

Abstract
Over 30 years ago, an intriguing posttranslational modification was found responsible for creating concanavalin A (conA), a carbohydrate-binding protein from jack bean (Canavalia ensiformis) seeds and a common carbohydrate chromatography reagent. ConA biosynthesis involves what was then an unprecedented rearrangement in amino-acid sequence, whereby the N-terminal half of the gene-encoded conA precursor (pro-conA) is swapped to become the C-terminal half of conA. Asparaginyl endopeptidase (AEP) was shown to be involved, but its mechanism was not fully elucidated. To understand the structural basis and consequences of circular permutation, we generated recombinant jack bean pro-conA plus jack bean AEP (CeAEP1) and solved crystal structures for each to 2.1 and 2.7 Å, respectively. By reconstituting conA biosynthesis in vitro, we prove CeAEP1 alone can perform both cleavage and cleavage-coupled transpeptidation to form conA. CeAEP1 structural analysis reveals how it is capable of carrying out both reactions. Biophysical assays illustrated that pro-conA is less stable than conA. This observation was explained by fewer intermolecular interactions between subunits in the pro-conA crystal structure and consistent with a difference in the prevalence for tetramerization in solution. These findings elucidate the consequences of circular permutation in the only posttranslation example known to occur in nature.
Revisiting the historically important plant protein concanavalin A and reconstituting its biosynthesis in vitro reveals the advantages of its unusual biosynthesis via circular permutation.
Introduction
Concanavalin A (conA) is a seed lectin of the jack bean plant (Canavalia ensiformis); it is a noncatalytic protein that binds specific carbohydrates (monomers and oligomers of mannose and glucose) reversibly and with high specificity and moderate affinity (Lis and Sharon, 1998). Since its discovery just over a century ago (Sumner, 1919), conA has gained much interest due to its carbohydrate-binding properties (Bernhard and Avrameas, 1971; Dwyer and Johnson, 1981; Goldstein et al., 1997; Lis and Sharon, 1998; Locke et al., 2014), and the unusual posttranslational circular permutation it undergoes to become the mature form. The carbohydrate-binding of conA has seen it widely adopted in chromatography where it is frequently immobilized on sepharose and used to purify glycosylated biomolecules bearing high-mannose type N-glycans, including glycoproteins, polysaccharides, and glycolipids (Ogata et al., 1975; Saleemuddin and Husain, 1991). Despite a depth of structural knowledge, evidenced by over 60 jack bean conA structures in complex with various ligands and metal ions, the properties of conA precursor (pro-conA) and its maturation are not fully understood.

The hypothesized biological roles of conA include involvement in seed storage and plant defense. These are based mostly on its ability to bind certain carbohydrates and inferred from conclusions drawn from studies with legume lectins having similar physicochemical properties (Sharon and Lis, 1990). Lectins have been suggested to function as packaging aids (Einhoff et al., 1986) or they might diffuse out of seedlings during imbibition to protect against pathogens (Peumans and Van Damme, 1995). Lectins generally interact with glycocomponents absent from plants, but found on microbial surfaces or the digestive tract of insects (Peumans and Van Damme, 1995; Lagarda-Diaz et al., 2017). Lectins can resist proteolysis and can be insecticidal (Melander et al., 2003; Macedo et al., 2007). With conA as 20% of jack bean seed storage protein (Dalkin and Bowles, 1983), it could also function as a defensive mechanism in the metabolically inactive seed, but provide amino acids and metal ions during germination.
The biosynthesis of conA involves a unique series of cleavages and a transpeptidation reaction on the carboxyl side of asparagine residues, resulting in conA circular permutation (Figure 1A). ConA is synthesized as an inactive glycoprotein precursor (pre-pro-conA; Herman et al., 1985), where the N-terminal signal sequence is cleaved off cotranslationally (Supplemental Figure S1). The N-linked glycan inhibits carbohydrate-binding activity by pre-pro-conA in the endoplasmic reticulum and appears to be required for transport of pro-conA out of the endoplasmic reticulum (Faye and Chrispeels, 1987). Its removal is catalyzed by either an N-glycanase or endoglycosidase H (Sheldon and Bowles, 1992; Ramis et al., 2001). Deglycosylation is the only modification necessary for pro-conA to acquire carbohydrate-binding ability (Min et al., 1992; Sheldon and Bowles, 1992; Ramis et al., 2001). For brevity, we refer to deglycosylated pro-conA simply as pro-conA. In the protein body compartment, an intervening 15-amino acid peptide (V120IRNSTTIDFNAAYN134) in the middle of the protein, where the glycan group was initially attached, is proteolytically excised by asparaginyl endopeptidase (AEP, sometimes referred to as vacuolar processing enzyme or legumain), creating new N- and C-termini (Figure 1B). A new peptide bond is formed between the original N- and C-termini via a postulated cleavage-coupled transpeptidation event, wherein nine amino acids (E253IPDIATVV261) at the original C-terminus are removed (Bowles and Pappin, 1988). The excision of the 15-amino acid intervening peptide involves three cleavage reactions (N119, N130, N134); two of which (N119, N130) usually occur before the transpeptidation event. The last cleavage reaction at N134 occurs much later after transpeptidation. It is not known why this seemingly complex sequence of processing events occurs in this order, as the cleavage and transpeptidation sites are on opposite ends of the globular conA protein and so should not sterically interfere with either reaction. The resulting circular permutation of a protein was at the time unprecedented (Carrington et al., 1985) and evocatively termed “protein carpentry” (Hendrix, 1991).
Figure 1.
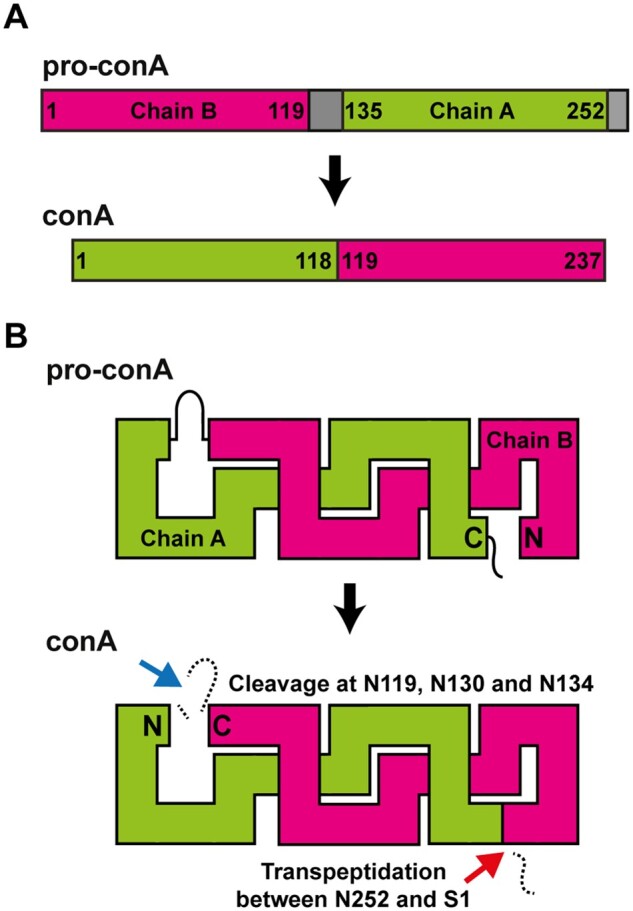
ConA maturation by circular permutation. A, During maturation, residue 1 of the N-terminal half (Chain B) of pro-conA becomes residue 119 of the C-terminal half of conA and vice versa residue 135 becomes residue 1 of conA. Grey segments are cleaved off during maturation. B, Peptide bond cleavages (blue arrow), and peptide bond formation, that is, transpeptidation (red arrow) within pro-conA are both required to make conA. The cleavage and transpeptidation events all occur on the carboxyl side of asparagine residues. The N-terminal ER signal of pre-pro-conA and the N-linked glycan, which are removed before circular permutation of pro-conA, are not shown.
Transpeptidation at the N- and C-termini appears inefficient as roughly half of conA in jack bean seeds are the two-chain form (Faye and Chrispeels, 1987) produced when the cleavage reaction is resolved by hydrolysis rather than aminolysis, that is, transpeptidation. In solution, conA exists in a pH- and temperature-dependent, dimer–tetramer equilibrium (McKenzie et al., 1972; Huet and Claverie, 1978; Senear and Teller, 1981). The conA tetramer is made up of a mixture of the hydrolyzed two-chain subunit and the transpeptidated full-length subunit. ConA dimers consisting of a mixture of the two-chain subunit and the full-length subunit are less competent at forming tetramers than purified conA consisting only of full-length subunits (Senear and Teller, 1981). Also, conA in the dimeric and tetrameric forms appear to have different biological activities on animal cells in vitro (Gunther et al., 1973).
After the discovery that conA was circularly permuted, protein engineers began to modify proteins or enzymes in a similar way (Goldenberg and Creighton, 1983) to gain insights into protein folding (Gebhard et al., 2006) or to modify biophysical properties to overcome limitations (Meister et al., 2011; Yu and Lutz, 2011; Bliven and Prlić, 2012). Many more examples of circularly permuted proteins have since been discovered, but all these natural examples are at the genetic level. ConA and close relatives from the Diocleinae subtribe remain the only proteins known to undergo posttranslational circular permutation (Cavada et al., 2018). The functional purpose for conA circular permutation remains elusive as few biochemical studies have been performed with pro-conA. Circular permutation is not required to induce carbohydrate-binding activity; the cleaved, two-chain form of conA is capable of carbohydrate-binding (Faye and Chrispeels, 1987), and homologs of pro-conA in other legumes do not undergo circular permutation (Cunningham et al., 1979; Carrington et al., 1985), making it unclear why circular permutation is needed.
The maturation of conA involves a protease (Abe et al., 1993; Min and Jones, 1994). This protease was the first enzyme discovered to be capable of forming peptide bonds posttranslationally within the backbone of protein substrates (Min and Jones, 1994). Interest in exploiting this function for biotechnological applications, on top of efforts to identify similar enzymes capable of peptide backbone transpeptidation, has led to the discovery and engineering of AEPs with varying efficiencies in peptide backbone cleavage, transpeptidation and macrocyclization, and AEPs with differing preferences for Asp and Asn residues (Nguyen et al., 2014; Yang et al., 2017; Haywood et al., 2018; Harris et al., 2019; James et al., 2019). However, our understanding of the AEP domains critical for determining hydrolase and transpeptidase efficiency is far from complete (Nonis et al., 2021).
Although recombinant jack bean AEP (CeAEP1) has been shown to be capable of carrying out transpeptidation on non-native substrates in vitro (Bernath-Levin et al., 2015), it has not been shown beyond doubt to be capable of conA circular permutation. Here, we reconstitute the biosynthesis of conA using recombinant pro-conA and CeAEP1, with structural evidence supporting cleavage-mediated transpeptidation in conA. The vast majority of studies on conA have characterized its mature form. Here, we focus on why pro-conA undergoes circular permutation and the structural features of CeAEP1 that facilitate this reaction. Although circular permutation does not cause structural changes in the conA carbohydrate-binding domain, it results in increased thermal and chemical stability of the protein. The differences observed in the oligomeric states of pro-conA and conA in solution and the lower number of atomic interactions between subunits observed in the crystal structure of pro-conA provide a plausible explanation for the difference in stability. The structure of the jack bean AEP is consistent with other related AEPs that carry out both cleavage and transpeptidation, explaining why only a single enzyme is required to circularly permutate pro-conA into conA.
Results
Pro-conA structure
To determine the structural differences between pro-conA and conA, we expressed recombinant pro-conA (residues 30–290; UniProt ID: P02866) in Escherichia coli (Supplemental Figure S2). We obtained a protein crystal which diffracted to 2.1 Å by X-ray diffraction. The crystal structure was solved by molecular replacement using conA [Protein Data Bank (PDB): 1JBC; Parkin et al., 1996] as the search model, yielding a homodimer in the asymmetric unit. Superposition of the two monomers yields a root-mean-square deviation (r.m.s.d.) of 0.4 Å over 225 Cα-atoms. Considerable chain mobility at the C-terminus (residues 250–260) of both pro-conA monomers is inferred from poor electron density in this region. The C-terminus is in close proximity to a solvent-exposed loop (residues 65–70) which are involved in different crystal contacts in the two monomers. Residues at the functional sites discussed below are clearly visible in electron density (Supplemental Figure S3).
Like other lectins, pro-conA is made up of two large β-pleated sheets consisting of a flat six-stranded antiparallel β-sheet and a curved seven-stranded antiparallel β-sheet (Figure 2). Pro-conA and the conA monomer (PDB 1JBC) are very similar, with an r.m.s.d. of 0.5 Å over 221 Cα-atoms (Supplemental Figure S4). For the 15-amino acid intervening peptide that gets cleaved off during conA maturation, there is an absence of electron density when contoured at 1σ. Crystals of pro-conA were therefore dissolved and run on a sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE) to confirm presence of full-length protein, demonstrating that the absence of electron density for the 15-amino acid intervening peptide is due to disorder rather than spurious cleavage by other enzymes during expression or purification (Supplemental Figure S2). The wider band observed for the dissolved pro-conA crystals is likely an artifact from the crystallization buffer. Considering the missing electron density at the termini of pro-conA when contoured at 1σ, indicating disorder, there is also a possibility that spurious trimming of several terminal residues may have occurred.
Figure 2.
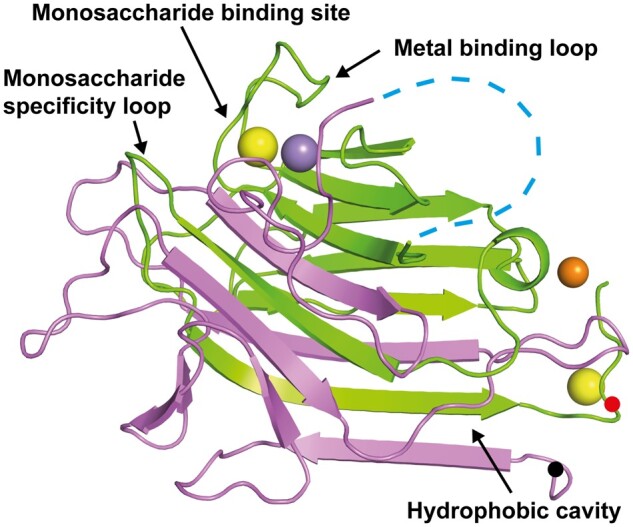
Pro-conA structure. Pro-conA consists of interleaved β-sheets from chain A (pink) and chain B (green). Transpeptidation occurs between the N-terminus (black dot) and a residue near the C-terminus (red dot). “Metal binding loop” and “monosaccharide specificity loop” are a single loop each, whereas the “monosaccharide binding site” consists of residues from three loops. The “hydrophobic cavity” consists of residues from two adjacent β-sheets and a nearby loop. Fifteen flexible residues linking chains A and B (blue dashed line); ions of calcium (yellow sphere); manganese (purple sphere); zinc (orange sphere). This tertiary structure is similar to conA (Supplemental Figure 4).
Structural features involved in conA activity
It is not known if conA circular permutation changes the structural domains involved in carbohydrate binding. Here we see that the monosaccharide binding sites of pro-conA and conA are very similar, with an r.m.s.d. of 0.2 Å for the three residues involved (Supplemental Figure S3A). The monosaccharide specificity loops of pro-conA and conA have an r.m.s.d. of 1.1 Å for the six residues involved (Supplemental Figure S3B). Differences in the side-chain positions of Leu233 and Tyr234 of the monosaccharide specificity loop, which has a nearby ethanediol molecule in the pro-conA structure, are unlikely to affect carbohydrate-binding as only their main chain atoms interact with carbohydrates (Kanellopoulos et al., 1996b; Hamodrakas et al., 1997). Other residues involved in carbohydrate recognition, including Tyr146–Ile151 (Tyr12-Ile17 in conA) and Thr108–Leu111 (Thr226–Leu229 in conA; Loris et al., 1998; Cavada et al., 2018), are also structurally unchanged after circular permutation.
ConA carbohydrate-binding activity is highly dependent on the binding of a transition metal, typically manganese, and a calcium ion (Sumner and Howell, 1936; Kalb and Levitzki, 1968; Shoham et al., 1973). The presence of transition metal ions and calcium has also been shown to improve conA structural stability (Blumberg and Tal, 1976; Doyle et al., 1976). Similar to conA, in pro-conA the manganese ion has six ligands in an octahedral coordination geometry, involving one nitrogen atom provided by His158, three carboxylate oxygen atoms from Glu142, Asp144, and Asp153, and two water molecules (Supplemental Figure S3C). Asp144 and Asp153 are also involved in the coordination of the calcium ion, which has seven oxygen ligands in a distorted octahedral geometry involving two carboxylate oxygen atoms from Asp144 and one from Asp153, an amide oxygen atom from Asn148, a backbone carbonyl from Tyr146, and two water molecules. The binding of these two metals has been shown to stabilize the active “locked” conformation in conA (Brown et al., 1977; Brewer et al., 1983; Bouckaert et al., 2000), which is observed here in metallated pro-conA as well (Supplemental Figure S3D). As proposed in the conA structure by Bouckaert and colleagues, in pro-conA the coordination of Asp144 and Tyr146 (Asp10 and Tyr12 in conA) to the calcium ion induces a bend in the β-sheet holding Thr145 (Thr11 in conA), causing a steric clash between Thr145 and Asp90 (Asp208 in conA) and inducing a trans-to-cis isomerization between Ala89 and Asp90 (Ala207 and Asp208 in conA). The key residues indicating a locked conformation in pro-conA is similar to those of conA, with an r.m.s.d. of 0.8 Å for the five key residues highlighted in Supplemental Figure S3D.
There is a small possibility that the transition-metal-binding site may be occupied by a nickel ion rather than a manganese ion. Although we cannot rule out the presence of nickel in the model, as nickel was used in pro-conA purification, the overall geometry of the metal-binding sites have been shown to be essentially independent of the nature of the transition metal (Emmerich et al., 1994). Zinc and calcium ions were modeled at four other locations in the homodimer and are not part of the metal-binding loop. There is potentially a mixture of metals in these locations, but these metals are unlikely to have a biological role and are likely present due to crystallization conditions.
A hydrophobic cavity conserved in conA-like lectins has been hypothesized by some to bind to secondary metabolites (Delatorre et al., 2007; Bezerra et al., 2011). An ethanediol (cryoprotectant) is observed in this cavity in the pro-conA structure, and the local structure appears unchanged when compared to a conA structure containing ethanediol in this position (PDB:4PF5; François-Heude et al., 2015), with an r.m.s.d. of 0.2 Å over three residues (Supplemental Figure S3E).
Pro-conA and conA interact differently in protein crystals
In contrast to the well-established dimer-of-dimers complex (i.e. tetramer) observed in the conA crystal structure (Figure 3A), pro-conA monomers in the crystal structure assemble in an atypical dimer-of-dimers complex (Figure 3B). The asymmetric unit contains a dimer, composed of inter-monomer interactions very similar to those observed in the principal dimer of conA (involving residues 3–21, 57–65, 99, 222–224, and 248–252 in pro-conA). Application of crystal symmetry to the coordinates, using the operator –x, y, −z−1, yields a possible tetrameric structure. PDBePISA (Krissinel and Henrick, 2007) was used to predict the free energy of assembly-dissociation (ΔGdiss) for pro-conA, indicating that pro-conA is likely capable of forming a stable dimer (ΔGdiss = 5.0 kcal·mol−1) and tetramer (ΔGdiss = 17.2 kcal·mol−1) in solution. These predicted complexes correspond to the atypical dimer-of-dimers complex observed in the crystal structure. Based on the parameterizations of the PISA program, positive values of ΔGdiss indicate that an external driving force should be applied to dissociate the assembly. Therefore, assemblies with ΔGdiss>0 are calculated to be thermodynamically stable. For comparison, high resolution structures of conA (e.g. PDB: 1JBC) also predict a stable dimer (ΔGdiss = 8.6 kcal mol−1) and tetramer (ΔGdiss = 9.6 kcal·mol−1) in solution. However, compared to conA, pro-conA has a smaller buried surface area and fewer interface residues, especially between the dimers (Supplemental Table S1). PDBsum (Laskowski et al., 2018) was used to analyze the number of noncovalent contacts with a distance cutoff of 4 Å, and this showed that pro-conA has fewer inter-dimer contacts than conA in the crystal structure (Figure 4). As shown in Figure 3B and Figure 4, A and C, the C-termini of the pro-conA subunits are wedged between the dimers and are therefore heavily involved in the inter-dimer interactions. These termini are cleaved off and shortened into a loop during circular permutation to form conA. Although the region spanning four β-sheets (residues 183–210) is involved in inter-dimer interactions in both pro-conA and conA, they have different interacting partners. The residues in this β-sheet region in pro-conA interact with the wedged C-termini, whereas in conA, they interact with the β-sheet region of the opposing subunit (Figure 4). Pro-conA also has substantially fewer interactions spanning residues 69–78.
Figure 3.
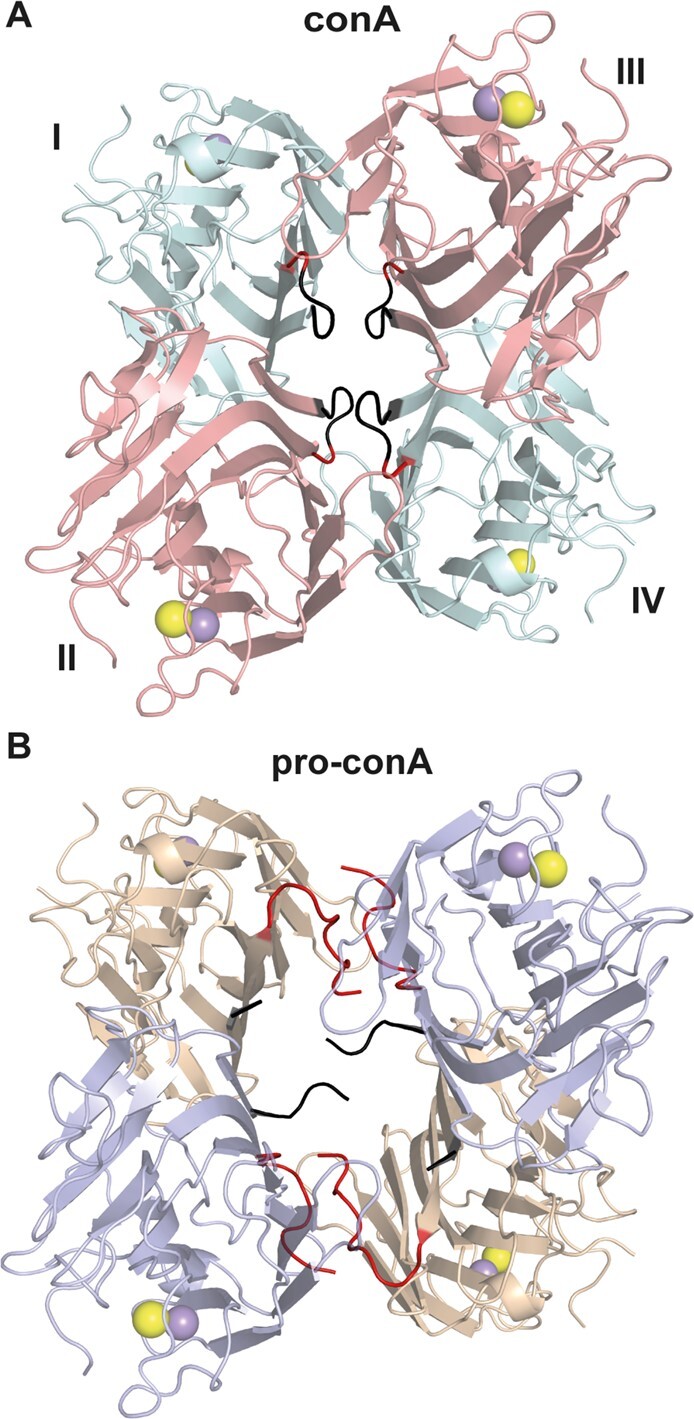
Pro-conA and conA have different tetrameric structures in protein crystals. (A) In conA, monomers I and II interact so that the flat six-stranded β-sheet from each monomer aligns to form a contiguous 12-stranded β-sheet. Monomers III and IV dimerizes in a similar manner. The two dimers then interact via the 12-stranded β-sheets to form a dimer-of-dimers. (B) Pro-conA adopts a dimer-of-dimers conformation with a larger cavity. The pro-conA termini interfere with the formation of the typical dimer-of-dimers. N- and C-termini of pro-conA monomers are shown in black and red, respectively. The equivalent residues in conA are colored similarly.
Figure 4.

The pro-conA tetramer has fewer inter-dimer contacts than the conA tetramer in crystal structures. Pro-conA (A and C) has 94 nonbonded contacts between dimers in a dimer-of-dimers complex. ConA (B and D) has 194 nonbonded contacts between dimers in a dimer-of-dimers complex. In pro-conA, the C-termini interfere with the inter-dimer interactions that would exist between the opposing β-sheets of conA. Residues involved in nonbonded contacts are shown as sticks. The “front” view shows all four subunits of the dimer-of-dimers. “Side” view is obtained with a 90° anti-clockwise turn when viewed from the top. The N- and C-termini are highlighted in black and red, respectively.
Pro-conA can form tetramers in solution
To determine if the observed pro-conA tetramer in the crystalline state is relevant to complex formation in aqueous solution, sedimentation velocity studies were performed in the analytical ultracentrifuge. Data at multiple time points generated at a rotor speed of 40,000 rpm were fitted to a continuous size distribution model. The resulting c(s) distributions for pro-conA at an initial concentration of 3.0 mg·mL−1 at pH values 4.5 and 6.8 at 20°C and 30°C were compared. At 20°C, the protein exists as a single species represented by a sharp peak spanning a sedimentation coefficient range of 3–4 S for both pH values (Figure 5A). This is indicative of a species of 50 kDa based on c(M) analyses, which is consistent with the formation of a dimer in solution. At 30°C, the presence of a broad peak spanning a sedimentation coefficient range of 3–5 S for both pH values suggests that pro-conA exists in a rapid dimer–tetramer equilibrium in solution. Similar analyses at 30°C and pH values 4.5 and 6.8 were also carried out with protein concentrations of 0.2 mg·mL−1 (Figure 5B). At 0.2 mg·mL−1, pro-conA exhibits a broad peak spanning a sedimentation coefficient range of 3–5 S for both pH values, but with a slight shift in equilibrium toward a smaller S value, and thus dimer formation. These results demonstrate that pro-conA can form larger oligomeric forms, most likely tetramers, and that tetramer formation is temperature- and concentration-dependent. However, unlike conA, pro-conA tetramerization is pH-independent between pH values 4.5 and 6.8; conA exists as a dimer below pH 5.5, with increasing tetramer formation in a rapid dimer–tetramer equilibrium corresponding to increasing pH up to 7.5 (Senear and Teller, 1981).
Figure 5.
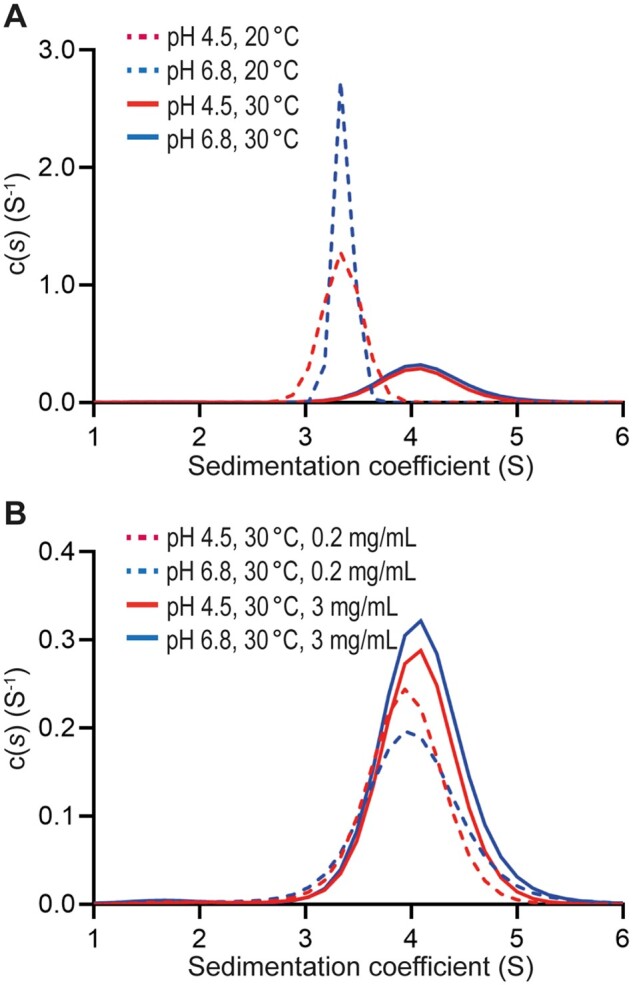
Pro-conA tetramer formation is temperature- and concentration-dependent. Sedimentation velocity analyses by analytical ultracentrifugation. A, Between pH 4.5 and 6.8, at 20°C pro-conA exists as a sharp peak spanning 3–4 S, which is consistent with the size of the pro-conA dimer of ∼50 kDa. In the same pH range but at 30°C, pro-conA exists as a broad peak spanning 3–5 S, which is consistent with a rapid dimer–tetramer equilibrium. B, In the same pH range and at 30°C, the dimer–tetramer equilibrium shifts toward tetramer formation at higher protein concentrations.
Reconstituting conA biosynthesis in vitro
To determine whether CeAEP1 alone can perform separate cleavage and cleavage-coupled transpeptidation reactions to make conA, we sought to reconstitute conA biosynthesis in vitro with recombinant pro-conA. Recombinant pro-conA (residue 30–290 of UniProt ID: P02866) was incubated with autoactivated CeAEP1 at a molar ratio of ∼20:1 pro-conA to activated CeAEP1. The pro-conA band was converted to several lower MW proteins after digestion (Figure 6A). The product of interest in the lane of the pro-conA digest was band I, which has a slightly smaller MW than pro-conA, consistent with a circularly permuted conA. This protein band was sequenced by N-terminal Edman degradation, and this yielded two N-terminal sequences: AAYNADTI and ADTIVAVE (Figure 6B;Supplemental Figure S5). These overlapping sequences obtained by Edman degradation both prove that the C-terminal half of conA has been circularly permuted to become the N-terminal half, and concurs with the slightly larger MW observed for band I when compared to the mature form (Figure 6A). In jack bean seeds, the removal of the AAYN tetrapeptide preceding the N-terminus of conA occurs very slowly (Bowles et al., 1986), explaining why in this in vitro experiment we observed both species. This AAYN tetrapeptide was also observed in conA extracted from immature jack bean seeds (Wang et al., 1971; Carrington et al., 1985). The bottom three MW bands (II, III, and IV) represent the cleaved products (Sheldon et al., 1996; Supplemental Figure S1). Performing the in vitro assay at a range of pH conditions (pH 4, 5, 6, and 7) showed that circular permutation of conA by CeAEP1 was most efficient at pH 5 (Supplemental Figure S6). Pro-conA was completely processed by CeAEP1-mediated transpeptidation or cleavage at pH 5 and pH 6. At pH 5, a higher proportion of pro-conA termini were transpeptidated to produce full-length conA (band I), whereas processing at pH 6 resulted in a higher proportion of cleaved products (bands III and IV). Processing at pH 4 and pH 7 produced an additional band (band II), indicating incomplete processing. ConA extracts from jack bean seeds also show that circular permutation does not occur with 100% efficiency (Carrington et al., 1985).
Figure 6.

In vitro reconstitution of conA maturation using auto-activated recombinant CeAEP1 and pro-conA. A, SDS-PAGE of recombinant pro-conA (Lane 1), conA (Sigma-Aldrich, Cat No. L7647; Lane 2), and pro-conA processed by CeAEP1 (Lane 3). N-terminal sequence of Band I (red box) was determined by Edman degradation. The slightly larger size than conA is due to the presence of the AAYN tetrapeptide, which is cleaved off at a slow rate, on the N-terminus of circularly permuted conA (Supplemental Figure 1). Bands III and IV are present in jack bean extract as well (Lane 2) and represent fragments that have undergone cleavage instead of transpeptidation (Sheldon et al., 1996). Band II likely represents cleaved fragment containing N-terminal AAYN tetrapeptide and/or the C-terminal EIPDIATVV peptide. B, Pre-pro-conA sequence with ER signal (black), chain B (pink), chain A (green), and residues removed during circular permutation (grey). To facilitate comparison with pro-conA structure and previous studies, residue numbering starts after N-terminal ER-signal, at S1. Pro-conA is synthesized with a glycosylated N123 residue. Deglycosylation is the first posttranslational modification. Cleavage by CeAEP1 occurs at N119, N130, and N134. N252 is either cleaved or transpeptidated to S1. The two N-terminal sequences from Edman degradation are underlined in blue and orange.
Thermal and pH stability of pro-conA and conA
Circular dichroism analysis showed that conA is more stable than pro-conA under heat stress at pH 6.5 and at various other pH conditions without heat stress, revealing a functional consequence of conA circular permutation. During heat stress at pH 6.5, where temperature was increased at a rate of 1°C·min−1, there was an increase in the magnitude of ellipticity at 218 nm for pro-conA at the 60–70°C range before decreasing from 70°C onwards (Figure 7A). In contrast, only a decrease in magnitude of ellipticity was observed for conA. This suggests that, under heat stress, pro-conA undergoes an increase in the amount of β-structure relative to conA. Controls showed no change in ellipticity for pro-conA and conA when no heat is applied (Supplemental Figure S7A). SDS–PAGE analysis was carried out to illustrate the precipitation observed for both pro-conA and conA during the heat analysis (Supplemental Figure S7B). A previous study combining circular dichroism (CD) analysis with scattered light intensity analysis showed that conA begins to aggregate before any conformational change occurs during heat stress (Maeda et al., 1989), which may explain why we do not see an increase in magnitude of ellipticity for conA in our CD analysis. Even though pro-conA and conA appear to react differently to heat stress, we cannot conclude if melting or precipitation occurs first with CD analysis. However, it is clear that conA has better heat tolerance as pro-conA begins to undergo changes in conformation and/or begins precipitating at a lower temperature than conA at pH 6.5, and also appears to approach complete precipitation at a lower temperature than conA (Figure 7A). The stability observed for conA agrees with a previous study that observed conA aggregation from 60°C onwards (Doyle et al., 1976).
Figure 7.
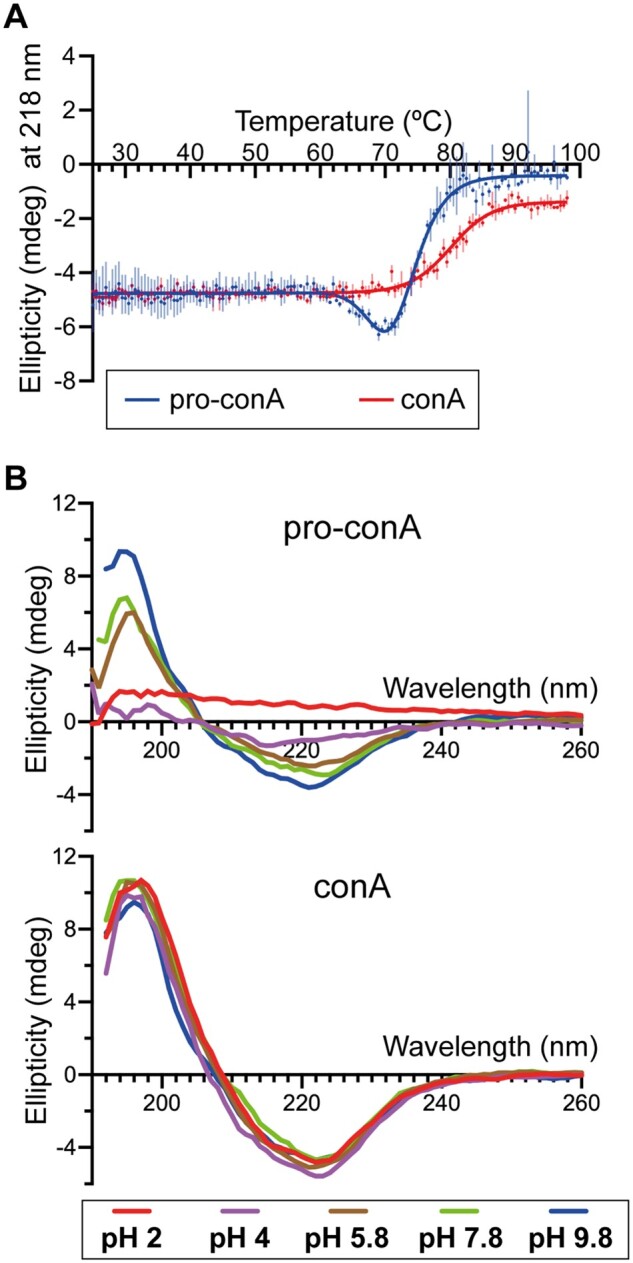
ConA is more stable than pro-conA. A, Melt curve generated from circular dichroism analysis of pro-conA and conA in water with a temperature slope of 1°C min−1. Pro-conA begins changing conformation and precipitates from solution (Supplemental Figure 7B) at a lower temperature than conA. The decrease in ellipticity for pro-conA but not conA indicates that these two proteins have different pathways of unfolding and/or aggregation. This experiment was done with one batch of protein in technical triplicate. B, Circular dichroism spectra of pro-conA and conA after 2-hour incubation in water at pH 2, 4, 5.7, 7.8, and 9.8. ConA remains stable from pH 2 to 9.8 whereas pro-conA is less stable at neutral and low pH.
To examine pH stability, pro-conA and conA were incubated at various pH conditions (pH 2.0–9.8) for 2 h before CD analysis at the respective pH. CD spectra analysis showed that conA maintains its general structure throughout the pH range tested for the 2-h period of the analysis, whereas pro-conA is more prone to precipitation at lower pH conditions (Figure 7B). Precipitation occurred similarly to what was observed during heat stress. Beyond the 2-h time frame, conA undergoes pH-dependent conformational changes as well (McCubbin et al., 1971; Zand et al., 1971).
It is not feasible to extract pro-conA from jack bean seeds as it is rapidly converted to conA. The high similarity of the recombinant pro-conA structure that we obtained to that of conA indicates that recombinant pro-conA folds into the correct conformation in our E. coli expression system. To ensure as much consistency as possible between preparation of pro-conA and conA, we dialyzed recombinant pro-conA into the same buffer that the dry commercial conA powder was dissolved in. It should be noted that conA (Sigma) contains a small, unquantified amount of the two-chain form (Figure 6A), representing the products that have undergone cleavage rather than transpeptidation.
Circular permutation has no effect on conA binding to methyl-α-d-mannose
To determine if conA circular permutation affects carbohydrate-binding, we investigated the ability for pro-conA and conA to bind to mannose, the preferred ligand in vitro, using isothermal titration calorimetry (ITC). The results were similar for pro-conA and conA, with association constants (KA) of 8270 ± 74.0 and 8240 ± 74.0 M−1, respectively, for methyl-α-d-mannose (Supplemental Figure S8). The KA we obtained for conA was in close agreement with that obtained by Loka et al. (2015) and Chervenak and Toone (1995), with both studies obtaining a KA of 7.6 × 103 M−1 for methyl-α-d-mannose.
Jack bean AEP structure
To understand the enzymatic basis for conA maturation, we expressed CeAEP1 (residues 36–475; UniProt ID: P49046) in E. coli (Supplemental Figure S9). We obtained a protein crystal which diffracts to 2.7 Å by X-ray diffraction. The crystal structure was solved by molecular replacement using Arabidopsis thaliana legumain γ (AtLegγ) as the search model, yielding a CeAEP1 homodimer in the asymmetric unit. The superposition of the two monomers yields an r.m.s.d. of 0.4 Å over 424 Cα-atoms. Residues at the catalytic site and the substrate-binding pockets are clearly visible in electron density (Supplemental Figure S10, A and B). Differences between monomers are only at solvent-exposed residues in or adjacent to loop regions. Electron density shows clear differences at residues 283–289 and 347–360, which can be attributed to different crystal contacts (Supplemental Figure S11). Residues 283–289 are part of an unstructured loop on the surface of the core domain and deviate from each other at the backbone. The biggest difference in this loop occurs at Asn285–Ser286, where the backbone deviates by about 4 Å and the side-chains point at different directions. Electron density at 1σ is poor for the side chains of Tyr287–Arg288 in monomer A. Residues 347–360 are part of a short loop and two adjoining α-helices in the cap domain. The differences are minor as the side chains of both monomers have similar orientations, and the backbone deviates at most by about 2.6 Å at Gln353. This deviation is likely also due to the inability to model residue 352 of monomer A due to the lack of electron density when contoured at 1σ. Electron density shows static disorder at residues 318–330, which contains the linker region that is cleaved off upon CeAEP1 autoactivation. Missing electron density at residues 323–329 when contoured at 1σ point to the highly flexible nature of this linker.
CeAEP1 is structurally similar to previously solved plant AEP structures: sunflower (Helianthus annuus) AEP1 or HaAEP1 6AZT (Haywood et al., 2018), AtLegγ 5NIJ (Zauner et al., 2018b), Oldenlandia affinis AEP1 or OaAEP1 5H0I (Yang et al., 2017), Viola yedoensis peptide asparaginyl ligase 2 or VyPAL2 6IDV (Hemu et al., 2019), and butelase1 6DHI (James et al., 2019) with an r.m.s.d. of 1.0–1.1 Å over 400–430 Cα-atoms between CeAEP1 and the other published structures (Supplemental Table S2). The inactive enzyme is made up of a “core” domain (Glu38–Asp313) linked to a C-terminal “cap” domain (Arg336–Ala474) via the flexible linker, which has weak electron density (Figure 8A). The core domain consists of a six-stranded β-sheet surrounded by five major α-helices, and is highly structurally conserved with a r.m.s.d. of 0.5–0.7 Å over 270–280 Cα-atoms between CeAEP1 and the other published structures (Supplemental Table S3). The cap domain, consisting of five α-helices, improves core domain stability, acts as the dimer interface and modulates enzymatic activity by occluding the active site in the inactive form (Zauner et al., 2018b).
Figure 8.
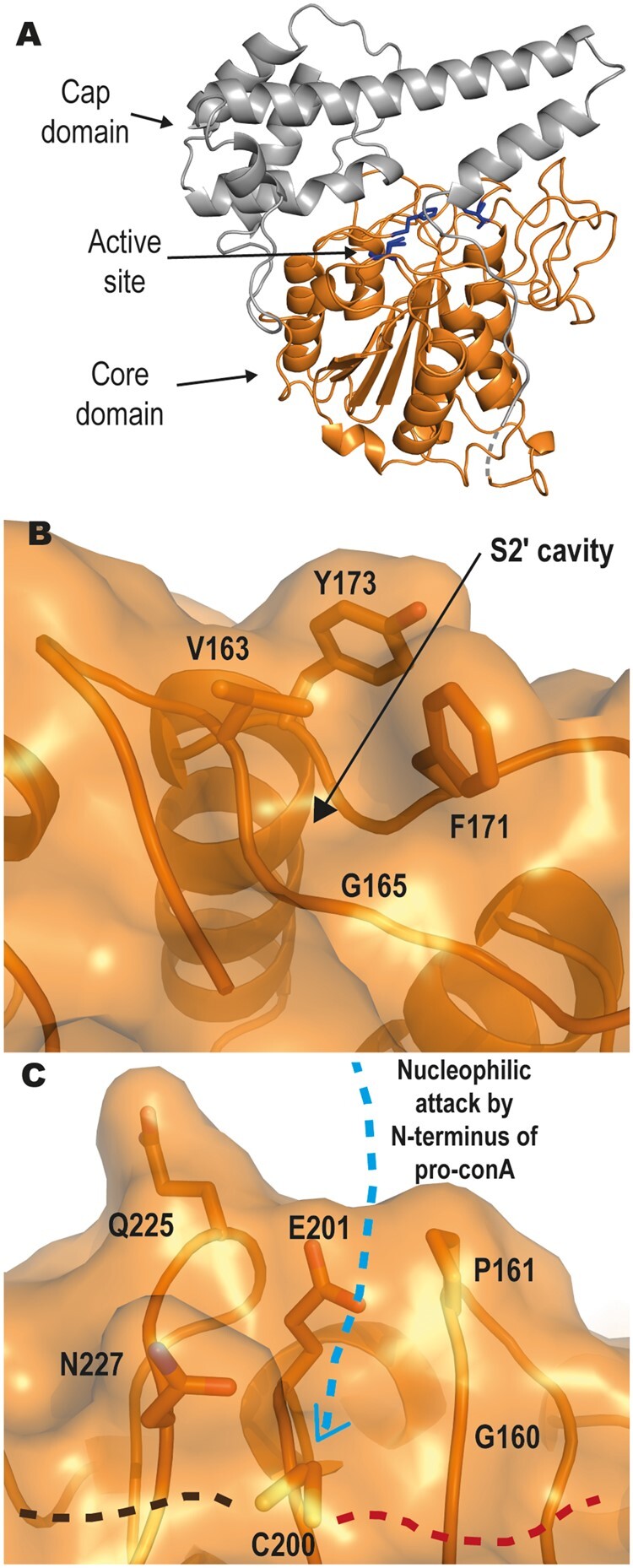
Jack bean asparaginyl endopeptidase 1 (CeAEP1) structure. A, Inactive CeAEP1. Core domain (orange); cap domain (grey); active site (blue residues); low electron density in linker region (dashed grey). B, Gly165 in S2′ pocket forms a deep cavity, possibly facilitating the binding of peptide substrate and preventing replacement with water molecules. C, Residues G160, P161, E201, Q225, and N227 likely interfere with incoming N-terminus of peptide substrate during nucleophilic attack, resulting in poor transpeptidase efficiency. C200 is the catalytic residue. Following the proposed mechanism of nucleophilic attack by an N-terminal nucleophile, the incoming N-terminus of pro-conA with nucleophilic serine (blue dashed arrow) attacks the acyl-enzyme intermediate at the catalytic cysteine; Residues N-terminal to cleavage site on pro-conA (brown dashed line); Residues C-terminal to cleavage site of pro-conA (red dashed line).
CeAEP1 active site residues are strictly conserved
The catalytic residues His158 and Cys200 form the catalytic dyad, with Asn53 sometimes included to make up a catalytic triad (Supplemental Figure S10A). These three residues are conserved across all AEPs (Supplemental Figure S12). The His158 imidazole likely acts as a general base in the catalytic mechanism (Elsässer et al., 2017; Nonis et al., 2021). Electron density at Cys200 indicates a dual conformation, which is also observed in the near-atomic resolution structure of a HaAEP1, likely representing conformational flexibility that can occur during catalysis (Haywood et al., 2018). Mutagenesis study on HaAEP1 shows that the asparagine residue of the catalytic triad (Asn53 in CeAEP1; Asn73 in HaAEP1) influences the ratio of cyclic to acyclic product, but is not a strict requirement for cleavage or transpeptidation (Haywood et al., 2018). We modeled a succinimide intermediate (SNN) at Asp157 to obtain a better fit with the electron density at this position (Supplemental Figure S10A). The presence of the SNN is consistent with all other plant structures except OaAEP1 (Yang et al., 2017; Haywood et al., 2018; Zauner et al., 2018b; Hemu et al., 2019; James et al., 2019), although OaAEP1 does have electron density suggesting that succinimide may in fact be present (James et al., 2019). Gln335, which is part of the cap domain, occupies the S1 pocket of the active site in the pro-enzyme (according to nomenclature by Schechter and Berger where P1, P2 and so on refer to residues N-terminal to the cleavage site, whereas P1′, P2′ and so on are C-terminal to it, and where the corresponding binding sites on the protease are termed S2, S1, S1′, S2′, etc.; Schechter and Berger, 1967; Supplemental Figure 10A). It was hypothesized that cleavage does not occur at Gln335 even though it is similar to asparagine because the glutamine backbone is kept further away from the catalytic cysteine by virtue of the longer side chain (4.9 Å in CeAEP1; Zauner et al., 2018a).
CeAEP1 substrate-binding pockets
With such high structural similarities amongst plant AEPs that have varying catalytic efficiencies, CeAEP1 catalytic preference and efficiency can be explained only by subtle variations in the substrate-binding pockets. For example, the CeAEP1 core domain is very similar to the sunflower AEP, HaAEP1, with an r.m.s.d. of 0.5 Å over 270 Cα-atoms (Supplemental Table S3), but it is more efficient at carrying out transpeptidation and cleavage than HaAEP1 (Bernath-Levin et al., 2015). The S1 pocket (Arg55, His56, Glu198, Ser228, and Asp250), as designated by a previous study on AtLegγ (Zauner et al., 2018a), is conserved (Supplemental Figure S10C) and accommodates the P1 asparagine residue at all pro-conA cleavage and transpeptidation sites. His158, Gly159, Gly160, Cys200, and Glu201 make up the S1′ pocket (Zauner et al., 2018a; Hemu et al., 2019) and is highly conserved with notable variability in side chain positioning of Glu201 (Supplemental Figure S10D). Val163, Gly165, Phe171, and Tyr173 make up the S2′ pocket, with only Gly165 strictly conserved (Supplemental Figure S10E). The vast majority of the residues in the S2′ pocket in CeAEP1 and other AEPs are otherwise hydrophobic. The Arg278–Met294 region, which was designated the Marker of Ligase Activity by Jackson et al. (2018), is generally hydrophilic in CeAEP1, similar to that of HaAEP1 (Supplemental Figure S12).
Discussion
It is just over 100 years since the discovery of conA. Despite conA being the subject of many studies and seeing daily use in laboratories worldwide, only now do we have a structure for pro-conA; this allows us for the first time to understand how circular permutation of pro-conA affects this long-studied protein.
Circular permutation influences conA tetramer formation
The crystal structure of pro-conA confirms the long-standing suspicion that circular permutation of conA occurs without any large changes in the tertiary structure (Carrington et al., 1985) (Supplemental Figure S4). We observed a difference, however, in the number of inter-dimer interactions between the pro-conA and conA crystal structures (Figure 4; Supplemental Table S1). Even though conA in the demetallated and the saccharide-bound form have different crystal packings, both forms adopt the typical dimer-of-dimers assembly (Kanellopoulos et al., 1996a). In pro-conA, the C-terminus is heavily involved in inter-dimer interactions of the crystalline state (Figures 3B; 4, A and C). During circular permutation, the N- and C-termini undergo cleavage-coupled transpeptidation and are therefore present as a shortened loop that does not interfere with the formation of the typical dimer-of-dimers assembly in the conA crystal structure (Figures 3A; 4, B and D). Likely, the inability for pro-conA to form the typical dimer-of-dimers assembly in the crystalline state is attributable solely to the presence of the termini. AUC analyses demonstrate that pro-conA is capable of forming tetramers in solution (Figure 5), with a potential atypical dimer-of-dimers observed in the crystal structure, in which PDBePISA analysis suggests is commensurate with tetramer formation in solution. On the basis of thermodynamic analyses in conA, it was proposed that the ionization of a histidine side chain, either His3 or His185 (His121 or His51 in conA, respectively) governs the pH-dependent association of the dimers to form a tetramer (Senear and Teller, 1981). Considering the differences observed in the number of contacts between the subunits, it is not surprising that the atypical dimer-of-dimer complex in pro-conA has different interactions at His3 and His185 when compared to conA (Supplemental Figure S13), which may explain why we do not observe a pH-dependent tetramerization of pro-conA in solution between pH 4.5 and pH 6.8.
Glycinin, a seed storage protein in soybean, exists as a trimer before cleavage at an asparagine residue by an AEP triggers hexamer formation (Jung et al., 1998; Adachi et al., 2001, 2003). Even though glycinin and conA are structurally different, in both cases AEP processing results in the dislocation of a peptide chain that appears to interfere with the formation of a higher oligomeric state. It would be interesting to see if the typical dimer-of-dimers conformation forms if pro-conA termini underwent AEP-mediated cleavage rather than transpeptidation.
Binding domains in pro-conA remain unchanged after circular permutation
The requirement for conA to be in the “locked” conformation to effectively bind carbohydrates has been explained in detail by in-depth structural analyses by Bouckaert et al. (2000). Here, we obtain a structure of pro-conA which is similar to the “locked” conformation (Supplemental Figure S3D), which explains why circular permutation is not required for carbohydrate-binding and that the binding of Mn2+ and Ca2+, even in pro-conA, is sufficient for carbohydrate-binding activity (Ramis et al., 2001). This was corroborated with ITC analysis showing no difference in association constant between pro-conA and conA when assayed with methyl-α-d-mannose. The similar conformation of both the monosaccharide specificity loop and the monosaccharide binding site between pro-conA and conA indicates that circular permutation does not affect carbohydrate specificity either. A conserved hydrophobic cavity, hypothesized by Bezerra et al. (2011) and Delatorre et al. (2007) to play a role in plant defense in their study of conA-like lectins, also remains unchanged by circular permutation.
Circular permutation improves conA stability
Artificial circular permutation has on only one occasion led to a slightly more stable version of protein (Topell et al., 1999); this explorative modification otherwise tends to result in proteins that are either less or equally stable as the parent protein (Heinemann and Hahn, 1995). Here, we discover a clear improvement in stability resulting from protein circular permutation. We demonstrate that conA circular permutation increases conA thermal and pH stability (Figure 7) without affecting in vitro binding to the carbohydrate methyl-α-d-mannose (Supplemental Figure S8). The exceptional pH stability of conA was indeed a useful property for its initial discovery, as conA was readily separated from other seed proteins using a protocol involving a range of pH (Sumner, 1919). A more comprehensive binding analysis with various carbohydrates of different oligomeric forms may be useful, but the carbohydrate-binding function of conA in vivo has so far only been supported by in vitro characterization rather than direct functional evidence in jack bean plants.
ConA circular permutation is carried out by CeAEP1
Our in vitro assay shows that CeAEP1 is capable of conA circular permutation. It should be noted that cDNA analysis suggests the presence of AEP isoenzymes with unknown relative abundance (Takeda et al., 1994), so there is possible redundancy or involvement of another dedicated protease/transpeptidase for conA circular permutation in vivo. The pH optimum for conA circular permutation by CeAEP1 is between pH 5 and 6, with higher efficiency closer to pH 5 (Supplemental Figure S6). The pH range observed in the protein storage vacuoles of A. thaliana, where seed storage proteins are likely processed by AEPs, is between pH 4.9 and 6.1 (Otegui et al., 2006). It appears that CeAEP1 has evolved to perform transpeptidation most efficiently at the pH range that it functions in.
CeAEP1 prefers serine over alanine as the nucleophile in conA circular permutation
AEP cleavage at the pro-conA intervening peptide at Asn130 and Asn134 generates new alanine N-termini (Ala131 and Ala135) which do not undergo transpeptidation with Asn119 despite their close proximity. This selectivity was also observed in Sunflower Trypsin Inhibitor-1 (SFTI-1), where transpeptidation occurs when the incoming N-terminal nucleophile in native SFTI-1 is glycine, but a Gly1 to Ala1 mutation inhibits transpeptidation (Bernath-Levin et al., 2015). In silico docking simulations performed by Zauner et al. (2018a) indicate that an ionic interaction may facilitate transpeptidation between the N-terminal nucleophile and Glu220 of AtLegγ (Glu201 in CeAEP1) in the S1′ pocket (Supplemental Figure S10D). Transpeptidation in pro-conA occurs with an incoming N-terminal serine, which is similar in size to alanine. It is therefore possible that the polar side chain of serine facilitates transpeptidation by CeAEP1 via ionic interactions, whereas the hydrophobic, nonpolar property of Ala131 and Ala135 in pro-conA hinders the formation of this ionic interaction, thereby preventing transpeptidation by CeAEP1.
CeAEP1 is predominantly a protease
The jack bean AEP structure not only provides a structural image of the enzyme responsible for conA circular permutation, but also improves our understanding on AEPs, a family of enzymes that are becoming recognized as a tool for protein engineering (Hemu et al., 2016; Yang et al., 2017; Tang et al., 2020). The structural features that make AEP favor hydrolysis or transpeptidation remains a matter of debate. Recent work by Du et al. (2020) uncovered a plant AEP with one of the highest transpeptidase activities despite predictions, based on previously published AEPs, that it would be a hydrolase. Here, we compared CeAEP1 structure to butelase1, an extremely efficient transpeptidase with efficiency up to 1,340,000 M−1 s−1, and to VyPAL2, OaAEP1, AtLegγ, and HaAEP1, which have intermediate to low transpeptidase efficiency. Enzymatic assays have shown that CeAEP1 favors hydrolysis over transpeptidation in a non-native substrate (Bernath-Levin et al., 2015). A few structural features of AEPs have been explored by several research groups, allowing us to better understand what makes CeAEP1 capable of transpeptidation, in addition to its proteolytic activity.
Residue 230 appears to be an important determinant for hydrolysis/transpeptidation. It belongs to a trio of residues (residue 229–331) that were designated Ligase-Activity Determinant 1 by Hemu et al. (2019; Supplemental Figure S12). CeAEP1, like HaAEP1 and AtLegγ, is an inefficient transpeptidase with a glycine in this position. Transpeptidation efficiency was successfully enhanced by a cysteine to alanine mutation in this position in OaAEP1 (Yang et al., 2017). In contrast, a cysteine to valine/isoleucine mutation at the equivalent position in OaAEP1 abolished transpeptidase activity (Yang et al., 2017) even though efficient transpeptidases butelase1 and VyPAL2 have valine and isoleucine respectively at this position. The effect of residue 230 on catalytic activity therefore appears to be dependent on a complex interplay with nearby residues. A glycine in this position, however, seems to be a good indicator of predominant protease activity. The same conclusion can be reached based on sequence comparison of other biochemically characterized AEPs (Hemu et al., 2019).
Hydrophobic S2′ pocket facilitates transpeptidation
The hydrophobic nature of both the P2′ residue of pro-conA (isoleucine) and the S2′ pocket of CeAEP1 (Val163, Phe171, Tyr173; Figure 8B) suggests that the peptide residues C-terminal of the cleavage site might bind to CeAEP1 through hydrophobic interactions. This would be crucial for preventing water molecules from entering the active site, therefore, facilitating aminolysis instead of hydrolysis (Bernath-Levin et al., 2015; Zauner et al., 2018a). Consistent with this hypothesis, hydrophobic residues are present in the S2′ site of HaAEP1 (Val183, Val193), AtLegγ (Val182, Tyr190, Tyr192), OaAEP1 (Val180, Tyr188, Tyr190), VyPAL2 (Tyr185, Tyr187), and butelase1 (Val170, Tyr178, Ala180), and at the P2′ residue of their native substrate, SFTI-1 in sunflower (P2′ leucine), kalata-B1 in O. affinis (P2′ leucine), V. yedoensis cyclotide precursors (P2′ leucine), and C. ternatea cyclotide precursors (P2′ valine). There is no known peptide/protein in A. thaliana that undergoes AEP-mediated transpeptidation, but AtLegγ has also been shown to perform transpeptidation on non-native substrates with valine as the P2′ residue (Zauner et al., 2018b). Molecular dynamic simulations performed on VyPAL2 also showed that the S2′ pocket favors hydrophobic P2′ residues (Hemu et al., 2019). Gly165 conservation in the S2′ site appears to be essential to maintain a cavity in this hydrophobic S2′ pocket (Figure 8B;Supplemental Figure S12) as a glycine to serine mutation in HaAEP1 was shown to severely affect catalytic activity (Haywood et al., 2018).
Facilitating N-terminal nucleophilic attack for transpeptidase activity
The backbone amino group of the N-terminal nucleophile is involved in AEP-mediated transpeptidation. It is possible that the side chain of the N-terminal serine residue acts as the nucleophile, which is observed in intein ligation. However, AEP-mediated transpeptidation has been demonstrated with an N-terminal glycine, which lacks a side chain (Bernath-Levin et al., 2015). Previous studies involving substrate-bound AEP structures have proposed a binding mode between plant AEPs and substrates (Haywood et al., 2018; Zauner et al., 2018a). From this we were able to identify residues around the catalytic cysteine that were likely to facilitate N-terminal nucleophilic attack in the transpeptidation reaction (Haywood et al., 2018). The residues involved include Glu201, Pro161, Gln225, and Asn227 of CeAEP1 (Figure 8C). Although in silico docking studies show that Glu201 forms an ionic bond with the incoming N-terminal nucleophile (Zauner et al., 2018a), this residue does not appear to be a major determinant of transpeptidation efficiency as glutamate is found in this position in AEPs that have differing transpeptidase efficiencies (Supplemental Figure S12). The nearby Gly160 (Figure 8C), which is one of the two residues designated as Ligase-Activity Determinant 2 by Hemu et al. (2019), is a highly conserved residue as most plant AEPs have a Gly or Ala at this residue (Supplemental Figure S12). At this position, a tyrosine to glycine mutation in VyPAL3 and a tyrosine to alanine mutation in Viola Canadensis AEP were shown to enhance the catalytic activity of transpeptidation. Any side chain bigger than alanine at residue 160 may therefore sterically interfere with the binding of peptide substrate residues C-terminal of the cleavage site, allowing water to enter to complete the cleavage reaction by hydrolysis.
Pro161, Gln225, and Asn227, on the other hand, have been hypothesized to sterically hinder the incoming N-terminus from interacting with Glu201 to initiate transpeptidation (Haywood et al., 2018). These three residues lining the active site are the same in CeAEP1 and HaAEP1, both of which are inefficient transpeptidases. In contrast, the smaller side chains in butelase1 (Gly167, Ala168, Gly232, Ser234) likely contribute to its high transpeptidase efficiency, and is also likely why it will accept most N-terminal amino acids for transpeptidation (Nguyen et al., 2014). The intermediate transpeptidation efficiency of AtLegγ, OaAEP1, and VyPAL2 relative to CeAEP1, HaAEP1 and butelase1 corroborates with the intermediate amino acid sizes (AtLegγ: Gly179, Pro180, Glu244, Ser246; OaAEP1: Ala177, Ala178, Thr242, Ser246; VyPAL2: Ala174, Pro175, Thr239, Gly241). Although Gln225 and Asn227 in CeAEP1 and HaAEP1 may sterically interfere with transpeptidation, their long, polar side chains may allow for redundancy in interacting with the N-terminal nucleophile, as was observed in the absence of Glu201 of HaAEP1 (Haywood et al., 2018).
Conclusion
Our investigations show that the mature form of conA is more stable than its precursor, pro-conA, revealing a functional consequence for the only known naturally-occurring circular permutation, which was discovered about 30 years ago. Structural evidence shows that no change occurs at the carbohydrate-binding site, and ITC analysis shows that monosaccharide binding is not affected. The difference in crystal packing between pro-conA and conA indicates that changes induced by circular permutation occur at the quaternary rather than the tertiary level. Analytical ultracentrifugation analyses are consistent with pro-conA forming tetramers in solution, and in silico analyses suggest that the atypical dimer-of-dimers conformation observed in the crystal structure occurs in aqueous solution as well. It is likely that the different quaternary conformation between pro-conA and conA is the basis for the difference in protein stability observed in circular dichroism analyses.
Although CeAEP1 has been shown to be predominantly a protease, this was determined with a non-native substrate (Bernath-Levin et al., 2015). Using purified recombinant proteins, we show that jack bean AEP is capable of carrying out both cleavage and transpeptidation reactions to circularly permutate pro-conA, with the highest circular permutation efficiency occurring at pH 5. Structural analysis and recently generated knowledge from other AEP structures allow us to understand how a protease like CeAEP1 is capable of transpeptidation.
Materials and methods
Pro-conA expression and purification
A synthetic DNA sequence encoding residues 31–290 of C. ensiformis pro-conA (UniProt ID: P02866) was codon optimized for E. coli (GenScript) and included a Gly–Ser linker (encoded by an in-frame BamHI site) between a six-His tag and tobacco etch virus (TEV) protease recognition site (Glu–Asn–Leu–Tyr–Phe–Gln–Ser) at the N-terminus. Ser30 of UniProt ID: P02866 was not included because TEV cleavage will leave a serine residue at the N-terminus of pro-conA, hence, reconstituting the native pro-conA protein sequence). This synthetic DNA sequence was subcloned into pQE30 (Qiagen) before being expressed in T7 SHuffle Express E. coli (New England Biolabs). Cultures were grown in lysogeny broth containing 100 µg·mL−1 ampicillin and 35 µg·mL−1 kanamycin at 30°C and allowed to cool to 16°C before inducing expression by adding 0.1 mM isopropyl β-d-1-thiogalactopyranoside at OD600 of 0.8–1.0. After 16 h, cultures were harvested by centrifugation and lysed by ultrasonication in MOPS-salt buffer at pH 6.8 [50-mM 3-(N-morpholino) propanesulfonic acid, 12.5-mM sodium acetate, 1-M sodium chloride, pH 6.8]. Lysed products were centrifuged and the supernatant incubated (batchwise) with Ni-NTA resin overnight at 4°C. The resin was washed with 100 mL of MOPS-salt buffer and 100 mL of MOPS-salt buffer containing 20-mM imidazole before the recombinant protein was eluted with MOPS-salt buffer containing 150-mM imidazole. Cleavage by TEV protease was then performed by first dialysing pro-conA into 50-mM sodium acetate, 500-mM sodium chloride, 1-mM dithiothreitol, pH 5.8 at 4°C for 2 h before centrifugation to remove precipitate that formed from the decrease in pH. His-tagged TEV protease was added (10% of pro-conA mass content) and the mixture incubated at 4°C for a further 30 h. To remove the TEV protease and the N-terminal tag cleaved from pro-conA, the mixture was dialyzed overnight in MOPS-salt buffer then incubated (batchwise) with Ni-NTA resin overnight. Purified pro-conA was obtained in flow-through from reverse His-tag purification before a final concentration of 1-mM manganese chloride and 1-mM calcium chloride was added to purified pro-conA.
Jack bean AEP expression and purification
A synthetic DNA sequence encoding residues 36–475 of C. ensiformis AEP (UniProt ID: P49046), including an N-terminal six-His tag and codon-optimized for E. coli (GeneArt), was subcloned into pQE30 (Qiagen) before being expressed in T7 SHuffle Express E. coli (New England Biolabs) containing the suppressor plasmid pREP4 (Qiagen). The first 35 residues were excluded as the N-terminal maturation point for CeAEP1, which involves an autocatalytic cleavage at residue 35, was known from Edman degradation analysis (Abe et al., 1993). Expression was performed as above and cultures were harvested and lysed by ultrasonication in Tris-salt buffer (50-mM Tris, pH 8.0, 100-mM sodium chloride) containing 0.1% Triton X-100. Lysed products were centrifuged and the supernatant incubated (batchwise) with Ni-NTA resin overnight at 4°C. The resin was washed with 50 mL of Tris-salt buffer and 50 mL of Tris-salt buffer containing 20-mM imidazole before the recombinant protein was eluted with 20 mL of Tris-salt buffer containing 300 mM imidazole. Nickel-purified CeAEP1 was further purified by anion-exchange chromatography (HiTrap Q HP 5 mL) with gradient of 0–500 mM sodium chloride in 50-mM Tris, pH 8.0 over 90 min, followed by size exclusion chromatography (HiLoad 16/600 Superdex 200) in 50-mM Tris, 50-mM sodium chloride.
Crystallization and data collection
For pro-conA, a 10 kDa molecular weight cutoff (M.W.C.O.) centrifugal filter (Amicon) was used to change buffer to 50-mM MOPS, 12.5-mM sodium acetate, 200-mM sodium chloride, 1-mM manganese chloride, and 1-mM calcium chloride. Protein was assessed for purity by SDS–PAGE and concentrated to 28.7 mg·mL−1. Purified protein was used immediately for crystallization. Crystal screening was performed using the sitting-drop vapor diffusion method with 60 µL of reservoir solution in 96-well Intelli-Plates at 16°C. Crystals were obtained from sitting drop containing 0.2-µL protein, 0.1-µL mother-liquor (10-mM zinc chloride, 100-mM HEPES, 20% PEG 6000 (w/v), pH 7.0) of the PACT Premier crystallization screen (Molecular Dimensions). Single crystals were soaked in mother-liquor supplemented with 25% ethanediol as a cryoprotectant prior to being flash frozen and stored in liquid nitrogen. Data collection was performed at 100 K on the Australian MX2 (microfocus) beamline (McPhillips et al., 2002) using a wavelength of 0.9537 Å and diffraction data was collected to 2.1 Å resolution (Table 1).
Table 1.
Summary of crystallographic data and refinement statistics for pro-conA and CeAEP1. Values in parentheses are for the highest resolution shell
| Data Collection | Pro-conA | CeAEP1 |
|---|---|---|
| Space group | I 1 2 1 | I 1 2 1 |
| Unit cell dimensions | ||
| a, b, c (Å) | 59.94, 90.42, 86.86 | 106.99, 88.88, 109.85 |
| α, β, γ (°) | 90.00, 91.13, 90.00 | 90.00, 111.72, 90.00 |
| Wavelength | 0.9536 | 0.9840 |
| Rmerge (%) | 12.0 (61.9) | 7.5 (76.5) |
| Rpim (%) | 7.7 (39) | 4.8 (47) |
| I/σI | 6.5 (1.8) | 9.6 (1.4) |
| Completeness (%) | 98.8 (92.6) | 98.8 (94.0) |
| Redundancy | 3.4 (3.3) | 3.4 (3.6) |
| CC1/2 | 0.994 (0.743) | 0.997 (0.662) |
| Refinement | ||
| Resolution (Å) | 48.88–2.10 | 45.57–2.69 |
| No. of reflections | 26,657 | 26,353 |
| Rwork/Rfree | 0.196/0.245 | 0.202/0.271 |
| No. of atoms | 3,845 | 6,794 |
| Protein | 3,697 | 6,762 |
| Ligand | 68 | 0 |
| Water | 80 | 32 |
| Wilson B (Å2) | 28.1 | 71.8 |
| Average B factor (Å2) | 33.0 | 75.0 |
| R.m.s.d.: | ||
| Bond lengths (Å) | 0.0079 | 0.0038 |
| Bond angles (°) | 1.5268 | 1.3552 |
| Ramachandran Analysis | ||
| Favored (%) | 97 | 91 |
| Allowed (%) | 3 | 7 |
| Outliers (%) | 0 | 2 |
| PDB code | 6XT6 | 6XT5 |
CeAEP1 was assessed for purity by SDS–PAGE. Purified protein was used immediately for crystallization. Low-resolution diffracting crystals were obtained initially using the sitting-drop vapor diffusion method with reservoir condition 100-mM sodium HEPES, 15% PEG 20,000 (w/v), pH 7 from the ProPlex crystallization screen (Molecular Dimensions). An additive screen was then performed using the sitting-drop vapor diffusion method with 30 µL of reservoir solution (125-mM HEPES, 12.5% PEG 20,000 (w/v), pH 7.5) in a MiTeGen In Situ 1 crystallization plate at 16°C. Crystals were obtained from a condition with 0.5 µL of 16.7 mg·mL−1 of protein, 0.4-µL reservoir solution, 0.1-µL additive (10-mM ethylenediaminetetraacetic acid disodium salt dihydrate) after 2 weeks. Single crystals were soaked in mother-liquor supplemented with 30% glycerol as a cryoprotectant prior to being flash frozen and stored in liquid nitrogen. Data collection was performed similarly to pro-conA crystals and diffraction data was collected to 2.7 Å resolution (Table 1).
Structural determination, refinement, and model building
For both pro-conA and CeAEP1, diffraction data were processed using XDS programme package (Kabsch, 2010) and scaled with AIMLESS from the CCP4 program suite (Winn et al., 2011). Pro-conA crystallized with space group I 1 2 1 and unit cell dimensions a = 59.94 Å, b = 90.42 Å, c = 86.86 Å, β = 91.13°. The structure of pro-conA was solved by molecular replacement using MOLREP, with conA (PDB: 1JBC; Parkin et al., 1996) as the search model. CeAEP1 crystallized with space group I 1 2 1 and unit cell dimensions a = 106.99 Å, b = 88.88 Å, c = 109.85 Å, β = 111.72°. The structure of CeAEP1 was solved by molecular replacement using MOLREP, with A. thaliana legumain (PDB: 5NIJ; Zauner et al., 2018b) as the search model. For both pro-conA and CeAEP1, manual building and refinement was performed in iterative cycles using COOT (Emsley et al., 2010) and REFMAC5 of the CCP4 program suite. Coordinates and structure factors were deposited into the PDB under accession code 6XT6 (pro-conA) and 6XT5 (CeAEP1). Figures illustrating both structures were generated using PyMol. PyMol was used to calculate r.m.s.d. values. For comparison of pro-conA and conA functional sites, only the residues involved in the respective functions were aligned. CheckMyMetal (http://csgid.org/metal_sites;Zheng et al., 2017) was used to evaluate the assignment of the metal binding sites in pro-conA.
Analytical ultracentrifugation (sedimentation velocity)
For AUC analyses, purified pro-conA in MOPS-salt buffer containing 1-mM manganese chloride and 1-mM calcium chloride was dialyzed extensively into either a pH 4.5 buffer or a pH 6.8 buffer (Table 2). Sedimentation velocity experiments were performed in a Beckman Coulter XL-A analytical ultracentrifuge as previously described (Soares da Costa et al., 2014, 2016, 2017), employing an 8-hole An50-Ti rotor and double-sector cells containing synthetic quartz windows. Cells were loaded with 380 µL of protein at 3.0 or 0.2 mg·mL−1 in either pH 4.5 buffer or pH 6.8 buffer and 400 µL of the respective buffer as reference. Centrifugation was performed at 40,000 rpm at either 20°C or 30°C, while data was collected in continuous mode without averaging at 280 nm with a radial step size of 0.003 cm. SEDNTERP software (Laue, 2001) was used to compute the estimated sample partial specific volume (0.732909 mL·g−1), solvent density and viscosity (Table 2). The continuous sedimentation (c(s)) and mass (c(M)) distributions were determined by fitting absorbance as a function of radial position to the Lamm equation using SEDFIT (Laue, 2001; Schuck et al., 2002).
Table 2.
Solvent properties for AUC analyses
| Buffer | Constituents | Density (g·mL−1) | Viscosity (cp) |
|---|---|---|---|
| pH 4.5 buffer | 50-mM sodium acetate, 0.5-M sodium chloride, 0.2-mM manganese chloride, 0.2-mM calcium chloride, pH 4.5 | 1.0211 | 0.0106284 |
| pH 6.8 buffer | 50-mM 3-(N-morpholino) propanesulfonic acid, 12.5-mM sodium acetate, 0.5-M sodium chloride, 0.2-mM manganese chloride, 0.2-mM calcium chloride, pH 6.8 | 1.0232 | 0.0108354 |
Confirming pro-conA maturation by CeAEP1 in vitro
CeAEP1 was first autoactivated by dialyzing purified CeAEP1 for 4 h in activation buffer (20-mM sodium acetate, 0.1-M sodium chloride, 1-mM ethylenediaminetetraacetic acid, 5-mM dithiothreitol, pH 4.0). Pro-conA maturation was performed by incubating 1 mg·mL−1 of pro-conA with 0.1 mg·mL−1 of activated CeAEP1 in MOPS storage buffer at pH 6.8 for 24 h. Concentrations of pro-conA and conA were measured by absorbance at 280 nm with a NanoDrop, with protein extinction coefficient of 33,920 M−1 cm−1 and 32,430 M−1 cm−1, respectively, and molecular weight of 28.22 and 25.57 kDa, respectively. Reaction was stopped by incubating in 5× sample loading buffer (20% (v/v) glycerol, 15% (w/v) sodium dodecyl sulfate, 312.5-mM Tris, 10-mM ethylenediaminetetraacetic acid disodium salt, 0.05% (v/v) β-mercaptoethanol, 0.05% (w/v) bromophenol blue, pH 6.9) at 37°C for 15 min. Samples were run on SDS–PAGE containing 10 mM reduced glutathione in the running buffer. Samples were loaded to have roughly similar band intensity for the band of interest to enable better estimation of relative protein size (recombinant pro-conA: 0.75 µg, conA (Sigma): 1.5 µg, CeAEP1-processed conA: 6 µg). Samples were electrophoresed on a Bolt 4–12% Bis-Tris Plus gel, electroblotted onto Immobilon-PSQ PVDF membrane in Towbin buffer (25-mM Tris, 192-mM glycine, 10% (v/v) methanol; Towbin et al., 1979) at 200 mA for 2 h in an ice water bath. Immobilon-PSQ PVDF membrane was then stained with 0.025% Coomassie Brilliant Blue R-250, 40% methanol. N-terminal Edman degradation of the first eight amino acids was performed by Proteomics International (Perth, Australia).
To determine pH-dependence of conA circular permutation by CeAEP1, the in vitro assay was also performed as above but at pH 4.0 (50-mM sodium acetate, 1-M sodium chloride, 1-mM manganese chloride, 1-mM calcium chloride, pH 4.0), pH 5.0 (50-mM sodium acetate, 1-M sodium chloride, 1-mM manganese chloride, 1-mM calcium chloride, pH 5.0), pH 6.0 (50-mM 2-(N-morpholino)ethanesulfonic acid, 1-M sodium chloride, 1-mM manganese chloride, 1-mM calcium chloride, pH 6.0), and pH 7.0 (50-mM 3-(N-morpholino) propanesulfonic acid, 1-M sodium chloride, 1-mM manganese chloride, 1-mM calcium chloride, pH 7.0) for 48 h.
Circular dichroism
Pro-conA was concentrated to 10 mg·mL−1 using 10 kDa M.W.C.O. centrifugal filter (Amicon) in MOPS-salt buffer. ConA purified from C. ensiformis (Type VI, lyophilized powder, Sigma-Aldrich, Cat No. L7647) was solubilized to 10 mg·mL−1 in MOPS-salt buffer with 1-mM manganese chloride and 1-mM calcium chloride. Proteins were then diluted 100 times in water to 0.1 mg·mL−1, pH 6.5 immediately before CD melt curve analysis. Melt curves were measured at 218 nm with a temperature slope of 1°C·min−1 from 25°C to 95°C, 4-s response time, 5 nm bandwidth. A biphasic curve (GraphPad Prism, version 8.00) was fitted to the data. No-heat controls for pro-conA and conA were performed by measuring ellipticity at 25°C for the same duration as the heat analysis. For pH stability analysis, pro-conA and conA were prepared in 20-mM Tris, pH 5.8, 1-M sodium chloride, 1-mM calcium chloride, 1-mM manganese chloride before diluting 100 times for CD analysis. CD measurements were performed in triplicate using JASCO J-810 spectropolarimeter with quartz cuvette of 1 mm path length, 100 millidegree sensitivity, 1 nm data pitch, 100 nm min−1 scanning speed, 2-s response time, 4 nm bandwidth, three accumulations, between 190 and 260 nm at room temperature (24°C).
Isothermal titration calorimetry
Microcal iTC200 from GE healthcare was used to perform ITC analysis. Pro-conA and conA were dialysed in 50-mM MOPS, 12.5-mM sodium acetate, 1-M sodium chloride, 1-mM calcium chloride, 1-mM manganese chloride, pH 5.8, and concentrated down to 449.4 µM and 439.5 µM, respectively. The ligand, methyl-α-d-mannose, was dissolved to 8 mM with the same buffer used in protein sample dialysis. Protein samples were placed in the sample cell (cell volume = 200 µL) and titrated with methyl-α-d-mannose. Titrations were performed at 25°C with a stirring speed of 750 rpm. Methyl-α-d-mannose was injected 76 times from a computer controlled syringe, at a volume of 0.5 µL over 1 s for each injection, with a spacing of 150 s between injections. Only 0.25 µL was injected for the first injection and ignored in analysis to minimize potential errors from preparation. Experimental data were fitted to a theoretical titration curve using the Origin software (version 2002, OriginLab Corporation). A “one site model” was used to generate the curve, with ΔH (enthalpy change), Ka (association constant), and the stoichiometry of the protein–ligand complex set as variable parameters.
Accession numbers
The structures and structure factors of pro-conA and CeAEP1 have been deposited in the PDB under accession numbers 6XT6 and 6XT5, respectively. The plasmids to express jack bean pro-conA (pQE30-TEV-pro-conA) and AEP1 (pQE30-CeAEP1) are available at Addgene under IDs 159527 and 159528, respectively.
Supplemental data
The following materials are available in the online version of this article.
Supplemental Figure S1 . Sequence from pre-pro-conA to conA and conA fragments.
Supplemental Figure S2 . Recombinant pro-conA purification and confirming full-length protein after crystal trials.
Supplemental Figure S3 . ConA functional sites remain unchanged after circular permutation.
Supplemental Figure S4 . Pro-conA and conA have similar structures.
Supplemental Figure S5 . N-terminal Edman degradation of pro-conA processed by recombinant CeAEP1.
Supplemental Figure S6 . ConA circular permutation by CeAEP1 is most efficient around pH 5.
Supplemental Figure S7 . Pro-conA and conA stability with and without heat treatment.
Supplemental Figure S8 . Circular permutation has no effect on conA binding to methyl-α-d-mannose.
Supplemental Figure S9 . Recombinant CeAEP1 purification and activation.
Supplemental Figure S10 . CeAEP1 residues involved in catalytic activity.
Supplemental Figure S11 . Different crystal contacts resulting in differences between CeAEP1 monomers in crystal structure.
Supplemental Figure S12 . AEP Alignment.
Supplemental Figure S13 . Pro-conA and conA have different interactions at His185 and His3.
Supplementary Material
Acknowledgments
The authors acknowledge the La Trobe University Comprehensive Proteomics Platform for providing infrastructure.
Funding
This work was supported in part by Australian Research Council (ARC; grant no. DP160100107 to J.S.M.). This research was undertaken in part using the MX2 beamline at the Australian Synchrotron, part of Australian Nuclear Science and Technology Organization, and made use of the Australian Cancer Research Foundation detector. J.H. was supported by an ARC Discovery Early Career Researcher Award (grant no. DE180101445). S.G.N. was supported by the Australian Research Training Program. E.R.R.M. is the recipient of a Research Training Program and a Grains Research and Development Corporation scholarship (grant no. 9176977). T.P.S.C. acknowledges the Australian Research Council for support through a DECRA Fellowship (grant no. DE190100806).
Conflict of interest statement. None declared.
J.S.M. and S.G.N. designed the research; S.G.N., J.H., J.W.S., and E.R.R.M. performed the research; S.G.N., J.W.S., E.R.R.M., T.P.S.C., C.S.B., and J.H. analyzed the data; S.G.N., J.S.M., and C.S.B. wrote the article with input from J.W.S and J.H.; J.S.M. acquired the funding for this study.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://academic.oup.com/plcell) is: Joshua S. Mylne (josh.mylne@curtin.edu.au).
References
- Abe Y, Shirane K, Yokosawa H, Matsushita H, Mitta M, Kato I, Ishii S (1993) Asparaginyl endopeptidase of jack bean seeds. Purification, characterization, and high utility in protein sequence analysis. J Biol Chem 268: 3525–3529 [PubMed] [Google Scholar]
- Adachi M, Takenaka Y, Gidamis AB, Mikami B, Utsumi S (2001) Crystal structure of soybean proglycinin A1aB1b homotrimer. J Mol Biol 305: 291–305 [DOI] [PubMed] [Google Scholar]
- Adachi M, Kanamori J, Masuda T, Yagasaki K, Kitamura K, Mikami B, Utsumi S (2003) Crystal structure of soybean 11S globulin: glycinin A3B4 homohexamer. Proc Natl Acad Sci USA 100: 7395–7400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernath-Levin K, Nelson C, Elliott AG, Jayasena AS, Millar AH, Craik DJ, Mylne JS (2015) Peptide macrocyclization by a bifunctional endoprotease. Chem Biol 22: 571–582 [DOI] [PubMed] [Google Scholar]
- Bernhard W, Avrameas S (1971) Ultrastructural visualization of cellular carbohydrate components by means of concanavalin A. Exp Cell Res 64: 232–236 [DOI] [PubMed] [Google Scholar]
- Bezerra EH, Rocha BA, Nagano CS, Bezerra Gde A, Moura TR, Bezerra MJ, Benevides RG, Sampaio AH, Assreuy AM, Delatorre P. , et al. (2011). Structural analysis of ConBr reveals molecular correlation between the carbohydrate recognition domain and endothelial NO synthase activation. Biochem Biophys Res Commun 408: 566–570 [DOI] [PubMed] [Google Scholar]
- Bliven S, Prlić A (2012) Circular permutation in proteins. PLoS Comp Biol 8: e1002445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumberg S, Tal N (1976) Effect of divalent metal ions on the digestibility of concanavalin a by endopeptidases. Biochim Biophys Acta 453: 357–364 [DOI] [PubMed] [Google Scholar]
- Bouckaert J, Dewallef Y, Poortmans F, Wyns L, Loris R (2000) The structural features of concanavalin A governing non-proline peptide isomerization. J Biol Chem 275: 19778–19787 [DOI] [PubMed] [Google Scholar]
- Bowles DJ, Pappin DJ (1988) Traffic and assembly of concanavalin A. Trends Biochem Sci 13: 60–64 [DOI] [PubMed] [Google Scholar]
- Bowles DJ, Marcus SE, Pappin DJ, Findlay JB, Eliopoulos E, Maycox PR, Burgess J (1986) Posttranslational processing of concanavalin A precursors in jackbean cotyledons. J Cell Biol 102: 1284–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer CF, Brown RD 3rd, Koenig SH (1983) Metal ion binding and conformational transitions in concanavalin A: a structure-function study. J Biomol Struct Dyn 1: 961–997 [DOI] [PubMed] [Google Scholar]
- Brown RD 3rd, Brewer CF, Koenig SH (1977) Conformation states of concanavalin A: kinetics of transitions induced by interaction with Mn2+ and Ca2+ ions. Biochemistry 16: 3883–3896 [DOI] [PubMed] [Google Scholar]
- Carrington DM, Auffret A, Hanke DE (1985) Polypeptide ligation occurs during post-translational modification of concanavalin A. Nature 313: 64–67 [DOI] [PubMed] [Google Scholar]
- Cavada BS, Pinto-Junior VR, Osterne VJS, Nascimento KS (2018) ConA-like lectins: high similarity proteins as models to study structure/biological activities relationships. Int J Mol Sci 20: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chervenak MC, Toone EJ (1995) Calorimetric analysis of the binding of lectins with overlapping carbohydrate-binding ligand specificities. Biochemistry 34: 7966–7966 [DOI] [PubMed] [Google Scholar]
- Cunningham BA, Hemperly JJ, Hopp TP, Edelman GM (1979) Favin versus concanavalin A: circularly permuted amino acid sequences. Proc Natl Acad Sci USA 76: 3218–3222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalkin K, Bowles DJ (1983) Analysis of inter-relationship of jackbean seed components by two-dimensional mapping of iodinated tryptic peptides. Planta 157: 536–539 [DOI] [PubMed] [Google Scholar]
- Delatorre P, Rocha BA, Souza EP, Oliveira TM, Bezerra GA, Moreno FB, Freitas BT, Santi-Gadelha T, Sampaio AH, Azevedo WF Jr. , et al. (2007) Structure of a lectin from Canavalia gladiata seeds: new structural insights for old molecules. BMC Struct Biol 7: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle RJ, Thomasson DL, Nicholson SK (1976) Stabilization of concanavalin A by metal ligands. Carbohydr Res 46: 111–118 [DOI] [PubMed] [Google Scholar]
- Du J, Yap K, Chan LY, Rehm FBH, Looi FY, Poth AG, Gilding EK, Kaas Q, Durek T, Craik DJ (2020) A bifunctional asparaginyl endopeptidase efficiently catalyzes both cleavage and cyclization of cyclic trypsin inhibitors. Nat Commun 11: 1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer JM, Johnson C (1981) The use of concanavalin A to study the immunoregulation of human T cells. Clin Exp Immunol 46: 237–249 [PMC free article] [PubMed] [Google Scholar]
- Einhoff W, Fleischmann G, Freier T, Kummer H, Rüdiger H (1986) Interactions between lectins and other components of leguminous protein bodies. Biol Chem Hoppe-Seyler 367: 15–25 [DOI] [PubMed] [Google Scholar]
- Elsässer B, Zauner FB, Messner J, Soh WT, Dall E, Brandstetter H (2017) Distinct roles of catalytic cysteine and histidine in the protease and ligase mechanisms of human legumain as revealed by DFT-based QM/MM simulations. ACS Catal 7: 5585–5593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmerich C, Helliwell JR, Redshaw M, Naismith JH, Harrop SJ, Raftery J, Kalb AJ, Yariv J, Dauter Z, Wilson KS (1994) High-resolution structures of single-metal-substituted concanavalin A: the Co,Ca-protein at 1.6 A and the Ni,Ca-protein at 2.0 A. Acta Crystallogr D 50: 749–756 [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Crystallogr D 66: 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faye L, Chrispeels MJ (1987) Transport and processing of the glycosylated precursor of concanavalin-a in jack-bean. Planta 170: 217–224 [DOI] [PubMed] [Google Scholar]
- François-Heude M, Méndez-Ardoy A, Cendret V, Lafite P, Daniellou R, Ortiz Mellet C, García Fernández JM, Moreau V, Djedaïni-Pilard F (2015) Synthesis of high-mannose oligosaccharide analogues through click chemistry: true functional mimics of their natural counterparts against lectins? Chemistry 21: 1978–1991 [DOI] [PubMed] [Google Scholar]
- Gebhard LG, Risso VA, Santos J, Ferreyra RG, Noguera ME, Ermácora MR (2006) Mapping the distribution of conformational information throughout a protein sequence. J Mol Biol 358: 280–288 [DOI] [PubMed] [Google Scholar]
- Goldenberg DP, Creighton TE (1983) Circular and circularly permuted forms of bovine pancreatic trypsin inhibitor. J Mol Biol 165: 407–413 [DOI] [PubMed] [Google Scholar]
- Goldstein JI, Winter CH, Poretz DR (1997) Plant lectins: tools for the study of complex carbohydrates. InMontreuil J, Vliegenthart JFG, Schachter H., eds, Glycoproteins II. Elsevier B.V., New York, pp 403–474 [Google Scholar]
- Gunther GR, Wang JL, Yahara I, Cunningham BA, Edelman GM (1973) Concanavalin A derivatives with altered biological activities. Proc Natl Acad Sci USA 70: 1012–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamodrakas SJ, Kanellopoulos PN, Pavlou K, Tucker PA (1997) The crystal structure of the complex of concanavalin A with 4'-methylumbelliferyl-alpha-D-glucopyranoside. J Struct Biol 118: 23–30 [DOI] [PubMed] [Google Scholar]
- Harris KS, Guarino RF, Dissanayake RS, Quimbar P, McCorkelle OC, Poon S, Kaas Q, Durek T, Gilding EK, Jackson MA. , et al. (2019) A suite of kinetically superior AEP ligases can cyclise an intrinsically disordered protein. Sci Rep 9: 10820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haywood J, Schmidberger JW, James AM, Nonis SG, Sukhoverkov KV, Elias M, Bond CS, Mylne JS (2018) Structural basis of ribosomal peptide macrocyclization in plants. eLife 7: e32955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann U, Hahn M (1995) Circular permutation of polypeptide chains: implications for protein folding and stability. Prog Biophys Mol Biol 64: 121–143 [DOI] [PubMed] [Google Scholar]
- Hemu X, Qiu Y, Nguyen GKT, Tam JP (2016) Total synthesis of circular bacteriocins by butelase 1. J Am Chem Soc 138: 6968–6971 [DOI] [PubMed] [Google Scholar]
- Hemu X, El Sahili A, Hu S, Wong K, Chen Y, Wong YH, Zhang X, Serra A, Goh BC, Darwis DA. , et al. (2019) Structural determinants for peptide-bond formation by asparaginyl ligases. Proc Natl Acad Sci USA 116: 11737–11746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix RW (1991). Protein carpentry. Curr Biol 1: 71–73 [DOI] [PubMed] [Google Scholar]
- Herman EM, Shannon LM, Chrispeels MJ (1985) Concanavalin A is synthesized as a glycoprotein precursor. Planta 165: 23–29 [DOI] [PubMed] [Google Scholar]
- Huet M, Claverie JM (1978) Sedimentation studies of reversible dimer-tetramer transition kinetics of concanavalin-A. Biochemistry 17: 236–241 [DOI] [PubMed] [Google Scholar]
- Jackson MA, Gilding EK, Shafee T, Harris KS, Kaas Q, Poon S, Yap K, Jia H, Guarino R, Chan LY. , et al. (2018). Molecular basis for the production of cyclic peptides by plant asparaginyl endopeptidases. Nat Commun 9: 2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James AM, Haywood J, Leroux J, Ignasiak K, Elliott AG, Schmidberger JW, Fisher MF, Nonis SG, Fenske R, Bond CS. , et al. (2019). The macrocyclizing protease butelase 1 remains autocatalytic and reveals the structural basis for ligase activity. Plant J 98: 988–999 [DOI] [PubMed] [Google Scholar]
- Jung R, Scott MP, Nam Y-W, Beaman TW, Bassüner R, Saalbach I, Müntz K, Nielsen NC (1998) The role of proteolysis in the processing and assembly of 11S seed globulins. The Plant Cell 10: 343–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010). XDS. Acta Crystallogr D 66: 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalb AJ, Levitzki A (1968) Metal-binding sites of concanavalin A and their role in the binding of alpha-methyl d-glucopyranoside. Biochem J 109: 669–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanellopoulos PN, Tucker PA, Pavlou K, Agianian B, Hamodrakas SJ (1996a) A triclinic crystal form of the lectin concanavalin A. J Struct Biol 117: 16–23 [DOI] [PubMed] [Google Scholar]
- Kanellopoulos PN, Pavlou K, Perrakis A, Agianian B, Vorgias CE, Mavrommatis C, Soufi M, Tucker PA, Hamodrakas SJ (1996b) The crystal structure of the complexes of concanavalin A with 4'-nitrophenyl-alpha-D-mannopyranoside and 4'-nitrophenyl-alpha-D-glucopyranoside. J Struct Biol 116: 345–355 [DOI] [PubMed] [Google Scholar]
- Krissinel E, Henrick K (2007) Inference of macromolecular assemblies from crystalline state. J Mol Biol 372: 774–797 [DOI] [PubMed] [Google Scholar]
- Lagarda-Diaz I, Guzman-Partida AM, Vazquez-Moreno L (2017) Legume lectins: proteins with diverse applications. Int J Mol Sci 18: 1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, Jabłońska J, Pravda L, Vařeková RS, Thornton JM (2018) PDBsum: structural summaries of PDB entries. Protein Sci 27: 129–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laue T (2001) Biophysical studies by ultracentrifugation. Curr Opin Struct Biol 11: 579–583 [DOI] [PubMed] [Google Scholar]
- Lis H, Sharon N (1998) Lectins: carbohydrate-specific proteins that mediate cellular recognition. Chem Rev 98: 637–674 [DOI] [PubMed] [Google Scholar]
- Locke AK, Cummins BM, Abraham AA, Coté GL (2014) PEGylation of concanavalin A to improve its stability for an in vivo glucose sensing assay. Anal Chem 86: 9091–9097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loka RS, McConnell MS, Nguyen HM (2015) Studies of highly-ordered heterodiantennary mannose/glucose-functionalized polymers and concanavalin A protein interactions using isothermal titration calorimetry. Biomacromolecules 16: 4013–4021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loris R, Hamelryck T, Bouckaert J, Wyns L (1998) Legume lectin structure. Biochim Biophys Acta 1383: 9–36 [DOI] [PubMed] [Google Scholar]
- Macedo ML, das Gracas Machado Freire M, da Silva MB, Coelho LC (2007) Insecticidal action of Bauhinia monandra leaf lectin (BmoLL) against Anagasta kuehniella (Lepidoptera: Pyralidae), Zabrotes subfasciatus and Callosobruchus maculatus (Coleoptera: Bruchidae). Comp Biochem Physiol A Mol Integr Physiol 146: 486–498 [DOI] [PubMed] [Google Scholar]
- Maeda H, Hattori H, Kanoh H (1989) Conformational change and aggregation of concanavalin A at high temperatures. Int J Biol Macromol 11: 290–296 [DOI] [PubMed] [Google Scholar]
- McCubbin WD, Oikawa K, Kay CM (1971) Circular dichroism studies on concanavalin A. Biochem Biophys Res Commun 43: 666–674 [DOI] [PubMed] [Google Scholar]
- McKenzie GH, Sawyer WH, Nichol LW (1972) The molecular weight and stability of concanavalin A. Biochim Biophys Acta 263: 283–293 [DOI] [PubMed] [Google Scholar]
- McPhillips TM, McPhillips SE, Chiu H-J, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP. , et al. (2002) Blu-Ice and the distributed control system: software for data acquisition and instrument control at macromolecular crystallography beamlines. J Synchrotron Radiat 9: 401–406 [DOI] [PubMed] [Google Scholar]
- Meister GE, Kanwar M, Ostermeier M (2011) Circular permutation of proteins. InLutz S, Bornscheuer UT, eds, Protein Engineering Handbook. Wiley-VCH Verlag GmbH & Co., Weinheim, New York, pp 453–471 [Google Scholar]
- Melander M, Åhman I, Kamnert I, Strömdahl AC (2003) Pea lectin expressed transgenically in oilseed rape reduces growth rate of pollen beetle larvae. Transgenic Res 12: 555–567 [DOI] [PubMed] [Google Scholar]
- Min W, Jones DH (1994) In vitro splicing of concanavalin A is catalyzed by asparaginyl endopeptidase. Nat Struct Mol Biol 1: 502–504 [DOI] [PubMed] [Google Scholar]
- Min W, Dunn AJ, Jones DH (1992) Non-glycosylated recombinant pro-concanavalin A is active without polypeptide cleavage. EMBO J 11: 1303–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen GKT, Wang S, Qiu Y, Hemu X, Lian Y, Tam JP (2014) Butelase 1 is an Asx-specific ligase enabling peptide macrocyclization and synthesis. Nat Chem Biol 10: 732–738 [DOI] [PubMed] [Google Scholar]
- Nonis SG, Haywood J, Mylne JS (2021) Plant asparaginyl endopeptidases and their structural determinants of function. Biochem Soc Trans 49: 965–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata S, Muramatsu T, Kobata A (1975) Fractionation of glycopeptides by affinity column chromatography on concanavalin A-sepharose. J Biochem 78: 687–696 [DOI] [PubMed] [Google Scholar]
- Otegui MS, Herder R, Schulze J, Jung R, Staehelin LA (2006) The proteolytic processing of seed storage proteins in Arabidopsis embryo cells starts in the multivesicular bodies. Plant Cell 18: 2567–2581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkin S, Rupp B, Hope H (1996) Atomic resolution structure of concanavalin A at 120 K. Acta Crystallogr D 52: 1161–1168 [DOI] [PubMed] [Google Scholar]
- Peumans WJ, Van Damme EJ (1995) Lectins as plant defense proteins. Plant Physiol 109: 347–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramis C, Gomord V, Lerouge P, Faye L (2001) Deglycosylation is necessary but not sufficient for activation of proconcanavalin A. J Exp Bot 52: 911–917 [DOI] [PubMed] [Google Scholar]
- Saleemuddin M, Husain Q (1991) Concanavalin A: a useful ligand for glycoenzyme immobilization–a review. Enzyme Microb Technol 13: 290–295 [DOI] [PubMed] [Google Scholar]
- Schechter I, Berger A (1967) On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun 27: 157–162 [DOI] [PubMed] [Google Scholar]
- Schuck P, Perugini MA, Gonzales NR, Howlett GJ, Schubert D (2002) Size-distribution analysis of proteins by analytical ultracentrifugation: strategies and application to model systems. Biophys J 82: 1096–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senear DF, Teller DC (1981) Thermodynamics of concanavalin A dimer-tetramer self-association: sedimentation equilibrium studies. Biochemistry 20: 3076–3083 [DOI] [PubMed] [Google Scholar]
- Sharon N, Lis H (1990) Legume lectins–a large family of homologous proteins. FASEB J 4: 3198–3208 [DOI] [PubMed] [Google Scholar]
- Sheldon PS, Bowles DJ (1992) The glycoprotein precursor of concanavalin A is converted to an active lectin by deglycosylation. EMBO J 11: 1297–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon PS, Keen JN, Bowles DJ (1996) Post-translational peptide bond formation during concanavalin A processing in vitro. Biochem J 320: 865–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoham M, Kalb AJ, Pecht I (1973) Specificity of metal ion interaction with concanavalin A. Biochemistry 12: 1914–1917 [DOI] [PubMed] [Google Scholar]
- Soares da Costa TP, Patel M, Desbois S, Gupta R, Faou P, Perugini MA (2017) Identification of a dimeric KDG aldolase from Agrobacterium tumefaciens. Proteins 85: 2058–2065 [DOI] [PubMed] [Google Scholar]
- Soares da Costa TP, Yap MY, Perugini MA, Wallace JC, Abell AD, Wilce MC, Polyak SW, Booker GW (2014) Dual roles of F123 in protein homodimerization and inhibitor binding to biotin protein ligase from Staphylococcus aureus. Mol Microbiol 91: 110–120 [DOI] [PubMed] [Google Scholar]
- Soares da Costa TP, Desbois S, Dogovski C, Gorman MA, Ketaren NE, Paxman JJ, Siddiqui T, Zammit LM, Abbott BM, Robins-Browne RM. , et al. (2016). Structural determinants defining the allosteric inhibition of an essential antibiotic target. Structure 24: 1282–1291 [DOI] [PubMed] [Google Scholar]
- Sumner BJ (1919) The globulins of the jack bean, Canavalia ensiformis. J Biol Chem 37: 137–142 [Google Scholar]
- Sumner JB, Howell SF (1936) Identification of hemagglutinin of jack bean with concanavalin A. J Bacteriol 32: 227–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda O, Miura Y, Mitta M, Matsushita H, Kato I, Abe Y, Yokosawa H, Ishii S-I (1994) Isolation and analysis of cDNA encoding a precursor of Canavalia ensiformis asparaginyl endopeptidase (legumain). J Biochem 116: 541–546 [DOI] [PubMed] [Google Scholar]
- Tang TMS, Cardella D, Lander AJ, Li XF, Escudero JS, Tsai YH, Luk LYP (2020) Use of an asparaginyl endopeptidase for chemoenzymatic peptide and protein labeling. Chem Sci 11: 5881–5888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topell S, Hennecke J, Glockshuber R (1999) Circularly permuted variants of the green fluorescent protein. FEBS Lett 457: 283–289 [DOI] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 76: 4350–4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JL, Cunningham BA, Edelman GM (1971) Unusual fragments in the subunit structure of concanavalin A. Proc Natl Acad Sci USA 68: 1130–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A. , et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr D 67: 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R, Wong YH, Nguyen GKT, Tam JP, Lescar J, Wu B (2017) Engineering a catalytically efficient recombinant protein ligase. J Am Chem Soc 139: 5351–5358 [DOI] [PubMed] [Google Scholar]
- Yu Y, Lutz S (2011) Circular permutation: a different way to engineer enzyme structure and function. Trends Biotechnol 29: 18–25 [DOI] [PubMed] [Google Scholar]
- Zand R, Agrawal BB, Goldstein IJ (1971) pH-dependent conformational changes of concanavalin A. Proc Natl Acad Sci USA 68: 2173–2176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zauner FB, Elsässer B, Dall E, Cabrele C, Brandstetter H (2018a) Structural analyses of Arabidopsis thaliana legumain gamma reveal the differential recognition and processing of proteolysis and ligation substrates. J Biol Chem 293: 8934–8946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zauner FB, Dall E, Regl C, Grassi L, Huber CG, Cabrele C, Brandstetter H (2018b) Crystal structure of plant legumain reveals a unique two-chain state with pH-dependent activity regulation. Plant Cell 30: 686–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Cooper DR, Porebski PJ, Shabalin IG, Handing KB, Minor W (2017) CheckMyMetal: a macromolecular metal-binding validation tool. Acta Crystallogr D Struct Biol 73: 223–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.