

Abstract
Breast cancer (BC) is the most common malignancy in women. Particular subtypes with aggressive behavior are major contributors to poor outcomes. Triple-negative breast cancer (TNBC) is difficult to treat, pro-inflammatory, and highly metastatic. We demonstrate that TNBC cells express TLR9 and are responsive to TLR9 ligands, and treatment of TNBC cells with chemotherapy increases the release of nucleic-acid-containing damage-associated molecular patterns (NA DAMPs) in cell culture. Such culture-derived and breast cancer patient-derived NA DAMPs increase TLR9 activation and TNBC cell invasion in vitro. Notably, treatment with the polyamidoamine dendrimer generation 3.0 (PAMAM-G3) behaved as a nucleic acid scavenger (NAS) and significantly mitigates such effects. In mice that develop spontaneous BC induced by polyoma middle T oncoprotein (MMTV-PyMT), treatment with PAMAM-G3 significantly reduces lung metastasis. Thus, NAS treatment mitigates cancer-induced inflammation and metastasis and represents a novel therapeutic approach for combating breast cancer.
Keywords: DAMPs, cfDNA, breast cancer, TNBC, nucleic acid scavengers, innate immunity, TLRs, metastasis, inflammation
Graphical abstract
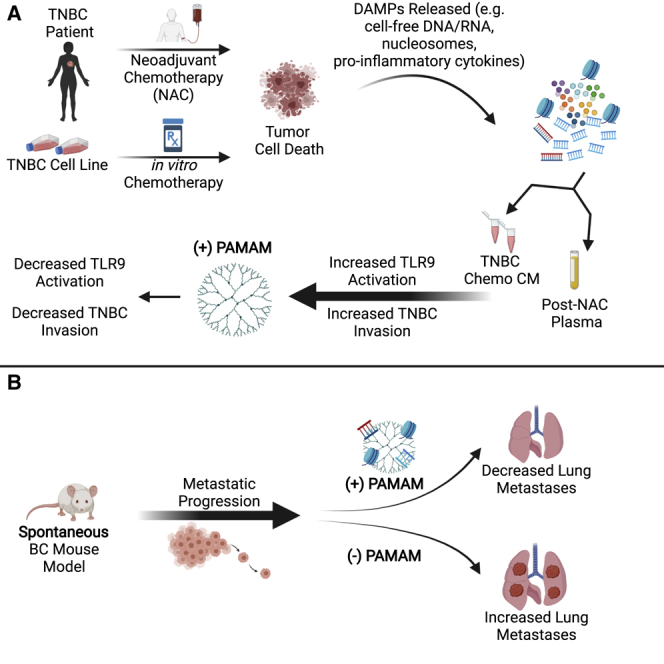
Using a nucleic acid scavenger, Eteshola et al. present an innovative way to slow down metastasis following standard-of-care therapy in difficult-to-treat malignancies such as triple-negative breast cancer. By capturing tumor-derived nucleic acid-containing DAMPs in vitro and in a spontaneous transgenic murine model, this strategy lays the groundwork for improving the clinical management of cancer by adding an adjuvant that limits metastatic spread.
Introduction
Breast cancer (BC) remains the most common malignancy in women worldwide and the second leading cause of cancer-related mortalities in the United States.1,2 Metastatic disease accounts for over 90% of cancer-related mortality in breast cancer.3, 4, 5, 6 Once metastasis has developed, treatment options are limited, leading to a decrease in patient overall survival. Thus, developing strategies to block metastasis is urgently needed to improve patient outcomes.5, 6, 7, 8 Significant progress has been made in the development of targeted therapy for breast cancers that are estrogen/progesterone receptor positive (ER+/PR+) and human epidermal growth factor receptor 2 positive (HER2+). Unfortunately, the current therapies are not as effective in the treatment of the most aggressive and metastatic-prone breast cancer subtype, termed triple-negative breast cancer (TNBC), which lacks the molecular targets ER, PR, and HER2.9, 10, 11 TNBCs are seen with higher frequency in younger patients and tend to have higher rates of local recurrence and distant metastasis when compared to other breast cancer subtypes.9,12 Standard of care for TNBC treatment includes a multi-agent chemotherapy regimen of taxane- and anthracycline-based therapeutics (e.g., nab-paclitaxel and doxorubicin) along with surgery.13, 14, 15 Despite recent advances in multimodality chemotherapy treatments, TNBC patient prognosis remains poor; for patients with advanced metastatic disease, there is a 3-year overall survival of 58% in TNBC patients compared to 80% in non-TNBC patients.16,17 Thus, an unmet clinical need exists for developing improved TNBC therapies.18,19
Originally designed for non-viral nucleic acid delivery,20, 21, 22, 23 our group identified a novel application for the cationic polymer polyamidoamine generation 3.0 (PAMAM-G3) as an extracellular nucleic acid scavenger (NAS). Since cell-free DNA (cfDNA) and other nucleic-acid-containing damage-associated molecular patterns (NA DAMPs) have been implicated in aberrant toll-like receptor (TLR) signaling in several inflammatory diseases,24, 25, 26, 27, 28 we and others have investigated the use of NASs in blocking pro-inflammatory signals through the binding of NA DAMPs in various disease models, including murine models of acute toxic shock syndrome, thrombosis, lupus, rheumatoid arthritis, sepsis, and pancreatic cancer (PC).29, 30, 31, 32, 33, 34, 35, 36 Tumor progression is associated with the production of numerous inflammatory mediators, including cytokines and chemokines, and the recruitment and activation and subsequent functional modulation of leukocytes, particularly mast cells, macrophages, and neutrophils.37,38 Inflammatory mediators are sensed by innate immune sensors, called pattern recognition receptors, such as TLRs, on immune cells in the tumor microenvironment, which amplifies the inflammatory responses by activating downstream molecular targets such as NF-κB and STAT3. These transcription factors activate genes that control cell survival, proliferation, pro-invasiveness, as well as cytokine and chemokine production (e.g., Bcl-xL, survivin, and c-Myc). Altered TLR expression has been associated with autoimmunity and chronic inflammatory diseases including atherosclerosis, liver disease, and cancer.39, 40, 41
Cancer treatment modalities such as surgery, chemotherapy, and radiotherapy can induce local and systemic inflammation due to direct tissue trauma and cancer cell death. Stressed and dying cells (apoptotic or necrotic) shed molecular motifs known as DAMPs, including cell-free nucleic acids (cfDNA, cfRNA, cf-miRNA), mitochondrial DNA (mtDNA), nucleosomes, and exosomes,42, 43, 44, 45, 46, 47, 48, 49 that elicit TLR-mediated pro-inflammatory responses. In addition to immune cells, cancer cells, including breast cancer cells, express TLR receptors.25,27,40,50 TLR signaling may facilitate metastasis by augmenting tumor cell adhesion and invasion and increasing vascular permeability;51,52 however, the complex signaling feedback in these axes means the role of TLRs in the events leading to cancer metastasis has yet to be fully elucidated. Overall, since it has been shown that NA DAMPs contribute to cancer progression, we believe the scavenging of these molecules can potentially limit these chronic inflammatory states.28,53, 54, 55
Previously, we demonstrated that PAMAM-G3 inhibits NA DAMP induction of TLR-mediated pancreatic cancer tumor cell invasion in vitro and reduces liver metastasis in an orthotopic immunocompetent syngeneic murine model.36 Most recently, we demonstrated that PAMAM-G3 is able to reduce lung metastasis in both an intravenous and orthotopic murine model of breast cancer.56 Here, PAMAM-G3 controlled systemic inflammation by decreasing NA DAMPs, inflammatory immune cell subtypes, and proinflammatory cytokine levels. Based on these observations, we hypothesized that the NAS-mediated approach to controlling cancer-associated inflammation, migration, and metastasis should be evaluated in the more clinically relevant samples and in a more relevant murine model of breast cancer. Therefore, in the present study, we investigate if PAMAM-G3 treatment limits the ability of chemotherapy-induced TNBC patient DAMPs to induce inflammatory responses and breast cancer cell migration ex vivo and mitigates metastasis in an aggressive, genetically engineered murine model of spontaneous breast cancer.
Results
Breast cancer cell lines strongly express functional TLR9
Several investigations have suggested that the DNA-sensing TLR9 may play a role in the invasive capability of TNBC due to dying tumor cells releasing DNA and activating pro-growth signaling cascades downstream of TLR9 in nearby breast cancer cells.25,37,50,57 Therefore, we screened a panel of breast cancer cell lines (ER+, TNBC, inflammatory breast cancer, and murine breast cancer cell lines) for TLR9 expression. As shown in Figure 1A, we determined via western blot analysis that several murine and human breast cancer cell lines express higher levels of the DNA-sensing TLR9 compared to the basal expression of TLR9 in HEK293 cells engineered to overexpress human murine TLR4 (HEK293-hTLR4 and HEK293-mTLR4).
Figure 1.

Breast cancer cell lines strongly express functional TLR9
(A) A panel of human and murine breast cancer lines were probed for TLR9 protein expression. The panel included hormone-receptor-positive (MCF-7), TNBC (MDA-MB-231), triple-negative inflammatory breast cancer (SUM149), and murine TNBC (4T1, E0771) cell lines. For comparison, a pancreatic cancer cell line (Panc-1) and the HEK293 TLR9 overexpressing reporter cell line (HEK293-hTLR9) were used as positive controls, while a HEK293 TLR4 reporter cell line was used as a baseline control. Similar protein loading was confirmed by probing for GAPDH. (B–D) Effect of vehicle (fresh growth media) or TLR9 agonist (ODN-2006 CpG, 1.0 μM) treatment with or without PAMAM-G3 (25 μg/mL) on nuclear translocation of NF-κB in (B) SUM149 cells, (C) MDA-MB-231 TNBC cells, and (D) HEK293-hTLR9 control cells (n = 3 for all conditions). Bar graphs depict mean ± SEM. ∗∗∗∗p < 0.0001; ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05; and N.S., not significant, by one-way ANOVA with a Holm-Šidák multiple comparison post-test.
To determine the functional consequences of these TLR9-expressing breast cancer cells and evaluate their response to TLR9 agonists, we analyzed activation by assessing the status of the downstream signaling mediator NF-κB.58, 59, 60, 61 To monitor NF-κB activation, nuclear cell lysates were generated from the TNBC cell lines SUM149 and MDA-MB-231 and compared to nuclear cell lysates from HEK293-hTLR9. These cells had been treated with or without a TLR9 agonist (CpG ODN-2006). As shown in Figures 1B–1D, addition of CpG DNA resulted in a significant increase in nuclear levels of NF-κB (measured via an ELISA probe of the RelA p65 subunit of the NF-κB protein complex), demonstrating that the TLR9 on the cancer cells responds to CpG DNA treatment. In addition, treatment with the NAS PAMAM-G3 mitigates the activation of TLR9 by CpG DNA (Figures 1B–1D). These results were recapitulated in both the TNBC (MDA-MB-231) and TLR9 reporter (HEK293-hTLR9) cell lines (Figures 1C and 1D). These observations indicate that PAMAM-G3 treatment leads to a reduction in downstream activation of NF-κB through the blocking of TLR9 signaling in these TNBC cells.
Chemotherapy treatment of breast cancer increases DNA-containing DAMP levels, and NAS treatment counteracts DAMP-mediated activation of TLR9
Next, we evaluated if TNBC TLR9-positive cells release their DNA following chemotherapeutic treatment. We quantified the amount of cfDNA generated by two TNBC cell lines, MDA-MB-231 and SUM149, that were treated with a chemotherapy regimen (Figure 2A). Such treatment caused the breast cancer cells to release significantly higher levels of cfDNA into the culture media after treatment with chemotherapy (4, 12, and 24 h post) as compared to untreated cancer cells (∼4–8 times as much; Figures 2B and 2C). Chemotherapy-treated (Figure 2B) MDA-MB-231 and (Figure 2C) SUM149 cells released high levels of cfDNA, as measured in chemotherapy-derived conditioned media (CM), so the 12-h-treated SUM149 CM and the 24-h MDA-MB-231 CM were used in subsequent experiments as a source of in vitro TNBC-derived NA DAMPs.
Figure 2.

Chemotherapy treatment of breast cancer increases DNA-containing DAMP levels, and NAS treatment counteracts DAMP-mediated activation of TLR9
(A) Schematic depicting treatment of TNBC cells with chemotherapy (doxorubicin and docetaxel) to obtain conditioned media (CM). (B and C) Triple-negative cell lines (MDA-MB-231 and SUM149) were treated with standard-of-care chemotherapy in vitro followed by cfDNA quantification via a PicoGreen dsDNA assay. (D–G) TLR stimulation was measured using a HEK293-TLR reporter assay. (D) Effects of PAMAM-G3 (50 μg/mL) and the TLR9 inhibitor (A151, 50 μM) treatments on TLR9 stimulation in the presence of a synthetic TLR9 agonist (ODN-2006 CpG, 10 μM). (E and F) Stimulation of DNA-sensing TLR9 activation by the synthetic TLR9 agonist (ODN-2006, 10 μM) and by 15% CM from chemotherapy-treated TNBC (E) MDA-MB-231 or (F) SUM149 cells, or the (G) activation of TLR9 reporter cells by four post-NAC patient plasma samples (2%) from representative breast cancer subtypes in combination with NAS PAMAM-G3 (50 μg/mL) (n = 5–6 replicate wells per condition). Experiments were repeated at least three times, and figures depict a single representative experiment. Bar graphs depict mean ± SEM. ∗∗∗∗p < 0.0001; ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05; and N.S., not significant, by one-way ANOVA with a Holm-Šidák multiple comparison post-test.
To determine if such samples activate TLR9, we employed human TLR9-overexpressing HEK293 reporter cells (HEK293-hTLR9). As expected, treatment of the reporter cells with a known TLR9 agonist, CpG (ODN 2006), led to an increase in TLR9 activation. This activation was reversed by PAMAM-G3 treatment to a similar degree as by a known synthetic oligonucleotide TLR9 inhibitor, ODN A151 (Figure 2D). Next, we evaluated the effects of PAMAM-G3 treatment on TLR9 signaling when the reporter cells were treated with DAMPs derived from TNBC chemotherapy-treated cells. As shown in Figure 2E (MDA-MB-231) and Figure 2F (SUM149), the TNBC-derived DAMPs are able to activate the TLR9 reporter cells also. Importantly, PAMAM-G3 treatment was able to significantly reduce TLR9 activation by the chemotherapy-derived NA DAMPs released from treated TNBC cells.
Lastly, we evaluated whether clinically relevant NA DAMPs present in post-neoadjuvant chemotherapy (post-NAC) plasma collected from breast cancer patients activate TLR9 and if NAS treatment can mitigate such activation. Again, using HEK293-hTLR9 reporter cells, we screened a small panel of four breast cancer post-NAC patient plasma samples to determine if they can activate TLR9. We observed that both post-NAC TNBC patient plasma and luminal B (ER+/PR−/HER2+) patient plasma strongly induced TLR9 signaling (Figure 2G; Figure S1). Next, we evaluated whether the NAS PAMAM-G3 could impede such activation. As shown in Figure 2G, in all four patients, PAMAM-G3 treatment reduced the ability of breast cancer patient plasma to activate the TLR9 pathway as measured by NF-κB activation. These results indicate that post-NAC patient plasma contains NA DAMPs that can induce TLR9 activation, and this proinflammatory process can be counteracted by PAMAM-G3 treatment.
Chemotherapy-derived CM and post-NAC breast cancer patient plasma induce TNBC invasion, and NAS treatment mitigates this pathological process
Once we determined that chemotherapy-derived TNBC CM and post-NAC breast cancer patient plasma were rich in NA DAMPs that activate TLR9 (Figure 2) and that TNBC cells lines such as SUM149 and MDA-MB-231 express functional TLR9 (Figure 1), we hypothesized that in vitro treatment of TNBC cells with a TLR9 agonist would induce a pro-invasive phenotype. Invasive potential was measured using a Transwell-Matrigel invasion assay as detailed in Figure 3A. We tested SUM149 and MDA-MB-231 cell invasion potential when incubated with CpG DNA or TNBC chemotherapy-derived CM in a Transwell-Matrigel invasion assay. As shown in Figures 3B and 3C (and the corresponding quantification in Figures 3E and 3F), treatment with either CpG DNA or TNBC chemotherapy CM enhanced breast cancer cell invasion. However, the significant increase in the number of invaded cells in the presence of TLR9 agonist and TNBC chemotherapy-derived CM was reversed to baseline by PAMAM-G3 treatment (Figures 3B and 3C and corresponding Figures 3E and 3F). Similar results were obtained using several other human and murine TNBC cell lines (Figure S2).
Figure 3.

Chemotherapy-derived CM and post-neoadjuvant chemotherapy (post-NAC) breast cancer patient plasma induce TNBC invasion, and NAS treatment mitigates this pathological process
(A) Schematic of Transwell-Matrigel invasion assay used to measure and quantify invasive potential. (B–D) Photographs of invaded cells after listed treatments. (E and F) Quantification of invasion of TNBC cell lines SUM149 and MDA-MB-231 after addition of a TLR9 agonist (ODN 2006, 10 μM) or TNBC chemo-derived CM (15%) with or without PAMAM-G3 (50 μg/mL). (G) Quantification of invasion of SUM149 cells after addition of a TLR9 agonist (ODN 2006, 10 μM), pooled normal human plasma (5%), or post-NAC breast cancer patient plasma (TNBC and luminal B, 5%) with or without PAMAM-G3 (50 μg/mL) (n = 3 replicate wells per condition). Experiments were repeated at least three times, and figures depict a single representative experiment. HPF, high-powered field (6–8 view fields). Inverted light microscope at 10× magnification. Bar graphs depict mean ± SEM. ∗∗∗∗p < 0.0001; ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05; and N.S., not significant, by one-way ANOVA with a Holm-Šidák multiple comparison post-test.
Next, we examined if TNBC post-NAC patient plasma induces a similar pro-invasive response in SUM149 cells. In addition, we wanted to evaluate the effect of PAMAM-G3 treatment in this setting. SUM149 cells were incubated in the presence of post-NAC TNBC patient plasma or pooled normal human plasma with or without PAMAM-G3, and invasive potential was measured using the Transwell-Matrigel invasion assay (Figures 3D and 3G). Post-NAC TNBC patient plasma induced cancer invasion, while normal plasma did not. PAMAM-G3 was once again able to largely abrogate the TNBC patient plasma-mediated increase in TNBC cell invasion. Similar results were obtained when using post-NAC luminal B patient plasma (Figures 3D and 3G). Thus, TLR9-positive TNBC cells are responsive to TLR9 agonists released by chemotherapy-treated TNBC cell lines and breast cancer patients, and NAS treatment can counteract such responses ex vivo.
NAS treatment reduces lung metastasis in a genetically engineered murine model of spontaneous breast cancer
Given the effects of PAMAM-G3 in reducing NA DAMPs signaling and cancer cell invasion, and since we had previously shown that the NAS could reduce lung metastatic burden in artificial orthotopic experimental murine models,56 we next sought to evaluate PAMAM-G3’s efficacy in a more clinically relevant spontaneous murine model of breast cancer. We utilized the aggressive mouse mammary tumor virus-polyoma middle T antigen mouse strain (MMTV-PyMT; FVB background), which contains the polyoma virus middle T-antigen as a transgene expressed from the MMTV LTR promoter. Hemizygous females undergo a course of mammary gland neoplasia that resembles aggressive human breast cancer pathogenesis62, 63, 64 and ultimately develop lung metastases (Figure S3). As shown in Figure 4A, hemizygous MMTV-PyMT females develop breast cancer in multiple mammary glands, but their transgene-negative sisters (identified via PCR genotyping as seen in Figure S4) do not. Hemizygous female mice were treated with either saline or PAMAM-G3 (2×/week, 20 mg/kg, intraperitoneal (i.p.) injections) from 5 weeks of age to 12 weeks of age, then sacrificed.
Figure 4.

NAS treatment reduces lung metastasis in a genetically engineered murine model of spontaneous breast cancer
(A) Representative image of mice splayed to reveal the mammary fat pads of a tumor-bearing hemizygous MMTV-PyMT female compared to an age-matched FVB female sibling at 10 weeks old. Yellow arrows indicate several tumor lesions. Mice (5 weeks old) were injected biweekly with saline (N = 14) or PAMAM-G3 (20 mg/kg; N = 13) intraperitoneally before sacrifice at 12 weeks. (B) Histological evaluation of metastatic lesions via hematoxylin and eosin (H&E) staining of lungs. Microscope scale at 5× magnification is 500 μm. (C) Cumulative weight of mammary glands at 12-week sacrifice. (D) Quantification of gross lung mass after saline or PAMAM-G3 treatment. Treatment groups included untreated FVB female mice (Normal), untreated MMTV-PyMT mice (PyMT), saline-treated and PAMAM-G3-treated MMTV-PyMT mice (PyMT + Saline and PyMT + PAMAM-G3, respectively). (E) Quantification of metastatic tumor burden in the lungs via total lesion surface area from saline and PAMAM-G3-treated mice. Bar graphs depict mean ± SEM. ∗∗∗∗p < 0.0001; ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05; and N.S., not significant, by an unpaired t test with a Welch’s post-test.
Primary tumor burden was evaluated by measuring the gross mass of excised mammary glands from each treated animal (Figure 4C). PAMAM-G3 treatment did have a modest but significant reductive effect on the primary tumor burden in the mammary glands. However, histological analyses revealed a marked reduction in metastatic lung lesions in the lungs of PAMAM-G3-treated mice compared to saline-treated mice (Figure 4B). This observation was confirmed by significantly lower lung masses and metastatic lung surface areas (Figures 4D and 4E) in PAMAM-G3-treated mice as compared to saline-treated mice.
Discussion
In this study, we found that treatment with chemotherapy leads to an increase in NA DAMP release from breast cancer cells in culture and into circulation in patients with breast cancer. Moreover, CM from TNBC cells treated with chemotherapy and plasma from breast cancer patients treated with chemotherapy robustly stimulate human TLR9 activation and signaling in reporter cells and induce a pro-invasive phenotype in human TNBC cells. Both of these NA DAMP-mediated effects can be mitigated by PAMAM-G3 treatment. Importantly, PAMAM-G3 treatment significantly decreased lung metastasis in a genetically engineered mouse model of spontaneous breast cancer. The decrease in lung metastasis observed after PAMAM-G3 treatment in such an aggressive breast cancer mouse model suggests that effective blocking of NA DAMP-induced inflammation with a NAS could be a valuable therapeutic approach to limit breast cancer metastasis, particularly when utilized in combination with the current standard-of-care chemotherapy, which, as demonstrated in this study, may itself promote metastatic spread. Use of a NAS (such as PAMAM-G3) as an early adjuvant therapy may be an effective approach to help limit breast cancer metastasis.
Systemic cancer therapies have been reported to induce aberrant inflammation32,65, 66, 67 and play a role in the enhanced metastatic behavior seen in the treatment-resistant clones that survive the initial therapeutic culling.68,69 Therefore, the characterization of disease-specific DAMPs that contribute to tumor progression and metastasis is of clinical importance (bearing in mind that a variety of DAMPs can activate TLR and other innate immune system signaling cascades such as HMGB-1 and histone proteins). Unfortunately, the redundant nature of DAMPs and pattern recognition receptors (PRRs) likely means that defining a complete mechanistic understanding of their actions in a complex tumor-immunocompetent setting will be challenging.70 Nevertheless, the potential to broadly block NA DAMP-induced inflammation with a NAS may be of significant therapeutic benefit, since some tumors tend to develop drug resistance via redundant signaling nodes, clonal heterogeneity, and immunomodulation—thus making their treatment unlikely to depend on one concomitant signaling pathway. Nonetheless, it is likely that a NAS-based protocol that targets inflammation-induced metastasis will require combination with other therapeutic agents for the clinical management of various malignancies.71, 72, 73
Previously, we have observed that PAMAM-G3 treatment can limit metastasis in syngeneic mouse models of pancreatic and breast cancer metastasis.36,56 The studies described herein build upon these studies that found that PAMAM-G3 treatment could limit artificial metastasis from the spleen to the liver (ectopic pancreatic cancer model) or mammary fat pad to the lung (orthotopic breast cancer model). Thus, these results significantly extend and corroborate prior studies and advance the field by moving it from artificial, non-spontaneous breast cancer animal models into a highly aggressive spontaneous cancer mouse model and moreover demonstrate that breast cancer patient samples induce similar NA DAMP-engendered activities in cell culture, which can be mitigated by NAS treatment. This study provides additional evidence about the importance of TLR9 signaling in mediating invasion and metastasis of breast cancer and describes an innovative chemical biology approach using nucleic-acid-binding polymers to scavenge such TLR9 agonists and mitigate such pathological effects. By targeting a phenotype that has been implicated in various cancer contexts, namely persistent nucleic acid DAMP-induced cell activation and inflammation, the NAS approach impedes an important signaling axis in difficult-to-treat inflammation-dependent malignancies. Given the lack of effective treatments for metastatic TNBC, this project highlights a potentially significant complementary treatment that could be utilized to reduce the mortality seen in this breast cancer patient population and facilitate the development of a novel class of anti-metastatic therapies using NASs.
Materials and methods
Reagents
PAMAM-G3 solution was purchased from Sigma Aldrich and used in the in vitro experiments. The PAMAM-G3 used for the treatment of mice was purchased from Dendritech. The TLR agonists CpG ODN-2006, Poly I:C, LPS, and TLR antagonist ODN-A151 were purchased from InvivoGen.
Cell culture
Human breast cancer cell lines SUM149 (triple-negative inflammatory breast cancer) and MDA-MB-231 (TNBC) were generously gifted by Dr. Neil Spector (Duke University School of Medicine) and purchased from ATCC, respectively. The following cell growth media were used for each cell line: SUM149 (HAM/F-12 plus 1% L-glutamine, 5 μg/mL insulin, 1 μg/mL hydrocortisone, 5% non-heat-inactivated fetal bovine serum [FBS]), and MDA-MB-231 (MEM plus 10% non-heat inactivated FBS).
Method details
TLR activation assays
HEK-Blue TLR 9 reporter cell lines were purchased from InvivoGen, and activation in response to control agonists or human plasma was determined according to the manufacturer’s instructions. These HEK293 cells have been engineered to stably co-express a TLR gene and an NF-κB-inducible SEAP (secreted embryonic alkaline phosphatase) reporter gene that can be monitored using SEAP detection media. Briefly, these cells were plated in 96-well, clear-bottom, flat-bottom plates at a density of 25,000 cells per well with at least 3–5 wells per condition. The cells were then treated for 18–24 h with either (1) growth media alone, (2) a TLR control agonist for each given TLR (LPS [0.1–1.0 μg/mL] for TLR4 and CpG ODN [0.25–5.0 μM] for TLR9), (3) TNBC patient plasma (2%), (4) pooled normal human plasma (2%), (5) PAMAM-G3 (25 or 50 μg/mL) alone, (6) TLR control agonist + PAMAM-G3 (25 or 50 μg/mL), (7) cancer patient plasma (2%) + PAMAM-G3 (25 or 50 μg/mL), or (8) normal patient plasma (2%) + PAMAM-G3 (25 or 50 μg/mL) in a final volume of 100 μL. After this incubation period, the cell supernatant was collected and mixed with Quanti-Blue (InvivoGen) at a 60:40 volumetric ratio and incubated for 2–6 h at 37°C in a new 96-well plate, after which the absorbance at 655 nm was measured using a Spectramax i3 plate reader (Molecular Devices).
Quantification of cfDNA levels
Total DNA was isolated from plasma using the DNA Blood Mini Kit (QIAGEN) or from therapy-treated CM supernatant from TNBC cell lines and quantified using the Quant-iT PicoGreen dsDNA Staining Kit (Thermo Fisher Scientific).
Analysis of nuclear NF-κB translocation
SUM149 cells were plated at ∼2 × 106 cells per plate in T-75 flasks overnight. The cells were then treated with serum-containing media alone, CpG ODN 2006 (10 μM), and PAMAM-G3 (50 μg/mL) for 4 h. Cell nuclear lysates were then isolated using a nuclear isolation kit (Abcam) according to the manufacturer’s guidelines. The protein content of each nuclear extract was quantified and standardized using a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). The nuclear extract was then used to quantify nuclear translocation of NF-κB using the p65 RelA subunit ELISA kit (Abcam) according to the manufacturer’s guidelines.
Western blot
Protein lysates were prepared by using Pierce RIPA lysis and extraction buffer (Thermo Fisher Scientific) containing Halt Protease and Phosphatase inhibitors (1×) on 80%–90% confluent T-75 flasks of each cell line. 250 μL of lysis buffer was added to the cells, and then they were scraped into clean microcentrifuge tubes and placed on ice for 10–15 min. Tubes were then agitated at 4°C for 15 min. Then the tubes were centrifuged for 30 min at 4°C. The supernatant was then transferred to fresh centrifuge tubes, and the protein content was quantified and standardized using a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). Immunoblotting was carried out using standard protocols with ≥30 μg of total protein mixed with Laemmli sample buffer (Bio-Rad) in each well of the Mini-PROTEAN TGX gels (Bio-Rad). Gels were run at 220–250 V for 30–40 min. Activate LF-PVDF membranes in high-performance liquid chromatography (HPLC)-grade methanol for 5 min. Gels were then transferred to LF-PVDF membranes using the Trans-Blot Turbo Transfer System (Bio-Rad). Antibodies in 3% w/v BSA in 1× TBST were then used to probe the membranes. The following membranes were purchased from Cell Signaling Technology (β-actin, GAPDH) or Santa Cruz Biotechnology (TLR4, TLR9). Blots were then visualized on a Bio-Rad ChemiDoc Imager.
Transwell-Matrigel invasion chamber assays
Transwell-Matrigel inserts and 24-well plates were purchased from Corning, and invasion assays were performed according to the manufacturer’s instructions. The invasion inserts were first incubated with serum-free media in the top and bottom chambers at 37°C for at least 2–4 h to allow the Matrigel to equilibrate and solidify before the addition of the TNBC cells and their various treatment conditions (CM, agonists, PAMAM-G3). The human TNBC SUM149 cell line was trypsinized, inactivated with serum-rich media, centrifuged for 5 min at 300 × g, and then resuspended in serum-free media at the correct dilution. After aspiration of serum-free media from the equilibrated invasion chambers, SUM149 cells were plated into the top chamber at a density of 35,000 cells/well (500 μL final volume) in the presence of serum-free media alone or various combinations of agonists and/or PAMAM-G3 (25 or 50 μg/mL). TLR agonists tested included CpG ODN-2006 (10 μM), TNBC patient plasma, or normal human plasma (25 μL). A total of 750 μL of complete growth media was added to the bottom chambers to create a nutrient gradient. All conditions were tested in at least duplicate. The plated invasion chambers were incubated at 37°C for 22–26 h, after which time media was aspirated from the top chambers.
The top chambers were then removed and placed into a new 24-well plate preloaded with 1 mL of 10% formaldehyde per well to fix the invaded cells on the bottom surface of the chamber. After fixation for 10 min, the top chambers were then removed from the formaldehyde and placed into a new 24-well plate preloaded with 1 mL of PBS (without divalent cations) per well for 1 min for washing purposes. The top chambers were then placed onto absorbent pads, and residual PBS was wiped from the inside of the top chambers using a cotton swab. 10 μL of crystal violet solution (5% w/v crystal violet, 25% v/v methanol) was added to the membranes and allowed to stain for 5–10 min. Crystal violet was purchased from Sigma-Aldrich. The chambers were then washed thoroughly in deionized water, and residual crystal violet was wiped from the inside of the chambers using a cotton swab. The membranes were then imaged using an inverted light microscope (Olympus IX50) at 10× magnification. A total of 6–8 random image view fields of each membrane were taken using a digital eyepiece camera (Dino-Eye AM7023B). The cells in the images were then counted using an ImageJ algorithm unique for each cell line (https://imagej.net/Particle_Analysis).
Genetically engineered breast cancer murine model
All animal experiments were performed in compliance with the Duke Institutional Animal Care and Use Committee (IACUC) protocols and housed in an animal facility at Duke University (Durham, NC, USA). Male MMTV-PyMT transgenic mice (stock no: 002374) and female FVB/NJ mice (stock no: 001800) were purchased from The Jackson Laboratory. A breeding colony was established and maintained by crossing male transgenics to congenic FVB female mice. PCR analysis of DNA prepared from a tail snip was used to identify carriers possessing the MMTV-PyMT transgene (Figure S4). A positive control fragment (200 bp) was amplified in all samples, and, as expected, a transgene fragment (556 bp) amplified in approximately half of the samples. Starting 5 weeks post birth, hemizygous MMTV-PyMT females were treated intraperitoneally twice per week with either PAMAM-G3 (20 mg/kg) or saline until mice were euthanized at 12 weeks of age. Initially, each treatment group contained 15 mice each, and there were no associated PAMAM-G3 deaths. One mouse from each treatment group was removed due to having outlier measurements beyond 3 standard deviations of the mean of said treatment group. An additional PAMAM-G3 mouse was excluded due to having red lungs of double the usual mass, and histological evaluation indicating evidence of a hemorrhage. Mammary glands and lungs were harvested from each mouse for assessment of gross organ mass. Lungs were fixed in formalin, and hematoxylin and eosin (H&E) staining was performed on three lung sections per animal, with 100 μm between slices (HistoWiz). With blinding as to treatment status, each metastatic lesion was identified, and then associated surface area determined based on the section where the lesion was largest using ImageJ.
Quantification and statistical analysis
For all assays and experiments, statistical comparison between treatment groups was first tested for normality via the Shapiro-Wilk test. If normal, the groups were then compared via a two-tailed Student’s t test or an ANOVA with an appropriate post hoc test. If the data were not normal, then they were log-transformed and re-tested for normality. If normal, data were analyzed by a two-tailed Student’s t test or an ANOVA with a Holm-Šídák or Tukey’s test. If not normal, the data were then tested with the non-parametric Fisher’s exact test. All data were plotted and statistically analyzed using GraphPad Prism (version 6.0 or 8.3) or JMP software. All data were presented as the mean ± standard error determined from 3–5 technical replicates. All biological experiments were performed at least twice.
Acknowledgments
This work was supported in part by the US National Institutes of Health (NIH) pre-doctoral fellowship award F31CA232394 (E.O.U.E.), the NIH Translation Research in Surgical Oncology award T32CA093245 (K.L.), the NIH Advanced Immunobiology Training Program for Surgeons award T32AI141342 (K.L.), the US Department of Defense (DoD) Breast Cancer Research Program awards W81XWH-16-1-0512 (S.K.N) and W81XWH-16-1-0513 (B.A.S), and the National Cancer Center post-doctoral award (I.A.N.). We thank Neil Spector, MD for providing the SUM149 inflammatory triple-negative breast cancer cell line.
Acknowledgments
Author contributions
Conceptualization of project and study resources was by S.K.N. and B.A.S. Overall study design and data review were by S.K.N. and B.A.S. E.O.U.E., K.L., and R.E.R. designed and performed the experiments and analysis. I.A.N. established the experimental protocols utilized in several experiments. E.O.U.E wrote the manuscript. Human breast cancer plasma samples were provided by E.S.H. and processed by K.L. Manuscript was edited by K.L., R.E.R., S.K.N., and B.A.S.
Declaration of interests
The authors declare no competing interests. However, Duke University has submitted patents on the applications of nucleic acid scavengers to combat inflammation and cancer.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.06.016.
Contributor Information
Smita K. Nair, Email: smita.nair@duke.edu.
Bruce A. Sullenger, Email: bruce.sullenger@duke.edu.
Supplemental information
References
- 1.American Cancer Society . 2020. Cancer statistics factsheets: Breast cancer. Cancer Statistics Center.https://cancerstatisticscenter.cancer.org/#/cancer-site/Breast [Google Scholar]
- 2.Vargo-Gogola T., Rosen J.M. Modelling breast cancer: one size does not fit all. Nat. Rev. Cancer. 2007;7:659–672. doi: 10.1038/nrc2193. [DOI] [PubMed] [Google Scholar]
- 3.Valastyan S., Weinberg R.A. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147:275–292. doi: 10.1016/j.cell.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klimov S., Rida P.C.G., Aleskandarany M.A., Green A.R., Ellis I.O., Janssen E.A.M., Rakha E.A., Aneja R. Novel immunohistochemistry-based signatures to predict metastatic site of triple-negative breast cancers. Br. J. Cancer. 2017;117:826–834. doi: 10.1038/bjc.2017.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steeg P.S. Targeting metastasis. Nat. Rev. Cancer. 2016;16:201–218. doi: 10.1038/nrc.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson R.L., Balasas T., Callaghan J., Coombes R.C., Evans J., Hall J.A., Kinrade S., Jones D., Jones P.S., Jones R., et al. Cancer Research UK and Cancer Therapeutics CRC Australia Metastasis Working Group A framework for the development of effective anti-metastatic agents. Nat. Rev. Clin. Oncol. 2019;16:185–204. doi: 10.1038/s41571-018-0134-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peinado H., Zhang H., Matei I.R., Costa-Silva B., Hoshino A., Rodrigues G., Psaila B., Kaplan R.N., Bromberg J.F., Kang Y., et al. Pre-metastatic niches: organ-specific homes for metastases. Nat. Rev. Cancer. 2017;17:302–317. doi: 10.1038/nrc.2017.6. [DOI] [PubMed] [Google Scholar]
- 8.Zardavas D., Baselga J., Piccart M. Emerging targeted agents in metastatic breast cancer. Nat. Rev. Clin. Oncol. 2013;10:191–210. doi: 10.1038/nrclinonc.2013.29. [DOI] [PubMed] [Google Scholar]
- 9.Foulkes W.D., Smith I.E., Reis-Filho J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010;363:1938–1948. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 10.Lehmann B.D., Jovanović B., Chen X., Estrada M.V., Johnson K.N., Shyr Y., Moses H.L., Sanders M.E., Pietenpol J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE. 2016;11:e0157368. doi: 10.1371/journal.pone.0157368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hartman Z.C., Poage G.M., den Hollander P., Tsimelzon A., Hill J., Panupinthu N., Zhang Y., Mazumdar A., Hilsenbeck S.G., Mills G.B., Brown P.H. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res. 2013;73:3470–3480. doi: 10.1158/0008-5472.CAN-12-4524-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lim B., Woodward W.A., Wang X., Reuben J.M., Ueno N.T. Inflammatory breast cancer biology: the tumour microenvironment is key. Nat. Rev. Cancer. 2018;18:485–499. doi: 10.1038/s41568-018-0010-y. [DOI] [PubMed] [Google Scholar]
- 13.American Cancer Society . 2016. Treatment of Inflammatory Breast Cancer.http://www.cancer.org/cancer/breast-cancer/treatment/treatment-of-inflammatory-breast-cancer.html [Google Scholar]
- 14.Volk-Draper L., Hall K., Griggs C., Rajput S., Kohio P., DeNardo D., Ran S. Paclitaxel therapy promotes breast cancer metastasis in a TLR4-dependent manner. Cancer Res. 2014;74:5421–5434. doi: 10.1158/0008-5472.CAN-14-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Echeverria G.V., Ge Z., Seth S., Zhang X., Jeter-Jones S., Zhou X., Cai S., Tu Y., McCoy A., Peoples M., et al. Resistance to neoadjuvant chemotherapy in triple-negative breast cancer mediated by a reversible drug-tolerant state. Sci. Transl. Med. 2019;11:eaav0936. doi: 10.1126/scitranslmed.aav0936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li X., Yang J., Peng L., Sahin A.A., Huo L., Ward K.C., O’Regan R., Torres M.A., Meisel J.L. Triple-negative breast cancer has worse overall survival and cause-specific survival than non-triple-negative breast cancer. Breast Cancer Res. Treat. 2017;161:279–287. doi: 10.1007/s10549-016-4059-6. [DOI] [PubMed] [Google Scholar]
- 17.Gonçalves H., Jr., Guerra M.R., Duarte Cintra J.R., Fayer V.A., Brum I.V., Bustamante Teixeira M.T. Survival Study of Triple-Negative and Non-Triple-Negative Breast Cancer in a Brazilian Cohort. Clin. Med. Insights Oncol. 2018;12 doi: 10.1177/1179554918790563. 1179554918790563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hudis C.A., Gianni L. Triple-negative breast cancer: an unmet medical need. Oncologist. 2011;16(Suppl 1):1–11. doi: 10.1634/theoncologist.2011-S1-01. [DOI] [PubMed] [Google Scholar]
- 19.Schmid P., Adams S., Rugo H.S., Schneeweiss A., Barrios C.H., Iwata H., Diéras V., Hegg R., Im S.-A., Shaw Wright G., et al. IMpassion130 Trial Investigators Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018;379:2108–2121. doi: 10.1056/NEJMoa1809615. [DOI] [PubMed] [Google Scholar]
- 20.Heidel J.D., Schluep T. Cyclodextrin-containing polymers: versatile platforms of drug delivery materials. J. Drug Deliv. 2012;2012:262731. doi: 10.1155/2012/262731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pack D.W., Hoffman A.S., Pun S., Stayton P.S. Design and development of polymers for gene delivery. Nat. Rev. Drug Discov. 2005;4:581–593. doi: 10.1038/nrd1775. [DOI] [PubMed] [Google Scholar]
- 22.Davis M.E., Chen Z.G., Shin D.M. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat. Rev. Drug Discov. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 23.Haensler J., Szoka F.C., Jr. Polyamidoamine cascade polymers mediate efficient transfection of cells in culture. Bioconjug. Chem. 1993;4:372–379. doi: 10.1021/bc00023a012. [DOI] [PubMed] [Google Scholar]
- 24.Liang H., Peng B., Dong C., Liu L., Mao J., Wei S., Wang X., Xu H., Shen J., Mao H.-Q., et al. Cationic nanoparticle as an inhibitor of cell-free DNA-induced inflammation. Nat. Commun. 2018;9:4291. doi: 10.1038/s41467-018-06603-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ilvesaro J.M., Merrell M.A., Li L., Wakchoure S., Graves D., Brooks S., Rahko E., Jukkola-Vuorinen A., Vuopala K.S., Harris K.W., Selander K.S. Toll-like receptor 9 mediates CpG oligonucleotide-induced cellular invasion. Mol. Cancer Res. 2008;6:1534–1543. doi: 10.1158/1541-7786.MCR-07-2005. [DOI] [PubMed] [Google Scholar]
- 26.Cook D.N., Pisetsky D.S., Schwartz D.A. Toll-like receptors in the pathogenesis of human disease. Nat. Immunol. 2004;5:975–979. doi: 10.1038/ni1116. [DOI] [PubMed] [Google Scholar]
- 27.Zemek R.M., De Jong E., Chin W.L., Schuster I.S., Fear V.S., Casey T.H., Forbes C., Dart S.J., Leslie C., Zaitouny A., et al. Sensitization to immune checkpoint blockade through activation of a STAT1/NK axis in the tumor microenvironment. Sci. Transl. Med. 2019;11:eaav7816. doi: 10.1126/scitranslmed.aav7816. [DOI] [PubMed] [Google Scholar]
- 28.Pandey S., Singh S., Anang V., Bhatt A.N., Natarajan K., Dwarakanath B.S. Pattern Recognition Receptors in Cancer Progression and Metastasis. Cancer Growth Metastasis. 2015;8:25–34. doi: 10.4137/CGM.S24314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jain S., Pitoc G.A., Holl E.K., Zhang Y., Borst L., Leong K.W., Lee J., Sullenger B.A. Nucleic acid scavengers inhibit thrombosis without increasing bleeding. Proc. Natl. Acad. Sci. USA. 2012;109:12938–12943. doi: 10.1073/pnas.1204928109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holl E.K., Shumansky K.L., Borst L.B., Burnette A.D., Sample C.J., Ramsburg E.A., Sullenger B.A. Correction for Holl et al., Scavenging nucleic acid debris to combat autoimmunity and infectious disease. Proc. Natl. Acad. Sci. USA. 2016;113:E6545. doi: 10.1073/pnas.1607011113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holl E.K., Bond J.E., Selim M.A., Ehanire T., Sullenger B., Levinson H. The nucleic acid scavenger polyamidoamine third-generation dendrimer inhibits fibroblast activation and granulation tissue contraction. Plast. Reconstr. Surg. 2014;134:420e–433e. doi: 10.1097/PRS.0000000000000471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee J., Jackman J.G., Kwun J., Manook M., Moreno A., Elster E.A., Kirk A.D., Leong K.W., Sullenger B.A. Nucleic acid scavenging microfiber mesh inhibits trauma-induced inflammation and thrombosis. Biomaterials. 2017;120:94–102. doi: 10.1016/j.biomaterials.2016.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee J., Sohn J.W., Zhang Y., Leong K.W., Pisetsky D., Sullenger B.A. Nucleic acid-binding polymers as anti-inflammatory agents. Proc. Natl. Acad. Sci. USA. 2011;108:14055–14060. doi: 10.1073/pnas.1105777108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dawulieti J., Sun M., Zhao Y., Shao D., Yan H., Lao Y.H., Hu H., Cui L., Lv X., Liu F., et al. Treatment of severe sepsis with nanoparticulate cell-free DNA scavengers. Sci. Adv. 2020;6:eaay7148. doi: 10.1126/sciadv.aay7148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng B., Liang H., Li Y., Dong C., Shen J., Mao H.Q., Leong K.W., Chen Y., Liu L. Tuned Cationic Dendronized Polymer: Molecular Scavenger for Rheumatoid Arthritis Treatment. Angew. Chem. Int. Ed. Engl. 2019;58:4254–4258. doi: 10.1002/anie.201813362. [DOI] [PubMed] [Google Scholar]
- 36.Naqvi I., Gunaratne R., McDade J.E., Moreno A., Rempel R.E., Rouse D.C., Herrera S.G., Pisetsky D.S., Lee J., White R.R., Sullenger B.A. Polymer-Mediated Inhibition of Pro-invasive Nucleic Acid DAMPs and Microvesicles Limits Pancreatic Cancer Metastasis. Mol. Ther. 2018;26:1020–1031. doi: 10.1016/j.ymthe.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tuomela J., Sandholm J., Karihtala P., Ilvesaro J., Vuopala K.S., Kauppila J.H., Kauppila S., Chen D., Pressey C., Härkönen P., et al. Low TLR9 expression defines an aggressive subtype of triple-negative breast cancer. Breast Cancer Res. Treat. 2012;135:481–493. doi: 10.1007/s10549-012-2181-7. [DOI] [PubMed] [Google Scholar]
- 38.Kim I.S., Gao Y., Welte T., Wang H., Liu J., Janghorban M., Sheng K., Niu Y., Goldstein A., Zhao N., et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat. Cell Biol. 2019;21:1113–1126. doi: 10.1038/s41556-019-0373-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Akira S., Takeda K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 40.Rakoff-Nahoum S., Medzhitov R. Toll-like receptors and cancer. Nat. Rev. Cancer. 2009;9:57–63. doi: 10.1038/nrc2541. [DOI] [PubMed] [Google Scholar]
- 41.Kawasaki T., Kawai T. Toll-like receptor signaling pathways. Front. Immunol. 2014;5:461. doi: 10.3389/fimmu.2014.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lemaitre B. The road to Toll. Nat. Rev. Immunol. 2004;4:521–527. doi: 10.1038/nri1390. [DOI] [PubMed] [Google Scholar]
- 43.Vogel S., Bodenstein R., Chen Q., Feil S., Feil R., Rheinlaender J., Schäffer T.E., Bohn E., Frick J.S., Borst O., et al. Platelet-derived HMGB1 is a critical mediator of thrombosis. J. Clin. Invest. 2015;125:4638–4654. doi: 10.1172/JCI81660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hawes M.C., Wen F., Elquza E. Extracellular DNA: A bridge to cancer. Cancer Res. 2015;75:4260–4264. doi: 10.1158/0008-5472.CAN-15-1546. [DOI] [PubMed] [Google Scholar]
- 45.Jorch S.K., Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017;23:279–287. doi: 10.1038/nm.4294. [DOI] [PubMed] [Google Scholar]
- 46.Leal A., van Grieken N.C.T., Palsgrove D.N., Phallen J., Medina J.E., Hruban C., Broeckaert M.A.M., Anagnostou V., Adleff V., Bruhm D.C., et al. White blood cell and cell-free DNA analyses for detection of residual disease in gastric cancer. Nat. Commun. 2020;11:525. doi: 10.1038/s41467-020-14310-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhong X.Y., Ladewig A., Schmid S., Wight E., Hahn S., Holzgreve W. Elevated level of cell-free plasma DNA is associated with breast cancer. Arch. Gynecol. Obstet. 2007;276:327–331. doi: 10.1007/s00404-007-0345-1. [DOI] [PubMed] [Google Scholar]
- 48.Razavi P., Li B.T., Brown D.N., Jung B., Hubbell E., Shen R., Abida W., Juluru K., De Bruijn I., Hou C., et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat. Med. 2019;25:1928–1937. doi: 10.1038/s41591-019-0652-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mouliere F., Chandrananda D., Piskorz A.M., Moore E.K., Morris J., Ahlborn L.B., Mair R., Goranova T., Marass F., Heider K., et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci. Transl. Med. 2018;10:1–14. doi: 10.1126/scitranslmed.aat4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tuomela J., Sandholm J., Kaakinen M., Patel A., Kauppila J.H., Ilvesaro J., Chen D., Harris K.W., Graves D., Selander K.S. DNA from dead cancer cells induces TLR9-mediated invasion and inflammation in living cancer cells. Breast Cancer Res. Treat. 2013;142:477–487. doi: 10.1007/s10549-013-2762-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tavora B., Mederer T., Wessel K.J., Ruffing S., Sadjadi M., Missmahl M., Ostendorf B.N., Liu X., Kim J.-Y., Olsen O., et al. Tumoural activation of TLR3-SLIT2 axis in endothelium drives metastasis. Nature. 2020;586:299–304. doi: 10.1038/s41586-020-2774-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McCoy M.G., Nascimento D.W., Veleeparambil M., Murtazina R., Gao D., Tkachenko S., Podrez E., Byzova T.V. Endothelial TLR2 promotes proangiogenic immune cell recruitment and tumor angiogenesis. Sci. Signal. 2021;14:5371. doi: 10.1126/scisignal.abc5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aswani A., Manson J., Itagaki K., Chiazza F., Collino M., Wupeng W.L., Chan T.K., Wong W.S.F., Hauser C.J., Thiemermann C., Brohi K. Scavenging circulating mitochondrial DNA as a potential therapeutic option for multiple organ dysfunction in trauma hemorrhage. Front. Immunol. 2018;9:891. doi: 10.3389/fimmu.2018.00891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nabet B.Y., Qiu Y., Shabason J.E., Wu T.J., Yoon T., Kim B.C., Benci J.L., DeMichele A.M., Tchou J., Marcotrigiano J., Minn A.J. Exosome RNA Unshielding Couples Stromal Activation to Pattern Recognition Receptor Signaling in Cancer. Cell. 2017;170:352–366.e13. doi: 10.1016/j.cell.2017.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chalmers S.A., Eidelman A.S., Ewer J.C., Ricca J.M., Serrano A., Tucker K.C., Vail C.M., Kurt R.A. A role for HMGB1, HSP60 and Myd88 in growth of murine mammary carcinoma in vitro. Cell. Immunol. 2013;282:136–145. doi: 10.1016/j.cellimm.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Holl E.K., Frazier V., Landa K., Boczkowski D., Sullenger B., Nair S.K. Controlling cancer-induced inflammation with a nucleic acid scavenger prevents lung metastasis in murine models of breast cancer. Mol. Ther. 2021;29:1772–1781. doi: 10.1016/j.ymthe.2020.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Merrell M.A., Ilvesaro J.M., Lehtonen N., Sorsa T., Gehrs B., Rosenthal E., Chen D., Shackley B., Harris K.W., Selander K.S. Toll-like receptor 9 agonists promote cellular invasion by increasing matrix metalloproteinase activity. Mol. Cancer Res. 2006;4:437–447. doi: 10.1158/1541-7786.MCR-06-0007. [DOI] [PubMed] [Google Scholar]
- 58.Grinberg-Bleyer Y., Ghosh S. A Novel Link between Inflammation and Cancer. Cancer Cell. 2016;30:829–830. doi: 10.1016/j.ccell.2016.11.013. [DOI] [PubMed] [Google Scholar]
- 59.Nomura A., Gupta V.K., Dauer P., Sharma N.S., Dudeja V., Merchant N., Saluja A.K., Banerjee S. NFκB-Mediated Invasiveness in CD133+ Pancreatic TICs Is Regulated by Autocrine and Paracrine Activation of IL1 Signaling. Mol. Cancer Res. 2018;16:162–172. doi: 10.1158/1541-7786.MCR-17-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 61.Pikarsky E., Porat R.M., Stein I., Abramovitch R., Amit S., Kasem S., Gutkovich-Pyest E., Urieli-Shoval S., Galun E., Ben-Neriah Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 62.Maglione J.E., Moghanaki D., Young L.J.T., Manner C.K., Ellies L.G., Joseph S.O., Nicholson B., Cardiff R.D., MacLeod C.L. Transgenic Polyoma middle-T mice model premalignant mammary disease. Cancer Res. 2001;61:8298–8305. [PubMed] [Google Scholar]
- 63.Lin E.Y., Jones J.G., Li P., Zhu L., Whitney K.D., Muller W.J., Pollard J.W. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am. J. Pathol. 2003;163:2113–2126. doi: 10.1016/S0002-9440(10)63568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guy C.T., Cardiff R.D., Muller W.J. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol. Cell. Biol. 1992;12:954–961. doi: 10.1128/mcb.12.3.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dutta P., Sarkissyan M., Paico K., Wu Y., Vadgama J.V. MCP-1 is overexpressed in triple-negative breast cancers and drives cancer invasiveness and metastasis. Breast Cancer Res. Treat. 2018;170:477–486. doi: 10.1007/s10549-018-4760-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Scaffidi P., Misteli T., Bianchi M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 67.Pidgeon G.P., Harmey J.H., Kay E., Da Costa M., Redmond H.P., Bouchier-Hayes D.J. The role of endotoxin/lipopolysaccharide in surgically induced tumour growth in a murine model of metastatic disease. Br. J. Cancer. 1999;81:1311–1317. doi: 10.1038/sj.bjc.6694369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee S.H., Hu W., Matulay J.T., Silva M.V., Owczarek T.B., Kim K., Chua C.W., Barlow L.J., Kandoth C., Williams A.B., et al. Tumor Evolution and Drug Response in Patient-Derived Organoid Models of Bladder Cancer. Cell. 2018;173:515–528.e17. doi: 10.1016/j.cell.2018.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Verma V., Shrimali R.K., Ahmad S., Dai W., Wang H., Lu S., Nandre R., Gaur P., Lopez J., Sade-Feldman M., et al. PD-1 blockade in subprimed CD8 cells induces dysfunctional PD-1+CD38hi cells and anti-PD-1 resistance. Nat. Immunol. 2019;20:1231–1243. doi: 10.1038/s41590-019-0441-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schoenfeld J.D. We Are All Connected: Modeling the Tumor-Immune Ecosystem. Trends Cancer. 2018;4:655–657. doi: 10.1016/j.trecan.2018.08.006. [DOI] [PubMed] [Google Scholar]
- 71.Ngwa W., Irabor O.C., Schoenfeld J.D., Hesser J., Demaria S., Formenti S.C. Using immunotherapy to boost the abscopal effect. Nat. Rev. Cancer. 2018;18:313–322. doi: 10.1038/nrc.2018.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Poggio M., Hu T., Pai C.-C., Chu B., Belair C.D., Chang A., Montabana E., Lang U.E., Fu Q., Fong L., Blelloch R. Suppression of Exosomal PD-L1 Induces Systemic Anti-tumor Immunity and Memory. Cell. 2019;177:414–427.e13. doi: 10.1016/j.cell.2019.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Binnewies M., Roberts E.W., Kersten K., Chan V., Fearon D.F., Merad M., Coussens L.M., Gabrilovich D.I., Ostrand-Rosenberg S., Hedrick C.C., et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018;24:541–550. doi: 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.