

Abstract
Bacillus cereus sensu lato is a group of Gram‐positive endospore‐forming bacteria with high ecological diversity. Their endospores are decorated with micrometer‐long appendages of unknown identity and function. Here, we isolate endospore appendages (Enas) from the food poisoning outbreak strain B. cereus NVH 0075‐95 and find proteinaceous fibers of two main morphologies: S‐ and L‐Ena. By using cryoEM and 3D helical reconstruction of S‐Enas, we show these to represent a novel class of Gram‐positive pili. S‐Enas consist of single domain subunits with jellyroll topology that are laterally stacked by β‐sheet augmentation. S‐Enas are longitudinally stabilized by disulfide bonding through N‐terminal connector peptides that bridge the helical turns. Together, this results in flexible pili that are highly resistant to heat, drought, and chemical damage. Phylogenomic analysis reveals a ubiquitous presence of the ena‐gene cluster in the B. cereus group, which include species of clinical, environmental, and food importance. We propose Enas to represent a new class of pili specifically adapted to the harsh conditions encountered by bacterial spores.
Keywords: Bacillus, Endospore, pilus, protein nanofiber, self‐assembly
Subject Categories: Microbiology, Virology & Host Pathogen Interaction; Structural Biology
Cryo‐EM reconstruction reveals a novel class of pili in Gram‐positive bacteria, ENdospore Appendages (Enas), with a unique architecture and two distinct morphologies.

Introduction
When faced with adverse growth conditions, some species belonging to the phylum Firmicutes can differentiate into the metabolically dormant endospore. These endospores exhibit extreme resilience toward environmental stressors due to their dehydrated nature and unique multilayered cellular structure, and can germinate into the metabolically active and replicating vegetative growth state even hundreds of years after their formation (Setlow, 2014). In this way, Firmicutes belonging to the classes Bacilli and Clostridia can withstand long periods of drought, starvation, high oxygen, or antibiotic stress.
Endospores typically consist of an innermost dehydrated core, which contains the bacterial DNA. The core is enclosed by an inner membrane surrounded by a thin layer of peptidoglycan that will become the cell wall of the vegetative cell that emerges during endospore germination (Fig 1A). Then follows a thick cortex layer of modified peptidoglycan that is essential for dormancy. The cortex layer is in turn surrounded by several proteinaceous coat layers (Atrih & Foster, 1999). In some Clostridium and most Bacillus cereus group species, the spore is enclosed by an outermost loose‐fitting paracrystalline exosporium layer consisting of (glyco)proteins and lipids (Stewart, 2015). The surface of Bacillus and Clostridium endospores can also be decorated with multiple micrometers long filamentous appendages, which show a great structural diversity between strains and species (Rode et al, 1971; Hachisuka & Kuno, 1976; Walker et al, 2007). Spores of species belonging to the B. cereus group are often covered with appendages which morphologically resemble pili of Gram‐negative and Gram‐positive bacteria when imaged by negative stain transmission electron microscopy (TEM) (Ankolekar & Labbe, 2010; Smirnova et al, 2013). The endospore appendages, hereafter called Enas, vary in number and morphology between B. cereus group strains and species, and some strains even simultaneously express Enas of different morphologies (Smirnova et al, 2013). Structures resembling the Enas have not been observed on the surface of the vegetative cells, suggesting that they may represent spore‐specific fibers.
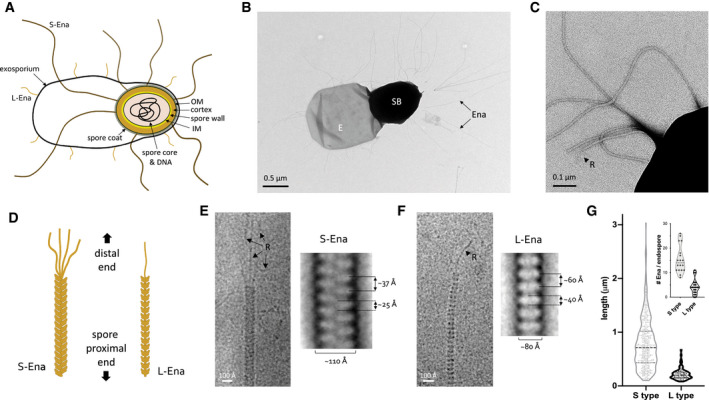
Figure 1. Bacillus cereus endospores carry S‐ and L‐Enas.
-
ASchematic representation of the B. cereus endospore. IM: inner membrane, OM: outer membrane. Together, spore core, IM, spore wall, cortex, OM, and spore coat make the spore body, surrounded by the exosporium. S‐ and L‐Ena represent staggered and ladder type endospore appendages – see this study.
-
B, CNegative stain TEM image of B. cereus NVH 0075‐95 endospore, showing spore body (SB), exosporium (E), and endospore appendages (Ena), which emerge from the endospore individually or as fiber bundles. At the distal end, Enas terminate in a single or multiple thin ruffles (R).
-
DSchematic drawing of S‐ and L‐Ena appearance and their orientation relative to the spore.
-
E, FcryoEM images (left) and negative stain 2D class averages of single S‐Ena (E) and L‐Ena (F) fibers. S‐Ena shows ˜100 Å diameter and a helical or staggered appearance with a ˜37 Å rise, whereas L‐Ena has a ˜80 Å diameter and appear as stacked disks of ˜40 Å height and ˜60 Å translation along the fiber. L‐ and S‐Ena show, respectively, a single or multiple ruffle(s) (black arrow, labeled “R”) at the distal end of fiber.
-
GLength distribution of S‐ and L‐Enas and number of Enas per endospore (inset), (n = 1,023 individual Enas, from 150 endospores, from 5 batches). Dashed and dotted lines show median and first (lower) and third (upper) quartile, resp. See also Fig EV1.
Although the presence of endospore appendages in species belonging to the B. cereus group was reported already in the ’60s, efforts to characterize their composition and genetic identity have failed due to difficulties to solubilize and enzymatically digest the fibers (Gerhardt & Ribi, 1964; DesRosier & Lara, 1981). Therefore, there is no genetic or structural information and very limited functional data available for endospore appendages.
Here, we isolate Enas from the food poisoning outbreak strain B. cereus NVH 0075‐95 and find proteinaceous fibers of two main morphologies: S‐ and L‐Enas. By using cryoEM and 3D helical reconstruction of S‐Enas, we show these to represent a novel class of Gram‐positive pili. A unique architecture of subunit stabilization by lateral β‐augmentation and longitudinal disulfide cross‐linking gives rise to pili that combine high flexibility with high resistance to heat, drought, and chemical damage. The molecular identity of the S‐Ena subunits was deduced from the cryoEM electron potential maps of fibers isolated directly from the endospores and confirmed by analysis of mutants lacking genes encoding potential Ena protein subunits. S‐Ena fibers are encoded by three associated genes which are present in most species of the B. cereus group. Remarkably, recombinant S‐Ena subunits spontaneously self‐assemble in vitro into protein nanofibers with native S‐Ena‐like properties and structure.
Results
Bacilluscereus NVH 0075‐95 show endospore appendages of two morphological types
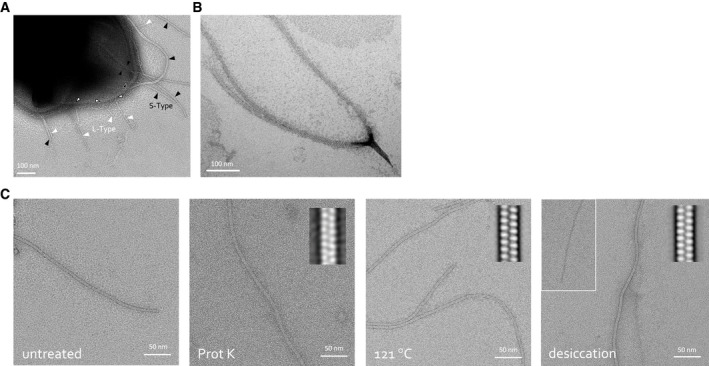
Negative stain EM imaging of B. cereus strain NVH 0075‐95 showed typical endospores with a dense core of ˜1 μm diameter, tightly wrapped by an exosporium layer that on TEM images emanates as a flat 2–3 μm long saclike structure from the endospore body (Fig 1A and B). The endospores showed an abundance of micrometer‐long appendages (Enas) (Fig 1B–F). The average endospore displayed 20–30 Enas ranging from 200 nm to 6 μm in length (Fig 1G), with a median length of approximately 600 nm. The density of Enas appeared highest at the pole of the spore body that lies near the exosporium. There, Enas seem to emerge from the exosporium as individual fibers or as a bundle of individual fibers that separates a few tens of nanometers above the endospore surface (Fig 1, EV1). Closer inspection revealed that the Enas showed two distinct morphologies (Fig 1C–F). The main or “Staggered‐type” (S‐Ena) morphology represents approximately 90% of the observed fibers. S‐Enas have a width of ˜110 Å and give a polar, staggered appearance in negative stain 2D classes, with alternating scales pointing down to the spore surface. At the distal end, S‐Enas terminate in multiple filamentous extensions or “ruffles” of 50–100 nm in length and ˜35 Å thick (Fig 1E). The minor or “Ladder‐like” (L‐Ena) morphology is thinner, ˜80 Å in width, and terminates in a single filamentous extension with dimensions similar to ruffles seen in S‐Ena fibers (Fig 1F). L‐Enas lack the scaled, staggered appearance of the S‐Enas, instead showing a ladder of stacked disk‐like units of ˜40 Å height. Whereas S‐Enas can be seen to traverse the exosporium and connect to the spore body, L‐Enas appear to emerge from the exosporium (Fig EV1A). Both Ena morphologies co‐exist on individual endospores (Fig EV1A). Neither Ena morphology is reminiscent of sortase‐mediated or type IV pili previously observed in Gram‐positive bacteria (Mandlik et al, 2008; Melville & Craig, 2013). In an attempt to identify their composition, shear force extracted and purified Enas were subjected to trypsin digestion for identification by mass spectrometry. However, despite the good enrichment of both S‐ and L‐Enas, no unambiguous candidates for Ena components were identified among the tryptic peptides, which largely contained contaminating mother cell proteins, EA1 S‐layer and spore coat proteins. Attempts to resolve the Ena monomers by SDS–PAGE were unsuccessful, including strong reducing conditions (up to 200 mM β‐mercaptoethanol), heat treatment (100°C), limited acid hydrolysis (1 h 1 M HCl), or incubation with chaotropes such as 8 M urea or 6 M guanidinium chloride. Ena fibers also retained their structural properties upon autoclaving, desiccation, or treatment with proteinase K (Fig EV1C).
Figure EV1. Ena morphology and robustness.
-
A, BNegative stain TEM of B. cereus NVH 0075‐95 endospore with indication of the two Ena morphologies: S‐type (black arrowheads) and L‐Enas (white arrowheads) (A), and closed‐up view of a dislodged S‐Ena bundle splitting into individual Ena fibers (B).
-
CNegative stain TEM images of isolated ex vivo S‐Ena. To test Ena stability under different stresses, samples were treated, from left to right, with: (1) untreated control, (2) 1 h of 1 mg/ml proteinase K, (3) autoclaving (i.e., 20 min at 121°C) or (4) a 4 h of desiccation at 43°C. Inset shows 2D class averages to assess the structural integrity of the treated Ena. S‐Enas are found to be resistant to Proteinase K treatment, autoclaving and desiccation at 43°C, although some fibers appear to lose subunit integrity upon desiccation (inset). Desiccation at 43°C may mimic conditions encountered by Bacillus spores during drought.
CryoEM of endospore appendages identifies their molecular identity
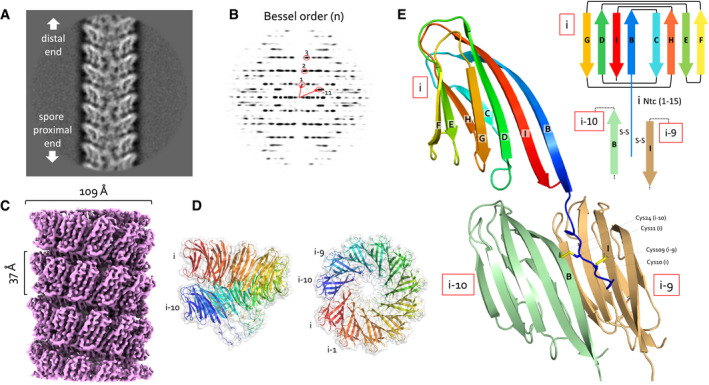
To further study the nature of the Enas, fibers purified from B. cereus NVH 0075‐95 endospores were imaged by cryogenic electron microscopy (cryoEM) and analyzed using 3D reconstruction. Isolated fibers showed a 9.4:1 ratio of S‐ and L‐Enas, similar to what was seen on endospores. Boxes with a dimension of 300 × 300 pixels (246 × 246 Å2) were extracted along the length of the fibers, with an inter‐box overlap of 21 Å, and subjected to 2D classification using RELION 3.0 (Zivanov et al, 2018). Power spectra of the 2D class averages revealed a well‐ordered helical symmetry for S‐Enas (Fig 2A and B), whereas L‐Enas primarily showed translational symmetry (Fig 1D). Based on a helix radius of approximately 54.5 Å, we estimated layer lines Z’ and Z" in the power spectrum of S‐Enas to have a Bessel order of −11 and 1, respectively (Fig 2A and B). In the 2D classes holding the majority of extracted boxes, the Bessel order 1 layer line was found at a distance of 0.02673 Å−1 from the equator, corresponding to a pitch of 37.4 Å, in good agreement with spacing of the apparent “lobes” seen also by negative stain (Figs 1C and 2B, and EV1). The correct helical parameters were derived by an empirical approach in which a systematic series of starting values for subunit rise and twist were used for 3D reconstruction and real space Bayesian refinement using RELION 3.0 (He & Scheres, 2017). Based on the estimated Fourier – Bessel indexing, input rise and twist were varied in the range of 3.05–3.65 Å and 29–35 degrees, respectively, with a sampling resolution of 0.1 Å and 1 degree between tested start values. This approach converged on a unique set of helical parameters that resulted in 3D maps with clear secondary structure and identifiable densities for subunit side chains (Fig 2C). The reconstructed map corresponds to a left‐handed 1‐start helix with a rise and twist of 3.22 Å and 31.03 degrees per subunit, corresponding to a helix with 11.6 units per turn (Fig 2D). After refinement and postprocessing in RELION 3.0, the map was found to be of resolution 3.2 Å according to the FSC0.143 criterion (Appendix Table S1, Fig EV2A).
Figure 2. CryoEM structure of S‐Enas.
-
A, B(A) Representative 2D class average of 300 × 300 pixel boxes covering ex vivo S‐Enas and (B) the corresponding power spectrum of the 2D class. The Bessel orders used to derive helical symmetry are indicated.
-
CReconstituted cryoEM electron potential map of ex vivo S‐Ena (3.2 Å resolution).
-
DSide and top view of a single helical turn of the de novo‐built 3D model of S‐Ena shown in ribbon representation and molecular surface. Adjacent Ena subunits are labeled i, i‐1, i‐2 to i‐10 (colored red to blue), where subunits i and i‐10 represent the upper and lower subunit in a single helical turn. Based on the orientation and low‐resolution features of 2D classes obtained on spore‐associated S‐Ena (Fig 1), subunits i and i‐10 would be oriented distal and proximal to the spore body, respectively.
-
ERibbon representation and topology diagram of the S‐Ena1B subunit (blue to red rainbow from N‐ to C‐terminus). The eight β‐strands making up the S‐Ena core domain are labeled B to I from N‐ to C‐terminus. The first 15 residues form an N‐terminal connector (Ntc) that for a subunit i, is in interaction with subunits i‐9 (sand) and i‐10 (green) through disulfide cross‐linking.
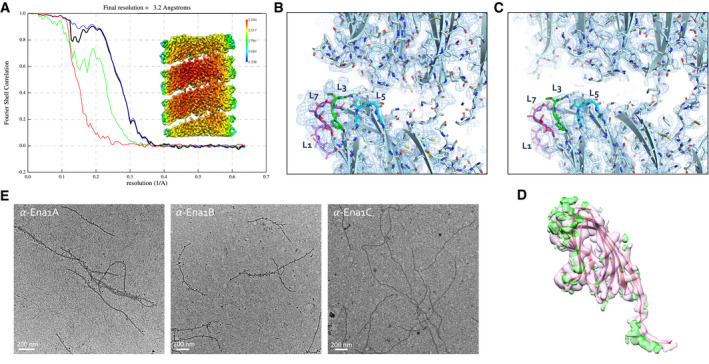
Figure EV2. S‐Ena is composed of both Ena1A and Ena1B subunits.
-
AFSC curve and local resolution heatmap (inset) of the recEna1B helical reconstruction, indicating a final resolution of 3.2 Å at a cutoff of 0.143. FSC curve and local resolution were calculated by postprocessing in RELION3.0 using a solvent mask consisting of 3 helical turns.
-
B, CSide‐by‐side comparison of cryoEM maps calculated from ex vivo (B) and recEna1B filaments (C), with the refined Ena1B model docked into the maps. The ex vivo Ena map shows features unaccounted for by the Ena1B model near loops 3 (L3) and 7 (L7), corresponding to regions of amino acid insertions in the Ena1A sequence (Fig EV2B).
- D
-
EImmunogold TEM of ex vivo S‐Enas, stained with, from left to right, anti‐Ena1A, anti‐Ena1B, and anti‐Ena1C sera, each with gold‐labeled (10 nm colloidal gold) anti‐rabbit IgG as secondary antibody. Specific staining with Ena1A and Ena1B sera confirms the presence of both subunits in native Enas. No staining was seen with Ena1C serum.
The resulting map showed well‐defined subunits comprising an 8‐stranded β‐sandwich domain of approximately 100 residues (Fig 2E). The side chain density was of sufficient quality to manually deduce a short motif with the sequence F‐C‐M‐V/T‐I‐R‐Y (Fig 3A). A search of the B. cereus NVH 0075‐95 proteome (GCA_001044825.1) identified KMP91698.1 as candidate Ena subunit, a 117 residue (12 kDa) hypothetical protein of unknown function encoded by TU63_02440 (Fig 3B). Further inspection of the electron potential map and manual model building of the S‐Ena subunit showed this to fit well with the full KMP91698.1 sequence. TU_63_02440 is located 15 bp downstream of TU63_02435, encoding KMP91697.1, a 126 residue (14 KDa) protein with 37% amino acid sequence identity, a shared domain of unknown function (DUF) 3992 and a similar Cys pattern compared with KMP91698.1 (Fig 3B). Downstream of TU_63_02440, on the minus strand, the locus TU63_0245 encodes a third DUF3992 containing hypothetical protein (KMP91699.1), of 160 amino acids and an estimated molecular weight of 17 kDa. As such, KMP91697.1, KMP91698.1, and KMP91699.1 are regarded as candidate Ena subunits, hereafter dubbed Ena1A, Ena1B, and Ena1C, respectively (Fig 3B and C).
Figure 3. S‐Ena structure determination and recombinant production.
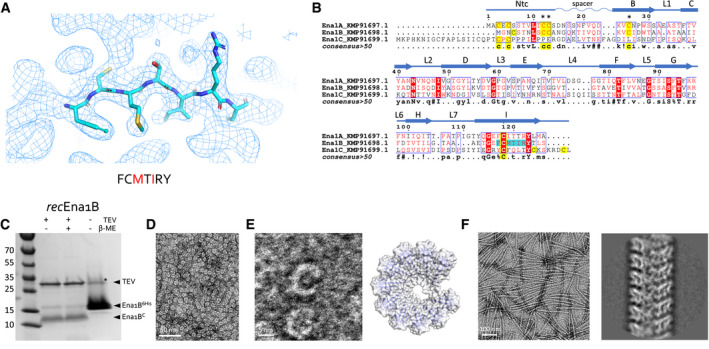
-
ARepresentative area of the 3D cryoEM potential map for ex vivo S‐Ena, at 3.2 Å resolution. An heptameric peptide with sequence FCMTIRY was deduced de novo from the cryoEM potential map (shown in sticks) and used for a BLAST search of the B. cereus NVH 0075‐95 genome. The red letters indicate amino acids that differ between KMP91697.1 and KMP91698.1 (see B).
-
BMultiple sequence alignment of 3 ORF’s (KMP91697.1, KMP91698.1, and KMP91699.1) corresponding to DUF3992 containing proteins. Sequence motifs corresponding or similar to that deduced from the EM potential map are shaded in cyan. Secondary structure and structural elements as determined from the built model (see Fig 2) are shown schematically above the sequences (Ntc: N‐terminal connecter; arrows correspond to β‐strands, labeled as in Fig 2). Contiguous areas of >50% similarity are boxed blue.
-
CSDS–PAGE of recEna1B, treated with β‐mercaptoethanol or TEV protease (to remove N‐terminal 6xHis tag) as indicated. Bands with apparent MW of ˜13 and ˜15 kDa correspond recEna1B monomer with (Ena1B6His) and without (Ena1BC) the 6xHis tag and TEV recognition site, respectively. In the uncleaved sample, a band running at ˜30 kDa (labeled *) corresponds to a non‐physiological disulfide bound S‐Ena1B dimer. Lower intensity in the cleaved recEna1B results from a loss of monomers to SDS‐resistant high‐molecular‐weight complexes stuck in the stacking gel.
-
DNegative stain TEM images of rec1Ena1B oligomers formed after refolding, but prior to TEV removal of the N‐terminal 6xHis tag.
-
EClose‐up view that shows recEna1B oligomers form open crescents similar in dimensions and shape to single helical turns or arcs found in the S‐Ena fiber (model – right). Steric hindrance by the 6xHis is thought to arrest recEna1B polymerization into single helical arcs.
-
FNegative stain image and 2D classification of S‐Ena‐like fibers formed after TEV digestion of recEna1B. Upon removal of the N‐terminal 6xHis tag, recEna1B readily assembles into fibers with helical properties closely resembling those found for ex vivo S‐Enas.
Ena1B self‐assembles into endospore appendage‐like nanofibers in vitro
To confirm the subunit identity of the endospore appendages isolated from B. cereus NVH0075‐95, we cloned a synthetic gene fragment corresponding to the coding sequence of Ena1B and an N‐terminal TEV protease cleavable 6xHis‐tag into a vector for recombinant expression in the cytoplasm of E. coli. The recombinant protein was found to form inclusion bodies, which were solubilized in 8 M urea before affinity purification. Removal of the chaotropic agent by rapid dilution resulted in the formation of abundant soluble crescent‐shaped oligomers reminiscent of a partial helical turn seen in the isolated S‐Enas (Fig 3D–E), suggesting the refolded recombinant Ena1B (recEna1B) adopts the native subunit–subunit β‐augmentation contacts (Fig 3E). We reasoned that recEna1B self‐assemble into helical appendages arrested at the level of a single turn due to steric hindrance by the 6xHis‐tag at the subunits N‐terminus. Indeed, proteolytic removal of the affinity tag readily resulted in the formation of fibers of 110 Å diameter and with helical parameters similar to S‐Enas, though lacking the distal ruffles seen in ex vivo fibers (Fig 3F). CryoEM data collection and 3D helical reconstruction were performed to assess whether in vitro recEna1B nanofibers were isomorphous with ex vivo S‐Enas. Real space refinement of helical parameters using RELION 3.0 converged on a subunit rise and twist of 3.43721 Å and 32.3504 degrees, respectively, approximately 0.2 Å and 1.3 degrees higher than found in ex vivo S‐Enas, and corresponding to a left‐handed helix with a pitch of 38.3 Å and 11.1 subunits per turn. Apart from the minor differences in helical parameters, the 3D reconstruction map of in vitro Ena1B fibers (estimated resolution of 3.2 Å; Fig EV2A and B) was near isomorphous to ex vivo S‐Enas in terms of size and connectivity of the fiber subunits (Fig EV2C and D). Closer inspection of the 3D cryoEM maps for recEna1B and ex vivo S‐Ena showed an improved side chain fit for Ena1B residues in the former (Fig EV2B–D) and revealed regions in the ex vivo Ena maps that showed partial side chain character of Ena1A, particularly in loops L1, L3, L5, and L7 (Figs 3B and EV2B and C). Although the Ena1B character of the ex vivo maps is dominant, this suggested that ex vivo S‐Enas consist of a mixed population of Ena1A and Ena1B fibers or that S‐Enas have a mixed composition comprising both Ena1A and Ena1B. Immunogold labeling using sera generated with recEna1A or recEna1B showed subunit‐specific labeling within single Enas, confirming these have a mixed composition of Ena1A and Ena1B (Figs EV2E and EV3A and B). No staining of S‐Enas was seen with Ena1C serum (Fig EV2E). No systematic patterning or molar ratio for Ena1A and Ena1B could be discerned from immunogold labeling or helical reconstructions with an asymmetric unit containing more than one subunit, suggesting the distribution of Ena1A and Ena1B in the fibers to be random. Apart from some side chain densities with mixed Ena1A and Ena1B character, the cryoEM electron potential maps of the ex vivo Enas showed a unique main chain conformation, indicating the Ena1A and Ena1B have near‐isomorphous folds.
Figure EV3. Ena1 sera and Ena1A–Ena1B overexpression.
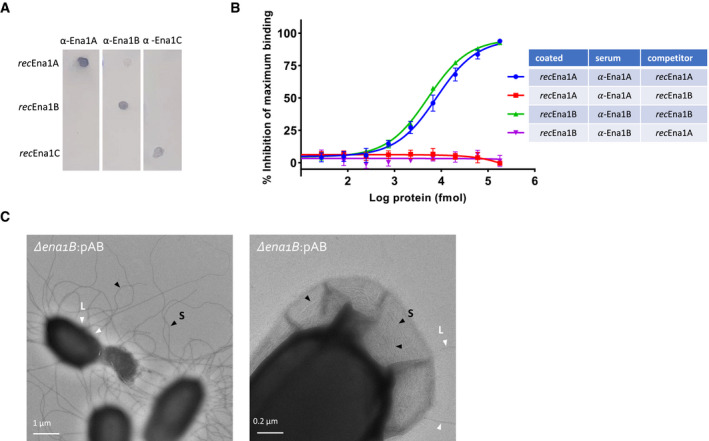
-
AEvaluation of residual cross‐reactivity in anti‐Ena1A, anti‐Ena1B, and anti‐Ena1C sera by dot blot. 100 ng of purified recEna1A, recEna1B, or recEna1C was coated on PVDF membrane, blocked, washed, and probed with the three different anti‐sera at 1:1,000 in TBST. Dot blot shows robust cognate binding with good selectivity against other Ena1 subunits.
-
BEvaluation of residual cross‐reactivity of anti‐Ena1A and anti‐Ena1B sera with recEnaB and recEnaA proteins, respectively, assayed using competitive ELISAs. The percentage inhibition of maximum binding was calculated using the formula (1‐(S‐B))/ (MA‐B) × 100, where S, B, and MA are the average absorbance of the sample (sera + recEna competitor added to the recEna‐coated wells), blank (only PBST added to the recEna‐coated wells), and maximum activity (only sera added to the recEna‐coated wells), respectively. The values presented are the averages of three independent experiments, and error bars depict ±SD of the averages. The lines are regression curves from three‐parameter logistic regression analyses (GraphPad prism version 9).
-
CNegative stain TEM images of endospores of NVH 0075‐95 mutant Δena1B complemented with pena1AB. Complementation with pena1AB results in aberrantly long and numerous S‐Ena (Fig 6). This overexpression of S‐Ena results in the frequent rupture of the exosporium (left panel), or the encapsulated of the S‐Ena into the exosporium (right panel). Selected S‐Ena and L‐Ena are labeled with black and with arrows, and labeled S and L, respectively.
Enas represent a novel family of Gram‐positive pili
Upon recognizing that native S‐Enas show a mixed Ena1A and Ena1B composition, we continued with 3D cryoEM reconstruction of recEna1B for model building. The Ena subunit consists of a typical jellyroll fold (Richardson, 1981) comprised of two juxtaposed β‐sheets consisting of strands BIDG and CHEF (Figs 2E and EV4). The jellyroll domain is preceded by a flexible 15‐residue N‐terminal extension hereafter referred to as N‐terminal connector (“Ntc”). In the helical turns, the side‐by‐side contact of Ena subunits occurs through β‐sheet augmentation, a protein interaction mechanism where an open‐edged β‐sheet is aligned and extended by a β‐sheet or β‐strand of the interaction partner (Remaut & Waksman, 2006). In S‐Ena, the sheet composed of strands BIDG of a subunit i is augmented with strands CHEF of the preceding subunit i‐1, and strands CHEF of subunit i are augmented with strands BIDG of the next subunit in row i + 1 (Figs 2E and EV4A and B). As such, the packing in the endospore appendages can be regarded as a slanted β‐propeller of 8‐stranded β‐sheets, with 11.6 blades per helical turn and an axial rise of 3.2 Å per subunit (Fig 2E). Subunit–subunit contacts in the β‐propeller are further stabilized by two complementary electrostatic patches on the Ena subunits (Fig EV4C). In addition to these lateral contacts, subunits across helical turns are also connected through the Ntc’s. The Ntc of each subunit i makes disulfide bond contacts with subunits i‐9 and i‐10 in the preceding helical turn (Figs 2E and EV4B). These contacts are made through disulfide bonding of Cys 10 and Cys 11 in subunit i, with Cys 109 and Cys 24 in the strands I and B of subunits i‐9 and i‐10, respectively (Figs 2E and EV4B). Thus, disulfide bonding via the Ntc results in a longitudinal stabilization of fibers by bridging the helical turns, as well as in a further lateral stabilization in the β‐propellers by covalent cross‐linking of adjacent subunits. The Ntc contacts lie on the luminal side of the helix, leaving a central void of approximately 1.2 nm diameter (Fig EV4D). Residues 12–17 form a flexible spacer region between the Ena jellyroll domain and the Ntc. Strikingly, this spacer region creates a 4.5 Å longitudinal gap between the Ena subunits, which are not in direct contact other than through the Ntc (Fig 4C). The flexibility in the Ntc spacer and the lack of direct longitudinal protein–protein contact of subunits across the helical turns create large flexibility and elasticity in the Ena fibers (Fig 4). 2D class averages of endospore‐associated fibers show longitudinal stretching, with a change in pitch of up to 8 Å (range: 37.1–44.9 Å; Fig 4D), and an axial rocking of up to 10 degrees per helical turn (Fig 4A and B).
Figure EV4. Inter‐subunit interactions in S‐Ena.
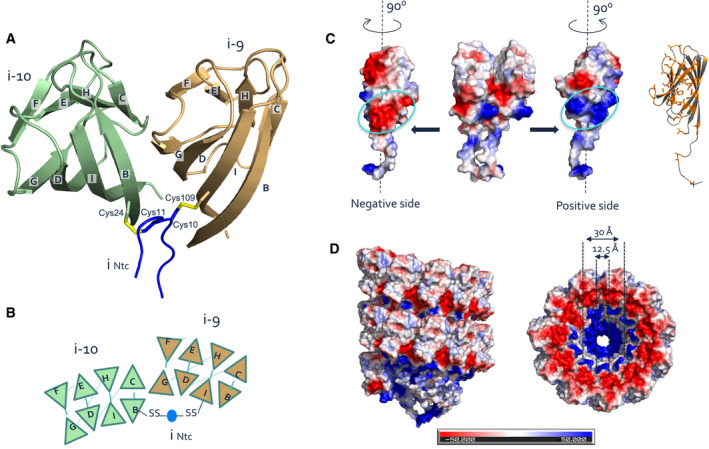
-
A, BRibbon (A) and schematic (B) representation of lateral subunit–subunit contacts in S‐Ena. Strand G of BIDG sheet of each subunit is augmented with strand C of CHEF β‐sheet of the succeeding subunit. Both subunits are covalently cross‐linked via the Ntc (blue) of a subunit located, respectively, 9 or 10 subunits above. Cys11 and Cys10 go into a disulfide bond with residues 24 in the B strand of subunit i‐10 and Cys109 in strand I of subunit i‐9.
-
C, DCoulomb potential maps (calculated in PyMOL) of two adjacent subunits (C) and two helical turns of the S‐Ena showing the distribution of charge on the atomic model surface. Each subunit possesses complementary positive and negatively charged patches of residues at the inter‐subunit surface that are responsible for electrostatic stabilizing interactions between the subunits. Similarly, stacked helical rings in the S‐Ena show a charge complementary interface (D).
Figure 4. Ntc linkers give high flexibility and elasticity to S‐Enas.
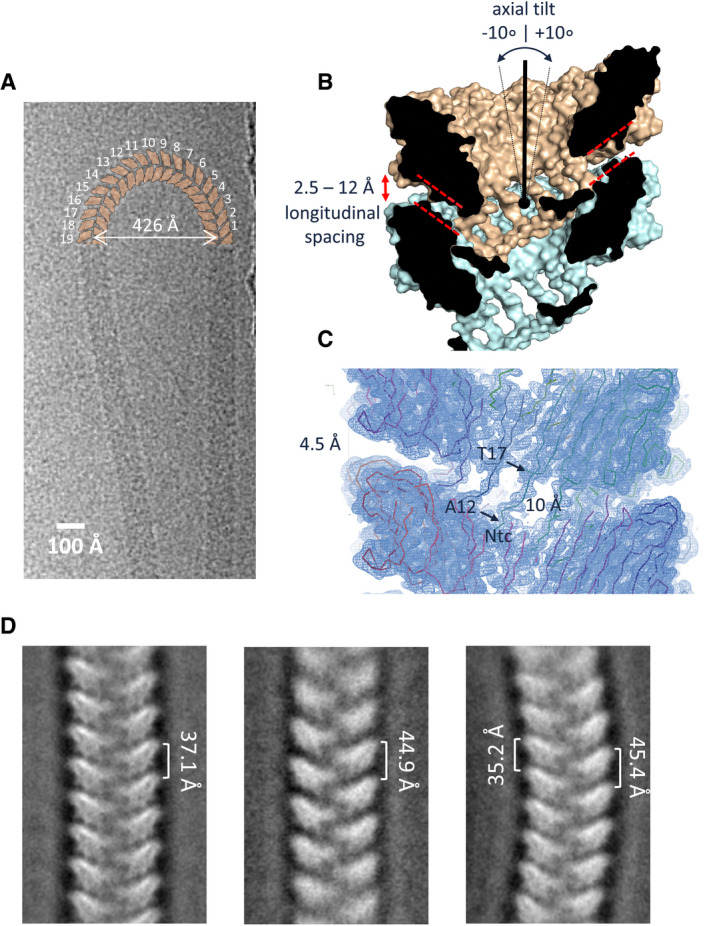
-
ACryoEM image of an isolated ex vivo S‐Ena making a U‐turn comprising 19 helical turns (shown schematically in orange), i.e., corresponding to a ˜10° axial tilt between consecutive helical turns.
-
B, CCross‐section of the molecular surface of the ex vivo S‐Ena model and the ex vivo S‐Ena 3D cryoEM electron potential map. Model and map reveal a 4.5 Å longitudinal spacing between Ena1B jellyroll domains (red dashed lines) as a result of the 10 Å long Ntc linker (residues 12–17).
-
DNegative stain 2D class averages of endospore‐associated S‐Enas show variation in pitch and axial curvature, likely as a result of strain exerted during deposition of spores on the grid surface. The observed range in pitch corresponds to a range in longitudinal spacing of 2.5 to 12 Å between helical turns (labeled red in panel B).
Thus, B. cereus endospore appendages represent a novel class of bacterial pili, comprising a left‐handed single start helix with non‐covalent lateral subunit contacts formed by β‐sheet augmentation, and covalent longitudinal contacts between helical turns by disulfide bonded N‐terminal connecter peptides, resulting in an architecture that combines extreme chemical stability (Fig EV1C) with high fiber flexibility (Fig 4).
The ena1 coding region for S‐Enas
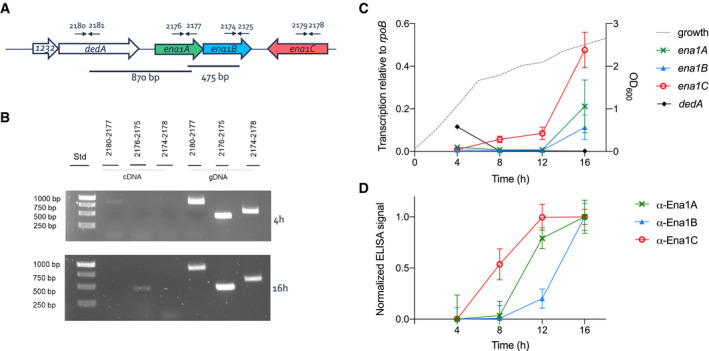
In B. cereus NVH 0075‐95, Ena1A, Ena1B, and Ena1C are encoded in a genomic region flanked upstream by dedA (genbank protein‐id: KMP91696.1) and a gene encoding a 93‐residue protein of unknown function (DUF1232, genbank: KMP91695.1) (Fig 5A). Downstream, the ena‐gene cluster is flanked by a gene encoding an acid phosphatase (TU63_02450). Within the ena‐gene cluster, ena1A and ena1B are found in forward, and ena1C is found in reverse orientation, respectively (Fig 5A). PCR analysis of NVH 0075‐95 cDNA made from mRNA isolated after 4 and 16 h of culture, representative for vegetative growth and sporulating cells, respectively, indicated that ena1A and ena1B are co‐expressed from a bicistronic transcript during sporulation but not during vegetative growth (Fig 5B). The ena1C transcript starts rising from about 4 h post‐inoculation (POI), followed by the ena1A and ena1B transcripts 4–8 h later (Fig 5C). ELISA on cell lysates shows a similar trend for protein levels, with Ena1C emerging at 4 h, and reaching a maximum at 12 h POI (Fig 5D). Again, Ena1A and Ena1B levels lag by approximately 4 h, starting to rise around 8 h POI and reaching a maximum at 16 h (Fig 5D). A weak amplification signal was observed in vegetative cells when the forward primer was located in dedA upstream of ena1A and the reverse primer within ena1A (Fig 5B, lane 2), suggesting that some enaA is co‐expressed with dedA. This was observed in vegetative cells or very early in sporulation but not during later sporulation stages and may represent a fraction of improperly terminated dedA mRNA. Reverse transcription quantitative PCR (RT–qPCR) analysis showed increased expression of ena1A, ena1B, and ena1C in sporulating cells compared with vegetative cells (Fig 5B and C).
Figure 5. ena is bicistronic and expressed during sporulation.
-
AChromosomal organization of the ena genes and primers used for transcript analysis (arrows).
-
BAgarose gel electrophoresis (1%) analysis of RT–PCR products using indicated primer pairs and cDNA made of mRNA isolated from NVH 0075‐95 after 4 and 16 h of growth in liquid cultures or genomic DNA as control.
-
CTranscription level of ena1A (x), ena1B (▲), ena1C (○), and dedA (♦) relative to rpoB determined by RT–qPCR during 16 h of growth of B. cereus strain NVH 0075‐95. The dotted line represents the bacterial growth measured by increase in OD600. Of note, the transcription of ena1C was surprisingly higher than ena1A and ena1B, the major components of the isolated S‐Ena (Fig EV2).
-
DExpression analysis of Ena1 subunits shown as normalized ELISA signal for α‐Ena1A, α‐Ena1B, or α‐Ena1C serum binding to 2,000 ng total protein of B. cereus strain NVH 0075‐95 cells lysed after 4, 8, 12, or 16 h of growth in sporulation medium.
Data information: Error bars represent standard deviation of three independent experiments, with two technical replicates each.
CryoEM maps and immunogold TEM analysis of ex vivo S‐Enas, using antibodies raised against recEna1A‐C, indicated these contain both Ena1A and Ena1B (Figs EV2E and Fig EV3A and B). To determine the relative contribution of Ena1 subunits to B. cereus Enas, we made individual chromosomal knockouts of ena1A, ena1B, and ena1C in strain NVH 0075‐95 and investigated their respective endospores by TEM (Fig 6A). All ena1 mutants made endospores of similar dimensions to WT and with intact exosporium (Fig 6B). Both the ena1A and ena1B mutants resulted in endospores completely lacking S‐Enas, in agreement with the mixed content of ex vivo fibers. The ena1C mutant also resulted in the loss of S‐Ena on the endospores (Fig 6A), even though staining with anti‐Ena1C serum did not identify the presence of the protein inside S‐Enas (Fig EV2E). All three mutants still showed the presence of L‐Enas, of similar size, and number density as WT endospores, although statistical analysis does not rule out L‐Enas to have a slight increase in length in the ena1B and ena1C mutants (length P = 0.003 and < 0.0001, respectively) (Fig 6B). Thus, Ena1A, Ena1B, and Ena1C are all required for in vivo S‐Ena assembly, but not for L‐Ena assembly. Complementation of the ena1A and ena1C mutants with a low copy plasmid (pHT315) containing the respective genes restored the expression of S‐Ena with an average length and a number density similar to the WT strain (Fig 6A and B). Transformation of the ena1B mutant with pHT315 containing ena1B as a single gene (pena1B; Appendix Table S3) repeatedly failed so that we resorted to complementation with a plasmid containing both ena1A and ena1B. Plasmid‐based expression of these subunits resulted in an average ˜2‐fold increase in the number of S‐Enas per spore, and a drastic increase in Ena length, now reaching several microns (Fig 6A and B). This is in contrast with the single gene complementation of ena1A or ena1C, where no such increase in Ena length or density was observed. Thus, the number and length of S‐Enas appear to depend on the relative concentration of both Ena1A and Ena1B subunits. Notably, several endospores overexpressing Ena1A and Ena1B appeared to lack an exosporium or showed the entrapment of S‐Enas inside the exosporium (Fig EV3C). This demonstrates that S‐Enas emanate from the spore body and that a disbalance in the concentration or timing of ena expression can result in mis‐assembly and/or mislocalization of endospore surface structures. Contrary to S‐Enas, close inspection of the WT and mutant endospores suggests that L‐Enas emanate from the surface of the exosporium rather than the spore body (Figs 6A and EV1A). The molecular identity of the L‐Ena, or the single or multiple terminal ruffles seen in L‐ and S‐Enas, respectively, was not determined in the present study.
Figure 6. Composition of S‐ and L‐Ena.
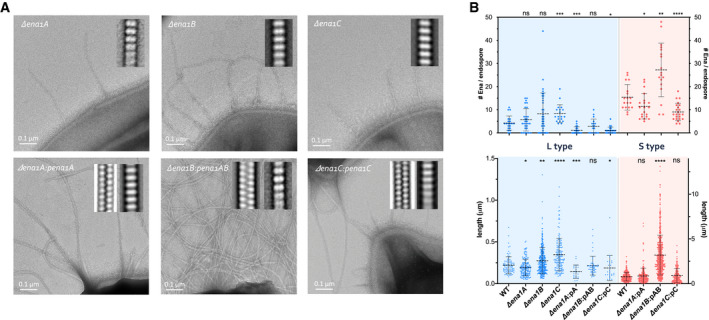
-
ARepresentative negative stain images of endospores of NVH 0075‐95 mutants lacking ena1A, ena1B, or ena1C, as well as endospores of strains complemented with the respective ena subunit expressed from plasmid (i.e., pA, pAB, and pC). The ena1B mutant was complemented with a plasmid carrying both ena1A and 1B (pAB) due to repeated failure to transform with a plasmid holding ena1B only. Inset are 2D class averages of Enas observed on the respective mutants. Knockout of ena1A, ena1B, or ena1C results in the loss of S‐Ena, a phenotype that is restored by plasmid‐based complementation.
-
BNumber (top) and length (bottom) of Enas found on WT, mutant (Δena1A, Δena1B, or Δena1C) and plasmid complemented (Δena1A:pena1A, Δena1B:pena1AB, or Δena1C:pena1C) NVH 0075‐95 endospores. Statistics: pairwise Mann–Whitney U‐tests against WT (n ≥ 18 spores; n ≥ 50 Enas; ns: not significant, *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001. —: mean ± s.d.)
Phylogenetic distribution of the ena1A‐C genes
To investigate the occurrence of ena1A‐C within the B. cereus s.l. group and other species belonging to the genus Bacillus, pairwise tBLASTn searches for homologs of Ena1A‐C were performed on a database containing all available closed, curated Bacillus spp. genomes, with the addition of scaffolds for species for which closed genomes were lacking (n = 735; Table EV1). Genes encoding proteins with high coverage (> 90%) and high amino acid sequence identity (>80%) to Ena1A or Ena1B of B. cereus NVH 0075‐95 were found in 48 strains including 11 of 85 B. cereus strains, 13 of 119 B. wiedmannii strains, 14 of 14 B. cytotoxicus strains, and also in B. luti, B. mobilis, B. mycoides, B. tropics, and B. paranthracis (Fig 7A and B).
Figure 7. Ena is widespread in pathogenic Bacilli.
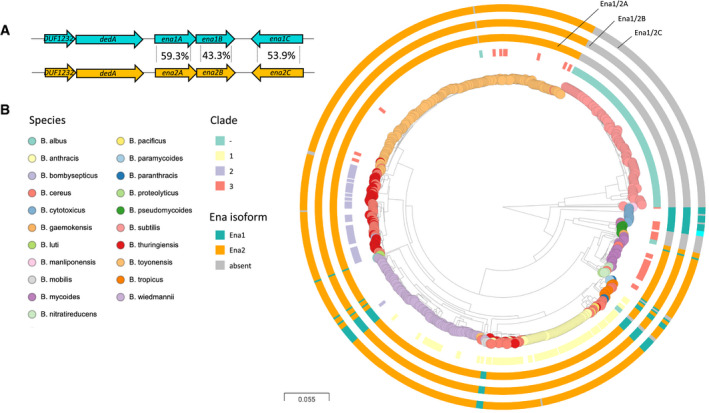
-
AEna1 and Ena2 loci with average amino acid sequence identity indicated between the population of EnaA‐C ortho‐ and homologs. Ena1C shows considerably more variation and is in B. cytotoxicus different from both Ena1C and Ena2C (see Fig EV5), while other genomes have enaC present at different loci (applies to two isolates of B. mycoides).
-
BDistribution of ena1/2A‐C among Bacillus species. Whole genome clustering of the B. cereus s.l. group and B. subtilis created by Mashtree (Katz et al, 2019; Ondov et al, 2016) and visualized in Microreact (Argimon et al, 2016). Rooted on B. subtilis. Traits for species (colored nodes), Bazinet clades and presence of ena are indicated on surrounding four rings in the following order from inner to outer: clade according to Bazinet, 2017 (when available) (Bazinet, 2017), and presence of enaA, enaB, and enaC, resp. (for all three, ena1: teal, ena2: orange, different locus: cyan). When no homo‐ or ortholog was found, the ring is gray. Interactive tree accessible at: https://microreact.org/project/vn2oWw7zM3cwejEFNoRGWA/0024f86c.
Upon searching for Ena1A‐C homologs in B. cereus group genomes, a candidate orthologous gene cluster encoding hypothetical EnaA‐C proteins was discovered. These three proteins had a shared gene synteny and an average of 59.3 ± 0.9%, 43.3 ± 1.6%, and 53.9 ± 2.2% amino acid sequence identity with Ena1A, Ena1B, and Ena1C of B. cereus NVH0075‐95, respectively (Fig 7A). The orthologous ena‐gene cluster was named ena2A‐C. Except for B. subtilis (n = 127) and B. pseudomycoides (n = 8), all genomes analyzed (n = 735) carried either the ena1 (n = 48) or the ena2 (n = 476) gene cluster. Ena1A‐C or the ena2A‐C were never present simultaneously, and no chimeric ena1A‐C/2A‐C clusters were discovered among the genomes analyzed (Fig 7B).
The ena2A‐C form was much more common among B. cereus group strains than the ena1A‐C form; all investigated B. toyonensis (n = 204), B. albus (n = 1), B. bombysepticus (n = 1), B. nitratireducens (n = 6), B. thuringiensis (n = 50) genomes and the majority of B. cereus (87%, 74/85), B. wiedmannii (105/119, 89.3%), B. tropicus (71%, 5/7, ), and B. mycoides (91%, 30/33) genomes carried the ena2A‐C form of the gene cluster (Figs 7B and EV5A–C). No ena genes were found outside the B. cereus group except for three misclassified Streptococcus pneumoniae genomes (GCA_001161325, GCA_001170885, and GCA_001338635) and one misclassified B. subtilis genome (GCA_004328845).
Figure EV5. Phylogenetic relationship between EnaA‐C protein sequences among Bacillus spp.
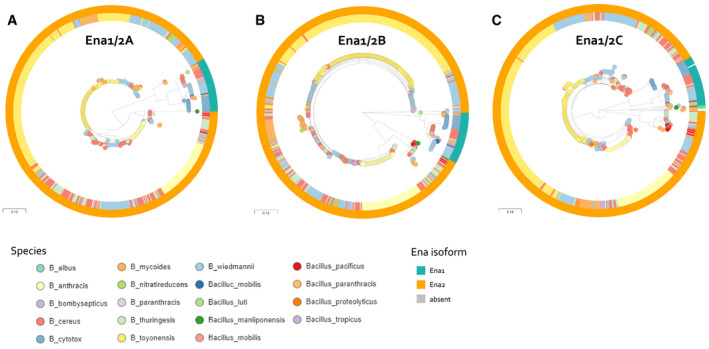
-
ARelationship between Ena1A and Ena2A isoforms of 593 isolates. Ena1A and Ena2A are defined as ortho‐ or homologues having >90% coverage and >80% and 50–65% sequence identity, respectively, with Ena1A_GCF_001044825; KMP91697.1 protein sequence defined in Appendix Table S5. Interactive tree accessible at: https://microreact.org/project/5UixxEY9vr2AVzXDVwa5t/1a8558fd.
-
BRelationship between Ena1B and Ena2B isoforms of 591 isolates. Ena1B, Ena1B_candidate, and Ena2B are defined as ortho‐ or homologues with > 90% coverage and > 80%, 60–80%, and 40–60% sequence identity to Ena1B_NM_Oslo protein sequence defined in Appendix Table S5, respectively. Interactive tree accessible at: https://microreact.org/project/jJ4pARvqf9gyT916sTar5u/1332f3b3.
-
CRelationship between Ena1C and Ena2C isoforms of 591 isolates. Ena1C, Ena1C_candidate, and Ena2C_candidates are defined as ortho‐ or homologues with > 90% coverage and > 80%, 60–80%, and 40–60% sequence identity to Ena1C_NM_Oslo protein sequence defined in Appendix Table S5, respectively. Furthermore, isolates in which an ortho‐ or homologue was found elsewhere in the genome than the usual EnaA‐B locus are colored cyan. Isolates that lacked an Ena1C homo‐ or orthologue are colored gray. Interactive tree accessible at: https://microreact.org/project/aQaqCUCJoj2mw55KQujbGY/099d7885.
Discussion
Endospores formed by Bacillus and Clostridium species frequently carry surface‐attached ribbon‐ or pilus‐like appendages (Driks, 2007), the role of which has remained largely enigmatic due to the lack of molecular annotation of the pathways involved in their assembly. Half a century following their first observation (Hodgikiss, 1971; Hachisuka & Kuno, 1976), we employ high‐resolution de novo structure determination by cryoEM to structurally and genetically characterize the appendages found on B. cereus spores. We found that B. cereus Enas come in two main morphologies: (i) staggered or S‐Enas that are several micrometers long and emerge from the spore body and traverses the exosporium, and (ii) smaller, less abundant ladder‐ or L‐Enas that appears to directly emerge from the exosporium surface. Our phylogenetic analyses of S‐type fibers reveal Ena subunits belonging to a conserved family of proteins encompassing the domain of unknown function DUF3992.
Covalent bonding in combination with the compact jellyroll fold results in high chemical and physical stability of the Ena fibers, withstanding desiccation, high‐temperature treatment, and exposure to proteases. The formation of linear filaments of multiple hundreds of subunits requires stable, long‐lived subunit–subunit interactions with high flexibility to avoid that a dissociation of subunit–subunit complexes results in pilus breakage. This high stability and flexibility are likely to be adaptations to the extreme conditions that can be met by endospores in the environment or during the infectious cycle.
Two molecular pathways are known to form surface fibers or “pili” in Gram‐positive bacteria: (i) sortase‐mediated pilus assembly, which encompasses the covalent linkage of pilus subunits by means of a transpeptidation reaction catalyzed by sortases (Ton‐That & Schneewind, 2004), and (ii) type IV pilus assembly, encompassing the non‐covalent assembly of subunits through a coiled‐coil interaction of a hydrophobic N‐terminal helix (Melville & Craig, 2013). Sortase‐mediated pili and type IV pili are formed on vegetative cells; however, to date, no evidence is available to suggest that these pathways are also responsible for the assembly of endospore appendages.
Until the present study, the only species for which the genetic identity and protein composition of spore appendages have been known is the non‐toxigenic environmental species Clostridium taeniosporum, which carry large (4.5 µm long, 0.5 µm wide, and 30 nm thick) ribbon‐like appendages, which are structurally distinct from those found in most other Clostridium and Bacillus species. C. taeniosporum lacks the exosporium, and the appendages seem to be attached to another layer, of unknown composition, outside the coat (Walker et al, 2007). The C. taeniosporum endospore appendages consist of four major components, three of which have no known homologs in other species and an ortholog of the B. subtilis spore membrane protein SpoVM (Walker et al, 2007). The appendages on the surface of C. taeniosporum endospores, therefore, represent distinct type of fibers than those found on the surface of spores of species belonging to the B. cereus group.
Our structural studies uncover a novel class of pili, where subunits are organized into helically wound fibers, held together by lateral β‐sheet augmentation inside the helical turns, and longitudinal disulfide cross‐linking across helical turns. Intramolecular disulfides are important for the structural integrity and assembly of most chaperone/usher, type IV, and several sortase‐mediated pili (Crespo et al, 2012; Bergeron & Sgourakis, 2015; Reardon‐Robinson et al, 2015). Intermolecular disulfides, however, have not previously been observed. Covalent cross‐linking of pilus subunits does form the assembly mechanism underlying sortase‐mediated pili in Gram‐positives, where the subunits’ C‐terminal LPXTG motif enables isopeptide bond formation with a conserved lysine in the succeeding subunit (Ton‐That & Schneewind, 2004). In Enas, subunit cross‐linking occurs through disulfide bonding of a conserved Cys‐Cys motif in the N‐terminal connector of a subunit i, to two single Cys residues in the core domain of the Ena subunits located at positions i‐9 and i‐10 in the helical structure. As such, the N‐terminal connectors form a covalent bridge across helical turns, as well as a branching interaction with two adjacent subunits in the preceding helical turn (i.e., i‐9 and i‐10). The use of N‐terminal connectors or extensions is also seen in chaperone‐usher pili and Bacteroides type V pili, but these systems employ a non‐covalent fold complementation mechanism to attain long‐lived subunit–subunit contacts, and lack a covalent stabilization (Sauer et al, 1999; Xu et al, 2016). Because in Ena, the N‐terminal connectors are attached to the Ena core domain via a flexible linker, the helical turns in Ena fibers have a large pivoting freedom and ability to undergo longitudinal stretching. These interactions result in highly chemically stable fibers, yet with a large degree of flexibility. Whether the stretchiness and flexibility of Enas carry a functional importance remains unclear. Of note, in several chaperone‐usher pili, a reversible spring‐like stretching provided by helical unwinding and rewinding of the pili has been found important to withstand shear and pulling stresses exerted on adherent bacteria (Fallman et al, 2005; Miller et al, 2006). Possibly, the longitudinal stretching seen in Ena may serve a similar role.
Typical Ena filaments have, to the best of our knowledge, never been observed on the surface of vegetative B. cereus cells, indicating that they are endospore‐specific structures. In support of that assumption, RT–qPCR and ELISA analyses of NVH 0075‐95 demonstrated increased ena1A‐C transcript during sporulation, compared to vegetative cells. A transcriptional analysis of B. thuringiensis serovar chinensis CT‐43 (CT43_CH0783‐785) at 7, 9, 13 h (30% of cells undergoing sporulation), and 22 h of growth has previously been performed (Wang et al, 2013). It is difficult to directly compare expression levels of ena1A‐C in B. cereus NVH 0075‐95 with their expression levels of these genes in B. thuringiensis serovar chinensis CT‐43 since the expression in the latter strain was normalized by converting the number of reads per gene into RPKM (Reads Per Kilo bases per Million reads) and analyzed by DEGseq software package, while the present study determines the expression level of the ena genes relative to the housekeeping gene rpoB. However, both studies indicate that enaA and enaB are only transcribed during sporulation. By searching a separate set of published transcriptomic profiling data, we found that ena2A‐C also are expressed in B. anthracis during sporulation (Bergman et al, 2006), although Enas have not previously been reported from B. anthracis spores. The complementation experiment suggests that overexpression of Ena1A and Ena1B results in entrapment of Ena fibers underneath the exosporium layer. Whether the entrapment relates to timing or abundance of expression of Ena protein subunits is an important line for further studies using techniques such as fluorescence time‐lapse microscopy, which can illustrate dynamic expression profiles and link them to events in the sporulation process.
Without knowledge on the function of Enas, we can only speculate about their biological role. The Enas of B. cereus group species resemble pili, which in Gram‐negative and Gram‐positive vegetative bacteria can play roles in adherence to living surfaces (including other bacteria) and non‐living surfaces, twitching motility, biofilm formation, DNA uptake (natural competence) and exchange (conjugation), secretion of exoproteins, electron transfer (Geobacter), and bacteriophage susceptibility (Proft & Baker, 2009; Lukaszczyk et al, 2019). Some bacteria express multiple types of pili that perform different functions. The most common function of pili‐fibers is adherence to a diverse range of surfaces from metal, glass, plastics rocks to tissues of plants, animals, or humans. In pathogenic bacteria, pili often play a pivotal role in colonization of host tissues and function as important virulence determinants. Similarly, it has been shown that appendages, expressed on the surface of C. sporogenes endospores, facilitate their attachment to cultured fibroblast cells (Panessa‐Warren et al, 2007). The Enas are, however, unlikely to be involved in active motility or uptake/transport of DNA or proteins as they are energy‐demanding processes that are not likely to occur in the endospore´s metabolically dormant state. Enas appear to be a widespread feature among spores of strains belonging to the B. cereus group (Fig 7), a group of closely related Bacillus species with a strong pathogenic potential (Ehling‐Schulz et al, 2019). For most B. cereus group species, the ingestion, inhalation, or contamination of wounds with endospores forms a primary route of infection and disease onset. Enas cover much of the spore surface so that they can be reasonably expected to form an important contact region with the endospore environment and may play a role in the dissemination and virulence of B. cereus species. Our phylogenetic analysis shows a widespread occurrence of Enas in pathogenic Bacilli, and a striking absence in non‐pathogenic species such as B. subtilis, a soil‐dwelling species, and gastrointestinal commensal that has functioned as the primary model system for studying endospores. Ankolekar et al, showed that all of 47 food isolates of B. cereus produced endospores with appendages (Ankolekar & Labbe, 2010). Appendages were also found on spores of ten out of twelve food‐borne, enterotoxigenic isolates of B. thuringiensis, which is closely related to B. cereus, and best known for its insecticidal activity (Ankolekar & Labbe, 2010).
The cryoEM images of ex vivo fibers showed 2–3 nm wide fibers (ruffles) at the terminus of S‐ and L‐Enas. No ruffles were observed on the in vitro assembled Ena1B fibers, suggesting that their formation requires additional components than the Ena1B subunits. Thus far, tryptic digests of ex vivo S‐Ena and mass spectrometry analysis have not allowed us to identify the molecular nature of the ruffles, nor have our attempts to add sub‐stoichiometric amounts of recEna1A or recEna1C to recEna1B fibers resulted in the formation of ruffles. The identity of the S‐Ena ruffles therefore remains unknown. The ruffles resemble tip fibrilla of P‐pili and type 1 pili seen in many Gram‐negative bacteria of the family Enterobacteriaceae (Proft & Baker, 2009). In Gram‐negative pilus filaments, these tip fibrilla are composed of specific low abundance (“minor”) subunits and connect a single copy of a terminal adhesive subunit (i.e., PagG or FimH, in P and type 1 pili, respectively) with a rigid pilus rod made of a single high abundance (“major” subunit). The fibrillae provide the adhesive tip protein additional conformational flexibility that enhances the interaction with receptors on mucosal surfaces (Mulvey et al, 1998). Whether S‐ and L‐Ena ruffles are important for Ena assembly and/or function is presently unknown.
We present the molecular identification of a novel class of spore‐associated appendages or pili widespread in pathogenic Bacilli. Future molecular and infection studies will need to determine whether and how Enas play a role in the virulence of spore‐borne pathogenic Bacilli. The advances in uncovering the genetic identity and the structural aspects of the Enas presented in this work now enable in vitro and in vivo molecular studies to tease out their biological role(s), and to gain insights into the basis for Ena heterogeneity among different Bacillus species.
Materials and Methods
Culture of B. cereus and appendage extraction
For extraction of Enas, the B. cereus strain NVH 0075‐95 was plated on blood agar plates and incubated at 37°C for 3 months. Upon maturation, the spores were resuspended and washed in Milli‐Q water three times (centrifugation 2,400 g at 4°C). To get rid of various organic and inorganic debris, the pellet was then resuspended in 3 ml of 20% Nycodenz (Axis‐Shield), a non‐ionic and metabolically inert universal density gradient medium. The spore suspension was layered on top of a gradient composed of 4 ml each of 45% and 47% (w/v) Nycodenz and subjected to centrifugation for 45 min at 4,500 g at 4°C. The pellet consisting only of the spores was then subsequently washed with 10 ml of Milli‐Q water, 18 ml of 1 M NaCl, TE buffer (50 mM Tris–HCl; 0.5 mM EDTA, pH 7.5) containing 0.1% SDS and TE buffer consecutively, and finally resuspended in 1–2 ml of Milli‐Q water. The spores were centrifuged at 2,500 g for 10 min at 4°C between the washing steps. To detach the appendages, the washed spores were sonicated at 20 kHz ± 50 Hz and 50 watts (Vibra Cell VC50T; Sonic & Materials Inc.; U.S.) for 30 s on ice followed by centrifugation at 4,500 g and appendages were collected from the supernatant. To further get rid of the residual components of spore and vegetative mother cells, n‐hexane was added and vigorously mixed with the supernatant in 1:2 v/v ratio. The mixture was then left to settle to allow phase separation of water and hexane. The hexane fraction containing the appendages was then collected and incubated at 55°C under pressured air for 1.5 h to evaporate the hexane. The appendages were finally resuspended in Milli‐Q water for further cryoEM sample preparation.
Construction of deletion mutants
The B. cereus strain NVH 0075‐95 was used as background for gene deletion mutants. The ena1A, ena1B, and ena1C genes were individually or together deleted in‐frame by replacing the reading frames with ATGTAA (5′–3′) using a markerless gene replacement method (Janes & Stibitz, 2006) with minor modifications. To create the deletion mutants the regions, upstream (primer A and B, Appendix Table S2) and downstream (primer C and D, Appendix Table S2) of the target ena genes were amplified by PCR. To allow assembly of the PCR fragments, primers B and C contained complementary overlapping sequences. An additional PCR step was then performed using the upstream and downstream PCR fragments as template and the A and D primer pair (Appendix Table S2). All PCRs were conducted using an Eppendorf Mastercycler gradient and high‐fidelity AccuPrime Taq DNA Polymerase (ThermoFisher Scientific) according to the manufacturer’s instructions. The final amplicons were cloned into the thermosensitive shuttle vector pMAD (Arnaud et al, 2004) containing an additional I‐SceI site as previously described (Lindback et al, 2012). The pMAD‐I‐SceI plasmid constructs were passed through One Shot™ INV110 E. coli (ThermoFisher Scientific) to achieve unmethylated DNA to enhance the transformation efficiency in B. cereus. The unmethylated plasmid was introduced into B. cereus NVH 0075‐95 by electroporation (Mahillon et al, 1989). After verification of transformants by PCR, the plasmid pBKJ233 (unmethylated), containing the gene for the I‐SceI enzyme, was introduced into the transformant strains by electroporation. The I‐SceI enzyme makes a double‐stranded DNA break in the chromosomally integrated plasmid. Subsequently, homologous recombination events lead to excision of the integrated plasmid resulting in the desired genetic replacement. The gene deletions were verified by PCR amplification using primers A and D (Appendix Table S2) and DNA sequencing (Eurofins Genomics).
Cloning, expression, and purification of Ena1 subunits
Synthetic open reading frames encoding Ena1A, Ena1B, and Ena1C were codon optimized for recombinant expression in E. coli, synthesized, and cloned into pET28a expression vector at Twist biosciences, resulting pMS‐BcE1B (Appendix Table S3). The inserts were designed to have an N‐terminal 6X histidine tag followed by a TEV protease cleavage site (ENLYFQG). Large‐scale recombinant expression of all Ena constructs was carried out in the T7 Express lysY/Iq E. coli strain from NEB. A single colony was inoculated into 200 ml of LB and grown at 37°C with shaking at 150 rpm overnight for primary culture. Next morning, 6 L of LB was inoculated with 20 ml/l of primary culture and grown at 37°C with shaking until the OD600 reached 0.8 after which protein expression was induced with 1 mM isopropyl β‐D‐1‐thiogalactopyranoside (IPTG). The culture was incubated for a further 3 h at 37°C and harvested by centrifugation at 5,000 rcf. The whole‐cell pellet was resuspended in lysis buffer (20 mM potassium phosphate, 500 mM NaCl, 10 mM β‐mercaptoethanol, 20 mM imidazole, 8 M Urea, pH 7.5). The lysate was centrifuged to pellet sacculi, and membrane envelopes at 40,000 rcf for 45 min. The cleared lysate was passed over a 5 ml HisTrap HP column (GE Healthcare) and eluted with 20 mM potassium phosphate, pH 7.5, 8 M urea, 250 mM imidazole in gradient mode (20–250 mM imidazole). Fractions were further analyzed using SDS–PAGE to check for purity.
Invitro assembly of recEna1B into S‐Ena
For in vitro recEna1B assembly, purified recEna1B was incubated with TEV in a 3:1 molar ratio, with the 8 M urea diluted down to 3 M, supplemented with 100 mM β‐mercaptoethanol and subsequently dialyzed against 20 mM Tris, pH 7.5, 50 mM NaCl overnight at 4°C. Removal of the 6xHis‐tag led to the assembly of the recEna1B into long Ena‐like filaments (Fig 3F).
Ena treatment experiments to test its robustness
Ex vivo Enas extracted from B. cereus strain NVH 0075‐95 (see above) were resuspended in deionized water, autoclaved at 121°C for 20 min, or subjected to treatment with buffer as indicated below and shown in Fig EV1. To determine Ena integrity upon the various treatments, samples were imaged using negative stain TEM and Enas were boxed and subjected to 2D classification as described below. To test protease resistance, ex vivo Ena was subjected to 1 mg/ml Ready‐to‐use Proteinase K digestion (Thermo Scientific) for 4 h at 37°C and imaged by TEM. To study the effects of desiccation on the appendages, ex vivo Ena were vacuum dried at 43°C using Savant DNA120 SpeedVac Concentrator (Thermo scientific) run for 2 h at a speed of 2k RPM.
Negative Stain Transmission Electron Microscopy (TEM)
For visualization of spores and recombinantly expressed appendages by negative stain TEM, formvar/carbon‐coated copper grids with 400‐hole mesh (Electron Microscopy Sciences) were glow discharged (ELMO; Agar Scientific) with a plasma current of 4 mA at vacuum for 45 s. 3 µl of sample was applied on the grids and allowed to bind to the support film for 1 min after which the excess liquid was blotted away with Whatman grade 1 filter paper. The grids were then washed three times in 15 µl drops of Milli‐Q followed by blotting of excess liquid. The washed grids were held in 15 µl drops of 2% uranyl acetate three times for, respectively, 10‐second, 2‐second, and 1‐minute durations, with a blotting step in between each dip. Finally, the uranyl acetate‐coated grids were blotted until dry. The grids were then imaged using a 120 kV JEOL 1400 microscope equipped with LaB6 filament and TVIPS F416 CCD camera. 2D classes of the appendages were generated in RELION 3.0 (Zivanov et al, 2018) as described below.
Preparation of cryo‐TEM grids and cryoEM data collection
QUANTIFOIL® holey Cu 400 mesh grids with 2‐µm holes and 1‐µm spacing were first glow discharged in vacuum using plasma current of 5 mA for 1 min (ELMO; Agar Scientific). 3 µl of 0.6 mg/ml graphene oxide (GO) solution was applied onto the grid and incubated 1 min for absorption at room temperature. Extra GO was blotted using a Whatman grade 1 filter paper and left to dry out. For cryo‐plunging, 3 µl of protein sample was applied on the GO‐coated grids at 100% humidity and room temperature in a Gatan CP3 cryo‐plunger. After 1 min of absorption, it was machine‐blotted with Whatman grade 2 filter paper for 5 s from both sides and plunge frozen into liquid ethane at 180°K. Grids were then stored in liquid nitrogen until data collection. Two datasets were collected for ex vivo and recEna1B appendages with slight changes in the collection parameters. High‐resolution cryoEM 2D micrograph movies were recorded on a JEOL Cryoarm300 microscope equipped with energy filter and a K2 or K3 direct electron detector run in counting mode. For the ex vivo Ena, the microscope was equipped with a K2 summit detector and had the following settings: 300 keV, 100 mm aperture, 30 frames/image, 62.5 e−/Å2, 2.315‐second exposure, and 0.82 Å/pxl. The recEna1B dataset was recorded using a K3 detector, at a pixel size of 0.782 Å/pxl, and an exposure of 64.66 e−/Å2 taken over 61 frames/image.
Image processing
MOTIONCORR2 (Zheng et al, 2017) implemented in RELION 3.0 (Zivanov et al, 2018) was used to correct for beam‐induced image motion, and averaged 2D micrographs were generated. The motion‐corrected micrographs were used to estimate the CTF parameters using CTFFIND4.2 (Rohou & Grigorieff, 2015) integrated in RELION 3.0. Subsequent processing used RELION 3.0. and SPRING (Desfosses et al, 2014). For both the datasets, the coordinates of the appendages were boxed manually using e2helixboxer from the EMAN2 package (Tang et al, 2007). Special care was taken to select micrographs with good ice and straight stretches of Ena filaments. The filaments were segmented into overlapping single‐particle boxes of dimension 300 × 300 pxl with an inter‐box distance of 21 Å. For the ex vivo Enas, a total of 53,501 helical fragments was extracted from 580 micrographs with an average of 2–3 long filaments per micrograph. For the recEna1B filaments, 100,495 helical fragments were extracted from 3,000 micrographs with an average of 4–5 filaments per micrograph. To filter out bad particles, multiple rounds of 2D classification were run in RELION 3.0. After several rounds of filtering, a dataset of 42,822 and 65,466 good particles of the ex vivo and recEna1B appendages was selected, respectively.
After running ˜50 iterations of 2D classification, well‐resolved 2D class averages were obtained. segclassexam of the SPRING package (Desfosses et al, 2014) was used to generate B‐factor enhanced power spectrum of the 2D class averages. The generated power spectrum had an amplified signal‐to‐noise ratio with well‐resolved layer lines (Fig 2B). To estimate crude helical parameters, coordinates and phases of the peaks in the layer lines were measured using the segclasslayer option in SPRING. Based on the measured distances and phases, possible sets of Bessel orders were deduced, after which the calculated helical parameters were used in a helical reconstruction procedure in RELION (He & Scheres, 2017). A featureless cylinder of 110 Å diameter generated using relion_helix_toolbox was used as an initial model for 3D classification. Input rise and twist deduced from Fourier–Bessel indexing were varied in the range of 3.05–3.65 Å and 29–35 degrees, respectively, with a sampling resolution of 0.1 Å and 1 degree between tested start values. Doing so, several rounds of 3D classification were run until electron potential maps with good connectivity and recognizable secondary structure features were obtained. Recentered particles were re‐extracted and subjected to 3D refinement using a 25 Å low‐pass filtered map generated from the 3D classification run as a starting map. To further improve the resolution, Bayesian polishing was performed in RELION. Finally, a solvent mask covering the central 50% of the helix z‐axis was generated in maskcreate and used for postprocessing and calculating the solvent‐flattened Fourier shell correlation (FSC) curve in RELION. After two rounds of polishing, maps of 3.2 Å resolution according to the FSC0.143 gold‐standard criterion as well as local resolution calculated in RELION were obtained (Fig EV2A).
Model building
Prior to model building, unfiltered maps for recEna1B calculated by Relion were masked down to three helical turns and used for cryoEM density modification as implemented in ResolveCryoEM (Terwilliger et al, 2020) from the PHENIX package (Afonine et al, 2018), resulting in a map of 3.05 Å final resolution (FSC0.143 criterium) for recEna1B. At first, the primary skeleton for a single asymmetric subunit from the density modified map was generated in Coot (Emsley et al, 2010). Primary sequence of Ena1B was manually threaded onto the asymmetric unit and fitted into the map. This manually built Ena1B model was then used to populate the remaining subunits within the helical map by means of rigid body fitting. The complete fiber model was then subjected to multiple rounds of real space structural refinement in PHENIX, and each residue was manually inspected after every round of refinement. Model validation was done in MolProbity (Davis et al, 2007) implemented in Phenix. All the visualizations and images for figures were generated in ChimeraX (Goddard et al, 2018), Chimera (Pettersen et al, 2004), or Pymol. CryoEM data and model statistics are summarized in Appendix Table S1.
Cell culturing and lysate preparation
One mL overnight culture of B. cereus NVH 0075‐95 or B. cereus NVH0075‐95 ΔenaABC in BHI broth was inoculated into 100 ml of sporulation medium [8 g/l Bacto nutrient broth (Difco), 1 µM FeSO4, 2.5 µM CuCl2, 12.5 µM ZnCl2, 66 µM MnSO4, 1 mM MgCl2, 5 mM (NH4)2SO4, 2.5 µM Na2MoO4, 2.5 µM CoCl2, 1 mM Ca(NO3)2, pH 7.6], and incubated for 16 h (37°C, 150 rpm). At the indicated time points, 20 ml of the cultures was withdrawn, centrifuged at 4,500 g (10 min, RT), and the cell pellets stored at −20°C until analyzed. Frozen cell pellets were thawed on ice and resuspended in 100 µl of lysis buffer [10 mM phosphate buffer (pH 7.2), 130 mM NaCl, 1 µM Triton X‐100 (Sigma), Complete EDTA‐free protease inhibitor cocktail (1 tablet per 10 ml, Sigma)]. The cell pellets from three independent cultures at each time point were pooled, and from this, 60 mg was weighed, and lysis buffer containing 1 mg/ml of lysozyme (L6876, Sigma) was added to a final weight of 1 g. The cell pellet‐lysozyme mixture was incubated for about 1 h at RT with shaking (225 rpm) and sonicated for a total of 1.5 min with 30‐second interval (output 40 kHz and 50 watts; Vibra Cell VC50T, Sonic & Materials Inc.). The lysates were finally clarified by centrifugation at 10,000 g (15 min, RT), and supernatants transferred to new Eppendorf tubes. The total protein concentrations (A280) were estimated using NanoDrop 1000 (Thermo Scientific).
ELISA and immuno‐labeling of the Enas
For antibody generation, recEna1A and recEna1C were cloned, expressed, and purified according to the method described above for recEna1B. Aliquots of purified recEna1A, recEna1B, and recEna1C were concentrated to 1 mg/ml in PBS for rabbit immunization (Davids Biotechnologie GmbH). Because of the high degree of conservation between Ena1A and Ena1B, the anti‐Ena1A and anti‐Ena1B sera were depleted of potentially cross‐reactive antibodies by incubation with 0.2 mg/ml recEna1B or recEna1A fibers added in 1:4 ratio in the respective sera, incubated overnight, and passed over a 0.22‐µm spin filter to remove the competing recombinant Ena. Dot blot and competition ELISA were performed to evaluate residual cross‐reactivity of the respective sera to the three different subunits (Fig EV3A and B). For this, 100 ng of recEna1A, recEna1B, and recEna1C was adsorbed onto a nitrocellulose membrane for 15 min, washed three times with TBST, and blocked by incubation in 5% milk at 37°C for 1 h. After a further 3 TBST washes, the membrane was incubated with the respective sera in a 1:1,000 dilution in TBST at 37°C for 1 h. After an additional three washes, the membrane was incubated with 1:2,000 anti‐rabbit alkaline phosphatase‐conjugated IgG as secondary antibody at 37°C for 1 h. The membrane was then developed using BCIP/NBT (Millipore) and imaged. For the competition ELISA, Corning® 96‐well flat bottom plates (CLS3370, Sigma) were coated with 0.78 pmol per well of purified recEna1A or recEna1B proteins in 100 µl PBS buffer (10 mM sodium phosphate, 130 mM NaCl, pH 7.2) at 4°C overnight. The wells were emptied and subsequently blocked with 1% ovalbumin (A5503, Sigma) in PBST buffer (PBS+0.05% (v/v) Tween‐20). Following washing the wells three times with PBST, 60 µl of threefold serial dilutions of recEna1A or recEna1B proteins (range, 180,000 fmol to 9.1 fmol) and 60 µl rabbit anti‐Ena1A sera (1:8,000) were added to the Ena1A‐coated wells. Similarly, threefold serial dilutions of recEna1A or recEna1B proteins and rabbit anti‐Ena1B sera (1:8,000) were added to the Ena1B coated wells. Maximum binding wells to which only sera (60 µl sera + 60 µl PBST) were added to the Ena‐coated wells and blank wells, to which only PBST (120 µl) was added to the Ena‐coated wells were also included in each experiment. The competition reaction proceeded for 1.30 h at room temperature with shaking (200 rpm). The plates were then washed with PBST, and 100 µl (1:1,000) anti‐rabbit IgG alkaline phosphatase antibody (A3687, Sigma) was added to all the wells, and incubated for 1 h at RT. Following three final washing steps, 100 µl of substrate (0.45 mg/ml 4‐nitrophenyl phosphate disodium salt hexahydrate (N9389, Sigma)) dissolved in diethanolamine buffer [9.7% (v/v) diethanolamine buffer, 0.5 mM MgCl2 (pH 9.8)] was added. The plates were incubated for 15 min at room temperature, and the absorbance was measured at 405 nm using plate reader (Infinite M200, Tecan). The average values of the blank wells were subtracted from all the reaction wells, and percentage inhibition of maximum binding was estimated relative to the maximum activity wells (only antibody wells).
For immunostaining EM imaging, 3 µl aliquots of purified ex vivo Enas were deposited on Formvar/Carbon grids (400 Mesh, Cu; Electron Microscopy Sciences), washed with 20 μL 1× PBS, and incubated for 1 h with 0.5% (w/v) BSA in 1× PBS. After additional washing with 1× PBS, separate grids were individually incubated for 2 h at 37°C with 1:1,000 dilutions in PBS of anti‐Ena1A, anti‐Ena1B, or anti‐Ena1C sera, respectively. Following washing with 1× PBS, grids were incubated for 1 h at 37°C with a 1:2,000 dilution of a 10 nm gold‐labeled goat anti‐rabbit IgG (G7277; Sigma Aldrich), washed with 1× PBS, and negative stained and imaged on a 120 kV JEOL 1400 microscope as described above.
Detection of Ena1A, Ena1B, and Ena1C proteins in cell lysates
Indirect ELISAs were carried out to detect Ena proteins using anti‐Ena1A, anti‐Ena1B, and anti‐Ena1C sera. Briefly, Corning® 96‐well flat bottom plates (CLS3370, Sigma) were coated with twofold serial dilutions of B. cereus lysates in PBS buffer [10 mM phosphate buffer (pH 7.2), 130 mM NaCl] overnight at 4°C. The microtiter plates were then blocked with 1.5% ovalbumin (w/v) in PBST buffer and subsequently incubated with 100 µl anti‐Ena rabbit sera in 1% ovalbumin (w/v) in PBST for 90 min at RT with shaking. Anti‐Ena1A, anti‐Ena1B, and anti‐Ena1C sera were used at 1:500, 1:500, and 1:250 dilutions, respectively. The wells were washed three times with PBST after blocking and after addition of the primary and secondary antibody. Detection of anti‐Ena antibodies and color development was done as described under competitive ELISA except that the secondary antibody was resuspended in 1% (w/v) ovalbumin in PBST. To account for potential non‐specific binding to cell lysate components, the absorbance values of the B. cereus ΔenaABC (16 h) lysate were subtracted from the values of the wild‐type B. cereus strain.
Quantitative RT–PCR
RT–qPCR experiments were performed on mRNA isolated from B. cereus cultures harvested from three independent Sporulation media cultures (37°C, 150 rpm) at 4, 8, 12, and 16 h post‐inoculation. RNA extraction, cDNA synthesis, and RT–qPCR analysis were performed essentially as described before (Madslien et al, 2014), with the following changes: pre‐heated (65°C) TRIzol Reagent (Invitrogen) and bead beating 4 times for 2 min in a Mini‐BeadBeater‐8 (BioSpec) with cooling on ice in between.
Each RT–qPCR analysis of the RNA samples was performed in triplicate, no template was added in the negative controls, and rpoB was used as internal control. Slopes of the standard curves and PCR efficiency (E) for each primer pair were estimated by amplifying serial dilutions of the cDNA template. For quantification of mRNA transcript levels, Ct (threshold cycle) values of the target genes and the internal control gene (rpoB), derived from the same sample in each RT–qPCR, were first transformed using the term E‐Ct. The expression levels of target genes were then normalized by dividing their transformed Ct‐values by the corresponding values obtained for the internal control gene (Pfaffl, 2001; Duodu et al, 2010; Madslien et al, 2014). The amplification was conducted by using StepOne PCR software V.2.0 (Applied Biosystems) with the following conditions: 50°C for 2 min, 95°C for 2 min, 40 cycles of 15 s at 95°C, 1 min at 60°C, and 15 s at 95°C. All primers used for RT–qPCR analyses are listed in Appendix Table S2.
Regular PCRs were performed on cDNA to confirm that ena1A and ena1B were expressed as an operon using the primers 2180/2177 and 2176/2175 and DreamTaq DNA polymerase (Thermo Fisher) amplified in an Eppendorf Mastercycler using the following program: 95°C for 2 min, 30 cycles of 95°C for 30 s, 54°C for 30 s, and 72°C for 1 min.
Search for orthologs and homologs of Ena1
Publicly available genomes of species belonging to the Bacillus s.l. group (Appendix Table S4) were downloaded from NCBI RefSeq database (n = 735, NCB (https://www.ncbi.nlm.nih.gov/refseq/; Table EV1). Except for strains of particular interest due to phenotypic characteristics (GCA_000171035.2_ASM17103v2, GCA_002952815.1_ASM295281v1, GCF_000290995.1_Baci_cere_AND1407_G13175) and species of which closed genomes were nonexistent or very scarce (n = 158), all assemblies included were closed and publicly available genomes from the curated database of NCBI RefSeq database. Assemblies were quality checked using QUAST (Gurevich et al, 2013), and only genomes of correct size (˜4.9–6 Mb) and a GC content of ˜35% were included in the downstream analysis. Pairwise tBLASTn searches were performed (BLAST+ v. 2.11.0 (Altschul et al, 1990), e‐value 1e‐10, max_hspr 1, default settings) to search for homo‐ and orthologs of the following query‐protein sequences from strain NVH 0075‐95: Ena1A, Ena1B, Ena1C (Appendix Table S5). The Ena1B protein sequences used as query originated from an in‐house amplicon sequenced product, while the Ena1A and Ena1C protein sequence queries originated from the assembly for strain NVH 0075‐95 (Accession number GCF_001044825.1, protein KMP91698.1, and KMP91699.1; Appendix Table S5). We considered proteins orthologs or homologs when a subject protein matched the query protein with high coverage (> 70%) and moderate sequence identity (> 30%).
Mashtree v. 0.57 was used to infer whole genome clustering for the B. cereus s.l. group and B. subtilis, using the accurate option and 100× bootstrap to estimate confidence of the tree (mashtree_bootstrap.pl,‐‐reps 100,‐‐ ‐‐min‐depth 0) (Katz et al, 2019; Ondov et al, 2016). Briefly, the algorithm creates a MinHash sketch of each isolate genome and computes the pairwise Jaccard distance to facilitate rapid comparison of the isolates. This eliminates the need for genome alignment, which is not an option for such a diverse group of bacteria. The relationship between the different isolates is visualized in a neighbor‐joining tree using Mashtree’s inbuilt Quicktree from the resulting distance matrix. The topology was confirmed with a separate phylogenetic approach: For a subset of the Clade 1 isolates which contained the B. cereus with Ena1 homologs, plasmids were first removed by an in‐house script (https://github.com/AdmiralenOla/filter‐plasmid‐contigs‐bacillus), followed by assembly annotation by Prokka (Seemann, 2014) and pangenome defined by the use of Roary v. 3.12.0 (Page et al, 2015). The output core genome alignment (sfastrict core genome, n = 1.788, > 99% of isolates) was aligned using Muscle v. 3.8.31 (Edgar, 2004), and default settings and approximately maximum‐likelihood phylogenies were created using FastTree v. 2.1.8 (Price et al, 2010).
Comparative genomics of the ena genes and proteins
Phylogenetic trees of the aligned Ena1A‐C proteins were constructed using approximately maximum likelihood by FastTree 2 v. 2.1.11 (Price et al, 2010) (default settings) for all hits resulting from the tBLASTn search. The amino acid sequences were aligned using mafft v.7.310 (Katoh et al, 2019), and approximately maximum‐likelihood phylogenetic trees of protein alignments were made using FastTree, using the JTT+CAT model (Price et al, 2010). All trees were visualized in Microreact (Argimon et al, 2016) and the metadata of species, and presence and absence for Ena1A‐C and Ena2A‐C overlaid the figures.
Author contributions
BP and MS performed TEM imaging, structural studies, and recombinant ena1B production and analysis. JL, TL, and KO produced endospores, performed TEM imaging, isolated Enas, and conducted genetic studies. EDZ designed and conducted competitive and indirect ELISA experiments. A‐KL and OB conducted the phylogenetic analysis. HR and MA designed and supervised experiments, and wrote the paper, with contributions from all authors.
Conflicts of interest
BP, MS, MA, and HR are named as inventor on a priority filing related to the production and use of recombinant Ena fibers.
Supporting information
Review Process File
Appendix
Expanded View Figures PDF
Table EV1
Acknowledgements
We thank Marcus Fislage and Adam Schröfel at the VIB‐VUB Facility for Bio Electron Cryogenic Microscopy (BECM) and for assistance in data collection and Jan Haug Anonsen at NORCE research, Norway / Department of Biosciences, University of Oslo and Norbert Roos at the EM laboratory for biological sciences, and University of Oslo for assistance with preliminary sample analysis. We are grateful to Ute Krengel (UiO) for the mentorship of J.L. and feedback on the manuscript. This work was funded by VIB, EOS Excellence in Research Program by FWO through grant G0G0818N to HR, the NMBU's talent development program to MA and travel grants from the national graduate school in infection biology and antimicrobials (IBA) through NFR grant 249069 to J.L.
The EMBO Journal (2021) 40: e106887.
Contributor Information
Marina Aspholm, Email: marina.aspholm@nmbu.no.
Han Remaut, Email: han.remaut@vub.be.
Data availability
The datasets produced in this study are available in the following databases:
cryoEM map: ex vivo S‐Ena, accession number EMD‐11592, https://www.ebi.ac.uk/pdbe/emdb/EMD‐11592
cryoEM map: in vitro S‐Ena, accession number EMD‐11591, https://www.ebi.ac.uk/pdbe/emdb/EMD‐11591
Structure coordinates RecEna1B: model of recEna1B, accession number 7A02, https://www.rcsb.org/structure/7A02
Primers and genetic constructs used in this study are listed in Appendix Tables S2 and S3, respectively. All genomes downloaded from the RefSeq database (n = 737) and their annotated species in addition to scaffolds /contigs are listed in Table EV1. All other data supporting the findings of this study are available within the article or its supplementary information or from the corresponding author upon reasonable request.
References
- Afonine PV, Poon BK, Read RJ, Sobolev OV, Terwilliger TC, Urzhumtsev A, Adams PD (2018) Real‐space refinement in PHENIX for cryo‐EM and crystallography. Acta Crystallogr D Struct Biol 74: 531–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215: 403–410 [DOI] [PubMed] [Google Scholar]
- Ankolekar C, Labbe RG (2010) Physical characteristics of spores of food‐associated isolates of the Bacillus cereus group. Appl Environ Microbiol 76: 982–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argimón S, Abudahab K, Goater RJE, Fedosejev A, Bhai J, Glasner C, Feil EJ, Holden MTG, Yeats CA, Grundmann Het al (2016) Microreact: visualizing and sharing data for genomic epidemiology and phylogeography. Microb Genom 2: e000093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaud M, Chastanet A, Debarbouille M (2004) New vector for efficient allelic replacement in naturally nontransformable, low‐GC‐content, gram‐positive bacteria. Appl Environ Microbiol 70: 6887–6891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atrih A, Foster SJ (1999) The role of peptidoglycan structure and structural dynamics during endospore dormancy and germination. Antonie Van Leeuwenhoek 75: 299–307 [DOI] [PubMed] [Google Scholar]
- Bazinet AL (2017) Pan‐genome and phylogeny of Bacillus cereus sensu lato. BMC Evol Biol 17: 176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron JR, Sgourakis NG (2015) Type IV pilus: one architectural problem, many structural solutions. Structure 23: 253–255 [DOI] [PubMed] [Google Scholar]
- Bergman NH, Anderson EC, Swenson EE, Niemeyer MM, Miyoshi AD, Hanna PC (2006) Transcriptional profiling of the Bacillus anthracis life cycle in vitro and an implied model for regulation of spore formation. J Bacteriol 188: 6092–6100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnley T, Palmer CM, Winn M (2017) Recent developments in the CCP‐EM software suite. Acta Crystallogr D Struct Biol 73: 469–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo MD, Puorger C, Scharer MA, Eidam O, Grutter MG, Capitani G, Glockshuber R (2012) Quality control of disulfide bond formation in pilus subunits by the chaperone FimC. Nat Chem Biol 8: 707–713 [DOI] [PubMed] [Google Scholar]
- Davis IW, Leaver‐Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, Snoeyink J, Richardson JSet al (2007) MolProbity: all‐atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35: W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desfosses A, Ciuffa R, Gutsche I, Sachse C (2014) SPRING ‐ an image processing package for single‐particle based helical reconstruction from electron cryomicrographs. J Struct Biol 185: 15–26 [DOI] [PubMed] [Google Scholar]
- DesRosier JP, Lara JC (1981) Isolation and properties of pili from spores of Bacillus cereus. J Bacteriol 145: 613–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driks A (2007) Surface appendages of bacterial spores. Mol Microbiol 63: 623–625 [DOI] [PubMed] [Google Scholar]
- Duodu S, Holst‐Jensen A, Skjerdal T, Cappelier JM, Pilet MF, Loncarevic S (2010) Influence of storage temperature on gene expression and virulence potential of Listeria monocytogenes strains grown in a salmon matrix. Food Microbiol 27: 795–801 [DOI] [PubMed] [Google Scholar]
- Edgar RC (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5: 113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehling‐Schulz M, Lereclus D, Koehler TM (2019) The Bacillus cereus group: Bacillus species with pathogenic potential. Microbiol Spectr 7: GPP3‐0032‐2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66: 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallman E, Schedin S, Jass J, Uhlin BE, Axner O (2005) The unfolding of the P pili quaternary structure by stretching is reversible, not plastic. EMBO Rep 6: 52–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farabella I, Vasishtan D, Joseph AP, Pandurangan AP, Sahota H, Topf M (2015) TEMPy: a Python library for assessment of three‐dimensional electron microscopy density fits. J Appl Crystallogr 48: 1314–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhardt P, Ribi E (1964) Ultrastructure of the Exosporium Enveloping Spores of Bacillus Cereus. J Bacteriol 88: 1774–1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, Ferrin TE (2018) UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci 27: 14–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich A, Saveliev V, Vyahhi N, Tesler G (2013) QUAST: quality assessment tool for genome assemblies. Bioinformatics 29: 1072–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hachisuka Y, Kuno T (1976) Filamentous appendages of Bacillus cereus spores. Jpn J Microbiol 20: 555–558 [DOI] [PubMed] [Google Scholar]
- He S, Scheres SHW (2017) Helical reconstruction in RELION. J Struct Biol 198: 163–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgikiss W (1971) Filamentous appendages on the spores and exosporium of certain Bacillus species, In Spore research Barker AN, Gould GW, Wolf J (eds.), pp 211–218., London and New York: Academic Press; [Google Scholar]
- Janes BK, Stibitz S (2006) Routine markerless gene replacement in Bacillus anthracis. Infect Immun 74: 1949–1953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Rozewicki J, Yamada KD (2019) MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform 20: 1160–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz LS, Griswold T, Morrison SS, Caravas JA, Zhang S, d.B.H C, Deng X, Carleton A (2019). Mashtree: a rapid comparison of whole genome sequence files. J Open Source Software 4: 1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindback T, Mols M, Basset C, Granum PE, Kuipers OP, Kovacs AT (2012) CodY, a pleiotropic regulator, influences multicellular behaviour and efficient production of virulence factors in Bacillus cereus . Environ Microbiol 14: 2233–2246 [DOI] [PubMed] [Google Scholar]
- Lukaszczyk M, Pradhan B, Remaut H (2019) The biosynthesis and structures of bacterial pili. Subcell Biochem 92: 369–413 [DOI] [PubMed] [Google Scholar]
- Madslien EH, Granum PE, Blatny JM, Lindback T (2014) L‐alanine‐induced germination in Bacillus licheniformis ‐the impact of native gerA sequences. BMC Microbiol 14: 101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahillon J, Chungjatupornchai W, Decock J, Dierickx S, Michiels F, Peferoen M, Joos H (1989) Transformation of Bacillus thuringiensis by electroporation. FEMS Microbiol Lett 60: 205–210 [Google Scholar]
- Mandlik A, Swierczynski A, Das A, Ton‐That H (2008) Pili in Gram‐positive bacteria: assembly, involvement in colonization and biofilm development. Trends Microbiol 16: 33–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melville S, Craig L (2013) Type IV pili in Gram‐positive bacteria. Microbiol Mol Biol Rev 77: 323–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller E, Garcia T, Hultgren S, Oberhauser AF (2006) The mechanical properties of E. coli type 1 pili measured by atomic force microscopy techniques. Biophys J 91: 3848–3856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvey MA, Lopez‐Boado YS, Wilson CL, Roth R, Parks WC, Heuser J, Hultgren SJ (1998) Induction and evasion of host defenses by type 1‐piliated uropathogenic Escherichia coli. Science 282: 1494–1497 [DOI] [PubMed] [Google Scholar]
- Ondov BD, Treangen TJ, Melsted P, Mallonee AB, Bergman NH, Koren S, Phillippy AM (2016) Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol 17: 132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MT, Fookes M, Falush D, Keane JA, Parkhill J (2015) Roary: rapid large‐scale prokaryote pan genome analysis. Bioinformatics 31: 3691–3693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panessa‐Warren BJ, Tortora GT, Warren JB (2007) High resolution FESEM and TEM reveal bacterial spore attachment. Microsc Microanal 13: 251–266 [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612 [DOI] [PubMed] [Google Scholar]
- Pfaffl MW (2001) A new mathematical model for relative quantification in real‐time RT‐PCR. Nucleic Acids Res 29: e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, Arkin AP (2010) FastTree 2–approximately maximum‐likelihood trees for large alignments. PLoS One 5: e9490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proft T, Baker EN (2009) Pili in Gram‐negative and Gram‐positive bacteria ‐ structure, assembly and their role in disease. Cell Mol Life Sci 66: 613–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon‐Robinson ME, Osipiuk J, Jooya N, Chang C, Joachimiak A, Das A, Ton‐That H (2015) A thiol‐disulfide oxidoreductase of the Gram‐positive pathogen Corynebacterium diphtheriae is essential for viability, pilus assembly, toxin production and virulence. Mol Microbiol 98: 1037–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remaut H, Waksman G (2006) Protein‐protein interaction through beta‐strand addition. Trends Biochem Sci 31: 436–444 [DOI] [PubMed] [Google Scholar]
- Richardson JS (1981) The anatomy and taxonomy of protein structure. Adv Protein Chem 34: 167–339 [DOI] [PubMed] [Google Scholar]
- Rode LJ, Pope L, Filip C, Smith LD (1971) Spore appendages and taxonomy of Clostridium sordellii. J Bacteriol 108: 1384–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohou A, Grigorieff N (2015) CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol 192: 216–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer FG, Futterer K, Pinkner JS, Dodson KW, Hultgren SJ, Waksman G (1999) Structural basis of chaperone function and pilus biogenesis. Science 285: 1058–1061 [DOI] [PubMed] [Google Scholar]
- Seemann T (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics 30: 2068–2069 [DOI] [PubMed] [Google Scholar]
- Setlow P (2014) Germination of spores of Bacillus species: what we know and do not know. J Bacteriol 196: 1297–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova TA, Zubasheva MV, Shevliagina NV, Nikolaenko MA, Azizbekian RR (2013) Electron microscopy of the surfaces of bacillary spores. Mikrobiologiia 82: 698–706 [PubMed] [Google Scholar]
- Stewart GC (2015) The exosporium layer of bacterial spores: a connection to the environment and the infected host. Microbiol Mol Biol Rev 79: 437–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ (2007) EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol 157: 38–46 [DOI] [PubMed] [Google Scholar]
- Terwilliger TC, Ludtke SJ, Read RJ, Adams PD, Afonine PV (2020). Improvement of cryo‐EM maps by density modification. Nat Methods 17: 923–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ton‐That H, Schneewind O (2004) Assembly of pili in Gram‐positive bacteria. Trends Microbiol 12: 228–234 [DOI] [PubMed] [Google Scholar]
- Walker JR, Gnanam AJ, Blinkova AL, Hermandson MJ, Karymov MA, Lyubchenko YL, Graves PR, Haystead TA, Linse KD (2007) Clostridium taeniosporum spore ribbon‐like appendage structure, composition and genes. Mol Microbiol 63: 629–643 [DOI] [PubMed] [Google Scholar]
- Wang J, Mei H, Zheng C, Qian H, Cui C, Fu Y, Su J, Liu Z, Yu Z, He J (2013) The metabolic regulation of sporulation and parasporal crystal formation in Bacillus thuringiensis revealed by transcriptomics and proteomics. Mol Cell Proteomics 12: 1363–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Shoji M, Shibata S, Naito M, Sato K, Elsliger M‐A, Grant J, Axelrod H, Chiu H‐J, Farr Cet al (2016) A distinct type of pilus from the human microbiome. Cell 165: 690–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, Agard DA (2017) MotionCor2: anisotropic correction of beam‐induced motion for improved cryo‐electron microscopy. Nat Methods 14: 331–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, Scheres SH (2018) New tools for automated high‐resolution cryo‐EM structure determination in RELION‐3. Elife 7: e42166 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Review Process File
Appendix
Expanded View Figures PDF
Table EV1
Data Availability Statement
The datasets produced in this study are available in the following databases:
cryoEM map: ex vivo S‐Ena, accession number EMD‐11592, https://www.ebi.ac.uk/pdbe/emdb/EMD‐11592
cryoEM map: in vitro S‐Ena, accession number EMD‐11591, https://www.ebi.ac.uk/pdbe/emdb/EMD‐11591
Structure coordinates RecEna1B: model of recEna1B, accession number 7A02, https://www.rcsb.org/structure/7A02
Primers and genetic constructs used in this study are listed in Appendix Tables S2 and S3, respectively. All genomes downloaded from the RefSeq database (n = 737) and their annotated species in addition to scaffolds /contigs are listed in Table EV1. All other data supporting the findings of this study are available within the article or its supplementary information or from the corresponding author upon reasonable request.