

Introduction
Chimeric oncogenic fusion genes can arise because of somatic structural events including chromosomal translocation, amplification, deletion, or inversions.1 A milestone in oncology research was the discovery of Philadelphia chromosome (Ph) in chronic myeloid leukemia with the identification of the oncogenic fusion kinase, breakpoint cluster region protein-ABL1 (BCR-ABL1), as a major tumorigenic factor of this disease and an important prognostic biomarker.2,3 Imatinib, a tyrosine kinase inhibitor (TKI), is a US Food and Drug Administration–approved targeted therapy for Ph-positive leukemia.4 Importantly, BCR-ABL1 is one of the most reported fusion genes related to hematologic malignancies; however, no study has reported on nonhematologic solid tumors.5
Glioblastomas are the most common primary malignant brain tumors. They are incurable largely because of their heterogeneity and highly dynamic genomic landscape.6 Therefore, identifying druggable oncogenic targets is critically important. Oncogenic fusion transcript is one of the most promising targets for glioblastomas, and up to 30%-50% of glioblastomas carry fusion transcripts, some of which are druggable, including neurotrophic tropomyosin receptor kinase (NTRK) fusions.7-9 In addition, the KIAA1549-Serine/threonine-protein kinase B-raf (BRAF) fusion that commonly occurs in the pilocytic astrocytoma, a low-grade glioma, has also been reported in glioblastoma and can be targeted using BRAF inhibitors.10 This study reports an unexpected finding of the actionable classical e1a2 BCR-ABL1 fusion transcript in a glioblastoma.
Case Report
The patient, a 61-year-old woman, was referred to the National Institutes of Health for consultation after a right frontal lobe glioblastoma resection, concomitant chemoradiation, and one cycle of adjuvant temozolomide treatment. During the initial visit, a nonenhancing lesion in the right temporal lobe was noted (Fig 1A). Because of the concern for a malignant process of the lesion, a pathologic diagnosis was recommended. A gross total resection of the temporal lobe lesion was subsequently performed. The pathologic diagnosis was consistent with high-grade astrocytoma (Fig 1B). The patient's blood and tumor samples were submitted for whole-exome sequencing (WES) and whole transcriptome analysis (RNA-seq). The tumor was found to harbor a wild-type isocitrate dehydrogenase (IDH) gene, but genomic alterations including phosphatase and tensin homolog (PTEN) deletion, telomerase reverse transcriptase (TERT) promotor mutation (c.-146C>T), and chromosome 10 loss, which are consistent with the molecular features of de novo glioblastoma.11,12 In addition, methylation profiling of the primary tumor sample (sample number V053) suggested that it was an IDH-wild-type glioblastoma of mesenchymal subclass (class assignment confidence calibrated score = 0.995; Fig 1C). The RNA-seq analysis revealed a classical BCR-ABL1 fusion, which has never been reported in glioblastoma. Sanger sequencing of the RT-PCR fusion amplicon validated the fusion transcript as an in-frame fusion with an intact tyrosine kinase domain on ABL1. The breakpoint was after exon 1 of BCR, resulting in exon 1 of BCR being juxtaposed to exon 2 of ABL1, coding for the classical e1a2 BCR-ABL1 fusion that usually occurs in acute lymphoblastic leukemia13 (Figs 1D and 1E). To rule out the possibility of hematologic malignancy, we performed both RT-PCR and RNA-seq assays on patient's blood RNA. BCR-ABL1 fusion transcript was not detected. The absence of BCR-ABL1 fusion transcript in the blood sample is consistent with the lack of clinical or laboratory evidence of hematologic malignancy.
FIG 1.
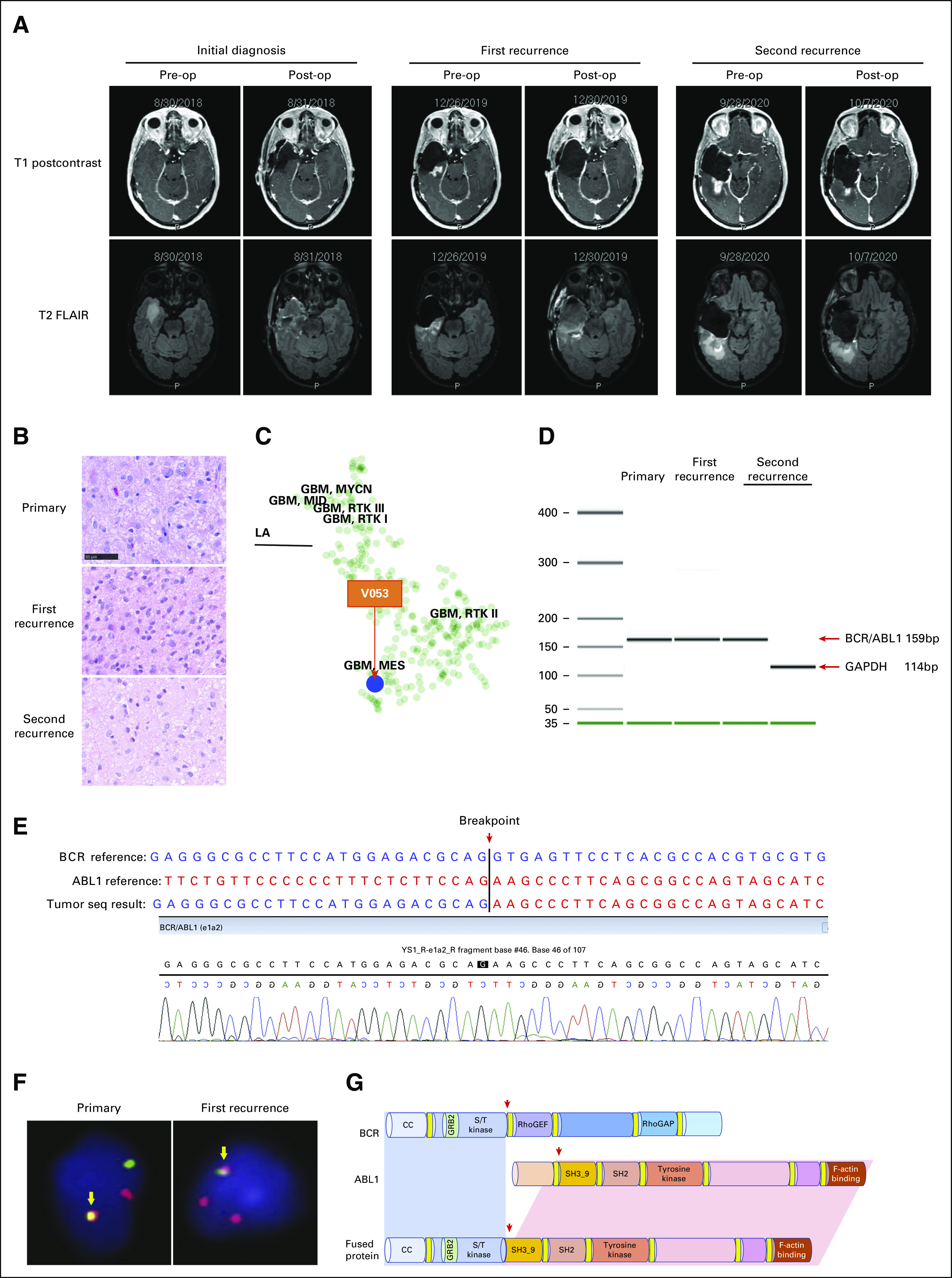
Case presentation: (A) serial brain MRIs of the patient with T1 postcontrast (upper panel) and T2 FLAIR (lower panel) images showing the right temporal lobe lesion before and after the surgical resections at the time of initial diagnosis, disease recurrent, and postimatinib treatment; (B) hematoxylin and eosin staining of tumor samples collected at initial diagnosis and subsequent recurrences; bar = 50 μm; imatinib was given after the first recurrence; (C) a t-distributed stochastic neighbor embedding plot showing clustering of the BCR-ABL1 fusion primary tumor sample with main glioblastoma cluster (V053, red arrow); (D) agarose gel separation of the BCR-ABL1 fusion–specific RT-PCR product from the patient's primary tumor, recurrent tumor, and blood samples; GAPDH was amplified as the positive control; (E) Sanger sequencing of BCR-ABL1 fusion–specific RT-PCR amplicons; (F) FISH assay showing BCR-ABL1 translocation in patient's primary and recurrent tumor samples (arrow); and (G) schematic representation of the BCR-ABL1 fusion transcript by detection. FISH, fluorescence in situ hybridization; FLAIR, fluid attenuated inversion recovery; GBM, glioblastoma; MRI, magnetic resonance imaging; RT-PCR, reverse transcription polymerase chain reaction; T1, T1-weighted image; T2, T2-weighted image.
Subsequently, the patient received concurrent chemoradiation therapy with temozolomide followed by six cycles of temozolomide after tumor resection. Several months after the completion of adjuvant temozolomide, an magnetic resonance imaging brain examination revealed a fast-growing enhancing lesion around the resection cavity to the right temporal lobe, suggesting tumor progression (Fig 1A). A second surgical resection was performed, confirming the recurrence of the tumor (Fig 1B). Methylation profiling of the recurrent tumor sample suggested it to be glioblastoma (sample number W879 and calibrated scores: 0.842), IDH-wild-type, subclass mesenchymal (Appendix Fig A1), which was similar to the primary tumor (sample number V053). BCR-ABL1 fusion was also detected from this recurrent tumor sample by RNA-seq and fluorescence in situ hybridization analysis (Figs 1D-1G), suggesting that the fusion gene might have contributed to the rapid disease progression. Given the well-known safety profile of imatinib, an US Food and Drug Administration–approved drug for BCR-ABL–positive leukemia, therapy with this drug was offered to the patient after a short course of radiation therapy. The patient was able to tolerate 400 mg once a day imatinib, and her condition remained stable for several months. Ten months after the surgical resection, a follow-up magnetic resonance imaging revealed a new enhancing lesion posterior to the resection cavity (Fig 1A, second recurrence). Since the new enhancing lesion area was in an area of overlap of the three previous radiation treatments, radiation necrosis was highly suspected. Following surgical resection, the pathologic examination revealed a hypercellular parenchyma with atypical glial cells, histiocytes, and gliosis. In addition, the actively proliferating cells, indicated by Ki-67 staining, decreased from 40% in the first recurrent tumor before imatinib treatment to only 6%-8% in the second recurrent tumors after imatinib treatment (Fig 1B, middle and lower panels), suggesting that the imatinib treatment dampened the rate of tumor progression. The WES of the second recurrent tumor sample post-treatment of imatinib did not reveal any imatinib-resistant mutation (at 150× sequencing coverage) within the kinase domain. Therefore, imatinib treatment was planned to be restarted after this third surgical resection. However, the patient was found to have disease relapse from a previous diagnosis of breast cancer during the perioperative time. The timely initiation of imatinib was complicated by the extensive diagnostic or staging tests and treatment for breast cancer. Unfortunately, patient's overall condition rapidly declined after < 1 month of imatinib treatment and the patient decided on hospice.
The patient gave informed consent to present information and/or images from the patient.
Discussion
To our knowledge, this study reports the first case of glioblastoma with the classical hematologic BCR-ABL1 fusion. The canonical fusion was confirmed in multiple tumor samples from the initial diagnosis and the two subsequent recurrences.
BCR-ABL1 tyrosine kinase fusion is caused by the reciprocal translocation t(9;22). On the basis of alternative breakpoints on the BCR gene, different BCR-ABL1 fusion proteins are formed, containing the ABL1 protein kinase domain. Since the presence of the BCR-ABL1 might have contributed to glioma pathogenesis in this patient, we prescribed imatinib, which is a first-generation inhibitor of BCR-ABL1 shown to significantly increase the overall survival of patients with chronic myeloid leukemia carrying this kinase fusion.14,15 Although imatinib is used to treat Ph+ leukemia, it has marginal effects in preventing or treating CNS leukemia when the blood-brain barrier (BBB) is intact.16 However, in the case of glioblastoma, the BBB within the tumor is usually significantly disrupted and the intratumoral concentration of imatinib can be comparable with the level in plasma.17,18 Therefore, we postulated that therapy with imatinib may be still beneficial. Compared with imatinib, second generation of TKIs (dasatinib and nilotinib) has improved BBB penetration and increased survival benefit.19,20 Unfortunately, the second generation of TKI was not available to this patient. As a second option for a TKI, imatinib was offered. Furthermore, one third of patients have developed resistance to imatinib, mostly because of point mutations within the BCR-ABL1 kinase domain, including T315I, Y253F/H, E255K/V, M351T, G250E, F359C/V, H396R/P, M244V, E355G, F317L, M237I, Q252H/R, D276G, L248V, and F486S. This has led to the development of second- and third-generation inhibitors to overcome the resistance.21 In our study, we did not find evidence of any BCR-ABL1 resistance mutation in the tumors by WES at a sequencing depth of 150× coverage (data not shown). Despite the aggressiveness of disease, a significantly lower proliferation index of tumor cells following radiation and imatinib treatment suggested a potential role of imatinib in control of tumor growth. Unfortunately, the evaluation of the imatinib response after the third tumor resection was challenging because of the complicated disease course. Although imatinib demonstrated some antiglioma activities in this patient, an alternative would have been the use of a second-generation TKI with a better BBB penetration. Despite the detection of BCR-ABL1 fusion in the tumor sample from the initial diagnosis, the patient was initially not offered imatinib because of the unavailability of the drug. However, the patient was able to receive imatinib after the second surgical resection. Although the potential antitumor effect of imatinib is suggested in this case report, the definitive treatment response to TKIs in glioblastoma with BCR-ABL fusion needs to be tested in a larger patient population. Although BCR-ABL1 fusion has never been reported in glioblastomas, it may be more common than appreciated in this patient population. However, a search of the literature and The Cancer Genome Atlas Program data from brain tumors failed to show evidence of the presence of BCR-ABL1.
In conclusion, our case study shows that BCR-ABL1 fusion should be considered as an albeit rare but druggable event in GBM. Thus, our report describes that the first BCR-ABL1 fusion gene in glioblastoma led to a targeted therapeutic option. The potential treatment response that resulted from targeting the BCR-ABL1 fusion oncoprotein underscores the importance of identifying oncogenic fusion genes, especially those that harbor intact druggable kinase domains. Importantly, we recommend BCR-ABL1 fusion be considered as an additional biomarker for clinical molecular testing panels to be further investigated in a larger cohort of patients with glioma.
ACKNOWLEDGMENT
We thank the patient for participating in the study and for her courage in fighting the disease.
Appendix
FIG A1.

A t-distributed stochastic neighbor embedding plot showing clustering of the BCR-ABL1 fusion in the first recurrent tumor sample with main glioblastoma cluster (W879, red arrow). GBM, glioblastoma.
Rosandra Kaplan
Research Funding: Lilly
Patents, Royalties, Other Intellectual Property: I have a patent with NCI
Kevin Camphausen
Employment: Fairfax Radiological Consultants (I)
Open Payments Link: https://openpaymentsdata.cms.gov/physician/535470
Matthias Holdhoff
Consulting or Advisory Role: Celgene, AbbVie, BTG, Newlink Genetics, DPClinical
Travel, Accommodations, Expenses: Arbor Pharmaceuticals
Javed Khan
Research Funding: Lentigen, Taiho Oncology
Patents, Royalties, Other Intellectual Property: Monoclonal antibody–based therapeutics targeting fibroblast growth factor receptor 4 (FGFR4) for potential treatment of human cancers expressing FGFR4
No other potential conflicts of interest were reported.
SUPPORT
Supported by the NIH Intramural Research Program and Lasker Clinical Research Scholars Program.
AUTHOR CONTRIBUTIONS
Conception and design: Ying Pan, Javed Khan, Jing Wu
Provision of study materials or patients: Ramya Antony, Lisa Boris, Orwa Aboud, David Kamson, Megan Mackey, Kevin Camphausen, Kareem Zaghloul, Terri S. Armstrong, Mark R. Gilbert, Kenneth Aldape, Matthias Holdhoff, Jing Wu
Collection and assembly of data: Ying Pang, Madison Butler, Sivasish Sindiri, Young K. Song, Jun S. Wei, Xinyu Wen, Hisen-Chao Chou, Martha Quezado, Svetlana Pack, Liqiang Xi, Zied Abdullaev, Ramya Antony, Lisa Boris, Orwa Aboud, David Kamson, Rosandra Kaplan, Megan Mackey, Kareem Zaghloul, Matthias Holdhoff, Javed Khan, Jing Wu
Data analysis and interpretation: Ying Pang, Guangyang Yu, Madison Butler, Sivasish Sindiri, Jun S. Wei, Xinyu Wen, Hisen-Chao Chou, Martha Quezado, Svetlana Pack, Olga Kim, Alice Ranjan, Mythili Merchant, Kevin Camphausen, Mark R. Gilbert, Kenneth Aldape, Matthias Holdhoff, Javed Khan, Jing Wu
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Rosandra Kaplan
Research Funding: Lilly
Patents, Royalties, Other Intellectual Property: I have a patent with NCI
Kevin Camphausen
Employment: Fairfax Radiological Consultants (I)
Open Payments Link: https://openpaymentsdata.cms.gov/physician/535470
Matthias Holdhoff
Consulting or Advisory Role: Celgene, AbbVie, BTG, Newlink Genetics, DPClinical
Travel, Accommodations, Expenses: Arbor Pharmaceuticals
Javed Khan
Research Funding: Lentigen, Taiho Oncology
Patents, Royalties, Other Intellectual Property: Monoclonal antibody–based therapeutics targeting fibroblast growth factor receptor 4 (FGFR4) for potential treatment of human cancers expressing FGFR4
No other potential conflicts of interest were reported.
REFERENCES
- 1.Dai X, Theobard R, Cheng H, et al. Fusion genes: A promising tool combating against cancer Biochim Biophys Acta Rev Cancer 1869149–1602018 [DOI] [PubMed] [Google Scholar]
- 2.Nowell PC.The minute chromosome (Phl) in chronic granulocytic leukemia Blut 865–661962 [DOI] [PubMed] [Google Scholar]
- 3.Heisterkamp N, Stephenson JR, Groffen J, et al. Localization of the c-ab1 oncogene adjacent to a translocation break point in chronic myelocytic leukaemia Nature 306239–2421983 [DOI] [PubMed] [Google Scholar]
- 4.Flynn JP, Gerriets V. Imatinib. Treasure Island, FL: StatPearls; 2020. [Google Scholar]
- 5.Price CM, Rassool F, Shivji MK, et al. Rearrangement of the breakpoint cluster region and expression of P210 BCR-ABL in a "masked" Philadelphia chromosome-positive acute myeloid leukemia Blood 721829–18321988 [PubMed] [Google Scholar]
- 6.Ostrom QT, Cioffi G, Gittleman H, et al. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2012-2016 Neuro Oncol 21v1–v1002019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim J, Lee Y, Cho HJ, et al. NTRK1 fusion in glioblastoma multiforme. PLoS One. 2014;9:e91940. doi: 10.1371/journal.pone.0091940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drilon A, Siena S, Ou SI, et al. Safety and antitumor activity of the multitargeted Pan-TRK, ROS1, and ALK inhibitor entrectinib: Combined results from two phase I trials (ALKA-372-001 and STARTRK-1) Cancer Discov 7400–4092017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shah N, Lankerovich M, Lee H, et al. Exploration of the gene fusion landscape of glioblastoma using transcriptome sequencing and copy number data. BMC Genomics. 2013;14:818. doi: 10.1186/1471-2164-14-818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Antonelli M, Badiali M, Moi L, et al. KIAA1549:BRAF fusion gene in pediatric brain tumors of various histogenesis Pediatr Blood Cancer 62724–7272015 [DOI] [PubMed] [Google Scholar]
- 11.Rogers TW, Toor G, Drummond K, et al. The 2016 revision of the WHO classification of central nervous system tumours: Retrospective application to a cohort of diffuse gliomas J Neurooncol 137181–1892018 [DOI] [PubMed] [Google Scholar]
- 12.Olar A, Aldape KD.Using the molecular classification of glioblastoma to inform personalized treatment J Pathol 232165–1772014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan LC, Karhi KK, Rayter SI, et al. A novel abl protein expressed in Philadelphia chromosome positive acute lymphoblastic leukaemia Nature 325635–6371987 [DOI] [PubMed] [Google Scholar]
- 14.Druker BJ, Tamura S, Buchdunger E, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells Nat Med 2561–5661996 [DOI] [PubMed] [Google Scholar]
- 15.O'Brien SG, Guilhot F, Larson RA, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia N Engl J Med 348994–10042003 [DOI] [PubMed] [Google Scholar]
- 16.Senior K. Gleevec does not cross blood-brain barrier. Lancet Oncol. 2003;4:198. doi: 10.1016/s1470-2045(03)01050-7. [DOI] [PubMed] [Google Scholar]
- 17.Sarkaria JN, Hu LS, Parney IF, et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data Neuro Oncol 20184–1912018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holdhoff M, Supko JG, Gallia GL, et al. Intratumoral concentrations of imatinib after oral administration in patients with glioblastoma multiforme J Neurooncol 97241–2452010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Porkka K, Koskenvesa P, Lundan T, et al. Dasatinib crosses the blood-brain barrier and is an efficient therapy for central nervous system Philadelphia chromosome-positive leukemia Blood 1121005–10122008 [DOI] [PubMed] [Google Scholar]
- 20.Pagan FL, Hebron ML, Wilmarth B, et al. Pharmacokinetics and pharmacodynamics of a single dose Nilotinib in individuals with Parkinson's disease. Pharmacol Res Perspect. 2019;7:e00470. doi: 10.1002/prp2.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Apperley JF.Part I: Mechanisms of resistance to imatinib in chronic myeloid leukaemia Lancet Oncol 81018–10292007 [DOI] [PubMed] [Google Scholar]