

Abstract
When compared with solid brain metastases from NSCLC, leptomeningeal disease (LMD) has unique growth patterns and is rapidly fatal. Patients with LMD do not undergo surgical resection, limiting the tissue available for scientific research. In this study we performed whole exome sequencing on eight samples of LMD to identify somatic mutations and compared the results with those for 26 solid brain metastases. We found that taste 2 receptor member 31 gene (TAS2R31) and phosphodiesterase 4D interacting protein gene (PDE4DIP) were recurrently mutated among LMD samples, suggesting involvement in LMD progression. Together with a retrospective review of the charts of an additional 44 patients with NSCLC LMD, we discovered a surprisingly low number of KRAS mutations (n = 4 [7.7%]) but a high number of EGFR mutations (n = 33 [63.5%]). The median interval for development of LMD from NSCLC was shorter in patients with mutant EGFR (16.3 months) than in patients with wild-type EGFR (23.9 months) (p = 0.017). Targeted analysis of recurrent mutations thus presents a useful complement to the existing diagnostic tool kit, and correlations of EGFR in LMD and KRAS in solid metastases suggest that molecular distinctions or systemic treatment pressure underpin the differences in growth patterns within the brain.
Keywords: Non–small cell lung cancer, Leptomeningeal metastases, Whole exome sequencing, KRAS, EGFR
Introduction
There are two distinct types of brain metastases: solid metastasis to the brain parenchyma and leptomeningeal metastasis (LMD) to the covering of the brain and cranial nerves. Although solid lung-to-brain metastases frequently respond well to surgery and radiation, LMD does not have a durable treatment.1,2 Because of the diffuse nature of LMD, surgical resection or biopsy is not an option in its treatment. The rarity of cases, rapid disease progression, and lack of available tissue for research have severely limited our understanding of the genetics and biology of LMD. The genetic underpinnings that differentiate LMD from solid tumor metastases are not clear; to our knowledge, a cohort analysis on recurrently mutated genes in lung cancer LMD has yet to be reported. Prior studies showing the genomic evolution of primary cancers and solid brain metastases3–5 have uncovered genes (such as fibroblast growth factor receptor 1 gene [FGFR1] and phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha gene [PIK3CA]) that appear to be important in brain metastases.6,7 Despite this, it is unknown whether specific mutations in lung cancer enhance its ability to thrive in the leptomeninges. We hypothesized that LMD harbors a distinct mutation profile compared with that of solid brain metastases, allowing these two metastatic subtypes to thrive in different brain niches. To this end, we compared our whole exome profiles of a cohort with NSCLC LMD with previously sequenced, publicly available solid lung-to-brain tumor metastases to discover differences in their mutational landscapes (Fig. 1).
Figure 1.
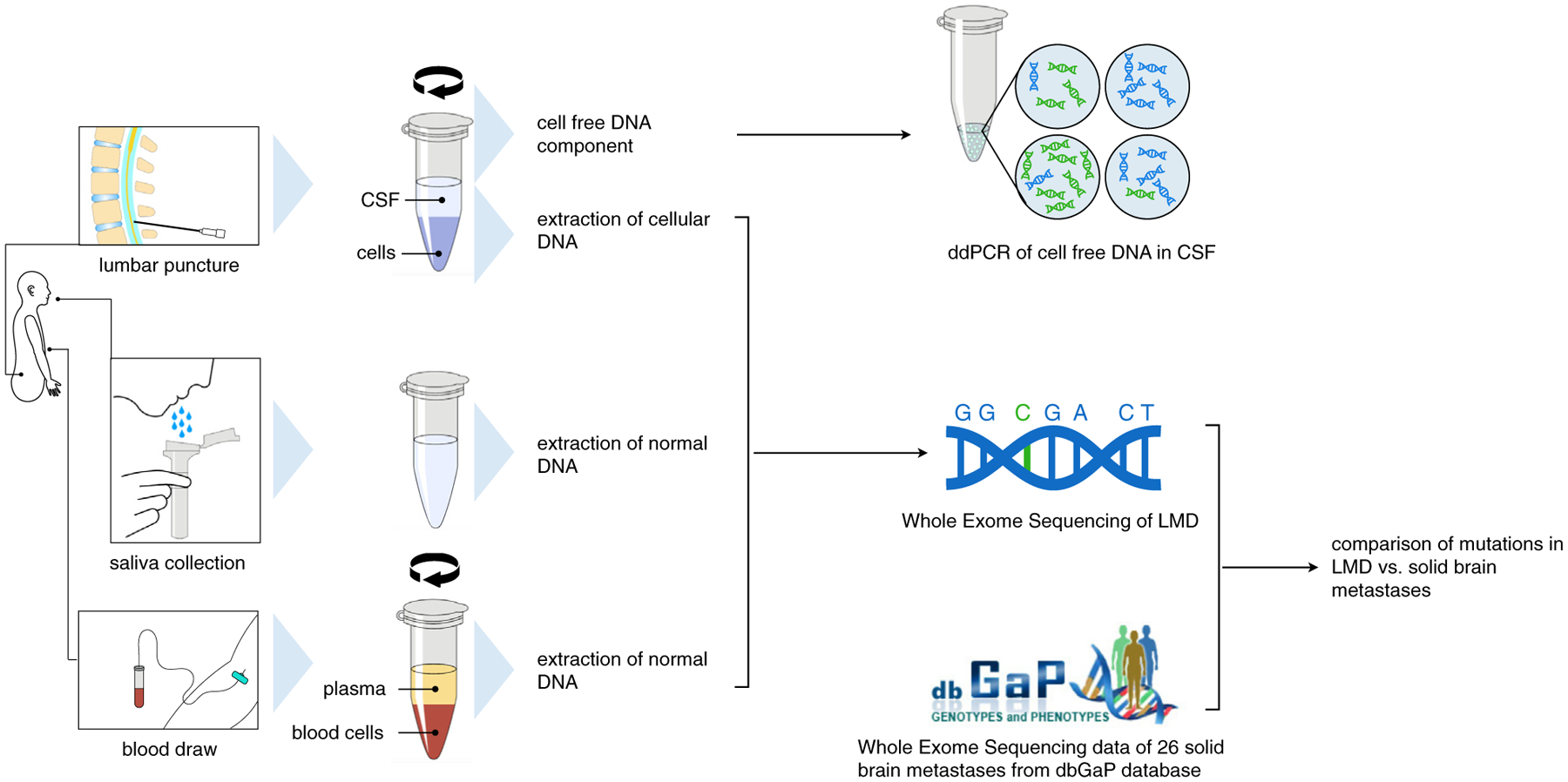
Workflow for comparing solid versus leptomeningeal NSCLC brain metastases. Patients undergoing clinical confirmation of leptomeningeal disease (LMD) (n = 8) have a DNA sample from a lumbar puncture compared with normal DNA from either a corresponding blood or saliva sample. After centrifugation and extraction of the DNA, the exome sequencing data were compared with data from the Database of Genotypes and Phenotypes (dbGaP) data on 26 solid NSCLC brain metastases. CSF, cerebrospinal fluid; ddPCR, digital droplet polymerase chain reaction.
Materials and Methods
Detailed methods can be found in the Supplementary Information. Briefly, cerebrospinal fluid (CSF) samples (Supplementary Table 1) were collected and centrifuged to separate the cellular and cell-free parts. Whole exome sequencing (WES) was performed, and the sequencing data were analyzed by using the Genome Analysis Toolkit (Broad Institute, Cambridge, MA). The same bioinformatic analysis was applied to previously published solid metastases WES data.5 The primary tumors from the eight patients with LMD were not available for WES, but those patients had previously undergone a clinical genetic test for a panel of canonical cancer mutations. Positive mutations are shown in Supplementary Table 1. A cohort of 44 patients with LMD who had been treated at Stanford Hospital between 2007 and 2017 were selected for retrospective chart review (Supplementary Fig. 1 and Supplementary Table 2).
Results
EGFR and TP53 Are Recurrently Mutated in NSCLC LMD
To identify recurrently mutated genes in LMD across all coding regions, we performed WES on eight LMD cases to an average sequencing depth of 86× (Supplementary Fig. 2). Mutations in EGFR were found in six patients (75% [Fig. 2A and B]). Deletions between residues 746 and 750 were found in two patients (LM4 and LM8). A similar deletion between residues 747 and 752 was found in patient LM6. Patient LM2 was known to have an EGFR exon 20 insertion in the primary tumor, and this mutation was identified by WES of CSF DNA (see Fig. 2A and B). A sensitizing recurrent mutation L858R was found in patients LM1 and LM5. Notably, the somatic mutation in patient LM5 was initially misidentified by WES owing to the low mutant allele fraction (see Fig. 2A [red dot]). We carried out an orthogonal validation with droplet digital PCR (ddPCR) on the cellular DNA and cell-free DNA (cfDNA) from CSF, both of which showed evidence for L858R mutation (Supplementary Fig. 3A and B). This point mutation was also present in patient LM1, and the level of the mutant allele fraction in CSF cfDNA was detected to be 40% by ddPCR (Supplementary Fig. 3C), which was higher than that identified by WES of cellular DNA (15 mutant allele reads out of the total of 68 reads [see Fig. 2A]). All mutations were within the tyrosine kinase domain that regulates protein activation (see Fig. 2B).8 We then compared the LMD EGFR mutation with the results of the clinical mutation panel analyses done on the patients’ primary tumors. No de novo mutation was found, indicating that all the metastatic mutations were inherited from the primary tumors.
Figure 2.
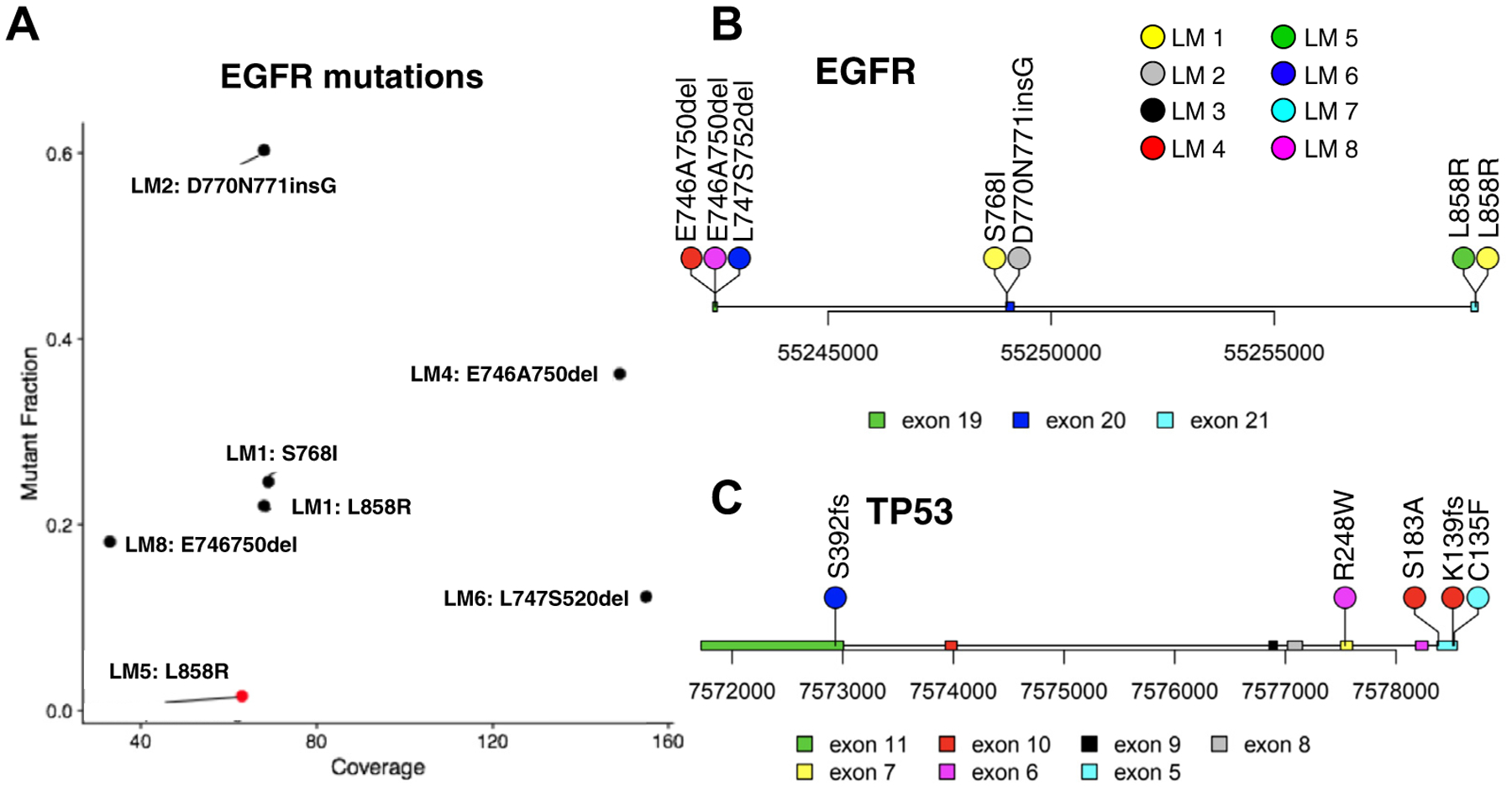
Common gene mutations in leptomeningeal disease identified by whole exome sequencing. (A) Mutation fraction and coverage of each EGFR mutation; LM5 was identified by droplet digital polymerase chain reaction (red). Mutation loci on the EGFR gene (B) and mutation loci on tumor protein p53 gene (TP53) (C). del, deletion; ins, insertion.
Tumor protein p53 gene (TP53) was mutated in four (50%) patients with LMD (Fig. 2C). Among all the TP53 mutations found in WES, only R248W was also covered in the clinical gene panel for primary tumors, and it was found in the primary tumor of patient LM8. The CSF cfDNA was also found to have this mutation by ddPCR (Supplementary Fig. 3D). As all the other TP53 mutations identified by WES were not in the clinical gene test, we were not able to determine whether they were de novo TP53 mutations during metastasis.
Additional NSCLC LMD Mutations
A missense mutation E545K in the proto-oncogene PIK3CA was found in patient LM8. The mutation was located within a highly conserved helical domain and has been shown to promote the catalytic activity of PIK3CA, resulting in enhanced downstream signaling.9 A missense mutation S140L in the proto-oncogene FGFR1 was found in patient LM7. A missense mutation H1180L in the receptor tyrosine kinase MET was found in patient LM5. Although we did not find any previous study on this mutation, it was highly conserved and predicted to be damaging by both Sorting Intolerant From Tolerant and Polymorphism Phenotyping v2 (Supplementary Fig. 4). Our data capture mutations overlooked by somatic cancer mutation panels. For instance, WES identified an S289I mutation in phosphodiesterase 4D interacting protein gene (PDE4DIP) for patient LM3, who did not have any other mutations in the clinical panels. A comprehensive list of all recurrent mutations that were called in LMD and solid brain metastasis patients is available in Supplementary Tables 1 and 2.
KRAS and EGFR Mutation Frequency Differs between LMD and Solid Metastases
We analyzed 26 publicly available NSCLC-derived solid brain metastases5 by using the same pipeline as used for our own LMD cohort. We compared the frequency of known recurrent mutations to identify shared and unique mutations across LMD and solid brain tumors. Mutations in TP53 were found in 50% of samples, making it a shared recurrent gene in both types. EGFR showed strong preference toward LMD cases (75% in LMD versus 3.8% in solid brain metastases [Fig. 3]). To confirm this distinction, we performed a retrospective chart review identifying 44 additional patients with NSCLC and LMD (see Supplementary Table 2). A total of 33 patients (63.5%) were positive for EGFR mutations, which is much higher than the mutation rate in solid brain tumors (Fisher’s exact test, p = 1.518e-07). A mutation in the KRAS was identified in 50% of solid brain metastases but not in any of the LMD cases (see Fig. 3), which was verified by sequencing coverage of KRAS gene (Supplementary Fig. 5). Additionally, through our retrospective case series, we found a remarkably low mutation rate in patients with lung cancer with LMD (four of 52), which is much lower than the mutation rate in solid brain metastasis (Fisher’s exact test, p = 4.85e-05). Likewise, given that the expected KRAS mutation frequency is around 30% in lung cancer,10 the lower frequency of KRAS mutations in LMD patients is notable.
Figure 3.
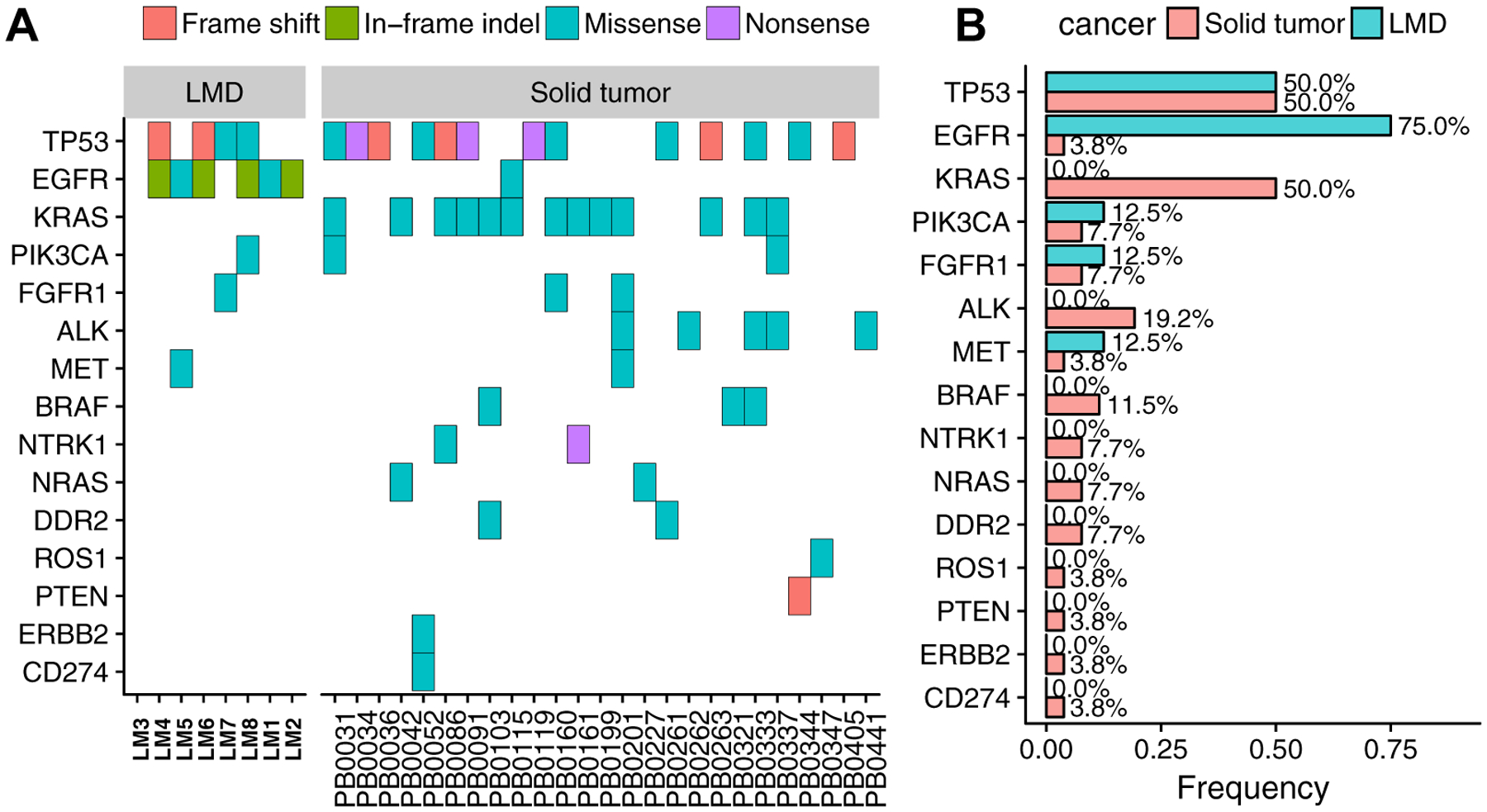
Canonical genes in leptomeningeal disease (LMD) and solid brain metastases. (A) Mutation types across all samples. (B) Mutation frequency in LMD and solid metastases. KRAS, Kirsten rat sarcoma viral oncogene homolog gene; PIK3CA, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha gene; FGFR1, fibroblast growth factor receptor 1 gene; ALK, ALK receptor tyrosine kinase gene; MET, MNNG HOS Transforming gene; NTRK1, neurotrophic receptor tyrosine kinase 1 gene; DDR2, discoidin domain receptor tyrosine kinase 2 gene; PTEN, phosphatase and tensin homolog erb-b2 receptor tyrosine kinase 2 gene; ERBB2, gene.
We then performed a Wilcoxon rank sum test to compare the time between diagnosis of the primary lung tumor and diagnosis of LMD. Patients with EGFR mutations had a median interval of 16.3 months compared with 23.9 months for patients with wild-type EGFR (p = 0.083 [Fig. 4A]). Kaplan-Meier curves on the interval between the diagnoses of primary lung tumor and LMD suggested that patients with EGFR mutation showed a faster progression toward LMD, although this result was not significant (p = 0.0863 [Fig. 4B]). This analysis is likely underpowered given the small numbers of patients in our cohort. It is known that patients with EGFR mutations benefit from tyrosine kinase inhibitor (TKI) treatment as first-line therapy.11 To correct for these potential confounding effects of differences in treatment, we used a Cox proportional hazard model with sex, race, age at primary diagnosis, KPS score, smoking history, and treatment (immunotherapy, brain radiation, targeted therapy, and TKIs) as covariates. EGFR mutation significantly increased the hazard (p = 0.017), whereas targeted therapy significantly decreased the hazard (p = 0.035).
Figure 4.
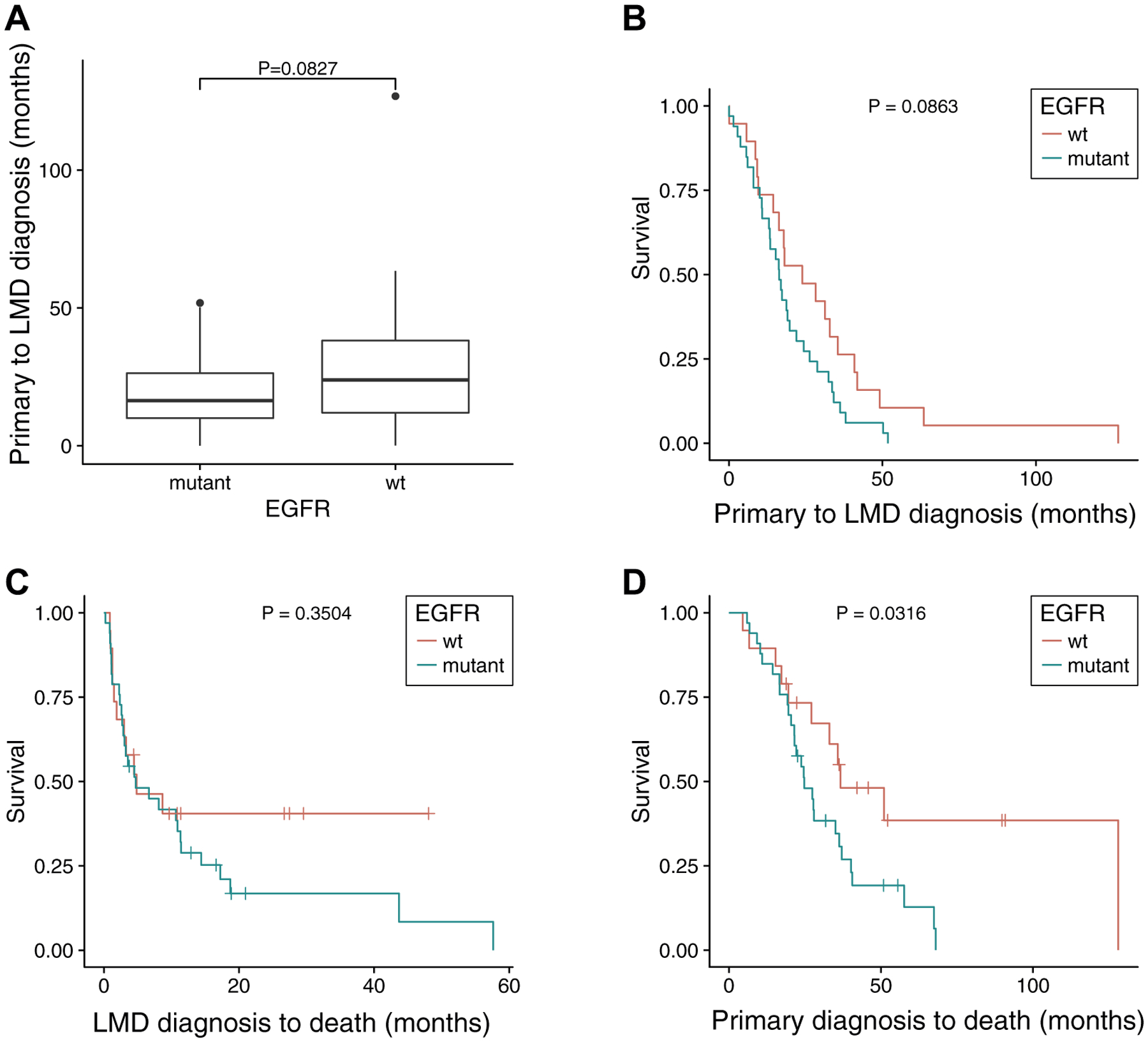
Correlation of EGFR mutation and progression of leptomeningeal disease (LMD). (A) Wilcoxon rank sum test of the time interval from diagnosis of the primary cancer to diagnosis of LMD (B) Kaplan-Meier plot of the interval from diagnosis of the primary cancer to diagnosis of LMD. (C) Patients’ survival from the point of diagnosis of LMD. (D) Patients’ survival from the point of diagnosis of the primary cancer. wt, wild type.
Discussion
The prominence of EGFR mutations and lack of KRAS mutations in LMD suggest intrinsic differences in tumor biology that allow these metastatic cells to thrive in distinct brain niches, or selection by progressive systemic treatments for subpopulations of cells from the primary tumors that are particular to a brain niche. One hypothesis was that patients with EGFR mutations treated with TKIs survived longer12 and thus had more time for development of LMD, whereas patients with KRAS mutations were not responsive to TKIs and typically had a worse prognosis.10 Data from our LMD cohort did not support this hypothesis, however, as patients with EGFR mutations either had similar survival time from the point of LMD diagnosis (Fig. 4C) or had shorter survival time from primary cancer diagnosis to death (Fig. 4D).
LMD developed faster in patients with EGFR mutation than in patients with wild-type EGFR (see Fig. 4B). EGFR mutations have been previously shown to occur early in NSCLC pathogenesis and to increase in frequency through cancer progression to advanced metastasis.13,14 Although the benefit of targeted therapeutics on overall survival is well established, its effect on the development of LMD and prognosis is still under investigation.15 One consideration is the relative lack of permeability to the blood-brain and blood-tumor barriers, which may allow for a haven for tumor cells in the leptomeninges. EGFR-mutated NSCLCs may metastasize within this protective space before treatment.
Our analysis also found several other genes not listed as recurrent genes in the current literature. For instance, taste 2 receptor member 31 gene (TAS2R31) was mutated in four of eight LMD samples (50%), and PDE4DIP was mutated in three LMD samples (37.5%). The significance of these mutations is difficult to ascertain, as they are not well described in the literature. TAS2R31 (3.8%) and PDE4DIP (19.2%) mutations were also found in the cohort with solid brain metastasis, albeit at lower frequency.
The paucity of data on LMD and its rare occurrence make recurrent mutation discovery challenging and difficult to interpret. We are committed to facilitating the sharing of our sequencing data with the greater scientific community, and we have deposited these results in a readily accessible website (www.LMDseq.org).
Supplementary Material
Acknowledgments
We acknowledge the generous donation of clinical samples by our patients, without whom this research would not have been possible and to whom we dedicate our work. Funding to Dr. Gephart included grants R21CA193046-01 and K08NS901527, and funding from the Hearst and Curci Foundations. Dr. Li is supported in part by the McCormick-Gabilan Award and a Stanford Child Health Research Institute fellowship. Mr. Liu is supported by a Stanford Center for Computational, Evolutionary, and Human Genetics fellowship and the National Key R&D Program of China (grant 2016YFD0400800). Mr. Connolly is supported by the Stanford Medscholars Grant. We thank Norma Neff and Gary Mantalas for sequencing support, and Tej Azad for providing two clinical samples.
Footnotes
Supplementary Data
Note: To access the supplementary material accompanying this article, visit the online version of the Journal of Thoracic Oncology at www.jto.org and at https://doi.org/10.1016/j.jtho.2018.03.018.
Disclosure: The authors declare no conflict of interest.
References
- 1.Steeg PS, Camphausen KA, Smith QR. Brain metastases as preventive and therapeutic targets. Nat Rev Cancer. 2011;11:352–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nagpal S, Riess J, Wakelee H. Treatment of leptomeningeal spread of NSCLC: a continuing challenge. Curr Treat Options Oncol. 2012;13:491–504. [DOI] [PubMed] [Google Scholar]
- 3.Ding L, Ellis MJ, Li S, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464:999–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Navin N, Kendall J, Troge J, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brastianos PK, Carter SL, Santagata S, et al. Genomic characterization of brain metastases reveals branched evolution and potential therapeutic targets. Cancer Discov. 2015;5:1164–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paik PK, Shen R, Won H, et al. Next-generation sequencing of stage IV squamous cell lung cancers reveals an association of PI3K aberrations and evidence of clonal heterogeneity in patients with brain metastases. Cancer Discov. 2016;5:610–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bos PD, Zhang XH-F, Nadal C, et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459:1005–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem. 2002;277:46265–46272. [DOI] [PubMed] [Google Scholar]
- 9.Kang S, Bader AG, Vogt PK. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci U S A. 2005;102:802–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marks JL, Broderick S, Zhou Q, et al. Prognostic and therapeutic implications of EGFR and KRAS mutations in resected lung adenocarcinoma. J Thorac Oncol. 2008;3:111–116. [DOI] [PubMed] [Google Scholar]
- 11.Li YS, Jiang BY, Yang JJ, Tu HY, Zhou Q, Guo WB, et al. Leptomeningeal metastases in patients with NSCLC with EGFR mutations. J Thorac Oncol. 2016;11: 1962–1969. [DOI] [PubMed] [Google Scholar]
- 12.Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. [DOI] [PubMed] [Google Scholar]
- 13.Tang X, Shigematsu H, Bekele BN, et al. EGFR tyrosine kinase domain mutations are detected in histologically normal respiratory epithelium in lung cancer patients. Cancer Res. 2005;65:7568–7572. [DOI] [PubMed] [Google Scholar]
- 14.Matsumoto S, Takahashi K, Iwakawa R, et al. Frequent EGFR mutations in brain metastases of lung adenocarcinoma. Int J Cancer. 2006;119:1491–1494. [DOI] [PubMed] [Google Scholar]
- 15.Lee SJ, Lee JI, Nam DH, et al. Leptomeningeal carcinomatosis in non-small-cell lung cancer patients: impact on survival and correlated prognostic factors. J Thorac Oncol. 2013;8:185–191. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.