

Abstract
As one of the largest families of angiosperms, the Orchidaceae family is diverse. Dendrobium represents the second largest genus of the Orchidaceae. However, an assembled high-quality genome of species in this genus is lacking. Here, we report a chromosome-scale reference genome of Dendrobium chrysotoxum, an important ornamental and medicinal orchid species. The assembled genome size of D. chrysotoxum was 1.37 Gb, with a contig N50 value of 1.54 Mb. Of the sequences, 95.75% were anchored to 19 pseudochromosomes. There were 30,044 genes predicted in the D. chrysotoxum genome. Two whole-genome polyploidization events occurred in D. chrysotoxum. In terms of the second event, whole-genome duplication (WGD) was also found to have occurred in other Orchidaceae members, which diverged mainly via gene loss immediately after the WGD event occurred; the first duplication was found to have occurred in most monocots (tau event). We identified sugar transporter (SWEET) gene family expansion, which might be related to the abundant medicinal compounds and fleshy stems of D. chrysotoxum. MADS-box genes were identified in D. chrysotoxum, as well as members of TPS and Hsp90 gene families, which are associated with resistance, which may contribute to the adaptive evolution of orchids. We also investigated the interplay among carotenoid, ABA, and ethylene biosynthesis in D. chrysotoxum to elucidate the regulatory mechanisms of the short flowering period of orchids with yellow flowers. The reference D. chrysotoxum genome will provide important insights for further research on medicinal active ingredients and breeding and enhances the understanding of orchid evolution.
Subject terms: Genome, Plant molecular biology, Gene regulation
Introduction
With more than 25,000 species, Orchidaceae is the largest angiosperm family1 and comprises 8–10% of flowering plants. Orchids are renowned for their specialized flowers, which have a very wide variety of growth forms, and have been successful colonizers of a wide variety of different habitats2. As one of the largest genera of Orchidaceae, Dendrobium encompasses ~1450 species with fleshy stems3. Many species of Dendrobium have high medicinal and commercial value, and the main medicinal active ingredients are in the stems4–9. Therefore, studying the molecular mechanism of these active ingredients and breeding cultivars with increased contents of natural products are the main objectives in Dendrobium scientific research and industrialization10.
Guchui Shihu (鼓槌石斛) Dendrobium chrysotoxum, a medicinal species, is listed in the Chinese Pharmacopoeia (2020, 2015, and 2010 edition) and contains an abundance of erianin, gigantol, polysaccharides, and fluorenones, among other compounds11–19 (Fig. 1). These compounds show antipyretic, analgesic, antihyperglycemic, and antioxidant effects and enhance immune function11–19. Recently, preliminary clinical study results suggested that gigantol could delay lens turbidity through the inhibition of aldose reductase and aldose reductase mRNA expression, which have good effects on diabetic cataracts16,17. Erianin has been demonstrated to exhibit metabolic inhibition20 and antitumor21, antiproliferative22, and antiangiogenic activity23. Moreover, it also inhibits high glucose-induced retinal angiogenesis12. Polysaccharides isolated from D. chrysotoxum have potential utility in enhancing antioxidation, immune function, and/or hypoglycemic activity11. The stems of D. chrysotoxum are fusiform and rich in active medicinal substances, which makes it a suitable species for scientific research and industrial applications in Dendrobium.
Fig. 1. A flowering D. chrysotoxum plant.

D. chrysotoxum is lithophytic on rocks or epiphytic on tree trunks with bright yellow flowers and fusiform, fleshy stems
With the improvement of sequencing technology and cost reduction, genome sequencing has become a necessary method for obtaining comprehensive genetic information and an effective method for screening candidate genes for specific traits, especially for identifying candidate genes involved in the biosynthesis pathways of medicinal compounds24–28. To date, only two Dendrobium spp. genomes have been sequenced, and some candidate genes involved in polysaccharide metabolic pathways have been identified in those two species24,29,30. However, these studies were largely limited due to their low-quality genome assemblies. Therefore, high-quality reference genomes and additional Dendrobium species need to be sequenced to better understand the molecular mechanisms underlying the production of medicinal compounds and enable the breeding of new varieties.
In this study, we used PacBio sequencing and Hi-C technology to generate a chromosome-level genome assembly. The specific genes of D. chrysotoxum were identified, which lays a foundation for further research on the functions of medicinal active ingredients, provides a reference for breeding new varieties and enhances the understanding of orchid evolution.
Results and discussion
Genome sequencing and characteristics
D. chrysotoxum has a karyotype of 2N = 2X = 38, with uniform chromosomes31. To completely sequence the D. chrysotoxum genome, 138.15 Gb of clean reads were generated by BGISEQ sequencing system (Supplementary Table 1). The estimated genome size was 1.38 Gb with 1.84% heterozygosity, as determined by K-mer analysis (Supplementary Fig. 1). To obtain a better assembly, PacBio technology was employed, and 132.64 Gb of PacBio sequencing data were generated (Supplementary Table 1). The assembly size was 1.37 Gb with a corresponding contig N50 value of 1.54 Mb (Supplementary Table 2). The BUSCO32 assessment indicated that the completeness of the gene set of the assembled genome was 90.3% (Supplementary Table 3). This indicates that the D. chrysotoxum genome assembly was complete and could be used for subsequent analysis. We further used 125.96 Gb of reads from the Hi-C library. The assembled scaffolds were ultimately clustered into 19 pseudomolecules, which represented the 19 chromosomes in the haploid genome of D. chrysotoxum (Fig. 2a). The lengths of the 19 pseudochromosomes ranged from 38.28 to 100.49 Mb with a scaffold N50 value of 67.80 Mb (Supplementary Tables 4 and 5). In addition, contigs with a length of 1.31 Gb were mapped onto the 19 pseudochromosomes at a 95.75% anchor rate (Supplementary Tables 4 and 5). The chromatin interaction data suggest that our Hi-C assembly is of high quality (Fig. 2b). Compared with those of other orchid genome assemblies, the contig N50 and scaffold N50 values of the D. chrysotoxum genome were much higher (Table 1), and the assembly completeness was higher than 90% (Table 1), suggesting high genome quality and completeness.
Fig. 2. Chromosomal features and intensity signal heat map of D. chrysotoxum chromosomes according to Hi-C output.

a From inside outward: chromosome (purple), gene density (red), DNA type repeat sequence density (green), copy density (blue), and gypsy density (orange). All the data are shown with sliding windows of 500 kb, and the inner lines (green indicates the positive direction, and red indicates the opposite direction) represent syntenic blocks on homologous chromosomes. b Heat map of the intensity of the Hi-C chromosome. The heat map represents the contact matrices generated by aligning the Hi-C data to the chromosome-scale assembly of the D. chrysotoxum genome. A higher value on the scale bar indicates a higher contact frequency
Table 1.
Genome statistics and comparisons among orchid species whose genome has been sequenced
| Species | Gene number | Contig N50 (bp) | Scaffold N50 (bp) | BUSCO assembly (%) | CEGMA assembly (%) |
|---|---|---|---|---|---|
| D. chrysotoxuma | 30044 | 1,540,953 | 67,798,029 | 90.30 | – |
| D. catenatum2 | 28910 | 51,736 | 1,055,340 | 92.46 | – |
| P. equestris33 | 29431 | 45,791 | 1,217,477 | 91.00 | – |
| A. shenzhenica2 | 21841 | 80,069 | 3,029,156 | 93.62 | – |
| D. officinale29 | 35567 | 25,122 | 76,489 | - | 91.50 |
aThis study
Gene prediction and annotation
In D. chrysotoxum genome, 30,044 protein-coding genes were annotated (see Materials and methods; Supplementary Table 6). The completeness of the genome was 95.64%, indicating that the D. chrysotoxum genome annotation was relatively complete (Supplementary Table 7).
In addition to a high number of genes, the average length of genes and introns was also larger in D. chrysotoxum than in Phalaenopsis equestris, Gastrodia elata, and D. catenatum24,33,34 and much higher than that in most other angiosperms (Supplementary Table 8). The average length of the coding DNA sequences (CDSs) in D. chrysotoxum was longer than those in other angiosperms, and a greater average intron length was also previously observed for P. equestris, G. elata, and D. catenatum24,33,34; thus, a relatively long CDS might be a unique characteristic of Orchidaceae (Supplementary Fig. 2; Supplementary Table 8). Regulatory elements are frequently present in introns, and alternative splicing events often occur among different introns and exons, diversifying the protein-coding aspect of the genome. All these factors might contribute to genome structure evolution, genome size, gene function diversification, and gene expression patterns35–38. For example, intron transcriptional delay in Drosophila is particularly important for proper development of the embryo39,40. Thus, this characteristic of orchids needs to be further analyzed and researched. Moreover, 80 microRNAs, 1281 transfer RNAs, 2275 ribosomal RNAs, and 882 small nuclear RNAs were identified in the D. chrysotoxum genome (Supplementary Table 9).
We estimated that the D. chrysotoxum genome comprised 62.81% repetitive sequences (Supplementary Figs. 3 and 4; Supplementary Table 10), the percentage of which was higher than 62% in P. equestris but lower than 78.1% in D. catenatum24,33. Transposable elements (TEs) are important forms of repeats and constitute a substantial part of the D. chrysotoxum genome (61.22%); TEs are the most abundant repeat subtypes in this species. In addition, repeats predicted de novo were much larger than those obtained based on Repbase11 database, suggesting that, compared with other plants species whose genome has been sequenced, D. chrysotoxum has many specific repeats (Supplementary Table 10). Long terminal repeats (LTRs) represented the highest proportion among all subtypes of repeats, accounting for ~53.15% of the genome, which was higher than the 46% for D. catenatum24 (Supplementary Table 11).
In addition, 27,575 (91.78%) predicted genes were functionally annotated (Supplementary Table 12). Among them, 27,268 (90.76%) and 26,808 (89.23%) genes were annotated to the TrEMBL and Nr databases, respectively (Supplementary Table 12). The numbers of annotated genes were 22,735 (75.67%), 19,185 (63.86%), and 18,666 (62.13%) in the InterPro, SwissProt, and KEGG databases, respectively (Supplementary Table 12).
Evolution of gene families
A high-confidence phylogenetic tree was constructed, and the divergence times were estimated based on 274 single-copy genes from 17 different plant species (Supplementary Fig. 5 and Supplementary Table 6). As expected, D. chrysotoxum was sister to D. catenatum, forming an Epidendroideae clade together with P. equestris, G. elata, and A. shenzhenica located at the bases of Orchidaceae branches (Supplementary Fig. 6). The Orchidaceae divergence was estimated to have occurred 123 Mya; the divergence between subfamily Apostasioideae and subfamily Epidendroideae occurred 80 Mya; the divergence between D. chrysotoxum and D. catenatum occurred 11 Mya; and the divergence between Dendrobium and Phalaenopsis occurred 38 Mya (Fig. 3). Then, the expansion and contraction of orthologous gene families were analyzed. According to the results, 140 and 1112 gene families expanded and contracted, respectively, in the lineage leading to Orchidaceae. In D. chrysotoxum, 953 gene families were expanded, as opposed to 783 in D. catenatum, 853 in P. equestris, 358 in G. elata, and 562 in A. shenzhenica. At the same time, 1335 gene families were contracted in D. chrysotoxum, as opposed to 644 in D. catenatum, 1009 in P. equestris, 2748 in G. elata, and 1615 in A. shenzhenica. A greater number of expanded gene families in D. chrysotoxum may lead to a larger genome size than that in other sequenced orchid species2,24,33,34.
Fig. 3. Phylogenetic tree showing divergence times and the evolution of gene families in D. chrysotoxum.

The green and red numbers represent the numbers of expanded gene families and contracted gene families, respectively. The blue portions of the pie charts show that the copy numbers of gene families are constant. Divergence times are represented by light blue bars at internodes; the divergence time is at a 95% confidence interval and shown by the range of these bars. The expansion and contraction of gene families are represented by members of branches (see Materials and methods and Supplementary Fig. 6). The orange part of the pie chart at the top left represents the ratio of 11,252 gene families found in the most recent common ancestor (MRCA) that expanded or contracted during recent differentiation events
The ancestral clade of Dendrobium had 464 expanded gene families and 216 contracted gene families. The D. chrysotoxum clade had 953 expanded gene families and 1335 contracted gene families. In the ancestral clade of Dendrobium, there were 19 significantly expanded gene families, including 236 genes from D. chrysotoxum. In the D. chrysotoxum clade, 107 gene families were significantly expanded, including 1048 genes, and 43 gene families were significantly contracted, including 59 genes. We also conducted Gene Ontology (GO) enrichment analysis for the expanded gene families, and the GO terms “cytoplasmic part” and “intracellular organelle” were found to be enriched (Supplementary Table 13). In addition, the bidirectional sugar transporter gene SWEET was identified (Supplementary Fig. 7), whose product plays important roles in sugar translocation between compartments41, phloem loading for long-distance translocation42, pollen nutrition43, and seed filling44. Further phylogenetic analysis showed that 17 genes were expanded in clade II (Supplementary Fig. 7), suggesting that these SWEET genes might be associated with a fleshy stem that is abundant in polysaccharides and other medicinal compounds.
Synteny analysis and whole-genome duplication (WGD)
Both the loss of a substantial fraction of genes and the increase in substitution rate complications were indicated by WGD in D. chrysotoxum, which is thought to have occurred among different orchid species2. WGD is evident in many lineages and is a practical method for genome expansion45. To determine the occurrence of WGDs in D. chrysotoxum, JCVI v0.9.1446 was used to analyze the protein sequences of D. chrysotoxum, P. equestris, P. aphrodite, and D. catenatum with the default parameters and obtain collinear gene pairs. There were 21,881 collinear gene pairs between D. chrysotoxum and P. equestris, 21,592 between D. chrysotoxum and P. aphrodite, 24,550 between D. chrysotoxum and D. catenatum, and 2800 between D. chrysotoxum and itself (Supplementary Table 14). Although D. chrysotoxum was assembled to the chromosome level, its self-collinearity was still very low compared to that of other sequenced orchid species. The collinearity between D. catenatum and D. chrysotoxum was fragmented, which may be the result of the quality of the D. catenatum genome, which was not at the chromosome level. The chromosomes of Dendrobium and Phalaenopsis showed a good corresponding relationship, indicating that after the divergence of Dendrobium and Phalaenopsis, the chromosomes were conserved, with few rearrangements. Syntenic figures show that the collinearity blocks were mainly in a 1:1 pattern, indicating that after the differentiation of D. chrysotoxum, no species-specific WGD events had occurred (Fig. 4; Supplementary Figs. 8–12).
Fig. 4. Self-collinearity map of D. chrysotoxum (Guchui).

The values on the X- and Y-axes are the numbers of cumulative genes on the 19 chromosomes
The distributions of synonymous substitutions per synonymous site (Ks) were estimated to infer polyploidization events that occurred in the D. chrysotoxum genome. There were two peaks in the distribution of Ks for paralogous D. chrysotoxum genes: Ks = 1.0 and 1.7–1.8 (Fig. 5). These results suggested that two polyploidization events occurred in D. chrysotoxum. To further verify the polyploidization events in D. chrysotoxum, its genome was compared with that of P. equestris, A. shenzhenica, and D. catenatum. The peaks in Ks values between both D. chrysotoxum/P. equestris and D. chrysotoxum/D. catenatum were less than 1.0, suggesting that the events occurred before the differentiation of these three species. There was a diverging peak in the Ks distribution of D. chrysotoxum and A. shenzhenica at Ks = 0.7–0.8, which was smaller than but close to the Ks peak of the Orchidaceae (Ks = 1), indicating that extant orchid species differentiated immediately after experiencing a shared WGD event. Based on the evolution of gene families, species differentiation mainly occurred through gene loss with little gene expansion, which confirmed that the WGD event occurred in the most recent common ancestor of extant orchid species. The second peak in the Ks distributions within D. chrysotoxum (1.7–1.8) indicated that the τ WGD had occurred in most monocot species45. Furthermore, the peak of the Ks distribution in D. chrysotoxum was smaller than 0.2, suggesting that it originated from background (tandem) duplications and likely did not signify additional recent WGDs2. Therefore, this study found that D. chrysotoxum experienced two polyploidization events: an early WGD event was shared among all extant orchid species, and a later event that was shared among most monocot species.
Fig. 5. Distribution of Ks values in the whole paranome of D. chrysotoxum.
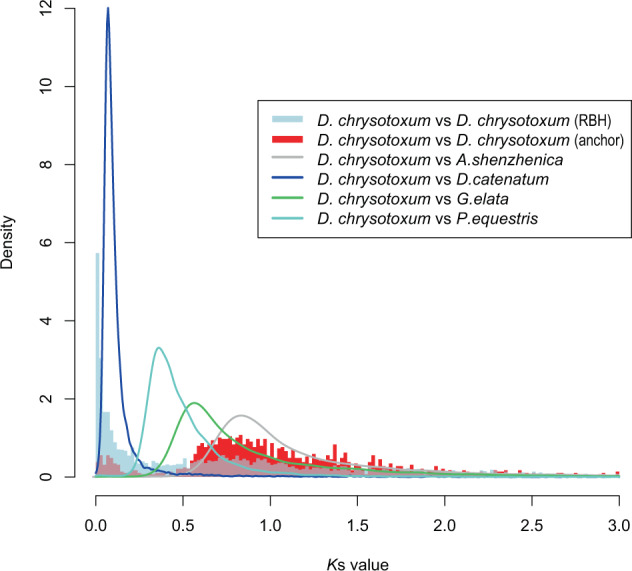
The Ks distributions of paralogs using the reciprocal best hit (RBH) and anchor are shown as light blue and red histograms, respectively. The Ks distribution for the one-to-one orthologs of D. chrysotoxum–G. elata, D. chrysotoxum–A. shenzhenica, D. chrysotoxum–D. catenatum, and D. chrysotoxum–P. equestris are shown as green, gray, blue, and cyan curves, respectively. RBH reciprocal best blast hit; “anchor” refers to colinear regions
MADS-box genes and the evolution of flowers
MADS-box genes are among the most important regulators of plant floral development and compose major class of regulators mediating floral transition. In total, the D. chrysotoxum genome encodes 58 putative functional MADS-box genes and 1 pseudogene (Table 2; Supplementary Table 15). Interestingly, the number of MADS-box genes was similar to that in other sequenced orchid species but smaller than that in most sequenced angiosperms2,24,33. D. chrysotoxum has 31 type II MADS-box genes, which is higher than that found in P. equestris (29) and A. shenzhenica (27), but smaller than that of D. catenatum (35)2,24,33. Phylogenetic analysis (Supplementary Fig. 13) showed that, except for those in the MIKC*, Bs, and OsMADS32 clades, most genes in the type II MADS-box clade were contracted. Bs genes are involved in the differentiation and development of ovules47. In D. chrysotoxum, there are four Bs members, more than the number found in other sequenced orchid species. The Bs genes had duplicated, as evidenced by higher seed production in D. chrysotoxum than in other sequenced orchid species. This must have been accompanied by duplication of the type I MADS-box gene Mα, as D. chrysotoxum has more Mα genes (19) than other sequenced orchid species (Table 2), ensuring seed development. In addition, there were no genes from the FLOWERING LOCUS C (FLC), AGL12, or AGL15 clades in the D. chrysotoxum genome or other sequenced orchid genomes. In comparison with genes in the AGL12 and AGL15 clades, which are present in both rice and Arabidopsis, orthologous genes of FLC, AGL12, and AGL15 might have been specifically lost in orchids. Although AGL12-like genes (XAL1 in A. thaliana) are necessary for root development and flowering48, D. chrysotoxum and P. equestris have varying mechanisms that perform the same function2, showing that D. chrysotoxum is not a terrestrial orchid but is an epiphytic orchid.
Table 2.
MADS-box genes in D. chrysotoxum, A. shenzhenica, P. equestris, D. catenatum, and Arabidopsis thaliana
| Category | P. equestris33 | D. catenatum24 | D. chrysotoxum* | A. shenzhenica2 | A. thaliana37 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Functional | Pseudo | Functional | Pseudo | Functional | Pseudo | Functional | Pseudo | Functional | Pseudo | |
| Type II (total) | 29 | 1 | 35 | 11 | 31 | 0 | 27 | 4 | 45 | 5 |
| MIKCc | 28 | 1 | 32 | 9 | 28 | 0 | 25 | 3 | 43 | 4 |
| MIKCa | 1 | 0 | 3 | 2 | 4 | 0 | 2 | 1 | 2 | 0 |
| Mδ | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 1 |
| Type I (total) | 22 | 8 | 28 | 1 | 26 | 1 | 9 | 0 | 62 | 36 |
| Mα | 10 | 6 | 15 | 1 | 19 | 1 | 5 | 0 | 20 | 23 |
| Mβ | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 17 | 5 |
| Mγ | 12 | 2 | 13 | 0 | 8 | 1 | 4 | 0 | 21 | 8 |
| Total | 51 | 9 | 63 | 12 | 58 | 1 | 36 | 4 | 107 | 41 |
aThis study
The D. chrysotoxum genome has 26 putative functional type I genes and 1 pseudogene (Table 2), which might have resulted in a lower expansion rate or a higher contraction rate compared with those of type II MADS-box genes in D. chrysotoxum (31 functional genes). Tandem gene duplication might play an important role in the increasing number of type I genes in the α group (type I Mα), suggesting that the type I genes have mainly been duplicated on a smaller scale from more-recent duplications49. Although members of the β group of type I MADS-box genes (type I Mβ) do exist in A. thaliana, poplar, and rice, they are absent in the D. chrysotoxum genome. Interactions among these type I MADS-box genes are essential for initiating endosperm development50; therefore, like in other sequenced orchids2,24,33, endosperm is also absent in D. chrysotoxum.
Floral color regulatory pathway in D. chrysotoxum
The flowering time of a single flower of D. chrysotoxum was ~10 days51,52, the limit of which might be associated with yellow flower color. All photosynthetic tissues in each of the biological kingdoms can produce carotenoids53. More than 1100 naturally occurring carotenoids (http://carotenoiddb.jp/) are involved in many of the red, orange, and yellow colors of flowers53. These compounds also play important roles in photosynthesis. Interestingly, carotenoids function as precursors for the biosynthesis of abscisic acid (ABA)53. Moreover, ethylene plays a role in senescing flowers54. Ethylene and ABA regulate plant growth and development55 synergistically or antagonistically. We therefore analyzed the network involving carotenoid, ABA, and ethylene biosynthesis and regulation (Fig. 6).
Fig. 6. Gene expression patterns among the interactions between carotenoids, the ABA and ethylene biosynthesis pathways, and regulatory mechanisms in D. chrysotoxum (adapted from Watkins and Pogson53, Yin et al.55, Nisar et al.101, Sun et al.102, and Finkelstein96).

Increased gene expression during the four developmental stages of flowers is shown by thick green arrows, while decreased gene expression is shown by thick red arrows. The enzymes or genes are indicated alongside the arrows. The dashed lines suggest that there are multiple steps. Carotenes are shown in orange, and light gold indicates xanthophylls. ABA-related genes are shown in yellow, and ethylene-related genes are shown in light green. CCDs (orange arrows) regulate carotenoid accumulation. Plastoglobule-localized metallopeptidase 48 (PGM48) is hypothesized to regulate CCD4. The orange protein regulates PSY activity, acting as a chaperone. ClpB3 (red arrow) regulates the activity of DXS, acting as an enzyme in the MEP pathway
Eighteen genes or gene family members in the carotenoid biosynthesis pathway and related regulatory mechanisms were identified (Supplementary Table 16). These genes encode phytoene synthase (PSY), orange protein, casein lytic proteinase B3 (ClpB3), deoxy-D-xylulose 5-phosphate synthase (DXS), phytoene desaturase, ζ-carotene isomerase, ζ-carotene desaturase, carotenoid isomerase (CRTISO), β-lycopene cyclase, ε-lycopene cyclase (LYCE), β-carotene hydroxylase, carotene ε-hydroxylase (CYP97C), zeaxanthin epoxidase (ZEP), violaxanthin de-epoxidase, neoxanthin synthase (NXS), xanthophyll acyl-transferase (XAT), plastoglobule-localized metallopeptidase 48, and carotenoid cleavage dioxygenase (CCD). The expression of these genes increased with flower development, except for LYCE, which is targeted for downregulation during biofortification, ZEP, NXS, and XAT (Supplementary Table 16; Fig. 6), suggesting that more carotenoids and fewer xanthophylls were produced during flowering to senescence.
The substrates used to produce ABA were neoxanthin and violaxanthin, and the process was regulated by nine-cis-epoxy carotenoid dioxygenases (NCEDs). The biosynthesis of ABA is catalyzed by the short-chain dehydrogenase/reductase-like (SDR1) enzyme abscisic aldehyde oxidase (AAO) and molybdenum cofactor (MoCo). The expression of NCED and SDR genes increased gradually with the development of flowers, while the expression of AAO and MoCo genes decreased gradually (Supplementary Table 17; Fig. 6). Furthermore, there were four AAO gene members detected in Arabidopsis, while there was only one gene detected in D. chrysotoxum (Supplementary Fig. 14). Taken together, these findings might indicate that relatively low amounts of ABA (C15) were produced, which might improve ethylene biogenesis.
For ethylene biogenesis, genes encoding three kinds of enzymes were identified. The expression of Maker79017, encoding S-AdoMet synthetase (SAMS), Maker75695 and Maker66290, encoding ACC synthase (ACS), and Maker29641, encoding ACC oxidase (ACO), increased gradually, suggesting that increased amounts of ethylene were produced during the development of flowers (Supplementary Table 18; Fig. 6). CONSTITUTIVE TRIPLE RESPONSE 1 and ETHYLENE INSENSITIVE 2 regulate the interaction between ethylene and the ABA pathway and are partially dependent on the MHZ5/CRTISO-mediated ABA pathway in rice55. Therefore, we also analyzed the expression patterns of the two genes in D. chrysotoxum, but there were no obvious differences in any of the four stages of flower development (Supplementary Table 18; Fig. 6).
In conclusion, carotenoid production increased gradually, and the content of xanthophylls decreased gradually in yellow D. chrysotoxum flowers during flowering to senescence. Less xanthophyll was degraded into less ABA, and less ABA led to more ethylene being produced. As a result, yellow flowers of D. chrysotoxum generally have a relatively short flowering period.
Identification of the terpene synthases (TPS) and Hsp90 gene families and adaptive evolution
Dendrobium spp., with epiphytic or lithophytic lifestyles, frequently experience adverse environmental conditions, such as chilling and water deficit56. During plant responses to environmental stresses, volatile terpenes play critical roles56. Moreover, terpenes also play an important role in the formation of orchid floral scents56. TPSs are the key enzymes involved in terpene biosynthesis57. Different sizes of TPS families and subfamilies in plant species have evolved to synthesize a specific set of terpene compounds57. There are seven subfamilies in the TPS family: TPS-a, TPS-b, TPS-c, TPS-d, TPS-e/f, TPS-g, and TPS-h57. Among them, TPS-a encodes a sesquiterpene synthase that is found in both dicotyledonous and monocotyledonous plants. Angiosperm-specific TPS-b encodes a monoterpene synthase with an R(R)X8W motif that catalyzes the isomerization cyclization reaction. TPS-c belongs to the ancestral clade and catalyzes the activity of copalyl diphosphate synthase. Gymnosperm-specific TPS-d performs several functions, such as those of diterpene, monoterpene, and sesquiterpene synthases. TPS-e/f encodes copalyl diphosphate/kaurene synthases, which are critical enzymes for gibberellic acid production. Another angiosperm-specific TPS, TPS-g, encodes monoterpene synthase enzymes that lack the R(R)X8W motif. TPS-h has been observed only in Selaginella moellendorffii58–61. Phylogenetic analysis of the TPS gene family members and their expression in bud formation and initial flower opening, blooming, and withering are shown in Fig. 7. In this study, the TPS gene number in D. chrysotoxum was 48, which was greater than that in D. catenatum (42) (Fig. 7a). Moreover, there were 14 and 21 genes in A. shenzhenica and P. equestris, respectively. The TPS-b subfamily can be divided into monocot and eudicot clades. More D. chrysotoxum TPS genes than D. catenatum ones clustered in the monocot A clade—14 (red gene ID) and 4 (blue gene ID), respectively (Fig. 7a). Fewer TPS genes were found in D. chrysotoxum than in D. catenatum in the monocot B clade—7 (red gene ID) and 10 (blue gene ID), respectively (Fig. 7a). The different distribution patterns might contribute to the difference in terpenoid compositions between these two species, which needs further validation.
Fig. 7. Analysis of TPS genes in D. chrysotoxum.

a Phylogenetic analysis of TPS genes in D. chrysotoxum, D. catenatum, A. shenzhenica, A. thaliana, and P. equestris. Ash A. shenzhenica, Maker D. chrysotoxum, Dca D. catenatum, Peq P. equestris, AT A. thaliana. b Expression patterns of TPS genes in buds and in the initial flower opening, blooming, and withering stages of D. chrysotoxum
To explore heat stress-related genes in D. chrysotoxum, we also analyzed heat stress-related gene families across orchid species. Only two Hsp90 genes (red gene ID) were identified (clustering in group III), with high expression during bud formation (Supplementary Fig. 15a, b). This number was lower than that for the other four species (six were identified in D. catenatum, seven in P. equestris, six in A. shenzhenica, and seven in A. thaliana). This large gene loss might be related to resistance to heat stress.
Conclusion
Although D. chrysotoxum has high ornamental and medicinal value, further molecular mechanism research and development of medicinal compounds have been limited by a lack of omics data. In this study, a chromosome-level reference genome of D. chrysotoxum with an assembled genome size of 1.37 Gb and 30,044 annotated protein-coding genes was obtained. Ks analysis suggested that two polyploidization events occurred in D. chrysotoxum: a recent WGD shared among other orchid species and an ancient polyploidization event shared among most monocots (τ event). Phylogenetic analysis of the SWEET gene family in D. chrysotoxum showed that gene expansion occurred in clade II of the SWEET gene family, which might be related to fleshy stems containing an abundance of polysaccharides. Floral color regulation analysis showed that fewer xanthophylls degraded into ABA, which led to more ethylene production, thus accelerating the senescence of D. chrysotoxum flowers. The analysis of D. chrysotoxum helped elucidate the mechanism through which fleshy stems produce an abundance of polysaccharides and other medicinal compounds, as well as flowering time regulation, which is critical for industrial development. Our results provide the first high-quality genome of Dendrobium and give important insights into the molecular mechanism underlying the production of medicinal active ingredients, breeding, and orchid evolution.
Materials and methods
DNA preparation and sequencing
Fresh leaves of wild D. chrysotoxum were collected for genome sequencing. A modified cetyltrimethylammonium bromide protocol was used to extract the genomic DNA. To estimate genome size and heterozygosity, 143.78 Gb of raw data from paired-end libraries (PE150) constructed from a MGISEQ-2000 sequencer were generated. After data filtering was carried out by SOAPnuke v1.6.5 software with the parameters -n 0.02 -l 20 -q 0.4 -Q 2 -i -G --seqType 0 –rmdup, clean data (138.15 Gb) were obtained (Supplementary Table 1). Then, a SMRTbell Template Prep Kit 1.0 (PacBio, Menlo Park, CA, USA) and a PacBio Sequel system were used to construct and sequence the DNA libraries, respectively, for PacBio long-read sequencing. A total of 132.64 Gb of sequencing data (coverage of 96.12%) were generated, with an N50 read length of 19.5 kb (Supplementary Table 1). Furthermore, all libraries with a 500 bp insert size were sequenced on a NovaSeq platform (2 × 150 bp). We ultimately produced 169.25 Gb of data and 125.96 Gb of clean data for Hi-C analysis. The transcriptomes of flowers of D. chrysotoxum were obtained from Huang’s doctoral thesis62 to assist gene annotation.
Genome assembly
Genome size and heterozygosity were measured using Jellyfish v.2.2.6 and GenomeScope (http://qb.cshl.edu/genomescope)63 based on a 17-K-mer distribution. Canu64 was used to assemble the PacBio sequencing reads, with the following parameters: minOverlapLength = 700; minReadLength = 1000; and corOutCoverage = 50. Then, Arrow software was used to polish the assembly, and Pilon v1.2365 was further used for correction of the assembly based on short reads, with the following parameters: fixed bases; mindepth 10; minqual 20; and diploid. Finally, the completeness and quality of the final assembled genome were evaluated with BUSCO v332.
Hi-C library construction and chromosome assembly
The raw reads produced by the NovaSeq sequencing platform were filtered by SOAPnuke66 (v1.6.5, https://github.com/BGI-flexlab/SOAPnuke) software with the following parameters: -n 0.02 -l 20 -q 0.4 -i –rmdup. Then, the obtained clean reads were compared with the preassembled contigs using Juicer67 software. After filtering the results and removing the misaligned reads, 3D-DNA68 software was used to preliminarily cluster, sequence, and direct the pseudochromosomes. Juicer-box was used to adjust, reset, and cluster the pseudochromosomes to improve the chromosome assembly quality. For the evaluation of the Hi-C assembly results, the final pseudochromosome assemblies were divided into 100 kb bins of equal lengths, and a heat map was used to visualize the interaction signals generated by the valid mapped read pairs between each bin.
Genome annotation
Repetitive sequences are an important part of a genome and are divided into two types, namely, tandem repeats and interspersed repeats. Two methods, de novo prediction and homology-based searches, were used to annotate repeat sequences in the genome. RepeatMasker v4.0.7 and RepeatProteinMask v4.0.7 software69 (http://www.repeatmasker.org) were used to identify repetitive sequences based on the Repbase v21.12 database69 (http://www.girinst.org/repbase). For de novo prediction, a repetitive sequence database was constructed using RepeatModeler v1.0.870 and LTR_FINDER v1.0671. RepeatMasker software and Tandem Repeats Finder v4.0972 were subsequently used to predict repeat sequences and identify tandem repeats in the genome, respectively. The annotation of high-quality protein-coding genes was carried out by integrating homology-based, de novo and transcriptome-based predictions. For homology-based prediction, protein sequences from six species (Arabidopsis thaliana, Oryza sativa, Sorghum bicolor, Zea mays, G. elata, and P. equestris) were used to align D. chrysotoxum genome sequences via Exonerate v2.2.073. Then, the complete sequences of 3000 genes from the homology-based prediction method were used to produce a training model through Augustus v3.2.374 and SNAP v2006-07-2875 software. The RNA-seq data of D. chrysotoxum were mapped to genome sequences through HISAT2 and StringTie software76,77. Finally, Maker v2.31.878 was used to annotate and integrate the results generated by the above methods. BUSCO v332 was then used to evaluate the completeness and quality of the gene models.
Functional annotation of the predicted gene models was carried out by BLAST v2.2.3179 software and aligned against the contents of the SwissProt80, TrEMBL (http://www.uniprot.org/), KEGG (http://www.genome.jp/kegg/), InterPro81, Nr, and GO (The Gene Ontology Consortium) databases82. For noncoding RNA annotations, tRNAscan-SE 1.3.1 (http://lowelab.ucsc.edu/tRNAscan-SE/)83 was used to annotate tRNA sequences. BLASTN79 was used to search for rRNA, and miRNA and snRNA sequences were predicted by Infernal 1.1 (http://infernal.janelia.org/) software84.
WGD analysis
Ks distribution analysis was used to infer the occurrence of WGD events in D. chrysotoxum and those between D. chrysotoxum and A. shenzhenica, D. catenatum, and P. equestris. BLASTP79 was used to search for putative paralogous and orthologous genes within and between genomes by alignment of each genome pair. MCScanX v1.5.285 was used to identify colinear regions, and then CodeML in the PAML package86 was used to calculate the Ks value of each salicoid duplicated gene pair. We used CAFÉ87 to evaluate the significance of each expanded and contracted gene family (P < 0.01).
SWEET gene family analyses
To identify SWEET proteins, proteomic datasets of four orchid species (D. chrysotoxum, A. shenzhenica, D. catenatum, and P. equestris) and A. thaliana were constructed. The MtN3_slv domain PF03083 model profile from the Pfam database88 was used for performing local searches of proteome datasets containing five species via the HMMER program89. The SWEET protein sequences were aligned with MAFFT90. The alignment was then used for phylogenetic tree reconstruction by PhyML 3.091–93 with the default parameters.
MADS-box gene family analysis
The sequences of the MADS-box proteins of A. thaliana and the HMM profile (PF00319) were obtained from the Arabidopsis information resource (TAIR) (https://www.arabidopsis.org/) and the Pfam database88, respectively. Then, the sequences of the MADS-box gene family members in the D. chrysotoxum genome were obtained using HMMER 3.2.1 software89 and BLASTP83 methods. The obtained amino acid sequences were used for TBLASTN79 analysis of the D. chrysotoxum transcriptomic assemblies. SMART94 was subsequently used to confirm the obtained sequences by domain analysis. MEGA X95 was then used for the alignment of the candidate genes, and the CIPRES website (https://www.phylo.org/portal2/) was used for phylogenetic tree construction. iTOL (https://itol.embl.de) was subsequently used to visualize the phylogenetic trees.
Identification of genes involved in the carotenoid, ABA, and ethylene biosynthesis pathways and regulatory mechanisms in D. chrysotoxum
The sequences of all 17 genes or gene family members involved in the carotenoid biosynthesis pathway and regulatory mechanisms in A. thaliana, Triticum aestivum, and Pantoea ananatis53 were used as queries to search against the protein database of D. chrysotoxum. The obtained amino acid sequences were aligned using MAFFT90. We then manually inspected the aligned sequences and removed any obviously inconsistent sequences.
Four genes or gene family members involved in the ABA biosynthesis pathway or regulatory mechanisms in Arabidopsis were obtained96. BLASTP79 was used to search for homologous genes by querying the protein database of D. chrysotoxum. After aligning the amino acid sequences with MAFFT90 software, we removed any obviously inconsistent sequences.
The sequences of genes encoding SAMS, ACS, and ACO, all of which are involved in the ethylene biosynthesis pathway, in Arabidopsis97 were used as queries for searching proteins by BLASTP79 software.
For gene families, a phylogenetic tree was constructed with PhyML98 based on the alignment of sequences from D. chrysotoxum, D. catenatum, A. shenzhenica, P. equestris, and A. thaliana. The tree was generated by the maximum likelihood method based on the Jones–Taylor–Thornton (JTT) matrix-based model99, and the fast likelihood-based method was used for phylogenetic tests with SH-like branch supports.
Gene expression analysis
Transcriptome data from flowers at four developmental stages (flower buds, initial flowering stage, blooming period, and withering flowers), stems, and leaves were obtained (BioProject PRJNA691441), and Salmon v1.3.0100 was used to quantify gene expression, with the default settings.
TPS and Hsp90 gene family identification
The HMM profiles for PF01397 (Terpene_synth) and PF03936 (Terpene_synth_C) were downloaded from the Pfam database (pfam.xfam.org/), and both profiles were used to carry out HMM searches against the information of the protein databases for five species (D. chrysotoxum, Dendrobium catenatum, P. equestris, Apostasia shenzhenica, and A. thaliana). The sequences aligned with MAFFT90 were used for phylogenetic tree construction through PhyML79. The tree was generated by the maximum likelihood method based on the JTT matrix-based model99 and the bootstrap method for phylogenetic tests with 1000 replications. Similarly, the HMM profile for PF00183 (Hsp90) was downloaded from the Pfam database (pfam.xfam.org/), and the subsequent steps were the same as those for TPS gene family identification.
Supplementary information
Acknowledgements
This project was supported by the Guangdong Innovation Research Team Fund (2014ZT05S078); National Natural Science Foundation of China (grants 31571252 and 31772322); Guangdong Special Support Program for Young Talents in Innovation Research of Science and Technology (2019TQ05N940); Shenzhen Peacock Grant (827/000189); Science and Technology Program of Guangdong Province, China (2019B121202006); Program of Forestry Administration of Guangdong Province (E036011002); Department for Wildlife and Forest Plant Protection of the National Forest and Grassland Administration (2019073010); National Key Research and Development Program of China (No. 2018YFD1000400); Special Research Foundation of Hebei Agricultural University (YJ201848); and Natural Science Foundation of Hebei Province (C2019204295).
Data availability
All the data from this study have been deposited in the NCBI database under BioProject ID PRJNA664445.
Conflict of interest
The authors declare no competing interests.
Contributor Information
Tengbo Huang, Email: tengbohuang@szu.edu.cn.
Shan-Ce Niu, Email: niushance@163.com.
Zhong-Jian Liu, Email: zjliu@fafu.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41438-021-00621-z.
References
- 1.Christenhusz MJM, Byng JW. The number of known plants species in the world and its annual increase. Phytotaxa. 2016;261:201–217. doi: 10.11646/phytotaxa.261.3.1. [DOI] [Google Scholar]
- 2.Zhang G, et al. The Apostasia genome and the evolution of orchid. Nature. 2017;549:379–383. doi: 10.1038/nature23897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pridgeon, A. M. et al. Genera Orchidacearum: Epidendroideae (Part three) (Oxford University, 2014).
- 4.Luo J-P, Deng Y-Y, Zha X-Q. Mechanism of polysaccharides from Dendrobium huoshanense. on streptozotocin-induced diabetic cataract. Pharm. Biol. 2008;46:243–249. doi: 10.1080/13880200701739397. [DOI] [Google Scholar]
- 5.Leitch IJ, et al. Genome size diversity in orchids: consequences and evolution. Ann. Bot. 2009;104:469–481. doi: 10.1093/aob/mcp003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ng TB, et al. Review of research on Dendrobium, a prized folk medicine. Appl. Microbiol. Biotechnol. 2012;93:1795–1803. doi: 10.1007/s00253-011-3829-7. [DOI] [PubMed] [Google Scholar]
- 7.Pan LH, et al. Comparison of hypoglycemic and antioxidative effects of polysaccharides from four different Dendrobium species. Int. J. Biol. Macromol. 2014;64:420–427. doi: 10.1016/j.ijbiomac.2013.12.024. [DOI] [PubMed] [Google Scholar]
- 8.Tian C-C, Zha X-Q, Luo J-P. A polysaccharide from Dendrobium huoshanense prevents hepatic inflammatory response caused by carbon tetrachloride. Biotechnol. Biotechnol. Equip. 2014;29:132–138. doi: 10.1080/13102818.2014.987514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang K, et al. Purification, characterization and biological activity of polysaccharides from Dendrobium officinale. Molecules. 2016;21:701. doi: 10.3390/molecules21060701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu J, et al. High-density genetic map construction and stem total polysaccharide content-related QTL exploration for Chinese endemic Dendrobium (Orchidaceae) Front. Plant Sci. 2018;9:398. doi: 10.3389/fpls.2018.00398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao Y, et al. Antioxidant and anti-hyperglycemic activity of polysaccharide isolated from Dendrobium chrysotoxum Lindl. J. Biochem. Mol. Biol. 2007;40:670–677. doi: 10.5483/bmbrep.2007.40.5.670. [DOI] [PubMed] [Google Scholar]
- 12.Li S, et al. Elution-extrusion counter-current chromatography separation of five bioactive compounds from Dendrobium chrysototxum Lindl. J. Chromatogr. A. 2011;1218:3124–3128. doi: 10.1016/j.chroma.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 13.Hu J, Fan W, Dong F, Miao Z, Zhou J. Chemical components of Dendrobium chrysotoxum. Chin. J. Chem. 2012;30:1327–1330. doi: 10.1002/cjoc.201100670. [DOI] [Google Scholar]
- 14.Yu Z, et al. Dendrobium chrysotoxum Lindl. alleviates diabetic retinopathy by preventing retinal inflammation and tight junction protein decrease. J. Diabetes Res. 2015;2015:518317. doi: 10.1155/2015/518317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu Z, et al. Erianin inhibits high glucose-induced retinal angiogenesis via blocking ERK1/2-regulated HIF-1alpha-VEGF/VEGFR2 signaling pathway. Sci. Rep. 2016;6:34306. doi: 10.1038/srep34306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu J, et al. Gigantol from Dendrobium chrysotoxum Lindl. binds and inhibits aldose reductase gene to exert its anti-cataract activity: an in vitro mechanistic study. J. Ethnopharmacol. 2017;198:255–261. doi: 10.1016/j.jep.2017.01.026. [DOI] [PubMed] [Google Scholar]
- 17.Lim V, Schneider E, Wu H, Pang IH. Cataract preventive role of isolated phytoconstituents: findings from adecade of research. Nutrients. 2018;10:1580. doi: 10.3390/nu10111580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ren ZY, et al. Functional analysis of a novel C-glycosyltransferase in the orchid Dendrobium catenatum. Hortic. Res. 2020;7:111. doi: 10.1038/s41438-020-0330-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robles-Rivera RR, et al. Adjuvant therapies in diabetic retinopathy as an early approach to delay its progression: the importance of oxidative stress and inflammation. Oxid. Med. Cell Longev. 2020;2020:1–23. doi: 10.1155/2020/3096470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gong Y, et al. Erianin induces a jnk/sapk-dependent metabolic inhibition in human umbilical vein endothelial cells. In Vivo. 2004;18:223–228. [PubMed] [Google Scholar]
- 21.Cushman M, et al. Synthesis and evaluation of stilbene and dihydrostilbene derivatives as potential anticancer agents that inhibit tubulin polymerization. J. Med. Chem. 1991;34:2579–2588. doi: 10.1021/jm00112a036. [DOI] [PubMed] [Google Scholar]
- 22.Ma G, et al. Iuhibitory effects of Dendrobium chrysotoxutm and its constiuents on the mouse HePA and ESC. J. China Pharm. Univ. 1994;25:188–189. [Google Scholar]
- 23.Gong YQ, et al. In vivo and in vitro evaluation of erianin, a novel anti-angiogenic agent. Eur. J. Cancer. 2004;40:1554–1565. doi: 10.1016/j.ejca.2004.01.041. [DOI] [PubMed] [Google Scholar]
- 24.Zhang G, et al. The Dendrobium catenatum Lindl. genome sequence provides insights into polysaccharide synthase, floral development and adaptive evolution. Sci. Rep. 2016;6:19029. doi: 10.1038/srep19029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu MJ, et al. Chromosome-scale assembly of the Kandelia obovata genome. Hortic. Res. 2020;7:75. doi: 10.1038/s41438-020-0300-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kang M, et al. A chromosome-scale genome assembly of Isatis indigotica, an important medicinal plant used in traditional Chinese medicine: an Isatis genome. Hortic. Res. 2020;7:18. doi: 10.1038/s41438-020-0240-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen F, et al. The sequenced angiosperm genomes and genome databases. Front. Plant Sci. 2018;9:418. doi: 10.3389/fpls.2018.00418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen F, et al. Genome sequences of horticultural plants: past, present, and future. Hortic. Res. 2019;6:112. doi: 10.1038/s41438-019-0195-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yan L, et al. The genome of Dendrobium officinale illuminates the biology of the important traditional Chinese orchid herb. Mol. Plant. 2015;8:922–934. doi: 10.1016/j.molp.2014.12.011. [DOI] [PubMed] [Google Scholar]
- 30.Si C, et al. DoRWA3 from Dendrobium officinale plays an essential role in acetylation of polysaccharides. Int. J. Mol. Sci. 2020;21:6250. doi: 10.3390/ijms21176250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng SG, et al. Genome-wide researches and applications on Dendrobium. Planta. 2018;248:769–784. doi: 10.1007/s00425-018-2960-4. [DOI] [PubMed] [Google Scholar]
- 32.Simão FA, et al. BUSCO online supplementary information: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 33.Cai J, et al. The genome sequence of the orchid Phalaenopsis equestris. Nat. Genet. 2015;47:65–72. doi: 10.1038/ng.3149. [DOI] [PubMed] [Google Scholar]
- 34.Yuan Y, et al. The Gastrodia elata genome provides insights into plant adaptation to heterotrophy. Nat. Commun. 2018;9:1615. doi: 10.1038/s41467-018-03423-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sena JS, et al. Evolution of gene structure in the conifer Picea glauca: a comparative analysis of the impact of intron size. BMC Plant Biol. 2014;14:95. doi: 10.1186/1471-2229-14-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De La Torre AR, et al. Insights into conifer giga-genomes. Plant Physiol. 2014;166:1724–1732. doi: 10.1104/pp.114.248708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Castillo-Davis CI, et al. Selection for short introns in highly expressed genes. Nat. Genet. 2002;31:415. doi: 10.1038/ng940. [DOI] [PubMed] [Google Scholar]
- 38.Keane PA, Seoighe C. Intron length coevolution across mammalian genomes. Mol. Biol. Evol. 2016;33:2682–2691. doi: 10.1093/molbev/msw151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Swinburne IA, Silver PA. Intron delays and transcriptional timing during development. Dev. Cell. 2008;14:324–330. doi: 10.1016/j.devcel.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Artieri CG, Fraser HB. Transcript length mediates developmental timing of gene expression across Drosophila. Mol. Biol. Evol. 2014;31:2879–2889. doi: 10.1093/molbev/msu226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin IW, et al. Nectar secretion requires sucrose phosphate synthases and the sugar transporter SWEET9. Nature. 2014;508:546–549. doi: 10.1038/nature13082. [DOI] [PubMed] [Google Scholar]
- 42.Chen LQ, et al. Sucrose efflux mediated by SWEET proteins as a key step for phloem transport. Science. 2012;335:207–211. doi: 10.1126/science.1213351. [DOI] [PubMed] [Google Scholar]
- 43.Sun MX, et al. Arabidopsis RPG1 is important for primexine deposition and functions redundantly with RPG2 for plant fertility at the late reproductive stage. Plant Reprod. 2013;26:83–91. doi: 10.1007/s00497-012-0208-1. [DOI] [PubMed] [Google Scholar]
- 44.Chen LQ, et al. A cascade of sequentially expressed sucrose transporters in the seed coat and endosperm provides nutrition for the Arabidopsis embryo. Plant Cell. 2015;27:607–619. doi: 10.1105/tpc.114.134585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiao Y, Li J, Tang H, Paterson AH. Integrated syntenic and phylogenomic analyses reveal an ancient genome duplication in monocots. Plant Cell. 2014;26:2792–2802. doi: 10.1105/tpc.114.127597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang, H. et al. jcvi: JCVI Utility Libraries (Zenodo, 2015). 10.5281/zenodo.31631.
- 47.Sang X, et al. CHIMERIC FLORAL ORGANS1, encoding a monocot-specific MADS box protein, regulates floral organ identity in rice. Plant Physiol. 2012;160:788–807. doi: 10.1104/pp.112.200980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tapia-López R, et al. An AGAMOUS-related MADS-box gene, XAL1 (AGL12), regulates root meristem cell proliferation and flowering transition in Arabidopsis. Plant Physiol. 2008;146:1182–1192. doi: 10.1104/pp.107.108647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Par̆enicová L, et al. Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: new openings to the MADS world. Plant Cell. 2003;15:1538–1551. doi: 10.1105/tpc.011544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Masiero S, et al. The emerging importance of type I MADS box transcription factors for plant reproduction. Plant Cell. 2011;23:865–872. doi: 10.1105/tpc.110.081737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Niu SC, et al. Morphological type identification of self-incompatibility in Dendrobium and its phylogenetic evolution pattern. Int. J. Mol. Sci. 2018;19:2595. doi: 10.3390/ijms19092595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu, R. H. Studies on the Germplasm Resources of Dendrobium in China and their Genetic Relationships. Doctoral dissertation, Chin. Acad. For. (2007).
- 53.Watkins, J. L. & Pogson B. J. Prospects for carotenoid biofortification targeting retention and catabolism. Trends Plant Sci.25, 501–512 (2020). [DOI] [PubMed]
- 54.Johnson PR, Ecker JR. The ethylene gas signal transduction pathway: a molecular perspective. Annu. Rev. Genet. 1998;32:227. doi: 10.1146/annurev.genet.32.1.227. [DOI] [PubMed] [Google Scholar]
- 55.Yin CC, et al. Ethylene responses in rice roots and coleoptiles are differentially regulated by a carotenoid isomerase-mediated abscisic acid pathway. Plant Cell. 2015;27:1061–1081. doi: 10.1105/tpc.15.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu Z, et al. Genome-wide identification and expression profile of TPS gene family in Dendrobium officinale and the role of DoTPS10 in linalool biosynthesis. Int. J. Mol. Sci. 2020;21:5419. doi: 10.3390/ijms21155419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jiang SY, Jin JJ, Sarojam R, Ramachandran S. A comprehensive survey on the terpene synthase gene family provides new insight into its evolutionary patterns. Genome Biol. Evol. 2019;11:2078–2098. doi: 10.1093/gbe/evz142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen F. The family of terpene synthases in plants: A mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 2011;66:212–229. doi: 10.1111/j.1365-313X.2011.04520.x. [DOI] [PubMed] [Google Scholar]
- 59.Tholl D. Biosynthesis and biological functions of terpenoids in plants. Adv. Biochem. Eng. Biotechnol. 2015;148:63–106. doi: 10.1007/10_2014_295. [DOI] [PubMed] [Google Scholar]
- 60.Bohlmann J, Meyer-Gauen G, Croteau R. Plant terpenoid synthases: molecular biology and phylogenetic analysis. Proc. Natl Acad. Sci. USA. 1998;95:4126–4133. doi: 10.1073/pnas.95.8.4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jiang SY, et al. Comprehensive survey on the terpene synthase gene family provides new insight into its evolutionary patterns. Genome Biol. Evol. 2019;11:2078–2098. doi: 10.1093/gbe/evz142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang, X. L. Research on molecular regulation mechanism of formation of floral color and floral fragrance of Dendrobium chrysotoxum based on transcriptome sequencing. Doctoral dissertation, Chin. Acad. For. (2019).
- 63.Ranallo-Benavidez TR, Jaron KS, Schatz MC. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 2020;11:1–10. doi: 10.1038/s41467-020-14998-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Koren S, et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–736. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Walker BJ, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE. 2014;9:e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen Y, et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. GigaScience. 2017;7:120. doi: 10.1093/gigascience/gix120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Durand NC, et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 2016;3:95–98. doi: 10.1016/j.cels.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dudchenko O, et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 2017;356:92–95. doi: 10.1126/science.aal3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jurka J, et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 70.Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics. 2005;21:i351–i358. doi: 10.1093/bioinformatics/bti1018. [DOI] [PubMed] [Google Scholar]
- 71.Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007;35:W265–W268. doi: 10.1093/nar/gkm286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Benson G, et al. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Slater GS, Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stanke M, et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006;34:W435–W439. doi: 10.1093/nar/gkl200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Johnson AD, et al. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24:2938–2939. doi: 10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pertea M, et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015;33:290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim D, et al. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019;37:907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Holt C, Yandell M. MAKER2: an annotation pipeline and genome database management tool for second generation genome projects. BMC Bioinform. 2011;12:491. doi: 10.1186/1471-2105-12-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kent WJ. BLAT—the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Boeckmann B, et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31:365–370. doi: 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jones P, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ashburner M, et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nawrocki EP, Eddy SR. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013;29:2933–2935. doi: 10.1093/bioinformatics/btt509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang Y, et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40:e49. doi: 10.1093/nar/gkr1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Bioinformatics. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- 87.De Bie T, et al. CAFE: a computational tool for the study of gene family evolution. Bioinformatics. 2006;22:1269–1271. doi: 10.1093/bioinformatics/btl097. [DOI] [PubMed] [Google Scholar]
- 88.El-Gebali S, et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019;47:D427–D432. doi: 10.1093/nar/gky995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Eddy SR. Accelerated profile HMM searches. PLoS Comput. Biol. 2011;7:e1002195. doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Guindon S, Lethiec F, Duroux P, Gascuel O. PHYML Online—a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res. 2005;33:W557–W559. doi: 10.1093/nar/gki352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wu SS, et al. The genome sequence of star fruit (Averrhoa carambola) Hortic. Res. 2020;7:95. doi: 10.1038/s41438-020-0307-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chen SP, et al. The Phoebe genome sheds light on the evolution of magnoliids. Hortic. Res. 2020;7:146. doi: 10.1038/s41438-020-00368-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Letunic I, Doerks T, Bork P. SMART: recent updates, new developments and status in 2015. Nucleic Acids Res. 2015;43:D257–D260. doi: 10.1093/nar/gku949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kumar S, et al. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018;35:1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Finkelstein RAbscisic. Acid synthesis and response. Arabidopsis Book. 2013;11:e0166–e0166. doi: 10.1199/tab.0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lin Z, Zhong S, Grierson D. Recent advances in ethylene research. J. Exp. Bot. 2009;60:3311–3336. doi: 10.1093/jxb/erp204. [DOI] [PubMed] [Google Scholar]
- 98.Guindon S, et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 99.Jones DT, Taylor WR, Thornton JM. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992;8:275–282. doi: 10.1093/bioinformatics/8.3.275. [DOI] [PubMed] [Google Scholar]
- 100.Patro R, et al. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nisar N, et al. Carotenoid metabolism in plants. Mol. Plant. 2015;8:68–82. doi: 10.1016/j.molp.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 102.Sun T, et al. Carotenoid metabolism in plants: the role of plastids. Mol. Plant. 2017;11:58–74. doi: 10.1016/j.molp.2017.09.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data from this study have been deposited in the NCBI database under BioProject ID PRJNA664445.