

Abstract
Non-coding RNAs play essential roles in breast cancer progression by regulating proliferation, differentiation, invasion, and metastasis. However, our understanding of most microRNAs (miRNAs) and long noncoding RNAs (lncRNAs) in breast cancer is still limited. miR-586 has been identified as an important factor in the progression of some types of cancer, but its exact function and relative regulation mechanisms in breast cancer development need to be further investigated. In this study, we showed miR-586 functioned as an oncogene by promoting breast cancer proliferation and metastasis both in vitro and in vivo. Meanwhile, miR-586 induced Wnt/β-catenin activation by directly targeting Wnt/β-catenin signaling antagonists SFRP1 and DKK2/3. Moreover, we demonstrated that LINC01189 functioned as a tumor suppressor and inhibited breast cancer progression through inhibiting an epithelial-mesenchymal transition (EMT)-like phenotype by sponging miR-586. In addition, β-catenin/TCF4 transactivated ZEB1, resulting in a transcriptional repression of LINC01189 expression. In conclusion, our data uncovered the LINC01189-miR-586-ZEB1 feedback loop and provided a novel mechanism participating in the regulation of Wnt/β-catenin signaling in breast cancer progression.
Keywords: breast cancer, Wnt/β-catenin signaling, miR-586, LINC01189, ZEB1
Graphical abstract
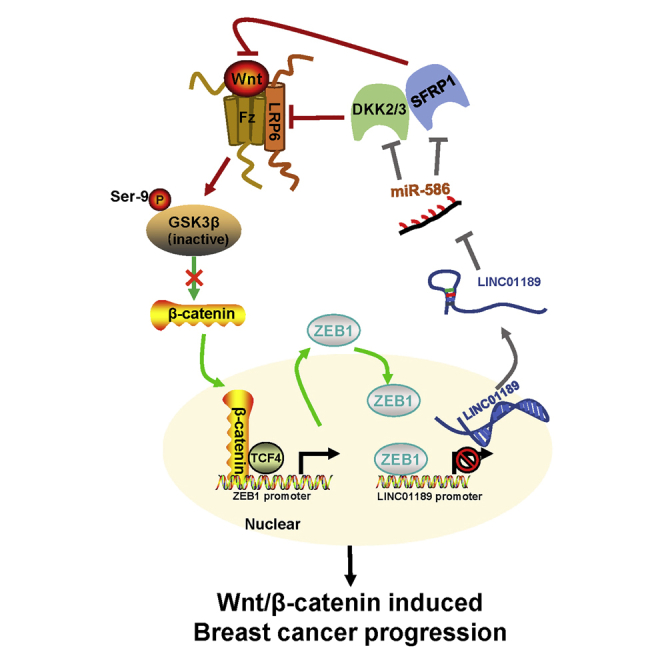
miR-586 activates Wnt/β-catenin signaling by downregulation of Wnt/β-catenin antagonists SFRP1 and DKK2/3. LINC01189 inhibits breast cancer progression and epithelial-mesenchymal transition-like phenotype by sponging miR-586. In addition, β-catenin/TCF4 transactivated ZEB1, resulting in a transcriptional repression of LINC01189. The LINC01189-miR-586-ZEB1 feedback loop is involved in regulation of Wnt/β-catenin signaling in breast cancer progression.
Introduction
Breast cancer is the most frequent malignant tumor and 2nd leading cause of cancer-associated mortality in females.1 Although diagnostic and therapeutic improvements have prolonged the overall survival of patients with breast cancer, recurrence and metastasis are still critical challenges in breast cancer therapy.2,3 Thus, it is essential to comprehensively explore the molecular mechanisms and genetic signatures underlying breast cancer tumorigenesis and progression, which can contribute to developing early diagnostic methods and improve the prognosis in patients with breast cancer.
Metastasis is a hallmark of malignant tumors in which cancer cells disseminate from primary tumor and form new colonies at distant sites, experiencing an invasion-metastasis cascade through a multi-step process, which includes growth of the primary tumor, local invasion, dissemination and intravasation, extravasation into a new microenvironment, and colonization in distant organs.4, 5, 6, 7, 8 Epithelial-mesenchymal transition (EMT), an indispensable metastasis-related process, endows cancer cells with migration and invasion abilities that lead to cancer progression.9 It is known that Wnt/β-catenin signaling is involved in multiple biological processes, including embryonic development, tissue regeneration, organogenesis, hematopoiesis, proliferation, differentiation, and stem cell renewal.10,11 Dysregulation of the Wnt/β-catenin signaling pathway drives carcinogenesis and progression in all human cancers, including breast cancer.12, 13, 14 Wnt/β-catenin signaling contributes to breast cancer progression through inducing EMT and cancer cell stemness.15,16 As a Wnt-regulated cell adhesion molecule, E-cadherin is observed to decrease during EMT. Three major EMT transcription factor families have been confirmed to repress E-cadherin expression, including TWIST, SNAI, and ZEB.17 Among them, ZEB1 is almost inversely associated with E-cadherin in all types of cancer.18,19 Although several studies revealed a relationship between the regulation of ZEB1 and the Wnt/β-catenin signaling pathway,20,21 the regulatory mechanism of ZEB1 in Wnt/β-catenin signaling still needs to be clarified during breast cancer progression.
MicroRNAs (miRNAs), a class of non-coding RNAs having a length of 18–23 nucleotides, were mainly directed to mRNAs for translational repression or degradation via binding to the 3′ untranslated regions (UTRs) of their target mRNA.22 Dysregulation of miRNAs is closely correlated with tumorigenesis and progression in all human cancers.23 miR-586 is located on chromosome 6p21.1 and was reported to regulate the dentin sialophosphoprotein after transcription during odontoblast differentiation.24 It has been identified as an oncogene in osteosarcoma, by which knockdown of miR-586 can inhibit tumor growth and metastasis.25,26 However, to the best of our knowledge, the role of miR-586 in breast cancer progression is still unknown.
Long non-coding RNAs (lncRNAs) are also a class of non-coding RNAs with transcripts of >200 nucleotides in length. lncRNAs can directly interact with proteins, DNA, and RNA to regulate protein functions, induce chromatin remodeling and histone modification, act as a scaffold, and modulate protein stability and DNA methylation.27 A broad spectrum of evidence demonstrates that aberrantly expressed lncRNAs are associated with many types of human cancer and can function as either oncogenes or tumor suppressors to exert great influence on breast cancer development and progression.28,29 Long intergenic non-protein coding RNA 1189 (LINC01189) belongs to intergenic lncRNAs originating from the region between protein-coding gene FGF7P6 and CNTNAP3P5 located on chromosome 9q13. Although LINC01189 was reported to be a tumor suppressor in hepatocellular carcinoma,30 the role of LINC01189 in other cancers remains unclear. Meanwhile, the relationship between the regulation of LINC01189 and the changes of the Wnt/β-catenin signaling pathway also needs to be further explored.
In the current study, we aimed to investigate the biological function and regulatory mechanism of miR-586 and LINC01189 in breast cancer progression. We observed an increased miR-586 expression in breast cancer, and its expression can predict prognosis in patients with breast cancer. miR-586 contributed to breast cancer progression by targeting Wnt antagonists DKK2/3 and SFRP1, inducing Wnt/β-catenin signaling activation. In addition, LINC01189 suppressed breast cancer progression by sponging miR-586, thereby leading to an inhibitory effect on Wnt/β-catenin signaling activation. Furthermore, ZEB1 transcriptionally repressed LINC01189 expression and can be transactivated by the β-catenin/TCF4 complex. Our data revealed the existence of a feedback loop between ZEB1 and miR-586 in the regulation of Wnt/β-catenin signaling during breast cancer progression.
Results
miR-586 functions as an oncogene in breast cancer progression
To explore the potential role of miR-586 in breast cancer progression, we first examined miR-586 expression in seven human breast cancer cell lines (MDA-MB-231, MDA-MB-468, BT549, SKBR3, BT474, T47D, and MCF7) and a normal breast cell line (MCF10A) by quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR). As shown in Figure 1A, the levels of miR-586 were significantly upregulated in breast cancer cell lines compared to MCF10A. Similarly, miR-586 expression levels were significantly higher in most of the breast cancer specimens compared to paired adjacent normal breast specimens (Figure 1B). Next, we analyzed the prognosis of patients with breast cancer between different miR-586 expression levels by KM-plotter and found that higher expression levels of miR-586 prominently correlated to greater overall decreased survival rate in patients with breast cancer (Figure 1C).
Figure 1.

miR-586 functioned as an oncogene in breast cancer
(A) qRT-PCR analysis of miR-586 expression in normal breast cell line MCF10A and breast cancer lines. (B) qRT-PCR analysis of miR-586 expression in breast cancer and the corresponding normal breast specimens. (C) The association between miR-586 expression and overall survival of patients with breast cancer analyzed by Kaplan-Meier. (D) Knockdown efficiency of miR-586 in stable miR-586-depleted MDA-MB-231 cells determined by qRT-PCR. (E and F), MTT (E) and colony-formation (F) analyses of the proliferation of the cells described in (D). (G) Transwell analysis measuring invasion ability of the cells described in (D). (H) Wound-healing assay measuring the migration ability of the cells described in (D). (I) Tumor growth curves of subcutaneous tumor made of miR-586-depleted MDA-MB-231 or control cells at the indicated times and dissected tumors photographed at the time of harvest. (J) Lung metastasis of mice injected with miR-586-depleted MDA-MB-231 or control cells via tail vein were evaluated by living imaging and quantification plot. ∗p < 0.05.
Next, we investigated whether miR-586 depletion can influence breast cancer progression. MDA-MB-231 was used to generate stable miR-586-depleted cells (231-anti-586) and control cells (231-control) via lentiviral infection (Figure 1D). Proliferation assays indicated that downregulation of miR-586 can inhibit MDA-MB-231 cell viability and colony formation ability in vitro by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Figure 1E) and colony formation (Figure 1F), respectively. Moreover, Transwell (Figure 1G) and wound-healing (Figure 1H) assays indicated that downregulation of miR-586 can suppress MDA-MB-231 cell migration and invasion abilities. Similar results were also observed in the experiments using the BT549 cell line (Figure S1). To evaluate the effect of miR-586 expression on tumor growth and metastasis in vivo, 231-control and 231-anti-586 cells were injected into mouse mammary fat pads (Figure 1I) or tail vein (Figure 1J), respectively. Tumor growth and metastasis were measured at 35 days post injection. As hypothesized, we observed that orthotopic tumor volume and lung metastasis were significantly decreased in the group of miR-586 downregulation compared to those in the control group. Together, these results elucidated that miR-586 serves as an oncogene in breast cancer.
miR-586 can induce Wnt/β-catenin signaling activation
To further explore the signaling pathways that miR-586 regulated, we investigated the effects of miR-586 overexpression on NF-κB, TGF-β, Wnt, Notch, and Hedgehog signaling activity. Overexpression of miR-586 notably induced Wnt signaling activation in both MCF7 and SKBR3 cell lines (Figure 2A). As accumulation of β-catenin in nucleus is a hallmark of activated Wnt/β-catenin signaling,31 we examined the effect of miR-586 expression on β-catenin nuclear translocation. As shown in Figure 2B, nuclear β-catenin was increased in miR-586-overexpressed MCF7 and SKBR3 cell lines according to immunofluorescence analysis. In addition, analysis of cytoplasmic and nuclear extracts by western blot showed that miR-586 overexpression significantly induced its nuclear translocation in both MCF7 and SKBR3 cell lines (Figure 2C, upper panel). However, miR-586 overexpression did not alter the total β-catenin expression (Figure 2C, lower panel). Correlation studies in 20 specimens from patients with breast cancer showed that miR-586 expression levels were remarkably associated with the expression of nuclear β-catenin (r = 0.408, p = 0.037; Figure 2D). We next assessed the effect of miR-586 expression on the interaction between β-catenin and TCF4, the most prominent TCF-LEF in breast cancer. We observed that miR-586 overexpression significantly enhanced the interaction between β-catenin and TCF4 in both MCF7 and SKBR3 cell linesby immunoprecipitation (IP) analysis (Figure 2E). Together, these data showed that miR-586 can induce Wnt/β-catenin signaling activation in breast cancer.
Figure 2.

miR-586 induced Wnt/β-catenin signaling activation in breast cancer
(A) Luciferase analysis of TGF-β, NF-κB, Wnt, Notch, and Hedgehog signaling activity in the indicated cells. (B) Effect of miR-586 on the nuclear localization of β-catenin in the indicated cells, as determined by immunofluorescence staining. (C) Subcellular fractionation and subsequent western blot assays were performed to reveal the effect of miR-586 on the subcellular distribution of β-catenin in the indicated cells. (D) Immunohistochemical staining analysis expression level of β-catenin in breast cancer tissues with high or low miR-586 expression. The correlation between expression of miR-586 and nuclear localization shown with percentage of specimens. (E) The effect of miR-586 on the interactions of β-catenin with TCF4 in the indicated cells, as determined by IP. ∗p < 0.05.
miR-586 can suppress multiple Wnt/β-catenin signaling antagonists
To elucidate the mechanisms underlying the regulation of miR-586 on Wnt/β-catenin signaling, we predicted the potential target molecules of miR-586 by using TargetScan. We found several antagonists of the Wnt/β-catenin signaling pathway, including SFRP1, DKK2/3, WIF1, NKD1, RPRD1A, and APC, were potential targets of miR-586 (Figure 3A). We next examined the expression of these antagonists by qRT-PCR and found that the expression of SFRP1, DKK2/3, and WIF1 were downregulated in miR-586-overexpressed MCF7 and SKBR3 cells (Figure 3B). However, miR-586 only specifically associated with SFRP1 and DKK2/3 in both MCF7 and SKBR3 cell lines according to micro ribonucleoprotein (miRNP) immunoprecipitation analysis (Figure 3C). miR-586 overexpression significantly decreased the luciferase activities driven by the 3′ UTRs of these transcripts (Figure 3D). We also found that the expression levels of SFRP1 and DKK2/3 were both decreased in miR-586-overexpressed MCF7 and SKBR3 cells and were both increased in miR-586-depleted MDA-MB-231 and BT549 cells (Figure 3E). Furthermore, individual overexpression of SFRP1 and DKK2/3 prominently repressed Wnt/β-catenin signaling activity in miR-586-overexpressed cells (Figure 3F). Overall, these results indicated that miR-586 can suppress multiple Wnt/β-catenin signaling antagonists.
Figure 3.

miR-586 suppressed multiple Wnt/β-catenin signaling antagonists
(A) The potential target gene of miR-586 predicted by TargetScan. (B) Expression of SFRP1, DKK2/3, WIF1, NKD1, RPRD1A, and APC in stable miR-586-overexpressed MCF7 and SKBR3, as well as the control cells by qRT-PCR. (C) miRNP immunoprecipitation analysis of the interaction between SFRP1, DKK2/3, and WIF1 mRNA and miRNP complexes following immunoprecipitation with Ago2. (D) Dual-luciferase reporter analysis was used to verify SFRP1 and DKK2/3 as the miR-586 targets. A 3′ UTR region containing the predicted miR-586 target site of SFRP1 or DKK2/3 and SFRP1 or DKK2/3 mutated reporter plasmids were constructed. The constructs were transfected into 293FT cells with or without miR-586 mimics, and the luciferase activity was measured at 48 h after transfection. (E) Western blot analysis of the expression levels of SFRP1 or DKK2/3 in miR-586-overexpressed MCF7 and SKBR3 or miR-586-depleted MDA-MB-231 and BT549 cells, as well as in control cells. (F) Dual-luciferase analysis of TCF/LEF transcriptional activity in SFRP1- or DKK2/3-expressing cells. ∗p < 0.05.
LINC01189 acted as a sponge for miR-586
Accumulating evidence indicates that lncRNAs could serve as competing endogenous RNAs (ceRNAs) by sponging miRNAs to relieve the repression of their target genes. We observed several potential lncRNAs had putative binding sites to miR-586 by DIANA-LncBase, including LINC01189, MAGI2-AS3, MIR4313, SOX2-OT, etc. (Figure 4A). We chose LINC01189 to test the association between LINC01189 and miR-586 based on the binding score (Figure 4B). To confirm whether LINC01189 can regulate miR-586 by ceRNA, we first assessed the subcellular localization of LINC01189. The results showed that LINC01189 mainly expressed in the cytoplasm in SKBR3 cells by fluorescence in situ hybridization (FISH) (Figure 4C) and qRT-PCR (Figure 4D). To further confirm whether miR-586 can bind to the LINC01189, we constructed the wild-type (LINC-wt) and miR-586 binding site mutant type (LINC-mut) LINC01189 luciferase reporters (Figure 4B) and then transfected them into 293FT cells with miR-586 or the corresponding control. The luciferase activity of LINC-wt, but not the LINC-mut, in miR-586-overexpressed 293FT cells was significantly reduced compared to that in control cells (Figure 4E). Additionally, an RNA immunoprecipitation assay indicated that LINC01189 and miR-586 were enriched in AGO-containing complexes, suggesting that both LINC01189 and miR-586 bound to AGO2 in breast cancer cells (Figure 4F). We next analyzed LINC01189 expression in human breast cancer cell lines and the MCF10A cell line by qRT-PCR. We observed that a dramatically higher expression level of LINC01189 was found in the MCF10A cell line, which was oppositely related to the miR-586 expression (Figures 4G and 1A). The expression of miR-586 was significantly decreased in LINC01189-overexpressed MCF7 and SKBR3 cells (Figure 4H). Finally, correlation analysis revealed that LINC01189 expression was negatively associated with miR-586 expression in 20 specimens of breast cancer tissues (Figure 4I). Collectively, these data showed that LINC01189 can function as a molecular sponge of miR-586 in breast cancer.
Figure 4.

LINC01189 acted as a sponge for miR-586 in cytoplasm
(A) DIANA-LncBase predicted the lncRNAs that sponge miR-586. (B) The predicted binding site of miR-586 with LINC01189. (C) Subcellular localization of LINC01189 (green) in SKBR3 cells by FISH analysis. DAPI was used to stain nuclei. (D) Relative LINC01189 expression levels in nuclear and cytosolic fractions of SKBR3 cells. U6 acted as nuclear control; GAPDH acted as cytosolic control. (E) The interaction between miR-586 and LINC01189 was validated by dual-luciferase reporter. (F) RNA immunoprecipitation analysis of endogenous AGO2 binding to RNA in SKBR3 cells; IgG acted as the negative control. The expression levels of LINC01189 and miR-586 were detected by qRT-PCR, and the results were shown with fold enrichment in AGO2 relative to input. (G) qRT-PCR analysis of LINC01189 expression in normal breast cell line and breast cancer cell lines. (H) Expression of miR-586 in LINC01189-overexpressed MCF7 and SKBR3, as well as the control cells, by qRT-PCR. (I) The correlation between miR-586 and LINC01189 expression analyzed in 20 cases of breast cancer specimens. ∗p < 0.05.
LINC01189 suppressed breast cancer progression
We next investigated the role of LINC01189 in breast cancer progression. We established the stable LINC01189-overexpressed MDA-MB-231 cells and control cells via lentiviral infection (Figure 5A). Both MTT and colony-formation assays showed that LINC01189 overexpression inhibited MDA-MB-231 cell proliferation in vitro (Figures 5B and 5C). Moreover, the Transwell (Figure 5D) and wound-healing (Figure 5E) assays indicated that overexpression of LINC01189 suppressed MDA-MB-231 cell migration and invasion. To examine the effect of LINC01189 expression on breast cancer progression in vivo, 231-LINC01189 and 231-control cells were injected into the mammary fat pads (Figure 5F) or the tail vein (Figure 5G) of severe combined immunodeficiency (SCID) mice. Tumor growth and metastasis were measured 35 days post injection. As hypothesized, we observed that tumor volume and lung metastasis were significantly reduced when LINC01189 was overexpressed compared to those in control cells (Figures 5F and 5G). We next compared the recurrence-free survival in patients with breast cancer at different LINC01189 expression levels and found that patients with high LINC01189 expression had a more favorable prognosis than those with low LINC01189 expression levels (Figure 5H).
Figure 5.

LIN01189 suppressed breast cancer progression
(A) Relative expression of LINC01189 in stable LINC01189-expressing MDA-MB-231 cells compared with the control cells determined by qRT-PCR. (B and C) MTT (B) and colony-formation (C) analyses of the proliferation of cells described in (A). (D) Transwell analysis of the invasion of cells described in (A). (E) wound-healing analysis of the migration of cells described in (A). (F) Tumor growth curves for subcutaneous tumor made of LINC01189-expressing MDA-MB-231 or control cells at the indicated times, and dissected tumors photographed at the time of harvest. (G) Bioluminescent imaging and quantification plot of mice harboring lung metastases after tail vein injection of LINC01189-expressing MDA-MB-231 or control cells. (H) Kaplan-Meier analysis of the recurrence-free survival of patients with different LINC01189 expression levels. (I) Morphology of cells as in (A). (J) Western blot was used to detect the expression of epithelial marker E-cadherin and mesenchymal marker vimentin in the cells described in (A). (K) The expression levels of epithelial marker E-cadherin and mesenchymal marker vimentin in the cells as in (A) were detected by immunofluorescence. (L) Immunohistochemical staining was performed to detect the expression of epithelial marker E-cadherin and mesenchymal marker vimentin in LINC01189-expressing MDA-MB-231 and control subcutaneous tumors. ∗p < 0.05.
To elucidate the role of LINC01189 on cancer cell EMT phenotype, we further investigated the morphological changes and vimentin expression using LINC01189-overexpressed MDA-MB-231. We observed that LINC01189-overexpressed MDA-MB-231 cells exhibited a cobblestone-like morphology, whereas 231-control cells retained their spindle-shaped and fibroblast-like morphology (Figure 5I). Furthermore, the expression of epithelial marker E-cadherin was raised, whereas the expression of mesenchymal marker vimentin was reduced in LINC01189-pverexpressing MDA-MB-231 cells by western blot (Figure 5J) and immunofluorescence (Figure 5K). Immunohistochemical staining further identified the downregulation of vimentin and upregulation of E-cadherin in tumors from 231-LINC01189 mice compared with those from 231-control mice (Figure 5L). Next, we transfected small interfering RNAs (siRNAs) targeting LINC01189 in SKBR3 cells with or without miR-586 inhibitor, and the LINC01189 or miR-586 knockdown efficiencies were confirmed by qRT-PCR (Figures S2A and S2B). We observed that knockdown of miR-586 eliminates LINC0065 depletion-induced breast cancer progression (Figures S2C–S2F) and EMT phenotype (Figure S2G). Together, these results suggested that LIN01189 can suppress breast cancer progression by regulation of miR-586.
ZEB1 transcriptionally repressed LINC01189 expression
EMT is mainly regulated by multiple transcription factors, including ZEB1/2, SNAI1/2, TWIST1/2, SOX4, and FOXQ1. We investigated whether these transcription factors can regulate LINC01189 expression. The expression of LINC01189 was downregulated in MCF7 cells overexpressing TWIST1, SNAI1, and ZEB1 compared with that in control cells by qRT-PCR (Figure 6A). Previous studies indicated that these EMT-related transcription factors can mutually regulate each other. To confirm the direct transcription factors that regulated LINC01189 expression, we conducted three groups of experiments. One of the three transcription factors was downregulated by corresponding siRNA, and one of the other two transcription factors was upregulated by overexpression plasmid. Results showed that the expression of LINC01189 was not affected by SNAI1 or TWIST1 in ZEB1-silenced cells (Figure 6B). We next analyzed the sequence of the LINC01189 promoter region and, as hypothesized, found a ZEB1 binding site on the LINC01189 promoter region (Figure 6C). Chromatin immunoprecipitation (ChIP) was used to immunoprecipitate ZEB1 or HA in MDA-MB-231 or MCF7 cells expressing HA-tagged ZEB1 and used primers to amplify the potential binding site of the LINC01189 promoter region. The occupancy by ZEB1 was detected in the LINC01189 promoter region in MDA-MB-231 and ZEB1-overexpressed MCF7 cells (Figure 6D). To further examine whether ZEB1 regulates LINC01189 promoter activity, we transfected the LINC01189 promoter luciferase reporter plasmid into 293FT cells with ZEB1 or the corresponding control. We observed a decreased luciferase activity after ZEB1 overexpression (Figure 6E). We next constructed a mutant clone on which the ZEB1 binding site was mutated from TCACACCTG to TAAAAAATG and used it to transfect the 293FT cells. We observed that this mutant exhibited no altered luciferase activity after transfection with ZEB1 (Figure 6E). Moreover, the correlation studies in 20 specimens of breast cancer tissues showed that LINC01189 expression levels were significantly associated with the expression of ZEB1 (r = −0.800, p < 0.001; Figure 6F). These findings indicated that ZEB1 transcriptionally repressed LINC01189 expression.
Figure 6.

ZEB1 transcriptionally repressed LINC01189 expression
(A) Expression of LINC01189 in MCF7 cells transfected with indicated EMT-related transcription factors, as well as the control cells, by qRT-PCR. (B) qRT-PCR analysis of expression of LINC01189 in indicated MDA-MB-231 cells. (C) Schematic depiction of the LINC01189 promoter. A potential ZEB1 binding site was located on the LINC01189 promoter region. (D) ChIP assay was used to validate interaction between ZEB1 and the LINC01189 promoter region in MDA-MB-231 or ZEB1-overexpressed MCF7 cells. (E) Dual-luciferase reporter analysis of LINC01189 promoter activity in 293FT cells transfected with or without ZEB1. The reporter plasmids of a fragment containing the predicted ZEB1 binding site and the mutated ZEB1 binding site fused upstream of the Luc gene were constructed. (F) Immunohistochemical staining of ZEB1 in breast cancer tissues with high or low LINC01189 expression. ∗p < 0.05.
β-catenin/TCF4 transactivates ZEB1 expression
The above results drove us to investigate the relationship between ZEB1 and β-catenin/TCF4. We first determined the expression of ZEB1 and nuclear β-catenin by immunohistochemical staining and observed that the expression of nuclear β-catenin was significantly associated with ZEB1 expression levels (r = 0.612, p = 0.002; Figure 7A). We next analyzed the promoter sequence of ZEB1 and found a binding site of TCF4 in breast cancer on the ZEB1 promoter region (Figure 7B). We then used ChIP assay to immunoprecipitate TCF4 or β-catenin in MDA-MB-231 and found the occupancy by TCF4 or β-catenin was detected in theZEB1 promoter region in MDA-MB-231 (Figure 7C, left panel). Furthermore, the occupancy by β-catenin was observed in the ZEB1 promoter region in TCF4-overexpressed MCF7 cells, but not in TCF4-depleted MCF7 cells (Figure 7C, left panel). Overexpression of TCF4 and β-catenin enhanced the ZEB1 promoter transcriptional activity, whereas depletion of TCF4 abolished the β-catenin-induced transcription in 293FT cells (Figure 7D, left panel). This effect, however, was not found in TCF4-binding site mutated ZEB1 promoter luciferase reporter (Figure 7D, right panel). The expression of ZEB1 was significantly reduced in TCF4- or β-catenin-depleted MDA-MB-231 cells and upregulated in TCF4- or β-catenin-overexpressed MCF7 cells compared with those in control cells by qRT-PCR (Figure 7E) and western blot (Figure 7F). Leptomycin B, an inhibitor of nuclear export, triggers a shift of β-catenin from cell cytoplasm to the nucleus.32 Accumulation of nuclear β-catenin in MCF7 cells significantly induced ZEB1 expression at both mRNA and protein levels by qRT-PCR and western blot (Figure 7G). Altogether, our results indicated that β-catenin/TCF4 can transactivate ZEB1 expression.
Figure 7.

β-catenin/TCF4 transactivated ZEB1 expression
(A) Immunohistochemical staining of β-catenin in breast cancer tissues with high or low ZEB1 expression. Percentage of specimens representing high or low ZEB1 expression in relation to nuclear localization of β-catenin. (B) Schematic depiction of the ZEB1 promoter. A potential TCF4 binding site was located on the ZEB1 promoter region. (C) ChIP assay was performed to verify the interaction between TCF4/β-catenin and the ZEB1 promoter region in indicated MDA-MB-231 or MCF7 cells. (D) Dual-luciferase reporter analysis of ZEB1 promoter activity in indicated 293FT cells. A fragment containing the predicted TCF4 binding site and the mutated TCF4 binding site was fused upstream of the Luc gene. (E and F) Expression of ZEB1 in TCF4- or β-catenin-depleted MDA-MB-231 cells and TCF4- or β-catenin-overexpressed MCF7 cells, as well as the control cells by qRT-PCR (E) or western blot (F). (G) Expression of ZEB1 in MCF7 cells after treatment with leptomycin B at the indicated times detected by qRT-PCR and western blot. ∗p < 0.05.
Discussion
In this study, we aimed to comprehensively explore the effect of miR-586 on breast cancer progression. We demonstrated that miR-586 can contribute to breast cancer progression and correlate with outcome in patients with breast cancer. miR-586 induced Wnt/β-catenin signaling activation by targeting Wnt/β-catenin signaling antagonists SFRP1 and DKK2/3. In terms of the biological mechanisms, we found that LINC01189 inhibited breast cancer progression and EMT by sponging miR-586. Furthermore, the β-catenin/TCF4 complex transactivated the ZEB1 expression, resulting in transcriptional repression of LINC01189. Overall, our results revealed a novel feedback loop between ZEB1, miR-586, and LINC01189 in regulation of Wnt/β-catenin signaling in breast cancer progression.
Dysregulation of miRNAs has been linked to development and progression in numerous cancers of humans.33,34 Although miR-586 has been identified as an oncogene in osteosarcoma, and knockdown of miR-586 can inhibit cell proliferation and invasion in vitro,26 the role of miR-586 in other types of cancer still remains unclear. Here we demonstrated that miR-586 is an oncogenic miRNA in breast cancer progression, and depletion of miR-586 suppressed breast cancer proliferation and metastasis both in vitro and in vivo. Aberrant activation of Wnt/β-catenin signaling is associated with multiple biological processes in cancer progression, including proliferation, apoptosis, migration, invasion, EMT, and cancer cell stemness.35,36 Since inhibition of Wnt antagonists is important for Wnt/β-catenin signaling activation, it might be possible that miRNAs activate Wnt/β-catenin signaling by targeting multiple Wnt antagonists at the post-transcriptional level.15,37,38 Our findings showed that miR-586 overexpression caused repression of Wnt antagonists DDK2/3 and SFRP1 and resulted in the stimulation of activity of Wnt/β-catenin activation, resulting in breast cancer progression.
As another class of non-coding RNAs, increasing evidence suggests that lncRNAs sponge specific miRNAs and therefore protect their target mRNAs from repression, named ceRNAs. The lncRNA-miRNA-mRNA network has been identified to widely participate during cancer initiation and progression.39 Previous study indicates that LINC01189 downregulation is associated with hepatitis C virus infection in hepatocellular carcinoma and functions as a tumor suppressor by sponging miR-155-5p.30 Consistent with their work, we provided evidence that LINC01189 functioned as a tumor suppressor in breast cancer, and its overexpression suppressed the EMT-like phenotype, resulting in an inhibition of breast cancer progression. Furthermore, LINC01189 was mainly expressed in the cytoplasm and served as a ceRNA to sponge miR-586, suggesting that LINC01189 suppresses breast cancer progression through inhibition of Wnt/β-catenin signaling by sponging miR-586.
ZEB1, a member of the zinc finger homeodomain transcription factor family, is a key inducer of EMT and is involved in regulation of DNA damage, cancer cell differentiation, and metastasis.40 Aberrant expression of ZEB1 has been observed in a variety of types of human cancers, including breast cancer. Moreover, ZEB1 expression is associated with advanced breast cancer, which indicates the relationship between ZEB1 induction of EMT and breast cancer progression.41 Here, we found that ZEB1 functions as a transcriptional repressor, which downregulates LINC01189 expression by directly binding on the LINC01189 promoter. ZEB1 expression is regulated by multiple signaling pathways triggering an EMT.40 In this study, we reported that ZEB1 was directly transactivated by the β-catenin/TCF4 complex, suggesting that ZEB1 was regulated by Wnt/β-catenin signaling and acted as an effector of this signaling in regulating downstream genes associated with breast cancer progression.
Conclusions
In conclusion, we reported that the LINC01189-miR-586-ZEB1 feedback loop was involved in regulation of Wnt/β-catenin signaling in breast cancer progression (Figure 8). Since Wnt/β-catenin signaling plays a pivotal role in cancer development and progression, miR-586 will be a promising target for cancer therapeutics.
Figure 8.
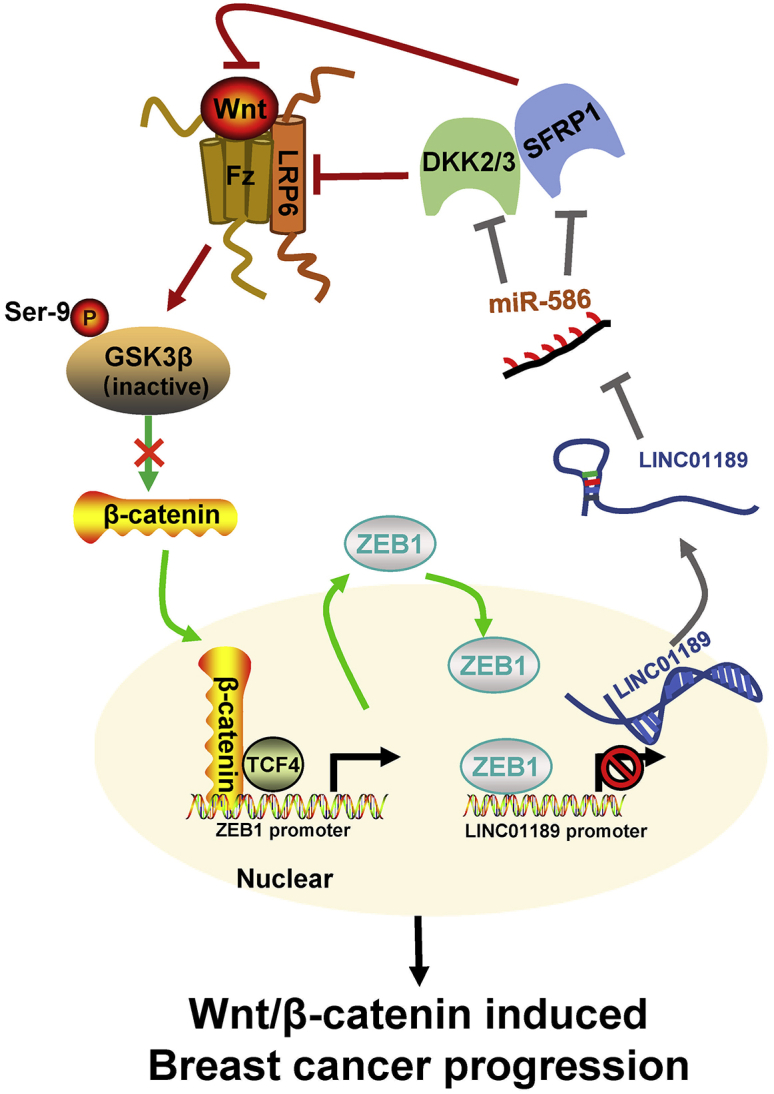
Schematic illustration of miR-586-ZEB1 feedback loop in regulating Wnt/β-catenin signaling during breast cancer progression
Materials and methods
Cell culture and clinical specimens
The T47D, BT474, MCF7, BT549, MDA-MB-468, MDA-MB-231, SKBR3, MCF10A, and 293FT cell lines obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) were cultured at 37°C in 5% CO2 as previously described.42
Tissue samples were obtained from Tianjin Medical University Cancer Institute and Hospital (TMUCIH). 20 cases of breast cancer and the paired normal tissue were included in this study, and related experiments were conducted under the methodology as previously described.43 This study was approved by the Institutional Review Board of TMUCIH, and all participants signed written informed consent.
Plasmids, miRNAs, and siRNAs
Full-length cDNAs of human LINC01189 and ZEB1 were synthesized and cloned into pcDNA3.0 expression vector (Life Technologies). The miR-586 mimic/inhibitor, siRNAs, and the corresponding controls were purchased from RiboBio (China) and are listed in Table S1. For the promoter luciferase reporter assay, the promoter regions of LINC01189 (−2,500 to +1) or ZEB1 (−1,000 to +1) and the promoter region with mutated ZEB1 or TCF4 binding site were synthesized and cloned into pGL3-basic luciferase reporter (Promega). For miRNA target gene luciferase reporter assays, target sequences containing predicted miRNA binding sites or corresponding mutants were synthesized and cloned into the psiCHEK2 luciferase vector.
qRT-PCR
Total RNA was isolated from tissues or cells by using mirVana miRNA Isolation Kit (Life Technologies, Grand Island, NY, USA) or TRIzol Reagent (Life Technologies) according to the standard protocol. For miRNA, reverse transcriptions were performed using the TaqMan miRNA Reverse Transcription Kit (Life Technologies), and the cDNA amplification was performed using the TaqMan miRNA Assay Kit (Life Technologies) according to the manufacturer’s instructions. The expression of mRNA was determined using the GoTaq qPCR Master Mix (Promega, Madison, WI, USA). GAPDH and U6 were used as the endogenous control. Gene expression fold changes were assessed using the 2−ΔCt method. The primers used are listed in Table S2. The median value was chosen as the cut-off value to categorize the samples as high and low miR-586 expression.
Transfection and generation of stable expressed cell lines
All transient transfections were performed using FuGENE HD Transfection Reagent (Promega) according to the instructions. To construct stable cell lines, cells were transfected with specific lentiviruses (RiboBio) for 48 h according to the instructions. Puromycin (2 μg/mL) was used to select the infected cells for 1 week.
MTT and colony-formation assays
For MTT assay, cells were seeded in 96-well plates (2 × 103 per well) after transfection for 48 h. Each well was incubated with 10 μL MTT for 4 h. Then, the medium was discarded, and the crystal violet was dissolved in 150 μL DMSO before the detection of absorbance at 570 nm using a micro-plate reader (Bio-Rad, Richmond, CA, USA). Cell viabilities were observed at indicated times.
For colony-formation assay, 500 cells were seeded in 6-well plates. After incubation at 37°C about 3 weeks, the colonies were fixed with 4% paraformaldehyde (PFA) for 30 min at room temperature and stained with 0.2% crystal violet for 15 min. The number of visible colonies were counted via Adobe Photoshop (version 2020).
Invasion assay
Matrigel-coated Transwell (BD Biosciences, San Diego, CA, USA) was used to evaluate the breast cancer cell invasion abilities in vitro. Briefly, 8 × 104 cells were seeded in the upper chamber with normal medium, and the lower chamber contained culture medium with 20% fetal bovine serum (Life Technologies). After being cultured at 37°C for 16∼24 h, cells in the upper chamber were fixed and dyed using three-step set (Thermo Scientific, Waltham, MA, USA). The invasive cells per field were photographed using an inverted microscope imaging system (Olympus). The number of invasive cells was calculated using Adobe Photoshop (version 2020).
Wound-healing assay
Cells were cultured and grown to 90% confluence in 6-well plates and then cultured overnight in serum-free medium. The cell wound was drawn by a 10 μL pipette tip in a straight line. After washed with PBS, wound healing images were taken immediately via an inverted microscope imaging system (Olympus). Then cells were cultured in medium containing 1% FBS for 24 h. The 24 h images were taken in the same way.
RNA FISH
FISH analysis was carried out using Fluorescent in situ Hybridization Kit (RiboBio) according to the instructions. Cells were washed with PBS three times and fixed in 4% PFA for 15 min. Then cells were permeabilized in ice-cold PBS containing 0.5% Triton X-100 for 30 min. After pre-hybridized in pre-hybridization buffer (Life Technologies) at 55°C for 30 min, hybridization was performed using fluorescence-conjugated LINC01189 probes (RiboBio) in a humidified chamber at 37°C overnight. The next day, the cells were incubated with 4′,6-diamidino-2-phenylindole (DAPI) for 15 min at room temperature. After washing with PBS three times, cells were imaged using a Zeiss fluorescence microscope (Germany). Probe information is listed in Table S3.
Immunofluorescence
Cells were seeded onto glass coverslips in 24-well plates and cultured overnight at 37°C with 5% CO2. The next day, cells were washed with PBS three times and fixed in 4% formaldehyde solution for 30 min. Then cells were permeabilized in ice-cold PBS containing 0.5% Triton X-100 for 15 min. After that, cells were blocked with 3% BSA for 1 h and were incubated with primary antibodies for 3 h, followed by incubation with tetramethylrhodamine-isothiocyanate (TRITC)- or fluorescein isothiocyanate (FITC)-conjugated secondary antibodies for 1 h at room temperature. DAPI was used to determine nuclear stain. Finally, coverslips were observed and imaged using a Zeiss fluorescence microscope.
ChIP-qPCR analysis
ChIP-qPCR analysis was carried out according to the manufacturer’s recommendations (Millipore, Bedford, MA, USA) using anti-ZEB1, anti-TCF4, anti-β-catenin, or an isotype control as previously described.44 Immunoprecipitated DNA was analyzed by qPCR using primers corresponding to the specific region (Table S3), and results were expressed as percentage of input.
Dual-luciferase reporter assay
293FT cells (5 × 104 per well) were seeded in 12-well plates. 200 ng of the indicated firefly luciferase reporter plasmid, 200 ng of expression plasmid, 50 nM siRNAs or miRNAs, and 20 ng of Renilla reporter were transfected into cells with FuGENE HD for 48 h. pRL-TK Renilla reporter was used as a normalization control. The luciferase activities were determined using Luciferase assay system (Promega) according to the instructions.
Western blot
Cells were lysed in RIPA buffer containing 1 mM PMSF (Solarbio, Beijing, China). NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific) were used for the nuclear and cytoplasmic extraction according to the instructions. The protein concentration was measured using BCA Protein Assay Kit (Thermo Fisher Scientific). Protein samples were boiled and then were separated on 8%–10% SDS-PAGE gels, followed by transfer on polyvinylidene fluoride (PVDF) membranes (Millipore). The membranes were blocked for 1 h in 5% (w/v) skimmed milk at room temperature and incubated at 4°C with primary antibody overnight. After washing with TBS-T three times, the membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h at room temperature. Blots were visualized with enhanced chemiluminescence (ECL) reagent (Millipore).
Immunoprecipitation
Cells were lysed in RIPA buffer containing 1 mM PMSF. 1 mg of the cell lysate was incubated at 4°C with primary antibody overnight. The immunoprecipitated proteins were collected using protein A/protein G agarose beads (Santa Cruz, Santa Cruz, CA, USA), washed, and resuspended in immunoprecipitation buffer (50 mmol/L Tris-Cl [pH 7.9], 50 mM NaCl, 0.1 mMEDTA). The boiled samples were detected by western blot as preciously described.
Immunohistochemistry
The breast cancer tissues or tumors of mice were formalin fixed and embedded in paraffin. The thick consecutive sections (4 μm) were obtained by using a rotatory microtome. After deparaffinization, hydration, antigen retrieval, and endogenous peroxidase blocking, each slide was incubated with specific primary antibody overnight at 4°C and then incubated with HRP-conjugated secondary antibody for 1 h at room temperature. Diaminobenzidine was added for coloration. The slides were counterstained with hematoxylin and visualized under an inverted microscope imaging system (Olympus). Two pathologists evaluated the expression of β-catenin, E-cadherin, vimentin, and ZEB1 in a blinded manner. The staining intensity of positive tumor cells was scored with 4 scales: 0 (no staining), 1 (weak staining), 2 (moderate staining), and 3 (strong staining). The percentage of positively stained tumor cells was scored as 0 (<10%), 1 (10%–25%), 2 (26%–50%), 3 (51%–75%), or 4 (>75%). The total immunostaining score was the multiplier of staining intensity and positive percentage, with a range from 0 to 12. A final staining score ≥4 was considered the high expression. For the staining results of β-catenin, nuclear and membranous immunostaining signals were evaluated independently. More than 10% of the tumor cells with nuclear staining were judged as a positive nuclear expression. Either no staining on the membrane or less than 10% of the tumor cells with membranous staining were considered as an abnormal membranous expression.
Xenograft
1 × 107 cells were inoculated subcutaneously into female NOD/SCID/IL2 receptor γ null (NSG) mice (4–6 weeks). The size of the tumor was measured with electronic vernier caliper every 5 days. Tumor volume was calculated by the following formula: tumor volume = (width × length)2/2. All the animals were executed after 35 days, and terminal volume and weight of tumor tissues were measured. NSG mice were injected with breast cancer cells (5 × 105 cells) at the lateral tail vein. The formation of metastasis was assessed every 7 days by bioluminescence imaging using in vivo bioluminescence imaging (Xenogen, Caliper Life Sciences, Hopkinton, MA, USA). All animal experiments were approved by the Animal Ethics Committee of TMUCIH and were performed according to the animal welfare guidelines in cancer research.
Statistical analysis
Data are shown as the mean with standard error. The one-way ANOVA or Student’s t test was used to determine differences between groups. p value < 0.05 was considered statistically significant, and calculations were performed with Statistical Package for Social Science (SPSS for Windows, version 22; SPSS, Chicago, IL, USA).
Acknowledgments
This study was supported by the National Natural Science Foundation of China (no. 82172827, no. 82172835, no. 81502518 and no. 81602926), Changzhou Sci & Tech Program (grant no. CJ20200058), and the Natural Science Foundation of Tianjin City (no. 17JCQNJC10400).
Author contributions
Y.Y. and W.X. designed the study; Z.D., L.Y., Z.P.J., Z.D., S.L., L.S.S., M.Y., and Z.H. performed the experiments and statistical analysis; Y.Y., L.X., W.X., and Z.D. wrote and revised the manuscript. All authors read and approved the final manuscript.
Declaration of Interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.06.007.
Contributor Information
Xin Wang, Email: wangxin@tjmuch.com.
Yue Yu, Email: yuyue@tmu.edu.cn.
Supplemental information
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2020. CA Cancer J. Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.The Lancet Breast cancer targeted therapy: successes and challenges. Lancet. 2017;389:2350. doi: 10.1016/S0140-6736(17)31662-8. [DOI] [PubMed] [Google Scholar]
- 4.Lambert A.W., Pattabiraman D.R., Weinberg R.A. Emerging Biological Principles of Metastasis. Cell. 2017;168:670–691. doi: 10.1016/j.cell.2016.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turajlic S., Swanton C. Metastasis as an evolutionary process. Science. 2016;352:169–175. doi: 10.1126/science.aaf2784. [DOI] [PubMed] [Google Scholar]
- 6.Talmadge J.E., Fidler I.J. AACR centennial series: the biology of cancer metastasis: historical perspective. Cancer Res. 2010;70:5649–5669. doi: 10.1158/0008-5472.CAN-10-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gupta G.P., Massagué J. Cancer metastasis: building a framework. Cell. 2006;127:679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 8.Fidler I.J. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat. Rev. Cancer. 2003;3:453–458. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 9.Bakir B., Chiarella A.M., Pitarresi J.R., Rustgi A.K. EMT, MET, Plasticity, and Tumor Metastasis. Trends Cell Biol. 2020;30:764–776. doi: 10.1016/j.tcb.2020.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schaefer K.N., Peifer M. Wnt/Beta-Catenin Signaling Regulation and a Role for Biomolecular Condensates. Dev. Cell. 2019;48:429–444. doi: 10.1016/j.devcel.2019.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perugorria M.J., Olaizola P., Labiano I., Esparza-Baquer A., Marzioni M., Marin J.J.G., Bujanda L., Banales J.M. Wnt-β-catenin signalling in liver development, health and disease. Nat. Rev. Gastroenterol. Hepatol. 2019;16:121–136. doi: 10.1038/s41575-018-0075-9. [DOI] [PubMed] [Google Scholar]
- 12.Galluzzi L., Spranger S., Fuchs E., López-Soto A. WNT Signaling in Cancer Immunosurveillance. Trends Cell Biol. 2019;29:44–65. doi: 10.1016/j.tcb.2018.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krishnamurthy N., Kurzrock R. Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat. Rev. 2018;62:50–60. doi: 10.1016/j.ctrv.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Schie E.H., van Amerongen R. Aberrant WNT/CTNNB1 Signaling as a Therapeutic Target in Human Breast Cancer: Weighing the Evidence. Front. Cell Dev. Biol. 2020;8:25. doi: 10.3389/fcell.2020.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ren L., Chen H., Song J., Chen X., Lin C., Zhang X., Hou N., Pan J., Zhou Z., Wang L. MiR-454-3p-Mediated Wnt/β-catenin Signaling Antagonists Suppression Promotes Breast Cancer Metastasis. Theranostics. 2019;9:449–465. doi: 10.7150/thno.29055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Z., Wu W., Huang Y., Xie L., Li Y., Chen H., Li W., Yin D., Hu K. RCC2 promotes breast cancer progression through regulation of Wnt signaling and inducing EMT. J. Cancer. 2019;10:6837–6847. doi: 10.7150/jca.36430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dongre A., Weinberg R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019;20:69–84. doi: 10.1038/s41580-018-0080-4. [DOI] [PubMed] [Google Scholar]
- 18.Schmalhofer O., Brabletz S., Brabletz T. E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009;28:151–166. doi: 10.1007/s10555-008-9179-y. [DOI] [PubMed] [Google Scholar]
- 19.Sánchez-Tilló E., Lázaro A., Torrent R., Cuatrecasas M., Vaquero E.C., Castells A., Engel P., Postigo A. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene. 2010;29:3490–3500. doi: 10.1038/onc.2010.102. [DOI] [PubMed] [Google Scholar]
- 20.Li L.Y., Yang C.C., Yang J.F., Li H.D., Zhang B.Y., Zhou H., Hu S., Wang K., Huang C., Meng X.M. ZEB1 regulates the activation of hepatic stellate cells through Wnt/β-catenin signaling pathway. Eur. J. Pharmacol. 2019;865:172787. doi: 10.1016/j.ejphar.2019.172787. [DOI] [PubMed] [Google Scholar]
- 21.Zhang M., Miao F., Huang R., Liu W., Zhao Y., Jiao T., Lu Y., Wu F., Wang X., Wang H. RHBDD1 promotes colorectal cancer metastasis through the Wnt signaling pathway and its downstream target ZEB1. J. Exp. Clin. Cancer Res. 2018;37:22. doi: 10.1186/s13046-018-0687-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pasquinelli A.E. MicroRNAs: heralds of the noncoding RNA revolution. RNA. 2015;21:709–710. doi: 10.1261/rna.049981.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y., Wang L., Chen C., Chu X. New insights into the regulatory role of microRNA in tumor angiogenesis and clinical implications. Mol. Cancer. 2018;17:22. doi: 10.1186/s12943-018-0766-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang X., Xu S., Gao J., Liu F., Yue J., Chen T., Wu B. miRNA expression profiling identifies DSPP regulators in cultured dental pulp cells. Int. J. Mol. Med. 2011;28:659–667. doi: 10.3892/ijmm.2011.721. [DOI] [PubMed] [Google Scholar]
- 25.Zhang P., Cao L., Fan P., Mei Y., Wu M. LncRNA-MIF, a c-Myc-activated long non-coding RNA, suppresses glycolysis by promoting Fbxw7-mediated c-Myc degradation. EMBO Rep. 2016;17:1204–1220. doi: 10.15252/embr.201642067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang L., Liu Z.M., Rao Y.W., Cui S.Q., Wang H., Jia X.J. Downregulation of microRNA-586 Inhibits Proliferation, Invasion and Metastasis and Promotes Apoptosis in Human Osteosarcoma U2-OS Cell Line. Cytogenet. Genome Res. 2015;146:268–278. doi: 10.1159/000441073. [DOI] [PubMed] [Google Scholar]
- 27.Pellecchia S., Sepe R., Decaussin-Petrucci M., Ivan C., Shimizu M., Coppola C., Testa D., Calin G.A., Fusco A., Pallante P. The Long Non-Coding RNA Prader Willi/Angelman Region RNA5 (PAR5) Is Downregulated in Anaplastic Thyroid Carcinomas Where It Acts as a Tumor Suppressor by Reducing EZH2 Activity. Cancers (Basel) 2020;12:235. doi: 10.3390/cancers12010235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goodall G.J., Wickramasinghe V.O. RNA in cancer. Nat. Rev. Cancer. 2021;21:22–36. doi: 10.1038/s41568-020-00306-0. [DOI] [PubMed] [Google Scholar]
- 29.Amelio I., Bernassola F., Candi E. Emerging roles of long non-coding RNAs in breast cancer biology and management. Semin. Cancer Biol. 2021;72:36–45.. doi: 10.1016/j.semcancer.2020.06.019. [DOI] [PubMed] [Google Scholar]
- 30.Yao Y., Shu F., Wang F., Wang X., Guo Z., Wang H., Li L., Lv H. Ann. Hepatol; 2020. Long noncoding RNA LINC01189 is associated with HCV-hepatocellular carcinoma and regulates cancer cell proliferation and chemoresistance through hsa-miR-155-5p. 22, 100269. [DOI] [PubMed] [Google Scholar]
- 31.Yu Y., Yin W., Yu Z.H., Zhou Y.J., Chi J.R., Ge J., Cao X.C. miR-190 enhances endocrine therapy sensitivity by regulating SOX9 expression in breast cancer. J. Exp. Clin. Cancer Res. 2019;38:22. doi: 10.1186/s13046-019-1039-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sánchez-Tilló E., de Barrios O., Siles L., Cuatrecasas M., Castells A., Postigo A. β-catenin/TCF4 complex induces the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proc. Natl. Acad. Sci. USA. 2011;108:19204–19209. doi: 10.1073/pnas.1108977108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petri B.J., Klinge C.M. Regulation of breast cancer metastasis signaling by miRNAs. Cancer Metastasis Rev. 2020;39:837–886. doi: 10.1007/s10555-020-09905-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crudele F., Bianchi N., Reali E., Galasso M., Agnoletto C., Volinia S. The network of non-coding RNAs and their molecular targets in breast cancer. Mol. Cancer. 2020;19:61. doi: 10.1186/s12943-020-01181-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lecarpentier Y., Schussler O., Hébert J.L., Vallée A. Multiple Targets of the Canonical WNT/β-Catenin Signaling in Cancers. Front. Oncol. 2019;9:1248. doi: 10.3389/fonc.2019.01248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wen X., Wu Y., Awadasseid A., Tanaka Y., Zhang W. New Advances in Canonical Wnt/β-Catenin Signaling in Cancer. Cancer Manag. Res. 2020;12:6987–6998. doi: 10.2147/CMAR.S258645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang N., Zou C., Zhu Y., Luo Y., Chen L., Lei Y., Tang K., Sun Y., Zhang W., Li S. HIF-1ɑ-regulated miR-1275 maintains stem cell-like phenotypes and promotes the progression of LUAD by simultaneously activating Wnt/β-catenin and Notch signaling. Theranostics. 2020;10:2553–2570. doi: 10.7150/thno.41120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lv C., Li F., Li X., Tian Y., Zhang Y., Sheng X., Song Y., Meng Q., Yuan S., Luan L. MiR-31 promotes mammary stem cell expansion and breast tumorigenesis by suppressing Wnt signaling antagonists. Nat. Commun. 2017;8:1036. doi: 10.1038/s41467-017-01059-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aprile M., Katopodi V., Leucci E., Costa V. LncRNAs in Cancer: From garbage to Junk. Cancers (Basel) 2020;12:3220. doi: 10.3390/cancers12113220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Drápela S., Bouchal J., Jolly M.K., Culig Z., Souček K. ZEB1: A Critical Regulator of Cell Plasticity, DNA Damage Response, and Therapy Resistance. Front. Mol. Biosci. 2020;7:36. doi: 10.3389/fmolb.2020.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu H.T., Zhong H.T., Li G.W., Shen J.X., Ye Q.Q., Zhang M.L., Liu J. Oncogenic functions of the EMT-related transcription factor ZEB1 in breast cancer. J. Transl. Med. 2020;18:51. doi: 10.1186/s12967-020-02240-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu Y., Luo W., Yang Z.J., Chi J.R., Li Y.R., Ding Y., Ge J., Wang X., Cao X.C. miR-190 suppresses breast cancer metastasis by regulation of TGF-β-induced epithelial-mesenchymal transition. Mol. Cancer. 2018;17:70. doi: 10.1186/s12943-018-0818-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu S.S., Li Y., Zhang H., Zhang D., Zhang X.B., Wang X., Yu Y. The ERα-miR-575-p27 feedback loop regulates tamoxifen sensitivity in ER-positive Breast Cancer. Theranostics. 2020;10:10729–10742. doi: 10.7150/thno.46297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nicolai S., Mahen R., Raschellà G., Marini A., Pieraccioli M., Malewicz M., Venkitaraman A.R., Melino G. ZNF281 is recruited on DNA breaks to facilitate DNA repair by non-homologous end joining. Oncogene. 2020;39:754–766. doi: 10.1038/s41388-019-1028-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.