

Abstract
Human AUTS2 mutations are linked to a syndrome of intellectual disability, autistic features, epilepsy, and other neurological and somatic disorders. Although it is known that this unique gene is highly expressed in developing cerebral cortex, the molecular and developmental functions of AUTS2 protein remain unclear. Using proteomics methods to identify AUTS2 binding partners in neonatal mouse cerebral cortex, we found that AUTS2 associates with multiple proteins that regulate RNA transcription, splicing, localization, and stability. Furthermore, AUTS2-containing protein complexes isolated from cortical tissue bound specific RNA transcripts in RNA immunoprecipitation and sequencing assays. Deletion of all major functional isoforms of AUTS2 (full-length and C-terminal) by conditional excision of exon 15 caused breathing abnormalities and neonatal lethality when Auts2 was inactivated throughout the developing brain. Mice with limited inactivation of Auts2 in cerebral cortex survived but displayed abnormalities of cerebral cortex structure and function, including dentate gyrus hypoplasia with agenesis of hilar mossy neurons, and abnormal spiking activity on EEG. Also, RNA transcripts that normally associate with AUTS2 were dysregulated in mutant mice. Together, these findings indicate that AUTS2 regulates RNA metabolism and is essential for development of cerebral cortex, as well as subcortical breathing centers.
Keywords: autism, epilepsy, hippocampus, respiration, RNA-binding
Development of the cerebral cortex involves cell-type-specific regulation of not only gene transcription, but also subsequent RNA metabolism by proteins such as splicing factors and RNA helicases (Zhang et al. 2016; Lennox et al. 2020). The present study identifies AUTS2 as a novel key regulator of cell-type-specific RNA metabolism that is particularly important for development of the dentate gyrus (DG), a cortical area for learning and memory, as well as the neocortex, and in regulation of respiratory rhythm generation. These findings help explain the human AUTS2 (autism susceptibility candidate 2) genetic syndrome of intellectual disability (ID), variably associated with microcephaly, epilepsy, autism spectrum disorder, and other phenotypes as previously described (Sultana et al. 2002; Beunders et al. 2013, 2016).
Intriguingly, AUTS2 has no close homologs, and AUTS2 protein structure and molecular functions, as well as the role of AUTS2 in neurodevelopment, have remained largely obscure. We previously reported (Bedogni et al. 2010b) that in mice, Auts2 mRNA is highly expressed in developing neocortex (including progenitor zones), hippocampus, thalamus, cerebellum, and some brainstem nuclei. Also, previous experiments (Bedogni et al. 2010b; Gao et al. 2014) have shown that AUTS2 protein localizes in the nuclei of neocortical neurons. One study has additionally described AUTS2 immunoreactivity in neuronal dendrites and axons (Hori et al. 2014). AUTS2 protein consists of 3 main isoforms: a full-length (AUTS2-FL) and 2 C-terminal isoforms. The mRNAs that encode the C-terminal isoforms are produced from transcription start sites (TSS) in exon 7, and exon 9, of 19 exons for AUTS2-FL in humans and mice (Beunders et al. 2013; Hori et al. 2014). The isoform beginning in exon 9, hereafter referred to as AUTS2-CT, appears to contain essential functions of AUTS2 as shown by Beunders et al. (2013, 2016), and the C-terminal isoforms are specifically upregulated in developing neocortex (Hori et al. 2014; Molyneaux et al. 2015). The 2 C-terminal isoforms differ only by a short initial stretch of 98 amino acids, which contains a portion of a Proline rich region and a Histidine–Glutamine repeat tract. Considering the overall similarity of the expressed regions and shared developmental expression pattern, the isoforms likely share the same functions. Additionally, interventions targeting the AUTS2-CT isoform will equally affect the exon 7 isoform, therefore only the AUTS2-CT isoform beginning in Exon 9 will be referred to in the present study.
The domain structure of AUTS2 is unknown. Its closest relatives by amino acid sequence are fibrosin (FBRS), a secreted lymphokine; and fibrosin-like-1 (FBRSL1), a protein implicated in mRNA-binding (Baltz et al. 2012). FBRS and FBRSL1 exhibit ~34–38% identity with amino acids 651–843 of AUTS2-FL (human), corresponding to amino acids 89–281 of AUTS2-CT. Other sequences in AUTS2 exhibit distant homology to tay bridge (Drosophila), mammalian topoisomerase, and Dwarfin-type MH1 domain, as shown in previous work (Sultana et al. 2002; Oksenberg and Ahituv 2013; Oksenberg et al. 2013; Huret 2014; Hori and Hoshino 2017). Additional motifs in AUTS2 include nuclear localization sequences (NLSs), proline-rich regions (PRRs), and histidine-rich regions (HRRs).
Previous studies have suggested that AUTS2 may have nuclear and cytoplasmic functions. In HEK293 cells, experiments demonstrated that AUTS2 interacted with polycomb repressive complex 1 (PRC1), and converted PRC1 from a transcriptional repressor to an activator by recruiting P300, a histone acetyltransferase (Gao et al. 2014). In separate experiments, also using HEK293 cells, Hori et al. (2014) found that AUTS2 interacted with P-REX1, a guanine nucleotide exchange factor, to activate RAC1 and modulate cytoskeletal organization. In yeast 2-hybrid experiments by Monderer-Rothkoff et al. (2021), both AUTS2-FL and -CT interacted with splicing factor SF3B1, while AUTS2-FL also interacted with PRC1. Moreover, AUTS2 has also been localized to chromatin, generally at actively transcribed genes (Gao et al. 2014; Oksenberg et al. 2014).
Neurodevelopmental functions of AUTS2 have been investigated in zebrafish and mice. In knockdown experiments using zebrafish by Beunders et al. (2013) and Oksenberg et al. (2014), auts2 knockdown caused apoptosis and microcephaly. In mice, Auts2 gene targeting experiments by Gao et al. (2014) and Hori et al. (2014, 2015) identified behavioral changes, but no cerebral malformations. Importantly, the previous mouse studies deleted Auts2 exon 7 in experiments by Gao et al. (2014) or exon 8 in experiments by Hori et al. (2014), leaving the exon 9 alternative TSS, and expression of functional AUTS2-CT, intact.
For the present study, we targeted mouse Auts2 exon 15 to disrupt the expression of all major AUTS2 isoforms, as occurs in the “complete” AUTS2 syndrome described in humans (Beunders et al. 2016). Using a combination of methods, we found that AUTS2 primarily regulates RNA-metabolism, and is critical for the neurogenesis of hilar mossy neurons (HMNs) and granule neurons during DG development. In addition, AUTS2 modulates the expression of transcripts involved in specifying cortical laminar and regional neuron identities, as well as cortical excitability, and is essential for the control of breathing rhythmogenesis by CNS neurons.
Materials and Methods
Mice
All animal experiments were approved by the Institutional Animal Care and Use Committees of Seattle Children’s Research Institute, and University of California San Diego, and followed guidelines in the NIH Guide for the Care and Use of Laboratory Animals. B6.129S2-Emx1tm1(cre)Krj/J (Emx1-Cre; Gorski et al. 2002) and Tg(Nes-Cre)1Kln (Nes11-Cre) mice were obtained from The Jackson Laboratory (Emx1-Cre JAX:005628; Nes11-Cre, JAX:003771). All mice were maintained on a C57BL6/J background. Animal ages were studied as indicated in respective figures; primary timepoints consisted of P0 (day of birth), P5, P7, and P52. Ages are given in postnatal days (days post birth). Animal subject sexes on P0.5 were not determined due to the difficulty of determining neonatal mouse gender; male and female animals were assessed at the remaining timepoints.
The Auts2 exon 15 floxed allele was generated by homologous recombination in embryonic stem (ES) cells, in collaboration with GenOway S.A. For this purpose, a targeting vector containing regions homologous to the genomic Auts2 sequences in C57BL/6 genetic background was constructed. The general features were: a long homology region of 6.2 kb, a short homology region of 1.6 kb, insertion of 2 loxP sites flanking exon 15, a positive selection neo gene flanked by FRT sites (removed in vivo with deleter mice), and a diphtheria toxin A negative selection marker to reduce the isolation of nonhomologous recombined ES cell clones. The homologous recombination event was detected by Southern blotting. PCR and Southern blot screening were used to detect and distinguish the recombined, Flp-excised, and Cre-excised alleles. The PCR primers used to distinguish wild-type, floxed, and Cre-excised alleles are given below in the section on PCR. The complete protocol of mouse generation, including results at each step is available upon request.
Bioinformatic Analyses of AUTS2 Structure and Function
Protein molecular weight, amino acid composition, and theoretical isoelectric point (pI) were calculated using ExPASy ProtParam tool (https://web.expasy.org/protparam/). NLSs were identified using cNLS Mapper (Kosugi et al. 2009). Domains were predicted using NCBI CD-Search (Marchler-Bauer et al. 2011; https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi); and using ProDom (Bru 2004; http://prodom.prabi.fr/prodom/current/html/home.php). To predict molecular function annotation of the identified domains, amino acid sequences were submitted to UniGOPred (Rifaioglu et al. 2018). UniGOPred is a machine learning methodology based on data from the UniProt knowledgebase which utilizes multiple modeling prediction to assign GO Molecular Function terms based on primary amino acid sequence and homology modeling.
Nuclear Complex Immunoprecipitation
C57Bl/6 mice were harvested at P0.5. The cortex was dissected in cold saline, weighed, and flash frozen to prevent degradation. Nuclei were prepared from 1.0-g pooled cortex and lysed for protein content in hypotonic buffer. Co-IP incubations were performed in accordance with the ActiveMotif Nuclear Complex Co-IP kit (Product Number 54001) for tissue using low stringency wash buffer to maximize candidate interactions and prevent loss of lightly bound complex members. AUTS2 was immunoprecipitated from pooled P0.5 cortices using rabbit polyclonal anti-AUTS2 HPA000390 (see Antibodies). Samples were run against negative control (blank) Protein A beads incubated with identical neuronal lysate. Whole eluted Co-IP reactions were trypsin digested and desalted for LTQ/mass-spectroscopy as described (Sharma et al. 2017). Additional Co-IP reactions were run on a denaturing acrylamide gel. The gel was Coomassie stained, bands were excised and trypsin-digested, and eluted for mass spectroscopy. Gel extraction was performed as previously described (Shevchenko et al. 1996; Sharma et al. 2017). Detection was performed on a ThermoScientific Linear Orbitrap Q Exactive mass spectrometer (LTQ-MS). Mass spectroscopy fragment database searches were performed by University of Washington core facilities. Total independent spectra for each protein were normalized to the length of the protein (S/L), and these enrichment values were used to determine the top 25% of proteins enriched in both replicates. This list was subsequently submitted for String and GO analysis. The top 20 enriched proteins (Fig. 2a) were ordered by S/L in replicate 1, excluding any proteins with very low detection (S/L < 0.1) in replicate 2.
Figure 2 .
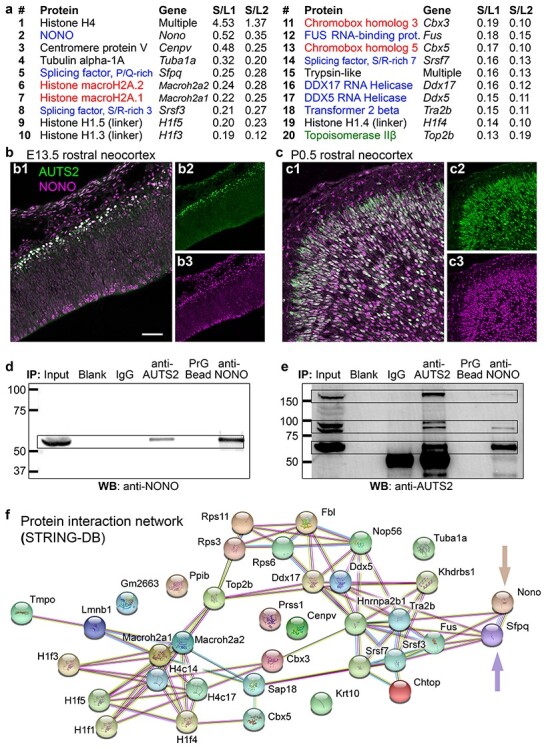
AUTS2-IPs and protein interaction network. (a) The top 20 AUTS2-IPs (ranked by S/L; see also Materials and Methods). RBPs in blue text; chromatin repressive proteins in red text; chromatin activating proteins in green text. (b, c) Double IF for AUTS2 (green) and NONO (magenta) in E13.5 (b) and P0.5 (c) neocortex. Sagittal sections. (d) IP-western blot (WB) of AUTS2 and NONO IPs, probed for NONO. (e) IP-WB of AUTS2 and NONO IPs, probed for AUTS2. (f) Interaction network of AUTS2-IPs as found by STRING-DB. One subnetwork (upper right) consisted of RBPs, including NONO and SFPQ (arrows); the other subnetwork (lower left) consisted of chromatin proteins. Scale bars: b1, 50 μm for b1, c1; 100 μm for b2, b3, c2, c3.
Western blotting for LTQ/MS validation was performed using 5 μg of either rabbit polyclonal anti-AUTS2 C-terminal, or goat polyclonal anti-NONO (see Antibodies), and 500-μg whole nuclear lysate. The nuclear lysate was prepared through the ActiveMotif nuclear isolation protocol from 3 pooled cortices per replicate. Co-IP was performed using included alternative ActiveMotif high stringency wash buffer and immunoprecipitated using Protein G Beads, as goat primary antibodies (such as the anti-NONO antibody used) are not selected with high fidelity by Protein A. Western blots were run on BioRad Mini-Protean TGX 4–20% gradient gels, double probed with Donkey Anti-Rabbit AlexaFluor 488 and donkey anti-goat AlexaFluor 647 diluted 1:5000 in blocking solution and imaged using the FluorChem R system (ProteinSimple).
Tissue Preparation for RNA-Seq, ISH, IF, and Western Blotting
For RNA-seq and western blotting, P0.5 neonates from Auts2 cKOctx litters were rapidly decapitated into cold saline. Frontal cortex (from eye socket forward) was dissected, meninges and olfactory tract were removed, and the frontal cortex (selected because of its high AUTS2 content) was flash frozen on dry ice. Tissue was stored at −80 °C prior to RNA or protein isolation.
For immunofluorescence (IF) or in-situ hybridization (ISH), pups were anesthetized prior to perfusion: neonates were cold-anesthetized, and postnatal mice were anesthetized with isoflurane. Animals were perfused with 4% paraformaldehyde in phosphate-buffered saline (PBS), pH 7.4. Brains were post-fixed by immersion in cold (4 °C) buffered 4% paraformaldehyde for 4–16 h. Brains were cryoprotected in progressive 10%, 20%, and 30% sucrose in 0.1-M sodium phosphate (pH 7.0), frozen in optimum cutting temperature (OCT) compound (Sakura Finetek), cryosectioned at 12 μm, and mounted on Superfrost Plus slides (Thermo Fisher Scientific). Slides were stored at −80 °C until needed.
Immunofluorescence
Methods for IF were as described by Englund et al. (2005). Briefly, antigen retrieval was performed by boiling in 0.01-M sodium citrate, pH 6.0. Sections were blocked for 30 min at room temperature in 10% serum (goat or donkey depending on primary antibody origin species), 3% BSA, and 0.1% Triton X-100 in PBS. Primary antibodies were diluted in blocking solution and incubated overnight at 4 °C. Sections were washed 3 × 5 min in PBS before secondary antibody application. AlexaFluor conjugated secondary antibodies were diluted 1:400 in blocking solution and incubated for 2 h at room temperature. Tissue was again washed 3× in PBS, incubated briefly in DAPI and mounted with SouthernBiotech Fluoromount-G and allowed to cure before imaging on a Zeiss LSM710 confocal microscope. Digital images were adjusted for brightness and contrast with Photoshop (Adobe).
Antibodies
The following antibodies were used: “AUTS2-106” rabbit polyclonal anti-AUTS2 (Sigma-Aldrich, HPA000390); “AUTS2-C17” rabbit polyclonal anti-AUTS2 (Hevner lab, PAS#19228; validated by IF expression pattern and absence of immunoreactivity in Auts2 cKO mice); “AUTS2-N17” rabbit polyclonal anti-AUTS2 (Hevner lab, PAS#19226; validated by IF expression pattern and absence of immunoreactivity in Auts2 cKO mice); goat polyclonal anti-NONO (Thermo Fisher, PA5-18514); rat monoclonal anti-CTIP2 (Abcam, AB18465); rabbit polyclonal anti-5HTT 602-622 (Millipore Sigma, PC177L); rat monoclonal anti-TBR2 (eBioscience, 14-4875-82); rabbit polyclonal anti-SOX9 (Millipore Sigma, ABE2868); rabbit polyclonal anti-calretinin (Swant, 7697); mouse monoclonal anti-calretinin (Millipore, MAB1568); mouse monoclonal anti-SATB1/2 (Abcam, AB51502); and rabbit anti-GFAP (Dako; 1:1000).
Cell Counting
Cell counting methodology was adapted from previous work (Hevner et al. 2004). Briefly, frozen microtome brain slices were prepared per the IF protocol. From each of 3 brains from control and Auts2 cKOctx neonates, single images of somatosensory cortex were taken using the 20× objective (Zeiss AxioZ1 Imager), and outlined in Photoshop to create a count box measuring 345-μm wide. Pial and layer 6 boundaries were traced and the section was gridded into 10 equal bins between them. Immunopositive cells were marked and cells were counted in each bin (Adobe Photoshop). Cell numbers were entered into Excel (Microsoft) and total counts were graphed, then compared statistically between control and mutant neocortex.
In-Situ Hybridization
Penk cDNA (Accession: BC049766) was subcloned into pBluescriptSK(+) (Stratagene). Digoxigenin-labeled riboprobes were prepared with Roche kit # 11175025910 as previously described (Elsen et al. 2013). The detailed hybridization protocol is available on OpenWetWare: https://openwetware.org/wiki/In_Situ_Hybridization_on_Cryosectioned_Tissue.
RNA Isolation and Sequencing (RNA-Seq)
RNA was prepared from individual dissected frontal cortices or hippocampi using the Qiagen RNAEasy Plus Universal Mini kit (Qiagen product number 73404), according to the manufacturer’s instructions with the exception that all reagent amounts were scaled to 1/3 of the indicated volume to account for small sample sizes. Samples were resuspended to 3500 ng RNA in 50-μL water (70 ng/μL) and submitted to the Northwest Genomics Core (NWGC) at the University of Washington Department of Genome Sciences for quality control, poly-A selection, Illumina TruSeq library preparation, and sequencing. For sequencing, 75-bp paired-end reads were obtained on an Illumina HiSeq 4000 from RNA samples isolated from P0.5 control and Auts2 cKOctx frontal cortex or hippocampus (n = 4 per group). Samples were sequenced to a depth of 30 million reads. Sequences were mapped to the reference genome by the NWGC using STAR (Dobin et al. 2013).
RNA Immunoprecipitation and Sequencing (RIP-seq)
Magna RIP RBP Immunoprecipitation Kit from Millipore Sigma (product number 17-700) was used for RNA-immunoprecipitation. Whole cortex was dissected from P0.5 C75BL/6J pups. Two cortices were pooled for each biological replicate. Two replicates (4 cortices total) were lysed for RNA immunoprecipitation according to the manufacturer’s instructions. After lysate preparation, RNA immunoprecipitation was performed according to the Millipore protocol. RNA samples were submitted to the NWGC for sequencing. Sequence libraries were prepared using the Clontech SMARTer Stranded Total RNA Sample Prep for RIP-Sequencing library preparation, performed as described (Zhang et al. 2008). Clontech SMARTer stranded libraries were run on an Illumina NextSeq. Reads were mapped to the mm10 mouse genome by the NWGC using STAR (Dobin et al. 2013).
Transcript Quantification and Differential Expression Analysis
Reference genome and transcriptome for mouse were obtained from the Gencode M14 annotation. Cuffdiff (Trapnell et al. 2013; Galaxy Version 2.2.1.3) was run to calculate transcript level differential expression, with the following deviations from default parameters: For RNA-seq: –library-type fr-firststrand -dispersion-method per-condition –no-effective-length-correction. For RIP-seq: sample enrichment was calculated in Cuffdiff with the following flags: –library-type fr-secondstrand -dispersion-method pooled –no-effective-length-correction.
As RIP-Seq does not fragment RNAs, we used CuffDiff 2.1 (Trapnell et al. 2013) to quantify enrichment at the whole transcript level, rather than peak calling methods which are more appropriate for defined regions of sharp sequence enrichment. RIP-sequencing analyses were run separately for each control type (IP vs., IgG in one run, IP vs., Input in a second run). Transcript differential expression testing files were joined, and only samples with a FDR q < 0.05 in both comparisons were considered significant hits. A blacklist file containing Gencode m14 annotations for rRNA, tRNA, tRNA pseudogenes, mitochondrial genes, and genes mapping to chromosome Y was also passed to the program for all analyses.
RIP-seq and RNA-seq datasets presented in this paper have been deposited in NCBI gene expression omnibus (GEO) under Accession Number GSE112792. These datasets were used in generating all or part of Figures 2 and 3; Supplementary Figure 5; Table 1; and see Supplementary Tables 1–9.
Figure 3 .
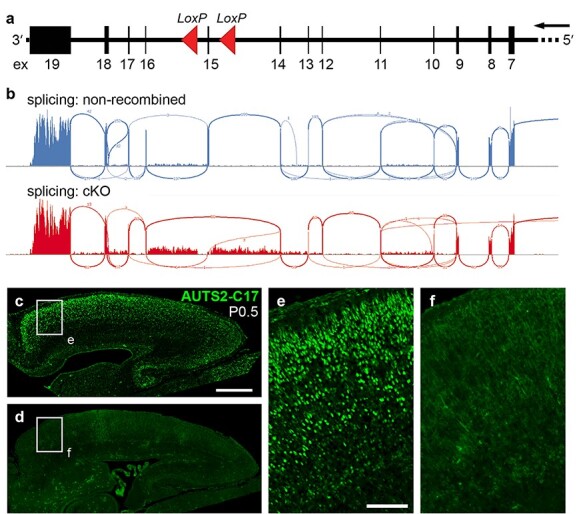
Auts2 exon 15 floxed allele and inactivation in Auts2 cKOctx cerebral cortex. (a) The Auts2 gene has 19 exons (numbered boxes). In the Auts2 floxed allele, LoxP sites (red arrowheads) flanked exon 15. Exons 1–6 are far upstream (not shown). Transcription is right to left, matching mouse genome build mm10. (b) “Sashimi” splicing plots of Auts2 mRNA in control (top) and Auts2 cKOctx (bottom) cerebral cortex. (c–f) IF to detect AUTS2-C17 in control (c, e) and Auts2 cKOctx (d, f) cortex. Boxed areas in (c) and (d) are shown at higher magnification in (e) and (f), respectively. Scale bars: c, 500 μm for c, d; and e, 100 μm for e, f.
Figure 5 .
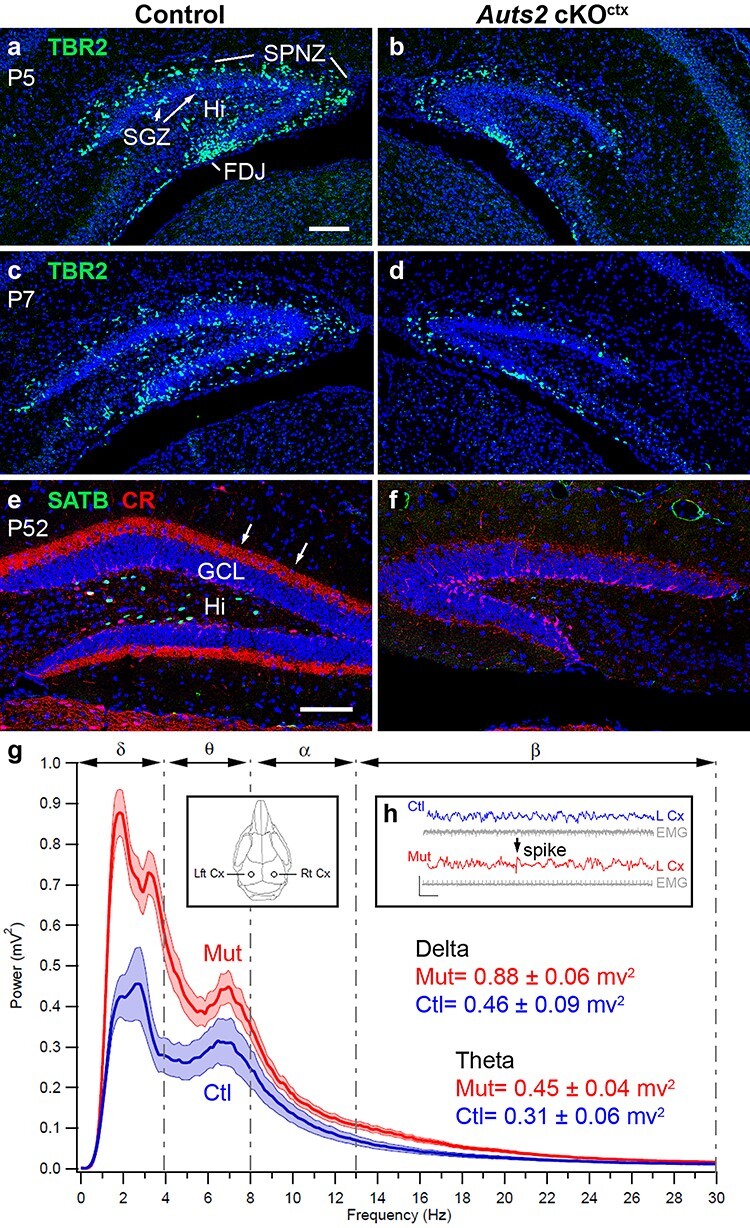
Cellular and functional defects in Auts2 cKOctx DG. (a–d) IF for TBR2 (green) in control (a, c) and mutant (b, d) DG at corresponding levels in P5 (a, b) and P7 (c, d) DG. (e, f) Double IF for SATB1/2 (green) and calretinin (red) in control (e) and mutant (f) DG at corresponding levels in adult (P52) DG. Arrows in (e) indicate calretinin+ HMN axons in the DG inner molecular layer. (g, h) EEG data from implanted electrodes in adult control (blue) and mutant (red) hippocampus. Inset in (g) shows the positions of electrodes. The mutant hippocampus displayed higher EEG power (g) at multiple frequency ranges, including delta (δ) and theta (θ). EEG traces showed abnormal spike discharges in the mutant hippocampus (arrow, h). Histology: coronal sections with DAPI (blue) counterstain. Scale bars: a, 100 μm for a–d; e, 100 μm for e, f.
Table 1.
| Gene | log2FC | Gene | log2FC |
|---|---|---|---|
| H1f2 (Hist1h1c) | −1.064 | Nav3 | −0.344 |
| Fn1 | −0.673 | Crtc3 | −0.336 |
| Per2 c | −0.649 | Rasgrf2 | −0.333 |
| Nkd2 | −0.617 | Tab1 | −0.332 |
| Myo10 | −0.523 | Ezr | −0.316 |
| Thoc2l (BC005561) | −0.516 | Nup160 | −0.313 |
| Plxna2 | −0.514 | Btaf1 | −0.309 |
| Fbn1 | −0.493 | Cramp1l | −0.309 |
| Abca1 | −0.492 | Jag2 | −0.305 |
| Prex1 | −0.484 | Kmt2d | −0.301 |
| Wscd1 | −0.480 | Zfp592 | −0.289 |
| 2900026A02Rik | −0.465 | Ep300 | −0.283 |
| Pcnt | −0.464 | Ino80 | −0.274 |
| Sdk2 | −0.457 | Ubn1 | −0.274 |
| Notch2 | −0.438 | Rsrp1 | +0.297 |
| Dmxl1 | −0.430 | Cacna2d3 | +0.299 |
| Dido1 | −0.427 | Jup | +0.302 |
| Cenpe | −0.402 | Lars | +0.323 |
| Celsr1 | −0.395 | Cachd1 | +0.332 |
| Bod1l | −0.391 | Frem2 | +0.340 |
| Taf3 | −0.389 | Snhg11 | +0.397 |
| Mga | −0.377 | Zbtb40 | +0.400 |
| Trip11 | −0.375 | Pcp4 | +0.426 |
| Tjp2 | −0.371 | Ldlr | +0.457 |
| Adamts3 | −0.366 | Kcnh4 | +0.513 |
| Usp36 | −0.355 | Snhg14 | +0.638 |
| Mki67 | −0.352 | Myo7a | +0.700 |
| Dennd4a | −0.351 | Penk | +1.430 |
Notes: aRIP-seq; see Supplementary Table 4R–AI, q < 0.05 and log2FC > 0.
bRNA-seq; see Supplementary Table 4J–Q, P < 0.01. Bold: q < 0.05, see Supplementary Table 4J–Q.
cChIP-seq peak within 5 kb of gene7.
Gene Ontology analysis
GeneSCF-v1.1-p2 (Subhash and Kanduri 2016) was used for GO analysis, the background parameter (–bg) was set using the total number of transcripts (for frontal cortex) or genes (for hippocampus) that passed Cuffdiff significance testing thresholds (status = OK).
Gene Set Enrichment Analysis
For analysis of hippocampal neuronal populations, A GMX formatted GeneSets database was constructed from the HippoSeq repository (Cembrowski et al. 2016). Gene sets consisted of expression lists filtered for genes which were enriched 2.5-fold in one neuronal population versus all other neuronal populations in the database. This resulted in the construction of 16 gene lists each uniquely identifying a neuronal population. A gene list filter threshold (Min Size: Exclude Smaller Sets) of 10 was set which excluded 2 of these datasets (which marked CA3 neurons) from the analysis. FPKM tables were extracted from the CuffDiff cummeRbund database produced from differential gene expression analysis of the Hippocampal RNA-sequencing study. Count tables were filtered to select only genes which passed the CuffDiff thresholds for determining expressed genes for statistical testing. This resulted in the selection of 14 900 expressed genes for gene set enrichment analysis (GSEA) for hippocampus. GSEA (Subramanian et al. 2005) was run in gene_set permutation type mode with the “weighted” enrichment statistic, the “Signal2Noise” ranking metric, and the “meandiv” normalization mode (default parameters).
Video-EEG–EMG Recording
One-month-old Auts2 cKOctx mice (n = 8) and controls (n = 7) underwent survival surgery to implant EEG and EMG electrodes. The mice were anesthetized with 2–3% isoflurane gas and given bupivacaine analgesia (0.1 mg/kg, s.c.). Using aseptic technique, a midline incision was made anterior to posterior to expose the cranium. Fine silver wires were placed through small cranial holes created with a fine cutting needle and fixed in place with cyanoacrylate glue. Bilateral EEG electrodes were placed at visually identified locations—2-mm posterior, 2-mm lateral from Bregma. EMG electrodes were placed in back muscles. A reference electrode was placed at the midline cerebellum and a ground electrode was placed subcutaneously over the back. Electrode impedances were typically < 25 kΩ. After electrode placement, the skin was closed with sutures and the mice were allowed to recover for at least 3 days to allow recovery from surgery and anesthetic effects. Simultaneous video-EEG–EMG records were collected in conscious mice on a PowerLab 8/35 data acquisition unit using LabChart 8.1 software (AD Instruments). All bio-electrical signals were acquired at 1-KHz sampling rate. The EEG signals were processed off-line with a 1–80-Hz bandpass filter and the EMG signals with a 3-Hz high pass filter. Video-EEG–EMG data were analyzed using LabChart software. The analysis was conducted on 1-h segments of the EEG when the animal was immobile. To control for circadian influence on the EEG all analyses were conducted on data collected between 1 and 3 pm.
Plethysmography
Whole-body plethysmography was performed on P0 pups. For studies of Nes11-Cre Auts2 cKO pups: n = 7 controls, n = 7 heterozygous, and n = 3 homozygous mutants. For studies of Emx1-Cre Auts2 cKOctx pups: n = 3 control, n = 3 heterozygous, and n = 3 homozygous mutants. Animals were placed in a custom-built barometric chamber and equilibrated to the chamber with open air flow. The chamber was sealed for a 60-s recording session, and the pressure difference was measured between the experimental and reference chamber with a differential pressure transducer. Only recording periods in which the animals were immobile were analyzed. Data were collected and analyzed using pCLAMP 10.5 software (Molecular Devices). All analyses were performed blinded to genotype.
ChIP-Seq Data Reprocessing
AUTS2 and control ChIP data (Gao et al. 2014) were downloaded from GEO (accession number GSE60411) and imported into Galaxy. Fastqsanger files were mapped to mm10 using Bowtie2 (Langmead et al. 2009; Galaxy Version 2.2.6.2) with the—very-sensitive-local presets (-D 20 -R 3 -N 0 -L 20 -i S,1,0.50).
Mapped reads were processed in accordance with the methods described in the Galaxy ChIP-seq tutorial for peak calling with MACS2 (Zhang et al. 2008), using Galaxy Version 2.1.1.20160309.0. Mapped reads were quality filtered for a MAPQ quality score > 20 using SAMTools (Li et al. 2009a). Additionally, reads overlapping regions in the ENCODE artifact blacklist (Amemiya et al. 2019), marked with the PCR duplicate or QCfail flags were removed with the BAMfilter tool. MFOLD parameters were set at 5, 100. Bandwidth was set using fragment size from size-selection where available (350). Extension size parameter for peak calling was obtained by running the MACS2 “predictd” component.
Binding and Expression Target Analysis
BETA Minus was used with the Gencode M14 transcriptome to identify targeted TSSs within 5 kb of AUTS2 ChIP peaks, in accordance with the identification of ±5kB of a TSS as the region of AUTS2 genomic binding by Gao et al. (2014). BETA 1.0.7 (Wang et al. 2013) binaries were retrieved from Cistrome.org and were configured on a local workstation. MACS2 peaks (bed) files were generated as described above. The inbuilt gene/transcript reference database corresponding to UCSC mm10 was replaced with an updated transcriptome file generated from the Gencode M14 release.
PCR Analysis of Auts2 Alleles and Exon Excision
For analysis of Auts2 genotypes, the primer trios 119467cre-HEV1, 119470cre-HEV1, and 119468cre-HEV1 were designed; these primers hybridize close to the loxP sites, upstream (119467cre-HEV1 and 119468cre-HEV1) and downstream (119470cre-HEV1) of the targeted exon 15. Using this primer combination, the wild-type (non-floxed) allele generated PCR products of 1440 and 148 bp; the recombined (floxed) allele generated products of 3180 (not detected under standard conditions) and 226 bp; and the Cre-excised allele generated a product of 457 bp. The PCR primer sequences (5′–3′) are:
119467cre-HEV1: TTGGCTGTCTGCTTCAAGTTGGACC
119470cre-HEV1: CCAATAGGACCTGTGCTGGCTGGT
119468cre-HEV1: TCAACGCTGAGAACACTGTGAGAATGAAG
The optimized protocol for PCR using these primers is available upon request.
Quantification and Statistical Analysis
For qualitative experiments representative images selected from 3 to 5 independent mice are shown. For both qualitative and quantitative analyses Auts2 mutant nice were analyzed alongside littermate controls. Transcripts from RNA-sequencing, and cognate genes or proteins, were selected for additional analysis using significance thresholds of P ≤ 0.01 or FDR adjusted q ≤ 0.05 where indicated. For geneSCF (Subhash and Kanduri 2016) GO analysis, a pathway was considered significant if it had a Benjamini–Hochberg FDR ≤ 0.05. For plethysmographic and EEG analysis at least 2 cohorts of animals were examined, and datasets were pooled. No outliers were excluded. Data collection and analysis were performed blinded to genotype. Data presentation for plethysmographic analysis was performed in GraphPad Prism. Graphs are presented as mean ± standard error of the mean (SEM). One-way analysis of variance was used for statistical analysis. All P < 0.05 were considered significant. Annotations for additional stringency of P values are indicated in figure legends. Video-EEG–EMG data were analyzed using LabChart software. Graphs are presented as mean ± SEM.
Results
AUTS2 Sequence Analysis Predicts Domains Linked to RNA-Binding and Processing
AUTS2-FL and AUTS2-CT display typical biochemical properties of nuclear proteins, which were previously described by Schwartz et al. (2001), including enrichment in Ser and Pro, and pI > 9.25. Both AUTS2 isoforms also exhibit the presence of at least one NLS (Fig. 1a). However, no experimental or theoretical data on AUTS2 domain structure are available. We used the bioinformatics tools Conserved Domain Search (CD-Search; Marchler-Bauer et al. 2011) and ProDom (Bru 2004), to scan for putative domains in human (NP_056385) and mouse (NP_001350409) AUTS2 sequences, and UniGOPred (Rifaioglu et al. 2018), to predict domain functions.
Figure 1 .
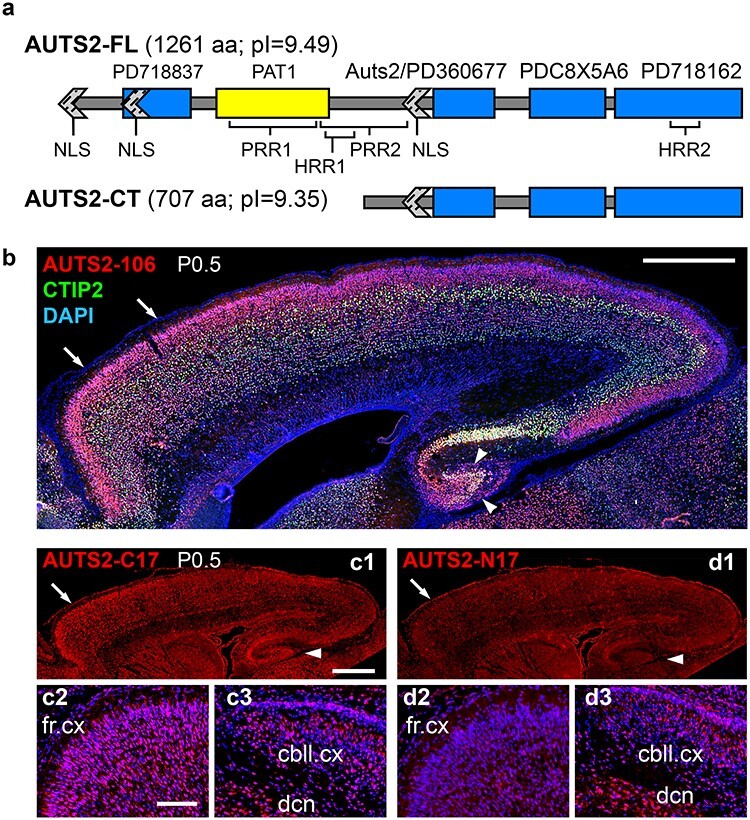
Mouse AUTS2 isoforms and expression in neonatal brain. (a) Putative domains and motifs in AUTS2-FL and AUTS2-CT. (b) Double IF for AUTS2-C (red) and CTIP2 (green), a marker of layer 5 and subplate. The AUTS2-106 antibody detects a 106-aa sequence present in AUTS2-FL and AUTS2-CT. Note high expression of AUTS2 in frontal cortex (arrows) and DG (arrowheads). (c1–c3) IF for AUTS2-C17 (C-terminal 17 aa), which recognizes AUTS2-FL and AUTS2-CT. (c1) Overview of cortex, showing high expression in frontal cortex (arrow) and DG (arrowhead). (c2) Frontal cortex at higher magnification. (c3) Cerebellum. (d1–d3) IF for AUTS2-N17 (N-terminal 17 aa), which recognizes only AUTS2-FL. (d1) Overview of cortex, showing low expression. (d2) Frontal cortex. (d3) Cerebellum. AUTS2-FL (specifically detected by AUTS2-N17 antibodies) was highly expressed in cerebellum, but not cerebral cortex. All sections sagittal (anterior left, dorsal up), P0.5. Abbreviations: fr.cx, frontal cortex; cbll.cx, cerebellar cortex; and dcn, deep cerebellar nuclei. Scale bars: b, 0.5 mm; c1, 0.5 mm for c1, d1; and c2, 50 μm for c2, c3, d2, d3.
CD-Search of mouse AUTS2 predicted 2 domains with high significance (P < 10−7): an “AUTS2 domain” (aa 644–849), present in both isoforms; and a PAT1 superfamily domain (aa 297–490), present in AUTS2-FL only (Fig. 1a). The AUTS2 domain represents sequence homology among AUTS2, FBRS, and FBRSL1. Interestingly, CD-Search of human AUTS2 also identified the AUTS2 domain, but not the PAT1 domain; the same region was instead identified as a putative viral protein domain. Since PAT1 is a eukaryotic protein, this homology is more likely relevant. PAT1 is an RNA-binding protein (RBP) with 2 mammalian homologs, PATL1 and PATL2, which perform multiple functions, including targeted mRNA degradation (Pilkington and Parker 2008). ProDom predicted 4 domains (E < 10−50). Among these, PD360677 largely overlapped the AUTS2 domain, while the other domains (PD718837, PDC8X5A6, and PD718162) did not overlap domains identified by CD-Search. Together, these algorithms predicted that AUTS2-FL has 5 nonoverlapping domains, 3 of which are present in AUTS2-CT (Fig. 1a).
To evaluate potential functions of the predicted domains, their sequences were analyzed using UniGOPred. Remarkably, RNA and/or DNA binding were high-scoring functions (prediction score ≥ 0.9) for 4 of the 5 predicted domains: PD718837, PAT1, AUTS2/PD360677, and PD718162 (see Supplementary Table 1). (The fifth predicted domain, PDC8X5A6, did not score highly for any functions.) Other high-scoring functions included cytoskeletal protein binding (PD718837), receptor binding (PAT1), and RNA polymerase II binding (PD718162). These results suggest that AUTS2 forms part of the machinery for RNA transcription and processing, and might potentially be an unconventional RBP that lacks canonical RNA-binding domains (Hentze et al. 2018).
Interestingly, the PAT1 and PD718837 domains, present only in AUTS2-FL, were linked to potential extranuclear functions (mRNA degradation and cytoskeletal protein binding, respectively). These findings may correlate with the previous report by Hori et al. (2014) that AUTS2-FL (but not AUTS2-CT) was detected in neuronal cytoplasm and exhibited functions related to cytoskeletal regulation.
AUTS2-CT is Highly Expressed in Neonatal Mouse Cortex
Previous immunoblot and RNA-sequencing (RNA-seq) assays (Hori et al. 2014; Molyneaux et al. 2015) suggested that AUTS2-CT is highly expressed in developing cerebral cortex. To further compare the expression of AUTS2-FL and -CT isoforms, we conducted IF experiments with antibodies that recognize different epitopes. Antibodies against 106-aa sequence in the C-terminal half of AUTS2 (“AUTS2-106” antibodies) demonstrated robust immunoreactivity in neonatal cerebral cortex, with highest AUTS2 levels in frontal cortex and hippocampus, especially DG (arrowheads, Fig. 1b). Since AUTS2-106 antibodies recognize both isoforms, this IF revealed total AUTS2 expression.
To semiquantitatively compare AUTS2-FL and AUTS2-CT expression, we generated antibodies against equal length (17-aa) N- and C-terminal AUTS2 peptides. The AUTS2-C17 epitope, which recognizes both isoforms, was highly expressed in cerebral cortex and cerebellum (Fig. 1c). In contrast, AUTS2-N17, while highly expressed in cerebellum, was detected at only very low levels in cerebral cortex (Fig. 1d). These results confirmed that in neonatal mouse cortex, AUTS2-CT is the predominant isoform.
AUTS2 Interacts with RBPs
To identify proteins that interact with AUTS2 in developing brain in vivo, cell nuclei were isolated from replicate lysates of neonatal mouse cerebral cortex and used for AUTS2 immunoprecipitation (using AUTS2-106 antibodies) followed by mass spectrometry (IP–MS). Proteins detected in both replicates (n = 231) were designated AUTS2-interacting proteins (AUTS2-IPs; see Supplementary Table 2). The top 20 AUTS2-IPs (ranked by spectra/length) consisted of 8 RBPs, 5 chromatin repressors, 1 chromatin activator, 4 conventional histones, 1 centromere protein, 1 microtubule protein, and 1 protease (Fig. 2a). The 8 RBPs were: NONO and SFPQ, which are DBHS family scaffolds previously described (Knott et al. 2016) as involved in RNA processing; splicing factors SRSF3, SRSF7, and TRA2β; RNA helicases DDX5 and DDX17; and FUS, a TET/FET family RNA- and DNA-binding protein.
To validate the IP–MS results, we further studied the interaction between AUTS2 and NONO. NONO was selected for further analysis as it was the highest enriched nonhistone protein, and second overall enriched candidate interactor of AUTS2 (Fig. 2a). First, we tested for colocalization of AUTS2 and NONO in developing neocortex by IF, and found that they were indeed colocalized in the nuclei of embryonic and neonatal cortical plate neurons (Fig. 2b, c). Next, we confirmed that each protein was co-immunoprecipitated by the other (Fig. 2d, e). These findings supported the conclusion that AUTS2 and NONO interact in developing cortex.
To determine if AUTS2-IPs are members of a known network of interacting proteins, the top 25% AUTS2-IPs were analyzed using STRING-DB (Szklarczyk et al. 2015). A significant network was identified, comprised of 2 subnetworks associated with chromatin regulation and RNA processing (Fig. 2f). The network of AUTS2-IPs was enriched (P < 10−4) for the gene ontology (GO) terms poly(A) RNA-binding, nucleic acid binding, and chromatin DNA binding (see Supplementary Table 3), as well as the KEGG spliceosome pathway (see Supplementary Table 3). Thus, the functions of AUTS2-IPs aligned with those predicted for the putative domains of AUTS2 identified computationally (Fig. 1a, see Supplementary Table 1). Together, the bioinformatics and protein interaction data indicated that AUTS2 is involved in RNA metabolism in addition to chromatin activity.
AUTS2 Complexes Bind RNA
To test the hypothesis that AUTS2 binds RNA (directly or indirectly through AUTS2-IPs), we used AUTS2-106 antibodies for RNA immunoprecipitation and sequencing (RIP-seq) of neonatal cortex. This method identified 1208 transcripts enriched in AUTS2-RIP relative to IgG and input control samples (q < 0.05 and log2FC > 0; see Supplementary Table 4R–AI). GO analysis using geneSCF (Subhash and Kanduri 2016) identified 27 Molecular Functions and 6 Biological Processes that were significantly associated with the transcripts identified by AUTS2-RIP (see Supplementary Table 5). The top 3 Biological Processes were mRNA processing, regulation of GTPase activity, and covalent chromatin modification. The top 3 Molecular Functions were microtubule binding, ATP binding, and helicase activity. (For comparison, highly expressed transcripts from the input control were most enriched for biogenesis, folding, and transport of proteins.) These data indicated that AUTS2 complexes bind transcripts that influence diverse neurodevelopmental processes, including RNA metabolism and chromatin regulation.
Conditional Excision of Auts2 Exon 15 Depletes Both AUTS2 Isoforms
To investigate how depletion of both AUTS2 isoforms impacts mouse brain development, we produced a novel conditional (floxed) Auts2 allele. Mouse, like human, Auts2 has 19 exons and 2 major TSSs. (Mouse genome build mm10 mistakenly shows 20 exons, due to a tiny intron erroneously mapped within exon 7.) The TSS upstream of exon 1 produces mRNA encoding AUTS2-FL; the second TSS, annotated in exon 9 (Beunders et al. 2013; Hori et al. 2014), produces mRNA for AUTS2-CT. Using homologous recombination in embryonic stem cells, we inserted loxP sites flanking exon 15 (Fig. 3a) to enable deletion of mRNAs containing the AUTS2-CT as well as AUTS2-FL sequence.
Homozygous cKO of Auts2 exon 15 throughout the CNS (Nes11-Cre) caused neonatal lethality, associated with severe breathing abnormalities (Supplementary Fig. 1a–f). Heterozygous and homozygous mutants showed strikingly erratic breathing, which was reflected in a significantly increased frequency irregularity (Supplementary Fig. 1b, c, f). They also exhibited large gasp- and sigh-like breaths, reflected in an overall increase in tidal volume as compared with controls (Supplementary Fig. 1b, c, e). Lethality was averted by cortex-specific Auts2 inactivation, restricted to cortical projection neurons (Emx1-Cre). These mice had much milder breathing problems at birth (Supplementary Fig. 1g–i). Because they did not generate the large amplitude gasp-like breaths, the tidal volume was not significantly different from controls. Importantly, these mice survived to adulthood and were fertile. Although AUTS2 disruptions are heterozygous in human AUTS2 syndrome, homozygous mice offer more robust phenotypes, especially when cKO is restricted to specific neuron types. Accordingly, all further studies were conducted using homozygous cortex-specific Auts2 cKO (Auts2 cKOctx) mice, with non-excised mice as controls.
RNA-seq of Auts2 cKOctx neonatal cortex confirmed that, as expected, Auts2 transcripts were aberrantly spliced from exon 14 to exon 16 or 17 (Fig. 3b). Both anomalous splice events were predicted to cause frameshift with premature stop codons and nonsense-mediated decay. Indeed, Auts2 mRNA levels were greatly reduced in mutant cortex, as determined by qPCR (log2FC = −2.59) and RNA-seq (log2FC = −1.69) of neonatal cortex. Absence of AUTS2-FL and AUTS2-CT proteins was confirmed by IF (Fig. 3c–f).
DG Hypoplasia in Auts2 cKOctx Mice
The brains of Auts2 cKOctx mice appeared externally normal at all ages. On histology, neocortex showed no overt malformation, but the DG evinced progressive hypoplasia, first apparent by standard histology (DAPI) on postnatal day (P) 5 (Supplementary Fig. 2). The granule cell layer (GCL) was thin and truncated, with the infrapyramidal blade appearing more severely affected than the suprapyramidal blade. This phenotype suggested that DG neurogenesis was impaired, and/or apoptosis was increased.
To assess if DG progenitor cells might be directly affected by AUTS2 deficiency, we scrutinized AUTS2 expression in normal developing DG. In neonatal DG, AUTS2 was expressed in the developing GCL, which contains postmitotic granule neurons (Fig. 4a), and in the dentate migration stream (DMS) and subpial neurogenic zone (SPNZ), where intermediate progenitors (INPs), a type of committed neurogenic progenitors, migrate and divide as shown by Nelson (Nelson et al. 2020). Double IF showed that AUTS2 colocalized with TBR2, an INP marker (Fig. 4b–c3). Thus, AUTS2 deficiency could potentially directly impact proliferating INPs, as well as neurons.
Figure 4 .
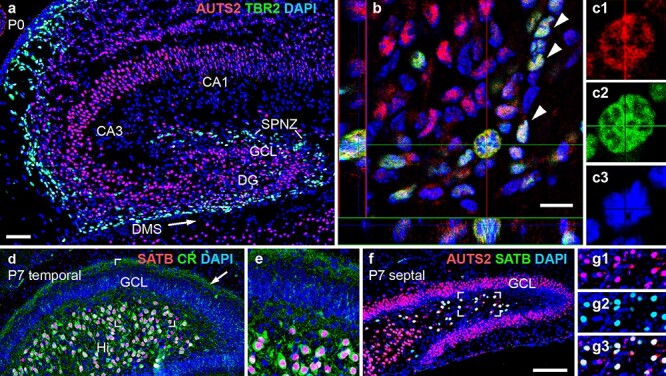
AUTS2 expression in neonatal hippocampus. (a–c3) Double IF for AUTS2 (red) and TBR2 (green) on P0. In (b), a confocal image of the DMS with orthogonal views (left and bottom) shows AUTS2 expression in TBR2+ INPs (arrowheads), including an M-phase INP (crosshairs) shown at higher magnification in (c1–c3). (d–e) Double IF for SATB1/2 (red) and calretinin (green) in P7 DG at temporal level. Calretinin+ recurrent axon collaterals from HMNs are indicated in the DG inner molecular layer (arrow). The boxed area in (d) is shown at higher magnification in (e). (f–g3) Double IF for SATB1/2 (green) and AUTS2 (red) in P7 DG at septal level. The boxed area of the Hi in (f) is shown at higher magnification in (g1–g3). Sagittal sections with DAPI (blue) counterstain. Scale bars: a, 50 μm for a, d, and 25 μm for e; b, 10 μm for b, and 5 μm for c1–c3; f, 100 μm for f, and 50 μm for g1–g3.
Besides granule neurons, HMNs described by Scharfman (2018), are another DG projection neuron type, located in the interior (hilus [Hi]) of the DG. Previous studies of adult mice by Huang et al. (2011) suggested that HMNs specifically express calretinin and SATB1, a chromatin regulator. Using IF, we verified that SATB1 is also expressed by HMNs during development (Fig. 4d, e), and moreover, AUTS2 is expressed by SATB1+ HMNs (Fig. 4f–g3). Thus, AUTS2 is expressed by both types of DG projection neurons (HMNs and granule neurons), and by the INPs that generate them.
To determine if DG hypoplasia is caused by decreased neurogenesis in Auts2 cKOctx mice, we compared the abundance of TBR2+ INPs, which have been shown in previous work (Hodge et al. 2012; Nelson et al. 2020) to produce most or all DG granule neurons, in control and Auts2 cKOctx mice. INPs were moderately decreased in Auts2 cKOctx DG as early as P0.5 (log2FC = −0.95, P = 0.05 by one-tailed wilcox test) (Supplementary Fig. 3a–c), and declined progressively in mutant DG during the first postnatal week (Fig. 5a–d). Among the multiple neurogenic niches of the postnatal DG (Nelson et al. 2020), INPs appeared moderately decreased in the fimbriodentate junction (FDJ) and SPNZ, and were virtually absent from the Hi and subgranular zone (SGZ). In contrast to TBR2+ INPs, SOX9+ neural stem cells did not appear significantly reduced in Auts2 cKOctx DG (data not shown). The migration of INPs into the DG is enabled by the interaction of Cajal–Retzius cells, and the trans-hilar glial scaffolding which were shown to be critical for DG morphogenesis in prior experiments (Frotscher et al. 2003; Li et al. 2009b; Hodge et al. 2013). We examined additional markers of this migratory pathway including Reelin, a marker of Cajal–Retzius cells (Supplementary Fig. 4e, f); and GFAP, a marker of the trans-hilar glial scaffolding (Supplementary Fig. 4g, h) at P0.5, P5, and P7, which indicate that the hilar scaffolding is unaffected in Auts2 cKOctx animals. (Supplementary Fig. 4e–h for P0.5; other ages not shown.) These results suggest that migration and/or proliferation of INPs specifically were impaired in Auts2 cKOctx brains. In contrast, no increase in cell death was observed by IF to detect activated caspase 3 (data not shown).
HMNs were also reduced in Auts2 cKOctx DG, and were in fact even more depleted than granule neurons. HMNs (SATB1+) and their axon collaterals (calretinin+) were virtually absent from the DG of adult Auts2 cKOctx mice (Fig. 5e, f). The loss of SATB1+ HMNs was evident even at birth (data not shown). Together, these results suggested that AUTS2 deficiency caused DG hypoplasia by reducing INP-mediated neurogenesis of HMNs and granule neurons.
EEG Abnormalities in Auts2 cKOctx Mice
To study the impact of AUTS depletion on cortical circuit activity, we used video-EEG–EMG with bilateral implanted EEG electrodes (Fig. 5g, inset). Recordings demonstrated several abnormalities in Auts2 cKOctx mice, including greater power in theta, delta, and beta frequencies (Fig. 5g), as well as abnormal spiking activity indicative of synchronous neuronal discharges (Fig. 5h). The abnormal spikes were similar to interictal discharges in epilepsy, although no overt seizures were observed. These anomalies may, however, indicate that Auts2 cKOctx mice have reduced threshold for seizure activity. Significantly, seizure disorders occur in a subset of patients with AUTS2 syndrome as demonstrated by Beunders et al. (2016).
Transcriptome Alterations in Auts2 cKOctx Hippocampus
To gain further insight into regulation of RNA metabolism by AUTS2, we dissected whole hippocampi from control and Auts2 cKOctx neonates for RNA-seq (see Supplementary Table 6). This comparison identified 57 dysregulated transcripts with high significance in Auts2 cKOctx hippocampus (q < 0.05; 11 increased, 46 decreased). One highly upregulated transcript was Penk (log2FC = +1.54), which encodes preproenkephalin. To validate this result, we used ISH. Penk mRNA was markedly increased in cells occupying the DG molecular layer and adjacent hippocampal marginal zone, locations consistent with Cajal–Retzius (C–R) neurons (Supplementary Fig. 3d–g). However, C–R cell transcriptomes were not globally dysregulated, as levels of Reln and Trp73 (general C–R neuron markers) were not significantly dysregulated. Together, these data suggest that the number of C–R neurons was unchanged in Auts2 cKOctx mice, while Penk expression was markedly upregulated in these cells.
To more broadly analyze the hippocampus RNA-seq data, we used bioinformatics approaches. GO analysis identified 19 Biological Processes and 4 Molecular Functions that were significantly represented (see Supplementary Table 7). Neuron migration and axon guidance were among the top 4 Biological Processes. We next assessed hippocampal cell types, using (GSEA; Subramanian et al. 2005) with gene sets constructed from HippoSeq (Cembrowski et al. 2016). The GSEA result revealed significant overall reduction of 3 cell types in Auts2 cKOctx hippocampus: HMNs, granule neurons, and ventral CA1 neurons (see Supplementary Table 8). (INPs are not represented in HippoSeq.) The most reduced cell type was HMNs (normalized enrichment score = −1.79, P = 0.000). Transcriptome profiling thus supported the conclusions reached by IF, that HMNs and granule neurons are deficient early in development of the Auts2 cKOctx DG (by P0.5).
Neocortical Abnormalities in Auts2 cKOctx Mice
On P7, when neocortical neurogenesis, cell migration, and axon pathway development are mostly complete, the Auts2 cKOctx neocortex appeared structurally normal, with well-defined layers and an unremarkable corpus callosum (Fig. 6a, b). Also, somatosensory barrels (modules for whisker sensation) developed appropriately in layer 4 (Fig. 6c, d). The latter result indicated that thalamocortical innervation and initial cortical plasticity were intact (Martini et al. 2018).
Figure 6 .
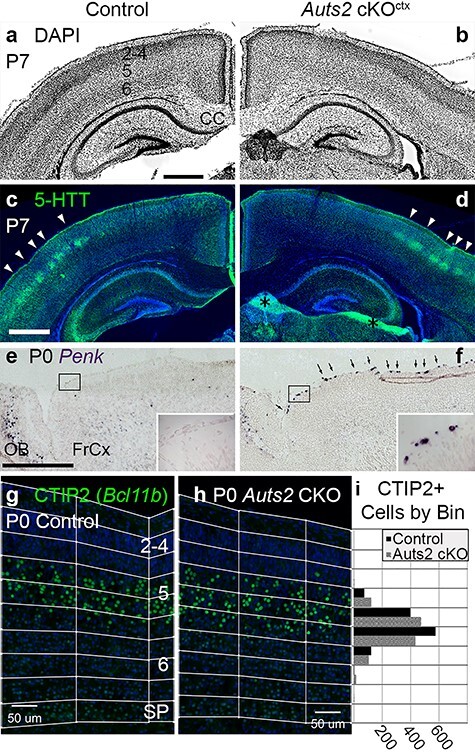
Normal structure but molecular defects in Auts2 cKOctx neocortex. (a, b) DAPI staining (inverted fluorescence) of P7 control (a) and Auts2 cKOctx (b) brains. Neocortical layers are numbered in (a). Coronal sections. Abbreviation: CC, corpus callosum. (c, d) IF to detect serotonin transporter (5-HTT) in same sections as (a, b). Arrowheads indicate 5-HTT+ layer 4 somatosensory barrels. Asterisks in (d): tissue artifacts. (e, f) ISH to detect Penk in P0 control (e) and Auts2 cKOctx (f) brains. Boxed areas are shown at higher magnification in insets. Arrowheads in (f) indicate cells with increased Penk expression, presumably C-R neurons. Sagittal sections. Abbreviations: FrCx, frontal cortex; OB, olfactory bulb. (g, h) IF to detect CTIP2 (gene Bcl11b) in P0.5 control (g) and Auts2 cKOctx (h) neocortex. Neocortical layers are numbered in (g). Pial to subplate binning is as indicated. Sagittal sections through somatosensory cortex. (i) Ctip2+ cells quantified across 9 binned images each for control and Auts2 cKO (wilcox-test P = 0.5321). Abbreviations: IZ, intermediate zone; SP, subplate. Scale bars: a, 500 μm for a–d; e, 500 μm for e, f; and g, 100 μm for g, h.
Transcriptome profiling of control and Auts2 mutant frontal cortex (P0.5) identified many differentially expressed transcripts (DETs; see Supplementary Table 4J–Q). Using the most stringent criteria (q < 0.05), 58 transcripts, representing 56 genes, were differentially expressed (14 upregulated, 42 downregulated). Excluding Auts2 (decreased due to cKO) and Xist (female expression only, reporting different sex ratios between control and mutant samples), 13 gene mRNAs were increased and 41 decreased. As in neonatal hippocampus (see Supplementary Table 6), one highly upregulated gene was Penk (log2FC = +1.43). This change was likewise validated by ISH, which showed increased presence of Penk positive neocortical marginal zone cells, consistent with C-R neurons (Fig. 6e, f).
Other notable dysregulated genes (q < 0.05) in frontal neocortex included Tshz2 (log2FC = +1.07), a regional marker of caudal cortex (Elsen et al. 2013); Wnt7b (log2FC = −0.64), a layer 6 marker (Hevner et al. 2001); and Pcdh20 (log2FC = −0.89), a layer 4 marker (Oishi et al. 2016). Using a less stringent statistical cutoff (P < 0.05), some laminar markers (Cux1, Satb2, and Tbr1) were not differentially expressed, but Bcl11b mRNA (which encodes CTIP2) was significantly increased (log2FC = +0.25). CTIP2 is a transcription factor and component of the BAF chromatin remodeling complex that regulates corticospinal projections (Arlotta et al. 2005). The transcriptomic data indicated that regional and laminar differentiation of neurons were perturbed in Auts2 cKOctx neocortex, with increased caudal and layer 5 features, decreased layer 4 and 6 features, and elevated expression of Penk in presumed Cajal–Retzius neurons.
To further investigate neocortical differentiation, we assessed laminar markers TBR1 (layer 6), CTIP2 (layer 5 and subplate), and CUX1 (layers 2–4) by IF in sections of P0.5 neocortex. These markers revealed no obvious alterations in cortical lamination (Fig. 6g, h; Supplementary Fig. 4a–d). Since CTIP2 (Bcl11b) mRNA was increased in Auts2 cKOctx neocortex as determined by RNA-Seq (see Supplementary Table 4J–Q), we conducted more detailed analysis of CTIP2 laminar distribution by bin analysis (Fig. 6g, h). However, no significant change in the distribution of CTIP2+ cells was observed (Fig. 6i). These observations agreed with those from previous studies, including Gao et al. (2014) and Hori et al. (2014), who (while suggesting that AUTS2 regulates cortical neuron migration) likewise reported no significant dyslamination in Auts2 mutant mice.
To more broadly evaluate the AUTS2-regulated transcripts in neocortex, we submitted the DETs with P < 0.01 (n = 383) for GO analysis, which identified 95 Biological Processes and 45 Molecular Functions associated with these transcripts (see Supplementary Table 9). Among the top Biological Processes were regulation of transcription, regulation of cell proliferation, and Notch signaling. Top Molecular Functions included protein binding, DNA binding, and calcium ion binding.
AUTS2-Regulated Transcripts Highly Correlate with AUTS2-Bound RNA
To investigate whether AUTS2 regulates mRNA levels primarily by binding chromatin and activating gene expression as previously proposed (Gao et al. 2014; Oksenberg et al. 2014), or by binding RNA in complexes with AUTS2-IPs (present study), we looked for correlations between DETs, RNA bound to AUTS2 complexes, and AUTS2 binding sites in chromatin.
Transcripts bound by AUTS2 complexes (RIP-seq) were tested for overlap with DETs in Auts2 mutant frontal cortex (RNA-seq). Of 383 DETs with P < 0.01 (see Supplementary Table 4J–Q), 56 (14.6%) were bound by AUTS2 complexes (Table 1). Among these, 42 (75%) had decreased expression in Auts2 mutant cortex, while 14 (25%) had increased expression (Table 1). Using the more stringent cutoff (q < 0.05 in RNA-seq), 54 genes (excluding Auts2 and Xist) had DETs, of which 11 (20.4%) were bound by AUTS2 complexes in RIP-seq (Table 1). In comparison, only 3.2% of the total transcriptome was bound by AUTS2 complexes, representing RIP-seq background. Interestingly, DETs bound and regulated by AUTS2 included Ep300 and Prex1, which encode P300 and P-REX1 (respectively), previously reported as AUTS2-IPs (Gao et al. 2014; Hori et al. 2014). Our results suggest those reported interactions may have been driven by AUTS2 overexpression, leading to P300 and P-REX1 overexpression as well (see Discussion).
To investigate whether the AUTS2-bound and -regulated transcripts encode a network of interacting proteins, we submitted them for analysis with STRING-DB (Szklarczyk et al. 2015). This algorithm identified an interaction network with 15 proteins (P = 0.010), including Notch signaling molecules NOTCH2 and JAG2 (Supplementary Fig. 5a). This STRING-DB result matched the GO analysis of DETs in Auts2 cKOctx frontal cortex, which also flagged Notch signaling (see Supplementary Table 9).
Using the same approaches, we also tested for correlations between AUTS2 chromatin binding sites and transcript regulation. We assessed genes within 5 kb of previously reported AUTS2 ChIP-seq peaks from P1 mouse brain (Gao et al. 2014). This analysis showed that the genes for DETs in Auts2 cKOctx frontal cortex were not significantly enriched above ChIP background. Also, STRING-DB did not identify any significant protein network encoded by DETs with ChIP-seq peaks, despite a similar number of targets (Supplementary Fig. 5b). Overall, these results indicate that transcripts regulated by AUTS2 in brain are better correlated with RNA bound to AUTS2, than with cognate genes bound by AUTS2.
Discussion
AUTS2 Functions Primarily to Regulate RNA Metabolism
The machinery for regulating RNA metabolism modulates processes from RNA transcription to splicing, editing, stabilization or destabilization, transport, and translation—a spectrum of activities substantially encompassed by the top AUTS2-IPs (Fig. 2a) described in experiments on these RNA interacting proteins’ molecular functions (Fuller-Pace 2013; Knott et al. 2016). Typically, regulation of RNA metabolism involves multiprotein complexes of variable, often cell-type-specific composition (Conlon and Manley 2017). Further, studies will be necessary to determine if AUTS2 binds RNA directly, or only through interacting proteins; and whether AUTS2 has enzymatic functions, or serves as a scaffold.
Previous studies suggested that AUTS2 functions primarily in chromatin activation (Gao et al. 2014; Oksenberg et al. 2014) and cytoskeletal reorganization (Hori et al. 2014). Our results do not exclude those functions, but rather point to RNA regulation as the primary function of AUTS2 in vivo. Indeed, chromatin regulation is part of the spectrum of RBP functions, although we found that AUTS2 can increase or decrease specific mRNA levels (Table 1) and thus is not solely an activator of chromatin. UniGoPred analysis of the PD718837 putative domain in AUTS2-FL implicated it in cytoskeletal binding (see Supplementary Table 1). However, in our hands and previous experiments by Bedogni et al. (2010b) AUTS2 protein is not significantly detected outside the nucleus, at least in neocortex (Figs 1 and 4).
The functions attributed to AUTS2 in previous studies were based in part on the identification of P300 and P-REX1 as AUTS2-IPs, after forced expression in HEK293 cells and the utilization of tagged constructs and shRNA silencing (Gao et al. 2014; Hori et al. 2014). Significantly, we found that Ep300 and Prex1 mRNAs are targets of AUTS2 binding and regulation, and were decreased in Auts2 cKOctx cortex (Table 1). These results suggest that AUTS2 overexpression could have also induced high levels of P300 and P-REX1, leading to nonphysiological interactions in the previous studies. Neither P300 nor P-REX1 was confirmed as AUTS2-IPs in vivo in the present study.
Cell-Type-Specific RNA Regulation by AUTS2
AUTS2 did not regulate mRNAs identically in all cortical neuron types. For example, Penk mRNA was upregulated in Auts2 cKOctx hippocampus and cortex, but only in presumed C–R neurons (Fig. 6e, f; Supplementary Fig. 3d–g). One way for such cell-type-specific mRNA regulation to occur would be if certain AUTS2-IPs are expressed only in C–R neurons. One likely candidate is DDX17, an AUTS2-IP (Fig. 2a) previously described (Fuller-Pace 2013) as a multifunctional regulator of RNA metabolism that appears to be specifically expressed in C-R neurons (Genepaint; https://gp3.mpg.de/viewer/setInfo/EH2654). These observations suggest that AUTS2 interacts with both Penk and DDX17 in C–R neurons, to reduce Penk mRNA levels. However, while expression of some genes was altered in C–R neurons, others, like Reelin which are involved in their canonical role of regulating lamination (Frotscher et al. 2003; Hodge et al. 2013), were unaffected. This implies that the impaired neurogenesis observed in the Auts2 cKOctx DG is occurring in a highly specific manner intrinsic to the affected neuronal populations.
Auts2 Mutant Mice and the Human AUTS2 Syndrome
The most consistent phenotype shown in previous studies of AUTS2 syndrome (Beunders et al. 2016) is ID (100%), which ranges from mild learning disability to severe cognitive impairment. If DG hypoplasia occurs early in human AUTS2 syndrome as in Auts2 cKOctx mice, this structural defect would probably impair learning and memory throughout life, because the DG has been shown in previous studies (Hainmueller and Bartos 2020) to mediate acquisition and retention of new memories. Disturbances of neocortical laminar and regional identity would presumably further impair cognitive as well as motor functions. In future studies, it will be interesting to study Auts2 cKOctx mice behaviorally for deficits of learning, memory, motor skills, and other neurological functions that may exhibit relevance for better understanding of the varying presentations of the human AUTS2 syndrome.
Previous studies of AUTS2 human subjects (Beunders et al. 2016) identified microcephaly in 60–80% of cases. While Auts2 cKOctx mice did not have small brains overall, DG hypoplasia was linked to decreased neurogenesis from INPs (Fig. 5a–d). Importantly, development of the DG has been shown (Nelson et al. 2020) to involve prolonged progenitor cell migrations and proliferation. In humans (but not mice), development of the neocortex likewise involves extensive progenitor cell migration and proliferation, to form distinct outer progenitor zones (Molnár et al. 2019). We hypothesize that the outer progenitor zones of neocortex may be deficient during fetal development of humans with AUTS2 mutations, leading to decreased neurogenesis and neocortical hypoplasia. Also, opioid signaling, which was potentially increased by Penk upregulation in Auts2 cKOctx mice, has been shown to impair cerebellar neuron progenitor proliferation in experiments by Hauser et al. (2000). Accordingly, some problems in AUTS2 syndrome might potentially be treatable by early intervention with anti-opioid drugs, such as naloxone, which has shown some benefit in ameliorating similar phenotypes (Hauser et al. 2000).
Epilepsy occurs in 7–15% of AUTS2 syndrome (Beunders et al. 2016). Auts2 cKOctx mice exhibited EEG spikes (Fig. 5h) that resemble discharges seen in seizure disorders (Staley and Dudek 2006), and are commonly used as a marker of epileptogenicity (Huneau et al. 2013). The EEG theta rhythm was also increased (Fig. 5g), which is relevant because theta activity is produced by the hippocampus, and has been shown in previous studies (Pernía-Andrade and Jonas 2014) to be indicative of hippocampal excitatory network activity. These EEG abnormalities might be related to HMN depletion (Fig. 5f; see Supplementary Table 8), which has been studied in previous work (Jinde et al. 2012) as a cause of hippocampal disinhibition. Normally, HMNs inhibit hippocampal discharges by activating GABAergic interneurons, which in turn suppress granule neuron firing (Scharfman 2018).
AUTS2 as a Candidate SIDS Gene
The most severe phenotype in Auts2 cKO mice, occurring after homozygous Auts2 inactivation throughout the CNS, was neonatal death associated with abnormalities of breathing rhythmogenesis (Supplementary Fig. 1). This phenotype is most likely related to Auts2 expression in the brainstem (Bedogni et al. 2010b), where nuclei that control breathing are located. In a previous mouse study (Hori et al. 2014), Auts2 exon 8 deletion (whole-body) also caused neonatal death, from unknown causes. Breathing problems have not been mentioned in human AUTS2 syndrome, but theoretically might manifest as infant death of unknown etiology. Indeed, there is abundant evidence that sudden infant death syndrome (SIDS) may be caused by abnormal brainstem and hippocampal circuits that heighten vulnerability to apnea (Kinney et al. 2009; Bright et al. 2018; Kinney and Haynes 2019). Increased opioid signaling from overproduction of enkephalins (Penk gene) might further contribute to respiratory depression. For these reasons, AUTS2 should be considered a candidate SIDS gene. Also, the feeding difficulties that commonly occur in AUTS2 syndrome (Beunders et al. 2016) may be related to disturbances in respiratory rhythmogenesis, which disrupt the coordination between swallowing and breathing.
Relation of AUTS2 to NONO and TBR1 Syndromes
The interaction of AUTS2 with NONO (Fig. 2) is interesting because NONO mutations cause an ID syndrome that overlaps strikingly with AUTS2 syndrome. Shared phenotypes include ID, seizure disorder, autistic features, shy behavior, feeding difficulties, motor and language delay, facial dysmorphisms, strabismus, hypotonia, short stature, kyphoscoliosis, cardiac atrial septal defect, and cryptorchidism (Beunders et al. 2016; Sewani et al. 2020). However, NONO syndrome is associated with macrocephaly, while AUTS2 subjects have microcephaly. This contrast may indicate that AUTS2 and NONO control the same developmental processes, albeit with sometimes antagonistic activities.
Another neurocognitive disorder that overlaps substantially with AUTS2 syndrome is TBR1 syndrome (Nambot et al. 2020). TBR1 is a T-box transcription factor that binds and activates Auts2 in developing cortex (Bedogni et al. 2010a). In TBR1 syndrome, mild to severe ID is variably accompanied by autistic features, epilepsy, and microcephaly. Moreover, homozygous Tbr1 KO mice have a hypoplastic DG, and perturbed regional and laminar identity in neocortex (Hevner et al. 2001; Bedogni et al. 2010a; Nambot et al. 2020).
Finally, since AUTS2 has also likely played an important role in human brain evolution (Oksenberg and Ahituv 2013), further studies of Auts2 mutant mice, of AUTS2 expression and functions in human cerebral organoids, and of structural brain phenotypes in AUTS2 syndrome, may further enrich our understanding of how RNA regulation influences human brain development, evolution, and disorders.
Supplementary Material
{kind=link}
Contributor Information
Anthony S Castanza, Department of Pathology, University of Washington, Seattle, WA 98195, USA; Center for Integrative Brain Research, Seattle Children’s Research Institute, Seattle, WA 98101, USA; Department of Pathology, University of California San Diego, La Jolla, CA 92093, USA.
Sanja Ramirez, Center for Integrative Brain Research, Seattle Children’s Research Institute, Seattle, WA 98101, USA.
Prem P Tripathi, Center for Integrative Brain Research, Seattle Children’s Research Institute, Seattle, WA 98101, USA.
Ray A M Daza, Center for Integrative Brain Research, Seattle Children’s Research Institute, Seattle, WA 98101, USA; Department of Pathology, University of California San Diego, La Jolla, CA 92093, USA.
Franck K Kalume, Center for Integrative Brain Research, Seattle Children’s Research Institute, Seattle, WA 98101, USA; Department of Neurological Surgery, University of Washington, Seattle, WA 98014, USA.
Jan-Marino Ramirez, Center for Integrative Brain Research, Seattle Children’s Research Institute, Seattle, WA 98101, USA; Department of Neurological Surgery, University of Washington, Seattle, WA 98014, USA.
Robert F Hevner, Department of Pathology, University of Washington, Seattle, WA 98195, USA; Center for Integrative Brain Research, Seattle Children’s Research Institute, Seattle, WA 98101, USA; Department of Pathology, University of California San Diego, La Jolla, CA 92093, USA; Department of Neurological Surgery, University of Washington, Seattle, WA 98014, USA.
Funding
National Institute of Neurological Disorders and Stroke at the National Institutes of Health (R01NS092339 and R01NS085081 to R.F.H., R01NS102796 to F.K.K); CURE Epilepsy Research Grant (2017 to F.K.K.).
Notes
We are grateful to Young Ah Goo at the University of Washington Neuroproteomics Core for technical support, instrument time, and running mass spectroscopy samples; Chris Frazar and the Northwest Genomics Core for providing technical support, library preparation, and sequencing of RNA samples.
References
- Amemiya HM, Kundaje A, Boyle AP. 2019. The ENCODE blacklist: identification of problematic regions of the genome. Sci Rep. 9:9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, MacKlis JD. 2005. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron. 45:207–221. [DOI] [PubMed] [Google Scholar]
- Baltz AG, Munschauer M, Schwanhäusser B, Vasile A, Murakawa Y, Schueler M, Youngs N, Penfold-Brown D, Drew K, Milek M et al. 2012. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell. 46:674–690. [DOI] [PubMed] [Google Scholar]
- Bedogni F, Hodge RD, Elsen GE, Nelson BR, Daza RAM, Beyer RP, Bammler TK, Rubenstein JLR, Hevner RF. 2010a. Tbr1 regulates regional and laminar identity of postmitotic neurons in developing neocortex. Proc Natl Acad Sci U S A. 107:13129–13134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedogni F, Hodge RD, Nelson BR, Frederick EA, Shiba N, Daza RA, Hevner RF. 2010b. Autism susceptibility candidate 2 (Auts2) encodes a nuclear protein expressed in developing brain regions implicated in autism neuropathology. Gene Expr Patterns. 10:9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beunders G, van de Kamp J, Vasudevan P, Morton J, Smets K, Kleefstra T, de Munnik SA, Schuurs-Hoeijmakers J, Ceulemans B, Zollino M, et al. 2016. A detailed clinical analysis of 13 patients with AUTS2 syndrome further delineates the phenotypic spectrum and underscores the behavioural phenotype. J Med Genet. 53:523–532. [DOI] [PubMed] [Google Scholar]
- Beunders G, Voorhoeve E, Golzio C, Pardo LM, Rosenfeld JA, Talkowski ME, Simonic I, Lionel AC, Vergult S, Pyatt RE, et al. 2013. Exonic deletions in AUTS2 cause a syndromic form of intellectual disability and suggest a critical role for the C terminus. Am J Hum Genet. 92:210–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright FM, Vink R, Byard RW. 2018. Neuropathological developments in sudden infant death syndrome. Pediatr Dev Pathol. 21:515–521. [DOI] [PubMed] [Google Scholar]
- Bru C. 2004. The ProDom database of protein domain families: more emphasis on 3D. Nucleic Acids Res. 33:D212–D215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cembrowski MS, Wang L, Sugino K, Shields BC, Spruston N. 2016. Hipposeq: a comprehensive RNA-seq database of gene expression in hippocampal principal neurons. Elife. 5:e14997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon EG, Manley JL. 2017. RNA-binding proteins in neurodegeneration: mechanisms in aggregate. Genes Dev. 31:1509–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsen GE, Hodge RD, Bedogni F, Daza RAM, Nelson BR, Shiba N, Reiner SL, Hevner RF. 2013. The protomap is propagated to cortical plate neurons through an Eomes-dependent intermediate map. Proc Natl Acad Sci U S A. 110:4081–4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englund C, Fink A, Lau C, Pham D, Daza RAM, Bulfone A, Kowalczyk T, Hevner RF. 2005. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci. 25:247–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frotscher M, Haas CA, Förster E. 2003. Reelin controls granule cell migration in the dentate gyrus by acting on the radial glial scaffold. Cereb Cortex. 13:634–640. [DOI] [PubMed] [Google Scholar]
- Fuller-Pace FV. 2013. The DEAD box proteins DDX5 (p68) and DDX17 (p72): multi-tasking transcriptional regulators. Biochim Biophys Acta. 1829:756–763. [DOI] [PubMed] [Google Scholar]
- Gao Z, Lee P, Stafford JM, Von Schimmelmann M, Schaefer A, Reinberg D. 2014. An AUTS2-Polycomb complex activates gene expression in the CNS. Nature. 516:349–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JLR, Jones KR. 2002. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci. 22:6309–6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hainmueller T, Bartos M. 2020. Dentate gyrus circuits for encoding, retrieval and discrimination of episodic memories. Nat Rev Neurosci. 21:153–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser KF, Houdi AA, Turbek CS, Elde RP, Maxson W. 2000. Opioids intrinsically inhibit the genesis of mouse cerebellar granule neuron precursors in vitro: differential impact of μ and δ receptor activation on proliferation and neurite elongation. Eur J Neurosci. 12:1281–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentze MW, Castello A, Schwarzl T, Preiss T. 2018. A brave new world of RNA-binding proteins. Nat Rev Mol Cell Biol. 19:327–341. [DOI] [PubMed] [Google Scholar]
- Hevner RF, Daza RAM, Englund C, Kohtz J, Fink A. 2004. Postnatal shifts of interneuron position in the neocortex of normal and reeler mice: evidence for inward radial migration. Neuroscience. 124:605–618. [DOI] [PubMed] [Google Scholar]
- Hevner RF, Shi L, Justice N, Hsueh YP, Sheng M, Smiga S, Bulfone A, Goffinet AM, Campagnoni AT, Rubenstein JLR. 2001. Tbr1 regulates differentiation of the preplate and layer 6. Neuron. 29:353–366. [DOI] [PubMed] [Google Scholar]
- Hodge RD, Garcia AJ, Elsen GE, Nelson BR, Mussar KE, Reiner SL, Ramirez JM, Hevner RF. 2013. Tbr2 expression in Cajal-Retzius cells and intermediate neuronal progenitors is required for morphogenesis of the dentate gyrus. J Neurosci. 33:4165–4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge RD, Nelson BR, Kahoud RJ, Yang R, Mussar KE, Reiner SL, Hevner RF. 2012. Tbr2 is essential for hippocampal lineage progression from neural stem cells to intermediate progenitors and neurons. J Neurosci. 32:6275–6287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori K, Hoshino M. 2017. Neuronal migration and AUTS2 syndrome. Brain Sci. 7:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori K, Nagai T, Shan W, Sakamoto A, Abe M, Yamazaki M, Sakimura K, Yamada K, Hoshino M. 2015. Heterozygous disruption of autism susceptibility candidate 2 causes impaired emotional control and cognitive memory. PLoS One. 10:e0145979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori K, Nagai T, Shan W, Sakamoto A, Taya S, Hashimoto R, Hayashi T, Abe M, Yamazaki M, Nakao K, et al. 2014. Cytoskeletal regulation by AUTS2 in neuronal migration and Neuritogenesis. Cell Rep. 9:2166–2179. [DOI] [PubMed] [Google Scholar]
- Huang Y, Zhang L, Song NN, Hu ZL, Chen JY, Ding YQ. 2011. Distribution of Satb1 in the central nervous system of adult mice. Neurosci Res. 71:12–21. [DOI] [PubMed] [Google Scholar]
- Huneau C, Benquet P, Dieuset G, Biraben A, Martin B, Wendling F. 2013. Shape features of epileptic spikes are a marker of epileptogenesis in mice. Epilepsia. 54:2219–2227. [DOI] [PubMed] [Google Scholar]
- Huret J. 2014. AUTS2 (autism susceptibility candidate 2). Atlas Genet Cytogenet Oncol Haematol. 18:306–310. [Google Scholar]
- Jinde S, Zsiros V, Jiang Z, Nakao K, Pickel J, Kohno K, Belforte JE, Nakazawa K. 2012. Hilar mossy cell degeneration causes transient dentate granule cell hsyperexcitability and impaired pattern separation. Neuron. 76:1189–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney HC, Haynes RL. 2019. The serotonin brainstem hypothesis for the sudden infant death syndrome. J Neuropathol Exp Neurol. 78:765–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney HC, Richerson GB, Dymecki SM, Darnall RA, Nattie EE. 2009. The brainstem and serotonin in the sudden infant death syndrome. Annu Rev Pathol Mech Dis. 4:517–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott GJ, Bond CS, Fox AH. 2016. The DBHS proteins SFPQ, NONO and PSPC1: a multipurpose molecular scaffold. Nucleic Acids Res. 44:3989–4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosugi S, Hasebe M, Tomita M, Yanagawa H. 2009. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc Natl Acad Sci U S A. 106:10171–10176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennox AL, Hoye ML, Jiang R, Johnson-Kerner BL, Suit LA, Venkataramanan S, Sheehan CJ, Alsina FC, Fregeau B, Aldinger KA, Moey C, Lobach I, Afenjar A, Babovic-Vuksanovic D, Büzieau S, Blackburn PR, Bunt J, Burglen L, Campeau PM, Charles P, Chung BHY, Cognü B, Curry C, D'Agostino MD, Di Donato N, Faivre L, Héron D, Innes AM, Isidor B, Keren B, Kimball A, Klee EW, Kuentz P, Küry S et al. 2020. Pathogenic DDX3X Mutations Impair RNA Metabolism and Neurogenesis during Fetal Cortical Development. Neuron. 106:404–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Kataoka H, Coughlin SR, Pleasure SJ. 2009b. Identification of a transient subpial neurogenic zone in the developing dentate gyrus and its regulation by Cxcl12 and reelin signaling. Development. 136:327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009a. The sequence alignment/map format and SAMtools. Bioinformatics. 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, et al. 2011. CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 39:D225–D229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini FJ, Moreno-Juan V, Filipchuk A, Valdeolmillos M, López-Bendito G. 2018. Impact of thalamocortical input on barrel cortex development. Neuroscience. 368:246–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnár Z, Clowry GJ, Šestan N, Alzu’bi A, Bakken T, Hevner RF, Hüppi PS, Kostović I, Rakic P, Anton ES, et al. 2019. New insights into the development of the human cerebral cortex. J Anat. 235:432–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molyneaux BJ, Goff LA, Brettler AC, Chen HH, Brown JR, Hrvatin S, Rinn JL, Arlotta P. 2015. DeCoN: genome-wide analysis of invivo transcriptional dynamics during pyramidal neuron fate selection in neocortex. Neuron. 85:275–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monderer-Rothkoff G, Tal N, Risman M, Shani O, Nissim-Rafinia M, Malki-Feldman L, Medvedeva V, Groszer M, Meshorer E, Shifman S. 2021. AUTS2 isoforms control neuronal differentiation. Mol Psychiatry. 26:666–681. [DOI] [PubMed] [Google Scholar]
- Nambot S, Faivre L, Mirzaa G, Thevenon J, Bruel AL, Mosca-Boidron AL, Masurel-Paulet A, Goldenberg A, Le Meur N, Charollais A, et al. 2020. De novo TBR1 variants cause a neurocognitive phenotype with ID and autistic traits: report of 25 new individuals and review of the literature. Eur J Hum Genet. 28:770–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson BR, Hodge RD, Daza RAM, Tripathi PP, Arnold SJ, Millen KJ, Hevner RF. 2020. Intermediate progenitors support migration of neural stem cells into dentate gyrus outer neurogenic niches. Elife. 9:e53777. 10.7554/eLife.53777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi K, Nakagawa N, Tachikawa K, Sasaki S, Aramaki M, Hirano S, Yamamoto N, Yoshimura Y, Nakajima K. 2016. Identity of neocortical layer 4 neurons is specified through correct positioning into the cortex. Elife. 5:e10907. 10.7554/eLife.10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksenberg N, Ahituv N. 2013. The role of AUTS2 in neurodevelopment and human evolution. Trends Genet. 29:600–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksenberg N, Haliburton GDE, Eckalbar WL, Oren I, Nishizaki S, Murphy K, Pollard KS, Birnbaum RY, Ahituv N. 2014. Genome-wide distribution of Auts2 binding localizes with active neurodevelopmental genes. Transl Psychiatry. 4:e431–e431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksenberg N, Stevison L, Wall JD, Ahituv N. 2013. Function and regulation of AUTS2, a gene implicated in autism and human evolution. PLoS Genet. 9:e1003221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernía-Andrade AJ, Jonas P. 2014. Theta-gamma-modulated synaptic currents in hippocampal granule cells in vivo define a mechanism for network oscillations. Neuron. 81:140–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilkington GR, Parker R. 2008. Pat1 contains distinct functional domains that promote P-body assembly and activation of decapping. Mol Cell Biol. 28:1298–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifaioglu AS, Doğan T, Saraç ÖS, Ersahin T, Saidi R, Atalay MV, Martin MJ, Cetin-Atalay R. 2018. Large-scale automated function prediction of protein sequences and an experimental case study validation on PTEN transcript variants. Proteins Struct Funct Bioinforma. 86:135–151. [DOI] [PubMed] [Google Scholar]
- Scharfman HE. 2018. Advances in understanding hilar mossy cells of the dentate gyrus. Cell Tissue Res. 373:643–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz R, Ting CS, King J. 2001. Whole proteome pl values correlate with subcellular localizations of proteins for organisms within the three domains of life. Genome Res. 11:703–709. [DOI] [PubMed] [Google Scholar]
- Sewani M, Nugent K, Blackburn PR, Tarnowski JM, Hernandez-Garcia A, Amiel J, Whalen S, Keren B, Courtin T, Rosenfeld JA, et al. 2020. Further delineation of the phenotypic spectrum associated with hemizygous loss-of-function variants in NONO. Am J Med Genet Part A. 182:652–658. [DOI] [PubMed] [Google Scholar]
- Sharma S, Mishra R, Bigham GE, Wehman B, Khan MM, Xu H, Saha P, Goo YA, Datla SR, Chen L, et al. 2017. A deep proteome analysis identifies the complete secretome as the functional unit of human cardiac progenitor cells. Circ Res. 120:816–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Mann M. 1996. Mass spectrometric sequencing of proteins from silver-stained polyacrylamide gels. Anal Chem. 68:850–858. [DOI] [PubMed] [Google Scholar]
- Staley KJ, Dudek FE. 2006. Interictal spikes and epileptogenesis. Epilepsy Curr. 6:199–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subhash S, Kanduri C. 2016. GeneSCF: a real-time based functional enrichment tool with support for multiple organisms. BMC Bioinformatics. 17:365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana R, Yu CE, Yu J, Munson J, Chen D, Hua W, Estes A, Cortes F, De La Barra F, Yu D, et al. 2002. Identification of a novel gene on chromosome 7q11.2 interrupted by a translocation breakpoint in a pair of autistic twins. Genomics. 80:129–134. [DOI] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP et al. 2015. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43:D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. 2013. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 31:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Sun H, Ma J, Zang C, Wang C, Wang J, Tang Q, Meyer CA, Zhang Y, Liu XS. 2013. Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat Protoc. 8:2502–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Chen MH, Wu X, Kodani A, Fan J, Doan R, Ozawa M, Ma J, Yoshida N, Reiter JF et al. 2016. Cell-Type-Specific Alternative Splicing Governs Cell Fate in the Developing Cerebral Cortex. Cell. 166:1147–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.