

Abstract
Tumors are complex cellular and acellular environments within which cancer clones are under continuous selection pressures. Cancer cells are in a permanent mode of interaction and competition with each other as well as with the immediate microenvironment. In the course of these competitive interactions, cells share information regarding their general state of fitness, with less‐fit cells being typically eliminated via apoptosis at the hands of those cells with greater cellular fitness. Competitive interactions involving exchange of cell fitness information have implications for tumor growth, metastasis, and therapy outcomes. Recent research has highlighted sophisticated pathways such as Flower, Hippo, Myc, and p53 signaling, which are employed by cancer cells and the surrounding microenvironment cells to achieve their evolutionary goals by means of cell competition mechanisms. In this review, we discuss these recent findings and explain their importance and role in evolution, growth, and treatment of cancer. We further consider potential physiological conditions, such as hypoxia and chemotherapy, that can function as selective pressures under which cell competition mechanisms may evolve differently or synergistically to confer oncogenic advantages to cancer.
Keywords: cancer, cell competition, chemotherapy, clonal selection, tumor heterogeneity
Subject Categories: Cancer; Cell Adhesion, Polarity & Cytoskeleton; Development & Differentiation
In this review, Rajan Gogna and colleagues integrate developmental and cancer perspectives on the role of cell competition mechanisms as drivers of malignancies at tissue level.

Introduction
Cell competition (CC) is an active cell selection mechanism that promotes elimination of the viable but suboptimal cells in the presence of more competitive neighbors. The primary observations of CC were drawn from experiments investigating the impact of a class of mutations in ribosomal protein (Rp) encoding genes termed Minutes in the developing Drosophila wing disk (Morata & Ripoll, 1975). Cells with Rp heterozygous mutations were viable on own but had lower division capacity due to reduced protein synthesis compared to their wild‐type (WT) counterparts. In genetic mosaic wing disks comprised of WT and MT cells, the wing disk is overtaken by the fast‐dividing WT clones (winners), suggesting clones with higher division potential grow at the expense of their growth‐deficient neighbors (losers) (Morata & Ripoll, 1975). Elimination of the weaker populations proceeds through apoptosis (Prober & Edgar, 2000; Moreno et al, 2002), specifically at the interface with their faster growing counterparts, suggesting CC is an active process enacted by short‐range cellular interactions (Moreno et al, 2002). The same phenomena of CC‐based elimination of growth‐deficient cells were demonstrated in mice (Oliver et al, 2004), evincing CC as a highly conserved and important mechanism employed by multicellular organisms. CC is now understood to act as a result of fitness comparisons, in which cellular fitness status is qualified by various parameters not limited to growth. Importantly, competition hinges upon relative and not absolute fitness and is determined by cues of the tissue microenvironment (Moreno et al, 2002; Rhiner et al, 2010). This is particularly highlighted by the demonstration of supercompetition, in which normal WT cells are outcompeted by highly competent neighbors. This facet of CC was identified in the fly wing by overexpression of the Drosophila homolog of Myc (dMyc), a master regulator of cell growth and division, which resulted in apoptosis of WT cells and compensatory proliferation of fast‐growing dMyc‐overexpressing supercompetitors (Moreno & Basler, 2004). CC as a mode of quality control plays important roles in the selection for the most robust cell populations during development (Claveria et al, 2013; Diaz‐Diaz et al, 2017; Hashimoto & Sasaki, 2019), organ size control (de la Cova et al, 2004), and maintenance of tissue homeostasis to regulate tissue regeneration (Oertel et al, 2006), and aging (Merino et al, 2015; Liu et al, 2019a). Studies on CC have offered important insights into how distinct cellular populations respond to each other and have provided a unique lens to explain various facets of cancer biology (Eichenlaub et al, 2016; Suijkerbuijk et al, 2016; Di Giacomo et al, 2017; Madan et al, 2019b; Moya et al, 2019). In this review, we discuss the basic mechanisms of homeostatic CC and how subversion of these mechanisms can result in cancer. We further focus on the recent discoveries that reveal the role of CC in a) CC between tumor and its microenvironment and b) intratumoral CC. In this review, we summarize the instances where CC is involved in the interactions between tumor and peritumoral cells, as they are crucial to the initial events of tissue transformation and subsequent tumor outgrowth and invasion. Additionally, we hypothesize that due to intratumoral CC, distinct cancer clones become more competent under selection pressures such as chemotherapy and this results in a more aggressive tumor with enhanced therapy resistance.
Cell competition general mechanisms
Since the original findings of CC identified in Drosophila, several additional studies have elaborated on key concepts such as how cells recognize fitness and trigger CC, genes and pathways involved in CC, and the various modes of elimination of the suboptimal cells (Table 1). The following mechanisms are discussed herein.
Table 1.
Summary of CC pathways and model systems.
| Gene | CC type | Animal model | Tissue type/organ | Described in cancer | Molecular mechanism | References |
|---|---|---|---|---|---|---|
| Dpp | Trophic factor theory | Drosophila | Wing disk | No | Rp‐MT are outcompeted by Rp‐WT owing to reduced Dpp‐mediated anti‐apoptotic signaling | Moreno et al (2002) |
| Il‐7 | Trophic factor theory | Mouse | Thymus | Yes (T‐ALL) | Old (loser) progenitor cells are replaced by young (winner) progenitor cells by competing for IL‐7 | Martins et al (2014) |
| Scribble | Unknown mechanism | Canine | MDCK Cells | No | Scribble‐deficient cells are engulfed by WT cells with JNK‐activated phagocytic pathways | Ohsawa et al (2011) |
| Scribble | Mechanical CC | Canine | MDCK Cells | No | ROCK/p38/p53 mediated elimination of Scribble knockdown cells by surrounding WT cells | Norman et al (2012) Wagstaff et al (2016) |
| RasV12 | Mechanical CC | Drosophila | Pupal notum midline | No | RasV12 cells induce compaction and elimination of WT cells through the downregulation of ERK signaling in loser cells | Moreno et al (2019) |
| RasV12 | Mechanical CC | Canine | MDCK cells | No | RasV12‐transformed cells undergo in Warburg‐like metabolic changes when surrounded by WT cell, which promote their elimination through apical extrusion | Kon et al (2017) |
| RasV12 | Mechanical CC | Mouse | Intestine epithelium | No | RasV12 cells harbor metabolic changes that promote their apical extrusion by winner cells | Kon et al (2017) |
| RasV12 | Mechanical CC | Canine | MDCK cells | No | Filamin and vimentin mediated extrusion of RasV12‐ and Src‐transformed loser cells by WT winner cells | Kajita et al (2014) |
| Src | Mechanical CC | Canine | MDCK cells | No | Filamin and vimentin mediated extrusion of RasV12‐ and Src‐transformed loser cells by WT winner cells | Kajita et al (2014) |
| Src | Mechanical CC | Zebrafish | Embryos | No | Filamin and vimentin mediated extrusion of RasV12‐ and Src‐transformed loser cells by WT winner cells | Kajita et al (2014) |
| Sas‐ PTP10D | Activation of receptor‐mediated cell death | Drosophila | Imaginal disks | No | Transactivation of Sas‐PTP10D signaling leads to Scribble MT cell elimination through JNK apoptotic signaling | Yamamoto et al (2017) |
| TRRs | Activation of receptor‐mediated cell death | Drosophila | Wing disk, imaginal disks | No | The secreted factor Spatzle activates TRRs in RpL14–/+ cells, leading to the activation of NF‐kB‐mediated apoptosis | Meyer et al (2014), Alpar et al (2018), Byun et al (2019), Katsukawa et al (2018) |
| dFwe | Fitness fingerprints | Drosophila | Imaginal disks | No | Fwe(Ubi) eliminates Fwe(Lose) cells via activation of Azot‐ and Hid‐mediated apoptosis | Rhiner et al (2010), Merino et al (2015) |
| hFWE | Fitness fingerprints | Human | Cancer cells, xenografts | Yes (multiple) | Fwe‐Win eliminates Fwe‐Lose cells via activation of apoptosis | Madan et al (2019) |
| SPARC | Fitness Fingerprints | Drosophila | Imaginal disks | Yes | SPARC transiently inhibits the activation of caspase in Flower‐Lose cells | Portela et al (2010) |
| dMYC | Unknown mechanism | Drosophila | Wing disk | No | MYC‐high cells eliminate through apoptosis neighbor MYC‐low cells | Moreno and Basler (2004) |
| c‐MYC | Unknown mechanism | Mouse | Embryo (epiblast) | Yes | MYC‐high cells eliminate through apoptosis neighbor MYC‐low cells | Claveria et al (2013) |
| c‐MYC | Unknown mechanism | Human | Cancer cells | Yes | MYC‐high cells eliminate through apoptosis neighbor MYC‐low cells | Di Giacomo et al (2017), Patel et al (2017) |
| Yki | Unknown mechanism | Drosophila | Wing disk | No | Yorkie (YAP homolog) expression activates dMyc promoting supercompetition causing the elimination of WT cells | Ziosi et al (2010) |
| YAP/TAZ | Unknown mechanism | Mouse | Glioblastoma | Yes | Glioblastoma cells with high expression of YAP outcompete cells with lower expression | Liu et al (2019) |
| YAP/TAZ | Unknown mechanism | Mouse | Liver | Yes | Peritumoral cells with high YAP/TAZ activation induce apoptosis in liver tumor cells with relatively less YAP/TAZ to suppress tumor outgrowth. High YAP/TAZ‐liver tumor cells can also outcompete low‐expressing normal hepatocytes and result in tumor growth. | Moya et al (2019) |
| YAP/TEAD | Unknown mechanism | Mouse | Embryo (epiblast and embryonic fibroblasts) | No | Embryonic cells with high YAP/TEAD activity eliminate their neighbors with lower YAP/TEAD activity | Hashimoto and Sasaki (2019), Mamada et al (2015) |
| Wingless | Unknown mechanism | Drosophila | Imaginal disks | No | Cells with Wingless (WNT homolog) overactivation eliminate surrounding cells with low Wingless activity | Vincent et al (2011) |
| WNT | Unknown mechanism | Mouse | iPSCs | No | During reprograming, cells derived from WNT‐expressing cells establish clone dominance | Shakiba et al (2019) |
| APC | Unknown mechanism | Drosophila | Intestine epithelium | Yes | Cells with loss of APC eliminate WT cells and cause growth of intestinal adenomas | Suijkerbuijk et al (2016) |
| APC | Unknown mechanism | Human and mouse | Intestinal 3D organoids; in vivo adenoma formation in mice | Yes | APC− / − intestinal stem cells secreted WNT antagonists that induced differentiation of nearby WT cells in organoids and resulted in tumor formation in mice. | Flanagan et al (2021), van Neerven et al (2021) |
| NOTCH | Unknown mechanism | Mouse | Esophageal epithelium | Yes | Cells with loss of NOTCH signaling outcompete their WT neighbors | Alcolea and Jones (2015) |
| TP53 | Unknown mechanism | Mouse | Embryo (epiblast) | No | p53 represses mTOR which induces loser cell elimination | Bowling et al (2018) |
| TP53 | Unknown mechanism | Mouse | Hematopoietic stem cell and progenitors (HSPCs) | No | In an irradiated niche, low‐expressing p53 HSPCs outcompete high‐expressing p53 HSPCs, which become senescent | Bondar and Medzhitov (2010) |
| TP53 | Mechanical CC | Drosophila | Wing disk | No | p53 acts as a sensor for cells with less resistance to apoptosis by mechanical stresses in overcrowded tissue | Wagstaff et al (2016) |
| TP53 | Unknown mechanism | Mouse | Irradiated epidermis and esophagus | Following radiation, epithelial Mutant p53 progenitors rapidly expand at the expense of wild‐type p53 progenitors | Fernandez‐Antoran et al (2019), Murai et al (2018) | |
| mTOR | Unknown mechanism | Mouse | Embryo (epiblast) | No | p53 represses mTOR which induces loser cell elimination | Bowling et al (2018) |
| BMP | Similar to trophic factor | Mouse | Embryo (epiblast) | No | Defective BMP cells express lower MYC and are eliminated from the epiblast | Sancho et al (2013) |
| EGFR | Unknown mechanism | Drosophila | Imaginal disk | Yes | EGFR‐overexpressing supercompetitors generate aggressive tumors and induce apoptosis in surrounding normal epithelium | Eichenlaub et al (2016) |
| JAK‐STAT | Unknown mechanism | Drosophila | Imaginal disk and eye | No | Cells with hyperactive STAT signaling function as supercompetitors that induce apoptosis in wild‐type cells | Rodrigues et al (2012) |
List of cell competition genes identified, their mechanism, the organism/tissue/cell type employed to study, role in cancer, and summary of key findings. Representative findings for each gene have been included to briefly summarize.
The trophic factor theory
This theory posits that homeostatic cell density is achieved by competition for the limited signaling cues in a tissue space. Herein, the more fit cells (winners) have a higher capacity to obtain growth/pro‐survival signals and thus proliferate compared to their less‐fit neighbors (losers), which undergo apoptosis. In the Drosophila wing, Rp‐WT cells outcompete their slower‐growing Rp‐MT neighbors for decapentaplegic (Dpp) survival factor, a crucial morphogen for cellular division, tissue growth and correct patterning. Deficient Dpp signaling in Rp‐MT cells resulted in lowered proliferation and activation of JNK‐induced apoptosis (Moreno et al, 2002) (Fig 1A). Rp‐MT cells display lowered protein translation rates (Moreno et al, 2002) and thus presumably reduced metabolic activity, which may account for diminished ability to capture Dpp ligands. In agreement, additional work has shown that loser cells displayed lower protein synthesis and a reliance on autophagy in association with downstream apoptosis via JNK signaling and Hid activation (Nagata et al, 2019). Mouse embryonic stem cells lacking the bone morphogenetic (BMP)‐receptor are non‐responsive to secreted BMPs, the mammalian homologs of Dpp, are deemed defective, and are outcompeted by WT neighbors. However, these losers did not display defects in autophagy (Sancho et al, 2013). Additionally, the dependency of extracellular growth cues to initiate competition is in question given the inconclusive role of competition for Dpp capture in dMyc‐mediated CC (Moreno et al, 2002; de la Cova et al, 2004). And secondly, indirect competition based on trophic theory does not account for compensatory hyper‐proliferation of winner cells subsequent to apoptosis of loser cells.
Figure 1. General mechanisms of cell competition.
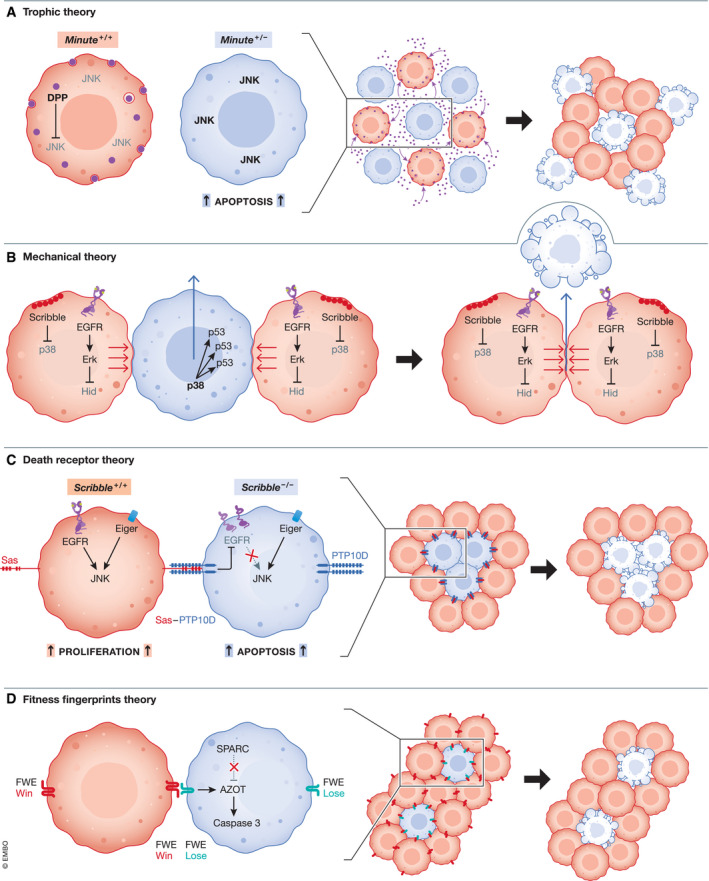
(A) Model of indirect CC based on trophic theory: Growth‐deficient ribosomal protein gene cells (RP+ / −) are outcompeted by wild‐type cells (Rp+/+) based on their reduced ability to capture trophic factors, such as Dpp, essential for their survival. Dpp allows cells to block the activation of apoptotic pathways and maintain a proliferative state. The lower capacity of growth‐deficient RP+ / − cells to obtain Dpp leads to the activation of JNK apoptotic signaling and ultimately their replacement by wild‐type cells that efficiently capture Dpp ligands. (B) Model of Mechanical theory: Mechanical‐based cell competition regulates cell growth and survival. Differential sensitivities to mechanical forces in epithelial tissues results in elimination of suboptimal cells by their healthy neighbors, and compensatory gain of winners to the tissue space. At the molecular level, in suboptimal cells, the high levels of stress and deformation downregulate ERK signaling and increase the expression of the apoptotic proteins p38, p53, and Hid. Suboptimal cells undergo apical extrusion, or apoptosis, and optimal cells expand. (C) Model of Death Receptor theory: Healthy cells can induce the elimination of suboptimal mutant neighbors by activation of cell surface ligand‐death receptor pairs. scribble (scrib)− / − MT cells expressing PTP10D receptor contact WT epithelial cells expressing surface ligand, Sas inducing transactivation of Sas‐PTP10D signaling and subsequent EGFR inactivation and JNK pro‐apoptotic signaling in scrib − / − loser cells, and compensatory proliferation of WT cells. Eiger (TNF superfamily member) activates JNK‐dependent cell death in scribble polarity mutants. (D) Model of cell competition via fitness fingerprints. Neighboring cells compare their fitness status through the expression of fitness fingerprints, such as transmembrane Flower proteins. Flower Win‐expressing cells recognize Flower Lose‐expressing suboptimal cells. Consequently, Flower Lose upregulates Azot, which induces caspase‐dependent apoptosis. In some cases, suboptimal cells can increase the expression of SPARC to antagonize Flower Lose‐mediated apoptosis and transiently prevent cell death.
One study showed just how homeostatic CC maintains tissue fitness status quo and suppresses dysfunction‐‐and specifically how this is achieved based on affinity for environmental cues. The ability to transduce IL‐7 signals was found to select youthful T‐cell progenitors at the expense of old progenitors in the mouse thymus. Lack of the Il‐7‐receptor on young progenitors obviates this mode of CC and results in accumulation of the old T‐cell progenitors, leading to the development of T‐cell acute lymphoblastic leukemia (T‐ALL) (Martins et al, 2014). This demonstrates that loser cells, by responding poorly to growth and survival factors present in the extracellular environment, are outcompeted by winner cells, independently of direct cell–cell interactions (Table 1).
Mechanical cell competition
Cells in any tissue space are members of a mechanical environment generated by forces from cell–cell contacts and the surrounding extracellular matrix. Slower or faster growing clones, relative to their surrounding neighbors, experience mechanical stress (Shraiman, 2005). Tissues have adopted various mechanisms to relieve this stress by balancing proliferation with elimination. Mechanical CC regulates the selection of the eliminated cells and, in some contexts, operates as a form of supercompetition (Levayer et al, 2016). In the Drosophila notum, cellular overcrowding, driven by conditional expression of activating RasV12 mutations, resulted in the compaction and subsequent outcompetition of neighboring WT cells via apoptosis, followed by delamination. Unlike other forms of CC described, this mode of mechanical supercompetition was not locally constrained and delamination was observed up to three cell diameters away from RasV12‐mutant clones (Levayer et al, 2016). Later work showed that mechanical CC eliminated the normal cells in between RasV12‐mutant clones via compaction‐driven downregulation of epidermal growth factor (EGFR)/extracellular‐regulated kinase (ERK) signaling. Tissue relaxation or densification also resulted in ERK downregulation, which suggests that ERK is not modulated simply by cellular density, but instead tissue strain rate (Moreno et al, 2019). In mammalian cells, polarity‐deficient MDCK cells generated by knockdown of the apico‐basal polarity and tumor suppressor gene, Scribble (Scrib KD), were hypersensitive to compaction and were eliminated via apoptosis upon cellular overcrowding. Compaction of Scrib KD relayed a mechanical stress response via Rho kinase (ROCK) and actin and myosin upregulation, in line with previous studies showing cytoskeletal proteins interpret and respond to mechanical stress in apoptotic epithelia (Rosenblatt et al, 2001). Eliminated MT cells displayed ROCK‐induced p38 stress signaling concomitant with high p53 expression, indicating that through compaction, mechanical CC supports removal of damaged cells (Wagstaff et al, 2016), potentially exploiting p53 as a sensor (Table 1). Altogether, these studies suggest that cells may have differential sensitivity to compaction that depends on their sensitivity to apoptosis. Mechanical CC seems to exploit these parameters to eliminate the relatively less‐fit population. Thus far, mechanical CC does not appear to hinge upon short‐range modes of molecular exchange of soluble signaling cues or membrane‐bound fitness fingerprints (Levayer et al, 2016; Wagstaff et al, 2016; Moreno et al, 2019).
Activation of receptor‐mediated cell death
This mode of CC proceeds through direct cell–cell contact in which a ligand–receptor system at the interface between competing cells drives elimination of losers or via ligands secreted by winner cells that directly activate cell death in loser cells. In Drosophila, this mode of CC can be observed in Scribble‐deficient (scrib − / −) epithelial cells that have loss of apico‐basal polarity (Table 1). When scrib − / − (MT) cells come in contact with WT cells, the ligand stranded at second (Sas) on WT and the surface receptor PTP10D on MT cells re‐localize laterally, resulting in transactivation of Sas‐PTP10D signaling (Yamamoto et al, 2017). As an outcome, EGFR signaling is inactivated, while JNK pro‐apoptotic pathway is induced in MT cells, thereby eliminating the polarity‐defective transformed cells (Fig 1C). Though these cells express both death ligands and receptors at their membranes, the activation of death signals is only observed in loser cells (Yamamoto et al, 2017). Elimination of polarity‐mutant losers in Drosophila epithelia was also shown to depend upon Eiger, the Drosophila tumor necrosis factor (TNF), and downstream JNK activation (Igaki et al, 2009).
Toll receptor signaling provides another compelling example by which winner cells eliminate adjacent loser cells by direct activation of cell death in losers. Current evidence suggests that this immunological process inhibits tissue growth and regulates cell fitness during infection (Germani et al, 2018). However, it is widely reported to be repurposed for non‐infectious cell selection (Meyer et al, 2014). In Drosophila wing disks, forced Myc expression or Minute‐induced competition in RpL14 –/+ cells leads to Toll‐related receptors (TRRs) activation by the secreted factor Spätzle, leading to NF‐kB mediated apoptosis (Meyer et al, 2014; Alpar et al, 2018). This behavior can also be mimicked in a cancer‐like invasive phenotype in Tai‐expressing Drosophila pupal wings, where its expression induces Spätzle‐Toll‐mediated apoptosis of loser cells and invasion by winner cells in the thoracic region (Byun et al, 2019). In contrast, in scrib − / − clones that are extruded (discussed later) from imaginal epithelia, TRR activation transforms these loser scrib − / − cells into winners by activation of Hippo signaling and prevention of cell death(Katsukawa et al, 2018) (Table 1).
Fitness fingerprints
Mechanistic detection of cellular fitness remains unclear. Thus far, the only fitness‐sensing mechanism identified is the Flower (Fwe) “fitness fingerprints”, which are highly conserved transmembrane proteins that directly communicate fitness status and drive competition (Table 1) (Rhiner et al, 2010; Madan et al, 2019b). In Drosophila, there are three alternately spliced Flower isoforms: Fweubi , FweLoseA , and FweLoseB . The ubiquitous form, FweUbi, is constantly expressed. Under competitive stress, Fweubi is downregulated and FweLoseA and FweLoseB are upregulated in loser cells that undergo apoptosis (Rhiner et al, 2010). Importantly, membrane FlowerLose presence does not autonomously induce apoptosis and FlowerLose cells are not eliminated when surrounded by FlowerLose neighbors (Rhiner et al, 2010). Secondly, FlowerLose cells can upregulate the expression of secreted protein acidic and cysteine‐rich protein (SPARC), which inhibits caspase induction of apoptosis as a transient defense against elimination (Portela et al, 2010). Thus, SPARC may serve as defense against unnecessary purging, particularly as means to counteract supercompetition. In contrast, loser cell upregulation of the gene, Azot, especially in weakened cells with diminished SPARC, ensures competitive elimination by apoptosis (Merino et al, 2015) (Fig 1D). Interestingly, expression of FlowerLose, Azot, and SPARC is impacted by various triggers of competition and supercompetition such as modulation of Dpp morphogen signaling or dMyc expression, and genetic mutation in genes encoding ribosomal proteins (Rp mutants) (Rhiner et al, 2010; Merino et al, 2015). This suggests the Flower code of fitness fingerprints and its downstream players are a common, tunable read‐out of cellular fitness likely functional in many tissue systems as means to continuously survey cellular status and rid suboptimal cells.
Flower is a highly conserved gene, and this fitness‐sensing program also occurs in mammals (Rhiner et al, 2010; Madan et al, 2019b). In humans, the Fwe locus generates four isoforms: two hFweWin and two hFweLose . Two Flower isoforms (hFWE2 and hFWE4) behave as Flower‐Win proteins, whereas the other isoforms (hFWE1 and hFWE3) behave as Flower‐Lose proteins. Like in Drosophila, hFwe‐Lose‐expressing cells are viable in a homotypic environment. However, when confronted by hFwe‐Win‐expressing cells, Lose cells are outcompeted and undergo apoptosis. Human Flower‐mediated competition requires contact between Win and Lose cells and was not dependent on soluble signal exchange (Madan et al, 2019b) (Table 1).
Other mechanisms
Several additional gene pathways have been implicated in CC, but do not fit the mechanisms as described above. We do not yet understand how interacting cells sense the imbalance in these pathways or whether these pathways fit within a larger scheme of signaling events underlying CC. In general, differential expression or activity of the following gene pathways result in differences in growth, proliferation, polarity, and/or survival that generates competitive interactions between the neighboring cells. These include Myc (de la Cova et al, 2004; Claveria et al, 2013; Di Giacomo et al, 2017; Diaz‐Diaz et al, 2017), Ras and Src (Kajita et al, 2010; Kajita et al, 2014), Scribble (Norman et al, 2012), Mahjong with Lgl (Tamori et al, 2010), TEAD and YAP/TAZ (Hashimoto & Sasaki, 2019; Moya et al, 2019), Wnt/Wg (Vincent et al, 2011; Suijkerbuijk et al, 2016; Akieda et al, 2019; Flanagan et al, 2021; van Neerven et al, 2021), Janus Kinase (JAK)‐signal transducer and activator of transcription (STAT) (Rodrigues et al, 2012), Notch (Alcolea & Jones, 2015), EGFR (Eichenlaub et al, 2016; Moreno et al, 2019), mTOR (Bowling et al, 2018), and p53 (Bondar & Medzhitov, 2010; de la Cova et al, 2014; Bowling et al, 2018; Murai et al, 2018; Fernandez‐Antoran et al, 2019) (Table 1). The most well studied example is CC induced by imbalances in the activity of Myc, a transcription factor that positively impacts proliferation and growth. High Myc‐expressing cells act as supercompetitors and outcompete low Myc‐expressing or wild‐type cells (de la Cova et al, 2004; Moreno & Basler, 2004; Claveria et al, 2013; Di Giacomo et al, 2017; Diaz‐Diaz et al, 2017). Myc‐mediated competition appears to require direct contact, but one study noted dMyc‐mediated competition across several cell diameters (de la Cova et al, 2004). Additionally, the role of molecule exchange or diffusible signal is under debate (Claveria et al, 2013; Sancho et al, 2013). Some evidence suggests dMyc‐mediated competition is through competition for Dpp capture; however, growth cues appear to be an incomplete mechanism (de la Cova et al, 2004). Thus, there are still many missing pieces in how cells infer fitness based on Myc status as well as the other pathways listed. Extensive crosstalk exists between these pathways—i.e., Myc and YAP/TAZ have been shown to cooperate to promote CC‐based elimination(Hashimoto & Sasaki, 2019), and p53 status has been identified as a sensor in various mechanisms of CC (de la Cova et al, 2014; Wagstaff et al, 2016; Bowling et al, 2018). Additionally, differential signaling in dMyc, WNT/Wg, and JAK‐STAT instigates CC via Flower fitness sensing (Rhiner et al, 2010).
Cell competition between tumor and its microenvironment
Surveillance and clearance of abnormal or transformed cells is an absolutely essential process for tissue maintenance and tumor prevention. Given its role in the detection and culling of aberrant cells, CC has been proposed to serve as a brake to tumorigenesis. In this section, we will discuss the mechanisms by which CC prevents neoplastic tumor development. We will also discuss how failure of homeostatic CC within the tissue microenvironment releases the break and facilitates clonal expansion of transformed cells into subsequent frank carcinoma. Secondly, we discuss how in some contexts cancer cells hijack CC mechanisms and act as supercompetitors resulting in tumor outgrowth at the expense of normal tissue. Moreover, we frame CC mechanisms in the context of tumors as complex ecosystems to hypothesize how CC may regulate intratumoral CC, particularly under hypoxia and following therapy.
Cell competition suppresses tumorigenesis
For the majority of epithelial cancers, a tumor develops from a single transformed cell. How the transformed cell and its early clonal expansion evade regulatory features imposed by its tissue microenvironment is not well understood. However, tissues appear to harbor intrinsic tumor suppressive features to rid mutant, pre‐tumoral cells. One process, epithelial defense against cancer (EDAC), is an active mechanism whereby WT cells sense and eliminate newly transformed cells (Kajita et al, 2010; Kajita et al, 2014; Kon et al, 2017). Importantly, this elimination of abnormal cells occurs only in the presence of normal cells and fails if an entire epithelium becomes comprised of mutants (Kon et al, 2017). This phenomenon is similar to the principles of CC. Using MDCK cells, a non‐transformed mammalian epithelial cell line, it was shown that Src‐activated or Rasv12‐transformed cells underwent apoptosis‐independent apical extrusion from the monolayer when co‐cultured with WT cells (Hogan et al, 2009; Kajita et al, 2010; Kajita et al, 2014; Kon et al, 2017). MT cell extrusion is an active process driven by accumulation of the cytoskeletal proteins, filamin and vimentin, within normal cells at their interface with the mutants (Kajita et al, 2014) (Fig 2A). Simultaneously, dysfunctional cells display notable differences in height, morphology, and elasticity accompanied by altered expression of cytoskeletal proteins and mechanotransduction cues required to extrude the cell (Kajita et al, 2010; Kajita et al, 2014). Additionally, normal cells can alter the metabolic profile of dysfunctional or transformed cells to further promote their extrusion (Kon et al, 2017). The efficiency of live‐cell extrusion to prevent tumorigenesis in different tissue types, including those without inherent clearance mechanisms (i.e., clearance of outcompeted cells with waste in the intestine), is for debate. On this note, clearance of outcompeted transformed cells may proceed through engulfment of live outcompeted cells or their corpses. For example, in Drosophila imaginal epithelia with scrib deletion, the WT cells were found to engulf the scrib − / − loser cells via non‐apoptotic JNK signaling, demonstrating that engulfment may be another mechanism by which the neoplastic loser cells are eliminated (Ohsawa et al, 2011) (Fig 2A‐b). Additionally, losers can undergo apoptosis prior to apical extrusion by active CC (Norman et al, 2012; Wagstaff et al, 2016). Polarity mutant cells generated by knockdown of Scribble (scribKD ) are outcompeted by their WT neighbors and undergo p38‐mediated apoptosis, followed by apical extrusion (Norman et al, 2012) (Fig 2C). ScribKD cells in contact with WT neighbors were hypersensitive to mechanical stress from tissue overcrowding, due to their elevated baseline p53 signaling, which marked them as losers. Contact with WT cells compacted scribKD into high‐density arrangements, with subsequent activation of p38 stress signaling and further upregulation of p53, driving their apoptotic elimination and extrusion from the epithelial monolayer (Wagstaff et al, 2016) (Fig 2A‐c).
Figure 2. Cell competition between newly transformed cells and normal neighbors.
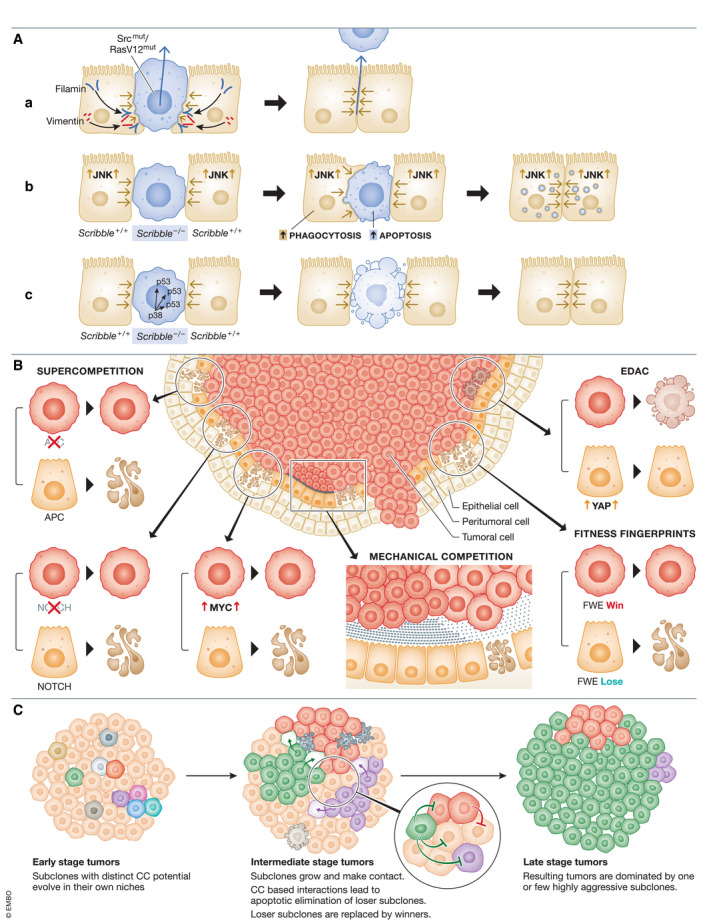
(A) (a). Epithelial defense against cancer (EDAC): a) EDAC supports removal of newly transformed cells with activated RasV12 or Src mutations by neighboring normal cells. Normal cells upregulate filamin and vimentin at border of transformed cell, inducing pressure on transformed cell. RasV12 or Src mutants undergo live‐cell apical extrusion. (b) Polarity‐defective Scribble − / − mutants undergo JNK‐mediated apoptosis in presence of wild‐type (WT) cells. Concurrently, WT cells activate JNK signaling resulting in activation of phagocytic pathways. WT cells engulf apoptotic Scribble − / − mutants, eliminating them from the tissue space. (c) In a separate mechanism, the presence of WT cells induces activation of p38 and p53 apoptotic signaling in Scribble − / − mutants, resulting in their apoptotic elimination from epithelia. (B) Cell competition between tumor and adjacent stromal cells. Tumor cells take advantage of competition mechanisms to promote their survival and eliminate stromal cells. Loss of APC and NOTCH or upregulation of fitness‐enhancing genes such as MYC in tumor cells generates supercompetitors that outcompete their wild‐type neighbors. Tumor cells expressing the fitness fingerprint Flower Win (Fwe‐Win) can sense and eliminate via apoptosis Fwe‐Lose‐expressing stromal cells, resulting in tumor outgrowth at the expense of normal tissue. Increased activation of YAP in peritumoral epithelial cells (red epithelial cells) leads to the apoptosis of tumor cells in a mode similar to EDAC to suppress tumor outgrowth. RasV12 mutants act as supercompetitors and outcompete surrounding normal cells via mechanical compression‐induced apoptosis. This can potentially facilitate the formation of tumors. (C) Tumor evolution based on cell competition principles: Clones evolve through advantageous driver mutations and competition mechanisms impact clonal dominance. During the early stages of tumor growth, distinct clones develop within their unique niches selected for by the microenvironment. As subclones engage in various forms of competition, this results in expansion of winner clones at the expense of loser clones (represented by empty cell‐outlines of dead loser cells being replaced by neighboring winner cells marked by arrows). A late stage tumor is dominated by select highly aggressive subclones that have outcompeted losers through rounds of cell competition.
These models of tumorigenesis using Drosophila epithelia had shown that the fate of mutant cells relies upon direct interactions between them and their normal neighbors. EDAC mechanisms have also been demonstrated using RasV12 mosaics within the mouse intestine, pancreas, and lung epithelia. Single RasV12 mutants confronted by an otherwise normal epithelium underwent apical extrusion, in support of similar RasV12 manipulations in vitro (Kon et al, 2017; Sasaki et al, 2018). MT cell extrusion blocked development of tumors. However, in a highly inflammatory microenvironment, which is a major driver of several malignancies, EDAC‐based CC fails and RasV12 mutants are retained, resulting in tumors (Sasaki et al, 2018). Crosstalk between a tumor and its microenvironment was investigated in depth further by genetic manipulation of the CC factors YAP/TAZ using a mouse model of liver cancer (Moya et al, 2019). Upregulation of YAP/TAZ is frequently observed in human cancers (Zhou et al, 2016) and high YAP/TAZ has previously been shown to generate supercompetitors (Hashimoto & Sasaki, 2019). Mouse liver tumors harboring high levels of active YAP/TAZ were generated and predictably grew at the expense of peritumoral tissue, which showed signs of apoptosis. However, when YAP was hyper‐activated in peritumoral normal hepatocytes, tumor growth was not only strongly suppressed, but tumor cells enclosed by YAP‐activated hepatocytes were apoptotic (Moya et al, 2019) (Fig 2B). These results suggest that supercompetitor normal cells can overtake oncogenic growth mechanisms of tumor cells and that a tissue microenvironment with high fitness has the capacity to restrain tumor growth.
Cell competition promotes mutant clonal expansion and tumor formation
Cancer cells are characterized by uncontrolled cellular division as they invade into the surrounding stroma. One possible mechanism by which the tumor manipulates its microenvironment to accomplish invasion is to exploit CC to create space by outcompeting and eliminating the non‐malignant neighbors. This phenomenon is particularly interesting given recent findings that CC can occur between different cell types, i.e., between epithelial cells and fibroblasts, which are a major cell type in the stroma of various tissues (Madan et al, 2019b). Cells with a supercompetitor status generate clones capable of expansion into fields of mutant cells, at the expense of normal cells, often with no changes in tissue morphology. This is very similar to a process known as field cancerization in which mutant clones expand into fields, at the expense of normal cells, often with no accompanied changes in tissue morphology. Overtime, these fields can develop into malignant tumors assuming additional accumulation of genetic mutations (Braakhuis et al, 2003). In a mechanism independent of fitness sensing or selection, oncogene‐activated cells exploited mechanical compression as a form of supercompetition to cause crowding‐induced cell death randomly in normal cells. In the Drosophila wing disk, oncogenic Ras clones rapidly expanded and sustained high growth despite compressive stress due to their resistance to apoptosis. To alleviate this stress and create space for their own growth, Ras MT clones instead compressed and activated apoptosis in neighboring WT cells, even up to several cell diameters away from the MT clonal boundaries (Levayer et al, 2016). Supercompetition resulting in field cancerization, or the vast expansion of MT clones, has also been shown in mammalian systems such as the normal mouse esophageal epithelium wherein loss of Notch signaling generates MT winners that rapidly expand and drives adjacent WT progenitors to differentiate (Alcolea & Jones, 2015) (Fig 2B) (Table 1). Similarly, UV‐exposure in the normal mouse epidermis (Murai et al, 2018), and ionizing radiation (IR) in the normal mouse esophageal epithelium drove clonal expansion of progenitors with GOF p53 mutations that outcompete and proliferate at the expense of their WT neighbors that terminally differentiate and slough off (Murai et al, 2018; Fernandez‐Antoran et al, 2019). These studies highlight how various stressors act as selective pressures that shift selection away from WT cells and skew toward MT clones. This may lower tissue fitness overtime, decrease EDAC‐like CC strength, and result in malignancy.
Many genes shown to generate supercompetitors are known tumor suppressors or oncogenes: Myc (Claveria et al, 2013; Di Giacomo et al, 2017; Diaz‐Diaz et al, 2017), JAK‐STAT (Rodrigues et al, 2012), Notch (Alcolea & Jones, 2015), YAP/TAZ (Hashimoto & Sasaki, 2019; Liu et al, 2019b; Moya et al, 2019), Ras (Moreno et al, 2019), APC (Flanagan et al, 2021; van Neerven et al, 2021; Yum et al, 2021) and p53 (Murai et al, 2018; Fernandez‐Antoran et al, 2019). Tumor cells behave as supercompetitors due to their growth, proliferation, and survival advantages. This was shown experimentally using tumorigenesis models in Drosophila epithelia by generating supercompetitors that were EGFR‐overexpressing (Eichenlaub et al, 2016), or that were hyperactive in Wnt signaling with increased YAP expression due to deletion in the tumor suppressor gene, APC (Suijkerbuijk et al, 2016) (Fig 2B). In both scenarios, mutant supercompetitors generated aggressive tumors, and tumor growth depended on apoptotic elimination of surrounding WT neighbors (Eichenlaub et al, 2016; Suijkerbuijk et al, 2016). EGFR‐overexpressing winner cells engulfed losers and resulted in tumor cells with additional polyploidization and faulty epithelial polarity. These tumor cells were more aggressive with enhanced metastatic capacity (Eichenlaub et al, 2016). Thus, engulfment may provide nutrients and metabolites to winners to sustain their high growth and proliferation demands. It is noteworthy that signs of entosis, or the engulfment of live cells, have been observed in human tumors (Sun, Luo et al, 2014).
APC−/−‐driven supercompetition was recently uncovered as an active mechanism in human intestinal stem cells to achieve mutant clonal expansion (Flanagan et al, 2021; van Neerven et al, 2021). Notably, the APC tumor suppressor gene is mutated in nearly 80% of human colon cancers (van Neerven et al, 2021). In corroboration with findings from Drosophila (Suijkerbuijk et al, 2016), APC − / − mutant intestinal stem cells derived from both mouse and human tissue acted as supercompetitors and outcompeted WT neighbors in 3D organoid cultures (van Neerven et al, 2021). APC − / − supercompetitors secreted WNT antagonists that decreased growth and induced differentiation of adjacent WT stem cells (as opposed to apoptosis in Drosophila). This supercompetition resulted in large intestinal adenomas in Apc− / − mice, but outcompetition was negated upon disruption of WT stem cell sensitivity to secreted WNT antagonists via pharmacologic or genetic intervention (Flanagan et al, 2021; van Neerven et al, 2021). Findings from these studies highlight a conserved mechanism of CC between mutant and wild‐type cells that dictates clonal selection and fuels tumorigenesis. In further support, intestinal stem cells carrying common colon cancer‐associated mutations in either Kras or Pik3ca were also shown to outcompete their surrounding normal neighbors in mouse intestinal crypts. MT Kras or Pik3ca crypts secreted BMP ligands that restrained stem cells and increased differentiation rates in WT crypts. MT crypts further manipulated their environments by inducing normally supportive proximal stromal cells to instead secret WNT inhibitors that additionally drove stem cell differentiation. Importantly, the MT cells were not as vulnerable to WNT inhibitors and maintained high turnover, cementing the competitive advantages of MT clones over WT neighbors (Yum et al, 2021). Thus, CC appears to underlie tissue remodeling efforts by MT cells to facilitate expansion and possibly, eventual tumor progression.
Additional data from human cancers suggest that supercompetitive Myc‐high tumor cells change the tumor microenvironment by inducing apoptosis in their neighboring stromal cells with relatively lower Myc expression (Di Giacomo et al, 2017). Myc‐overexpressing cells outcompete and eliminate low‐Myc cells via apoptosis (Moreno & Basler, 2004; Claveria et al, 2013). Human cancers bear a striking resemblance to this mechanism as cancer cells frequently express higher levels of c‐Myc compared to stromal cells, which correlates with increased cleaved caspase in the stroma (Di Giacomo et al, 2017) (Fig 2B). There is additional evidence that tumor cells can manipulate stromal cell fate by taking advantage of CC fitness fingerprint mechanisms (Madan et al, 2019b). Various human cancers were shown to express high levels of hFweWin isoforms in the tumor tissue and hFweLose isoforms in the stroma, which displayed signs of apoptosis (Madan et al, 2019b) (Fig 2B). In vivo testing of fitness fingerprints in tumorigenesis by implantation of hFweWin‐expressing human breast cancer cells into hFweLose‐expressing mammary tissue in mice generated large, aggressive tumors. In contrast, implantation of hFweLose cells into a hFweWin tissue background failed to develop into tumors. These findings align with those obtained from YAP/TAZ mediated CC in liver cancer (Moya et al, 2019), supporting that tissue fitness and CC between the tumor and the surrounding microenvironment directly impacts tumorigenesis. Additionally, the stroma of many cancers shows high expression of SPARC, a protein upregulated in loser cells in response to CC, including in Flower‐fitness mechanisms (Petrova et al, 2011). This further indicates active CC occurs between confronted cancer and normal cells, and outcomes of this competition may very well alter the fate of the cancer.
Tissue architecture in the tumor microenvironment presents opportunities of mechanisms involving or resembling mechanical CC to affect oncogenic development (Fiore et al, 2020). One characteristic of aggressive cancers is their ability to grow uncontrollably and invade neighboring tissues, which is only possible when tumors overcome spatial constraints, potentially due to mechanical CC between tumors and their surrounding tissue. As an example, basal cell carcinomas (BCCs), a slow growing, typically non‐metastatic cancer (Crowson, 2006), grow in “buds”, possibly due to softening and remodeling of the basement membrane, whereas invasive squamous cell carcinomas (SCCs) grow in bidirectional “folds”, possibly due to membrane stiffening. Thus, the differential biophysical and biomechanical properties of the basement membrane that is positioned directly below the layer of basal epidermal progenitors that give rise to BCCs and SCCs can act differently in these cancers resulting in different architectures (Fiore et al, 2020) (Fig 2B). These different mechanical forces pervasive throughout the stratified epidermis act on the tumor cells and impact the invasive potential of epidermal progenitors at the tumor‐stroma border.
Intratumoral competition
Tumors are heterogeneous masses containing distinct cancer and non‐cancer cells that must compete for space and resources under the selective pressures exerted by their environments (Lu et al, 2012). While early tumors consist of relatively homogenous cell populations, intrinsic genomic instability of cancer cells along with various microenvironment and “host” defense factors introduce widespread heterogeneity of cancer clones (Greaves & Maley, 2012). In heterogeneous tumors, after an initial phase of uniform growth of various subclones, a phase of rapid expansion follows where one or two clones emerge at the cost of others, indicating that the selection of dominant subclones is a time‐dependent process (Gao et al, 2016). We hypothesize that CC between tumor cells results in the selection of the distinct clones that harbor distinct fitness advantages (Fig 2C). For example, human tumors show heterogeneous expression of Myc (Gupta et al, 2017) and human cancer cells with high c‐Myc outcompete cancer cells with low c‐Myc (Patel et al, 2017). Similarly, it was recently shown that YAP protein expression is highly heterogeneous in glioblastoma. Differential YAP expression between glioma cells results in clonal dominance of high‐YAP cells and apoptosis of low‐YAP cells and an overall more aggressive tumor (Liu et al, 2019b). These findings directly show how CC between different clones can potentially exacerbate oncogenic growth by dominance of more competitive clones.
The primary intrinsic driver of intratumoral heterogeneity (ITH) is the stepwise accumulation of somatic mutations in parental clones, followed by subclonal branching, a term that defines clones with additional mutations that are different from the parental clone (Nowell, 1976). ITH also results from the various stromal cell types including immune cells as well as the extracellular matrix (Ramon et al, 2020). Next generation sequencing technologies and mathematical modeling have shown that principles of population genetics also apply to the evolving tumor, whereby multiple clones may emerge from an initial genetic event, similar to the allelic variation in a population (Hu et al, 2017). Therefore, neutral drift and stochastic processes (Mao & Wheldon, 1995; Ling et al, 2015; Williams et al, 2016)‐terms that define the gradual emergence of new variants in a population and may cause new clones to emerge, some of which have altered CC potential. This is in contrast with a Darwinian model of tumor evolution where mutations that are beneficial to the tumor are selected for and are often a result of selection bottlenecks such as chemotherapy, hypoxia and other microenvironment factors (Gerlinger & Swanton, 2010; Polyak, 2014; Lloyd et al, 2016). Regardless of the genetic principles that lead to the emergence of new clones, the dominance of the resulting tumor by cells bearing winner mutations or gene‐expression changes suggests that CC‐based selection of clones occurs during tumor evolution.
The variable access to space and nutrients by cancer clones introduces competition between subclones based on spatial partitioning of the clone within the tumor. In addition, tumor cells must adapt to selective pressures such as hypoxia, and chemotherapy (Ramon et al, 2020). We have previously speculated that many of these hypoxia‐induced metabolic and transcriptional changes that result in gain of aggressive phenotype have potential to crosstalk with CC pathways and thus fuel clonal evolution and heterogeneity (Madan et al, 2020). In one example of crosstalk, hypoxic zones frequently harbor mutant p53 clones that boost tumor aggressiveness (Madan et al, 2019a), meanwhile MT p53 status has been associated with a winner phenotype in various epithelia (Murai et al, 2018; Fernandez‐Antoran et al, 2019). Possibly, hypoxic MTp53 clones act as winners that outcompete WTp53 clones, further manipulating clonal evolution and tumor aggression.
In the following subsection, we discuss potential CC pathways that may be activated as a result of chemotherapy and confer winner status to cancer clones that successfully activate these pathways. We propose CC mechanisms play an essential role in clonal selection by constantly sensing and replacing clones not conducive to thrive in the harsh environments generated from therapy.
Cell competition and cancer therapy
Therapy is an artificial selection pressure that acts on tumor cells and can lead to the expansion of rare clones due to intrinsic or adaptive‐resistant features (Diaz et al, 2012). Several studies have shown that cells within the relapse‐driven clone are often present as a low‐frequency subclone prior to treatment, indicating these subclones are selected during therapy (Diaz et al, 2012; Morrissy et al, 2016). Resistance to ibrutinib, an inhibitor of the Bruton’s tyrosine kinase (BTK), is a compelling example of this where whole‐exome sequencing and kinetic analysis identified that ibrutinib therapy led to the expansion of clones with mutations in TRAIL‐R that were initially present in low numbers, favoring the pathways which are similar to the receptor‐mediated cell death mechanism of CC in winner cells (Burger et al, 2016). More generally, platinum agent‐based chemotherapy provides another instance of this phenomenon. Platinum agents are used in a wide number of cancers in combination with other chemotherapeutic drugs (e.g., FOLFOX in the colon and pancreatic cancer) (Grothey & Venook, 2018) or alone (e.g., in ovarian cancer) (Ledermann et al, 2018) to induce DNA damage, thus leading to cell apoptosis (Diaz Osterman et al, 2019) (Fig 3A). Secondly, these agents are genotoxic, and thus, cells surviving treatment are likely to accumulate additional mutations enabling them to regenerate a more aggressive tumor (Fig 3B). Therefore, mechanisms underlying therapy resistance and growth of clones following treatment‐induced selective sweeps must be understood and accounted for in therapy rationale.
Figure 3. Cell competition in resistance to chemotherapy.
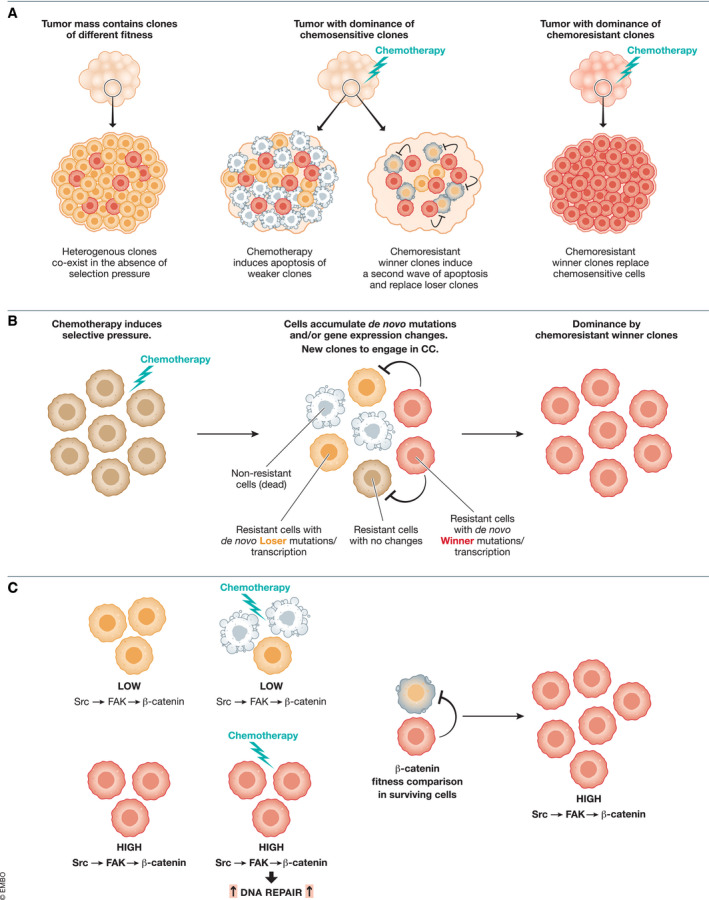
(A) Tumors harbor therapy‐resistant subclones. Chemotherapy results in massive cell death, which frees space for resistant clones to grow. Resistant cells now engage in competition‐based elimination of less‐fit cells and generate a more aggressive, “superfit” tumor. (B) Chemotherapy regimens induce large‐scale mutations and gene‐expression changes that can eliminate most non‐chemotherapy‐resistant cells. However, in a small population of surviving chemotherapy‐resistant cells, de‐novo mutations and gene‐expression changes pertinent to cell competition occur as a result of chemotherapy‐induced selection pressure. These distinct populations can now engage in cell competition, and a single population is expected to be clonally dominant in the relapsed tumor. (C) An example of selection of a chemoresistant population with greater fitness. Src‐induced FAK/β‐catenin activity results in chemotherapy resistance. Upon chemotherapy, chemosensitive cells are eliminated and remaining cells may undergo cell competition based on β‐catenin activity, in which cells with higher β‐catenin activity function as winners. This ultimately would generate a tumor with strong chemotherapy resistance.
Studies have reported that focal adhesion kinase (FAK) activity leads to platinum resistance (Diaz Osterman et al, 2019). FAK is fully activated by forming a signaling complex with SRC (Brunton et al, 2005) and promotes nuclear accumulation of β‐catenin (Chen et al, 2012), which is associated with CC mechanisms (Enomoto & Igaki, 2013; Akieda et al, 2019). Upon platinum therapy, subclones harboring higher activity of FAK have higher activity of SRC (Mitra & Schlaepfer, 2006) and increased β‐catenin transcriptional activity (Diaz Osterman et al, 2019) (Fan et al, 2019) (Fig 3C). Similarly, subclones with GOF mutations in genes such as KRAS, NRAS and BRAF have constitutive activation of EGFR/RAS/MAPK pathway, and can bypass EGFR blockade and are thus resistant to cetuximab therapy (anti‐EGFR therapy) (Lievre et al, 2006). Additionally, enhanced EGFR/RAS/MAPK signaling confers a competitive advantage (Moreno et al, 2019) and would enable these clones to outcompete their surrounding WT neighbors, ultimately resulting in a more aggressive, and therapeutically resistant tumor. Similarly, increased nuclear translocation of the CC effector, YAP, which can also be induced by hypoxia, leads to chemoresistance (Dai et al, 2016) again highlighting how CC pathways potentially converge with molecular mechanisms that drive enhanced oncogenic properties of tumors such as hypoxia and drug resistance. Chemotherapy eliminates sensitive cells leaving behind clones with intrinsic resistance, or those that adapted to survive. We hypothesize that these alterations confer resistant cells different competitive status. Thus, CC mechanisms can be involved in parallel with chemotherapy, by selecting winner resistant clones that will establish their dominance over loser resistant clones. Lastly, it would be of interest to better predict the effects of therapy on competition and design therapeutic strategies that harness CC mechanisms to shift outcomes in favor of normal cells, and not repopulation of resistant oncogenic clones. In vitro modeling showed that treatment of normal human squamous cells with antioxidant or growth factors enhanced their fitness and they obtained the ability to outcompete Barrett’s esophagus cells (Merlo et al, 2011). Similarly, antioxidant loading prior to irradiation of the mouse esophagus thwarted outcompetition of normal cells by MT‐p53 progenitors and instead promoted normal cell proliferation (Fernandez‐Antoran et al, 2019). Moreover, disruption of supercompetitor oncogenic mutant clonal expansion and tumor growth was achieved by reversion of loser status of surrounding WT neighbors through the disabling of competition by secreted factors (Flanagan et al, 2021; van Neerven et al, 2021). In summary, we propose that cancer therapy could potentially provoke unchecked CC and generate more aggressive tumors. However, there is potential to tilt the fate of CC by manipulation of the microenvironment and herein lies the power of CC that we argue should be taken into account in therapeutic rationale.
Summary
In the current review, we discuss the classical and novel pathways involved in cell competition and their roles in cancer. Many CC genes such as TP53, Myc, Hippo signaling components YAP/TAZ, and others are repurposed by cancers to attain growth and survival advantages. These models elucidate key pathways such as cancer growth (Di Giacomo et al, 2017), cell death (Moreno et al, 2002), and loss of polarity (Merino et al, 2015) in CC‐driven oncogenesis. Intratumoral competition is an inevitable facet of tumor biology, and results in enhanced tumor aggressiveness and complicates therapy. But lessons learned from CC studies may provide important insights into how cancer cells compete with each other and normal cells to overtake tissue space, particularly under the pretense of various microenvironment pressures. Recent studies presented in this review led us to postulate that tumor heterogeneity, and chemotherapy manipulate CC mechanisms to select for mutations that confer a supercompetitor phenotype and result in a more aggressive tumor. A better understanding of CC directly in drug resistance, which plague clinical interventions, is therefore highly important to better our understanding of cancer biology. But first, many fundamental questions in CC remain, such as fitness recognition mechanisms, alteration of cell fitness, and more comprehensive studies of how these homeostatic mechanisms are altered during cancer initiation and growth.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgements
This work is supported by the ERC CoG and Champalimaud Foundation research funds to EMo; La Caixa Junior Group Leader Funding to EMa; FCT, Swiss Cancer League, NIH/NCI, SNSF (RG); The Novo Nordisk Foundation grant number NNF17CC0027852 (KJW); National Institutes of Health, National Cancer Institute grants R01 CA244993 and R01 CA259599; and the National Foundation for Cancer Research (NFCR) (PBF). We thank Luni Emdad (VCU) for assistance in editing of manuscript.
The EMBO Journal (2021) 40: e107271.
This article is part of the Cancer Reviews 2021 series.
Contributor Information
Esha Madan, Email: esha.madan@research.fchampalimaud.org.
Eduardo Moreno, Email: eduardo.moreno@research.fchampalimaud.org.
Rajan Gogna, Email: rajangogna@gmail.com, Email: rajan.gogna@bric.ku.dk, Email: rajan.gogna@research.fchampalimaud.org.
References
- Akieda Y, Ogamino S, Furuie H, Ishitani S, Akiyoshi R, Nogami J, Masuda T, Shimizu N, Ohkawa Y, Ishitani T (2019) Cell competition corrects noisy Wnt morphogen gradients to achieve robust patterning in the zebrafish embryo. Nat Commun 10: 4710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcolea MP, Jones PH (2015) Cell competition: winning out by losing notch. Cell Cycle 14: 9–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alpar L, Bergantinos C, Johnston LA (2018) Spatially restricted regulation of spatzle/toll signaling during cell competition. Dev Cell 46: 706–719 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondar T, Medzhitov R (2010) p53‐mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell 6: 309–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowling S, Di Gregorio A, Sancho M, Pozzi S, Aarts M, Signore M, D. Schneider M, Martinez‐Barbera JP, Gil J, Rodríguez TA (2018) P53 and mTOR signalling determine fitness selection through cell competition during early mouse embryonic development. Nat Commun 9: 1763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braakhuis BJ, Tabor MP, Kummer JA, Leemans CR, Brakenhoff RH (2003) A genetic explanation of Slaughter's concept of field cancerization: evidence and clinical implications. Cancer Res 63: 1727–1730 [PubMed] [Google Scholar]
- Brunton VG, Avizienyte E, Fincham VJ, Serrels B, Metcalf CA 3rd, Sawyer TK, Frame MC (2005) Identification of Src‐specific phosphorylation site on focal adhesion kinase: dissection of the role of Src SH2 and catalytic functions and their consequences for tumor cell behavior. Cancer Res 65: 1335–1342 [DOI] [PubMed] [Google Scholar]
- Burger JA, Landau DA, Taylor‐Weiner A, Bozic I, Zhang H, Sarosiek K, Wang L, Stewart C, Fan J, Hoellenriegel Jet al (2016) Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition. Nat Commun 7: 11589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun PK, Zhang C, Yao B, Wardwell‐Ozgo J, Terry D, Jin P, Moberg K (2019) The Taiman transcriptional coactivator engages toll signals to promote apoptosis and intertissue invasion in Drosophila . Curr Biol 29: 2790–2800.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XL, Nam JO, Jean C, Lawson C, Walsh CT, Goka E, Lim ST, Tomar A, Tancioni I, Uryu Set al (2012) VEGF‐induced vascular permeability is mediated by FAK. Dev Cell 22: 146–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claveria C, Giovinazzo G, Sierra R, Torres M (2013) Myc‐driven endogenous cell competition in the early mammalian embryo. Nature 500: 39–44 [DOI] [PubMed] [Google Scholar]
- de la Cova C, Abril M, Bellosta P, Gallant P, Johnston LA (2004) Drosophila Myc regulates organ size by inducing cell competition. Cell 117: 107–116 [DOI] [PubMed] [Google Scholar]
- de la Cova C, Senoo‐Matsuda N, Ziosi M, Wu DC, Bellosta P, Quinzii CM, Johnston LA (2014) Supercompetitor status of Drosophila Myc cells requires p53 as a fitness sensor to reprogram metabolism and promote viability. Cell Metab 19: 470–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowson AN (2006) Basal cell carcinoma: biology, morphology and clinical implications. Mod Pathol 19: S127–S147 [DOI] [PubMed] [Google Scholar]
- Dai XY, Zhuang LH, Wang DD, Zhou TY, Chang LL, Gai RH, Zhu DF, Yang B, Zhu H, He QJ (2016) Nuclear translocation and activation of YAP by hypoxia contributes to the chemoresistance of SN38 in hepatocellular carcinoma cells. Oncotarget 7: 6933–6947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giacomo S, Sollazzo M, de Biase D, Ragazzi M, Bellosta P, Pession A, Grifoni D (2017) Human cancer cells signal their competitive fitness through MYC activity. Sci Rep 7: 12568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz LA Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MAet al (2012) The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 486: 537–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz Osterman CJ, Ozmadenci D, Kleinschmidt EG, Taylor KN, Barrie AM, Jiang S, Bean LM, Sulzmaier FJ, Jean C, Tancioni Iet al (2019) FAK activity sustains intrinsic and acquired ovarian cancer resistance to platinum chemotherapy. Elife 8: e47327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz‐Diaz C, Fernandez de Manuel L, Jimenez‐Carretero D, Montoya MC, Claveria C, Torres M (2017) Pluripotency surveillance by Myc‐driven competitive elimination of differentiating cells. Dev Cell 42: 585–599.e4 [DOI] [PubMed] [Google Scholar]
- Eichenlaub T, Cohen SM, Herranz H (2016) Cell competition drives the formation of metastatic tumors in a Drosophila model of epithelial tumor formation. Curr Biol 26: 419–427 [DOI] [PubMed] [Google Scholar]
- Enomoto M, Igaki T (2013) Src controls tumorigenesis via JNK‐dependent regulation of the Hippo pathway in Drosophila . EMBO Rep 14: 65–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Z, Duan J, Wang L, Xiao S, Li L, Yan X, Yao W, Wu L, Zhang S, Zhang Yet al (2019) PTK2 promotes cancer stem cell traits in hepatocellular carcinoma by activating Wnt/beta‐catenin signaling. Cancer Lett 450: 132–143 [DOI] [PubMed] [Google Scholar]
- Fernandez‐Antoran D, Piedrafita G, Murai K, Ong SH, Herms A, Frezza C, Jones PH (2019) Outcompeting p53‐mutant cells in the normal esophagus by redox manipulation. Cell Stem Cell 25: 329–341.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiore VF, Krajnc M, Quiroz FG, Levorse J, Pasolli HA, Shvartsman SY, Fuchs E (2020) Mechanics of a multilayer epithelium instruct tumour architecture and function. Nature 585: 433–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan DJ, Pentinmikko N, Luopajärvi K, Willis NJ, Gilroy K, Raven AP, Mcgarry L, Englund JI, Webb AT, Scharaw Set al (2021) NOTUM from Apc‐mutant cells biases clonal competition to initiate cancer. Nature 594: 430–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao R, Davis A, McDonald TO, Sei E, Shi X, Wang Y, Tsai PC, Casasent A, Waters J, Zhang Het al (2016) Punctuated copy number evolution and clonal stasis in triple‐negative breast cancer. Nat Genet 48: 1119–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlinger M, Swanton C (2010) How Darwinian models inform therapeutic failure initiated by clonal heterogeneity in cancer medicine. Br J Cancer 103: 1139–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germani F, Hain D, Sternlicht D, Moreno E, Basler K (2018) The Toll pathway inhibits tissue growth and regulates cell fitness in an infection‐dependent manner. Elife 7: e39939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves M, Maley CC (2012) Clonal evolution in cancer. Nature 481: 306–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grothey A, Venook AP (2018) Optimizing adjuvant therapy for localized colon cancer and treatment selection in advanced colorectal cancer. J Natl Compr Canc Netw 16: 611–615 [DOI] [PubMed] [Google Scholar]
- Gupta N, Jung K, Wu C, Alshareef A, Alqahtani H, Damaraju S, Mackey JR, Ghosh S, Sabri S, Abdulkarim BSet al (2017) High Myc expression and transcription activity underlies intra‐tumoral heterogeneity in triple‐negative breast cancer. Oncotarget 8: 28101–28115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto M, Sasaki H (2019) Epiblast formation by TEAD‐YAP‐dependent expression of pluripotency factors and competitive elimination of unspecified cells. Dev Cell 50: 139–154.e5 [DOI] [PubMed] [Google Scholar]
- Hogan C, Dupre‐Crochet S, Norman M, Kajita M, Zimmermann C, Pelling AE, Piddini E, Baena‐Lopez LA, Vincent JP, Itoh Yet al (2009) Characterization of the interface between normal and transformed epithelial cells. Nat Cell Biol 11: 460–467 [DOI] [PubMed] [Google Scholar]
- Hu Z, Sun R, Curtis C (2017) A population genetics perspective on the determinants of intra‐tumor heterogeneity. Biochim Biophys Acta Rev Cancer 1867: 109–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaki T, Pastor‐Pareja JC, Aonuma H, Miura M, Xu T (2009) Intrinsic tumor suppression and epithelial maintenance by endocytic activation of Eiger/TNF signaling in Drosophila . Dev Cell 16: 458–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajita M, Hogan C, Harris AR, Dupre‐Crochet S, Itasaki N, Kawakami K, Charras G, Tada M, Fujita Y (2010) Interaction with surrounding normal epithelial cells influences signalling pathways and behaviour of Src‐transformed cells. J Cell Sci 123: 171–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajita M, Sugimura K, Ohoka A, Burden J, Suganuma H, Ikegawa M, Shimada T, Kitamura T, Shindoh M, Ishikawa Set al (2014) Filamin acts as a key regulator in epithelial defence against transformed cells. Nat Commun 5: 4428 [DOI] [PubMed] [Google Scholar]
- Katsukawa M, Ohsawa S, Zhang L, Yan Y, Igaki T (2018) Serpin facilitates tumor‐suppressive cell competition by blocking toll‐mediated Yki activation in Drosophila . Curr Biol 28: 1756–1767.e6 [DOI] [PubMed] [Google Scholar]
- Kon S, Ishibashi K, Katoh H, Kitamoto S, Shirai T, Tanaka S, Kajita M, Ishikawa S, Yamauchi H, Yako Yet al (2017) Cell competition with normal epithelial cells promotes apical extrusion of transformed cells through metabolic changes. Nat Cell Biol 19: 530–541 [DOI] [PubMed] [Google Scholar]
- Ledermann JA, Raja FA, Fotopoulou C, Gonzalez‐Martin A, Colombo N, Sessa C, Group EGW (2018) Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow‐up. Ann Oncol 29: iv259 [DOI] [PubMed] [Google Scholar]
- Levayer R, Dupont C, Moreno E (2016) Tissue crowding induces caspase‐dependent competition for space. Curr Biol 26: 670–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lievre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, Cote JF, Tomasic G, Penna C, Ducreux Met al (2006) KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res 66: 3992–3995 [DOI] [PubMed] [Google Scholar]
- Ling S, Hu Z, Yang Z, Yang F, Li Y, Lin P, Chen Ke, Dong L, Cao L, Tao Yet al (2015) Extremely high genetic diversity in a single tumor points to prevalence of non‐Darwinian cell evolution. Proc Natl Acad Sci USA 112: E6496–E6505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Matsumura H, Kato T, Ichinose S, Takada A, Namiki T, Asakawa K, Morinaga H, Mohri Y, De Arcangelis Aet al (2019a) Stem cell competition orchestrates skin homeostasis and ageing. Nature 568: 344–350 [DOI] [PubMed] [Google Scholar]
- Liu Z, Yee PP, Wei Y, Liu Z, Kawasawa YI, Li W (2019b) Differential YAP expression in glioma cells induces cell competition and promotes tumorigenesis. J Cell Sci 132: jcs225714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd MC, Cunningham JJ, Bui MM, Gillies RJ, Brown JS, Gatenby RA (2016) Darwinian dynamics of intratumoral heterogeneity: not solely random mutations but also variable environmental selection forces. Cancer Res 76: 3136–3144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P, Weaver VM, Werb Z (2012) The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol 196: 395–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan E, Parker TM, Pelham CJ, Palma AM, Peixoto ML, Nagane M, Chandaria A, Tomas AR, Canas‐Marques R, Henriques Vet al (2019a) HIF‐transcribed p53 chaperones HIF‐1alpha. Nucleic Acids Res 47: 10212–10234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan E, Peixoto ML, Dimitrion P, Eubank TD, Yekelchyk M, Talukdar S, Fisher PB, Mi QS, Moreno E, Gogna R (2020) Cell competition boosts clonal evolution and hypoxic selection in cancer. Trends Cell Biol 30: 967–978 [DOI] [PubMed] [Google Scholar]
- Madan E, Pelham CJ, Nagane M, Parker TM, Canas‐Marques R, Fazio K, Shaik K, Yuan Y, Henriques V, Galzerano Aet al (2019b) Flower isoforms promote competitive growth in cancer. Nature 572: 260–264 [DOI] [PubMed] [Google Scholar]
- Mamada H, Sato T, Ota M, Sasaki H (2015) Cell competition in mouse NIH3T3 embryonic fibroblasts controlled by Tead activity and Myc. J Cell Sci 128: 790–803 [DOI] [PubMed] [Google Scholar]
- Mao JH, Wheldon TE (1995) A stochastic model for multistage tumorigenesis in developing and adult mice. Math Biosci 129: 95–110 [DOI] [PubMed] [Google Scholar]
- Martins VC, Busch K, Juraeva D, Blum C, Ludwig C, Rasche V, Lasitschka F, Mastitsky SE, Brors B, Hielscher Tet al (2014) Cell competition is a tumour suppressor mechanism in the thymus. Nature 509: 465–470 [DOI] [PubMed] [Google Scholar]
- Merino MM, Rhiner C, Lopez‐Gay JM, Buechel D, Hauert B, Moreno E (2015) Elimination of unfit cells maintains tissue health and prolongs lifespan. Cell 160: 461–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlo LM, Kosoff RE, Gardiner KL, Maley CC (2011) An in vitro co‐culture model of esophageal cells identifies ascorbic acid as a modulator of cell competition. BMC Cancer 11: 461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer SN, Amoyel M, Bergantinos C, de la Cova C, Schertel C, Basler K, Johnston LA (2014) An ancient defense system eliminates unfit cells from developing tissues during cell competition. Science 346: 1258236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra SK, Schlaepfer DD (2006) Integrin‐regulated FAK‐Src signaling in normal and cancer cells. Curr Opin Cell Biol 18: 516–523 [DOI] [PubMed] [Google Scholar]
- Morata G, Ripoll P (1975) Minutes: mutants of Drosophila autonomously affecting cell division rate. Dev Biol 42: 211–221 [DOI] [PubMed] [Google Scholar]
- Moreno E, Basler K, Morata G (2002) Cells compete for decapentaplegic survival factor to prevent apoptosis in Drosophila wing development. Nature 416: 755–759 [DOI] [PubMed] [Google Scholar]
- Moreno E, Basler K (2004) dMyc transforms cells into super‐competitors. Cell 117: 117–129 [DOI] [PubMed] [Google Scholar]
- Moreno E, Valon L, Levillayer F, Levayer R (2019) Competition for space induces cell elimination through compaction‐driven ERK downregulation. Curr Biol 29: 23–34.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrissy AS, Garzia L, Shih DJH, Zuyderduyn S, Huang Xi, Skowron P, Remke M, Cavalli FMG, Ramaswamy V, Lindsay PEet al (2016) Divergent clonal selection dominates medulloblastoma at recurrence. Nature 529: 351–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya IM, Castaldo SA, Van den Mooter L, Soheily S, Sansores‐Garcia L, Jacobs J, Mannaerts I, Xie J, Verboven E, Hillen Het al (2019) Peritumoral activation of the Hippo pathway effectors YAP and TAZ suppresses liver cancer in mice. Science 366: 1029–1034 [DOI] [PubMed] [Google Scholar]
- Murai K, Skrupskelyte G, Piedrafita G, Hall M, Kostiou V, Ong SH, Nagy T, Cagan A, Goulding D, Klein AMet al (2018) Epidermal tissue adapts to restrain progenitors carrying clonal p53 mutations. Cell Stem Cell 23: 687–699.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata R, Nakamura M, Sanaki Y, Igaki T (2019) Cell competition is driven by autophagy. Dev Cell 51: 99–112.e4 [DOI] [PubMed] [Google Scholar]
- Norman M, Wisniewska KA, Lawrenson K, Garcia‐Miranda P, Tada M, Kajita M, Mano H, Ishikawa S, Ikegawa M, Shimada Tet al (2012) Loss of Scribble causes cell competition in mammalian cells. J Cell Sci 125: 59–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowell PC (1976) The clonal evolution of tumor cell populations. Science 194: 23–28 [DOI] [PubMed] [Google Scholar]
- Oertel M, Menthena A, Dabeva MD, Shafritz DA (2006) Cell competition leads to a high level of normal liver reconstitution by transplanted fetal liver stem/progenitor cells. Gastroenterology 130: 507–520. quiz 590 [DOI] [PubMed] [Google Scholar]
- Ohsawa S, Sugimura K, Takino K, Xu T, Miyawaki A, Igaki T (2011) Elimination of oncogenic neighbors by JNK‐mediated engulfment in Drosophila . Dev Cell 20: 315–328 [DOI] [PubMed] [Google Scholar]
- Oliver ER, Saunders TL, Tarle SA, Glaser T (2004) Ribosomal protein L24 defect in belly spot and tail (Bst), a mouse minute. Development 131: 3907–3920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel MS, Shah HS, Shrivastava N (2017) c‐Myc‐dependent cell competition in human cancer cells. J Cell Biochem 118: 1782–1791 [DOI] [PubMed] [Google Scholar]
- Petrova E, Soldini D, Moreno E (2011) The expression of SPARC in human tumors is consistent with its role during cell competition. Commun Integr Biol 4: 171–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak K (2014) Tumor heterogeneity confounds and illuminates: a case for Darwinian tumor evolution. Nat Med 20: 344–346 [DOI] [PubMed] [Google Scholar]
- Portela M, Casas‐Tinto S, Rhiner C, Lopez‐Gay JM, Dominguez O, Soldini D, Moreno E (2010) Drosophila SPARC is a self‐protective signal expressed by loser cells during cell competition. Dev Cell 19: 562–573 [DOI] [PubMed] [Google Scholar]
- Prober DA, Edgar BA (2000) Ras1 promotes cellular growth in the Drosophila wing. Cell 100: 435–446 [DOI] [PubMed] [Google Scholar]
- Ramon YCS, Sese M, Capdevila C, Aasen T, De Mattos‐Arruda L, Diaz‐Cano SJ, Hernandez‐Losa J, Castellvi J (2020) Clinical implications of intratumor heterogeneity: challenges and opportunities. J Mol Med 98: 161–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhiner C, Lopez‐Gay JM, Soldini D, Casas‐Tinto S, Martin FA, Lombardia L, Moreno E (2010) Flower forms an extracellular code that reveals the fitness of a cell to its neighbors in Drosophila . Dev Cell 18: 985–998 [DOI] [PubMed] [Google Scholar]
- Rodrigues AB, Zoranovic T, Ayala‐Camargo A, Grewal S, Reyes‐Robles T, Krasny M, Wu DC, Johnston LA, Bach EA (2012) Activated STAT regulates growth and induces competitive interactions independently of Myc, Yorkie, Wingless and ribosome biogenesis. Development 139: 4051–4061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblatt J, Raff MC, Cramer LP (2001) An epithelial cell destined for apoptosis signals its neighbors to extrude it by an actin‐ and myosin‐dependent mechanism. Curr Biol 11: 1847–1857 [DOI] [PubMed] [Google Scholar]
- Sancho M, Di‐Gregorio A, George N, Pozzi S, Sanchez JM, Pernaute B, Rodriguez TA (2013) Competitive interactions eliminate unfit embryonic stem cells at the onset of differentiation. Dev Cell 26: 19–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki A, Nagatake T, Egami R, Gu G, Takigawa I, Ikeda W, Nakatani T, Kunisawa J, Fujita Y (2018) Obesity suppresses cell‐competition‐mediated apical elimination of RasV12‐transformed cells from epithelial tissues. Cell Rep 23: 974–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakiba N, Fahmy A, Jayakumaran G, McGibbon S, David L, Trcka D, Elbaz J, Puri MC, Nagy A, van der Kooy Det al (2019) Cell competition during reprogramming gives rise to dominant clones. Science 364: eaan0925 [DOI] [PubMed] [Google Scholar]
- Shraiman BI (2005) Mechanical feedback as a possible regulator of tissue growth. Proc Natl Acad Sci USA 102: 3318–3323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suijkerbuijk SJ, Kolahgar G, Kucinski I, Piddini E (2016) Cell competition drives the growth of intestinal adenomas in Drosophila . Curr Biol 26: 428–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Luo T, Ren Y, Florey O, Shirasawa S, Sasazuki T, Robinson DN, Overholtzer M (2014) Competition between human cells by entosis. Cell Res 24: 1299–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamori Y, Bialucha CU, Tian AG, Kajita M, Huang YC, Norman M, Harrison N, Poulton J, Ivanovitch K, Disch Let al (2010) Involvement of Lgl and Mahjong/VprBP in cell competition. PLoS Biol 8: e1000422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Neerven SM, de Groot NE, Nijman LE, Scicluna BP, van Driel MS, Lecca MC, Warmerdam DO, Kakkar V, Moreno LF, Vieira Braga FAet al (2021) Apc‐mutant cells act as supercompetitors in intestinal tumour initiation. Nature 594: 436–441 [DOI] [PubMed] [Google Scholar]
- Vincent JP, Kolahgar G, Gagliardi M, Piddini E (2011) Steep differences in wingless signaling trigger Myc‐independent competitive cell interactions. Dev Cell 21: 366–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagstaff L, Goschorska M, Kozyrska K, Duclos G, Kucinski I, Chessel A, Hampton‐O'Neil L, Bradshaw CR, Allen GE, Rawlins ELet al (2016) Mechanical cell competition kills cells via induction of lethal p53 levels. Nat Commun 7: 11373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MJ, Werner B, Barnes CP, Graham TA, Sottoriva A (2016) Identification of neutral tumor evolution across cancer types. Nat Genet 48: 238–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Ohsawa S, Kunimasa K, Igaki T (2017) The ligand Sas and its receptor PTP10D drive tumour‐suppressive cell competition. Nature 542: 246–250 [DOI] [PubMed] [Google Scholar]
- Yum MK, Han S, Fink J, Wu SS, Dabrowska C, Trendafilova T, Mustata R, Chatzeli L, Azzarelli R, Pshenichnaya Iet al (2021) Tracing oncogene‐driven remodelling of the intestinal stem cell niche. Nature 594: 442–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Huang T, Cheng AS, Yu J, Kang W, To KF (2016) The TEAD family and its oncogenic role in promoting tumorigenesis. Int J Mol Sci 17: 138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziosi M, Baena‐López LA, Grifoni D, Froldi F, Pession A, Garoia F, Trotta V, Bellosta P, Cavicchi S, Pession A (2010) dMyc functions downstream of yorkie to promote the supercompetitive behavior of hippo pathway mutant cells. PLoS Genet 6: e1001140 [DOI] [PMC free article] [PubMed] [Google Scholar]