

X连锁铁粒幼细胞贫血(XLSA)是遗传性铁粒幼细胞贫血(CSA)最常见类型(约占40%)[1],由位于Xp11.21染色体上的红系特异性5′-氨基乙酰丙酸合酶2(ALAS2)基因突变引起并表现出X连锁遗传模式[2]。XLSA特征为骨髓中无效红细胞生成、大量的环形铁粒幼红细胞、小细胞低色素性贫血以及血清铁和铁蛋白水平升高[3]。目前,已鉴定出104种不同的ALAS2突变。本研究中,我们报道了一个新发现的ALAS2基因V110Afs*2移码突变并进行文献复习如下。
病例资料
患者,男,16岁,因“反复头昏、乏力2年”于2019年10月15日就诊我院。患者2年前开始反复出现头晕、乏力,当地医院查血常规提示小细胞低色素性贫血并予对症治疗,症状无改善,需反复输血,为求进一步诊治来我院。查体:重度贫血貌,全身皮肤黏膜无黄染及出血,浅表淋巴结未触及肿大,胸骨无压痛,心肺未见明显异常,肝脾肋缘下未触及,双下肢无水肿。既往史及家族史无特殊。血常规:WBC 5.58×109/L、RBC 2.5×1012/L、HGB 51 g/L、平均红细胞体积(MCV)70 fl、平均血红蛋白含量(MCH)20.4 pg、平均血红蛋白浓度(MCHC)291 g/L、红细胞分布宽度(RDW)38.8%、网织红细胞绝对计数(Ret)3.3×109/L、PLT 858×109/L;铁蛋白:>2000 µg/L;血清铁:34.1 mmol/L;外周血涂片:无核红细胞形态大小不一,中心淡染区扩大,椭圆、泪滴、棒状、碎片等异形红细胞可见(图1A);骨髓象:增生活跃,粒系占0.280,红系占0.675,粒红比值为0.41,红系增生活跃,以中晚幼红细胞增生为主,形态可见体积小、胞质蓝染、核固缩,淋巴细胞占4.5%,全片见巨核细胞48个,环状铁粒幼红细胞增多(69%),细胞内铁81%,细胞外铁(++),红细胞形态提示小细胞低色素,考虑环形铁粒幼细胞贫血(图1B)。骨髓活检:增生性骨髓象(红系比例增高)。流式免疫分型:粒系减低,红系比例增高,可见CD71减弱。FISH:5号、7号、8号、20号、性染色体未见异常。染色体核型:46,XY[10]。
图1. 遗传性铁粒幼细胞贫血患者外周血涂片及骨髓铁染色结果.
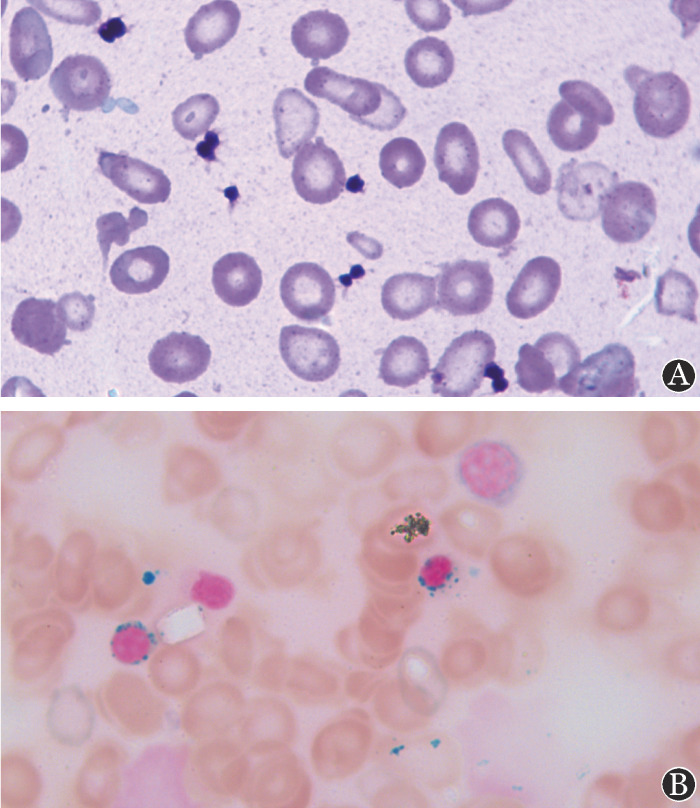
A:外周血涂片示无核红细胞形态大小不一,中心淡染区扩大,可见椭圆、泪滴、棒状、碎片等异形红细胞;B:骨髓铁染色可见红细胞成串排列及大量环形铁粒幼细胞
根据患者的临床表现及相关实验室检查结果,经患儿父母知情同意,采集患儿外周静脉血行基因测序,结果显示,患儿ALAS2基因第4外显子的329-332位点TCAG缺失(c.329-332delTCAG),相应的氨基酸残基110处的缬氨酸变为丙氨酸,并在112个氨基酸终止编码(p.V110Afs*2)(图2A)。在人类基因突变数据库(The Human Gene Mutation Database,HGMD)中未见相关报道。Polyphen 2(http://genetics.bwh.harvard.edu/pph2/)软件预测中得分为0.00,该突变无害;SIFT(http://sift.jcvi.org)软件预测值为0.033,该突变对蛋白质功能影响不确定,然而本例患儿却有重度贫血,该变异不属于多态性位点,在人群中发生频率较低。采集其母亲外周血进行测序显示,患儿母亲为相应位点同样的杂合突变(图2B),患儿的上述变异遗传自母亲,可推断为致病性变异。结合遗传学、临床表现及病理学,综合分析诊断为XLSA。予维生素B6(100 mg/d)口服,治疗1个月后HGB水平明显升高(HGB 76 g/L、MCV 92.5 fl、MCH 22.7 pg、MCHC 245 g/L、RDW 24.8%),目前HGB已恢复正常。
图2. 遗传性铁粒幼细胞贫血患者及其母亲ALAS2基因测序结果.
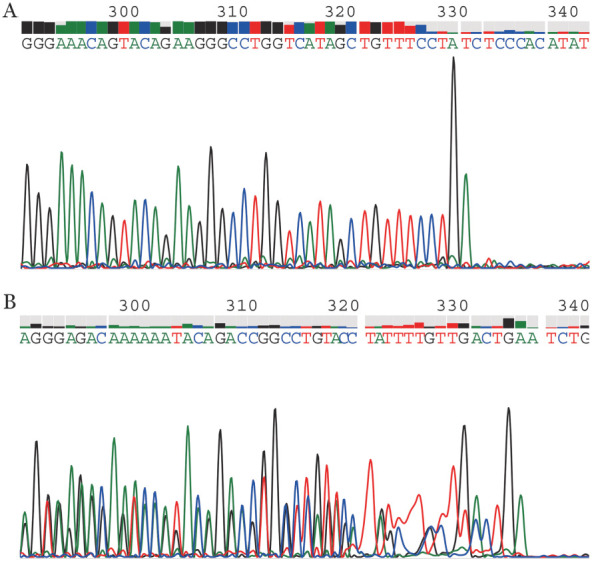
A:患者ALAS2基因第4外显子c.329-332delTCAG(p.V110Afs*2),为半合子;B:患者母亲ALAS2基因第4外显子c.329-332delTCAG(p.V110Afs*2),为杂合子
讨论及文献复习
CSA是一组极为罕见的异质性疾病,由Cooley[4]于1945年首次报道,1992年Cotter等[5]在XLSA患者中首次发现ALAS2基因突变,并揭示了XLSA的致病基因。临床上,XLSA患者通常为半合子男性,发病年龄多<40岁,初诊断为不同程度的小细胞性贫血[3],然而,也有中年XLSA病例的报道,多诊断为铁过载[6]。但也有少数男性不会发病[7]。大多数杂合子女性没有贫血的临床表现,因为正常的ALAS2蛋白可维持必需数量的红细胞产生,已报道的少量女性病例和家族性病例,可能是偏斜的X染色体失活和后天因素影响了ALAS2正常等位基因表达[8]–[9]。目前报道的104种ALAS2突变中,尚未见V110Afs*2。本研究为首次发现XLSA家系ALAS2基因V110Afs*2突变。
在血红素生物合成途径的第一步中,线粒体基质中的甘氨酸和琥珀酰辅酶A缩合成5-氨基乙酰丙酸(ALA),此反应由ALA合成酶催化,在反应过程中吡哆醛5′-磷酸(PLP,维生素B6)作为关键辅助因子,通过稳定琥珀酰部分的碳基和甘氨酸α-碳的相互位置,从而增强ALA合成酶的活性。随后,ALA被转运到细胞质中,参与血红素的合成[3]。Bailey等[10]报告了人类ALAS2晶体结构,ALAS突变后可引起ALAS蛋白构象发生改变,其C端延伸折叠到催化核心顶部形成loop结构,降低了对底物琥珀酰辅酶A的结合,从而影响ALA合成效率。此外,人类ALAS2与PLP采用一组高度保守的相互作用,主要涉及催化核心的中心子结构域。因此,我们推测PLP能够结合ALAS突变后形成的loop结构,抑制其进入ALAS酶活性中心顶部,由此增加ALAS酶与琥珀酰辅酶A的结合,从而增加ALA合成效率。人类ALA合成酶由ALAS2基因编码,当ALAS2基因发生功能丧失性突变时,ALAS2的酶活性降低,ALA不能有效生成,而转铁蛋白受体仍然稳定,因此摄入铁不能有效利用并积累在线粒体中。
XLSA患者的ALAS2基因突变通常是保守氨基酸的错义突变,通过改变ALAS2构象而导致功能丧失[3],[11]–[12]。在近一半的病例中,PLP治疗效果良好,从而恢复酶活性并改善小细胞低色素性贫血[10]。但仍有部分患者对吡哆醇治疗反应欠佳,ALAS2基因突变位点不同影响治疗反应:产生过早终止密码子的无义突变和使ALAS2蛋白失去稳定性的突变均会导致维生素B6治疗无效;启动子突变导致ALAS2基因转录调控受损会导致维生素B6疗效不佳[13]–[14]。对ALAS2基因突变的检测有助于准确诊断CSA及判断维生素B6疗效。维生素B6常用评价标准:①完全缓解:贫血治愈或接近治愈,HGB≥110 g/L;②部分缓解:贫血得到改善、血红蛋白水平升高>10 g/L或症状缓解;③无效:血红蛋白水平没有升高或升高≤10 g/L[15]。对于维生素B6难治性SA患者,可进行异基因造血干细胞移植,但疗效仍不确切[16]–[17]。
迄今为止,HGMD(http://www.hgmd.cf.ac.uk/ac/index.php)中登记的104种ALAS2突变中大多数与XLSA相关,且多位于第5~11外显子的高度保守区[6],主要影响ALAS2的催化活性或者与PLP、甘氨酸或sCoA的结合[18]。在本病例中检测到的突变c.329-332delTCAG(p.V110Afs*2)位于第4外显子,该外显子突变的报道罕见。突变导致第110个氨基酸由甘氨酸变成丙氨酸,并在112个氨基酸终止编码。由于以前没有报道密码子110发生突变并且Val110不是已知的PLP结合位点,尚不清楚该突变如何影响PLP依赖性ALAS2活性,因此我们使用了SIFT和Polyphen-2来预测这种缺失突变的重要性,结果表明,SIFT评分为0.033,Polyphen-2评分为0。尽管预测分数较低,但V110A可能会产生功能改变的突变蛋白,从而引起XLSA发病,这种氨基酸取代的影响可能是间接的,但它可使蛋白质不稳定或影响酶催化作用[19]。患者ALAS2基因c.329-332delTCAG变异遗传来自其母亲,其母亲该位点为杂合子,符合X连锁遗传方式,为患者发病的致病性变异。患者母亲未发病,大多数女性携带者因ALAS2基因表达正常而不发病,但若出现X染色体失活,可出现X连锁疾病临床表现。由于患者采用维生素B6治疗有效,因此我们认为该突变可能影响ALAS2与PLP的相互作用。
Funding Statement
基金项目:重庆市技术创新与发展专项面上项目(cstc2019jscx-msxmX0147)
References
- 1.Camaschella C. Recent advances in the understanding of inherited sideroblastic anaemia[J] Br J Haematol. 2008;143(1):27–38. doi: 10.1111/j.1365-2141.2008.07290.x. [DOI] [PubMed] [Google Scholar]
- 2.May A, Bishop DF. The molecular biology and pyridoxine responsiveness of X-linked sideroblastic anaemia[J] Haematologica. 1998;83(1):56–70. [PubMed] [Google Scholar]
- 3.Fleming MD. Congenital sideroblastic anemias: iron and heme lost in mitochondrial translation[J] Hematology Am Soc Hematol Educ Program. 2011;2011:525–531. doi: 10.1182/asheducation-2011.1.525. [DOI] [PubMed] [Google Scholar]
- 4.Cooley TB. A severe type of hereditary anemia with elliptocytosis. Interesting sequence of splenectomy[J] Am J Med Sci. 1945;209:561–562. [Google Scholar]
- 5.Cotter PD, Baumann M, Bishop DF. Enzymatic defect in “X-linked” sideroblastic anemia: molecular evidence for erythroid delta-aminolevulinate synthase deficiency[J] Proc Natl Acad Sci U S A. 1992;89(9):4028–4032. doi: 10.1073/pnas.89.9.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Méndez M, Moreno-Carralero MI, Morado-Arias M, et al. Sideroblastic anemia: functional study of two novel missense mutations in ALAS2[J] Mol Genet Genomic Med. 2016;4(3):273–282. doi: 10.1002/mgg3.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cazzola M, May A, Bergamaschi G, et al. Absent phenotypic expression of X-linked sideroblastic anemia in one of 2 brothers with a novel ALAS2 mutation[J] Blood. 2002;100(12):4236–4238. doi: 10.1182/blood-2002-03-0685. [DOI] [PubMed] [Google Scholar]
- 8.Cazzola M, May A, Bergamaschi G, et al. Familial-skewed X-chromosome inactivation as a predisposing factor for late-onset X-linked sideroblastic anemia in carrier females[J] Blood. 2000;96(13):4363–4365. [PubMed] [Google Scholar]
- 9.Sankaran VG, Ulirsch JC, Tchaikovskii V, et al. X-linked macrocytic dyserythropoietic anemia in females with an ALAS2 mutation[J] J Clin Invest. 2015;125(4):1665–1669. doi: 10.1172/JCI78619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bailey HJ, Bezerra GA, Marcero JR, et al. Human aminolevulinate synthase structure reveals a eukaryotic-specific autoinhibitory loop regulating substrate binding and product release[J] Nat Commun. 2020;11(1):2813. doi: 10.1038/s41467-020-16586-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bottomley SS, Fleming MD. Sideroblastic anemia: diagnosis and management[J] Hematol Oncol Clin North Am. 2014;28(4):653–670. v. doi: 10.1016/j.hoc.2014.04.008. [DOI] [PubMed] [Google Scholar]
- 12.Furuyama K, Kaneko K. Iron metabolism in erythroid cells and patients with congenital sideroblastic anemia[J] Int J Hematol. 2018;107(1):44–54. doi: 10.1007/s12185-017-2368-0. [DOI] [PubMed] [Google Scholar]
- 13.Bekri S, May A, Cotter PD, et al. A promoter mutation in the erythroid-specific 5-aminolevulinate synthase (ALAS2) gene causes X-linked sideroblastic anemia[J] Blood. 2003;102(2):698–704. doi: 10.1182/blood-2002-06-1623. [DOI] [PubMed] [Google Scholar]
- 14.Kaneko K, Furuyama K, Fujiwara T, et al. Identification of a novel erythroid-specific enhancer for the ALAS2 gene and its loss-of-function mutation which is associated with congenital sideroblastic anemia[J] Haematologica. 2014;99(2):252–261. doi: 10.3324/haematol.2013.085449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu G, Guo S, Kang H, et al. Mutation spectrum in Chinese patients affected by congenital sideroblastic anemia and a search for a genotype-phenotype relationship[J] Haematologica. 2013;98(12):e158–160. doi: 10.3324/haematol.2013.095513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Imataki O, Uchida S, Uemura M, et al. Graft failure after reduced-intensity stem cell transplantation for congenital sideroblastic anemia[J] Pediatr Hematol Oncol. 2019;36(1):46–51. doi: 10.1080/08880018.2019.1578844. [DOI] [PubMed] [Google Scholar]
- 17.Kim MH, Shah S, Bottomley SS, et al. Reduced-toxicity allogeneic hematopoietic stem cell transplantation in congenital sideroblastic anemia[J] Clin Case Rep. 2018;6(9):1841–1844. doi: 10.1002/ccr3.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.陈 昌明, 丁 秋兰, 陆 晔玲, et al. 一个X连锁遗传性铁粒幼细胞贫血家系的基因诊断及文献复习[J] 中华血液学杂志. 2016;37(2):154–156. doi: 10.3760/cma.j.issn.0253-2727.2016.02.015. [DOI] [Google Scholar]
- 19.Astner I, Schulze JO, van den Heuvel J, et al. Crystal structure of 5-aminolevulinate synthase, the first enzyme of heme biosynthesis, and its link to XLSA in humans[J] EMBO J. 2005;24(18):3166–3177. doi: 10.1038/sj.emboj.7600792. [DOI] [PMC free article] [PubMed] [Google Scholar]