells in the NHP model. In addition, these findings showed that reducing the target cell count by more than 10-fold and solely editing CD90+CD45RA– population achieved comparable in vivo editing relative to the current clinical gold standard targeting the entire CD34+ cell population.
Quantification of circulating F-cells and hemoglobin expression in PB of transplanted animals
The long-term persistence of HBG-edited cells in the transplanted animals is expected to result in PB HbF reactivation. We first measured the HbF production in all animals by quantifying circulating F-cells in PB. In contrast to control transplant recipients that demonstrated a rapid and transient increase in F-cells, which returned to a frequency of less than 1.5% at 250 days post-treatment (15), all CRISPR/Cas9-edited animals showed an F-cell response that stabilized at frequencies of 6% to 18% in PB. This response was sustained for more than one year after transplant (Fig. 3A) and very strongly correlated (R2=0.96, P=0.0005) with the extent of in vivo PB HBG editing (Fig. 3B).
Fig. 3. HbF response in transplanted animals.

(A) Longitudinal measurement of F-cell frequency in PB of transplanted animals from CD34+ (blue) and CD90+ (red) cohorts as compared to historical transplant controls (gray) and one untransplanted control (black). (B) Longitudinal HPLC measurement of γ-globin protein expression (calculated as γ/(γ+β) globin) in the same animals as in panel A. Globin expression was calculated from the area of each eluted peak in HPLC chromatograms. (C) Linear correlation analysis of F-cell frequency (y-axis, red) or γ-globin protein expression (y-axis, purple) and HBG editing frequency (x-axis, determined by TIDE) at 200 to 300 days post-transplant.
To quantify hemoglobin (Hb) protein expression in PB of all transplanted animals, we used high-performance liquid chromatography (HPLC). Due to the divergence in globin polypeptide sequences between human and rhesus Hb (fig. S9), we initially optimized this assay for comparing the HPLC profiles of rhesus adult blood to cord blood, which has a high content of HbF. Cord blood showed two late peaks that were absent in adult blood and were suggestive of the two γ-globin chains (figs. S10, S11A). In addition, an early peak showed much greater amplitude in adult blood as compared to cord blood, consistent with β-globin expression (fig. S11A). The identity of each globin species was further validated by elution of individual HPLC peaks followed by mass spectrometry analysis, which confirmed our initial predictions that the β-globin chain was eluted first, followed by two α-globin chains, and lastly by the two γ-globin chains (fig. S11B). In contrast to humans, who produce two identical α-globin polypeptides from the genes HBA1 and HBA2, rhesus α-globin polypeptides diverge by a single amino acid (fig. S10B), resulting in distinct elution times and allowing for separate measurements.
HPLC analysis of PB in CRISPR/Cas9-treated animals showed expression of the two γ-globin peaks as early as 1 month after transplant even though they were undetectable before transplantation, indicative of HbF reactivation (fig. S12). Longitudinal quantification of γ-globin protein expression relative to all β-like globins (γ/(γ+β)) showed an overall response that was comparable to our measurement of PB F-cells (Fig. 3A). γ-globin protein expression peaked at 1- to 2-months after transplant and stabilized at up to 5% of β-like globins for more than 1 year (Fig. 3C, fig. S12). In addition, γ-globin expression correlated very strongly (R2=0.97, P=0.0004) with the extent of in vivo PB editing, similar to our findings from F-cell measurements (Fig. 3B). Reverse transcription quantitative PCR measurement of PB globin transcripts also corroborated findings of increased γ-globin expression in all transplanted animals as compared to control transplant animals (fig. S13). In summary, these multiple independent measurements of Hb expression confirm the occurrence of HbF reactivation in transplanted animals at rates that strongly correlate with measurements of in vivo HBG editing frequencies.
Multilineage HBG-editing and reconstitution of the BM compartment in transplanted animals
Life-long HbF production requires the engraftment of HBG-edited HSPCs in the BM stem cell niche, with subsequent differentiation into mature Hb-producing erythrocytes in PB. We sampled the BM of transplanted animals infused with CRISPR/Cas9-modified CD34+ and CD90+CD45RA– cells approximately 6 months after treatment, performed comprehensive flow-cytometric analysis, and obtained functional read outs. Immunophenotypic characterization of BM samples demonstrated normal distribution of phenotypically defined HSPC subsets that were not affected by our editing or transplant strategy (Fig. 4A, fig. S14A,B). In addition, FACS-sorted HSPC subsets were introduced into CFC assays, demonstrating robust erythro-myeloid differentiation potential (Fig. 4B), similar to freshly isolated and non-modified BM cells (figs. S4B, S5, S6B). Editing efficiency (Fig. 4C) and deletion signatures (Fig. 4D) were comparable between bulk CD34+ and CD90+CD45RA– cells in both animal cohorts, and matched those detected in the PB at similar time points (Fig. 2A,F,G, fig. S8).
Fig. 4. BM analysis from treated animals at 6 months post-transplant.
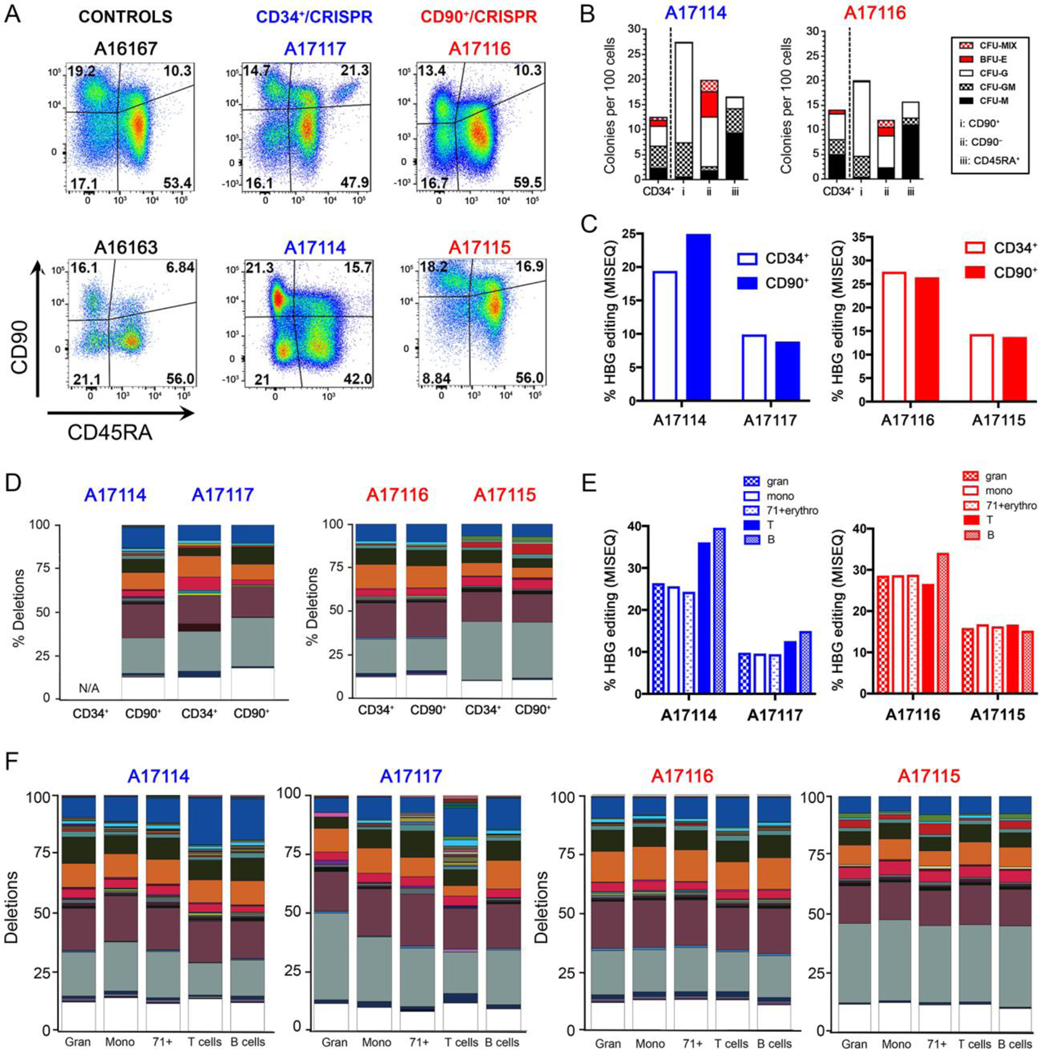
(A) Immunophenotypic characterization of CD34+ subsets from two CD34+ animals (middle) and two CD90+ animals (right) as compared to untransplanted animal controls (left). (B) Representative CFC assays obtained from BM of transplanted animals from panel A. CFU-M=macrophages, CFU-G=granulocytes, CFU-GM=granulocyte/macrophage, BFU-E=erythroid. (C) HBG editing in BM-sorted populations from two CD34+ animals (left) and two CD90+ animals (right). (D) Deletion profile in different HSPC subsets of BM from the same animals as in (C). N/A=not available. (E) HBG editing measured in different cell lineages of BM from the same animals as in (C). (F) Deletion profile in different cell lineages of BM from the same animals as in (C). In panels D, F, all deletion frequencies were normalized to 100%; colored boxes show distinct deletions, and the white portion shows all combined deletions contributing less than 1%. The 13-nt deletion is on top in the dark blue box.
To confirm that our approach effectively edited multipotent HSCs with multilineage differentiation potential, we sort-purified lymphoid (T cells, B cells, NK cells) and myeloid (granulocytes, monocytes) lineages and nucleated erythroid precursors from BM white blood cells (fig. S14C,D). All lineages revealed substantial amounts of editing, with a consistent increase in frequency for all deletions, including the 13-nt HPFH deletion, in B- and T-lymphocyte populations from the CD34+ transplanted animals (Fig. 4E, F). In conclusion, gene-edited CD34+ population as well as HSC-enriched CD34+CD90+CD45RA– cells are capable of homing and repopulating the BM stem cell compartment to generate a balanced output of gene-edited lymphoid, myeloid, and erythroid blood cells.
Safety of HBG-directed CRISPR/Cas9 gene editing
The transplanted rhesus animals have so far been monitored for up to 1.5 years, and no adverse effects on the counts or composition of mature blood lineages have been detected (fig. S7). All CRISPR/Cas9-induced HBG deletions have been maintained at stable frequencies, indicating no selective advantage for any particular mutation during the long-term follow-up (Fig. 2G,H, fig. S8). To further evaluate safety, the off-target activity of our CRISPR/Cas9 approach was measured by next-generation sequencing analysis of 35 potential off-target (OT) sites determined in the rhesus macaque reference genome. These sites differ by 1, 2, or 3 nts as compared to the 20 bases proximal to the protospacer adjacent motif (PAM) sequence of the CRISPR target site (OT1-OT23) or contain a 1-nt insertion or deletion with or without an additional mismatch (OT24-OT34) and a site (OT35) that contains a ‘NAG’ PAM sequence in addition to a single nt mismatch (table S2). Cells from the infusion product (day 0) as well as PB sampled at about 150 days post-transplant were sequenced in two animals from each experimental cohort. Out of 35 OT sites that were tested, only 2 showed mutations that were slightly above the detection limit of 0.1% (Fig. 5A,B). Considering the low number of sequencing reads obtained (about 10-fold fewer as compared to other sites) and the limit of detection of this methodology, the intergenic site OT5 showed a frequency of mutations of 0.23% for A17117 infusion product and 0.12% for A16116 PB at 150 days. Similarly, the intergenic site OT27 showed a signal above background only for A17114 infusion product, albeit with a low number of sequencing reads.
Fig. 5. Safety of HBG-directed CRISPR/Cas9 gene editing.

(A-B) Frequencies of predicted off-target (OT) sites determined by next generation sequencing analysis of infusion products (CD34+ or CD90+ cells) and of PB sampled at about 150 days post-transplant in CD34+/CRISPR animals (A17117/A17114) (A) and in CD90+/CRISPR animals (A17115/A17116) (B). (C) Schematic representation of CRISPR/Cas9-induced 4.8 kbp deletion with position of primers and probes. (D) Droplet digital PCR (ddPCR) quantification of the 4.8 kbp deletion in the infusion product (CD34+ or CD90+ cells, left) and from PB of the indicated animals at different time points post-transplant. WBC = white blood cells. Results show means and standard deviations from 2 or 3 technical replicates.
Because the HBG CRISPR target site is present in the promoter regions of both HBG1 and HBG2 genes, it is expected that a large chromosomal deletion (~4.9 kbp) encompassing the entire HBG2 gene would occur when both sites are cut simultaneously as described previously (6, 16) (Fig. 5C). We used a droplet digital PCR approach to quantify loss of signal that occurs when the HBG2-specific probe cannot bind to the deleted fragment in CRISPR-treated samples relative to untreated cells. As shown in Fig. 5D, a frequency of 15% to 30% HBG deletion was detected in the infused cells of all 6 animals in the CD34+ and CD90+CD45RA– cohorts. The frequency of this particular deletion was reduced to less than 10% in PB of transplanted animals after just 25 days and was detected at a frequency of less than 2% at 300 and 450 days post-treatment (Fig. 5D). We confirmed these results by long-range PCR analysis, where amplicons for both the intact and deleted alleles were detected in the infusion product, and the frequency of the deleted allele declined after transplant (fig. S15). This semi-quantitative method suffers, however, from the preferential amplification of the deleted (shorter) allele, resulting in an overestimation of deletion frequency. In summary, these results confirm the occurrence of a large chromosomal deletion encompassing the HBG2 gene and a large portion of the HBG1 promoter in edited NHP HSPCs at the time of infusion; the frequency of this deletion decreases rapidly post-transplant and is detected long term at low rates in only a fraction of the animals tested.
Discussion
This study describes the long-term engraftment of HSPCs edited at a previously validated CRISPR/Cas9 target site (6) to generate an array of potentially therapeutic indels that would disrupt BCL11A binding site and reactivate HbF. We provide proof of concept that edited bulk CD34+ as well as HSC-enriched CD34+CD90+CD45RA– cells engraft and are maintained at stable frequencies after transplant in NHPs. In addition, engraftment of edited cells resulted in sustained HbF reactivation with up to 18% F-cells produced in PB for more than 1 year so far. The high frequency of CRISPR/Cas9 in vivo editing reported here in a large animal model of autologous transplantation with no evidence for off-target or any other adverse effects demonstrates feasibility of HSPC-based editing approaches for a number of genetic disorders.
Ongoing clinical gene therapy trials using autologous HSPCs modified by means of viral vectors expressing β-like globin transgenes are showing promising results in β-thalassemia and SCD patients (17–19). However, the potential for genotoxic complications due to the semi-random chromosomal integration of the provirus remains. Moreover, practical limitations in large-scale vector production are a widely-recognized bottleneck to clinical implementation (20). The genome editing approach described here has the potential to offer a curative option for patients with hemoglobinopathies by introducing targeted, naturally occurring HPFH mutations within the γ-globin promoters. Allogeneic HSPC transplant studies indicated that a donor-host chimerism of 20% is required to reverse the sickle cell phenotype (21–23). Although the CRISPR/Cas9-edited cells produced in our study may not have the same therapeutic potential as fully corrected cells, the frequency of in vivo editing achieved, reaching as much as 27% in PB, is likely to provide some clinical benefits for hemoglobinopathies as well as for other genetic diseases. Editing in the BM was detected in all lineages and was comparable to that measured in PB at similar time points. Interestingly, editing frequencies were increased in B- and T-lymphocyte lineages in the CD34+ animals, possibly reflecting the long-lived nature of these cells, which may have been generated from HSPCs soon after transplantation at a time when editing efficiency was greater.
The animals with the highest rates of in vivo editing showed about 5% γ-globin protein expression (of total β-like globin) at 400 days post-treatment. While these percentages of γ-globin are below the reported therapeutic threshold ranging from 10% to 20% depending on the severity of the hemoglobinopathy (24–26), these results are in healthy, normal animals without any selective advantage of F-cells. In SCD, however, the lifespan of F-cells is expected to increase by 4- to 5-fold in the context of the disease, as demonstrated by studies using the humanized sickle mouse model (27), and thus our results may already be sufficient to ameliorate the disease phenotype in SCD. Nevertheless, the survival advantage of F-cells generated through CRISPR/Cas9-editing will have to be investigated further because they may not be equivalent to F-cells generated under hydroxyurea treatment or in the context of natural HPFH. Importantly, the NHP model enables long-term measurement of hemoglobin production in PB, which is not possible in humanized mouse models where human erythrocyte differentiation is severely limited (28). Our findings demonstrating stable and high frequencies of in vivo editing will be valuable for other editing strategies aimed at HbF reactivation, such as knocking down the HbF repressor BCL11A exclusively in the erythroid lineage using lentiviral delivery of shRNA (29), or disrupting the BCL11A intronic enhancer motif using site-specific nucleases (13, 30, 31).
These results represent a substantial improvement in engraftment of gene-edited HSPCs as compared to our previous investigations using bulk CD34+ cells treated with zinc finger (32) or TALE nucleases (15) in the NHP model. Beyond distinct nuclease platforms and the mRNA-based delivery used, these previous studies targeted different genomic loci and used different culture conditions, both of which may also explain the differences in engraftment. Similar to our previous studies, engraftment of CRISPR/Cas9-edited CD34+ HSPCs in a recently published study in the same rhesus macaque transplantation model was only in the 3 to 6% range (33). Although myeloablative conditioning by TBI was performed in our study to facilitate engraftment of edited cells, clinical translation of this approach will require reduced-intensity conditioning regimens to minimize toxicity without compromising engraftment. For example, antibody-drug conjugates (ADCs) offer a nongenotoxic strategy to specifically target HSCs in the BM niche, and recent studies using anti-CD117 ADCs have demonstrated feasibility of this approach (34, 35).
Our approach resulted in equivalent editing efficiency in bulk CD34+ and in the HSC-enriched CD34+CD90+CD45RA- cells. The predominant edits were small deletions, all of which overlapped with the recently characterized BCL11A binding site (9, 10), but also included a high frequency of the naturally occurring 13-nt HPFH deletion. This deletion, first described in SCD patients (5), was successfully recapitulated with site-specific nucleases in human CD34+ HSPCs (Traxler et al., (6) and Lux et al. (7)) and repair of the resulting double-strand DNA break by the MMEJ pathway, which is most active in cycling cells (36). We found the frequency of this 13-nt deletion to be reduced in CD34+CD90+CD45RA– cells relative to bulk CD34+, supporting the interpretation that this subset is enriched for primitive HSCs, which are generally found in a state of quiescence (37). The 13-nt HPFH deletion was stably maintained in vivo for more than 1 year post-transplant, albeit at reduced frequency as compared to input cells, consistent with findings from editing of the BCL11A enhancer locus (13). Importantly, no substantial difference in in vivo editing was observed between the two experimental groups, further indicating that the CD34+CD90+CD45RA– subset is the critical target cell population that contributes to multilineage long-term engraftment and confirming our previous finding (11). This HSC-enriched population decreases the number of target cells by over 10-fold thus circumventing issues associated with scale-up and considerably reducing the need for editing reagents without impacting hematopoietic recovery, engraftment, or HbF reactivation. These results also corroborate earlier attempts to enrich for a phenotypically-defined, HSC-enriched CD34+ subpopulation in humans dating back to the late 1990s (38–40). These autologous stem cell transplantation studies evaluated flow-sorting-based HSC-enrichment strategies for the treatment of myeloma, breast cancer, and non-Hodgkin lymphoma patients, using lin–CD34+CD90+ or CD34+CD90+ cell fractions that are enriched for primitive long-term engrafting HSCs, while phenotypically depleting CD90- malignant cells. The rapid and sustained hematopoietic engraftment of HSC-enriched cell fractions (38–40) demonstrated that this approach was technically possible and safe. Nevertheless, the engraftment kinetics and multilineage potential of these purified cells could not be assessed because of a lack of marking.
A potential limitation of our approach is the presence of the CRISPR/Cas9 target site in the promoters of both HBG1 and HBG2, which generates a deletion encompassing the entire HBG2 gene and part of the HBG1 promoter upon simultaneous cleavage. This deletion was detected in the NHP infusion product at a frequency of 15 to 27%, consistent with data reported in human cells (7). Nevertheless, we observed a rapid decline in this deletion post-transplant, which was detectable at frequencies of less than 2% in only a fraction of all animals after 1 year. This result suggests that cells bearing this deletion are at a selective disadvantage for engraftment in our model, but the underlying biological mechanism remains unknown. Safety of our approach was further validated by rapid reconstitution of all blood cell lineages with counts that remained within normal range during the entire course of the study. Our query of 35 predicted off-target sites in NHP cells by next-generation sequencing before and after transplant showed two sites with low activity in some of the animals tested, but these results were likely skewed by low sequencing reads. Future work to verify the absence of off-target activity without making any sequence assumption will need to apply a genome-wide and unbiased approach such as GUIDE-seq (41) or CIRCLE-seq (42). Additionally, any occurrence of other large deletion or chromosomal rearrangement that may occur at the target site as previously described (43) will have to be investigated.
In conclusion, the extent of in vivo gene editing achieved in our study using bulk CD34+ or the CD34+CD90+CD45RA- subpopulation may be within a therapeutically relevant range for a number of genetic diseases. The conservation of the CD34+CD90+CD45RA- phenotype and the HBG CRISPR/Cas9 gRNA target site between NHP and human, combined with the use of a highly clinically relevant large animal model for stem cell gene therapy and transplantation, should facilitate the direct translation of this approach to patients.
Materials and Methods
Study Design
The purpose of the current study was to assess engraftment of CRISPR/Cas9-edited CD34+ HSPCs in the autologous setting and compare results with editing and transplantation of an HSC-enriched CD34+CD90+CD45RA- subpopulation. Healthy rhesus macaques were enrolled in the study, and 3 animals were randomly assigned to each experimental strategy to account for animal to animal variation within each group. Animal caretakers and investigators processing and analyzing specimens were blinded for the animal allocation to each group.
Nonhuman primate animal housing, care and transplantation
Healthy juvenile pigtailed macaques and juvenile rhesus macaques were housed at the UW National Primate Research Center (WaNPRC) under conditions approved by the American Association for the Accreditation of Laboratory Animal Care. All experimental procedures performed were reviewed and approved by the Institutional Animal Care and Use Committee of the Fred Hutchinson Cancer Research Center (Fred Hutch) and University of Washington (UW; Protocol #3235–01). This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (“The Guide”), and monkeys were randomly assigned to the study. and monkeys were randomly assigned to the study. This study included at least twice-daily observation by animal technicians for basic husbandry parameters (for example food intake, activity, stool consistency, and overall appearance), as well as daily observation by a veterinary technician and/or veterinarian. Animals were housed in cages approved by “The Guide” and in accordance with Animal Welfare Act regulations. Animals were fed twice daily and were fasted for up to 14 hours before sedation. Environmental enrichment included grouping in compound, large activity, or run-through connected cages, perches, toys, food treats, and foraging activities. If a clinical abnormality was noted by WaNPRC personnel, standard WaNPRC procedures were followed to notify the veterinary staff for evaluation and determination for admission as a clinical case. Animals were sedated by administration of ketamine HCl and/or telazol and supportive agents for balanced anesthesia (such as diazepam, midazolam) before all procedures. After sedation, animals were monitored according to WaNPRC standard protocols. WaNPRC surgical support staff are trained and experienced in the administration of anesthetics and have monitoring equipment available to assist with electronic monitoring of heart rate, respiration, and blood oxygenation; audible alarms and LCD readouts; monitoring of blood pressure, temperature, etc. For minor procedures, the presence or absence of deep pain was tested by the toe-pinch reflex and the absence of response (leg flexion) to this test indicated adequate anesthesia. In cases of general anesthesia, similar monitoring parameters were used and anesthesia was tested by the loss of palpebral reflexes (eye blink). Analgesics (generally buprenorphine with meloxicam or buprenorphine SR) were provided as prescribed by the Clinical Veterinary staff for at least 48 hours after the procedures and could be extended at the discretion of the clinical veterinarian, based on clinical signs. Autologous NHP transplantation, priming (mobilization), collection of cells, and genetic engineering were conducted consistent with our previously published protocols (44). In parallel to cell processing, macaques were conditioned with myeloablative total body irradiation (TBI) of 1020 cGy from a 6 MV x-ray beam of a single-source linear accelerator located at the Fred Hutch South Lake Union Facility (Seattle, Washington, USA); irradiation was administered as a fractionated dose over the 2 days before cell infusion. During irradiation, animals were housed in a specially modified cage that provided unrestricted access for the irradiation while simultaneously minimizing excess movement. The dose was administered at a rate of 7 cGy/min delivered as a midline tissue dose. G-CSF was administered daily from the day of cell infusion until the animals began to show onset of neutrophil recovery. Supportive care, including antibiotics, electrolytes, fluids, and transfusions, was given as necessary, and blood counts were analyzed daily to monitor hematopoietic recovery. Cytomegalovirus (CMV) monitoring was performed by quantitative PCR as described previously (45). Briefly, DNA was isolated from whole blood using Qiagen DNA Blood Mini Kit (Qiagen) using manufacturer’s instructions. Isolated DNA was then used for quantitative Real-Time PCR analysis on an Applied Biosystems QuantStudio Real-Time PCR system (12K Flex) using CMV-specific primers (Forward: ATC CGC GTT CCA ATG CA; Reverse: CGG AGG AGC ACC ATA GAA GGT) and a TaqMan Probe (6FAM CCT TCC CGG CTA TGG MGBNFQ). Each sample was run in triplicate for 40 cycles along with positive controls. Copy numbers were calculated by comparing to a standard curve, and the viral load was reported as CMV copies/ml of whole blood.
CD34+ enrichment and in vitro culture
NHP primed BM was harvested, enriched, and cultured as previously described (44, 46). Briefly, before enrichment of CD34+ cells, red cells were lysed in ammonium chloride lysis buffer, and white blood cells were incubated for 20 minutes with the 12.8 immunoglobulin-M anti-CD34 antibody, then washed and incubated for another 20 minutes with magnetic-activated cell-sorting anti-immunoglobulin-M microbeads (Miltenyi Biotech). The cell suspension was run through magnetic columns enriching for CD34+ cell fractions with a purity of 60% to 80% confirmed by flow cytometry. Human CD34+ cells were harvested and enriched from mobilized PB as previously described (46). Enriched CD34+ cells were cultured in SFEMII media (Stemcell Technologies) supplemented with 100 U/ml penicillin and streptomycin (Gibco by Life Technologies) and SCF (Peprotech), TPO (Thrombopoietin, Peprotech), and FLT3-L (Fms-related tyrosine kinase 3 ligand, Miltenyi Biotec) (100 ng/ml for each cytokine). For CFC assays, 1000–1200 sorted cells were seeded into 3.5 ml ColonyGEL 1402 (ReachBio). Hematopoietic colonies were scored after 12–14 days. Arising colonies were identified as colony-forming unit- (CFU-) granulocyte (CFU-G), macrophage (CFU-M), granulocyte-macrophage (CFU-GM), and burst-forming unit-erythrocyte (BFU-E). Colonies consisting of erythroid and myeloid cells were scored as CFU-MIX. In vitro erythroid differentiation of NHP CD34+ cells was carried out as described in Humbert et al. (15).
Flow cytometric analysis and FACS
Antibodies used for FACS (fluorescence-activated cell sorting) analysis and sorting of NHP cells are listed in table S3. Dead cells and debris were excluded by FSC/SSC gating. Flow cytometric analysis was performed on an LSR IIu, FACSAria IIu and Canto II (BD). Cells for in vitro assays as well as autologous NHP stem cell transplants were sorted right after CD34+ enrichment using a FACSAria IIu cell sorter (BD) and sort purity assessed by recovery of sorted cells. p53 expression was assessed by intracellular flow cytometric staining after fixation with the BD Perm/Wash kit (BD). Annexin V/7-AAD staining was conducted to assess apoptosis and cell death following manufacturer specifications (BD).
Fetal hemoglobin measurements
HbF was measured by flow cytometry after fixation and permeabilization of unlysed blood and by quantitative PCR of globin transcripts using previously described procedures (15). For HPLC quantification of globin subtypes, cells were lysed in HPLC grade water (Sigma-Aldrich) and centrifuged at 4°C for 20 min at 20,000 g (Centrifuge 5430R, Eppendorf). Supernatant was injected onto an Aeris 3.6 μm Widepore C4 250 × 4.6 mm column (Phenomenex). Mobile phases used were: A: Water 0.1% TFA (trifluoroacetic acid), B: Acetonitrile 0.08% TFA at a flow rate of 0.8 ml/min. A gradient from 39% to 50% B was run over a 75-minute timed program. The column oven temperature was 40°C and the sample tray was at kept at 4°C. The peaks were detected at 220 nm, characterized by their retention time relative to a reference control, and were formally identified by liquid chromatography mass spectrometry (LC-MS). In LC-MS, eluted fractions were taken to dryness by speed-vacuum and resuspended in a smaller (50 μl) volume of 50 mM ammonium bicarbonate. Each sample was digested overnight in trypsin at 37°C and acidified with 9 μl of 0.2% trifluoroacetic acid (Sigma Aldrich). A 2-μl aliquot of each sample was diluted with 18 μl of 98/2/0.1% water/acetonitrile/formic acid solution (v/v) and loaded onto a 100-micron capillary trap packed with Magic C18 aQ packing material. The samples were then eluted off of a 25-cm long 75-micron i.d. capillary column (also packed with Magic C18 aQ material) connected to a Thermo Easy-nLC II HPLC (Thermo Fisher Scientific) with a gradient going from 7% mobile phase B to 35% mobile phase B in 60 minute time at 400 nl/minute. Analysis was conducted on a Thermo-Electron Orbitrap Elite mass spectrometer (Thermo Fisher Scientific) using a resolution of 240K at 200 m/z for the MS1 scans and performing MS/MS on the top 20 most abundant ions in the MS1 spectrum using collisional induced dissociation (CID). The data were searched against a Uniprot macaca-mulatta database modified to include entries from the common Repository of Adventitious Proteins (ver 061217) using Proteome Discoverer (ver 2.2) with a false-discovery rate of 1%.
CRISPR/Cas9 gene editing
Cas9 purified protein was obtained from ThermoFisher Scientific (TrueCut Cas9 protein v2, 5 μg/μl, # A36499), and chemically-modified gRNAs (2’-O-methyl analogs and 3’ phosphorothioate internucleotide linkages at the first three 5’ and 3’ terminal RNA residues) were custom-ordered from Synthego. Lyophilized gRNAs were resuspended in nuclease-free water at a concentration of 200 pmol/μl and stored as frozen aliquots at −80°C. For the editing of rhesus CD34+ or CD90+ subsets, cells were first cultured overnight after enrichment and/or sorting. CRISPR/Cas9 RNPs were formed by combining 180 pmol Cas9 protein with 1800 pmol gRNA at room temperature for 10 min and used for the electroporation of 3 million cells in 2 mm cuvettes following manufacturer instructions (Harvard Apparatus). Edited cells were recovered in medium overnight at 37°C before infusion in the animals.
Measurement of on- and off-target editing efficiency
Editing at the HBG locus was measured either by TIDE analysis using Sanger sequencing (47) or by next-generation sequencing using Illumina barcoded, 2×150 bp pair-end Miseq primers for complete sequencing (Illumina). Primers are described in table S4, and bioinformatic processing of sequencing data was conducted using an in-house-built pipeline (15). Potential off-target sites were bioinformatically identified using the webtool COSMID (48). Off-target loci were amplified by two rounds of PCR using 2 × 250 bp pair-end Miseq primers. Indels at potential off-target sites were quantified as previously described (49). Sequence data are available for download through the National Center for Biotechnology Information (BioProject ID: PRJNA544343).
Quantification of large deletion at HBG locus
For the digital droplet PCR, HBG1 and HBG2 specific probes were designed and used in combination with forward and reverse primers that amplify both HBG1 and HBG2, resulting in amplicons of 959 bp and 972 bp, respectively. The droplet PCR reaction was set up with 50 ng of template gDNA, 900 nM primers, 250 nM probes (both HBG1 FAM or HBG2 HEX probe), and ddPCR supermix for probes (no dUTP), digested with EcoRV-HF for 10 minutes at 37oC; droplets were generated at room temperature using a QX200 Droplet Generator (Biorad). PCR amplification was done on a thermal cycler using the following program: 1) 95oC 10 min; 2) 95oC 30 s; 3) 61.3oC 1 min; 4) 72oC 1 min; 5) repeat steps 2–4 for 49 cycles; 6) 98 oC 10 min. Fluorescence intensity was measured on the QX200 Droplet reader TM (Biorad), and the data were analyzed using Quantasoft software (Biorad). Results were normalized to number of copies/μl, and the frequency of G1–G2+ events was calculated for each sample and compared to frequency of all positive events. The deletion rate was then calculated as the increase in frequency of G1–G2+ in the edited sample compared to the mock-electroporated sample. For the long-range PCR, primers designed 5’ of HBG2 and 3’ of HBG1 (table S4) were used for PCR amplification using LongAmp Taq DNA polymerase (New England Biolabs) following manufacturer instructions. Resulting amplicons were measured semi quantitatively by densitometry analysis.
RT-qPCR quantification of globin chains and p21 mRNA expression
For quantification of p21 (CDKN1A) and globin mRNA, whole RNA was isolated from treated cells at the defined time points with the RNAeasy Mini Kit (Qiagen) and converted to cDNA using the Superscript III first strand synthesis kit (Thermo Fisher Scientific). Taqman reaction was run following the manufacturer’s protocol using the Applied Biosystems 7500 Real-Time PCR system (Thermo Fisher Scientific). mRNA expression was measured with established assays (CDKN1A Rh02822707_m1; HbA Rh02828921_qH, HbB Rh02809243_m1, and HbG Rh02576538_gH) and normalized to GAPDH (GAPDH Rh02621745_g1) by the comparative Ct method.
Statistics
Statistical analysis of data was performed using GraphPad Prism version 7. Significance analyses were performed with the two-tailed unpaired Student’s t test. Correlations were calculated using Spearman’s rank correlation coefficient (correlation coefficient R2 values of 0.0 to 0.19 = very weak, 0.20 to 0.39 = weak, 0.4 to 0.59 = moderate, 0.6 to 0.79 = strong, and 0.8 to 1.0 = very strong).
Supplementary Material
Fig. S1. Optimization of HBG editing in NHP HSPCs.
Fig. S2. Induction of the p53/p21 DNA damage response in human and rhesus HSPCs treated with CRISPR/Cas9
Fig. S3. CRISPR/Cas9-induced cell death and apoptosis in human and rhesus HSPCs
Fig. S4. Immunophenotypic and functional characterization of infusion products of CD34 transplants.
Fig. S5. In vitro validation of the CD90+ sorting/editing approach Fig. S6. Immunophenotypic and functional characterization of infusion products of CD90 transplants.
Fig. S7. Complete blood cell count in transplanted animals.
Fig. S8. Tracking of deletion profiles in transplanted animals by Miseq analysis.
Fig. S9. Conservation of globin polypeptide sequences between human and rhesus macaque.
Fig. S10. Conservation of globin polypeptide homologs in rhesus macaque. Fig. S11. Validation of reverse phase-HPLC approach for the analysis of hemoglobin from NHP blood.
Fig. S12. Representative HPLC profiles of PB from transplanted animals.
Fig. S13. Quantitative PCR measurement of globin transcripts from PB of transplanted animals.
Fig. S14. Flow cytometry validation of sorting approach of HPSC subsets and cell lineages from 6-month BM of transplanted animals.
Fig. S15. Long-range PCR analysis of large HBG deletion
Table S1. PCR-based detection of cytomegalovirus (CMV) DNA from PB of transplanted animals
Table S2. Sequence and chromosomal location of predicted off target (OT) sites for the given HBG CRISPR/Cas9
Table S3. List of antibodies used for flow cytometric analysis
Table S4. List of primers
Data file S1. Legend for tracking of CRISPR/Cas9-induced deletion signatures
Data file S2: Original data
Acknowledgments:
We thank Veronica Nelson, Erica Curry, Kelvin Sze, and Mallory Llewellyn for excellent support in our macaque studies and in processing biological specimens, and are grateful to Helen Crawford for her help with manuscript and figure preparation. This work has been supported by grant R01 HL136135 from the NIH, Bethesda, MD. H.P.K. is a Markey Molecular Medicine Investigator and received support as the inaugural recipient of the José Carreras/E. Donnall Thomas Endowed Chair for Cancer Research and the Fred Hutch Endowed Chair for Cell and Gene Therapy. G.B. acknowledges the support from the Cancer Prevention and Research Institute of Texas (RR14008 and RP170721).
Footnotes
Competing interests: H.P.K is a consultant to and has ownership interests with Rocket Pharma and Homology Medicines. H.P.K. is a consultant to CSL Behring and Magenta Therapeutics. S.R., J.E.A. and H.-P.K. are inventors on patent applications (#62/351,761, #62/428,994 and #PCT/US2017/037967) submitted by the Fred Hutchinson Cancer Research Center that covers the selection and use of cell populations for research and therapeutic purposes as well as strategies to assess and/or produce cell populations with predictive engraftment potential. A.M.S is co-founder and equity holder of Umoja Biopharma. A.M.S. receives equity compensation and is SAB chair of Alpine Immune Sciences. A.M.S. receives equity compensation as well as salary and is CSO of Casebia Therapeutics. The other authors declare that they have no competing interests.
Data and materials availability: All data associated with this study are present in the paper or Supplementary Materials.
References and Notes:
- 1.Modell B, Darlison M, Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ 86, 480–487 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leonard A, Tisdale JF, Stem cell transplantation in sickle cell disease: therapeutic potential and challenges faced. Expert Rev Hematol 11, 547–565 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Esrick EB, Bauer DE, Genetic therapies for sickle cell disease. Semin Hematol 55, 76–86 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Forget BG, Molecular basis of hereditary persistence of fetal hemoglobin. Ann N Y Acad Sci 850, 38–44 (1998). [DOI] [PubMed] [Google Scholar]
- 5.Gilman JG, Mishima N, Wen XJ, Stoming TA, Lobel J, Huisman TH, Distal CCAAT box deletion in the A gamma globin gene of two black adolescents with elevated fetal A gamma globin. Nucleic Acids Res 16, 10635–10642 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Traxler EA, Yao Y, Wang YD, Woodard KJ, Kurita R, Nakamura Y, Hughes JR, Hardison RC, Blobel GA, Li C, Weiss MJ, A genome-editing strategy to treat beta-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat Med 22, 987–990 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lux CT, Pattabhi S, Berger M, Nourigat C, Flowers DA, Negre O, Humbert O, Yang JG, Lee C, Jacoby K, Bernstein I, Kiem HP, Scharenberg A, Rawlings DJ, TALEN-Mediated Gene Editing of HBG in Human Hematopoietic Stem Cells Leads to Therapeutic Fetal Hemoglobin Induction. Mol Ther Methods Clin Dev 12, 175–183 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martyn GE, Quinlan KGR, Crossley M, The regulation of human globin promoters by CCAAT box elements and the recruitment of NF-Y. Biochim Biophys Acta 1860, 525–536 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Liu N, Hargreaves VV, Zhu Q, Kurland JV, Hong J, Kim W, Sher F, Macias-Trevino C, Rogers JM, Kurita R, Nakamura Y, Yuan GC, Bauer DE, Xu J, Bulyk ML, Orkin SH, Direct promoter repression by BCL11A controls the fetal to adult hemoglobin switch. Cell 173, 430–442 e417 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martyn GE, Wienert B, Yang L, Shah M, Norton LJ, Burdach J, Kurita R, Nakamura Y, Pearson RCM, Funnell APW, Quinlan KGR, Crossley M, Natural regulatory mutations elevate the fetal globin gene via disruption of BCL11A or ZBTB7A binding. Nat Genet 50, 498–503 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Radtke S, Adair JE, Giese MA, Chan YY, Norgaard ZK, Enstrom M, Haworth KG, Schefter LE, Kiem HP, A distinct hematopoietic stem cell population for rapid multilineage engraftment in nonhuman primates. Sci Transl Med 9, [Epub ahead of print; doi: 10.1126/scitranslmed.aan1145] (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bak RO, Dever DP, Porteus MH, CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nat Protoc 13, 358–376 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu Y, Zeng J, Roscoe BP, Liu P, Yao Q, Lazzarotto CR, Clement K, Cole MA, Luk K, Baricordi C, Shen AH, Ren C, Esrick EB, Manis JP, Dorfman DM, Williams DA, Biffi A, Brugnara C, Biasco L, Brendel C, Pinello L, Tsai SQ, Wolfe SA, Bauer DE, Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat Med 25, 776–783 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schiroli G, Conti A, Ferrari S, Della Volpe L, Jacob A, Albano L, Beretta S, Calabria A, Vavassori V, Gasparini P, Salataj E, Ndiaye-Lobry D, Brombin C, Chaumeil J, Montini E, Merelli I, Genovese P, Naldini L, Di Micco R, Precise gene editing preserves hematopoietic stem cell function following transient p53-mediated DNA damage response. Cell Stem Cell 24, 551–565 e558 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Humbert O, Peterson CW, Norgaard ZK, Radtke S, Kiem HP, A nonhuman primate transplantation model to evaluate hematopoietic stem cell gene editing strategies for beta-hemoglobinopathies. Mol Ther Methods Clin Dev 8, 75–86 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li C, Psatha N, Sova P, Gil S, Wang H, Kim J, Kulkarni C, Valensisi C, Hawkins RD, Stamatoyannopoulos G, Lieber A, Reactivation of gamma-globin in adult beta-YAC mice after ex vivo and in vivo hematopoietic stem cell genome editing. Blood 131, 2915–2928 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sii-Felice K, Giorgi M, Leboulch P, Payen E, Hemoglobin disorders: lentiviral gene therapy in the starting blocks to enter clinical practice. Exp Hematol 64, 12–32 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Thompson AA, Walters MC, Kwiatkowski J, Rasko JEJ, Ribeil JA, Hongeng S, Magrin E, Schiller GJ, Payen E, Semeraro M, Moshous D, Lefrere F, Puy H, Bourget P, Magnani A, Caccavelli L, Diana JS, Suarez F, Monpoux F, Brousse V, Poirot C, Brouzes C, Meritet JF, Pondarre C, Beuzard Y, Chretien S, Lefebvre T, Teachey DT, Anurathapan U, Ho PJ, von Kalle C, Kletzel M, Vichinsky E, Soni S, Veres G, Negre O, Ross RW, Davidson D, Petrusich A, Sandler L, Asmal M, Hermine O, De Montalembert M, Hacein-Bey-Abina S, Blanche S, Leboulch P, Cavazzana M, Gene therapy in patients with transfusion-dependent beta-thalassemia. N Engl J Med 378, 1479–1493 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Ribeil JA, Hacein-Bey-Abina S, Payen E, Magnani A, Semeraro M, Magrin E, Caccavelli L, Neven B, Bourget P, El Nemer W, Bartolucci P, Weber L, Puy H, Meritet JF, Grevent D, Beuzard Y, Chretien S, Lefebvre T, Ross RW, Negre O, Veres G, Sandler L, Soni S, de Montalembert M, Blanche S, Leboulch P, Cavazzana M, Gene therapy in a patient with sickle cell disease. N Engl J Med 376, 848–855 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Merten OW, Hebben M, Bovolenta C, Production of lentiviral vectors. Mol Ther Methods Clin Dev 3, 16017 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fitzhugh CD, Cordes S, Taylor T, Coles W, Roskom K, Link M, Hsieh MM, Tisdale JF, At least 20% donor myeloid chimerism is necessary to reverse the sickle phenotype after allogeneic HSCT. Blood 130, 1946–1948 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abraham A, Hsieh M, Eapen M, Fitzhugh C, Carreras J, Keesler D, Guilcher G, Kamani N, Walters MC, Boelens JJ, Tisdale J, Shenoy S, National Institutes of H, Center for International B, Marrow Transplant R, Relationship between mixed donor-recipient chimerism and disease recurrence after hematopoietic cell transplantation for sickle cell disease. Biol Blood Marrow Transplant 23, 2178–2183 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walters MC, Patience M, Leisenring W, Rogers ZR, Aquino VM, Buchanan GR, Roberts IA, Yeager AM, Hsu L, Adamkiewicz T, Kurtzberg J, Vichinsky E, Storer B, Storb R, Sullivan KM, Multicenter D. Investigation of Bone Marrow Transplantation for Sickle Cell, Stable mixed hematopoietic chimerism after bone marrow transplantation for sickle cell anemia. Biology of Blood and Marrow Transplantation 7, 665–673 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Steinberg MH, Chui DH, Dover GJ, Sebastiani P, Alsultan A, Fetal hemoglobin in sickle cell anemia: a glass half full? Blood 123, 481–485 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, Klug PP, Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Eng J Med 330, 1639–1644 (1994). [DOI] [PubMed] [Google Scholar]
- 26.Noguchi CT, Rodgers GP, Serjeant G, Schechter AN, Levels of fetal hemoglobin necessary for treatment of sickle cell disease. N Engl J Med 318, 96–99 (1988). [DOI] [PubMed] [Google Scholar]
- 27.Franco RS, Yasin Z, Palascak MB, Ciraolo P, Joiner CH, Rucknagel DL, The effect of fetal hemoglobin on the survival characteristics of sickle cells. Blood 108, 1073–1076 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu Z, Van Rooijen N, Yang YG, Macrophages prevent human red blood cell reconstitution in immunodeficient mice. Blood 118, 5938–5946 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brendel C, Guda S, Renella R, Bauer DE, Canver MC, Kim YJ, Heeney MM, Klatt D, Fogel J, Milsom MD, Orkin SH, Gregory RI, Williams DA, Lineage-specific BCL11A knockdown circumvents toxicities and reverses sickle phenotype. J Clin Invest 126, 3868–3878 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Canver MC, Smith EC, Sher F, Pinello L, Sanjana NE, Shalem O, Chen DD, Schupp PG, Vinjamur DS, Garcia SP, Luc S, Kurita R, Nakamura Y, Fujiwara Y, Maeda T, Yuan GC, Zhang F, Orkin SH, Bauer DE, BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 527, 192–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vierstra J, Reik A, Chang KH, Stehling-Sun S, Zhou Y, Hinkley SJ, Paschon DE, Zhang L, Psatha N, Bendana YR, O’Neil CM, Song AH, Mich AK, Liu PQ, Lee G, Bauer DE, Holmes MC, Orkin SH, Papayannopoulou T, Stamatoyannopoulos G, Rebar EJ, Gregory PD, Urnov FD, Stamatoyannopoulos JA, Functional footprinting of regulatory DNA. Nat Methods 12, 927–930 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peterson CW, Wang J, Norman KK, Norgaard ZK, Humbert O, Tse CK, Yan JJ, Trimble RG, Shivak DA, Rebar EJ, Gregory PD, Holmes MC, Kiem HP, Long-term multilineage engraftment of autologous genome-edited hematopoietic stem cells in nonhuman primates. Blood 127, 2416–2426 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim MY, Yu KR, Kenderian SS, Ruella M, Chen S, Shin TH, Aljanahi AA, Schreeder D, Klichinsky M, Shestova O, Kozlowski MS, Cummins KD, Shan X, Shestov M, Bagg A, Morrissette JJD, Sekhri P, Lazzarotto CR, Calvo KR, Kuhns DB, Donahue RE, Behbehani GK, Tsai SQ, Dunbar CE, Gill S, Genetic inactivation of CD33 in hematopoietic stem cells to enable CAR T cell immunotherapy for acute myeloid leukemia. Cell 173, 1439–1453 e1419 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kwon HS, Logan AC, Chhabra A, Pang WW, Czechowicz A, Tate K, Le A, Poyser J, Hollis R, Kelly BV, Kohn DB, Weissman IL, Prohaska SS, Shizuru JA, Anti-human CD117 antibody-mediated bone marrow niche clearance in non-human primates and humanized NSG mice. Blood 133, 2104–2108 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Czechowicz A, Palchaudhuri R, Scheck A, Hu Y, Hoggatt J, Saez B, Pang WW, Mansour MK, Tate TA, Chan YY, Walck E, Wernig G, Shizuru JA, Winau F, Scadden DT, Rossi DJ, Selective hematopoietic stem cell ablation using CD117-antibody-drug-conjugates enables safe and effective transplantation with immunity preservation. Nat Commun 10, 617 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Truong LN, Li Y, Shi LZ, Hwang PY, He J, Wang H, Razavian N, Berns MW, Wu X, Microhomology-mediated end joining and homologous recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc Natl Acad Sci U S A 110, 7720–7725 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pietras EM, Warr MR, Passegue E, Cell cycle regulation in hematopoietic stem cells. J Cell Biol 195, 709–720 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Michallet M, Philip T, Philip I, Godinot H, Sebban C, Salles G, Thiebaut A, Biron P, Lopez F, Mazars P, Roubi N, Leemhuis T, Hanania E, Reading C, Fine G, Atkinson K, Juttner C, Coiffier B, Fiere D, Archimbaud E, Transplantation with selected autologous peripheral blood CD34+Thy1+ hematopoietic stem cells (HSCs) in multiple myeloma: impact of HSC dose on engraftment, safety, and immune reconstitution. Experimental Hematology 28, 858–870 (2000). [DOI] [PubMed] [Google Scholar]
- 39.Negrin RS, Atkinson K, Leemhuis T, Hanania E, Juttner C, Tierney K, Hu WW, Johnston LJ, Shizurn JA, Stockerl-Goldstein KE, Blume KG, Weissman IL, Bower S, Baynes R, Dansey R, Karanes C, Peters W, Klein J, Transplantation of highly purified CD34+Thy-1+ hematopoietic stem cells in patients with metastatic breast cancer. Biology of Blood and Marrow Transplantation 6, 262–271 (2000). [DOI] [PubMed] [Google Scholar]
- 40.Vose JM, Bierman PJ, Lynch JC, Atkinson K, Juttner C, Hanania CE, Bociek G, Armitage JO, Transplantation of highly purified CD34+Thy-1+ hematopoietic stem cells in patients with recurrent indolent non-Hodgkin’s lymphoma. Biology of Blood and Marrow Transplantation 7, 680–687 (2001). [DOI] [PubMed] [Google Scholar]
- 41.Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV, Thapar V, Wyvekens N, Khayter C, Iafrate AJ, Le LP, Aryee MJ, Joung JK, GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol 33, 187–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsai SQ, Nguyen NT, Malagon-Lopez J, Topkar VV, Aryee MJ, Joung JK, CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat Methods 14, 607–614 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kosicki M, Tomberg K, Bradley A, Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol 36, 765–771 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trobridge GD, Beard BC, Gooch C, Wohlfahrt M, Olsen P, Fletcher J, Malik P, Kiem HP, Efficient transduction of pigtailed macaque hematopoietic repopulating cells with HIV-based lentiviral vectors. Blood 111, 5537–5543 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zheng HB, Watkins B, Tkachev V, Yu S, Tran D, Furlan S, Zeleski K, Singh K, Hamby K, Hotchkiss C, Lane J, Gumber S, Adams AB, Cendales L, Kirk AD, Kaur A, Blazar BR, Larsen CP, Kean LS, The knife’s edge of tolerance: inducing stable multilineage Mixed chimerism but with a significant risk of CMV reactivation and disease in rhesus macaques. Am J Transplant 17, 657–670 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adair JE, Waters T, Haworth KG, Kubek SP, Trobridge GD, Hocum JD, Heimfeld S, Kiem HP, Semi-automated closed system manufacturing of lentivirus gene-modified haematopoietic stem cells for gene therapy. Nat Commun 7, 13173 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brinkman EK, Chen T, Amendola M, van Steensel B, Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42, e168–e168 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cradick TJ, Qiu P, Lee CM, Fine EJ, Bao G, COSMID: A Web-based Tool for Identifying and Validating CRISPR/Cas Off-target Sites. Molecular therapy. Nucleic acids 3, e214 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee CM, Cradick TJ, Bao G, The Neisseria Meningitidis CRISPR-Cas9 System enables specific genome editing in mammalian cells. Mol Ther 24, 645–654 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Optimization of HBG editing in NHP HSPCs.
Fig. S2. Induction of the p53/p21 DNA damage response in human and rhesus HSPCs treated with CRISPR/Cas9
Fig. S3. CRISPR/Cas9-induced cell death and apoptosis in human and rhesus HSPCs
Fig. S4. Immunophenotypic and functional characterization of infusion products of CD34 transplants.
Fig. S5. In vitro validation of the CD90+ sorting/editing approach Fig. S6. Immunophenotypic and functional characterization of infusion products of CD90 transplants.
Fig. S7. Complete blood cell count in transplanted animals.
Fig. S8. Tracking of deletion profiles in transplanted animals by Miseq analysis.
Fig. S9. Conservation of globin polypeptide sequences between human and rhesus macaque.
Fig. S10. Conservation of globin polypeptide homologs in rhesus macaque. Fig. S11. Validation of reverse phase-HPLC approach for the analysis of hemoglobin from NHP blood.
Fig. S12. Representative HPLC profiles of PB from transplanted animals.
Fig. S13. Quantitative PCR measurement of globin transcripts from PB of transplanted animals.
Fig. S14. Flow cytometry validation of sorting approach of HPSC subsets and cell lineages from 6-month BM of transplanted animals.
Fig. S15. Long-range PCR analysis of large HBG deletion
Table S1. PCR-based detection of cytomegalovirus (CMV) DNA from PB of transplanted animals
Table S2. Sequence and chromosomal location of predicted off target (OT) sites for the given HBG CRISPR/Cas9
Table S3. List of antibodies used for flow cytometric analysis
Table S4. List of primers
Data file S1. Legend for tracking of CRISPR/Cas9-induced deletion signatures
Data file S2: Original data