

Abstract
Sparse population activity is a well-known feature of supragranular sensory neurons in neocortex. The mechanisms underlying sparseness are not well understood because a direct link between the neurons activated in vivo, and their cellular properties investigated in vitro has been missing. We used two-photon calcium imaging to identify a subset of neurons in layer L2/3 (L2/3) of mouse primary somatosensory cortex that are highly active following principal whisker vibrotactile stimulation. These high responders (HRs) were then tagged using photoconvertible green fluorescent protein for subsequent targeting in the brain slice using intracellular patch-clamp recordings and biocytin staining. This approach allowed us to investigate the structural and functional properties of HRs that distinguish them from less active control cells. Compared to less responsive L2/3 neurons, HRs displayed increased levels of stimulus-evoked and spontaneous activity, elevated noise and spontaneous pairwise correlations, and stronger coupling to the population response. Intrinsic excitability was reduced in HRs, while we found no evidence for differences in other electrophysiological and morphological parameters. Thus, the choice of which neurons participate in stimulus encoding may be determined largely by network connectivity rather than by cellular structure and function.
Keywords: barrel cortex, patch clamp, somatosensory, sparse coding, two-photon imaging
Introduction
The perception of the world around us with its rich sensory information depends on the selective activation of neurons in the brain. While the activation of too many neurons is associated with cognitive impairment and pathological states like epilepsy, the activation of too few neurons can lead us to miss critical information from our environment. When neurons are activated, they fire action potentials, or spikes, which represent the primary mechanism of information transmission in the brain. However, it is still an open question how and under which conditions neurons are recruited into active spiking ensembles. Two-photon imaging provides a key method to observe large populations of identified active and silent neurons, thereby enabling us to shed light on the principles of neuronal coding.
Mounting evidence indicates that sensory information is encoded by only a small number of active neurons in neocortex (Olshausen and Field 2004; Wolfe et al. 2010; Sachdev et al. 2012)—a feature that is referred to as “sparse coding.” Such sparse coding scheme has also been described for excitatory neurons in L2/3 of the primary somatosensory (barrel) cortex of rodents, where a small fraction of excitatory neurons has been found to fire the majority of single whisker stimulation evoked spikes (de Kock et al. 2007; Kerr et al. 2007; O’Connor et al. 2010; Crochet et al. 2011; Margolis et al. 2012; Ranjbar-Slamloo and Arabzadeh 2017).
Why only a subset of neurons is activated under these circumstances is a question under active investigation. It has been suggested that such sparseness may arise either from high trial-to-trial variability in the fraction of activated neurons or from a small subset of neurons that is consistently recruited by sensory stimulation (Barth and Poulet 2012). The latter possibility has been supported by the finding that a distinct set of L2/3 neurons is consistently activated by passive whisker stimulation over the course of days and months (Margolis et al. 2012; Mayrhofer et al. 2015). In addition, sparseness might be related to specific stimulus parameters as other studies have described more variable responses (Kerr et al. 2007; Ranjbar-Slamloo and Arabzadeh 2017).
If specific stimuli activate only a small subset of neurons, it is possible that these cells show specific morphological features (Oberlaender et al. 2012), physiological properties (Crochet et al. 2011) and/or synaptic input connectivity (Benedetti et al. 2013) that render them more responsive compared to other neurons. However, studies that have attempted to address these questions have failed to provide a direct link between the neurons activated in vivo by whisker stimulation and the cellular and circuit properties investigated in vitro (Yassin et al. 2010; Elstrott et al. 2014). To overcome this limitation, we have chosen a combination of in vivo and in vitro methods (Fig. 1): We used two-photon calcium imaging to identify neurons in L2/3 of the mouse primary somatosensory cortex that are strongly activated by vibrotactile stimulation of the principal whisker. These high responders (HRs) were then tagged using light activation of the photo-convertible green fluorescent protein (PAGFP; Lien and Scanziani 2011). The tagged neurons showed strong fluorescence so that they could be identified in the brain slice and selected for intracellular patch-clamp recordings and biocytin staining (Marx et al. 2012). The combination of techniques made it possible to perform a detailed characterization of identified HRs and their physiological and morphological properties that may be causally related to their elevated activity levels. During in vivo imaging, HRs exhibited strong, fast, and reliable activation by whisker stimulation as well as increased levels of spontaneous activity. Compared to less active neurons, HR activity was more correlated during both stimulation and non-stimulation epochs and showed stronger coupling to the population response. Furthermore, in vitro intracellular measurements of tagged neurons revealed reduced intrinsic excitability as well as larger and more frequent excitatory network inputs for HRs compared to control neurons. Both groups displayed similar passive cell properties and similar morphological characteristics pertaining to different excitatory subclasses. Our findings support the view that a subset of excitatory L2/3 neurons are preferentially targeted by excitatory network inputs, leading to their increased activation both in the presence and absence of sensory stimulation and to a compensatory down-regulation of intrinsic excitability in this population. We conclude that the choice of which neurons participating in stimulus coding may largely depend on local network connectivity, rather than on the specific morphological and physiological characteristics of the neurons.
Figure 1 .

Combined in vivo and in vitro approach for the functional and structural characterization of neurons with known stimulus–response properties. The method comprised four main experimental steps (A-D): (A) Intrinsic imaging for localizing whisker-evoked response and injection site, followed by injections of two viruses and a chronic window implant. The viruses comprised a red calcium indicator (jRGECO1a or RCaMP2) and a photo-activatable green fluorescent protein (H2B-PAGFP). (B) Two-photon imaging of whisker-evoked responses and neuronal tagging by light-activation of H2B-PAGFP-postive neurons. (C) Intracellular recordings of neurons tagged in vivo and simultaneous dye injection (biocytin). (D) Neuronal staining and reconstruction of neuronal morphology.
Materials and Methods
Animals
Animal procedures were approved by the local authorities (animal facility at University Hospital Aachen and the Northrhine-Westphalian Landesamt für Natur, Umwelt und Verbraucherschutz) and were carried out in accordance with the German Animal Welfare Act, the European Directive on the Protection of Animals used for Scientific Purposes, as well as guidelines by the Federation of European Laboratory Animal Science Associations (FELASA). Thirty-five C57/BL6J adult, male mice were used for the procedures. Mice were obtained from Charles River and kept under a 12-h light–dark cycle, with food and water available ad libitum.
Surgery
At the time of surgery, animals were aged between P45 and P105. Mice were anesthetized with isoflurane (AbbVie, Germany; induction: 4%, maintenance: ~ 2%) and placed in a stereotactic frame (Kopf Instruments, USA). They were injected with buprenorphine (Temgesic; Reckitt Benckiser, UK; 0.02 mg/kg, s.c.) for pain management and their eyes were protected from desiccation by applying ointment. The fur was removed over the skull, the skin was disinfected with betadine (Mundipharma, Germany), and bupivacaine (Actavis, Germany; 0.25%, 0.04 ml) was injected subcutaneously for local analgesia. The skull was exposed and the C1 barrel in the left hemisphere was identified using intrinsic optical imaging and piezo-controlled whisker stimulation (Margolis et al. 2012; Mayrhofer et al. 2015). A 3 × 3-mm craniotomy was performed and two viruses were injected sequentially in L2/3 of the C1 and adjacent barrel columns (Figs 1 and 2): One virus for expressing histone-associated, photo-activatable green fluorescent protein (pACAGW-H2B-PAGFP-AAV was a gift from Massimo Scanziani [Addgene plasmid # 33000; http://n2t.net/addgene:33000; RRID:Addgene_33 000]; AAV9 was produced by Viral Core Facility Charite Berlin, Germany) that was used for photo-tagging cells (Lien and Scanziani 2011), and a red calcium indicator for two-photon imaging of neuronal activity (Inoue et al. 2015; Dana et al. 2016). The red indicator was either jRGECO1a (23 animals; AAV1.Syn.NES.jRGECO1a.WPRE.SV40; Dana et al. 2016) obtained from Penn Vector Core, USA, or RCaMP2 (9 animals; AAV-hsyn-RCaMP2) produced by the Viral Core Facility of the Charite Berlin, Germany (details about AAV-hsyn-RCaMP2 virus generation can be found in Supplementary Methods S1). Virus stocks showed high titers (H2B-PAGFP: 5.36e12 GC/ml; jRGECO1a: 2.08e13 GC/ml; RCaMP2: 5,01e12 VG/ml) and were diluted 1:1 with mannitol (Mannit 15%; Serag, Germany) for injections. To avoid tissue damage, small volumes of virus were injected (H2B-PAGFP: 400–450 nl; jRGECO1a: 600–700 nl; RGeco: 600–650 nl) at slow rates (~50 nl/min). Injections were performed using beveled glass pipettes (Drummond, USA) with a diameter of 13–20 μm. The craniotomy was then covered with a glass window (3 × 3 mm; UQG Optics Ltd, UK). Dental cement (DE Healthcare Products, UK) was applied to keep the window in place and to form a head cap holding a custom-made head-post made of aluminum or titanium. Throughout surgery, body temperature was maintained at 37°C with a feedback-controlled heating pad (Thorlabs, USA). After surgery, mice received buprenorphine injections for pain management (Temgesic; Reckitt Benckiser, UK; 0.02 mg/kg s.c. every 12 h, 3 d postoperatively). Antibiotic treatment was administered 1 day before surgery until the end of the experiment by supplementing the drinking with enrofloxacin (Baytril, 1 ml/l; Bayer, Germany). Post-operative pain management was carried out using buprenorphine (Temgesic; Reckitt Benckiser, UK; 0.02 mg/kg, s.c.; 2 × per day for 1–1.5 days). Mice were single-housed for the rest of the experimental procedures to avoid potential damage to the implant.
Figure 2 .

In vivo whisker stimulation, two-photon imaging and neuronal tagging. (A) Intrinsic imaging was performed prior to each two-photon imaging session to identify barrel location (white circle). Vasculature under green light illumination (left) and change in reflected red light RD/R0 during single whisker stimulation (right). (B) Mice (n = 35) were kept under light isoflurane anesthesia during two-photon imaging in barrel cortex L2/3 (green square; left) and piezo-electric stimulation of single whiskers at 90 Hz (right). (C) Co-expression of red calcium indicator for two-photon imaging (here jRGECO1a) and photo-activatable green fluorescent protein (H2B-PAGFP) for photo-tagging cells. Overlay of green and red channels before (top) and after photo-activation of H2B-PAGFP (bottom). (D) Single-neuron calcium transients Δf/f0 (black) in response to whisker stimulation (blue). HRs #1–7 showed large average responses (red curves), while LRs #8–14 showed small average responses (blue curves). (E-G) Criteria for defining HRs (red) and LRs (blue): Peak calcium signals (Δf/f0) averaged across all trials (E), fraction of trials with significant activation (F), and response latency (G). In (G), only neurons for which the response latency could be calculated were included (100% of all HRs, 99% of MR and 65% of LRs). Response latencies take discrete values from the sampling time of calcium imaging at our sampling rate (11.4 Hz). Neurons that did not match these criteria were categorized as medium responders.
In Vivo Two-Photon Imaging
Two-Photon Calcium Imaging Data Acquisition
Two-photon imaging was carried out after a minimum of 2 weeks following surgery to allow for sufficient viral expression. Functional images were acquired continuously using bidirectional galvanometer scanning mounted in a custom-built two-photon microscope (Mayrhofer et al. 2015). The imaging rate was set to either 8.5 or 11.4 Hz with an image resolution of 256 × 256 or 256 × 220 pixels, respectively, spanning 204 × 204 or 245 × 210 μm. Prior to two-photon calcium imaging, intrinsic optical imaging (IOI) was performed to identify the single whisker representation in neocortex (details of the IOI procedure can be found in Supplementary Methods S2). The calcium imaging field of view (FOV) was placed on the center of the IOI signal. The genetically encoded red calcium indicator (jRGECO1a or RCaMP2) was excited with a Ti:sapphire laser (1040 nm, Chameleon Ultra II; Coherent, USA) using 25–60 mW depending on the recording depth. Emitted light was recorded through a 16×, 0.8 NA objective (Nikon, Japan) and detected with two Hamamatsu photomultiplier modules (H10770PA-40) with the following filter settings: band pass filter 540/40 nm and 617/73 nm for green and red channels, respectively, and a 750-nm short pass filter (Semrock, USA) for each channel. Two-photon laser scanning and image acquisition was performed using version r3.8 or 2017 of the ScanImage software (Vidrio technology, USA; Pologruto et al. 2003).
Animals remained anesthetized during the whole procedure with isoflurane (AbbVie, Germany; induction: 4%, maintenance: ~0.5–1.5%) and head-fixed. Body temperature was maintained at 37°C with a feedback-controlled heating-pad and respiratory rate was monitored by means of a piezoelectric sensor placed under the animal’s trunk and a custom-written LabView program (National Instruments, USA). Images were acquired when the respiratory rate was above 120 breaths per minute and whiskers did not move, as assessed with a camera placed underneath the animal’s head. One target whisker at a time (B1, C1, D1, C2, or D2) was placed into a capillary glass tube that was mounted on a piezo element and positioned 3 mm away from the whisker pad. Whiskers other than the stimulated one were either trimmed below 3 mm length or were located at sufficient distance to not be in contact with the whisker stimulation apparatus. The stimulus consisted of a periodic rostro-caudal deflection of the target whisker with a prototypic pulse (120 Hz sine wave, 700°·s−1, ~0.5 mm amplitude) that was delivered at 90 Hz for 1 s, as previously described (Mayrhofer et al. 2015). This was repeated 60 to 100 times with an ITI of 6.2 s. Multiple FOVs could be acquired sequentially in the same animal, either regularly spaced in the z-dimension (1–5 different depths, at 100–300 μm from the pial surface), and/or in several whiskers barrel fields. In a subset of 21 FOVs from seven animals with jRGECO1a, extra epochs of spontaneous neuronal activity lasting between 175 and 1000 s were recorded immediately following the stimulation session. No sensory stimulation was delivered to the whiskers during these epochs. Immediately after imaging a FOV, the calcium imaging data was analyzed, and target cells were selected and photo-converted.
Two-Photon Calcium Imaging Data Processing
Movies were first motion-corrected using the registration module from suite2p (Pachitariu et al. 2016). We then manually selected region of interests (ROIs) corresponding to individual neurons using a custom MATLAB software (MathWorks, USA). Selection and segmentation was based on three average frames: The first frame was an average of all frames in the red channel to see expression of the calcium indicator. The second frame was an average of all frames in the green channel to visualize nuclei through H2B-PAGFP expression. The third frame was a Δf/f0 heat map based on the pixel-wise response to the stimulation in the red channel, which was computed as
 |
(1) |
where f0 was the average pixel value in frames during the baseline (1 s prior to stimulus start), and fstim was the average pixel value in frames during the 1 s stimulation.
Immediately after calcium imaging acquisition in a FOV, only cells that met the criteria for photo-conversion were selected. For detailed analyses of calcium imaging data presented in Figs 2–4 (carried out on the subset of 21 FOVs from n = 7 mice with extra epochs of spontaneous activity), ROIs were selected for every “visible neuron.” A visible neuron was defined as a distinguishable ring of red calcium indicator surrounding a nucleus of green H2B-PAGFP, based on averaged green and red frames over the session. Neurons’ individual fluorescence time series were obtained by averaging pixel values in the corresponding ROI. Local, out-of-focus fluorescence (neuropil) was measured by averaging pixel fluorescence values in a ring of ~40 μm diameter surrounding the ROI, excluding pixels from other ROIs (Peron et al. 2015). Slow baseline fluctuations were removed individually from each neuronal and neuropil time series by subtracting the 10th percentile in a 30 s wide moving window. Neuropil subtraction was then scaled for each individual ROI to the slope α according to the following equation:
 |
(2) |
where froi represents the variation of fluorescence observed in the ROI, fc denotes fluorescence originating from the cellular soma, α is the scaling factor of neuropil contamination and fpil is the neuropil fluorescence. Most values of α were found in a range of 0.45–0.9 with a robust linear regression (Driscoll et al. 2017). Values below and above this limit were set to a minimum of 0.45 and a maximum of 0.9, respectively. We used this corrected fluorescence trace as a common basis for the computation of Δf/f0 time series, z-scored time series and spiking estimates. The Δf/f0 time series was computed individually for each trial as
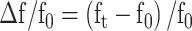 |
(3) |
where f0 was the average fluorescence value during the baseline (1 s prior to stimulus start) and ft was the average fluorescence in frame t. The peak of Δf/f0 was the maximal value within the stimulation period after averaging Δf/f0 across trials. Z-score time series were computed as
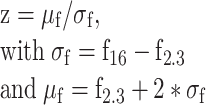 |
(4) |
where f16 is the 16th percentile of all fluorescence values in the time series and f2.3 is the 2.3th percentile. These values were picked because the distance between them represents roughly 1 standard deviation (SD) in Gaussian distributions. Gaussian noise statistics were computed on the negative hillside of the fluorescence histogram (representing the non-active period of the neuron), such that z-score time series were normalized to the noise level independent of the cellular activity level. Therefore, z-scores could be used to detect calcium events as follows: When two consecutive time points had a z-score above 2.5, it was considered as a calcium event. Rather than retrieving precise spike timing, this analysis focused on estimating key statistical parameters to reliably detect periods of activity. These calcium events were later used in binary form over 1-s time bins to study neuronal co-activation in the recorded population ensemble. Finally, a constrained deconvolution was applied on the corrected fluorescence trace (Pachitariu et al. 2016) to obtain a continuous spiking estimate. This spiking estimate was used for later computation of pairwise correlations. These methods and criteria were optimized from the freely available jRGECO1a dataset from the CRCNS website (Dana et al. 2016; Mohar et al. 2016).
Figure 4 .

Network-dependent response profiles during stimulation and spontaneous activity. Data comprised 193 HRs, 608 MRs and 1299 LRs measured in 21 FOVs from 7 mice. Neuronal activity was analyzed with respect to changes in the fraction of active neurons (“ensemble size”, A-C), correlations among cell classes (D-J), and the degree of coupling to the population activity (K-P). (A) Probability of ensemble activation (% of neural population activated by stimulus) under shuffled (dashed line) and experimental conditions (continuous line). For each neuron, activity was shuffled across trials while keeping the total activity constant. Data (n = 1454 trials) were pooled across FOVs. Error shades represent shuffled data from 1st to 99th percentile. (B) Stimulus-induced changes in ensemble composition (top) and sub-ensemble recruitment (bottom) as a function of responding ensemble size. (C) Changes in response amplitude of different cell classes as a function of the size of the responding ensemble. The response amplitude was defined as the area under the curve of the z-scored calcium transient. The ensemble size was normalized by the maximum ensemble size per FOV across trials. Inset: Slope of the linear regression of the amplitude on the responding ensemble size (Friedman test with Dunn–Sidàk’s correction). (D-F) Pairwise correlations during stimulation. Correlations were quantified using Pearson r, which was averaged across 69 ± 10 trials (mean ± SD) separately for each FOV (n = 21 FOVs). Shown are the average r between pairs of neurons during stimulation across FOVs (D), the corresponding boxplots with the median (midline), interquartile ranges and 2.7 SD (E) (Friedman test with Dunn–Sidàk’s correction), and the cumulative probability distributions of single pairwise correlations with n = 1135 HRs/HRs, n = 9498 MRs/MRs, n = 41 200 LRs/LRs (F). (G-J) Pairwise correlations during spontaneous activity. Pearson r was calculated based on 258 ± 199 s (mean ± SD) of spontaneous activity. Displayed are the average r between pairs of neurons during baseline across FOVs (G), the corresponding boxplots (H), and the cumulative probability distributions of single pairwise correlations (I). Average spontaneous correlations for neuronal pairs (n = 891 HRs/HRs, n = 891 MRs/MRs, n = 891 LRs/LRs) with matched geometric mean firing rates (GMFR > 0.05 Hz) are shown in J (Kruskall–Wallis test with Dunn–Sidàk’s correction). P-values: * < 0.05; ** < 0.01; *** < 0.001. (K) Population coupling as a function of response probability during stimulation. Across all neurons (n = 2100), population coupling and response probability were positively correlated (Spearman rank correlation). (L) A model based on population coupling predicts pairwise correlations similar to those measured. Shown are the pairwise correlations for all pairs of neurons in an example FOV, with a comparison of the predicted (upper left triangle) and the experimentally observed pairwise correlations (lower right triangle). Neurons are sorted by increasing population coupling. (M) Cumulative distribution of residual correlations during spontaneous activity in HRs, MRs and LRs with matching GMFR above 0.05 Hz. (n = 623 HRs/HRs, n = 623 MRs/MRs, n = 623 LRs/LRs, (P < 0.001 HRs versus LRs, Mann–Whitney U test). (N) Average residual correlations during stimulation (top) and spontaneous activity (bottom) for all neuronal pairs (n = 1135 HRs/HRs, n = 9498 MRs/MRs, n = 41 200 LRs/LRs, Kruskall–Wallis test with Dunn–Sidàk’s correction). Error bars represent 95% confidence intervals (C, J, N).
Target Cell Selection
Neurons were classified as HRs when they met three criteria: First, their average Δf/f0 peak across trials had to cross a threshold of 25% during the stimulation period. Second, these neurons had to show significant activation in at least 25% of the trials. This was assessed by comparing fluorescence between the baseline epoch (1 s pre-stimulus) and the stimulus epoch (1 s stimulus) with a two-sample t-test. Therefore, at a sampling rate of N, N values of baseline fluorescence were compared to N values of fluorescence during stimulation. Third, a significant increase in fluorescence had to occur in the two imaging frames following the onset of stimulation (at either sampling rate). For this analysis, only active trials (N values) were selected, in accordance with criterion 2. Using a two-sample t-test, the baseline fluorescence averaged for each trial (N values) was compared to the fluorescence in the first frame after stimulus onset for each trial (N values). This procedure was repeated iteratively for each time point. The first significant test was reported as the time of significant activation of the neuron. Only neurons with first significant activation in either the first or the second frame were categorized as HRs (at either sampling rate). Neurons were classified as low responders (LRs) if their average Δf/f0 peak was below a threshold of 10% and if they showed significant activation in less than 10% of the trials. Neurons that did not meet criteria for either LRs or HRs were classified as medium responders (MRs). The average baseline fluorescence in HRs (98.1 ± 7.3, mean ± standard error to the mean (SEM)) and LRs (93.1 ± 7.2, mean ± SEM) did not differ statistically between the groups (P > 0.05, Wilcoxon signed-rank test, n = 21 FOVs; Supplementary Figure 1D).
Photo-Conversion of the H2B-PAGFP
Once the target ROIs were selected, the corresponding H2B-PAGFP expressing cell nuclei were identified at 850 nm excitation wavelength (imaging power ranging from 4–10 mW depending on the recording depth). The precise position of the focal plane in the z-axis was adjusted with μm precision using a piezo actuator controlled through ScanImage. This strategy was needed to compensate for a shift in the focal depth of ~10 μm induced by the change in excitation wavelength from 1040 to 850 nm. Using a ScanImage feature for sub-FOV imaging, we manually drew rectangles on individual target nuclei with a resolution of 16 × 16 pixels (~4 μm2). Target ROIs of 16 × 16 pixels (~4 μm2) with a line period of 512 ns were illuminated at 750 nm for 2 s in order to photo-convert H2B-PAGFP. The laser power was adjusted ranging from 5–15 mW depending on cell depth. Each cell was individually and sequentially imaged in order to photo-convert the H2B-PAGFP. Photo-conversion efficiency was assessed at either 950 (4–12 mW) or 1040 nm (25–60 mW). Due to two-photon photo-conversion of PAGFP, the tagging was highly specific to individual neurons (Fig. 2A; Supplementary Figure 1A, B). The fluorescence of tagged neurons was on average 18 times higher than their brightest neighboring cell (Supplementary Figure 1C).
Population Coupling and Correlations
Trial-to-trial correlations, also called noise correlations, were calculated using the Pearson’s correlation coefficient between two neuronal responses to repeated presentations of the same stimulus (Cohen and Kohn 2011). We used spiking estimates of each cell, excised period of baseline, and averaged the spike estimate within 1-s whisker stimulation period, generating one activity vector per cell. All correlations between pairs of cells were calculated from this activity vector. To measure spontaneous correlations during ongoing activity, we adopted the same approach: Spike estimates were averaged in 1-s long, non-overlapping time bins over the entire period of spontaneous activity.
Correlations between population rate and single cell activity were assessed in the same FOV. The fraction of the population that was active in a given trial (also termed “ensemble size”) was calculated as the number of neurons detected active divided by the total number of neurons in the FOV. To allow pooling of ensemble sizes from all FOVs, we sorted trial-by-trial ensemble sizes in ascending order and distributed them in six bins, each containing the same number of trials (from 1 to 6, smallest active ensembles to largest active ensembles).
Population coupling per cell ci could be measured using the following formula (Okun et al. 2015):
 |
(5) |
f represents the binary vector of activity, μ its mean event rate and ||fi|| its norm (i.e., the total sum of events). Couplings were pooled across sessions by z-scoring data separately in every FOV. Details of the random generative model used to generate pairwise correlations related to population coupling can be found in Supplementary Methods S3 (Okun et al. 2015).
To test whether the strength of the pairwise noise correlations could be explained from the change in the population’s firing rate, rather than a specific one-to-one relationship, we subtracted the model-based correlations from experimentally observed correlations to obtain residual correlations (Okun et al. 2015). This procedure was conducted separately for noise correlations and spontaneous correlations. For each pair of neurons, the subtracted model-based prediction was computed as the average of Pearson’s correlations across shuffles. Residuals were then compared between the three groups of neurons; first all included, then with the matched level of activity during spontaneous activity (Fig. 4).
To avoid spurious relationships between the event rates and correlations when comparing different classes of neurons, we matched geometrical mean event rates (GMFRs) across classes of responders during epochs of spontaneous activity (Fig. 4). It was computed as
 |
(6) |
μi being the mean spike estimate of neuron i (Fig. 4D-J) or the mean event rate (Fig. 4M and N). Next, we distributed all GMFRs from the three responder’s classes in bins of 0.01 Hz width. From each bin we drew the same number of samples for the three classes. Thus, the numbers of pairs and their distributions of GMFRs were matching across the three responder classes with a resolution of 0.01 Hz.
Brain Slice Preparation
After in vivo two-photon imaging, mice aged P72 to P213 were anesthetized with isoflurane and decapitated. The brain was quickly removed and placed in 4°C cold artificial cerebrospinal fluid (ACSF) containing 206 mM sucrose, 2.5 mM KCl, 1.25 mM NaH2PO4, 3 mM MgCl2, 1 mM CaCl2, 25 mM NaHCO3, 12 mM N-acetyl-L-cysteine, and 25 mM glucose (325 mOsm, pH 7,45; aerated with 95% O2 and 5% CO2). Then the brain was shortly placed in a custom-made chamber with a ramp (10° in the rostral-caudal direction) to make an initial cut with a razor blade at an angle of 45° relative to the midline. The brain was then quickly transferred to a vibratome (HM 650 V, Thermo Fisher Scientific, Germany) filled with ASCF and cut in 300-μm thick slices. The thickness and slicing angle were optimized for preserving neuronal morphology, which is important for maximizing the number of intact neurons in the slice and for high-quality morphological reconstructions. After cutting, slices were incubated (30 min at 33°C and then at room temperature) in ACSF containing 125 mM NaCl, 25 mM NaHCO3, 2.5 mM KCl, 1.25 mM NaH2PO4, 5 mM MgCl2, 1 mM CaCl2, 25 mM glucose, 3 mM Myo-Inositol, 2 mM Na-pyruvate, 12 mM N-acetyl-L-cysteine, and 0.4 mM vitamin C (300 mOsm; 95% O2, 5% CO2). This protocol was adapted from previous studies (Ting et al. 2014; van Aerde and Feldmeyer 2015) and modified to obtain healthy slices from adult mice (P30 up to 8 months). Using this approach, we were able to record for about an hour in the slice before the cell quality would begin to deteriorate (as determined based on changes in, for example, series resistance; see also Materials and Methods subsection: Analysis of patch clamp data).
In Vitro Patch Clamping
Intracellular recordings were performed at room temperature. This deviation from standard protocols was necessary to maintain the health of the adult slices, presumably because oxygen saturation is higher at lower temperatures (Jiang et al. 1991). During recordings, slices were perfused at 5 ml/min with an extracellular ACSF containing 125 mM NaCl, 25 mM NaHCO3, 2.5 mM KCl, 25 mM glucose, 1.25 mM NaH2PO4, 2 mM CaCl2, and 1 mM MgCl2 (300 mOsm; 95% O2, 5% CO2). The intracellular solution contained 135 mM K-gluconate, 4 mM KCl, 10 mM HEPES, 10 mM phosphocreatine-Na, 4 mM ATP-Mg, and 0.3 mM GTP (pH 7.3; 290 mOsm). Biocytin (Sigma Aldrich, Germany) was added to the intracellular solution at a concentration of 3 mg/ml, and cells were filled during the recordings. Glass pipettes were pulled from borosilicate glass capillaries (2 mm outer diameter, 1 mm inner diameter) with a P-1000 Flaming/Brown micropipette puller (Sutter Instruments, USA) and had a resistance of 5–8 MΩ. Slices were observed under infrared differential interference contrast (IR-DIC) and fluorescent illumination (BP 500/25, FT 515, BP 535/30; Zeiss, Germany) with an upright microscope (Axio Examiner D1 with 5×/0.16 plan and 40×/1.0 DIC immersion objectives; Zeiss, Germany). Signals were acquired with an EPC10 amplifier (HEKA, Germany), sampled at 10 kHz and filtered at 2.9 kHz using Patchmaster software (HEKA, Germany). Offline analysis was carried out using custom made Igor Pro software (Wavemetrics, USA).
Histology
After patch recordings, brain slices were immersed in fixative (4% paraformaldehyde, 100 mM phosphate-buffered saline; pH 7.4) and stored at 4°C for a minimum of 48 h. Following a protocol published previously (Marx et al. 2012), biocytin-filled neurons were visualized using avidin-biotinylated horseradish peroxidase complex reaction (ABC-Elite; Camon, Germany) and 3,3-diaminobenzidine (Sigma Aldrich, Germany) as a chromogen. After dehydration and embedding in Eukitt (Marienfeld Laboratory Glassware, Germany), neurons were reconstructed in 3D using Neurolucida® software (MBF Bioscience, USA) under an Olympus BX61 optical microscope with a final magnification of 1000×.
Analysis of Patch Clamp Data
Series resistance was measured in whole-cell, voltage-clamp mode at −60 mV with a − 10 mV square pulse applied once a second for 300 ms (10 repetitions), compensated by 80% and monitored throughout the experiment. Neurons were excluded from the analysis if their series resistance was larger than 60 MΩ or changed by more than 10% during the experiment. Because the majority of identified HRs were excitatory neurons (16/21), only these were included in the analysis. Excitatory neurons were distinguished from GABAergic interneurons based on their firing pattern and morphology. Resting membrane potential (Vrest) was measured immediately after breakthrough.
The following passive membrane properties were measured in current-clamp mode by injecting 1 s-long current in steps of 10 pA starting at −100 pA: Input resistance (Rinput) was calculated as the slope of the linear fit of the current–voltage (IV) curve measured close to Vrest in a window of −50 to 50 pA. The membrane time constant τ was calculated with a mono-exponential fit of the voltage onset response to a −50 pA current. The voltage sag was defined as the percentage difference between the initial and sustained response to current injection. The voltage sag was measured in two different ways: First, the voltage sag amplitude was measured when the neuron was hyperpolarized by approximately −7.5 mV (“sag at −7.5 mV”). Second, the voltage sag amplitude was measured as the maximum voltage excursion independent of the level of hyperpolarization or current injection during the stimulation protocol (“max. sag”). The rheobase was determined as the minimum current amplitude required for eliciting an action potential (AP). The first elicited spike was used for quantifying AP properties, including the threshold (second derivative of membrane potential exceeding 3× SD of its baseline value before spike generation), amplitude (difference between peak and threshold membrane potentials), half-width (measured at half-amplitude), and the amplitude and timing of the afterhyperpolarization (AHP; difference between spike threshold and the most negative membrane potential measured during the AHP).
The following active properties were determined in current-clamp mode by injecting 1-s-long current steps of 25 pA starting at −20 pA: The firing frequency per 100 pA current injected, the maximum firing frequency, and the slope of the adaptation measured as the ratio between the 2nd and 9th inter-spike intervals (ISI). In addition, a sweep with a 10-spike train was chosen for every cell to measure doublet spiking (1st ISI) and adaptation (ratio of 2nd and 9th ISIs). Since not all cells fired exactly 10 spikes, a range of 9–12 spikes was chosen (if only 9 spikes were available, then the 2nd and the 8th ISI was chosen to calculate adaptation).
Spontaneous excitatory postsynaptic currents (sEPSCs) were measured in voltage-clamp at −70 mV holding potential for periods of 70–90 s. Importantly, inhibitors such as tetrodotoxin or gabazine have not been applied in order to maintain spontaneous network activity. In a first step, the mean baseline activity and SD of the noise were quantified in a 1-s time window without obvious spontaneous events. Subsequently, a threshold-crossing detection algorithm implemented in the SpAcAn (G. P. Dugué & C. V. Rousseau) extension for Igor Pro was used for detecting sEPSCs: For every point N of the trace, the algorithm calculated the difference between the baseline segment (8 ms window centered on N) and a peak segment (0.3 ms window). Baseline and peak were separated by a rise time segment (0.8 ms window). Whenever the difference exceeded a certain threshold, the algorithm then identified a local maximum in a 3-ms peak detection window. The threshold was set for every cell individually at three times the SD of the recording noise. All automatically detected peaks were verified by visual inspection.
Analysis of Morphological Data
Neurons with clear labeling and minimal background staining were chosen for further morphological analysis. Care was taken to include only neurons with minimal to no dendritic truncations as assessed based on the number of dendrites at the slice surface with “blebs” resulting from biocytin spill. Neuronal morphology was reconstructed in 3D using an Olympus BX61 microscope fitted with a 100× oil immersion objective (NA 1.4) and a 10× eye piece. Reconstructions of neurons, pial and layer borders were corrected for tissue shrinkage by a factor of 1.1 in the x- and y-dimension and 2.1 in the z-dimension (Neurolucida software, MBF Bioscience, USA). Thick- and thin-tufted excitatory neurons were distinguished based on the ratio of the apical and basal dendritic field spans. Field spans were measured parallel to the pia as the maximal spread of the dendritic branches. Neurons with apical/dendritic field span ratios >1 were defined as thick-tufted, while neurons with ratios <1 were classified as thin-tufted (Feldmeyer et al. 2006; Lübke and Feldmeyer 2010; van Aerde and Feldmeyer 2015). Spine counts were obtained from 2nd-order branches of apical and basal dendrites located close to the surface for better visibility. Spines were counted on stretches of approximately 100 μm, whereby all types of spines were taken into account and pooled for analysis. Parameters of dendritic morphology were analyzed using Neuroexplorer software (MBF Bioscience, USA). Dendritic complexity was calculated as the sum of the terminal orders and the number of terminals, multiplied with the ratio of the total dendritic length and the number of primary dendrites (Pillai et al. 2012).
Statistical Tests
Unless stated otherwise, the non-parametric Wilcoxon signed rank test and Mann–Whitney U tests were used for paired and unpaired comparisons, respectively. Pairwise noise and spontaneous correlations were assessed using Pearson’s correlation coefficient. Correlations between two variables were evaluated using the non-parametric Spearman’s rank correlation test, unless stated otherwise. All statistical tests were two-sided and were performed with either XLSTAT (Addinsoft, USA) or MATLAB (MathWorks, USA). Statistical tests are specified in the figure legends and P-values are indicated using asterisks: * < 0.05; ** < 0.01; *** < 0.001.
Due to the number of steps and complexity of the experimental protocol, calcium imaging data, electrophysiological data, and morphological data could not be obtained and matched for all animals. The numbers of FOVs, cells and animals used are specified in each statistical test and graphic.
Results
High- and low-Responding Neurons Identified by In Vivo Whisker Stimulation and Two-Photon Imaging
We characterized whisker-evoked responses in L2/3 neurons of mouse barrel cortex using two-photon imaging of a genetically encoded calcium indicator (Fig. 2). L2/3 neurons were chosen based on 1) the depth of their soma relative to the cortical surface (100–300 μm below the pia) and 2) on the visual assessment of the density of cell bodies in L2/3, which is substantially higher compared to L1. In addition, targeted neurons in L2/3 could be visually distinguished from L4 neurons since neither of the two calcium indicators (jRGECO1a, RCaMP2) was expressed in L4.
To ensure that the imaging field of view (FOV) was centered on the target barrel, intrinsic optical imaging was performed prior to each two-photon imaging session (Fig. 2A). Depending on the area in which the calcium indicator was expressed, we imaged between one and five barrels per animal. During imaging, the animal was kept under light isoflurane anesthesia and the principal whisker was stimulated with 1 s pulses at 90 Hz (Fig. 2B). This whisker stimulation protocol was chosen because it has been shown to evoke sparse and long-term stable responses from a subset of highly responsive neurons (Mayrhofer et al. 2015). Under passive vibrotactile stimulation conditions, high-frequency stimulation evokes the strongest responses compared to lower frequencies with the same individual pulse kinematic parameters (Mayrhofer et al. 2015).
Neurons were found to show various degrees of responsiveness to whisker stimulation—with some showing high levels of activity (Fig. 2C, top, and 2D, cells #1–7) and others low levels (cells #8–14). To define high- and low-responding neurons quantitatively, we used three criteria (Fig. 2E-G): First, neurons were defined as HRs if they showed peak calcium signals (Δf/f0) at or above 25% averaged across all trials. Second, HRs displayed significant activation relative to baseline in at least 25% of the recorded trials. Third, response latencies of HRs did not exceed 200 ms following stimulus onset. In contrast, neurons were categorized as LRs if their responses showed low amplitudes (≤10% peak Δf/f0) and low reliability (≤ 10% trials with significant activity), irrespective of latency.
Co-expression of a histone-bound, photo-activatable green fluorescent protein (PAGFP; Lien and Scanziani 2011) enabled us to selectively tag single neurons (Fig. 2C, bottom) for later identification in the brain slice and targeted intracellular recordings. Per animal, tagged neurons were classified as either HR or LR, but never mixed, so that either class could be identified unequivocally in the brain slice used in patch experiments.
A gradient of Neuronal Activity in Response to Whisker Stimulation
Supporting the notion of sparse coding in barrel cortex, only a small subset of highly active neurons was recruited during whisker stimulation, while the majority of neurons showed little if any activity (Fig. 3A). The analysis included 2100 manually selected neurons that co-expressed the red calcium indicator (jRGECO1a or RCaMP2) and the H2B-PAGFP. Approximately, 30% of all neurons recorded showed no activity in any trial. By applying our criteria, we categorized neurons as HRs (Fig. 3A, top row, red lines) and LRs (blue lines). Neurons that did not fit these categories were classified as medium responders (MRs; gray lines), displaying an intermediate level of responsiveness. On average, HRs and LRs showed large and small response amplitudes, respectively (Fig. 3B). Taking a closer look at the distribution of response amplitudes, latency, and fraction of trials active, a gradient of responses emerged with HRs and LRs at the opposite ends of the spectrum (Fig. 3C). This finding supports earlier results that neurons with high and low levels of activity are not separate cell clusters, but rather two extremes observed in the neuronal population (Elstrott et al. 2014).
Figure 3 .

A gradient of neuronal activity in response to whisker stimulation. We recorded from 193 HRs, 608 MRs and 1299 LRs in 21 FOVs from 7 mice. (A) Neuronal calcium responses during trials, ranked according to their average z-scores. Top row identifies neurons as HR (red), MR (gray) or LR (blue). (B) Response amplitudes of HRs (left) and LRs (right). (C) Fraction of active trials as a function of the stimulus-evoked peak amplitude (left) and the response latency (right). Only neurons for which the response latency could be calculated were included (right panel; 100% of all HRs, 99% of MR and 65% of LRs). Each dot represents one neuron. (D) Discrete events were extracted from continuous two-photon recordings using a deconvolution algorithm. Shown are z-scored events during spontaneous (top) and stimulus-evoked activity (bottom) for a representative set of 100 neurons recorded in one FOV. Each arrow indicates stimulus onset. (E) Left-to-right: Average distribution of HRs (red), MRs (gray) and LRs (blue) in the population (n = 21 FOVs), probability of calcium events during stimulation (69 ± 10 trials per FOV, mean ± SD) and during spontaneous activity (262 ± 199 s per FOV, mean ± SD)). Statistical comparisons were performed using Friedman test with Dunn–Sidàk’s correction. (F) Average event rates across cell classes during stimulus-evoked and spontaneous activity (n = 21 FOVs, Friedman test with Dunn–Sidàk’s correction). (G) Relationship between the frequency of stimulus-induced and spontaneous calcium events across all neurons (Spearman rank correlation). Trend line (black) based on least squares fit. P-values: * < 0.05; ** < 0.01; *** < 0.001.
To assess the contribution of each responder category to the total population activity, we applied a deconvolution algorithm to the recorded calcium signals (see Materials and Methods) and extracted discrete spiking-related calcium events during stimulation that were significantly different from baseline levels (Fig. 3D). On average, stimulation increased the event rate in the entire neuronal population by a factor of three compared to baseline levels. Although HRs represented only a minority of neurons in the population (9.2%), they contributed more than 40% of whisker-evoked spikes (Fig. 3E). In contrast, LRs constituted the majority of neurons in the population, but fired a minority of spikes in response to whisker stimulation. Again, MRs displayed an intermediate pattern of activity. Of note, the number of HRs per FOVs did not depend on the calcium indicator used (RCaMP2: mean ± SEM, 4.07 ± 0.88, n = 14 FOVs; jRGECO1a: 4.69 ± 0.32, n = 71 FOVs; P > 0.05; Mann–Whitney U test; number of HRs quantified across all available FOVs with either calcium indicator). During epochs with spontaneous firing, HRs also contributed more spikes than one would expect based on their low prevalence in the population, while LRs contributed less than expected. With respect to average firing rates, HRs showed higher spontaneous and stimulus-evoked spiking activity than LRs (Fig. 3F). While average firing rates were higher during stimulation compared to spontaneous activity for HRs (Wilcoxon signed-rank test; P < 0.001) and MRs (P < 0.001), firing in LRs was similar under both conditions (P > 0.05, n = 21 FOVs). Furthermore, we found a positive correlation between stimulus-evoked and spontaneous spiking activity, with HRs scoring high on both measures, and MRs and LRs showing lower levels of activity in both conditions (Fig. 3G). These observations support earlier findings (Kerr et al. 2007; Clancy et al. 2015) and suggest that L2/3 neurons do not only show sparse coding, but more generally sparse population activity that is independent of stimulus parameters.
HRs Show Elevated Coupling to the Network and to Each Other
While stimulation led to an overall increase in calcium responses in the neuronal population, we observed a large trial-to-trial variability in the ensemble size of the recruited neurons, i.e., the proportion of cells responding to stimulation in single trials. This proportion of recruited neurons ranged from 0 to 37.5% of the total population. To explore the possibility that HR and LRs were recruited during different states of network activation, we analyzed their activity profiles in more detail (Fig. 4A-C). First, we tested whether random, independent firing of neurons in the FOV could account for the variability in responding ensemble size and shuffled the activity of each neuron across trials. (Kerr et al. 2007; see Materials and Methods). If single neurons were independent from overall network activity, shuffling the trials where they become active should not affect the trial-to-trial variability of the population response. Compared to the shuffled data, the actual experimental data showed a higher probability of trials with either very small responding ensembles (0–5% of the total population) or very large responding ensembles (>25% of the total population; Fig. 4A). The discrepancy between shuffled and experimental data indicates that the recruitment of neurons did indeed covary with the network activity.
We then asked how the composition of the responding ensemble changed (i.e., which responder category was recruited) as a function of the size of the responding ensemble. Data from all FOVs were pooled after normalization to the most active ensemble recruited per FOV. Largely independent of the responding ensemble size, HRs and MRs dominated the population of activated neurons, while LRs contributed very little to the stimulus-evoked activity (Fig. 4B, top). In addition, we measured the fraction of HRs, LRs, and MRs that were recruited as the size of the responding ensemble changed (Fig. 4B, bottom). We found a monotonic increase for all responder types, with HRs being recruited faster than the others. When the responding ensemble size was large, roughly 70% of all HRs were active, but less than 10% of neurons in the LR ensemble.
Knowing that the size of the responding ensemble is related to the types of responders that become activated, we investigated whether ensemble size also affects the neurons’ response amplitudes. Indeed, we found that HRs increased their response amplitudes with increasing size of the responding ensemble (Fig. 4C). Compared to HRs, LR and MR cells showed lower response amplitudes and lower dependence of the latter on network activation states (Fig. 4C, inset).
If network activity affects the recruitment of LRs and HRs, the question arises which neurons in the population tend to be activated together. To evaluate this, we computed pairwise noise correlations and spontaneous correlations using Pearson‘s correlation coefficient r. During stimulation, the highest average pairwise correlations were measured among HRs, while correlations among LRs were less strong (Friedman test with Dunn–Sidàk correction, P < 0.001, n = 21 FOVs; Fig. 4D-F). The analysis of average pairwise correlations during epochs of spontaneous activity yielded the same conclusion (Fig. 4G-I). Besides, the measure of correlations between two neurons can be critically underestimated when the firing rate of at least one neuron is very small (effect reviewed in Cohen and Kohn 2011). This error in the estimated correlations is best tracked by the measure of geometrical mean firing rate (GMFR), i.e., the product of the mean firing rates of two neurons. We thus compared correlations between selected pairs of neurons with closely matching GMFR (see Materials and Methods). The result of this analysis confirmed increased levels of correlations among HRs relative to LRs (Fig. 4J). The finding of enhanced correlations in HRs during both stimulus-evoked and spontaneous epochs further supports the notion of stimulus-independent properties that differentiate high from low responding neurons.
It is conceivable that the increased correlations in HR activity result from shared, non-specific synaptic inputs from the local network. In line with this notion, a previous study in visual cortex showed that neurons with enhanced coupling to the firing of the population receive more excitatory inputs from neighboring neurons, independent of sensory tuning (Okun et al. 2015). Thus, population coupling was found to predict a large portion of spontaneous pairwise correlations, and was a better predictor than stimulus tuning (Okun et al. 2015). Following the same approach, we first measured population coupling and found a positive correlation between population coupling and response probability (linear fit, r = 0.42, P < 0.001). HRs scored highest on population coupling and response probability, while LRs were found at the opposite end of the spectrum (Fig. 4K). We then asked whether enhanced pairwise correlations among HRs could result from stronger population coupling. We, therefore, compared the experimental data to a simple random generative model (Okun et al. 2015) that matched the experimental data in terms of the coupling of single neurons to the population, the mean firing rate of the neurons and the population rate (see Materials and Methods). During spontaneous network activity, the observed pairwise correlations were predicted to a large degree by the model (linear fit, r = 0.48, P < 0.001, Fig. 4L). More specifically, the model explained the largest variance in spontaneous correlations for neurons with low activity (GMFRs <0.05 Hz; Supplementary Figure 2A, B). To avoid potential bias due to the dependence of the model’s performance on the firing rate, we repeated the analysis using pairs of neurons with matching GMFR. For neurons with a GMFR above 0.05 Hz, the model predicted approximately 40% of the spontaneous correlations among HRs, MRs, and LRs. After subtraction of predicted from observed spontaneous correlations, residuals for HRs remained significantly positive, while residuals for MRs and LRs were on average close to zero (Fig. 4M). This analysis was repeated for pairs of neurons during stimulus-evoked activity and the results showed again increased residuals for HRs (Fig. 4N). This suggests that population coupling cannot fully explain the strength of pairwise correlations in the population of HRs. Rather than being indistinct recipients of inputs from neighboring neurons, HRs appear to share more specific, one-to-one correlations, thus creating a functional subnetwork.
High Responders Show Reduced Intrinsic Excitability
We first aimed to test whether L2/3 pyramidal neurons that were highly responsive to whisker stimulation had electrophysiological properties different from those of less responsive cells. For this, we performed in vitro patch clamp recordings on cells previously imaged in vivo. Thalamocortical brain slices showed basal, low fluorescent H2B-PAGFP-labeling of neurons in layers 1, 2/3, 5, and 6 of the barrel cortex. H2B-PAGFP is bound to the histone protein H2B and is thus expressed in the nucleus (Lien and Scanziani 2011). As a result, tagged HRs and LRs appeared as distinctly bright spots in L2/3 and could be targeted selectively for recordings (Fig. 5A and B).
Figure 5 .

Electrophysiological and morphological characterization of an example high responder (HR) and an example low responder (LR) in barrel cortex L2/3. (A) Example of a patched HR. Thalamocortical slice and pipette position under infrared-differential interference contrast (IR-DIC; A1) to visualize barrels (white), and under fluorescent illumination to visualize H2B-PAGFP-labeled cells (A2). HRs previously tagged in vivo appear brighter than non-tagged neurons (A2, inset). Fluorescent micrograph (A3) of patched neuron with H2B-PAGFP-labeled nucleus (green circle) and the biocytin-filled patch pipette (white line). (B) Example of a patched LR. Thalamocortical slice (IR-DIC, B1) with brightly tagged LRs (H2B-PAGFP; B2, B3). Same conventions as in A. (C,D) Minimum current required to elicit spiking in HR (160 pA; C) and LR (100 pA; D). (E,F) Sustained firing pattern in HR (330 pA; E) and LR (205 pA; F). Insets display the first three spikes from the trace (gray shading). (G,H) Number of spikes fired as a function of current injected in HR (G) and LR (H). Data points are indicated by crosses and the fitting curve by a gray line. (I,J) Histological processing and morphological reconstruction of biocytin-filled HR (I) and LR (J). Left panel: Micrograph of neuron (top) with enlarged view of apical spines (AS; bottom left) and basal spines (BS; bottom right; scale bar: 10 μm). Right panel: 3D neuronal reconstruction with dendrites (HR: red, LR: blue), axons (HR: orange, LR: cyan), layer borders in gray and barrels in black.
Based on previous findings (Yassin et al. 2010; Elstrott et al. 2014), we hypothesized that the excitability of HR and LR L2/3 pyramidal neurons is different. Using in vitro patch clamp recordings and post-hoc histological reconstructions (see Materials and Methods), we identified excitatory neurons based on their firing pattern and morphological features. Among all tagged HRs, 76.2% (16/21) were pyramidal neurons, while 23.8% (5/21) were fast-spiking interneurons. Of the tagged LRs, 70.6% (12/17) were pyramidal neurons, 17.6% (3/17) were fast-spiking interneurons and 11.8% (2/17) were non-fast spiking interneurons. Figure 5 shows an example HR (left column) and LR (right column) with clear fluorescent labeling (A, B), typical firing properties (C-H) and classical pyramidal neuron morphology (I, J). To detect changes in excitability, we measured input resistance (Rinput), resting membrane potential (Vrest), rheobase, firing frequency during current injections steps, and spike threshold in 16 HR pyramidal neurons (n = 12 mice) and 12 LR pyramidal neurons (n = 9 mice). In addition, we quantified the initial doublet spike burst as the first inter-spike interval (ISI1) in a 10-spike train. We found that HRs fired fewer spikes per 100 pA current injected compared to LRs (HRs: mean ± SD 8.4 ± 2.6, LRs: 11.7 ± 4.6; Mann–Whitney U = 45, P < 0.05), indicating reduced excitability in this population. Other measures of excitability were, however, not significantly different between the two groups (Fig. 6).
Figure 6 .

Electrophysiological parameters of neuronal excitability. HR neurons (n = 16 cells, 12 mice) and LR neurons (n = 12 cells, 9 mice) were compared in terms of firing frequency per 100 pA injected, input resistance (Rinput), resting membrane potential (Vrest), rheobase current, spike threshold, and first inter-interspike interval (ISI1 in a 10-spike train). Statistical comparisons were performed using a Mann–Whitney U test. Box plots show the median (central horizontal bar), mean (+), as well as the first and third quartiles (lower and upper limits).
To further characterize HRs and LRs, we measured passive and active electrophysiological properties. Since excitatory neurons in L2/3 of the barrel cortex show similar electrophysiological properties (Avermann et al. 2012), we did not expect to see differences between the groups of excitatory HRs and LRs. Indeed, passive membrane properties were comparable in both groups, as demonstrated by similar membrane time constants and sag amplitudes (Table 1). Moreover, no differences were found in AP amplitude or half-width, and in the amplitude and timing of the AHP. Other active properties, including the maximum firing frequency and adaptation of the inter-spike intervals were also comparable between HRs and LRs (Table 1).
Table 1.
Electrophysiological parameters in tagged HR and LR pyramidal neurons. Comparison between HRs (n = 16) and LRs (n = 12) were assessed using Mann–Whitney U tests. Bonferroni-adjusted significance level: P = 0.0045
| Parameter | HR mean | HR SD | LR mean | LR SD | U | P |
|---|---|---|---|---|---|---|
| Tau at −50 pA (ms) | 23.69 | 5.87 | 24.29 | 6.90 | 95.0 | 0.982 |
| Sag at −7.5 mV (%) | 6.04 | 6.61 | 7.40 | 9.65 | 90.0 | 0.802 |
| Max. Sag (%) | 8.45 | 5.54 | 11.34 | 8.66 | 77.0 | 0.397 |
| AP amplitude (mV) | 85.00 | 8.80 | 86.73 | 7.08 | 88.0 | 0.732 |
| AP half-width (ms) | 1.53 | 0.22 | 1.38 | 0.17 | 133.0 | 0.090 |
| AHP amplitude (mV) | 21.54 | 7.83 | 22.35 | 8.78 | 83.0 | 0.568 |
| AHP time (ms) | 499.27 | 118.02 | 452.24 | 101.34 | 124.0 | 0.205 |
| Max. Firing freq (Hz) | 43.75 | 15.59 | 60.83 | 28.60 | 58.5 | 0.084 |
| Adaptation ratio ISI2/9 (10-spike train) | 0.56 | 0.18 | 0.62 | 0.15 | 65.0 | 0.159 |
| Adaptation slope/100 pA | −0.08 | 0.24 | −0.10 | 0.25 | 101.0 | 0.837 |
| Recording depth | −58.50 | 17.36 | −56.67 | 15.24 | 89.5 | 0.811 |
High Responders Receive Larger and More Frequent Excitatory Network Inputs
It is possible that decreased excitability in HRs is a result of homeostatic adaptation to increased inputs (Turrigiano 2012) provided by the network of highly connected HRs. We therefore examined the timing and magnitude of spontaneous excitatory postsynaptic currents (sEPSCs) in high and low responding neurons. Cells were clamped at −70 mV to minimize contamination by inhibitory postsynaptic potentials. Importantly, blockers of spiking activity were not added to the extracellular solution to maintain spontaneous network activity and avoid hyperexcitability. In total, we recorded 700 s of spontaneous activity in HRs (n = 8 neurons; 5 mice; 3092 sEPSCs; see example in Figure 7A) and 720 s in LRs (n = 8 neurons; 6 mice; 3145 sEPSCs; see example in Fig. 7B). For each group, we randomly selected 150 sEPSCs per cell, pooled them across neurons and found a significant increase in sEPSC amplitude in HRs (mean ± SD, −9.29 ± 6.12 pA, n = 1200) compared to LRs (−8.26 ± 4.27 pA, n = 1200; Mann–Whitney U test, P < 0.001; Fig. 7C and D). Differences between the two groups were particularly notable with respect to high amplitude sEPSCs (<−20 pA), which were more common in HRs (4.7% of all sEPSCs) than LRs (2.6% of all EPSCs; Fisher’s exact test, P < 0.01; Fig. 7C, left panel, inset). These rare, high-amplitude events may play a decisive role in determining overall network activity and may thus be of key importance for stimulus coding (Lefort et al. 2009). The increase in HR sEPSC amplitude cannot be explained by changes in sEPSC properties as the 20–80% rise time was comparable across groups (HRs: 0.84 ± 0.55 ms, n = 1200; LRs: 0.8 ± 0.54 ms, n = 1200; Mann–Whitney U test, P > 0.05). There was also no significant difference in inter-event-intervals of sEPSCs between HRs (240.4 ± 254.1 ms, n = 1200) and LRs (250.7 ± 298.1 ms, n = 1200; Mann–Whitney U test, P > 0.05; Fig. 7C and D). Overall, the observed increase in spontaneous excitatory inputs in HRs suggests a presynaptic mechanism for the increased activity level of HRs and lends further support to the idea that HRs form a network of highly active and strongly interconnected neurons.
Figure 7 .

Spontaneous excitatory postsynaptic currents (sEPSCs) in HRs and LRs. (A) Left panel: HR example raw trace with sEPSCs (market by asterisks). Right panel: Enlarged view of example sEPSC (marked by gray asterisk in raw trace) with the 20–80% rise time indicated by green crosses and the peak indicated by a gray bar. (B) LR example raw trace (left) and enlarged sEPSC (right). Same conventions as in A. (C) Scattergrams of sEPSC amplitudes as a function of inter-event intervals (left) and rise times (right) with respective marginal histograms (50 bins). (D) Comparison of amplitude (left), inter-event interval (middle) and 20–80% rise time (right) between HR sEPSCs (n = 1200; 8 cells in 5 mice) and LR sEPSCs (n = 1200; 8 cells in 6 mice) using Mann–Whitney U tests. 150 randomly selected events were included from each neuron and pooled for each group. Same conventions for box plots as in Figure 6.
Morphological Characteristics Are Similar in HRs and LRs
To pinpoint possible structural differences between high and low responsive cells in L2/3 of the barrel cortex, we filled patched HRs and LRs with biocytin and reconstructed their morphology in 3D subsequently (see Materials and Methods). In total, morphological features were obtained from 19 tagged excitatory HRs (n = 15 mice) and 10 tagged excitatory LRs (n = 7 mice). In agreement with earlier studies (Feldmeyer et al. 2006; Lübke and Feldmeyer 2010; van Aerde and Feldmeyer 2015), neurons in both groups could be divided in three sub-categories of L2/3 excitatory neurons based on the structure of the apical dendrite: horizontal, broad-tufted and thin-tufted pyramidal neurons (Fig. 8A-C). Horizontal pyramidal neurons were observed in upper L2/3 close to the border with L1 (mean ± SD, 128.7 ± 20.0 μm below pia; n = 9) and were characterized by an apical dendrite with an atypical, oblique orientation relative to the pial surface. The remaining neurons were classified based on morphological characteristics of the apical dendrites: Neurons for which the field span of the apical dendrites was larger than that of the basal dendrites (see Materials and Methods) were termed “broad-tufted,” whereas neurons with the inverse relationship were termed “thin-tufted.” In line with previous reports (Lubke et al. 2003; Feldmeyer et al. 2006; Oberlaender et al. 2012; van Aerde and Feldmeyer 2015), broad-tufted neurons were mainly located at an intermediate depth of L2/3 (166 ± 37.8 μm below pia; n = 13), and thin-tufted neurons were more frequent in deeper regions of L2/3 (186 ± 23.5 μm below pia; n = 7; Fig. 8D). The proportion of these sub-types among HRs and LRs were not different.
Figure 8 .

Dendritic morphology of excitatory HRs and LRs. Pyramidal HR neurons (red; n = 19 cells, 15 mice) and LR neurons (blue; n = 10 cells, 7 mice) could be subdivided into three categories based on the structure of the apical dendrite: horizontal, thin-tufted and broad-tufted. (A) Pyramidal neurons with a horizontally oriented apical dendrite. Layer borders are indicated by gray dashed lines. (B) Pyramidal neurons with a thin-tufted apical dendrite. (C) Pyramidal neurons with a thick-tufted apical dendrite. There is no difference of this sub-type proportion between the two groups (P = 0.43, Fisher exact test). (D) Subpial depth of reconstructed cells. Left-to-right: Infrared-DIC micrograph of layers 1–3, fluorescent image of tagged cells in layers 1–3, and depth distribution of cells included in morphological analysis (H: horizontal, B: broad-tufted, T: thin-tufted apical dendrites).
Various morphological parameters were examined and compared between HRs and LRs, including the number, length, layer distribution, complexity, and spine density of apical and basal dendrites. In addition, the dendritic branching frequency as well as the ratio of apical and basal dendritic length was analyzed. Since no significant differences were found when comparing individual sub-types of excitatory HRs and LRs, data were pooled and only comparisons at the group level are shown here. However, excitatory HRs and LRs did not differ in any of the analyzed morphological parameters (Table 2). The recording depth relative to the pial surface was comparable between the two groups, as the patched neurons were located at similar depths in L2/3 (Table 2, Fig. 8D). Our results indicate that morphological characteristics are unlikely to account for the observed differences in activity in HRs and LRs.
Table 2.
Morphological parameters in tagged HR and LR pyramidal neurons. Statistical comparisons were performed with Mann–Whitney U tests. Bonferroni-adjusted significance level: P = 0.0036
| Parameter | HR n | HR mean | HR SD | LR n | LR mean | LR SD | U | P | |
|---|---|---|---|---|---|---|---|---|---|
| Basal dendrites | Number | 19 | 7.32 | 1.60 | 10 | 6.80 | 1.40 | 114.5 | 0.372 |
| Total length | 19 | 2601.43 | 635.50 | 10 | 2519.06 | 1010.16 | 106.0 | 0.636 | |
| L1 length (% total) | 19 | 12.22 | 20.40 | 10 | 11.14 | 14.39 | 92.0 | 0.903 | |
| L2/3 length (% total) | 19 | 84.69 | 21.33 | 10 | 84.40 | 14.11 | 106.0 | 0.626 | |
| Complexity | 19 | 37288.37 | 15363.20 | 10 | 49369.52 | 40354.79 | 85.0 | 0.668 | |
| Spines/100 um | 14 | 51.43 | 14.50 | 9 | 62.37 | 16.13 | 38.0 | 0.124 | |
| Apical dendrite | Total length | 19 | 2077.47 | 794.02 | 10 | 1944.24 | 612.07 | 105.0 | 0.668 |
| L1 Length (% total) | 19 | 51.94 | 26.01 | 10 | 52.32 | 31.22 | 94.0 | 0.982 | |
| L2/3 length (% total) | 19 | 47.77 | 26.38 | 10 | 46.34 | 32.22 | 99.0 | 0.875 | |
| Complexity | 19 | 334794.95 | 249281.69 | 10 | 312926.49 | 242597.45 | 97.0 | 0.946 | |
| Spines/100 um | 14 | 55.28 | 17.59 | 9 | 61.78 | 15.63 | 44.0 | 0.250 | |
| Basal + apical dendrites | Branching frequency (nodes/100 μm) | 19 | 1.02 | 0.44 | 10 | 0.96 | 0.13 | 72.0 | 0.308 |
| Ratio total dendritic length (apical/basal) | 19 | 0.84 | 0.32 | 10 | 0.88 | 0.35 | 87.0 | 0.735 | |
| Cell body | Soma-pia distance (μm) | 19 | 156.25 | 37.76 | 10 | 165.48 | 35.96 | 77.0 | 0.530 |
Discussion
Our study sheds light on how L2/3 neurons are recruited into active spiking ensembles during passive whisker touch and establishes for the first time a direct link between highly active and less active neurons observed in vivo and their characteristics studied in vitro. In summary, we found that a small subset (9.2%) of highly active neurons accounted for a large portion (41%) of the overall spiking activity measured during passive whisker stimulation. These HRs displayed large, fast and reliable activity following stimulation. HRs tended to be co-activated and were more strongly coupled to the population response compared to LRs. In vitro recordings of HRs previously identified in vivo revealed an increased amplitude and frequency of sEPSCs compared to LRs. The increased excitatory drive in HRs was accompanied by a reduction in intrinsic excitability, as measured by a reduced number of spikes in response to increasing current injection. Both HRs and LRs displayed morphological features typical for pyramidal cells, including horizontal, thin- or broad-tufted apical dendrites.
In response to principal whisker deflection, excitatory neurons in barrel cortex normally fire only very few APs (Simons 1978; Brecht et al. 2003; Wilent and Contreras 2005; de Kock et al. 2007; Kerr et al. 2007; Sato et al. 2007), even when optimal stimulation parameters are chosen (Ramirez et al. 2014). This form of sparseness has been referred to as “population sparseness” and is related to a high trial-to-trial variability in spiking output for a given neuron. Another type of sparseness is “life-time sparseness” where a small and fixed subset of neurons responds faithfully to stimulation (Wolfe et al. 2010; Barth and Poulet 2012; Sachdev et al. 2012). This type of sparseness is especially prevalent in L2/3 of the barrel cortex where studies have identified a neuronal subpopulation that shows elevated stimulus-evoked and spontaneous responses (Kerr et al. 2007; Margolis et al. 2012; Mayrhofer et al. 2015). Our data provide a link between these two concepts of sparseness: Compared to LRs, HRs show larger and more reliable responses (Figs 2E and F and 3A), and contribute more to the stimulus-evoked activity despite fluctuations in the responding ensemble size (i.e., trial-to-trial variability; Figs 3E and 4B). Thus, lifetime and population sparseness are not mutually exclusive principles and both can be observed in L2/3 of the barrel cortex.
Apart from being highly responsive to stimuli, HRs also showed more correlated evoked responses compared to LRs (Fig. 4D-F), suggesting that they tended to be co-activated during stimulation. Thus, not only were HRs active during more trials, but they tended to be active during the same trials. This shared variability in trial-to-trial activation could originate from either direct synaptic connections or from common inputs (Shadlen and Newsome 1998; Cohen and Kohn 2011). The observation that HR activity is more correlated not only during stimulation but also outside periods of stimulation (Fig. 4G-J) indicates that shared connections, rather than stimulus-driven bottom-up input, may be responsible for their co-activation. In line with this reasoning, correlated spontaneous activity in barrel cortex has previously been shown to emerge from synchronized intracortical inputs, rather than from thalamic inputs (Cohen-Kashi Malina et al. 2016). Furthermore, HRs displayed increased coupling to the population activity (Fig. 4K), which has previously been associated with an increased amount of non-specific excitatory connections with neighboring neurons (Okun et al. 2015). Population coupling predicted a substantial amount of the observed spontaneous correlations in all types of responders (Fig. 4L), but left positively tailed correlation residuals only in HR pairs (Fig. 4M and N). In V1, such residuals were shown to be mainly dependent on the similarity of the neurons’ tuning curves (Okun et al. 2015). Most importantly, it has been shown that enhanced pairwise correlations, especially in neurons with similar tuning, directly relate to an increased synaptic connection strength (Ko et al. 2011; Cossell et al. 2015). Taken together, these findings suggest that HRs form a subnetwork of neurons that are highly interconnected (Song et al. 2005).
Increased spontaneous activity and correlated spontaneous events in HRs indicate that their intrinsic or morphological properties might be key determinants for their recruitment during stimulation. Previous studies addressing this issue in L2/3 of the barrel cortex identified HRs in vitro based on elevated expression of the immediate early gene c-fos as an indicator of neuronal activity (Yassin et al. 2010; Benedetti et al. 2013) or based on electrical stimulation of L4 (Elstrott et al. 2014). In agreement with our results (Fig. 6), these studies found a reduction in intrinsic excitability in HRs as evidenced by a shallower slope of the F-I curve, while resting membrane potential and input resistance did not differ from LRs (Yassin et al. 2010; Elstrott et al. 2014). However, the studies disagreed on potential differences in rheobase current and AP threshold between HR and LRs. Our study design allowed us to address this issue by identifying HRs in vivo and measuring their properties in vitro, thereby establishing a direct link between in vivo and in vitro measurements and thus avoiding possible methodological confounds in previous research. Our results indicate that rheobase current and AP threshold (as indicators of neuronal excitability) are unaltered in HRs (Fig. 6).
If HRs are less excitable than LRs, then more inputs may explain their enhanced responsiveness. In line with previous in vitro research (Yassin et al. 2010), we found that HRs show a higher frequency of spontaneous sEPSCs. In addition, our results also revealed larger amplitudes of sEPSCs in HRs (Fig. 7). These findings indicate that HRs may down-regulate their intrinsic excitability in response to elevated excitatory inputs in order to maintain their homeostatic balance (Lambo and Turrigiano 2013; Gainey and Feldman 2017), as previously suggested (Yassin et al. 2010; Elstrott et al. 2014). The highly skewed distribution of sEPSC amplitudes, with many small-amplitude and few large-amplitude events, is in close agreement with previous data on excitatory postsynaptic potentials (EPSPs) in barrel cortex L2/3 (Holmgren et al. 2003; Feldmeyer et al. 2006; Lefort et al. 2009). Based on experimental observations and computational models, it has been argued that rare large-amplitude events may be more important than small ones in determining the overall network activity and may be fundamental to sparse stimulus coding (Poulet and Petersen 2008; Lefort et al. 2009). It is, therefore, interesting to note that the relative frequency of large amplitude sEPSCs was higher in HRs compared to LRs (Fig. 7C, left panel, inset), indicating that HRs may function as “hubs” in processing sensory information. The observed enhanced correlations among HRs in vivo (Fig. 4D-J) support the notion that large-amplitude sEPSCs may arise as a result of correlated activity of local pre- and postsynaptic neurons (Hebb 1961; Markram et al. 1997; Feldman 2000; Sjostrom et al. 2001). In contrast, low-amplitude sEPSCs may provide opportunities for plasticity in the neural network, as previously suggested (Lefort et al. 2009).
An increase in excitatory inputs to HRs may arise from enhanced bottom-up, top-down or intracortical drive. In our preparation of thalamocortical brain slices, inputs from longer-range connections (even outside S1) cannot be completely excluded but are rather unlikely due to the truncation of axons during the slicing process. It is more likely that the increased inputs to HRs arose from local L4-to-L2/3 or L2/3-to-L2/3 connections because of the high density of presynaptic L4 and L2/3 axons and postsynaptic L3 dendrites, which results in a large number of potential synaptic connections (Feldmeyer et al. 2002; Brecht et al. 2003; Holmgren et al. 2003; Feldmeyer et al. 2006). Previous studies discovered that c-fos-labeled HRs in L2/3 were more likely to be connected to each other (Yassin et al. 2010) and received a larger number of L4 inputs compared to LRs (Benedetti et al. 2013). Similarly, studies in visual cortex revealed that if L2/3 pyramidal cells shared common input from L4 and within L2/3, they were more likely to be reciprocally connected (Yoshimura et al. 2005). These findings support the idea that enhanced inputs to HRs arise from L4 as well as L2/3, in particular from other connected L2/3 HRs.
As differences in neuronal function may also relate to differences in morphological features (Oberlaender et al. 2012), we set out to characterize the dendritic structure of HR and LRs by means of biocytin staining and 3D morphological reconstructions. Our study showed for the first time that HRs span all three subcategories of pyramidal cell morphologies, including thick-tufted, thin-tufted and horizontal pyramidal neurons (Fig. 8). Similar to an earlier study (Elstrott et al. 2014), HRs and LRs did not show differences in the spine density or total length of basal or apical dendrites. In addition, we found no differences in the complexity, layer-length distribution and branching frequency of basal and apical dendrites. While the statistical power of our study may not be sufficient to detect subtle differences in morphology, we believe that the similarity of morphology between HR and LR pyramidal neurons suggests that differences in responsiveness may largely be due to functional characteristics related to connectivity and enhanced inputs.
An often-debated issue is whether the observed sparseness of responses is a result of anesthesia, during which activity in the brain is generally suppressed (Barth and Poulet 2012), and/or a result of specific stimulation conditions (Barth and Poulet 2012; Ranjbar-Slamloo and Arabzadeh 2017; Ranjbar-Slamloo and Arabzadeh 2019). Although we cannot exclude this possibility, we believe that anesthesia-related effects did not play a significant role in our study since the concentration of administered isoflurane was kept to a minimum in order to observe large stimulus-evoked responses. Several studies have previously demonstrated sparse activity of L2/3 excitatory neurons both during anesthetized and awake states (de Kock and Sakmann 2009; O’Connor et al. 2010; Crochet et al. 2011; Margolis et al. 2012). Regarding the role of stimulation, current literature suggests that dense evoked responses are restricted to very specific stimulation conditions and anesthetic agents used, i.e., passive, high velocity whisker deflections during urethane anesthesia (Ranjbar-Slamloo and Arabzadeh 2017). Together, these findings indicate that sparse responses are a general feature of L2/3 neurons and not exclusively associated with the anesthetized brain state or specific stimuli (Poulet and Petersen 2008; O’Connor et al. 2010; Mayrhofer et al. 2015; Peron et al. 2015).
Sparse population responses are not unique to the barrel cortex, but occur across different brain regions and in different species (Vinje and Gallant 2000; Hromadka et al. 2008; Poo and Isaacson 2009; Ko et al. 2011; Okun et al. 2015). It has been argued that the advantage of sparse coding is the reduced metabolic cost associated with a reduced number of firing neurons (Laughlin 2001; Laughlin and Sejnowski 2003; Lennie 2003). However, it has been shown that AP generation is not as metabolically costly as has been previously thought (Alle et al. 2009). In addition, maintaining a large number of non-responsive neurons in a resting state is also energy-expensive. Thus, there might be other, more important benefits associated with a sparse code, e.g., that it allows for transmitting signals with a higher information content (Olshausen and Field 2004), that it maximizes the number of patterns of activity robustly stored in the network (Brunel 2016), or that it represents a salient signal in the brain (Wolfe et al. 2010). Indeed, the firing of a few L2/3 barrel cortex neurons has been shown to be behaviorally detectable by the animal (O’Connor et al. 2010). In the future, it will be interesting to determine the effects of activating or de-activating HRs under different behavioral conditions, e.g., when the animal is solving a discrimination task, and to track HR activity under the influence of plasticity, e.g., after whisker removal. Optogenetic tools in combination with two-photon holographic stimulation (Chaigneau et al. 2016) can help to shed light on these questions.
Supplementary Material
Contributor Information
C S Barz, Institute of Neuroscience and Medicine, INM-10, Research Centre Jülich, 52425 Jülich, Germany; Department of Psychiatry, Psychotherapy and Psychosomatics, Medical School, RWTH Aachen University, 52074 Aachen, Germany; Jülich-Aachen Research Alliance - Translational Brain Medicine, 52074 Aachen, Germany; IZKF Aachen, Medical School, RWTH Aachen University, 52074 Aachen, Germany.
P M Garderes, IZKF Aachen, Medical School, RWTH Aachen University, 52074 Aachen, Germany; Department of Neuropathology, Medical School, RWTH Aachen University, 52074 Aachen, Germany; Department of Ophthalmology, Medical School, RWTH Aachen University, 52074 Aachen, Germany; Unit of Neural Circuits Dynamics and Decision Making, Institut Pasteur, 75015 Paris, France.
D A Ganea, IZKF Aachen, Medical School, RWTH Aachen University, 52074 Aachen, Germany; Department of Neuropathology, Medical School, RWTH Aachen University, 52074 Aachen, Germany; Department of Ophthalmology, Medical School, RWTH Aachen University, 52074 Aachen, Germany; Biomedical Department, University of Basel, 4056 Basel, Switzerland.
S Reischauer, Medical Clinic I, (Cardiology/Angiology) and Campus Kerckhoff, Justus-Liebig-University Giessen, 35390 Giessen Germany; Department of Developmental Genetics, Max Planck Institute for Heart and Lung Research, 61231 Bad Nauheim, Germany; Cardio-Pulmonary Institute (CPI), 35392 Giessen, Germany.
D Feldmeyer, Institute of Neuroscience and Medicine, INM-10, Research Centre Jülich, 52425 Jülich, Germany; Department of Psychiatry, Psychotherapy and Psychosomatics, Medical School, RWTH Aachen University, 52074 Aachen, Germany; Jülich-Aachen Research Alliance - Translational Brain Medicine, 52074 Aachen, Germany.
F Haiss, IZKF Aachen, Medical School, RWTH Aachen University, 52074 Aachen, Germany; Department of Neuropathology, Medical School, RWTH Aachen University, 52074 Aachen, Germany; Department of Ophthalmology, Medical School, RWTH Aachen University, 52074 Aachen, Germany; Unit of Neural Circuits Dynamics and Decision Making, Institut Pasteur, 75015 Paris, France.
Author Contributions
Conceptualization, F.H.; Methodology, C.B., P.M.G. and F.H.; Software, C.B., P.M.G. and D.G.; Formal Analysis, C.B. and P.M.G.; Investigation, C.B. and P.M.G; Resources, D.G., S.R., D.F. and F.H.; Writing—Original Draft, C.B.; Writing—Review & Editing, C.B., P.M.G., D.G., S.R., D.F. and F.H.; Visualization, C.B. and P.M.G.; Supervision, D.F. and F.H.; Project Administration, C.B.; Funding Acquisition, D.F. and F.H.
Data Availability
Source data files of raw fluorescence traces and MATLAB code pertaining to Figs 2-4 are provided under the following link: https://doi.org/10.5061/dryad.cnp5hqc2q. Other data supporting the findings of this study are available from the corresponding author upon reasonable request.
Funding
The Minerva Stiftung Gesellschaft für die Forschung mbH (to D.G.); Deutsche Forschungsgemeinschaft SFB-1213 Project B01 (to S.R.); the Helmholtz Society (to D.F.); Deutsche Forschungsgemeinschaft Research Unit ``Barrel Cortex Function'' (Grant 472/4-2 to D.F.); the EU's Horizon 2020 Research and Innovation Programme (Grant Agreement No. 720270, HBP SGA1 to D.F.); Interdisziplinäres Zentrum für Klinische Forschung, Medical Faculty of RWTH Aachen University, Germany (to F.H.).
Notes
The RCaMP2 construct has been kindly provided by Haruhiko Bito (University of Tokyo, Graduate School of Medicine, Tokyo, Japan). pAAV.Syn.NES-jRGECO1a.WPRE.SV40 was a gift from Douglas Kim & GENIE Project and pACAGW-H2B-PAGFP-AAV was a gift from Massimo Scanziani. We thank Werner Hucko (Research Centre Jülich, Jülich, Germany) for the histological processing of the brain slices and Axel Honné (Medical Faculty, RWTH Aachen University, Aachen, Germany) for technical support. Furthermore, we thank Björn Michael Kampa (RWTH Aachen University, Aachen, Germany), Charly Rousseau and Christoph Schmidt-Hieber (Institut Pasteur, Paris, France) for helpful discussions on the manuscript. Conflict of Interest. No competing interests are declared.
References
- Alle H, Roth A, Geiger JR. 2009. Energy-efficient action potentials in hippocampal mossy fibers. Science. 325:1405–1408. [DOI] [PubMed] [Google Scholar]
- Avermann M, Tomm C, Mateo C, Gerstner W, Petersen CC. 2012. Microcircuits of excitatory and inhibitory neurons in layer 2/3 of mouse barrel cortex. J Neurophysiol. 107:3116–3134. [DOI] [PubMed] [Google Scholar]
- Barth AL, Poulet JF. 2012. Experimental evidence for sparse firing in the neocortex. Trends Neurosci. 35:345–355. [DOI] [PubMed] [Google Scholar]
- Benedetti BL, Takashima Y, Wen JA, Urban-Ciecko J, Barth AL. 2013. Differential wiring of layer 2/3 neurons drives sparse and reliable firing during neocortical development. Cereb Cortex. 23:2690–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brecht M, Roth A, Sakmann B. 2003. Dynamic receptive fields of reconstructed pyramidal cells in layers 3 and 2 of rat somatosensory barrel cortex. J Physiol. 553:243–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunel N. 2016. Is cortical connectivity optimized for storing information? Nat Neurosci. 19:749–755. [DOI] [PubMed] [Google Scholar]
- Chaigneau E, Ronzitti E, Gajowa MA, Soler-Llavina GJ, Tanese D, Brureau AYB, Papagiakoumou E, Zeng H, Emiliani V. 2016. Two-photon holographic stimulation of ReaChR. Front Cell Neurosci. 10:234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy KB, Schnepel P, Rao AT, Feldman DE. 2015. Structure of a single whisker representation in layer 2 of mouse somatosensory cortex. J Neurosci. 35:3946–3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MR, Kohn A. 2011. Measuring and interpreting neuronal correlations. Nat Neurosci. 14:811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Kashi Malina K, Mohar B, Rappaport AN, Lampl I. 2016. Local and thalamic origins of correlated ongoing and sensory-evoked cortical activities. Nat Commun. 7:12740–12740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossell L, Iacaruso MF, Muir DR, Houlton R, Sader EN, Ko H, Hofer SB, Mrsic-Flogel TD. 2015. Functional organization of excitatory synaptic strength in primary visual cortex. Nature. 518:399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crochet S, Poulet JF, Kremer Y, Petersen CC. 2011. Synaptic mechanisms underlying sparse coding of active touch. Neuron. 69:1160–1175. [DOI] [PubMed] [Google Scholar]
- Dana H, Mohar B, Sun Y, Narayan S, Gordus A, Hasseman JP, Tsegaye G, Holt GT, Hu A, Walpita D, et al. 2016. Sensitive red protein calcium indicators for imaging neural activity. Elife. 24:12727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kock CP, Bruno RM, Spors H, Sakmann B. 2007. Layer- and cell-type-specific suprathreshold stimulus representation in rat primary somatosensory cortex. J Physiol. 581:139–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kock CP, Sakmann B. 2009. Spiking in primary somatosensory cortex during natural whisking in awake head-restrained rats is cell-type specific. Proc Natl Acad Sci U S A. 106:16446–16450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll LN, Pettit NL, Minderer M, Chettih SN, Harvey CD. 2017. Dynamic reorganization of neuronal activity patterns in parietal cortex. Cell. 170:986–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elstrott J, Clancy KB, Jafri H, Akimenko I, Feldman DE. 2014. Cellular mechanisms for response heterogeneity among L2/3 pyramidal cells in whisker somatosensory cortex. J Neurophysiol. 112:233–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman DE. 2000. Timing-based LTP and LTD at vertical inputs to layer II/III pyramidal cells in rat barrel cortex. Neuron. 27:45–56. [DOI] [PubMed] [Google Scholar]
- Feldmeyer D, Lubke J, Sakmann B. 2006. Efficacy and connectivity of intracolumnar pairs of layer 2/3 pyramidal cells in the barrel cortex of juvenile rats. J Physiol. 575:583–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmeyer D, Lubke J, Silver RA, Sakmann B. 2002. Synaptic connections between layer 4 spiny neurone-layer 2/3 pyramidal cell pairs in juvenile rat barrel cortex: physiology and anatomy of interlaminar signalling within a cortical column. J Physiol. 538:803–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainey MA, Feldman DE. 2017. Multiple shared mechanisms for homeostatic plasticity in rodent somatosensory and visual cortex. Philos Trans R Soc Lond B Biol Sci. 372:20160157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebb DO. 1961. The Organization of Behavior. New York: Science Editions. Inc., 77:416–426. [Google Scholar]
- Holmgren C, Harkany T, Svennenfors B, Zilberter Y. 2003. Pyramidal cell communication within local networks in layer 2/3 of rat neocortex. J Physiol. 551:139–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hromadka T, Deweese MR, Zador AM. 2008. Sparse representation of sounds in the unanesthetized auditory cortex. PLoS Biol. 6:0060016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Takeuchi A, Horigane S, Ohkura M, Gengyo-Ando K, Fujii H, Kamijo S, Takemoto-Kimura S, Kano M, Nakai J, et al. 2015. Rational design of a high-affinity, fast, red calcium indicator R-CaMP2. Nat Methods. 12:64–70. [DOI] [PubMed] [Google Scholar]
- Jiang C, Agulian S, Haddad GG. 1991. O2 tension in adult and neonatal brain slices under several experimental conditions. Brain Res. 568:159–164. [DOI] [PubMed] [Google Scholar]
- Kerr JN, de Kock CP, Greenberg DS, Bruno RM, Sakmann B, Helmchen F. 2007. Spatial organization of neuronal population responses in layer 2/3 of rat barrel cortex. J Neurosci. 27:13316–13328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko H, Hofer SB, Pichler B, Buchanan KA, Sjostrom PJ, Mrsic-Flogel TD. 2011. Functional specificity of local synaptic connections in neocortical networks. Nature. 473:87–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambo ME, Turrigiano GG. 2013. Synaptic and intrinsic homeostatic mechanisms cooperate to increase L2/3 pyramidal neuron excitability during a late phase of critical period plasticity. J Neurosci. 33:8810–8819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughlin SB. 2001. Energy as a constraint on the coding and processing of sensory information. Curr Opin Neurobiol. 11:475–480. [DOI] [PubMed] [Google Scholar]
- Laughlin SB, Sejnowski TJ. 2003. Communication in neuronal networks. Science (New York, NY). 301:1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefort S, Tomm C, Floyd Sarria JC, Petersen CC. 2009. The excitatory neuronal network of the C2 barrel column in mouse primary somatosensory cortex. Neuron. 61:301–316. [DOI] [PubMed] [Google Scholar]
- Lennie P. 2003. The cost of cortical computation. Curr Biol. 13:493–497. [DOI] [PubMed] [Google Scholar]
- Lien AD, Scanziani M. 2011. In vivo labeling of constellations of functionally identified neurons for targeted in vitro recordings. Frontiers in Neural Circuits. 5:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubke J, Roth A, Feldmeyer D, Sakmann B. 2003. Morphometric analysis of the columnar innervation domain of neurons connecting layer 4 and layer 2/3 of juvenile rat barrel cortex. Cereb Cortex. 13:1051–1063. [DOI] [PubMed] [Google Scholar]
- Lübke JHR, Feldmeyer D. 2010. The axon of excitatory neurons in the neocortex: projection patterns and target specificity. In: Feldmeyer D, Lübke JHR, editors. New Aspects of Axonal Structure and Function. Boston, MA: Springer US, pp. 157–178. [Google Scholar]
- Margolis DJ, Lutcke H, Schulz K, Haiss F, Weber B, Kugler S, Hasan MT, Helmchen F. 2012. Reorganization of cortical population activity imaged throughout long-term sensory deprivation. Nat Neurosci. 15:1539–1546. [DOI] [PubMed] [Google Scholar]
- Markram H, Lubke J, Frotscher M, Sakmann B. 1997. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 275:213–215. [DOI] [PubMed] [Google Scholar]
- Marx M, Gunter RH, Hucko W, Radnikow G, Feldmeyer D. 2012. Improved biocytin labeling and neuronal 3D reconstruction. Nat Protoc. 7:394–407. [DOI] [PubMed] [Google Scholar]
- Mayrhofer JM, Haiss F, Haenni D, Weber S, Zuend M, Barrett MJP, Ferrari KD, Maechler P, Saab AS, Stobart JL, et al. 2015. Design and performance of an ultra-flexible two-photon microscope for in vivo research. Biomed Opt Express. 6:4228–4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohar B, Dana H, Kim DS, Svoboda K. 2016. jRGECO1a and jRCaMP1a characterization in the intact mouse visual cortex, using AAV-based gene transfer, 2-photon imaging and loose-seal cell attached recordings, as described in Dana et al 2016. 10.6080/K0W37T87. [DOI]
- Oberlaender M, de Kock CP, Bruno RM, Ramirez A, Meyer HS, Dercksen VJ, Helmstaedter M, Sakmann B. 2012. Cell type-specific three-dimensional structure of thalamocortical circuits in a column of rat vibrissal cortex. Cereb Cortex. 22:2375–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor DH, Peron SP, Huber D, Svoboda K. 2010. Neural activity in barrel cortex underlying vibrissa-based object localization in mice. Neuron. 67:1048–1061. [DOI] [PubMed] [Google Scholar]
- Okun M, Steinmetz N, Cossell L, Iacaruso MF, Ko H, Bartho P, Moore T, Hofer SB, Mrsic-Flogel TD, Carandini M, et al. 2015. Diverse coupling of neurons to populations in sensory cortex. Nature. 521:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshausen BA, Field DJ. 2004. Sparse coding of sensory inputs. Curr Opin Neurobiol. 14:481–487. [DOI] [PubMed] [Google Scholar]
- Pachitariu M, Stringer C, Schröder S, Dipoppa M, Rossi LF, Carandini M, Harris KD. 2016. Suite2p: beyond 10,000 neurons with standard two-photon microscopy. In: bioRxiv, p. 061507. doi: 10.1101/061507. [DOI]
- Peron SP, Freeman J, Iyer V, Guo C, Svoboda K. 2015. A cellular resolution map of barrel cortex activity during tactile behavior. Neuron. 86:783–799. [DOI] [PubMed] [Google Scholar]
- Pillai AG, de Jong D, Kanatsou S, Krugers H, Knapman A, Heinzmann JM, Holsboer F, Landgraf R, Joels M, Touma C. 2012. Dendritic morphology of hippocampal and amygdalar neurons in adolescent mice is resilient to genetic differences in stress reactivity. PLoS One. 7:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pologruto TA, Sabatini BL, Svoboda K. 2003. ScanImage: flexible software for operating laser scanning microscopes. Biomed Eng Online. 2:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poo C, Isaacson JS. 2009. Odor representations in olfactory cortex: “sparse” coding, global inhibition, and oscillations. Neuron. 62:850–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulet JF, Petersen CC. 2008. Internal brain state regulates membrane potential synchrony in barrel cortex of behaving mice. Nature. 454:881–885. [DOI] [PubMed] [Google Scholar]
- Ramirez A, Pnevmatikakis EA, Merel J, Paninski L, Miller KD, Bruno RM. 2014. Spatiotemporal receptive fields of barrel cortex revealed by reverse correlation of synaptic input. Nat Neurosci. 17:866–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjbar-Slamloo Y, Arabzadeh E. 2017. High-velocity stimulation evokes “dense” population response in layer 2/3 vibrissal cortex. J Neurophysiol. 117:1218–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjbar-Slamloo Y, Arabzadeh E. 2019. Diverse tuning underlies sparse activity in layer 2/3 vibrissal cortex of awake mice. J Physiol. 597:2803–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachdev RN, Krause MR, Mazer JA. 2012. Surround suppression and sparse coding in visual and barrel cortices. Front Neural Circuits. 6:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato TR, Gray NW, Mainen ZF, Svoboda K. 2007. The functional microarchitecture of the mouse barrel cortex. PLoS Biol. 5:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadlen MN, Newsome WT. 1998. The variable discharge of cortical neurons: implications for connectivity, computation, and information coding. J Neurosci. 18:3870–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons DJ. 1978. Response properties of vibrissa units in rat SI somatosensory neocortex. J Neurophysiol. 41:798–820. [DOI] [PubMed] [Google Scholar]
- Sjostrom PJ, Turrigiano GG, Nelson SB. 2001. Rate, timing, and cooperativity jointly determine cortical synaptic plasticity. Neuron. 32:1149–1164. [DOI] [PubMed] [Google Scholar]
- Song S, Sjöström PJ, Reigl M, Nelson S, Chklovskii DB. 2005. Highly nonrandom features of synaptic connectivity in local cortical circuits. PLoS Biol. 3:e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting JT, Daigle TL, Chen Q, Feng G. 2014. Acute brain slice methods for adult and aging animals: application of targeted patch clamp analysis and optogenetics. Methods Mol Biol. 1183:221–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G. 2012. Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb Perspect Biol. 4:a005736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Aerde KI, Feldmeyer D. 2015. Morphological and physiological characterization of pyramidal neuron subtypes in rat medial prefrontal cortex. Cereb Cortex. 25:788–805. [DOI] [PubMed] [Google Scholar]
- Vinje WE, Gallant JL. 2000. Sparse coding and decorrelation in primary visual cortex during natural vision. Science. 287:1273–1276. [DOI] [PubMed] [Google Scholar]
- Wilent WB, Contreras D. 2005. Dynamics of excitation and inhibition underlying stimulus selectivity in rat somatosensory cortex. Nat Neurosci. 8:1364–1370. [DOI] [PubMed] [Google Scholar]
- Wolfe J, Houweling AR, Brecht M. 2010. Sparse and powerful cortical spikes. Curr Opin Neurobiol. 20:306–312. [DOI] [PubMed] [Google Scholar]
- Yassin L, Benedetti BL, Jouhanneau JS, Wen JA, Poulet JF, Barth AL. 2010. An embedded subnetwork of highly active neurons in the neocortex. Neuron. 68:1043–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura Y, Dantzker JL, Callaway EM. 2005. Excitatory cortical neurons form fine-scale functional networks. Nature. 433:868–873. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data files of raw fluorescence traces and MATLAB code pertaining to Figs 2-4 are provided under the following link: https://doi.org/10.5061/dryad.cnp5hqc2q. Other data supporting the findings of this study are available from the corresponding author upon reasonable request.