

Abstract
Intravenous (IV) infusion of bone marrow‐derived mesenchymal stem/stromal cells (MSCs) stabilizes the blood‐spinal cord barrier (BSCB) and improves functional recovery in experimental models of spinal cord injury (SCI). Although IV delivered MSCs do not traffic to the injury site, IV delivered small extracellular vesicles (sEVs) derived from MSCs (MSC‐sEVs) do and are taken up by a subset of M2 macrophages. To test whether sEVs released by MSCs are responsible for the therapeutic effects of MSCs, we tracked sEVs produced by IV delivered DiR‐labelled MSCs (DiR‐MSCs) after transplantation into SCI rats. We found that sEVs were released by MSCs in vivo, trafficked to the injury site, associated specifically with M2 macrophages and co‐localized with exosome markers. Furthermore, while a single MSC injection was sufficient to improve locomotor recovery, fractionated dosing of MSC‐sEVs over 3 days (F‐sEVs) was required to achieve similar therapeutic effects. Infusion of F‐sEVs mimicked the effects of single dose MSC infusion on multiple parameters including: increased expression of M2 macrophage markers, upregulation of transforming growth factor‐beta (TGF‐β), TGF‐β receptors and tight junction proteins, and reduction in BSCB permeability. These data suggest that release of sEVs by MSCs over time induces a cascade of cellular responses leading to improved functional recovery.
Keywords: blood‐spinal cord barrier, exosomes, macrophages, mesenchymal stem/stromal cells, small extracellular vesicles, spinal cord injury, transforming growth factor‐beta
1. INTRODUCTION
Traumatic spinal cord injury (SCI) results in persistent functional deficits below the level of the injury. The pathophysiology of SCI is complex and includes neuronal death, axon severing, loss of oligodendrocytes which myelinate axons and secondary changes which affect repair of the damaged CNS tissue and influence recovery. Reactive astrocytes form a glial scar and produce chondroitin sulfate proteoglycans (CSPGs) that can inhibit axonal growth (Bradbury et al., 2002) as do other endogenous inhibitory molecules that cause growth cone collapse (Schwab & Strittmatter, 2014). Pericytes detach from endothelial cells and migrate into the scar area (Goritz et al., 2011; Matsushita et al., 2015), disrupting the integrity of the blood‐spinal cord barrier (BSCB), increasing vascular permeability altering the local microenvironment and facilitating entry of peripheral immune cells into the CNS parenchyma (Beck et al., 2010).
Macrophages, in particular, dominate the lesion core within a week after injury and play both positive and negative roles in tissue repair and recovery (Beck et al., 2010; Milich et al., 2019). Macrophages represent heterogeneous populations of cells with an array of different subtypes based on gene expression (Chen et al., 2019). However, macrophages are typically classified as either M1, a classically Th1‐activated proinflammatory macrophage, or as M2, an alternatively Th2‐activated anti‐inflammatory macrophage (Kigerl et al., 2009). M1 macrophages are characterized by expression of iNOS and release of tumour necrosis factor‐alpha (TNF‐α), while M2 anti‐inflammatory macrophages are characterized by expression of CD206 and release of transforming growth factor‐beta (TGF‐β). M2 macrophages are also highly phagocytic. The persistence of a proinflammatory M1 macrophage phenotype in spinal cord lesions (Kigerl et al., 2009), which is typically not observed at injury sites with more complete wound repair and healing (Krzyszczyk et al., 2018), suggests that pro‐inflammatory M1 macrophages may inhibit recovery after SCI, while an M2 phenotype may promote wound healing (Mantovani et al., 2013).
Numerous studies have demonstrated that intravenous (IV) infusion of mesenchymal stem/stromal cells (MSCs) derived from bone marrow can improve locomotor recovery in experimental models of SCI (Chopp et al., 2000; Honmou et al., 2012; Oliveri et al., 2014; Osaka et al., 2010; Sasaki et al., 2009). MSC infusion reduced lesion volume (de Almeida et al., 2015; Osaka et al., 2010; Urdzikova et al., 2014), accelerated restoration of the BSCB (Matsushita et al., 2015), increased neovascularization (Morita et al., 2016; Quertainmont et al., 2012), conferred neuroprotection (Sasaki et al., 2009; Spejo et al., 2013) and increased axon sprouting (Quertainmont et al., 2012; Sasaki et al., 2009). Importantly, the therapeutic effects of intravenous MSC delivery have been observed after both acute (Matsushita et al., 2015; Osaka et al., 2010) and chronic (Morita et al., 2016) SCI. Clinical trials of bone marrow‐derived MSC treatment for both spinal cord injury (Honmou et al., 2021) and stroke (Honmou et al., 2011) have been carried out. However, we (Matsushita et al., 2015) and others (Quertainmont et al., 2012) have found that IV delivered MSCs do not appreciably target the lesion site, lodging instead in the lungs where they survive for a few days, arguing that therapeutic actions of MSCs may be mediated by release of cellular products which travel to the injury site.
MSCs produce a wide array of trophic (Chen et al., 2002; Hamano et al., 2000; Mayer et al., 2005; Whone et al., 2012) and anti‐inflammatory factors (Prockop & Oh, 2012), and also are known to secrete protein and RNA‐containing small extracellular vesicles (sEVs) or exosomes (Chen et al., 2010; Witwer et al., 2019; Yeo et al., 2013) which have therapeutic effects in rodent models of very acute SCI (Huang et al., 2017; Ji et al., 2019; Lu et al., 2019; Wang et al., 2018). We previously demonstrated that IV delivery of sEVs characterized as exosomes from bone marrow‐derived MSCs (MSC‐sEVs) traffic to the injury site and target M2‐type macrophages in the injured spinal cord (Lankford et al., 2018). We hypothesized that secretion and circulatory transport of sEVs produced by MSCs lodged in the lungs contribute to the therapeutic efficacy of MSCs (Lankford et al., 2018; Matsushita et al., 2015). Since sEVs are smaller and more stable and storable (Sokolova et al., 2011) than living cells, infusion of MSC‐derived sEVs could obviate many potential problems associated with cell infusion (Volarevic et al., 2018).
Here we report that sEVs are secreted in vivo by MSCs and travel to the lesion site, as evidenced by the presence of membrane‐labelled (DiR) sEVs within M2 macrophages in the SCI lesion site after IV delivery of DiR‐labeled MSCs. A second key finding is that fractionated dosing of the MSC‐sEVs (1/3 total dosage on three consecutive days, F‐sEVs), designed to mimic release of sEVs from MSCs entrapped in the lungs for 2–3 days after IV infusion, was more therapeutically effective than delivery of the same quantity of sEVs in one single dose (1‐sEVs). Furthermore, both MSC and F‐sEV infusion resulted in increased expression of M2‐type macrophages markers, TGF‐β, TGF‐β receptors, and key microvascular tight junction proteins, as well as a reduction in vascular permeability.
Our results support a model of therapeutic action of MSCs in which uptake of MSC‐sEVs by M2 macrophages at SCI lesion sites leads to their increased expression of TGF‐β and subsequent upregulation of TGF‐β receptors and a number of BSCB‐related microvasculature proteins, ultimately leading to stabilization of the BSCB. This improvement in BSCB function may contribute to functional recovery by producing an environment more conducive to tissue repair and neuronal function. Given that sEVs are more stable and storable than living cells (Sokolova et al., 2011), MSC‐sEVs may represent an advantageous alternative to MSC therapy, which would allow for greater flexibility in treatment of SCI.
2. MATERIALS AND METHODS
2.1. Induction of contusive SCI and assessment of functional recovery
All experiments were carried out in accordance with National Institutes of Health guidelines for the care and use of laboratory animals and the VA Connecticut Healthcare System Institutional Animal Care and Use Committee (IACUC) approved all animal protocols.
To induce contusive spinal cord injuries, dorsal laminectomies (T9) were performed on adult male Sprague–Dawley rats (185–215 g) under isoflurane gas anaesthesia, followed immediately by a delivery of 22.5 Newton impact (equal to 225 kilodynes) with a 2.5 mm tip using the Infinite Horizon (IH) impactor (Precision Systems & Instrumentation, Lexington KY). Appropriate postoperative care, including twice‐daily bladder expression for up to 7 days, antibiotic treatment (enrofloxacin 0.05 mg/kg/day SQ), and pain relief (buprenorphine 0.05 mg/kg/day SQ) for 48 h, was provided for all animals. Open‐field locomotor function was assessed using the Basso‐Beattie‐Bresnahan (B‐B‐B) score (Basso et al., 1996) by a tester blinded to the treatment. Rats were scored 2‐day presurgery, 3‐day post‐SCI, 1‐week post‐SCI, and at weekly intervals thereafter until the time of sacrifice. To assure the consistency of lesions, animals were included in the study only if they had B‐B‐B scores of 0 on 3‐day and 0.5 on 7‐day post‐SCI. A total of 182 surgical animals were used for these experiments with an additional 12 unoperated controls.
2.2. MSC culture, MSC‐sEVs isolation and characterization
MSCs were isolated from the bone marrow of young adult Sprague‐Dawley rats (150‐200 g) or green fluorescent protein (GFP) transgenic rats (CZ‐004, SD‐Tg (Act‐ EGFP) CZ‐004Osb; SLC, Shizuoka, Japan) cultured and passaged 4 times as previously described, (Osaka et al., 2010) before detaching for cell transplantation or changing to serum‐free media (Dulbecco's Modified Eagle Medium (DMEM) with glutamate and penicillin/streptomycin) for collection of sEVs. No differences were observed between the efficacy of MSC‐sEVs from GFP rats and nontransgenic littermates.
MSC‐sEVs were isolated from 2‐day serum‐free conditioned media from MSC cultures using the differential centrifugation method (Lankford et al., 2018; Thery et al., 2006). Protein content was assessed using a Bradford protein assay kit (Thermo Fisher Scientific Inc., Waltham, MA, USA) and Synergy HT plate reader (Bio‐Tek Instruments Inc., Winooski, VT, USA). For visualization of MSC‐sEVs, a sample of the sEV preparation was examined with transmission electron microscopy as described in Lankford et al. (2018). The composition of exosome fractions was also evaluated in the two samples by nanoparticle tracking analysis (NTA) employing SALD‐7500nano (Shimadzu Co., Kyoto, Japan) using triton detergent extraction methods (Osteikoetxea et al., 2015). Briefly, Triton‐X100 (Sigma‐Aldrich) was added to MSC‐sEVs containing samples with different final concentrations (0.025% and 0.075%), vortexed for 30 s, and analysed by SALD‐7500nano. All steps of detergent lysis were conducted at room temperature. Protein markers of exosomes were assessed in three samples of MSC‐sEVs and corresponding MSCs by western blotting (see the section of Western blots). Gene expression of miRNA‐21 was also analysed using three MSC‐sEVs samples (see the section of Real‐time quantitative PCR).
To trace trafficking of MSC‐sEVs from IV infused MSCs, dissociated non‐GFP expressing MSCs were labelled with the fluorescence lipophilic tracer, DilC18(7);1,1′‐dioctadecyl‐3,3,3′,3′‐tetramethylindotricbocyanine iodide (DiR) (Molecular Probes, Grand Island, NY, USA) for 10 min, and washed three times with DMEM before infusion. To observe MSC‐sEVs uptake in vitro, attached MSCs were labelled with DiR for 10 min, allowed to recover for 4 h in control media, and washed three times with phosphate‐buffered saline (PBS) before changing to serum‐free collection media for 48 h and isolation of MSC‐sEVs from the conditioned media.
2.3. Infusion of MSCs and MSC‐sEVs
On 7‐day post‐SCI, MSCs or MSC‐sEVs suspended in 1 ml DMEM, or DMEM alone, were infused via the femoral vein of isoflurane‐anesthetized rats over the course of 1 min, with bupivacaine liposome (5.3 mg/kg SQ) administered post‐surgery for pain relief. On 8‐ and 9‐day post‐SCI, 0.2 ml DMEM alone or 1/3 dose MSC‐sEVs in 0.2 ml DMEM was infused via the saphenous vein. The total dosage of sEVs for both the one‐ dose (1‐sEVs) and fractioned dose (F‐sEVs) conditions represented the quantity of sEVs isolated as above from 2‐day serum‐free conditioned media from approximately 2 × 106‐MSCs (approximately 4.6 ± 0.5 μg protein or 2.5 × 109‐MSC‐sEVs as estimated by protein assay and NanoSight LM10, NanoSight Ltd., Minton Park, Amesbury, UK). For the F‐sEVs condition, sEVs were delivered as 3 doses of 1/3 each of the quantity of MSC‐sEVs from the same MSC‐sEVs isolation preparation (8.3 × 108‐MSC‐sEVs). Thus, SCI rats were randomized into one of four conditions, DMEM vehicle only infusion (Vehicle), a single MSC infusion followed by two DMEM infusions (MSC), one single MSC‐sEVs infusion comparable to the maximum amount of sEVs that might be expected to be produced by transplanted MSCs over a 2–3 day period followed by two DMEM infusions (1‐sEV), or infusion of 3 fractionated doses of sEVs at 1/3 each of the single sEVs infusion on three consecutive days (F‐sEVs). See Figure 1. The experimenter performing infusions was naïve with respect to transplant contents and all subsequent analysis was performed blinded until the data was collected.
FIGURE 1.
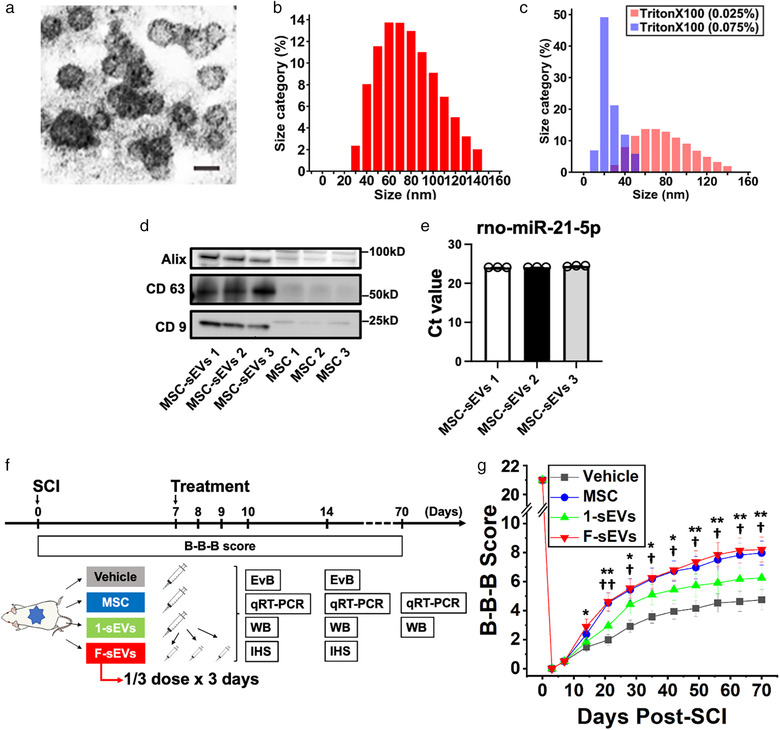
Experimental protocol, characterization of MSC‐sEVs, and effects of MSC‐derived sEVs infusion on functional recovery. a, Representative electron microscopic image of an sEV preparation showing the sizes and morphology of particles. Scale bar = 100 nm b, Histogram of the size distribution of sEVs. c, Histograms of size distribution of sEVs exposed with 0.025% Triton X‐100 (red) and 0.075% Triton X‐100 (blue). d, Expressions of CD 9, CD 63, and Alix in sEVs derived from MSCs were evaluated by western blotting and compared to equal concentrations of proteins from their MSCs. e, Micro RNA 21 was detected within MSC‐sEVs tested with primers targeting for rno‐miR‐21‐5p. The threshold cycle (Ct value) was evaluated using three exosome samples. f, Experimental protocol. Animals were randomly assigned to one of 4 treatment groups: 1) vehicle (Vehicle), 2) MSCs (MSC), 3) MSC‐sEVs with a single dosing protocol (1‐sEVs), and 4) MSC‐sEVs‐infused rats with fractionated dosing protocol over the course of 3 days (F‐sEVs). g, B‐B‐B locomotor scores of contused rats infused with Vehicle (black squares), MSCs (blue circles), or MSC‐derived sEVs delivered in a single dose (1‐sEVs green upward triangles) or three fractionated doses over 3 days (F‐sEVs red downward triangles). Values are presented as means ± SEM. Repeated‐ measures two‐way ANOVA followed by Sidak post hoc tests were conducted. *: P < .05 between F‐sEVs group and Vehicle group, **: P < .01 between F‐sEVs group and Vehicle group, †: P < .05 between MSC group and Vehicle group, ††; P < .01 between MSC group and Vehicle group. MSCs: mesenchymal stem/stromal cells, B‐B‐B score: Basso–Beattie–Bresnahan locomotor scale score, EvB: Evans blue leakage analysis, qRT‐PCR: quantitative reverse transcription‐polymerase chain reaction, WB: Western blotting, IHS: Immunohistochemical staining. Particle sizes, stability to detergent degradation, and tetraspanin expression were consistent with sEVs. MSC: mesenchymal stem/stromal cell, sEVs: small extracellular vesicles, MSC‐sEVs: small extracellular vesicles derived from MSCs
To assess trafficking of sEVs from transplanted cells, DiR‐labeled MSCs were administered at five times the standard dose (5 × 106 DiR‐labeled MSCs) and animals were sacrificed after 48 h for histological examination.
2.4. Histological analysis
Rats were deeply anesthetized with sodium pentobarbital, perfused with saline, followed by 4% paraformaldehyde in 0.1 M phosphate buffer, and processed for standard frozen sectioning. Coronal (20 μm) sections taken at 1.5 mm from the centre of each block were stained with two or more of the primary antibodies (Supplementary Table 1) diluted in 0.01% Triton X‐100, 5% fishskin gelatin (0.1 M PBS) blocking buffer, visualized with species‐specific secondary antibodies (Supplementary Table.1) counterstained with 4′,6‐diamidino‐2‐phenylindole (DAPI) mounting media (Vectashield, Vector Laboratories, Burlingame, CA, USA) and photographed with a Nikon A1R multiphoton confocal microscope with NIS Elements software (Nikon, Tokyo, Japan).
2.5. In vitro uptake of MSC‐sEVs by macrophages
Macrophages were isolated from bone marrow of 6 weeks old SD rats as described in Ying et al. (Ying et al., 2013), plated on 8 well multichambered slides (Falcon 354108) at 100,000 cells per well in macrophage growth media consisting of IDMEM without HEPES (Sigma‐Aldrich I3390), with 10% heat‐inactivated foetal calf serum and 10 ng/ml colony‐stimulating factor (CSF) (Sigma‐Aldrich, # SRP3332) and fed every 2–3 days. After 7–10 days, growth media was replaced with M1 or M2 induction media containing either 100 ng/ml LPS (Sigma‐Aldrich, L2630) plus 50 ng/ml interferon‐gamma (Sigma‐Aldrich, I3257) or 10 ng/ml interleukin 4 (Sigma‐Aldrich, I3650) respectively without CSF, at normal pH or a pH of 6. Two days after transfer to M1 or M2 phenotype induction media, 1 μl of a central myelin enriched fraction (Lankford et al., 2017) was added to half of the wells to induce phagocytosis. DiR‐labeled MSC‐sEVs were added to wells three days after transfer to induction media. After 24 h, cultures were washed 3 times with plain IDMEM, fixed with 4% paraformaldehyde and stained with antibodies directed against CD206 and iNOS, visualized with fluorescent 488 and 594 wavelength secondary antibodies, counterstained with DAPI mounting media, examined, and photographed with a Nikon A1R multiphoton confocal microscope with NIS Elements software as above (Supplementary Table.1).
2.6. Western blots
Animals were sacrificed under deep anaesthesia with sodium pentobarbital. Spinal cords were removed and stored ‐70°C, and total protein and RNA were purified from a homogenized 8 mm (30 mg wet weight) segment of spinal cord tissue centred around the impact sites using the AllPrep DNA/RNA/Protein Mini Kit (QIAGEN, Venlo, The Netherlands). Protein was quantified using a BCA protein assay (Thermo Fisher Scientific Inc), and equal amounts were loaded and run on 10% SDS–polyacrylamide gels (10ug protein/lane), transferred to polyvinylidene difluoride membranes, probed with appropriate primary antibodies (Supplementary Table 1) overnight at 4°C, followed by secondary antibody staining and visualization with an enhanced chemiluminescence detection system (Pierce Thermo Scientific) using Luminescent Image Analyzer (ChemiDoc MP Imaging System, Bio‐Rad) and band density analysis by ImageJ software (NIH, Bethesda, MA, USA). Protein expression of sEVs was also quantified using a BCA protein assay by loading equal amounts of protein (10ug protein/lane). Polyclonal rabbit anti‐rat CD9, CD63, and Alix antibodies (Supplementary Table 1) were used.
2.7. Real‐time quantitative PCR
Total purified RNA from above described samples were quantified with a spectrophotometer (NanoDrop, Technologies Inc., Wilmington, DE, USA), and reverse transcription of RNA was performed with an iScript cDNA Synthesis Kit (Bio‐Rad, Hercules, CA, USA). Quantitative RT‐PCR analysis was performed in triplicate using TaqMan® Universal Master Mix II with Uracil‐N glycosylase (UNG) and specific sets of primers and TaqMan probes (Thermo Fisher Scientific Inc.) (Supplementary Table 2) with a CFX96TM real‐time PCR detection system (Bio‐Rad), using approximately 20 ng total mRNA for each sample. Thermal cycling was carried out at 50°C for 2 min and 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Delta cycle threshold (Ct) (ΔCT) was calculated against the endogenous control (glyceraldehyde 3‐phosphate dehydrogenase), and delta‐delta Ct (ΔΔCT) was calculated against the ΔCT of the sham. Fold change (FC) was calculated using the comparative Ct method (Schmittgen & Livak, 2008).
To assess gene expression of miRNA 21 in MSC‐sEVs, three samples of MSC‐sEVs were analysed. Purified RNA was extracted from MSC‐sEVs samples and reverse transcription of RNA was performed with a miRNeasy Serum/Plasma Advanced Kit (Qiagen). Quantitative RT‐PCR analysis was performed in triplicate using TaqMan Fast Advanced Master Mix and a specific primer for miRNA 21 (rno481342_mir) (Thermo Fisher Scientific Inc) with a CFX96TM real‐time PCR detection system. Thermal cycling was carried out at 95°C for 20 s, followed by 40 cycles of 95°C for 3 s and 60°C for 30 s. Cycle threshold was measured.
2.8. Evans blue (EvB) dye extravasation
On 10‐ and 14‐day post‐SCI, 4% EvB in saline (2.5 mg/kg; Sigma) was administered via the femoral vein of isoflurane‐anesthetized rats. Two hours after the EvB infusion, rats were deeply anesthetized with sodium pentobarbital, transcardially perfused with ice‐cold PBS, and 1 cm segments of spinal cords centred around the impact were removed, weighed, homogenized in 50% (w/v) trichloroacetic acid, centrifuged, and the absorbance of the supernatant measured at 620 nm (Synergy HT multilabel plate reader) to calculate dye concentrations from a standard curve(Matsushita et al., 2015; Morita et al., 2016; Nakazaki et al., 2017).
2.9. Statistical analysis
All statistical analyses were performed using GraphPad Prism software (version 9.0) and R (version 4.0.0). Repeated‐measures two‐way ANOVA followed by Sidak post hoc tests were conducted for multiple comparisons of B‐B‐B scores. Continuous data were analysed by 1‐way ANOVA or the Kruskal‐Wallis test. The Tukey‐Kramer test or Steel‐Dwass test were used to compare the subgroups. All P values < .05 were considered statistically significant. All values shown here are expressed as mean ± SEM. All methods and data were reported with consideration of guidelines provided by Animals in Research: Reporting in Vivo Experiments (ARRIVE)(Kilkenny et al., 2010) and Minimum Information about a Spinal Cord Injury Experiment (MIASCI) (Lemmon et al., 2014).
3. RESULTS
3.1. Characterization of MSC‐sEVs
Transmission electron microscopy of sEV preparations revealed large numbers of vesicles in the 30–100 nm diameter size range characteristic of exosomes (Figure 1a). Consistent with previous characterizations (Jeppesen et al., 2019), particle sizes in our MSC‐sEVs samples, measured by nanoparticle tracking analysis, ranged from 25 to 140 nm (mean 83.0 ± 14.1 nm; n = 3 samples) (Figure 1b). Particle numbers were not significantly reduced by exposure to 0.025% Triton X‐100, which has been demonstrated to lyse and degrade other types of vesicles, including microvesicles and apoptotic bodies (Osteikoetxea et al., 2015), but particles larger than 60 nm in diameter were effectively eliminated by 0.075% Triton X‐100, which disrupts exosome membranes (Figure 1c) (Osteikoetxea et al., 2015). Western blots showed that three characteristic surface marker proteins of exosomes, CD63, CD9, and Alix, were all greatly enriched in the sEVs samples compared to equal concentrations of proteins from their MSC sources (Figure 1d). Quantification of miR‐21 microRNAs confirmed the MSC‐sEVs preparations had significant levels of this microRNA, which is known to represent a significant fraction of the micro‐RNA in MSC‐derived exosomes (Figure 1e). These results are consistent with the samples designated as sEVs in this study containing large numbers of exosomes (Jeppesen et al., 2019).
3.2. Both MSC infusion and fractionated MSC‐sEVs promote functional recovery after SCI
To assess the relative therapeutic efficacy of MSCs and MSC‐sEVs on functional recovery after SCI, rats were randomly assigned to one of four treatment groups at 1‐week post‐contusion: Vehicle group, MSC group (1 × 106 MSCs delivered in one dose), 1‐sEVs group (MSC‐sEVs delivered as one dose treatment with a quantity of MSC‐sEVs intended to approximate the greatest quantity of sEVs which might be released by MSCs surviving transiently post‐infusion), and F‐sEVs group (MSC‐sEVs of the same quantity as the one dose condition delivered in 3 fractions of 1/3 total on each of three successive days) (Figure 1f).
Weekly blinded evaluations of locomotor function showed improvement in the B‐B‐B locomotor rating scale (Basso et al., 1996) in the F‐sEVs group (n = 14), relative to the Vehicle group (n = 17) beginning 2‐week post‐contusion (1‐week after onset of treatment) and in the MSC group (n = 14) beginning 3‐week post‐SCI (Figure 1 g). At 10‐week post‐SCI, animals in the MSC and F‐sEVs groups exhibited average B‐B‐B scores of 8.54 ± 0.86 and 7.96 ± 0.83, respectively, which were greater than the Vehicle group (4.74 ± 0.67). MSC and F‐sEVs B‐B‐B scores differed from controls at P < .05 and P < .01, respectively. Animals in the 1‐sEVs group (n = 14) showed only a trend towards improved functional scores and did not differ from controls at any timepoint, with an average B‐B‐B score of 6.25 ± 0.77 at 10‐week post‐SCI.
3.3. sEVs are released by IV infused MSCs and are taken up by M2 macrophages at the SCI site
To test the hypothesis that IV infused MSCs release sEVs which can traffic to SCI lesion sites, we intravenously delivered DiR‐labeled MSCs (5 × 106) 1‐week post‐contusion and examined the spinal cord 2 days after cell infusion (Figure 2a). No DiR‐labeled MSCs were detected at the SCI lesion sites in animals infused with DiR‐MSCs, as evidenced by a lack of cells exhibiting DiR wavelength fluorescence in the lesion (Figure 2b). However, large numbers of punctate ‘hotspots’ of DiR wavelength fluorescence were detected at SCI sites. Consistent with our previous observations of DiR hotspot distribution within lesions in animals infused with DiR‐labeled sEVs. DiR hotspots in DiR‐MSC infused animals were localized predominantly to cells along the edges of the lesions (Figure 2b‐f). Co‐staining with antibodies directed against the M2 marker CD206 and the exosome marker CD63 showed that the majority of these DiR hotspots were localized within CD206+ M2 macrophages and also stained with CD63 (Figure 2c‐f), and this intracellular localization could be confirmed when z stack images for individuals cells were rotated (Figure 2c1‐f1 ). These findings support the hypothesis that IV infused MSCs, which do not themselves reach the injury site, release exosomes that can be taken up by M2‐type macrophages at the lesion site.
FIGURE 2.
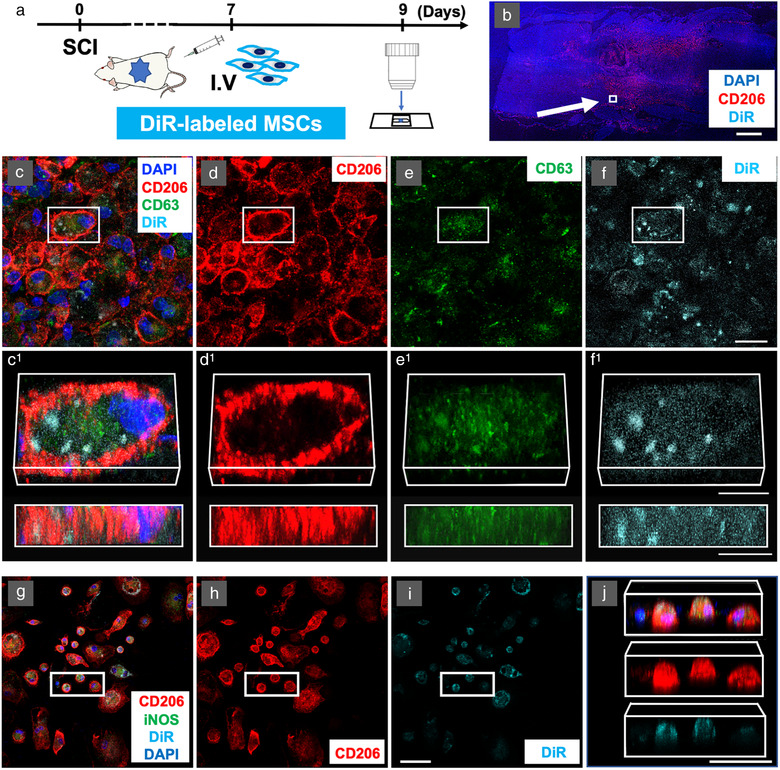
M2 macrophages take up DiR‐labeled MSC‐sEVs in vivo at the lesion site after intravenous infusion of DiR‐labeled MSCs and in vitro after exposure to DiR‐labeled MSC‐sEVs. a, Experimental protocol for tracking the MSC‐sEVs released from IV infused DiR‐labeled MSCs in vivo. b, Confocal micrographs of a representative region of a frozen sectioned contused spinal cord harvested 48 h after IV infusion of DiR‐labeled MSCs, immunostained with antibodies directed against M2 macrophage marker CD206 (red) and DAPI (blue). Scale bars indicate 100 μm. c‐f, Images of a representative region of the white square in the figure. c, immunostained with antibodies directed against CD206 (red) and exosomes marker CD63 (green) counterstained with DAPI (blue) with DiR visualized as cyan. Images from left to right show the same area showing fluorescence channels for c, CD206, CD63, DAPI, & DiR, d, CD206, e, CD63, and f, DiR. Images in c1‐f1 show enlarged images of the boxed area above rotated and illustrated in 3D. Note the strong co‐localization of DiR fluorescence with CD206, suggesting that the majority of these DiR hotspots were localized within CD206+ M2 macrophages. Scale bars in c‐f and c1‐f1 indicate 20 μm and 10 μm, respectively. Note that although the labelled MSCs themselves were not detected at the lesion site DiR‐ labelled hotspots co‐staining with exosome markers were. g‐j Confocal micrographs of a representative region of rat bone marrow macrophage cultures stimulated to induce a phagocytic M2 phenotype with IL‐4 and central myelin enriched fraction and fixed 24 h after the addition of DiR‐labeled MSC‐sEVs at pH = 6. Cultures were stained with antibodies directed against CD206 (red), iNOS (green), and counterstained with DAPI (blue) with DiR fluorescence visualized as Cyan. Images from left to right show: (g) CD206, iNOS, DAPI, & DiR, (h) CD206 & DiR, (i) DiR only, and (j) rotated 3D reconstructions of the boxed area indicated in (g‐j) containing 3 CD206+ presumptive M2 macrophages and one CD206– and iNOS– presumptive M0 macrophage (left). Note that the two cells in the centre of the group appear largely filled with DiR fluorescence hotspots, while both the cells on the left and on the right do not appear to have taken up significant quantities of exosomes. Scale bars in (i) and (j) indicate 50 μm. MSC: mesenchymal stem/stromal cell, MSC‐sEVs: small extracellular vesicles derived from MSCs, DiR: DilC18(7);1,1′‐dioctadecyl‐3,3,3′,3′‐tetramethylindotricbocyanine iodide, DAPI: 4′,6‐diamidino‐2‐phenylindole, iNOS: inducible nitric oxide synthase
In vitro studies confirmed that bone marrow macrophages could take up labelled sEVs under certain conditions. When DiR‐labeled MSC‐sEVs were added to IL‐4 stimulated, predominantly M2‐type macrophages, in pH6 media with a central myelin enriched fraction (CMEF) added to induce phagocytosis, hotspots of DiR wavelength fluorescence were readily apparent in CD206+ macrophages 24 h later (Figure 2 g‐j). Three‐dimensional rotation showed that the “clouds” of DiR fluorescence were within the macrophage cell bodies and not on the cell surface (Figure 2j). Cells stimulated to an M1 phenotype, cultured at a pH7 or at pH6 without the addition of CMEF, did not show a detectable uptake of DiR‐labeled exosomes (not shown), suggesting that MSCexos uptake was specific to M2 macrophages and was dependent on local environmental factors.
3.4. Both infusion of MSCs and fractionated dosing of MSC‐sEVs promote M2 macrophage polarization after SCI
Immunostaining of macrophages at lesion sites and quantification of macrophage subtype specific genes and proteins revealed an increase in M2 specific markers relative to M1 specific markers in both the MSC and F‐sEVs groups 1‐week post‐treatment. Large numbers of macrophages were detected at lesion sites for all treatment groups 7‐day post‐treatment (14‐day post‐SCI). However, while the proportion of macrophages staining positive for the M1 marker iNOS and the M2 marker CD206 appeared approximately equal in the Vehicle and one dose 1‐sEVs groups, we observed a strong predominance of cells staining positive for the M2 marker CD 206 relative to the M1 marker iNOS in the MSC and fractioned dosing F‐sEVs groups, (Figure 3a‐d).
FIGURE 3.
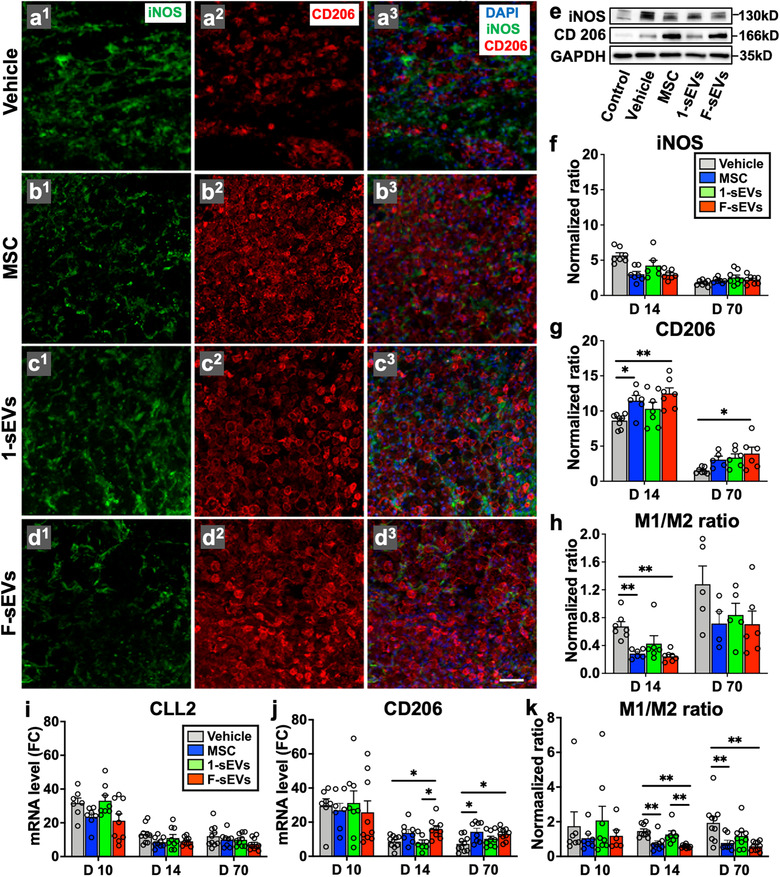
Intravenous infusion of MSCs and fractionated dosing of MSC‐sEVs promote M2 macrophage polarization in the injured spinal cord. a1‐3 ‐d1‐3, Confocal micrographs of a representative region of a frozen sectioned contused spinal cord harvested 7 days after the treatment (14‐day post‐SCI), immunostained with antibodies directed against Type M1 macrophage marker iNOS (green), Type M2 macrophage marker CD206 (red), and counterstained with DAPI (blue). Scale bar in a indicates 50 μm. e, Representative images of western blot of the lesion site harvested at 7 days after the treatment (14‐day post‐SCI), immunostained with antibodies directed against Type M1 macrophage marker iNOS, Type M2 macrophage marker CD206, and GAPDH as a control. f and g, Graphs of quantitative density analysis for western blot results for iNOS (f) and CD 206 (g) in each treatment condition normalized to control spinal cords. h, M1/M2 macrophage protein expression ratios calculated using quantitative density analysis of western blot data for iNOS and CD206. i and j, qRT‐PCR analysis of relative mRNA expression levels for M1 macrophage marker CLL2 (i) and M2 macrophage marker CD206 (j). [ΔCT was calculated against the endogenous control (GAPDH), and ΔΔCT (mRNA level) was calculated against the ΔCT of the control.] k, M1/M2 macrophage mRNA ratios calculated by using qRT‐PCR results for CLL2 and CD206. Values are presented as means ± SEM. A 1‐way ANOVA followed by the Tukey‐Kramer test or the Kruskal‐Wallis test followed by the Steel‐Dwass test was conducted. *: P < .05, **: P < .01, SCI: spinal cord injury, MSC: mesenchymal stem/stromal cell, MSC‐sEVs: small extracellular vesicles derived from MSCs, iNOS: inducible nitric oxide synthase, DAPI: 4′,6‐diamidino‐2‐phenylindole, GAPDH: Glyceraldehyde 3‐phosphate dehydrogenase, CCL2: C–C motif chemokine ligand 2, ΔCT: delta‐cycle threshold, qRT‐PCR: quantitative reverse transcription‐polymerase chain reaction, FC: fold change
Consistent with gross visual impressions of staining for macrophage subtypes, western blot analysis showed increases in macrophage markers in all contused spinal cords relative to intact controls, as well as increases in M2 markers, relative to M1 markers, in both MSC and F‐sEVs treatment conditions, when compared with the control or 1‐sEV treated conditions. Both iNOS (Figure 3e, f) and CD206 (Figure 3e, g) protein levels were increased in all lesion conditions (n = 6‐7/group), relative to intact controls, at 1‐week post‐treatment (14 post‐SCI), and modestly elevated at 70‐day post‐contusion. However, 1‐week post‐treatment, the levels of iNOS were significantly lower and CD206 levels were significantly higher in the MSC and F‐sEVs groups, compared to Vehicle, while the 1‐sEVs group showed a similar trend but did not differ statistically from the Vehicle group. This resulted in a statistically significant decrease in the M1/M2 macrophage ratios in both the MSC and F‐sEVs treatment groups relative to Vehicle treated controls, while the 1‐sEVs group showed only a trend in this direction (Figure 3h).
Gene expression analysis revealed no statistically significant differences between the levels of mRNA for the M1 marker CLL2 at any timepoint studied (n = 7‐9/group) (Figure 3i) and no differences in mRNA expression levels for the M2 marker CD206 at 3‐day post‐treatment (10‐day post‐SCI) (Figure 3i). However, levels of CD206 mRNA were significantly increased in the MSC and F‐sEVs conditions at both 14‐ and 70‐day post‐SCI (Figure 3j). This resulted in statistically significant reductions in the M1/M2 mRNA ratios in the MSC and F‐sEVs treatment conditions at both 14‐day and 70‐day post‐SCI (Figure 3k). Gene expression levels of the M2 marker arginase‐1 were also significantly increased in both the MSC and F‐sEVs group, compared to the Vehicle group (Supplementary Figure 1a), whereas the gene expression level of the M1‐related cytokine, TNF‐α, showed no statistically significant difference between treatment groups at 14‐day post‐SCI (Supplementary Figure 1b).
3.5. Both infusion of MSCs and fractionated dosing of MSC‐sEVs upregulate TGF‐β and TGF‐β receptors in the injured spinal cord
As M2 macrophages are well known to secrete TGF‐β (Freytes et al., 2013; Hu et al., 2015), we attempted to assess whether the TGF‐β signalling pathway was upregulated by MSCs or MSC‐sEVs infusion in this model. Examination of immunostained sections 7‐day post‐treatment revealed that TGF‐β1 staining was low within macrophages at lesion sites of animals in the Vehicle treated group (Figure 4a‐c), while noticeably stronger TGF‐β1 staining was observed within CD206+ M2 macrophages at lesion sites in the F‐sEV treated group (Figure 4d‐f). Although staining for TGF‐β1 was stronger within CD206+ cells in the F‐sEVs treatment group (Figure 4d‐f) than in the Vehicle group (Figure 4a‐c), no apparent differences were observed for TGF‐β1 staining intensity in astrocytes or other cells in the region, which exhibited low levels of staining under all treatment conditions (not shown). Consistent with the increased number of macrophages observed at the lesion sites throughout the study period and the apparent high levels of TGF‐β1 within macrophages, levels of both TGF‐β1 and TGF‐β2 mRNA were increased in all lesion conditions, relative to intact control spinal cords at all timepoints studied (Figure 4 g, h). Furthermore, three days after the onset of treatment (10‐day post‐SCI), the expression of TGF‐β1 mRNA was significantly increased in both the MSC and 1‐sEVs groups, compared to the Vehicle group (Figure 4 g, n = 7‐10/group). At this timepoint, there was no statistically significant increase in TGF‐β1 or TGF‐β2 in the fractionated treatment F‐sEVs group relative to the Vehicle group (Figure 4 g, h), although TGF‐β levels showed a trend towards an increase at this time. At 1 week post‐treatment (14‐day post‐SCI), however, the pattern of TGF‐β gene expression changed. At 14‐day post‐SCI, levels of TGF‐β1 mRNA were significantly increased in the MSC and F‐sEV treated conditions compared to Vehicle, while the 1‐sEVs group did not differ from the Vehicle treatment (Figure 4 g). Notably, levels of TGF‐β1 mRNA were three‐fold higher in the F‐sEVs condition than in the 1‐sEVs and Vehicle conditions at 14‐day post‐SCI. This pattern was repeated for TGF‐β2 (Figure 4h) gene expression, which was significantly increased in the F‐sEVs treatment condition, compared to all other treatment groups at 14‐day post‐SCI. By 70‐day post‐SCI, levels of TGF‐β and TGF‐β receptor genes did not differ across treatments.
FIGURE 4.
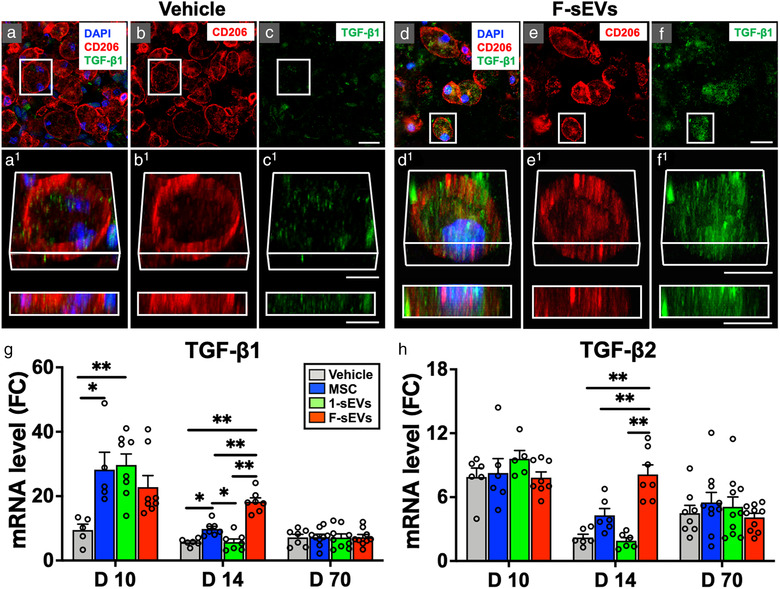
Both infusion of MSCs and fractionated dosing of MSC‐sEVs upregulate TGF‐β in the injured spinal cord. a‐f, Confocal micrographs of representative regions of frozen sectioned contused spinal cord harvested 14‐day post‐SCI and 7 days after the treatment with Vehicle (a‐c, left) or fractioned MSC‐sEVs (d‐f, right), immunostained with antibodies directed against Type M2 macrophage marker CD206 (red), and TGF‐β1 (green), and counterstained with DAPI (blue). Images in each group (a‐c, and d‐f) from left to right show the same area with fluorescence channels for CD206, TGF‐β1, & DAPI (a and f), CD206 (b and e), and TGF‐β1 (c and f). Note that TGF‐β1 staining is strongly expressed and localized within macrophages staining positive for the M2 type marker in the F‐sEVs group, while such staining is negligible in the Vehicle group. Scale bars indicate 20 μm (c and f) and 10 μm (c1 and f1). g and h, Graphs illustrating the qRT‐PCR analysis of the relative expression of mRNAs for TGF‐β1 (g) and TGF‐β2 (h). [The ΔCT was calculated against the internal control (GAPDH), and ΔΔCT (mRNA level) was calculated against the ΔCT of the control.] Note the transient upregulation of TGF‐β1 at 10‐day post‐ SCI in the 1‐sEVs treatment group, compared with the upregulation of TGF‐β1 and TGF‐β2 expression in both the MSC and the F‐sEVs treatment groups at 14‐day post‐SCI. Values are presented as means ± SEM. A 1‐way ANOVA followed by the Tukey‐Kramer test or the Kruskal‐Wallis test followed by the Steel‐Dwass test was conducted. *: P < .05, **: P < .01, TGF: transforming growth factor, MSC: mesenchymal stem/stromal cell, MSC‐sEVs: small extracellular vesicles derived from MSCs, TGF‐β: transforming growth factor‐beta, DAPI: 4′,6‐diamidino‐2‐phenylindole, qRT‐PCR: quantitative reverse transcription‐polymerase chain reaction, ΔCT: delta‐cycle threshold, SCI: spinal cord injury, FC: fold change
Immunostaining, mRNA analysis, and protein expression levels also supported an upregulation of TGF‐β receptors at the lesion site in the MSC and F‐sEVs treatment conditions, which was potentially linked to the microvasculature. Immunohistochemistry of lesions showed that strong TGF‐βR2 immunostaining was observed along capillary vessels of spinal cords in the F‐sEVs group (Figure 5a‐d), while such staining was rarely observed in the Vehicle group (not shown). Quantitative PCR showed that the levels of TGF‐βR1 mRNA (Figure 5e) and TGF‐βR2 mRNA (Figure 5f) were increased in all lesion conditions relative to intact control cords at almost all timepoints. As with TGF‐β1 and TGF‐β2, the levels of TGF‐βR1 and TGF‐βR2 mRNA did not differ between Vehicle and other treatment groups at 3 days after the onset of treatment (10‐day post‐SCI) (Figure 5e, f; n = 7‐10/group). At 14‐day post‐contusion, however, the levels of TGF‐βR1 and TGF‐βR2 mRNA were almost four‐ and five‐fold higher in the fractionated treatment F‐sEVs group than the one dose 1‐sEVs and Vehicle groups, respectively (Figure 5e, f). Western blots also showed a 1.8‐fold increase in signal intensities of TGF‐βR2 protein in both the MSC and F‐sEVs groups relative to Vehicle at 14‐day post‐SCI, while the 1‐sEVs group did not show an increase in TGF‐βR2 protein (Figure 5 g, h, n = 8‐10/group). No statistically significant differences in TGF‐βR2 protein were observed between treatment conditions at 70‐day post‐SCI (Figure 5h).
FIGURE 5.
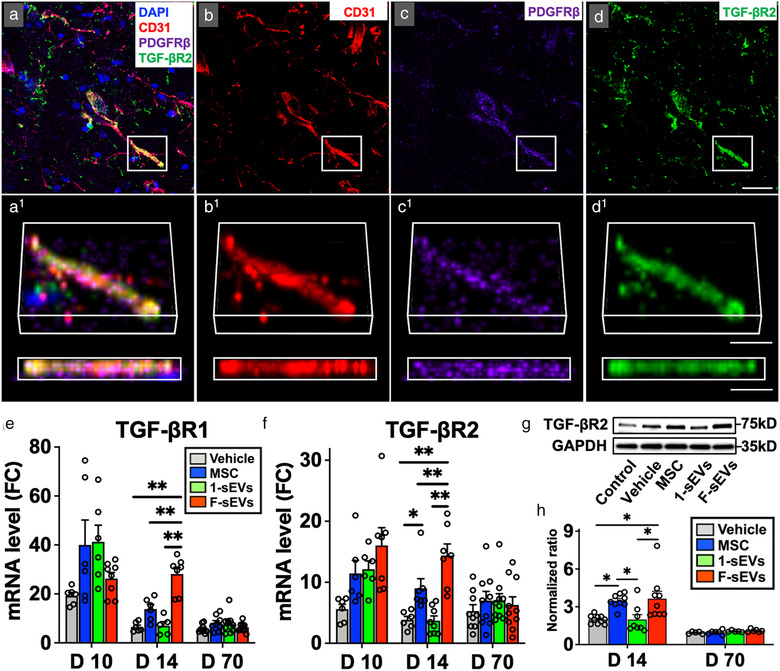
Both infusion of MSCs and fractionated dosing of MSC‐sEVs upregulate TGF‐β receptors in the injured spinal cord. a‐d, Confocal micrographs of a representative region of a frozen sectioned contused spinal cord harvested 7 days after the treatment with fractioned MSC‐sEVs (14‐day post‐SCI), immunostained with antibodies directed against endothelial cell marker CD31 (red), PDGFRβ (purple), and TGF‐βR2 (green), and counterstained with DAPI (blue). Images in each group from left to right show the same area with fluorescence channels for CD31, PDGFRβ, TGF‐βR2, & DAPI (a), CD31 (b), PDGFRβ (c), and TGF‐βR2 (d). Note that TGF‐βR2 is localized along capillaries. Scale bar indicates 20 μm (d) and 10 μm (d1). e and f, Graphs illustrating qRT‐PCR analysis of the relative expression of mRNAs for TGF‐βR1(e) and TGF‐βR2(f). [The ΔCT was calculated against the internal control (GAPDH), and ΔΔCT (mRNA level) was calculated against the ΔCT of the control.] g, Representative images of western blots from lesioned tissue harvested 7 days after the onset of treatment (14‐day post‐SCI), showing levels of TGF‐βR2 protein. h, Quantitative density analysis of western blot results for TGF‐βR2. The normalized ratio was calculated relative to intact controls. Note the upregulation of TGF‐βR1, TGF‐βR2, and increased TGF‐βR2 protein expression in both the MSC and F‐sEVs treatment groups at 14‐day post‐SCI. Values are presented as means ± SEM. A 1‐way ANOVA followed by the Tukey‐Kramer test or the Kruskal‐Wallis test followed by the Steel‐Dwass test was conducted. *: P < .05, **: P < .01, MSC: mesenchymal stem/stromal cell, MSC‐sEVs: small extracellular vesicles derived from MSCs, SCI: spinal cord injury, PDGFRβ: platelet‐derived growth factor receptor‐beta, TGF‐βR: transforming growth factor‐beta receptor, ΔCT: delta‐cycle threshold, GAPDH: Glyceraldehyde 3‐phosphate dehydrogenase, DAPI: 4′,6‐diamidino‐2‐phenylindole, FC: fold change
Overall, SCI lesions in the MSC and F‐sEVs groups showed increases in TGF‐β production and upregulation of TGF‐β receptors over a 1 week period post‐treatment. In contrast, we did not observe evidence of acute upregulation of TGF‐β signally in organs including lungs, kidney, liver and spleen at 10‐day post‐SCI in animals infused with either control vehicle, MSCs or MSC‐sEVs (n = 3 each group, Supplementary Figure 2a‐d). We did observe a trend towards an increase in TGF‐β and TGF‐β receptors in the brain after the MSC or MSC‐sEVs infusion (Supplementary Figure 2e), which would be consistent with the degradation of ascending axons, but the TGF‐β signalling response observed in the brain was much smaller than that in the spinal cord (1.5‐2.0 fold increase in TGF‐βR2 in the brain compared to 20–30 fold increase in the spinal cord). Likewise, we saw no evidence of upregulation of mRNAs for the fibrotic markers collagen type 1 alpha 1 (Col1a1) or fibronectin 1 (FN1) in lungs or other organs after infusion of MSCs or MSC‐sEVs. Rather, we observed a trend towards slight decreases in fibrotic markers after SCI in lungs, kidney, liver and spleen in SCI animals under all treatment conditions (Supplementary Figure 2a‐e). These observations argue that MSC‐derived sEVs specifically targeted TGF‐β signalling in the M2‐type macrophages at the lesion site, and not resident macrophages or TGF‐β producing cells in general, and that neither infusions of MSC or sEVs induced significant fibrotic responses in organs which might cause safety concerns.
3.6. Both infusion of MSCs and fractionated dosing of MSC‐sEVs reduce BSCB leakage
To assess whether the MSC‐sEVs‐induced upregulation of the TGF‐β pathway correlated with restoration of BSCB integrity after SCI, the effects of MSCs and MSC‐sEVs treatments on dye extravasation in the injured spinal cord were evaluated. Even by the naked eye, intense extravasation of EvB dye could readily be observed at impact sites under all lesion conditions at 10‐ and 14‐day post‐SCI (Figure 6a). However, while no visible differences were apparent between dye extravasation levels for different treatment conditions at 10‐day post‐SCI, dye at the lesion site was noticeably diminished in the MSC and F‐sEV treated conditions compared to the 1‐sEVs and Vehicle treatment conditions 14‐day post‐SCI (Figure 6a).
FIGURE 6.

Both infusion of MSCs and fractionated dosing of MSC‐sEVs reduce BSCB leakage in the injured cord. a, Photographs illustrating the typical appearances of spinal cords of Evans blue injected animals from left to right for the Vehicle‐treated group, MSC‐treated group, 1‐sEV‐treated group, and F‐sEV‐treated group harvested at 10‐day post‐SCI (upper row) and 14‐day post‐SCI (lower row). Note that while no visible differences were apparent between dye extravasation levels for different treatment conditions at 10‐day post‐SCI, blue dye at the lesion site was noticeably diminished in both the MSC and F‐sEV treated conditions compared to the 1‐sEVs or Vehicle treatment conditions at 14‐day post‐SCI. Scale bar indicates 3 mm. b and c, Quantification of EvB concentrations (b) and total dye content (c) of the entire 1 cm segment surrounding the lesion at 10‐ and 14‐day post‐SCI. Note: EvB concentration and total dye content of lesioned tissue were significantly reduced in both the MSC treated and F‐sEV treated compared to the Vehicle treated condition at 14‐day post‐SCI. Values are presented as means ± SEM. A 1‐way ANOVA followed by the Tukey‐Kramer test or the Kruskal‐Wallis test followed by the Steel‐Dwass test was conducted. *: P < .05, **: P < .01, MSC: mesenchymal stem/stromal cell, MSC‐sEVs: small extracellular vesicles derived from MSCs, SCI: spinal cord injury, EvB: Evans blue
Quantitative analysis of EvB content in a 1 cm segment surrounding the lesion confirmed that dye leakage levels were greatly increased in all lesion conditions (n = 5‐8/group), compared to intact controls, at 10‐day post‐SCI (Figure 6b, c) and the dye concentration (Figure 6b) and total dye content (Figure 6c) did not differ significantly between treatment conditions at this time. At 14‐day post‐SCI, however, EvB concentration (Figure 6b) and total dye content (Figure 6c) in lesioned tissue were significantly reduced in the MSC and F‐sEV treated compared to the Vehicle group, (MSC and F‐sEVs values differ from Vehicle group at P < .001). Values for the 1‐sEVs group did not differ significantly from the Vehicle group but did differ from the MSC‐ and F‐sEVs groups (1‐sEVs values differ from MSC and F‐sEVs groups at P < .05, and P < .01, respectively.)
Importantly, MSC‐sEVs‐induced reductions in BSCB permeability were much greater when MSC‐sEVs were delivered in three fractions over the course of three consecutive days (F‐sEVs) than when the same dose of MSC‐sEVs was delivered in a single treatment (1‐sEV).
3.7. Both infusion of MSCs and fractionated MSC‐sEVs dosing promote increases of tight and adherens junction proteins
Analyses of expression of both mRNA and protein levels for critical blood‐brain barrier/blood‐spinal cord barrier (BBB/BSCB) proteins indicated an upregulation of tight and adherens junction proteins in MSC and F‐sEV treated conditions 14‐day post‐SCI, but not in the 1‐sEVs group.
Gene expression analysis showed that levels of vascular proteins ZO‐1 (Figure 7a), occludin (Figure 7b), and N‐cadherin (Figure 7c) mRNA were increased in all SCI treatment groups (n = 7‐9/group), relative to the intact condition at 10‐, 14‐ and 70‐day post‐SCI, with no significant differences observed between treatment groups at 10‐day post‐SCI or 70‐day post‐SCI (Figure 7a‐c). At 14‐day post‐SCI, however, the gene expression levels of ZO‐1 (Figure 7a) and occludin (Figure 7b) began to fall from their 10‐day post‐SCI levels in the Vehicle and 1‐sEVs group, and N‐cadherin (Figure 7c) mRNA remained unchanged, while in the MSC and F‐sEVs groups, levels of ZO‐1 and occludin held steady and N‐cadherin levels increased. This resulted in statistically significant increases in ZO‐1 (Figure 7a), occludin (Figure 7b), and N‐cadherin (Figure 7c) mRNA in the MSC and F‐sEVs groups relative to the Vehicle groups at 14‐day post‐SCI (Figure 7a‐c). The most highly enriched tight junction protein in the BBB/BSCB, Claudin‐5 (Greene et al., 2019), did not show differences in expression levels between treatment groups (Supplementary Figure 3a‐b).
FIGURE 7.
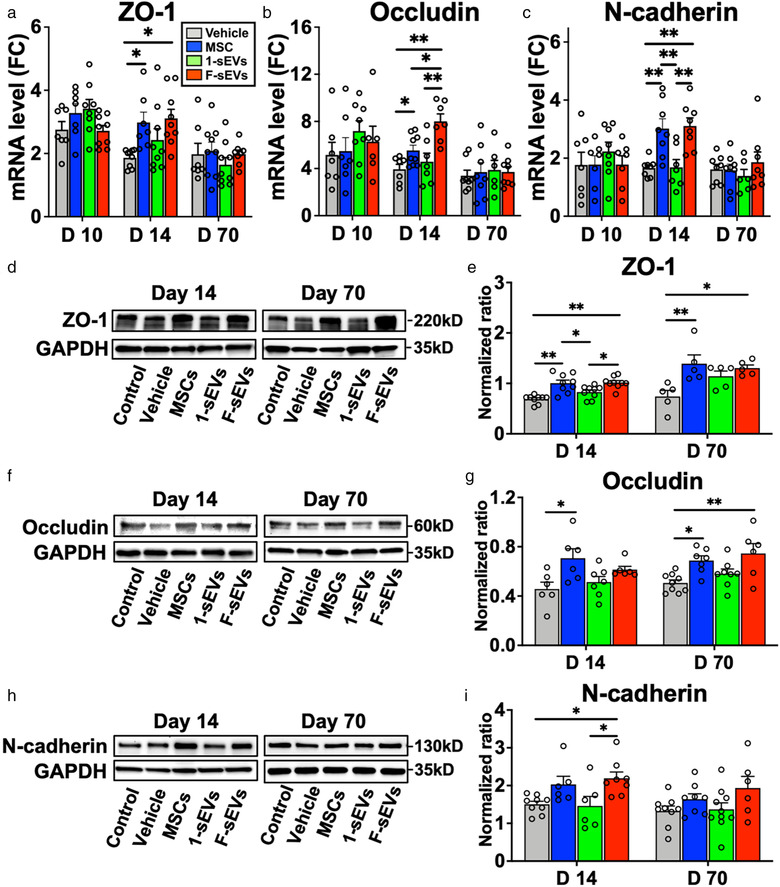
Both infusion of MSCs and fractionated MSC‐sEVs dosing promote increases of tight and adherens junction proteins. a‐c, Graphs showing qRT‐PCR analysis of relative mRNA expression levels of ZO‐1 (a), occludin (b), and N‐cadherin (c). [ΔCT was calculated against GAPDH control, and ΔΔCT (mRNA level) was calculated against the ΔCT of the control.] Note significant increases were observed in ZO‐1, occludin, and N‐cadherin mRNA in both the MSC and F‐sEVs treatment groups relative to the Vehicle groups at 14‐day post‐SCI. d‐i, Representative images of western blots and quantitative density analysis of the lesion sites harvested at 14‐ and 70‐day post‐SCI, showing levels of ZO‐1 (d and e), occludin (f and g), and N‐cadherin (h and i). Note that levels ZO‐1, occludin, and N‐cadherin were all significantly increased in the MSC treated condition and levels of ZO‐1 and N‐cadherin levels were significantly increased in the F‐sEVs treatment condition relative to the Vehicle treated condition at 14‐day post‐SCI. Animals in both the MSC and F‐sEVs treatment conditions also showed increases in ZO‐1 and occludin protein levels at 70‐day post‐SCI. Values are presented as means ± SEM. A 1‐way ANOVA followed by the Tukey‐Kramer test or the Kruskal‐Wallis test followed by the Steel‐Dwass test was conducted. *: P < .05, **: P < .01, MSC: mesenchymal stem/stromal cell, MSC‐sEVs: small extracellular vesicles derived from MSCs, ΔCT: delta‐cycle threshold, SCI: spinal cord injury, GAPDH: Glyceraldehyde 3‐phosphate dehydrogenase, FC: fold change
Western blot analysis supported the notion that increased gene expression led to increased production of tight and adherens junctions in MSC and F‐sEV treated animals. At 14‐day post‐SCI, the levels of ZO‐1 (Figure 7d, e), occludin (Figure 7f, g), and N‐cadherin (Figure 7h, i) were all significantly increased in the MSC treated condition, and the levels of ZO‐1 (Figure 7d, e), and N‐cadherin (Figure 7h, i) were significantly increased in the F‐sEVs treatment condition relative to the Vehicle group. Furthermore, both MSC and F‐sEVs treatments resulted in sustained increases in ZO‐1 (Figure 7d, e) and occludin (Figure 7f) protein levels at 70‐day post‐SCI.
4. DISCUSSION
Systemic delivery of mesenchymal stem/stromal cells has therapeutic effects in experimental models of SCI (Honmou et al., 2012) and ongoing clinical studies are evaluating MSC therapy in SCI patients (Honmou et al., 2021; Kabat et al., 2020). The results of this study provide evidence that IV‐infused MSCs, which do not reach the lesion site (Matsushita et al., 2015; Quertainmont et al., 2012), promote recovery after SCI by releasing sEVs that are taken up by M2 macrophages at the lesion site. Importantly, when MSC‐sEVs were injected in fractionated doses, the therapeutic effect was greater than when the same quantity of MSC‐sEVs was delivered in a single dose. We found that fractioned MSC‐sEVs delivery was as therapeutically effective as MSC infusion. The results of this study suggest a model of action (Figure 8) in which MSC‐sEVs uptake by M2‐type macrophages at the SCI site leads to upregulation of TGF‐β and stimulates a TGF‐β signalling pathway at the lesion that leads to increased expression of key BSCB‐associated proteins, accelerating restoration of BSCB integrity and promotion of functional recovery.
FIGURE 8.
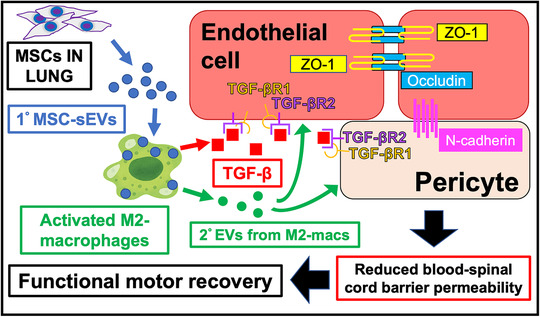
Schematic representation of the proposed mechanisms underlying the therapeutic effects of MSC‐sEVs on the blood‐spinal cord barrier (BSCB) in spinal cord injury. Circulating MSC‐sEVs (1° MSC‐sEVs: blue circles) produced either by release of sEVs from IV infused MSCs trapped in the lungs or by repeated IV dosing of MSC‐sEVs are taken up by M2 macrophages in the lesion. sEVs uptake by M2 macrophages promotes sustained M2 macrophage polarization and increased TGF‐β (red squares) production. TGF‐β or TGF‐β surface expressing second order EVs (2° EVs) released by the MSC‐sEV‐stimulated M2 macrophages (green circles) bind to TGF‐βR2 on endothelial cells and/or pericytes, to complex with TGF‐βR1 with thereby enhance avidity as a surface array to activate downstream pathways, which lead to upregulation of junctional proteins ZO‐1, occludin, and N‐cadherin contributing to restoration of BSCB integrity. The restored BSCB provides a more favourable environment for neuronal functioning and promotes greater recovery of locomotor functioning. MSCs: mesenchymal stem/stromal cells, MSC‐sEVs: small extracellular vesicles derived from MSCs, M2‐macs: M2 macrophages, 1° MSC‐sEVs: first order MSC‐sEVs, 2° EVs: second order extracellular vesicles released from activated M2 macrophages, TGF‐β: transforming growth factor‐beta, TGF‐βR1 or 2: transforming growth factor‐beta receptor 1 or 2, BSCB: blood spinal cord barrier
Consistent with our previous findings (Matsushita et al., 2015), we found no detectable engraftment of labelled MSCs at the lesion site after infusion of a large number of DiR‐labeled MSCs 1‐week post‐SCI. However, many DiR‐labeled “hotspots” stained with antibodies to the exosome marker CD63 were identified within M2 macrophages at the lesion site, indicating that IV infused MSCs, previously found to lodge transiently in the lungs (Matsushita et al., 2015), do in fact release sEVs that target M2‐type macrophages at the SCI site.
Previous studies reported therapeutic efficacy with both a high single dose (Huang et al., 2017; Ji et al., 2019) and multiple dosing (Lu et al., 2019; Wang et al., 2018) of MSC‐sEVs delivered acutely immediately after SCI induction. In the present study, when treatment with MSC‐sEVs was delayed 1‐week post‐SCI, functional recovery was observed only when MSC‐sEVs were delivered in fractionated doses over 3 days. The statistically significant improvement in functional recovery after a single infusion of MSCs, or fractionated delivery of MSC‐sEVs, but not after infusion of the same quantity of MSC‐sEVs delivered in a single dose, suggests that the efficacy of IV delivered MSC results from release of sEVs over time. Since MSCs that were IV infused into rats with SCI were detected within lungs for up to 3 days post‐infusion (Matsushita et al., 2015), IV infused MSCs might release sEVs into the circulation for as long as 3 days post‐infusion. Previously, we observed that IV delivery of a large dose of DiR‐labeled MSC‐sEVs resulted in transient appearance of DiR fluorescence within the kidneys at 2–4 h, but not at 24 h, suggesting that MSC‐sEVs may be rapidly cleared from the system (Lankford et al., 2018). These observations suggest that the therapeutic efficacy of MSC‐sEVs for the degree of injury in our severe SCI model may require more prolonged stimulation of the targeted M2 macrophages than can be achieved by short‐term exposure. It is possible that microRNAs transferred to targeted cells via exosomes are quickly degraded after uptake and exert only short term effects on gene expression unless additional exosomes are taken up. Moreover, an earlier study revealed that the injured spinal cord environment promotes polarization of M1 cytotoxic macrophages, especially from 7‐ to 14‐day post‐SCI, although both M1 and M2 macrophages are present in the lesion (Kigerl et al., 2009). It is plausible that prolonged stimulation by MSC‐sEVs in the injured spinal cord lesion is required for M2 macrophages to stabilize the M2 phenotype or reduce their death. In animals with less severe lesions or earlier timepoints of treatment, a single dose of sEVs may be efficacious, particularly if the dose is large enough to maintain reasonably high circulating levels of vesicles even with systemic clearance.
The observed upregulation of TGF‐β1 in SCI lesions at the 3‐day timepoint after a single dose of MSC‐sEVs (1‐sEVs), but not at the 1‐week timepoint for this treatment condition, suggests that short‐term exposure to MSC‐sEVs may only induce short lived changes in gene expression. In contrast, the increased expression of TGF‐β mRNA and TGF‐βR2 protein, as well as increased levels of BSCB‐related mRNA and protein, at 1‐week post‐treatment in both MSC and F‐sEVs groups, argues that a prolonged presence of MSC‐sEVs can induce sustained changes in gene regulation. Further studies will be necessary to determine whether extending the treatment period with additional MSC‐sEVs injections or osmotic pump delivery may promote even greater recovery.
Although both MSC and fractionated MSC‐sEVs dosing were associated with increased expression of M2 markers at the lesion site 7 days after treatment, it is not clear how MSC‐sEVs affect macrophages. Further in vitro studies will be needed to understand whether and how sEVs may affect macrophage polarization. Others have reported that MSC‐sEVs increased the percentage of macrophages expressing M2 markers in models of spinal cord ischemia (Sun et al., 2018) and wound healing (He et al., 2019), suggesting that MSC‐sEVs may promote polarization of macrophages to an M2 phenotype. In this study, the increase in expression of M2 markers relative to M1 markers in both the MSC and F‐sEV treatment groups argue that MSC‐sEV uptake may affect macrophage polarity to favour an M2 phenotype. However, we did not observe DiR hotspots outside of CD206 expressing macrophages in animals infused with DiR‐labeled MCSs in this study nor in a prior study (Lankford et al., 2018). The later study extensively examined spinal cord sections in DiR‐labeled sEVs infused animals and failed to identify DiR fluorescence in any iNOS expressing macrophages within the lesion. Furthermore, our in vitro studies here confirm that MSC‐sEVs are only taken up by cells which had been polarized to an M2 phenotype with IL‐4 treatment. Our lack of detection of DiR‐MSC‐sEVs within cells exhibiting M1 markers or resting M0 phenotype in vivo or in vitro argues for a preferential receptivity of some M2‐type macrophages for MSC‐sEV uptake, rather than an MSC‐sEV‐induced shift from an M1 or M0 phenotype to an M2 phenotype. An alternative explanation may be that MSC‐sEV uptake stabilizes the M2 phenotype or reduces their death, resulting in an increase in the relative proportion of M2 macrophages in the lesion.
It is noteworthy that not all CD206 expressing macrophages appeared to take up DiR‐MSC‐sEVs, either in vivo or in vitro, implying that expression of this aspect of an M2 phenotype alone is not sufficient for MSC‐sEV uptake. It is not clear why DiR‐labeled MSC‐sEVs seem to be taken up only by a subset of macrophages expressing M2 markers. We hypothesize that such exosome uptake may be an active process induced in some M2 macrophages by the SCI environment and reflected in a certain signature of receptive ligands for binding specific sEVs. Since all cell types produce and release exosomes and these nanovesicle structures remain stable in a wide range of environments, macrophages may selectively bind and then take up specific subsets of MSC‐derived exosomes only after they have been primed in some way to express a certain surface signature and ‘expect’ congruity from the corresponding surface signature of this particular class of exosomes. Based on our in vitro work, low pH and the presence of phagocytosis‐inducing cell debris, as well as anti‐inflammatory cytokines, may be part of this priming process which causes exposed subsets M2 macrophages versus all macrophages at the site of the contusive spinal cord lesions to selectively bind and then take up surface congruent subsets of the MSC‐sEVs.
Since low pH is associated with inflammatory environments, the preferential uptake of MSC‐sEVs under low pH conditions versus neutral pH also implies that the likely site of sEV uptake by macrophages is within the lesion itself, rather than sEVs being taken up by circulating macrophages. This would be consistent with the observations in our previous study (Lankford et al., 2018), showing that the sizes of the DiR hot spots increased in all DiR containing macrophages at 24 h after DiR‐MSC‐sEVs infusion, compared with cells examined 2 h and 4 h post‐infusion. If macrophages took up DiR labelled sEVs in the periphery and ceased accumulation after entering the CNS parenchyma, one would expect macrophages exhibiting a broader range of hot spot sizes at the later time point, which was not observed. The lack of increase in TGF‐β expression in lung, liver, kidney and spleen after MSC or sEV infusion further supports the notion that macrophages within the spinal cord lesion site were the targets of the MSC‐derived sEVs.
The mechanism of therapeutic action of sEVs in this model has not yet been fully established. MSC‐sEVs are known to contain a wide range of RNA and protein cargos, including cytokines and microRNAs, which can alter cell function (Ferguson et al., 2018; Toh et al., 2018). The regulatory microRNA‐21 is one of the 10 most highly expressed microRNAs in MSC‐sEVs. It is a key mediator of the anti‐inflammatory response in macrophages within tissue (Sheedy, 2015) and we have confirmed the presence of miR‐21 within our MSC‐sEVs (Figure 1e). Recent reports found that increased miR‐21expression in exosomes from primed MSCs correlated with increased M2 polarization in vitro and reduced sepsis in vivo (Yao et al., 2020), while a reduction in miR‐21 expression in exosomes from MSCs derived from obese animals correlates with reduced therapeutic efficacy of MSC‐sEVs for acute SCI (Ji et al., 2019). Taken together, these findings suggest that miR‐21 cargos in MSC‐sEVs taken up by macrophages at the lesion site could play a key role in the therapeutic response. Further experiments using siRNAs are needed to evaluate whether miR‐21 or other specific microRNA cargos may play critical roles in the effects of MSC‐sEVs on SCI.
The observation of substantial uptake of MSC‐sEVs by M2 macrophages 48 h after MSC infusion argues that uptake of MSC‐sEVs by macrophages is likely responsible for initiating at least some of the therapeutic effects of MSCs. It is well known that macrophages expressing classic M2 markers play crucial roles in suppressing inflammation by secreting anti‐inflammatory cytokines, including TGF‐β (Freytes et al., 2013; Hu et al., 2015), thereby increasing tissue sparing and providing a more conducive environment for axon regeneration and neuronal functioning after SCI (Hu et al., 2015; Nakajima et al., 2012). In the present study, MSC and F‐sEV treatment not only resulted in increased expression of M2 specific markers but also increases in TGF‐β mRNA and protein. These findings are consistent with reports that MSC‐sEVs induce high levels of anti‐inflammatory TGF‐β transcripts in monocyte lineage cells in vitro and modulate immunological activities related to graft rejection with repeated injections in vivo (Zhang et al., 2014) and argue that uptake of MSC‐sEVs by macrophages may directly result in increased TGF‐β production by these cells. Although other cells, including neurons and astrocytes, can produce TGF‐β, we did not detect differences in TGF‐β staining in those cells between the different experimental groups. Further in vitro studies are planned to investigate whether MSC‐sEVs or MSC‐sEV treated macrophages alter TGF‐β production in astrocytes or other potential CNS targets.
Further investigations of tissue responses to treatments indicated that MSC and F‐sEVs infusions resulted in changes consistent with the targeting of the microvasculature. Both MSC and F‐sEVs infusions resulted in overall increases in TGF‐β receptor expression in the lesion and an apparent increase in TGF‐β receptor 2 staining of the microvasculature. Furthermore, expression of the BSCB proteins ZO‐1, occludin, and N‐cadherin and their mRNAs were increased and BSCB leakage was reduced following MSC and F‐sEV treatment. Since DiR‐MSC‐sEVs were not detected within endothelial cells or pericytes, the microvasculature may be an indirect target of MSC‐sEV action, with increased production of TGF‐β (presumably by macrophages which take up MSC‐derived exosomes) stimulating changes in endothelial cells and pericytes (Shelke et al., 2019; Wada et al., 2010). However, since individual sEVs are below the detection threshold for our imaging, we cannot rule out the possibility that MSC‐sEVs may have been directly taken up by endothelial cells or pericytes at levels too low to detect. Alternatively, according to the study by Shelke et al. (2019), it is possible that the primary targeted M2 macrophages go on to produce secondary extracellular vesicles which stimulate the neighboring microvasculature to induce synthesis of the downstream microvascular proteins we observed (Figure 8).
The molecular mechanism for TGF‐β influence on the BSCB is yet to be elucidated. Initially, TGF‐β binds to TGF‐βR2 dimers and activates the intracellular kinase domain of TGF‐βR1, initiating phosphorylation cascades in both SMAD (Heldin et al., 1997; Massague, 1996) and SMAD‐independent signalling pathways. The SMAD independent pathways include PI3K mediated phosphorylation of AKT (Lamouille & Derynck, 2007; Qureshi et al., 2007; Zhang et al., 2013) which promotes transcription of many gene products (Seoane et al., 2004), some of which play key roles in regulating tight junction proteins in the CNS (Li et al., 2015; Sun et al., 2019). Further analysis will be necessary to assess which downstream SMAD or non‐SMAD pathways may be responsible for the observed changes in tight junction and adherens junction proteins.
The results from this study indicate that sEVs are released by IV delivered MSCs and have a role in their therapeutic action in SCI. Our data support a model of action in which exosomes released by MSCs into the general circulation are taken up by M2 macrophages at the lesion site stimulating them to increase the production of TGF‐β, which in turn upregulates TGF‐β receptors on pericytes or endothelial cells and induces a cascade of molecular responses leading to repair of the microvasculature and restoration of BSCB integrity. This restoration of the BSCB provides an environment more conducive to neuronal repair and improved function (Figure 8). Clinical studies using MSC therapy for SCI (Kabat et al., 2020) and stroke (Honmou et al., 2011) have established safety and we are evaluating the efficacy in an ongoing clinical study (Honmou et al., 2021). While a single dose of MSCs has been shown to be therapeutic in experimental models of SCI and stroke, the data presented here suggest that multiple dosing protocols may be necessary for optimal clinical efficacy using MSC‐sEVs. Unlike MSCs which could pose a risk of lung emboli with multiple dosing, the small size and clearance of excess MSC‐sEVs suggest that multiple dosing of MSC‐sEVs would be safer. sEVs are more stable and storable than living cells (Sokolova et al., 2011), thus allowing greater flexibility in treatment protocols.
5. CONCLUSION
Intravenously infused MSCs release sEVs in vivo over time and target M2 macrophages to increase TGF‐β and induce TGF‐β signalling pathways to mediate therapeutic effects in SCI. Intravenous delivery of MSC‐sEVs may represent an important noncellular therapeutic approach for SCI treatment.
CONFLICT OF INTERESTS
The authors report no competing interests.
AUTHOR CONTRIBUTIONS
Masahito Nakazaki, Tomonori Morita, Karen L. Lankford, Jeffery D. Kocsis and Philip W Askenase designed the study. Karen L. Lankford, Masahito Nakazaki, Tomonori Morita and Jeffery D. Kocsis wrote the manuscript. Masahito Nakazaki, Tomonori Morita and Karen L. Lankford performed experiments. Masahito Nakazaki, Tomonori Morita, Karen L. Lankford and Jeffery D. Kocsis, analysed data.
Supporting information
Supplementary Figure 1. Both MSC infusion and fractioned MSC‐sEVs dosing upregulate Arg1 expression, but do not decrease TNF‐α gene expression. a and b, Gene expression analysis (qRT‐PCR) for the M2 macrophage maker, Arg1 (a) and M1 macrophage‐related cytokine, TNF‐α (b). The ΔCT was calculated against the endogenous control (GAPDH), and ΔΔCT (mRNA level) was calculated against the ΔCT of the control. Note the gene expression levels of TNF‐α and Arg1 were increased in all treatment groups at day14 post‐SCI, respectively. However, the level of Arg1 in both the MSC infusion and F‐sEVs treatment group showed a significant increase compared to the Vehicle treatment group. Values are presented as means ± SEM. A 1‐way ANOVA followed by the Tukey‐Kramer test was conducted. *: P < .05, **: P < .01, MSC: mesenchymal stem/stromal cell, MSC‐sEVs: small extracellular vesicles derived from MSCs, Arg1: Arginase 1, TNF: Tumour necrosis factor, qRT‐PCR: quantitative reverse transcription‐polymerase chain reaction, ΔCT: delta‐cycle threshold, GAPDH: Glyceraldehyde 3‐phosphate dehydrogenase, FC: fold change.
Supplementary Figure 2. Gene expression analysis shows that neither MSC infusion nor MSC‐sEVs upregulate TGF‐β or TGF‐β receptors in off‐target organs or increase expression of fibrotic markers in lung, spleen, kidney, liver, or brain at 10 days post‐SCI. a‐e, Graphs illustrating the results of qRT‐PCR analysis for the relative gene expression of TGF‐β1, TGF‐β2, TGF‐βR1, TGF‐βR2, Col1a1, and FN1 in lung (a), spleen (b), kidney (c), liver (d), and brain (e). The samples were collected at day 3 after the infusion (day 10 post‐SCI). Three SCI rats per group were analysed. Total purified RNAs from each organ were quantified with a spectrophotometer (NanoDrop, Technologies Inc., Wilmington, DE, USA). qRT‐PCR analysis was performed in triplicate using TaqMan® Universal Master Mix II with Uracil‐N glycosylase (UNG) and specific sets of primers and TaqMan probes (Thermo Fisher Scientific Inc.) (Supplementary Table 2) with a CFX96TM real‐time PCR detection system (Bio‐Rad), using approximately 20 ng total mRNA for each sample. The ΔCT was calculated against the internal control (GAPDH), and ΔΔCT (mRNA level) was calculated against the ΔCT of the control. Note that a trend towards an increase in TGF‐β and TGF‐β receptors after the MSC or MSC‐sEVs infusion in the brain (e), but the response was much smaller than that in the spinal cord (1.5‐2.0 fold increase in TGF‐βR2 in the brain compared to 20–30 fold increase in the spinal cord) and no effect on other lungs, kidney, liver, or spleen (a‐d). We also saw no evidence of upregulation of fibrotic markers (Col1a1 and Fn1) in lungs or other organs for any of the tissue by the infusion of MSCs or MSC‐sEVs, but rather a trend towards a slight decrease in the fibrotic markers after SCI in lungs, kidney, liver and spleen in both Vehicle, MSCs and MSC‐sEVs infused conditions. qRT‐PCR: quantitative reverse transcription‐polymerase chain reaction, TGF: transforming growth factor, TGF‐βR: transforming growth factor‐beta receptor, Col1a1: collagen, type 1, alpha 1, Fn1: fibronectin 1, GAPDH: glyceraldehyde 3‐phosphate dehydrogenase, SCI: spinal cord injury, MSC: mesenchymal stem/stromal cell, MSC‐sEVs: small extracellular vesicles derived from MSCs, ΔCT: delta‐cycle threshold, FC: fold change.
Supplementary Figure 3. Gene expression analysis shows no effect of MSC infusion or MSC‐sEVs on Claudin‐5 expression at the lesion site at 14 days post‐SCI. a, Relative level of Claudin‐5, ZO‐1, occludin, and N‐cadherin mRNA expression (calculated compared to GAPDH using qRT‐PCR) at 14 days after SCI, 7 days post‐treatment for vehicle treatment, MSC, 1‐sEVs, and F‐sEVs treatment conditions. Note that mRNA for Claudin‐5 (the most abundant tight junction protein at the blood‐spinal cord barrier) was expressed at much higher levels at the injury site than mRNA for ZO‐1, occluding, or N‐cadherin under all treatment conditions. b, Relative levels of Claudin‐5 mRNA expression at lesion sites 10 days and 14 days post‐SCI, (3 days and 7 days after onset of Vehicle, MSC, 1‐sEVs, or F‐sEVs treatments (qRT‐PCR). The ΔCT was calculated against the endogenous control (GAPDH), and ΔΔCT (mRNA level) was calculated against the ΔCT of the control. Note that although the expression levels of Claudin‐5 mRNA showed a trend towards increasing values in the MSCs, 1‐sEVs, and F‐sEVs treatment groups compared to the Vehicle treatment at day 10, these differences were not statistically significant. Values are presented as means ± SEM. A 1‐way ANOVA followed by the Tukey‐Kramer test was conducted. *: P < .05, **: P < .01, MSC: mesenchymal stem/stromal cell, MSC‐sEVs: small extracellular vesicles derived from MSCs, SCI: spinal cord injury, GAPDH: Glyceraldehyde 3‐phosphate dehydrogenase, qRT‐PCR: quantitative reverse transcription‐polymerase chain reaction, ΔCT: delta‐cycle threshold, FC: fold change.
Supplementary Table 1. Antibodies used in specific applications
Supplementary Table 2. PCR primer information used in this study
ACKNOWLEDGEMENTS
We thank Christopher Borelli for his technical assistance. This work was supported by the RR&D and BLR&D Services of U.S. Department of Veterans Affairs (B7335R, B9260L). M.N. and T.M received fellowships from Nipro Corp., Japan. M.N. received a research fellowship from the Uehara Memorial Foundation.
Nakazaki, M., Morita, T., Lankford, K. L., Askenase, P. W., & Kocsis, J. D. (2021). Small extracellular vesicles released by infused mesenchymal stromal cells target M2 macrophages and promote TGF‐β upregulation, microvascular stabilization and functional recovery in a rodent model of severe spinal cord injury. Journal of Extracellular Vesicles, 10, e12137. 10.1002/jev2.12137
Masahito Nakazaki and Tomonori Morita contributed equally to this study.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Basso, D. M., Beattie, M. S., & Bresnahan, J. C. (1996). Graded histological and locomotor outcomes after spinal cord contusion using the NYU weight‐drop device versus transection. Experimental Neurology, 139:244–256. [DOI] [PubMed] [Google Scholar]
- Beck, K. D., Nguyen, H. X., Galvan, M. D., Salazar, D. L., Woodruff, T. M., & Anderson, A. J. (2010). Quantitative analysis of cellular inflammation after traumatic spinal cord injury: Evidence for a multiphasic inflammatory response in the acute to chronic environment. Brain, 133, 433–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury, E. J., Moon, L. D., Popat, R. J., King, V. R., Bennett, G. S., Patel, P. N., Fawcett, J. W., & McMahon, S. B. (2002). Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature, 416, 636–640. [DOI] [PubMed] [Google Scholar]
- Chen, C., Skog, J., Hsu, C. H., Lessard, R. T., Balaj, L., Wurdinger, T., Carter, B. S., Breakefield, X. O., Toner, M., & Irimia, D. (2010). Microfluidic isolation and transcriptome analysis of serum microvesicles. Lab on A Chip, 10, 505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, H. J., Li Yim, A. Y. F., Griffith, G. R., de Jonge, W. J., Mannens, M., Ferrero, E., Henneman, P., & de Winther, M. P. J. (2019). Meta‐analysis of in vitro‐differentiated macrophages identifies transcriptomic signatures that classify disease macrophages in vivo. Frontiers in Immunology, 10, 2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, X., Katakowski, M., Li, Y., Lu, D., Wang, L., Zhang, L., Chen, J., Xu, Y., Gautam, S., Mahmood, A., & Chopp, M. (2002). Human bone marrow stromal cell cultures conditioned by traumatic brain tissue extracts: Growth factor production. Journal of Neuroscience Research, 69, 687–691. [DOI] [PubMed] [Google Scholar]
- Chopp, M., Zhang, X. H., Li, Y., Wang, L., Chen, J., Lu, D., Lu, M., & Rosenblum, M. (2000). Spinal cord injury in rat: Treatment with bone marrow stromal cell transplantation. Neuroreport, 11, 3001–3005. [DOI] [PubMed] [Google Scholar]
- de Almeida, F. M., Marques, S. A., Ramalho Bdos, S., Massoto, T. B., & Martinez, A. M. (2015). Chronic spinal cord lesions respond positively to transplants of mesenchymal stem cells. Restorative Neurology and Neuroscience 33, 43–55. [DOI] [PubMed] [Google Scholar]
- Ferguson, S. W., Wang, J., Lee, C. J., Liu, M., Neelamegham, S., Canty, J. M., & Nguyen, J. (2018). The microRNA regulatory landscape of MSC‐derived exosomes: A systems view. Scientific Reports, 8, 1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freytes, D. O., Kang, J. W., Marcos‐Campos, I., & Vunjak‐Novakovic, G. (2013). Macrophages modulate the viability and growth of human mesenchymal stem cells. Journal of Cellular Biochemistry, 114, 220–229. [DOI] [PubMed] [Google Scholar]
- Goritz, C., Dias, D. O., Tomilin, N., Barbacid, M., Shupliakov, O., & Frisen, J. (2011). A pericyte origin of spinal cord scar tissue. Science, 333, 238–242. [DOI] [PubMed] [Google Scholar]
- Greene, C., Hanley, N., & Campbell, M. (2019). Claudin‐5: Gatekeeper of neurological function. Fluids Barriers CNS, 16, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamano, K., Li, T. S., Kobayashi, T., Kobayashi, S., Matsuzaki, M., & Esato, K. (2000). Angiogenesis induced by the implantation of self‐bone marrow cells: A new material for therapeutic angiogenesis. Cell transplantation, 9, 439–443. [DOI] [PubMed] [Google Scholar]
- He, X., Dong, Z., Cao, Y., Wang, H., Liu, S., Liao, L., Jin, Y., Yuan, L., & Li, B. (2019). MSC‐derived exosome promotes M2 polarization and enhances cutaneous wound healing. Stem Cells International, 2019, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldin, C. H., Miyazono, K., & ten Dijke, P. (1997). TGF‐beta signalling from cell membrane to nucleus through SMAD proteins. Nature, 390, 465–471. [DOI] [PubMed] [Google Scholar]
- Honmou, O., Onodera, R., Sasaki, M., Waxman, S. G., & Kocsis, J. D. (2012). Mesenchymal stem cells: Therapeutic outlook for stroke. Trends in Molecular Medicine, 18, 292–297. [DOI] [PubMed] [Google Scholar]
- Honmou, O., Houkin, K., Matsunaga, T., Niitsu, Y., Ishiai, S., Onodera, R., Waxman, S. G., & Kocsis, J. D. (2011). Intravenous administration of auto serum‐expanded autologous mesenchymal stem cells in stroke. Brain, 134, 1790–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honmou, O., Yamashita, T., Morita, T., Oshigiri, T., Hirota, R., Iyama, S., Kato, J., Sasaki, Y., Ishiai, S., Ito, Y. M., Namioka, A., Namioka, T., Nakazaki, M., Kataoka‐Sasaki, Y., Onodera, R., Oka, S., Sasaki, M., Waxman, S. G., & Kocsis, J. D. (2021). Intravenous infusion of auto serum‐expanded autologous mesenchymal stem cells in spinal cord injury patients: 13 case series. Clinical Neurology and Neurosurgery, 203, 106565. [DOI] [PubMed] [Google Scholar]
- Hu, X., Leak, R. K., Shi, Y., Suenaga, J., Gao, Y., Zheng, P., & Chen, J. (2015). Microglial and macrophage polarization‐new prospects for brain repair. Nature reviews Neurology, 11, 56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, J. H., Yin, X. M., Xu, Y., Xu, C. C., Lin, X., Ye, F. B., Cao, Y., & Lin, F. Y. (2017). Systemic administration of exosomes released from mesenchymal stromal cells attenuates apoptosis, inflammation, and promotes angiogenesis after spinal cord injury in rats. Journal of Neurotrauma, 34, 3388–3396. [DOI] [PubMed] [Google Scholar]
- Jeppesen, D. K., Fenix, A. M., Franklin, J. L., Higginbotham, J. N., Zhang, Q., Zimmerman, L. J., Liebler, D. C., Ping, J., Liu, Q., Evans, R., Fissell, W. H., Patton, J. G., Rome, L. H., Burnette, D. T., & Coffey, R. J. (2019). Reassessment of exosome composition. Cell, 177, 428–445 e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji, W., Jiang, W., Li, M., Li, J., & Li, Z. (2019). miR‐21 deficiency contributes to the impaired protective effects of obese rat mesenchymal stem cell‐derived exosomes against spinal cord injury. Biochimie, 167, 171–178. [DOI] [PubMed] [Google Scholar]
- Kabat, M., Bobkov, I., Kumar, S., & Grumet, M. (2020). Trends in mesenchymal stem cell clinical trials 2004–2018: Is efficacy optimal in a narrow dose range? Stem Cells Translational Medicine, 9, 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigerl, K. A., Gensel, J. C., Ankeny, D. P., Alexander, J. K., Donnelly, D. J., & Popovich, P. G. (2009). Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. The Journal of neuroscience, 29, 13435–13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny, C., Browne, W., Cuthill, I. C., Emerson, M., Altman, D. G., & Group NCRRGW (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzyszczyk, P., Schloss, R., Palmer, A., & Berthiaume, F. (2018). The role of macrophages in acute and chronic wound healing and interventions to promote pro‐wound healing phenotypes. Frontiers in Physiology, 9, 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamouille, S., & Derynck, R. (2007). Cell size and invasion in TGF‐beta‐induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. Journal of Cell Biology, 178, 437–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lankford, K. L., Arroyo, E. J., & Kocsis, J. D. (2017). Chronic TNFalpha exposure induces robust proliferation of olfactory ensheathing cells, but not schwann cells. Neurochemical Research, 42, 2595–2609. [DOI] [PubMed] [Google Scholar]
- Lankford, K. L., Arroyo, E. J., Nazimek, K., Bryniarski, K., Askenase, P. W., & Kocsis, J. D. (2018). Intravenously delivered mesenchymal stem cell‐derived exosomes target M2‐type macrophages in the injured spinal cord. Plos One, 13, e0190358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon, V. P., Ferguson, A. R., Popovich, P. G., Xu, X. M., Snow, D. M., Igarashi, M., Beattie, C. E., Bixby, J. L., & Consortium, M. (2014). Minimum information about a spinal cord injury experiment: A proposed reporting standard for spinal cord injury experiments. Journal of Neurotrauma, 31, 1354–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z., Liang, G., Ma, T., Li, J., Wang, P., Liu, L., Yu, B., Liu, Y., & Xue, Y. (2015). Blood‐brain barrier permeability change and regulation mechanism after subarachnoid hemorrhage. Metabolic Brain Disease, 30, 597–603. [DOI] [PubMed] [Google Scholar]
- Lu, Y., Zhou, Y., Zhang, R., Wen, L., Wu, K., Li, Y., Yao, Y., Duan, R., & Jia, Y. (2019). Bone mesenchymal stem cell‐derived extracellular vesicles promote recovery following spinal cord injury via improvement of the integrity of the blood‐spinal cord barrier. Frontiers in Neuroscience, 13, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani, A., Biswas, S. K., Galdiero, M. R., Sica, A., & Locati, M. (2013). Macrophage plasticity and polarization in tissue repair and remodelling. Journal of Pathology, 229, 176–185. [DOI] [PubMed] [Google Scholar]
- Massague, J. (1996). TGFbeta signaling: Receptors, transducers, and Mad proteins. Cell, 85, 947–950. [DOI] [PubMed] [Google Scholar]
- Matsushita, T., Lankford, K. L., Arroyo, E. J., Sasaki, M., Neyazi, M., Radtke, C., & Kocsis, J. D. (2015). Diffuse and persistent blood‐spinal cord barrier disruption after contusive spinal cord injury rapidly recovers following intravenous infusion of bone marrow mesenchymal stem cells. Experimental Neurology, 267, 152–164. [DOI] [PubMed] [Google Scholar]
- Mayer, H., Bertram, H., Lindenmaier, W., Korff, T., Weber, H., & Weich, H. (2005). Vascular endothelial growth factor (VEGF‐A) expression in human mesenchymal stem cells: Autocrine and paracrine role on osteoblastic and endothelial differentiation. Journal of Cellular Biochemistry, 95, 827–839. [DOI] [PubMed] [Google Scholar]
- Milich, L. M., Ryan, C. B., & Lee, J. K. (2019). The origin, fate, and contribution of macrophages to spinal cord injury pathology. Acta Neuropathologica, 137, 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita, T., Sasaki, M., Kataoka‐Sasaki, Y., Nakazaki, M., Nagahama, H., Oka, S., Oshigiri, T., Takebayashi, T., Yamashita, T., Kocsis, J. D., & Honmou, O. (2016). Intravenous infusion of mesenchymal stem cells promotes functional recovery in a model of chronic spinal cord injury. Neuroscience, 335, 221–231. [DOI] [PubMed] [Google Scholar]
- Nakajima, H., Uchida, K., Guerrero, A. R., Watanabe, S., Sugita, D., Takeura, N., Yoshida, A., Long, G., Wright, K. T., Johnson, W. E., & Baba, H. (2012). Transplantation of mesenchymal stem cells promotes an alternative pathway of macrophage activation and functional recovery after spinal cord injury. Journal of Neurotrauma, 29, 1614–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazaki, M., Sasaki, M., Kataoka‐Sasaki, Y., Oka, S., Namioka, T., Namioka, A., Onodera, R., Suzuki, J., Sasaki, Y., Nagahama, H., Mikami, T., Wanibuchi, M., Kocsis, J. D., & Honmou, O. (2017). Intravenous infusion of mesenchymal stem cells inhibits intracranial hemorrhage after recombinant tissue plasminogen activator therapy for transient middle cerebral artery occlusion in rats. Journal of Neurosurgery, 127, 917–926. [DOI] [PubMed] [Google Scholar]
- Oliveri, R. S., Bello, S., & Biering‐Sorensen, F. (2014). Mesenchymal stem cells improve locomotor recovery in traumatic spinal cord injury: Systematic review with meta‐analyses of rat models. Neurobiology of Disease, 62, 338–353. [DOI] [PubMed] [Google Scholar]
- Osaka, M., Honmou, O., Murakami, T., Nonaka, T., Houkin, K., Hamada, H., & Kocsis, J. D. (2010). Intravenous administration of mesenchymal stem cells derived from bone marrow after contusive spinal cord injury improves functional outcome. Brain Research, 1343, 226–235. [DOI] [PubMed] [Google Scholar]
- Osteikoetxea, X., Sodar, B., Nemeth, A., Szabo‐Taylor, K., Paloczi, K., Vukman, K. V., Tamasi, V., Balogh, A., Kittel, A., Pallinger, E., & Buzas, E. I. (2015). Differential detergent sensitivity of extracellular vesicle subpopulations. Organic & Biomolecular Chemistry, 13, 9775–9782. [DOI] [PubMed] [Google Scholar]
- Prockop, D. J., & Oh, J. Y. (2012). Mesenchymal stem/stromal cells (MSCs): Role as guardians of inflammation. Molecular Therapy, 20, 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quertainmont, R., Cantinieaux, D., Botman, O., Sid, S., Schoenen, J., & Franzen, R. (2012). Mesenchymal stem cell graft improves recovery after spinal cord injury in adult rats through neurotrophic and pro‐angiogenic actions. Plos One, 7, e39500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi, H. Y., Ahmad, R., Sylvester, J., & Zafarullah, M. (2007). Requirement of phosphatidylinositol 3‐kinase/Akt signaling pathway for regulation of tissue inhibitor of metalloproteinases‐3 gene expression by TGF‐beta in human chondrocytes. Cellular Signalling, 19, 1643–1651. [DOI] [PubMed] [Google Scholar]
- Sasaki, M., Radtke, C., Tan, A. M., Zhao, P., Hamada, H., Houkin, K., Honmou, O., & Kocsis, J. D. (2009). BDNF‐hypersecreting human mesenchymal stem cells promote functional recovery, axonal sprouting, and protection of corticospinal neurons after spinal cord injury. The Journal of Neuroscience, 29, 14932–14941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen, T. D., & Livak, K. J. (2008). Analyzing real‐time PCR data by the comparative C(T) method. Nature Protocols, 3, 1101–1108. [DOI] [PubMed] [Google Scholar]
- Schwab, M. E., & Strittmatter, S. M. (2014). Nogo limits neural plasticity and recovery from injury. Current Opinion in Neurobiology, 27, 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoane, J., Le, H. V., Shen, L., Anderson, S. A., & Massague, J. (2004). Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell, 117, 211–223. [DOI] [PubMed] [Google Scholar]
- Sheedy, F. J. (2015). Turning 21: Induction of miR‐21 as a Key Switch in the Inflammatory Response. Frontiers in Immunology, 6, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelke, G. V., Yin, Y., Jang, S. C., Lasser, C., Wennmalm, S., Hoffmann, H. J., Li, L., Gho, Y. S., Nilsson, J. A., & Lotvall, J. (2019). Endosomal signalling via exosome surface TGFbeta‐1. Journal of Extracellular Vesicles, 8, 1650458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolova, V., Ludwig, A. K., Hornung, S., Rotan, O., Horn, P. A., Epple, M., & Giebel, B. (2011). Characterisation of exosomes derived from human cells by nanoparticle tracking analysis and scanning electron microscopy. Colloids and Surfaces. B, Biointerfaces, 87, 146–150. [DOI] [PubMed] [Google Scholar]
- Spejo, A. B., Carvalho, J. L., Goes, A. M., & Oliveira, A. L. (2013). Neuroprotective effects of mesenchymal stem cells on spinal motoneurons following ventral root axotomy: Synapse stability and axonal regeneration. Neuroscience, 250, 715–732. [DOI] [PubMed] [Google Scholar]
- Sun, G., Li, G., Li, D., Huang, W., Zhang, R., Zhang, H., Duan, Y., & Wang, B. (2018). hucMSC derived exosomes promote functional recovery in spinal cord injury mice via attenuating inflammation. Materials Science & Engineering. C, Materials for Biological Applications, 89, 194–204. [DOI] [PubMed] [Google Scholar]
- Sun, M. L., Shinoda, Y., & Fukunaga, K. (2019). KY‐226 protects blood‐brain barrier function through the Akt/FoxO1 signaling pathway in brain ischemia. Neuroscience, 399, 89–102. [DOI] [PubMed] [Google Scholar]
- Thery, C., Amigorena, S., Raposo, G., & Clayton, A. (2006). Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Current Protocols in Cell Biology, Chapter 3:Unit 3.22. [DOI] [PubMed] [Google Scholar]
- Toh, W. S., Lai, R. C., Zhang, B., & Lim, S. K. (2018). MSC exosome works through a protein‐based mechanism of action. Biochemical Society Transactions, 46, 843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urdzikova, L. M., Ruzicka, J., LaBagnara, M., Karova, K., Kubinova, S., Jirakova, K., Murali, R., Sykova, E., Jhanwar‐Uniyal, M., & Jendelova, P. (2014). Human mesenchymal stem cells modulate inflammatory cytokines after spinal cord injury in rat. International Journal of Molecular Sciences, 15, 11275–11293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volarevic, V., Markovic, B. S., Gazdic, M., Volarevic, A., Jovicic, N., Arsenijevic, N., Armstrong, L., Djonov, V., Lako, M., & Stojkovic, M. (2018). Ethical and Safety Issues of Stem Cell‐Based Therapy. International Journal of Medical Sciences, 15, 36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada, J., Onishi, H., Suzuki, H., Yamasaki, A., Nagai, S., Morisaki, T., & Katano, M. (2010). Surface‐bound TGF‐beta1 on effusion‐derived exosomes participates in maintenance of number and suppressive function of regulatory T‐cells in malignant effusions. Anticancer Research, 30, 3747–3757. [PubMed] [Google Scholar]
- Wang, L., Pei, S., Han, L., Guo, B., Li, Y., Duan, R., Yao, Y., Xue, B., Chen, X., & Jia, Y. (2018). Mesenchymal stem cell‐derived exosomes reduce A1 astrocytes via downregulation of phosphorylated NFkappaB P65 subunit in spinal cord injury. Cellular Physiology and Biochemistry, 50, 1535–1559. [DOI] [PubMed] [Google Scholar]
- Whone, A. L., Kemp, K., Sun, M., Wilkins, A., & Scolding, N. J. (2012). Human bone marrow mesenchymal stem cells protect catecholaminergic and serotonergic neuronal perikarya and transporter function from oxidative stress by the secretion of glial‐derived neurotrophic factor. Brain Research, 1431, 86–96. [DOI] [PubMed] [Google Scholar]
- Witwer, K. W., Van Balkom, B. W. M., Bruno, S., Choo, A., Dominici, M., Gimona, M., Hill, A. F., De Kleijn, D., Koh, M., Lai, R. C., Mitsialis, S. A., Ortiz, L. A., Rohde, E., Asada, T., Toh, W. S., Weiss, D. J., Zheng, L., Giebel, B., & Lim, S. K. (2019). Defining mesenchymal stromal cell (MSC)‐derived small extracellular vesicles for therapeutic applications. Journal of Extracellular Vesicles, 8, 1609206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, M., Cui, B., Zhang, W., Ma, W., Zhao, G., & Xing, L. (2020). Exosomal miR‐21 secreted by IL‐1beta‐primed‐mesenchymal stem cells induces macrophage M2 polarization and ameliorates sepsis. Life Sciences, 118658. [DOI] [PubMed] [Google Scholar]
- Yeo, R. W., Lai, R. C., Zhang, B., Tan, S. S., Yin, Y., Teh, B. J., & Lim, S. K. (2013). Mesenchymal stem cell: An efficient mass producer of exosomes for drug delivery. Advanced Drug Delivery Reviews, 65, 336–341. [DOI] [PubMed] [Google Scholar]
- Ying, W., Cheruku, P. S., Bazer, F. W., Safe, S. H., & Zhou, B. (2013). Investigation of macrophage polarization using bone marrow derived macrophages. Journal of visualized experiments: JoVE, 76, E50323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, B., Yin, Y., Lai, R. C., Tan, S. S., Choo, A. B., & Lim, S. K. (2014). Mesenchymal stem cells secrete immunologically active exosomes. Stem Cells and Development, 23, 1233–1244. [DOI] [PubMed] [Google Scholar]
- Zhang, L., Zhou, F., & ten Dijke, P. (2013). Signaling interplay between transforming growth factor‐beta receptor and PI3K/AKT pathways in cancer. Trends in Biochemical Sciences, 38, 612–620. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Both MSC infusion and fractioned MSC‐sEVs dosing upregulate Arg1 expression, but do not decrease TNF‐α gene expression. a and b, Gene expression analysis (qRT‐PCR) for the M2 macrophage maker, Arg1 (a) and M1 macrophage‐related cytokine, TNF‐α (b). The ΔCT was calculated against the endogenous control (GAPDH), and ΔΔCT (mRNA level) was calculated against the ΔCT of the control. Note the gene expression levels of TNF‐α and Arg1 were increased in all treatment groups at day14 post‐SCI, respectively. However, the level of Arg1 in both the MSC infusion and F‐sEVs treatment group showed a significant increase compared to the Vehicle treatment group. Values are presented as means ± SEM. A 1‐way ANOVA followed by the Tukey‐Kramer test was conducted. *: P < .05, **: P < .01, MSC: mesenchymal stem/stromal cell, MSC‐sEVs: small extracellular vesicles derived from MSCs, Arg1: Arginase 1, TNF: Tumour necrosis factor, qRT‐PCR: quantitative reverse transcription‐polymerase chain reaction, ΔCT: delta‐cycle threshold, GAPDH: Glyceraldehyde 3‐phosphate dehydrogenase, FC: fold change.
Supplementary Figure 2. Gene expression analysis shows that neither MSC infusion nor MSC‐sEVs upregulate TGF‐β or TGF‐β receptors in off‐target organs or increase expression of fibrotic markers in lung, spleen, kidney, liver, or brain at 10 days post‐SCI. a‐e, Graphs illustrating the results of qRT‐PCR analysis for the relative gene expression of TGF‐β1, TGF‐β2, TGF‐βR1, TGF‐βR2, Col1a1, and FN1 in lung (a), spleen (b), kidney (c), liver (d), and brain (e). The samples were collected at day 3 after the infusion (day 10 post‐SCI). Three SCI rats per group were analysed. Total purified RNAs from each organ were quantified with a spectrophotometer (NanoDrop, Technologies Inc., Wilmington, DE, USA). qRT‐PCR analysis was performed in triplicate using TaqMan® Universal Master Mix II with Uracil‐N glycosylase (UNG) and specific sets of primers and TaqMan probes (Thermo Fisher Scientific Inc.) (Supplementary Table 2) with a CFX96TM real‐time PCR detection system (Bio‐Rad), using approximately 20 ng total mRNA for each sample. The ΔCT was calculated against the internal control (GAPDH), and ΔΔCT (mRNA level) was calculated against the ΔCT of the control. Note that a trend towards an increase in TGF‐β and TGF‐β receptors after the MSC or MSC‐sEVs infusion in the brain (e), but the response was much smaller than that in the spinal cord (1.5‐2.0 fold increase in TGF‐βR2 in the brain compared to 20–30 fold increase in the spinal cord) and no effect on other lungs, kidney, liver, or spleen (a‐d). We also saw no evidence of upregulation of fibrotic markers (Col1a1 and Fn1) in lungs or other organs for any of the tissue by the infusion of MSCs or MSC‐sEVs, but rather a trend towards a slight decrease in the fibrotic markers after SCI in lungs, kidney, liver and spleen in both Vehicle, MSCs and MSC‐sEVs infused conditions. qRT‐PCR: quantitative reverse transcription‐polymerase chain reaction, TGF: transforming growth factor, TGF‐βR: transforming growth factor‐beta receptor, Col1a1: collagen, type 1, alpha 1, Fn1: fibronectin 1, GAPDH: glyceraldehyde 3‐phosphate dehydrogenase, SCI: spinal cord injury, MSC: mesenchymal stem/stromal cell, MSC‐sEVs: small extracellular vesicles derived from MSCs, ΔCT: delta‐cycle threshold, FC: fold change.
Supplementary Figure 3. Gene expression analysis shows no effect of MSC infusion or MSC‐sEVs on Claudin‐5 expression at the lesion site at 14 days post‐SCI. a, Relative level of Claudin‐5, ZO‐1, occludin, and N‐cadherin mRNA expression (calculated compared to GAPDH using qRT‐PCR) at 14 days after SCI, 7 days post‐treatment for vehicle treatment, MSC, 1‐sEVs, and F‐sEVs treatment conditions. Note that mRNA for Claudin‐5 (the most abundant tight junction protein at the blood‐spinal cord barrier) was expressed at much higher levels at the injury site than mRNA for ZO‐1, occluding, or N‐cadherin under all treatment conditions. b, Relative levels of Claudin‐5 mRNA expression at lesion sites 10 days and 14 days post‐SCI, (3 days and 7 days after onset of Vehicle, MSC, 1‐sEVs, or F‐sEVs treatments (qRT‐PCR). The ΔCT was calculated against the endogenous control (GAPDH), and ΔΔCT (mRNA level) was calculated against the ΔCT of the control. Note that although the expression levels of Claudin‐5 mRNA showed a trend towards increasing values in the MSCs, 1‐sEVs, and F‐sEVs treatment groups compared to the Vehicle treatment at day 10, these differences were not statistically significant. Values are presented as means ± SEM. A 1‐way ANOVA followed by the Tukey‐Kramer test was conducted. *: P < .05, **: P < .01, MSC: mesenchymal stem/stromal cell, MSC‐sEVs: small extracellular vesicles derived from MSCs, SCI: spinal cord injury, GAPDH: Glyceraldehyde 3‐phosphate dehydrogenase, qRT‐PCR: quantitative reverse transcription‐polymerase chain reaction, ΔCT: delta‐cycle threshold, FC: fold change.
Supplementary Table 1. Antibodies used in specific applications
Supplementary Table 2. PCR primer information used in this study
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.