

Abstract
Inflammation plays an important role in the pathogenesis of Alzheimer's disease (AD). Some evidence suggests that misfolded protein aggregates found in AD brains may have originated from the gut, but the mechanism underlying this phenomenon is not fully understood. C/EBPβ/δ‐secretase signaling in the colon was investigated in a 3xTg AD mouse model in an age‐dependent manner. We applied chronic administration of 1% dextran sodium sulfate (DSS) to trigger gut leakage or colonic injection of Aβ or Tau fibrils or AD patient brain lysates in 3xTg mice and combined it with excision/cutting of the gut–brain connecting vagus nerve (vagotomy), in order to explore the role of the gut–brain axis in the development of AD‐like pathologies and to monitor C/EBPβ/δ‐secretase signaling under those conditions. We found that C/EBPβ/δ‐secretase signaling is temporally activated in the gut of AD patients and 3xTg mice, initiating formation of Aβ and Tau fibrils that spread to the brain. DSS treatment promotes gut leakage and facilitates AD‐like pathologies in both the gut and the brain of 3xTg mice in a C/EBPβ/δ‐secretase‐dependent manner. Vagotomy selectively blunts this signaling, attenuates Aβ and Tau pathologies, and restores learning and memory. Aβ or Tau fibrils or AD patient brain lysates injected into the colon propagate from the gut into the brain via the vagus nerve, triggering AD pathology and cognitive dysfunction. The results indicate that inflammation activates C/EBPβ/δ‐secretase and initiates AD‐associated pathologies in the gut, which are subsequently transmitted to the brain via the vagus nerve.
Keywords: Alzheimer’s disease, C/EBPβ, fibrils, inflammation, δ‐secretase
Subject Categories: Immunology, Neuroscience
Activation of the C/EBPβ/δ‐secretase pathway in the gut of patients and mouse models of Alzheimer's disease (AD) gives rises to misfolded protein aggregates that can via the vagus nerve spread into the brain to trigger AD‐associated pathology.
Introduction
Alzheimer's disease is a neurodegenerative disorder associated with the progressive decline in cognitive functions. AD is characterized by several pathological hallmarks, including extracellular β‐amyloid (Aβ) plaques, intraneuronal neurofibrillary tangles (NFTs), chronic neuroinflammation, and neuronal loss. Aβ pathology arises from the improper sequential cleavage of the transmembrane amyloid precursor protein (APP) by BACE1 and γ‐secretase, resulting in Aβ peptides that aggregate into oligomers and eventually into Aβ fibrils and plaques. NFTs are mainly composed of hyperphosphorylated and truncated tau, a protein that stabilizes microtubules (Holtzman et al, 2011). Although approximately 1% of AD cases arise from rare causative mutations in APP or γ‐secretase, most AD is sporadic, with ApoE4 as the most well‐characterized genetic risk factor (Mahley, 2016). While age is the most critical factor for AD, epidemiological studies show that some diseases and lifestyle factors also increase the risk of developing AD, including traumatic brain injury, diabetes, hypertension, obesity, and other metabolic syndromes, which are all associated with inflammation. Enhanced inflammation can be detected in the cerebrospinal fluid (CSF) and blood of AD patients (Kauwe et al, 2014; Monson et al, 2014). In blood, cognitively impaired patients with brain amyloidosis show higher levels of pro‐inflammatory cytokines compared with patients without brain amyloidosis and control subjects. A lower level of the anti‐inflammatory cytokine IL‐10 is observed in brain amyloidosis positive patients than in negative patients (Cattaneo et al, 2017).
During aging, both the gastrointestinal tract epithelium and the blood–brain barrier become more permeable to small molecules, increasing the contribution of various microbiota metabolites to amyloid formation and dissemination (Marques et al, 2013; Montagne et al, 2015; Shoemark & Allen, 2015). Western diets high in saturated fats and sugars promote gut inflammation (Reichardt et al, 2017) and exacerbate brain neuropathological and associated behavioral deficits in animal models of AD and PD (Maesako et al, 2012; Rotermund et al, 2014; Busquets et al, 2017; Walker et al, 2017). Diarrhea, which is associated with gut inflammation, often affects older people and those with AD, and rapidly progressive dementia is one of the features in inflammatory bowel disease (IBD; Papathanasiou et al, 2014). Bacteria populating the gut microbiome produce amyloids, lipopolysaccharides (LPS), and other immunogenic compounds (Syed & Boles, 2014; Zhao et al, 2015). Enhanced inflammation, as a consequence of alterations in gut microbiota composition, is implicated in the initiation of α‐synuclein (α‐Syn) misfolding (Olanow et al, 2014), a hallmark of Parkinson’s disease (PD). Notably, the young 5xFAD mice which have not yet developed AD neuropathology do not have inflammation in GALT (gut‐associated lymphoid tissues), and GALT of 5xFAD mice mirror the disease progression and reflect inadequate immune surveillance in the gut and lead to enhanced AD pathology (Saksida et al, 2018).
Recently, we have reported that asparagine endopeptidase (AEP, gene name: LGMN) acts as a δ‐secretase that cleaves both APP and Tau, promoting Aβ and Tau aggregation in AD brains. δ‐secretase cuts APP at the N373 and N585 residues on the extracellular domain, facilitating BACE1 to produce Aβ more efficiently (Zhang et al, 2015). It also cleaves Tau at the N255 and N368 sites, accelerating Tau hyperphosphorylation and subsequent accumulation into NFTs (Zhang et al, 2014). Interestingly, δ‐secretase cleavage also produces a truncated α‐Synuclein (α‐Syn) at N103 and promotes Lewy body formation in PD (Zhang et al, 2017). We have recently shown that α‐Syn N103/Tau N368 fibrils spread from the gut into the brain via the vagus nerve, initiating PD pathogenesis (Ahn et al, 2019). δ‐secretase is upregulated in the control and AD patient brains in an age‐dependent manner. Notably, we identified that C/EBPβ, an inflammation‐activated transcription factor (Magalini et al, 1995; Poli, 1998), dictates LGMN mRNA transcription and escalates δ‐secretase abundance during aging (Wang et al, 2018b). Importantly, we showed that the C/EBPβ/δ‐secretase pathway spatiotemporally mediates AD‐like neuropathologies in 3xTg mice (Wang et al, 2018a). C/EBPβ mediates the learning and memory (Taubenfeld et al, 2001) and is implicated and upregulated in inflammation in various neurodegenerative diseases including AD (Li et al, 2004; Lukiw, 2004; Ejarque‐Ortiz et al, 2007).
In AD, poorly myelinated projection neurons with long axons are particularly prone to developing NFTs (Braak & Braak, 1996) and neurons affected by Tau pathology appear to be anatomically connected (Hyman et al, 1984). Intracellular Tau fibrils are directly released into the medium and can be taken up by co‐cultured cells, and internalized Tau aggregates induce fibrillization of intracellular Tau in the naive recipient cells (Kfoury et al, 2012). The spread of Tau pathology between interconnected neurons is thought to occur via exosomes (Wang et al, 2017). Inoculation of Tau aggregates induces time‐dependent spreading of Tau pathology from the inoculation site to brain regions with synaptic connection in transgenic mice overexpressing human tau or even in wild‐type mice (de Calignon et al, 2012; Liu et al, 2012; Iba et al, 2015). Likewise, intracerebral injection of diluted, Aβ‐containing brain extracts from AD patients or APP transgenic mice induces cerebral β‐amyloidosis and associated pathology in APP transgenic mice in a time‐ and concentration‐dependent manner (Kane et al, 2000; Meyer‐Luehmann et al, 2006). Hence, both Aβ and Tau fibrils spread via prion‐like mechanisms in AD (Walker, 2018).
In the current study, we tested the hypothesis that inflammation triggers C/EBPβ/δ‐secretase signaling, which initiates the onset of Aβ and Tau pathologies in the gut. The aggregated fibrils subsequently spread along the vagus nerve into the brain, initiating AD pathogenesis and cognitive defects.
Results
C/EBPβ/δ‐secretase is age‐dependently activated in the gut and brain, mediating AD pathologies
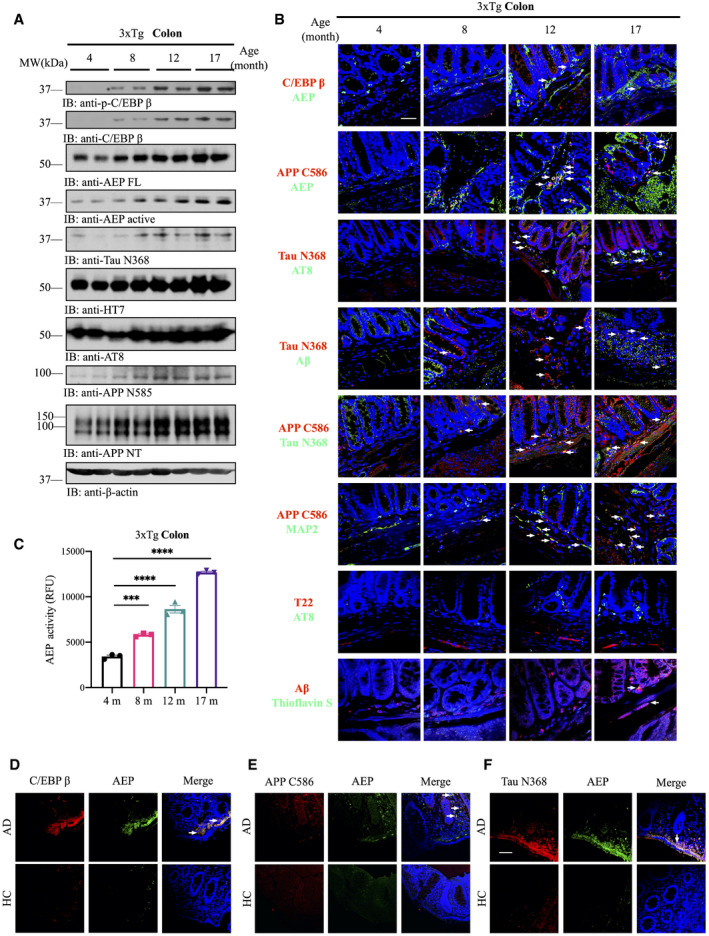
The C/EBPβ/δ‐secretase axis is activated in an age‐dependent manner in different brain regions of the 3xTg AD mouse model, elevating δ‐secretase‐truncated APP N585 and Tau N368 and promoting senile plaques and NFT formation (Wang et al, 2018a). Moreover, δ‐secretase also initiates α‐Syn N103/Tau N368 aggregates spreading from the gut into the brain, triggering PD pathogenesis (Ahn et al, 2019). To explore whether this pathway is also activated in the gut and mediates AD‐like pathology in the enteric nervous system, we conducted immunoblotting and monitored C/EBPβ/δ‐secretase signaling and the downstream APP and Tau fragmentation. We found that p‐C/EBPβ, a marker for its activation, and its total protein levels, as well as the total protein levels of δ‐secretase and its cleaved form, which indicated its activation, were increased in the gut of 3xTg mice in an age‐dependent manner (Fig 1A, top‐4th panels). Consequently, the abundance of δ‐secretase‐truncated Tau and APP, two suggested downstream effectors of C/EBPβ (Kfoury & Kapatos, 2009), was also elevated (Fig 1, 4, 4th‐bottom panels). Immunofluorescent (IF) co‐staining with the gut tissues also demonstrated that expression of C/EBPβ and δ‐secretase increased with age, mirroring the temporal appearance of APP C586 and Tau N368, which were distributed in MAP2‐positive enteric neurons. In addition, hyperphosphorylated Tau and oligomeric Tau were evident as assessed by AT8 and T22 immunoreactivity, respectively. Notably, Aβ co‐localized with Tau N368 and Thioflavin S (ThS) (Fig 1B). Enzymatic assay with gut lysates revealed that δ‐secretase activity gradually increased over time (Fig 1C). Similar results were obtained using 3xTg brain lysates (Appendix Fig S1A and B). Thus, C/EBPβ/δ‐secretase signaling is gradually activated in an age‐dependent manner in both the gut and the brain of 3xTg mice, promoting AD pathogenesis. To examine whether the similar events take place clinically, we conducted IF co‐staining in gut biopsy samples from AD patients. C/EBPβ and δ‐secretase were detected in AD samples but not those from healthy control (HC) (Fig 1D). Consequently, truncated APP C586 and Tau N368 fragments were co‐localized with δ‐secretase in AD gut tissues (Fig 1 E and F), indicating that the C/EBPβ/δ‐secretase pathway is activated in AD patient gut tissues, stimulating APP and Tau proteolytic cleavage and AD pathology.
Figure 1. C/EBPβ/δ‐secretase (AEP) is age‐dependently activated in the gut, mediating AD pathologies.
-
AImmunoblot showing p‐C/EBPβ, C/EBPβ, AEP, APP, and Tau expression and proteolytic processing in mouse colon.
-
BImmunofluorescent staining of C/EBPβ (red) and AEP (green), cleaved APP C586 (red) and AEP (green), cleaved Tau N368 (red) and AT8 (green), cleaved Tau N368 (red) and Aβ (green), cleaved APP C856 (red) and cleaved Tau N368 (green), cleaved APP C586 (red) and MAP2 (green), T22 (red) and AT8 (green), Aβ (red) and Thioflavin S (green) in colons of 3xTg mice. Scale bar: 20 μm.
-
CAEP activity assay with colon lysates from age‐dependent 3xTg mice. Data represent the mean ± SEM; representative data of three samples; ***P = 0.0001, ****P < 0.0001 compared with control, one‐way ANOVA.
-
DC/EBPβ and AEP immunofluorescent staining of colons from age‐matched healthy control and AD patients. White arrows indicate that C/EBPβ and AEP were co‐localized in AD patient’s colon.
-
EAEP and cleaved APP C586 fragment immunofluorescent staining of colons from age‐matched healthy control and AD patient. White arrows indicate that AEP and cleaved APP C586 were co‐localized in AD patient’s colon.
-
FAEP and cleaved Tau N368 fragment immunofluorescent staining of colons from age‐matched healthy control and AD patient. Scale bar: 20 μm. White arrows indicate that APP C586 and AEP were co‐localized in AD patient’s colon.
Source data are available online for this figure.
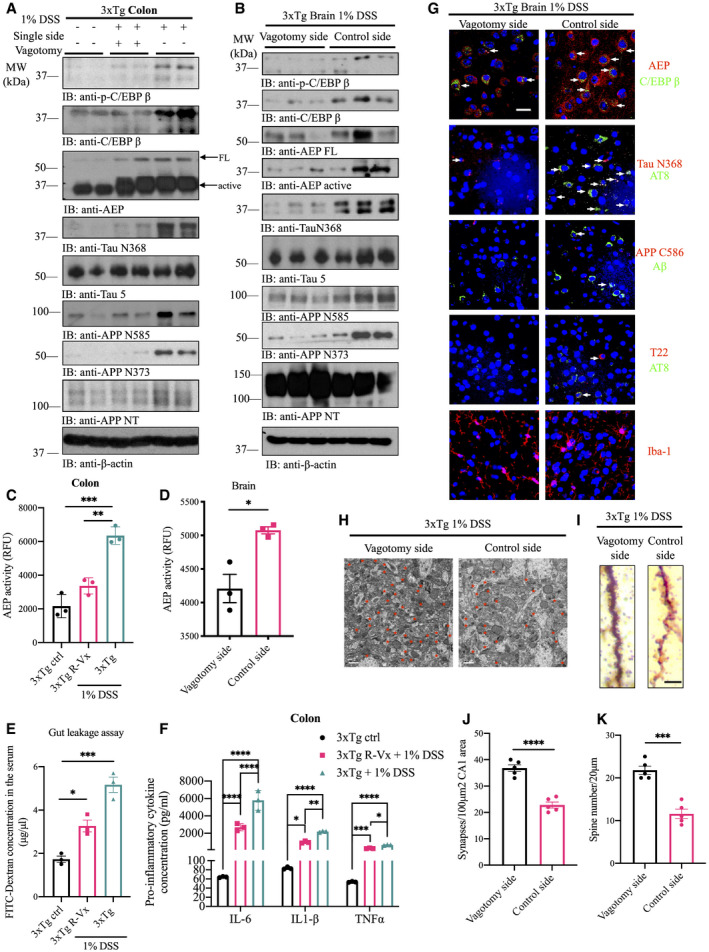
Figure 4. Vagotomy attenuates DSS‐elicited AD pathologies in 3xTg mice.
-
AImmunoblot showing p‐C/EBPβ, C/EBPβ, AEP, APP, and Tau expression and processing in mouse colon of 3xTg mice with neither DSS treatment nor single side vagotomy, with both DSS treatment and single side vagotomy, or with DSS treatment but no single side vagotomy.
-
BImmunoblot showing p‐C/EBPβ, C/EBPβ, AEP, APP, and Tau expression and proteolytic processing in mouse brain.
-
CAEP activity assay in colon lysates from pre‐vagotomy DSS‐treated, DSS‐treated, and vehicle‐treated 3xTg mice. DSS treatment escalated AEP activity in colon of 3xTg mice, which was reversed by vagotomy. Data represent the mean ± SEM; representative data of three samples; ***P = 0.0002, **P = 0.0013 compared with control, one‐way ANOVA.
-
DAEP activity assay with brain lysates from different brain hemispheres of single side pre‐vagotomy DSS‐treated 3xTg mice. Data represent the mean ± SEM; representative data of three samples; *P = 0.0162 compared with control, one‐way ANOVA.
-
EGastrointestinal permeability barrier defect as determined by FITC‐dextran translocation in pre‐vagotomy DSS‐treated, DSS‐treated, and vehicle‐treated 3xTg mice. Data represent the mean ± SEM; representative data of three samples; *P = 0.0126, ***P = 0.0002 compared with control, one‐way ANOVA.
-
FPro‐inflammatory cytokines IL‐6, IL‐1β, and TNFα concentrations in colon lysates of vehicle‐treated 3xTg mice, DSS‐treated single side vagotomy 3xTg and DSS‐treated 3xTg, respectively. Representative data of three samples; data are shown as mean ± SEM. *P = 0.0372, **P = 0.0050, ***P = 0.0008, ****P < 0.0001 compared with control, two‐way ANOVA.
-
GImmunofluorescent staining of AEP (red) and C/EBPβ (green), cleaved Tau N368 (red) and AT8 (green), cleaved APP C856 (red) and Aβ (green), T22 (red) and AT8 (green) and Iba‐1(red) of the cerebral cortex in different brain hemispheres of pre‐vagotomy DSS‐treated 3xTg mice. Scale bar: 20 μm. White arrows indicate that C/EBPβ and AEP, Tau N368 and AT8, and T22 and AT8 were co‐localized in mouse brain, respectively.
-
HRepresentative electron microscopy of the synaptic structures in hippocampus. Red stars indicate the synapses (scale bar: 1 μm).
-
IThe dendritic spines from the apical dendritic layer of the cerebral cortex region were analyzed by Golgi staining (scale bar: 5 μm).
-
JQuantitative analysis of the synaptic densities in DSS‐treated mice. Representative data of five samples, data are shown as mean ± SEM. ****P < 0.0001, unpaired t‐tests.
-
KQuantitative analysis of the spine density. Representative data of five samples, data are shown as mean ± SEM. ***P = 0.0001, unpaired t‐tests.
Source data are available online for this figure.
DSS induces gut inflammation and C/EBPβ/δ‐secretase pathway activation in 3xTg mice
C/EBPβ is potently activated by pro‐inflammatory cytokines, reactive oxygen species (ROS), or Aβ (Strohmeyer et al, 2014). To investigate whether the gut inflammation triggered by dextran sodium sulfate (DSS), a broadly utilized chemical that induces gut leakage, elicits C/EBPβ/δ‐secretase signaling activation in the mouse gut tissues, we added 1% DSS to the drinking water of wild‐type and 3xTg mice for 1 month. In wild‐type mice, DSS robustly increased MPO (myeloperoxidase) and its truncation in the colon, confirming the gut inflammation. Total and p‐C/EBPβ, as well as δ‐secretase, and its active truncated form were augmented upon DSS stimulation. APP and Tau were also augmented, as were δ‐secretase‐cleaved APP and Tau fragments in the colon (Appendix Fig S2A). Similar but slighted less robust effects of DSS treatment were observed in wild‐type mouse brains (Appendix Fig S2B). We observed even more prominent effects of DSS in both the gut and the brain of 3xTg mice, suggesting that DSS‐induced gut leakage and inflammation strongly agitates C/EBPβ/δ‐secretase signaling activation, resulting in augmented APP and Tau proteolytic fragmentation by active δ‐secretase (Appendix Fig S2C and D). δ‐secretase enzymatic assays with both gut and brain lysates corroborated its strong activation by DSS (Appendix Fig S2E and F). In alignment with the robust MPO activation, quantitative analysis with ELISA revealed that IL‐6, TNFα, and IL‐1β in the colon, but not the brain, were highly increased (Appendix Fig S2G). IF staining demonstrated that DSS treatment stimulated δ‐secretase in the guts of both wild‐type and 3xTg mice, but only elevated Aβ and p‐Tau in the colon of 3xTg mice, and Aβ was detected in the cortex of young 3xTg mice but not wild‐type mice. It is worth noting that Aβ amount in the hippocampus was substantially lower than the amount found in the cortex of 3xTg mice (Appendix Fig S2H–K). Thus, DSS‐elicited gut inflammation strongly activates the C/EBPβ/δ‐secretase pathway in the gut and brain of 3xTg mice, provoking AD pathology onset in the gut.
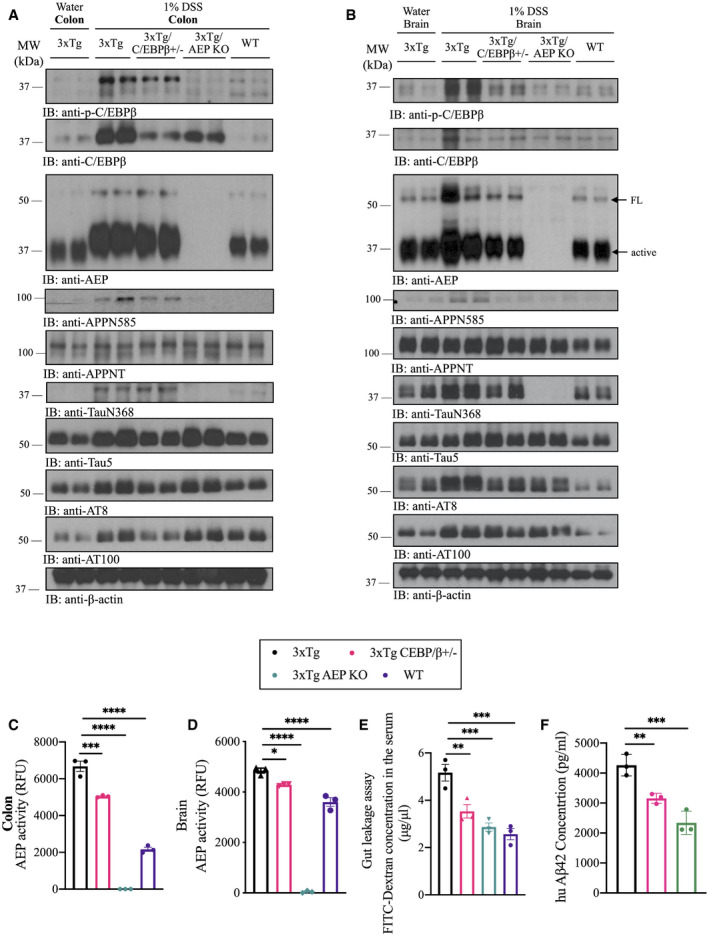
To explore whether the C/EBPβ/δ‐secretase pathway is responsible for the biological effects triggered by chronic DSS treatment (4 months), we employed 3xTg/C/EBPβ+/− and 3xTg/AEP−/− mice, as well as wild‐type mice as control. Depletion of C/EBPβ or δ‐secretase from 3xTg mice strongly attenuated DSS‐provoked C/EBPβ/δ‐secretase activation and its downstream effects in both the colon and the brain. Knockout of δ‐secretase completely abolished APP N585 and Tau N368 cleavage and mitigated AT8 hyperphosphorylation (Fig 2A and B). As a result, DSS‐stimulated δ‐secretase enzymatic activity was significantly reduced in 3xTg/C/EBPβ+/− and below the level of detection in 3xTg/AEP−/− mice compared to 3xTg mice and wild‐type mice (Fig 2C and D). Gut leakage assay with FITC‐Dextran showed that DSS‐elicited gut permeability was gradually reduced from 3xTg/C/EBPβ+/− to 3xTg/AEP−/− mice and wild‐type mice as compared to 3xTg mice (Fig 2E). In addition, human Aβ42 concentration in the brain of 3xTg animals was decreased in a similar pattern (Fig 2F).
Figure 2. DSS induces gut inflammation and C/EBPβ/δ‐secretase pathway activation in 3xTg mice.
-
AImmunoblot showing p‐C/EBPβ, C/EBPβ, AEP, APP, and Tau expression and proteolytic processing in mouse colon.
-
BImmunoblot showing p‐C/EBPβ, C/EBPβ, AEP, APP, and Tau expression and proteolytic processing in mouse brain.
-
CAEP activity assay with colon lysates from DSS‐treated mice. Data represent the mean ± SEM; representative data of three samples; ***P = 0.0002, ****P < 0.0001 compared with control, one‐way ANOVA.
-
DAEP activity assay with brain lysates from DSS‐treated mice. Data represent the mean ± SEM; representative data of three samples; *P = 0.0103, ****P < 0.0001 compared with control, one‐way ANOVA.
-
EGastrointestinal permeability barrier defect as determined by FITC‐dextran translocation in DSS‐treated mice. Data represent the mean ± SEM; representative data of three samples; **P = 0.0071, ***P = 0.0009 (3xTg vs. 3xTg AEP KO), ***P = 0.0004 (3xTg vs. WT) compared with control, one‐way ANOVA.
-
FHuman Aβ1‐42 concentration in brain lysates from DSS‐treated mice. Data represent the mean ± SEM; representative data of three samples; **P = 0.0097, ***P = 0.0006 compared with control, one‐way ANOVA.
Source data are available online for this figure.
IF co‐staining revealed that DSS stimulated demonstrable C/EBPβ/δ‐secretase signaling in the brains of 1% DSS‐treated 3xTg mice at the age of 6 months, which were decreased in 3xTg/C/EBPβ+/− and 3xTg/AEP−/− mice and wild‐type mice. Accordingly, Tau N368/AT8, APP C586/Aβ, and T22/AT8 co‐staining were demonstrable in 3xTg mice, and these effects were significantly abrogated in 3xTg/C/EBPβ+/− and 3xTg/AEP−/− mice and wild‐type mice (Fig 3A–E). Thus, the C/EBPβ/δ‐secretase pathway is indispensable for chronic DSS‐induced AD‐like neuropathology in the brain of young 3xTg mice. Electronic microscopic (EM) analysis revealed that synapse loss triggered by DSS was significantly rescued in 3xTg/C/EBPβ+/− and 3xTg/AEP−/− mice and wild‐type mice as compared to 3xTg mice (Fig 3F and H). Golgi staining displayed that dendritic spine reduction evoked by DSS in 3xTg mice was also substantially restored in 3xTg/C/EBPβ+/−, 3xTg/AEP−/−, and wild‐type mice (Fig 3G and I). Hence, inactivation of C/EBPβ/δ‐secretase pathway alleviates DSS‐induced AD pathologies in young 3xTg mice.
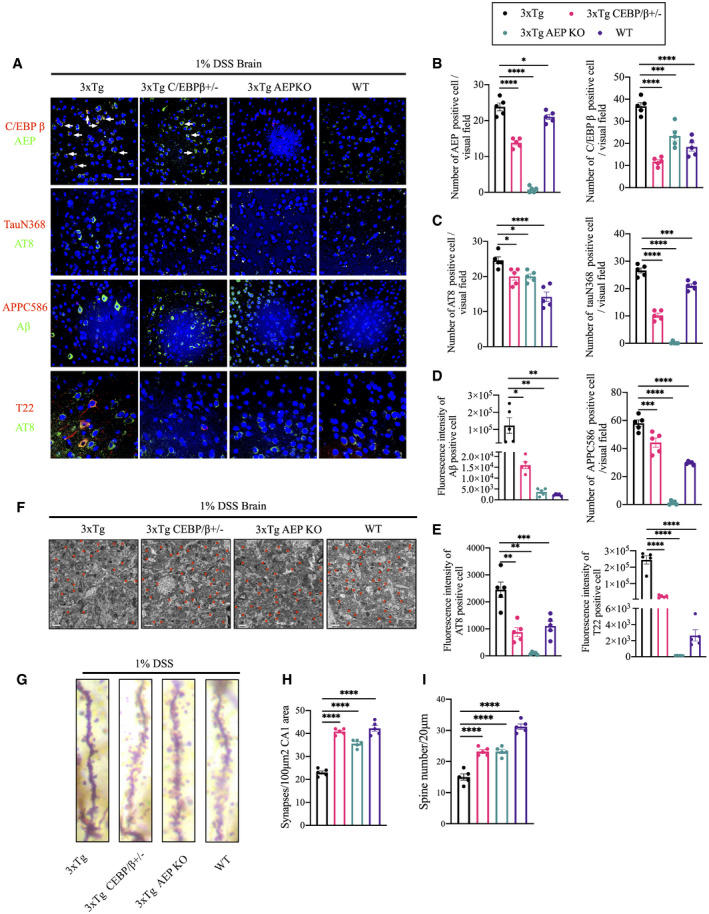
Figure 3. DSS triggers C/EBPβ/δ‐secretase activation in 3xTg mice, leading to synaptic degeneration.
-
AImmunofluorescent staining of C/EBPβ (red) and AEP (green), Iba‐1 (red) and AEP (green), cleaved Tau N368 (red) and AT8 (green), cleaved APP C856 (red) and Aβ (green), T22 (red) and AT8 (green) in cerebral cortex of brains from DSS‐treated mice. Scale bar: 20 μm. White arrows indicate that C/EBPβ and AEP were co‐localized in mouse brain.
-
BQuantitative analysis of AEP‐positive cells and C/EBPβ‐positive cells, respectively. The density of both AEP‐positive cells and C/EBPβ‐positive cells was significantly decreased through knocking down C/EBPβ or knocking out AEP in mice. Representative data of five samples, data are shown as mean± SEM. *P = 0.0390, ***P = 0.0002, ****P < 0.0001, one‐way ANOVA.
-
CQuantitative analysis of AT8‐positive cells (left) and TauN368‐positive cells (right), respectively. The density of both AT8‐positive cells and TauN368‐positive cells was significantly decreased through knocking down C/EBPβ or knocking out AEP in mice. n = 5 in each group, data are shown as mean± SEM. *P = 0.0194, ***P = 0.0001, ****P < 0.0001, one‐way ANOVA.
-
DQuantitative analysis of Aβ‐positive cells (left) and APPC586‐positive cells (right), respectively. The fluorescence intensity of Aβ‐positive cells and the density APPC586‐positive cells were significantly decreased through knocking down C/EBPβ or knocking out AEP in mice. Representative data of five samples, data are shown as mean± SEM. *P = 0.0107, **P = 0.0048 (3xTg vs. 3xTg AEP KO), **P = 0.0044 (3xTg vs. WT), ***P = 0.0006, ****P < 0.0001, one‐way ANOVA.
-
EQuantitative analysis of AT8‐positive cells (upper) and T22‐positive cells (lower), respectively. The fluorescence intensity of both AT8‐positive cells and T22‐positive cells was significantly decreased through knocking down C/EBPβ or knocking out AEP in mice. Representative data of five samples, data are shown as mean± SEM. **P = 0.0014 (3xTg vs. 3xTg C/EBPβ+/−), **P = 0.0030 (3xTg vs. 3xTg AEP KO), ***P = 0.001, ****P < 0.0001, one‐way ANOVA.
-
FRepresentative electron microscopy of the synaptic structures in hippocampus of brains from DSS‐treated mice. Red stars indicate the synapses (scale bar: 1 μm).
-
GThe dendritic spines from the apical dendritic layer of the cerebral cortex region were analyzed by Golgi staining (scale bar: 5 μm).
-
HQuantitative analysis of the synaptic densities in DSS‐treated mice. Representative data of five samples, data are shown as mean ± SEM. ****P < 0.0001, one‐way ANOVA.
-
IQuantitative analysis of the spine density. Representative data of five samples, data are shown as mean ± SEM. ****P < 0.0001, one‐way ANOVA.
Vagotomy attenuates DSS‐elicited AD pathologies in the brain of 3xTg mice
DSS‐elicited gut inflammation stimulates AD‐like neuropathology in the gut, and it has been proposed that the pathology might get transmitted to the brain via the vagus nerve (Fulling et al, 2019). To test this possibility, we resected the right side of vagus nerve in 2‐month‐old 3xTg mice, followed by chronic 1% DSS treatment for 4 months. Immunoblotting analysis showed DSS‐induced upregulation of C/EBPβ/δ‐secretase pathway and its downstream proteolytic effects on APP and Tau by DSS treatment (Fig 4A and B). DSS‐triggered AEP activities in the colon and the brain were abrogated by vagotomy (Fig 4C and D). Strikingly, the effects of DSS on C/EBPβ/δ‐secretase pathway, Aβ and tau fragmentation and aggregation, gut leakage, neuroinflammation, and synapse/spine loss in both the colon and the brain were robustly attenuated in vagotomized mice (Fig 4E–K). These data strongly support a model in which DSS‐induced AD pathologies originate in the gut and then are propagated into the brain via the vagus nerve. To further explore this possibility, we compared AD pathologies in colons from 3xTg mice with either reduced C/EPBPβ or δ‐secretase, or vagotomy. DSS‐induced Tau hyperphosphorylation and aggregation in the colon was significantly and similarly mitigated by vagotomy and in 3xTg/C/EBPβ+/− and 3xTg/AEP−/− mice (Appendix Fig S3).
Vagotomy or reduction of the C/EBPβ/δ‐secretase pathway rescues the cognitive dysfunctions in DSS‐treated 3xTg mice
To assess whether vagotomy or reduction of the C/EBPβ/δ‐secretase pathway ameliorates DSS‐induced cognitive impairment in 3xTg mice, we conducted a fear‐conditioning test to assess learning and memory. Cued fear‐conditioning tests showed that all of the groups displayed comparable baseline freezing time prior to conditioning. After tone‐shock pairing, DSS‐treated 3xTg mice spent less freezing time in both cued (Fig 5A) and contextual (Fig 5B) fear tests compared to control 3xTg mice after CS (conditioned stimulus), indicating impaired associative memory. However, DSS‐treated 3xTg/C/EBPβ+/− or DSS‐treated vagotomized 3xTg mice showed increases in freezing time in both cued (Fig 5A) and contextual (Fig 5B) fear tests as compared to DSS‐treated 3xTg mice after CS stimulation training, suggesting improved associative memory. Notably, vagotomized 3xTg mice showed higher survival rate and maintained their body weights better compared with 3xTg, 3xTg/C/EBPβ+/−, and 3xTg/AEP−/− mice under 1% DSS chronic treatment (Fig 5C and D).
Figure 5. DSS‐induced cognitive dysfunction in 3xTg mice is rescued through knocking out C/EBPβ/δ‐secretase or vagotomy.
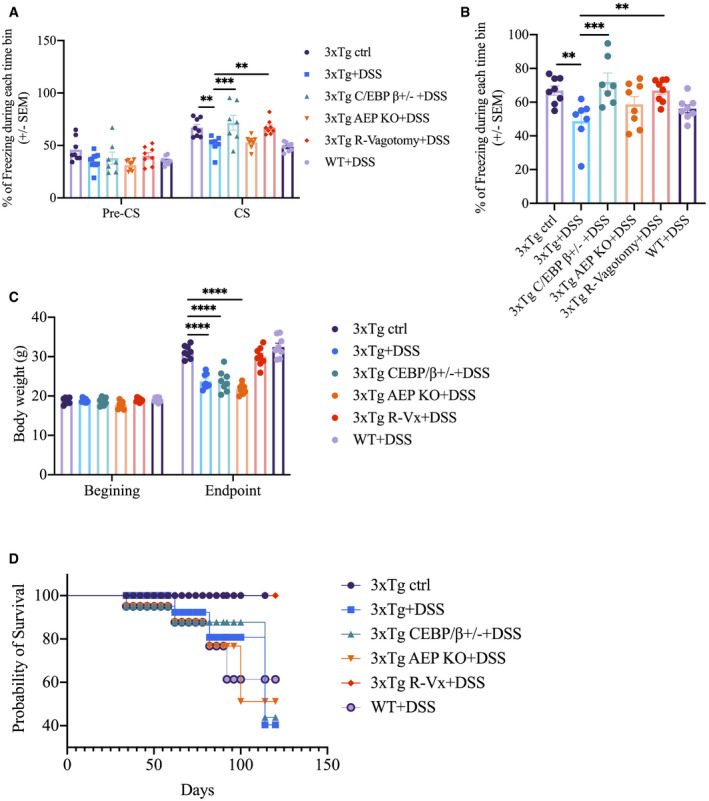
-
A, BCued and contextual fear‐conditioning tests. Data represent the mean ± SEM of n = 6–8 mice per group; **P = 0.0067 (3xTg + DSS vs. 3xTg ctrl), **P = 0.0059 (3xTg + DSS vs. 3xTg R‐vagotomy + DSS), ***P = 0.0009, two‐way ANOVA (A); **P = 0.0086 (3xTg + DSS vs. 3xTg ctrl), **P = 0.0085 (3xTg + DSS vs. 3xTg R‐vagotomy + DSS), ***P = 0.0005, one‐way ANOVA (B).
-
CBody weight changes of the experimental mice during the experimental paradigm. Data represent the mean± SEM of n = 8 mice per group; ****P < 0.0001, two‐way ANOVA.
-
DSurvival rate curve of the experimental mice during the experimental paradigm.
Vagotomy alleviates DSS‐induced C/EBPβ/δ‐secretase pathway activation in the brain of wild‐type mice
To further validate the hypothesis that vagus nerve is the path for pathology transmission to the brain from colon, we resected the right side of vagus nerve in 2‐month‐old wild‐type mice, followed by chronic 1% DSS treatment for 4 months, and then we assessed whether vagotomy alleviates DSS‐induced C/EBPβ/δ‐secretase pathway activation and cognitive impairment in wild‐type mice. Immunoblotting analysis showed that vagotomy substantially alleviated the DSS‐induced upregulation of C/EBPβ/δ‐secretase pathway and its downstream proteolytic effects on APP and Tau by DSS treatment (Appendix Fig S4A). DSS‐stimulated δ‐secretase enzymatic activity was also significantly reduced by vagotomy (Appendix Fig S4B). The fear‐conditioning test, which assesses the learning and memory functions, was also conducted on both groups of mice. Cued fear‐conditioning tests showed that both groups displayed comparable baseline freezing time prior to conditioning (Appendix Fig S4C). After tone‐shock pairing, DSS‐treated wild‐type mice spent less freezing time in both cued (Appendix Fig S4C) and contextual (Appendix Fig S4D) fear tests compared to DSS‐treated vagotomized wild‐type mice after CS (conditioned stimulus), indicating vagotomy rescuing the impaired associative memory.
Recombinant Aβ and Tau N368 fibrils spread from the gut into the brain along the vagus nerve
The above data demonstrate that DSS strongly stimulates AD pathologies in the gut and the brain of 3xTg mice, and vagotomy markedly abolishes these events. These findings prompted us to test whether accumulated Aβ and Tau pathologies originating in the gut propagate into the brain along the vagus nerve. We generated Aβ and Tau N368 fibrils in vitro by constant shaking recombinant Aβ1‐42 peptide and Tau N368 proteins for 7 days and then we conjugated them with fluorescent dye ATTO 550, respectively. Aβ and Tau N368 fibrils were validated by Thioflavin T binding assay and electron microscopy (EM) analysis, respectively (Appendix Fig S5B and C). The fluorescent‐labeled fibrils were colonoscopically injected into the colon wall of wild‐type mice, and the vagus nerve was isolated 4 and 7 days post‐injection (Appendix Fig S5D and E). ATTO 550 injected alone was not detected on vagus nerve at either time point, but ATTO 550‐labeled Aβ fibrils or Tau N368 aggregates were readily detectable at both time points (Fig 6A), in alignment with our previous report that Tau N368 fibrils spread along the vagus nerve (Ahn et al, 2019). To investigate whether these exogenously injected fibrils transport into the brain along the vagus nerve, we examined the fluorescent signals in different brain regions 3 months after colonic injection. Again, IF co‐staining revealed no detectable ATTO 550 alone signals in the dorsal motor vagus nerve (DMVN) or the locus coeruleus (LC) in the brainstem, or in the hippocampus. By contrast, we observed prominent Aβ‐ATTO 550 and Tau N368‐ATTO 550 signals in these regions. It is worth noting that Aβ fibrils were mainly enriched in the DMVN and the hippocampus, whereas Tau N368 fibrils preferentially accumulated in the LC and the hippocampus (Fig 6B).
Figure 6. Colonic‐injected Aβ PFFs, Tau N368 PFFs, AD patient brain extracts, or 5xFAD brain extracts travel along the vagus nerve to brain stem in WT mice.
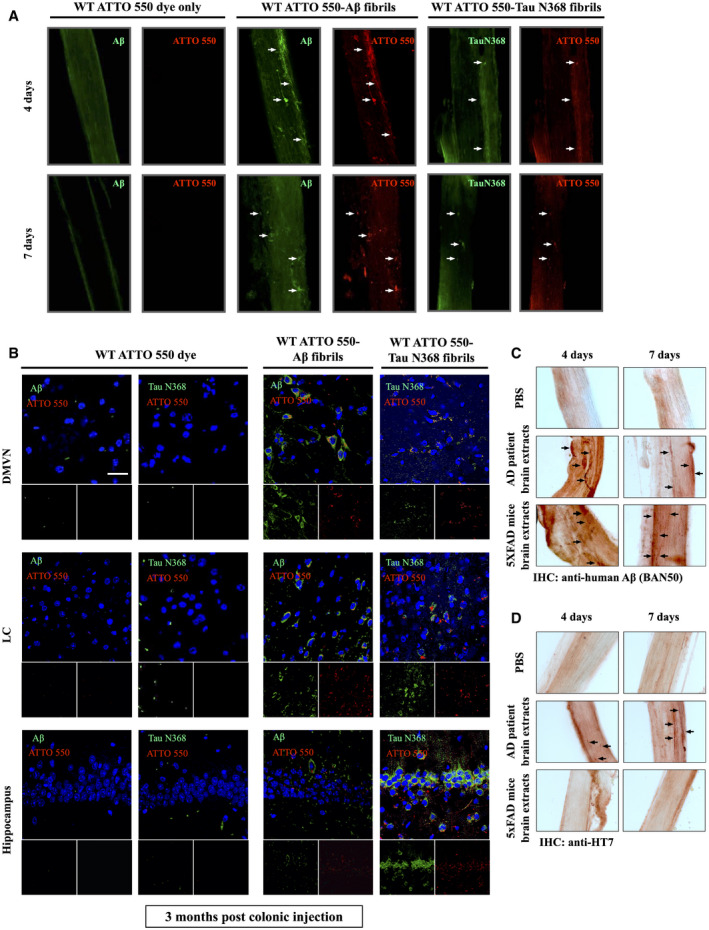
-
AImmunofluorescent staining showed ATTO 550‐labeled Aβ PFFs (pre‐formed fibrils) and Tau N368 PFFs traveling along the vagus nerve in WT mice at different time points. White arrows shows that ATTO 550 signals and Aβ PFFs (pre‐formed fibrils) signals, and ATTO 550 signals and Tau N368 PFFs were co‐localized in the vagus nerve, indicating the positive signals were from the injected exogenous pre‐form fibrils.
-
BImmunofluorescent staining showed ATTO 550‐labeled Aβ PFFs and Tau N368 PFFs spread into different brain regions in WT mice 3 months after colonic injection. Scale bar: 20 μm.
-
CImmunohistochemistry staining of human Aβ propagating along the vagus nerve from WT mice colonic‐injected with PBS, AD brain extracts, or 5xFAD brain extracts, using human‐specific Aβ antibody (BAN50). Black arrows indicate the human Aβ in mouse vagus nerve.
-
DImmunohistochemistry staining of human Tau propagating along the vagus nerve from WT mice colonic‐injected with PBS or AD brain extracts, using human Tau‐specific antibody (HT7). Black arrows indicate the human Tau in mouse vagus nerve.
Source data are available online for this figure.
Next, we extended our study to brain extracts from AD patients and 5xFAD mice. The soluble fractions from the brains were examined using immunoblotting by anti‐Aβ and various Tau antibodies including human HT7, AT8, and δ‐secretase‐truncated Tau N368. Both Aβ and Tau were aggregated in human AD brain lysates, whereas 5xFAD brain extracts only contained Aβ but not Tau aggregates. Protein samples were also validated by Coomassie blue staining (Appendix Fig S5F and H). Human Aβ40 and Aβ42 concentrations in these soluble fractions were quantitatively analyzed, confirming that the human AD brain possesses much more abundant Aβ40 in the soluble fractions (Appendix Fig S5G). Colonic injection of the human AD brain soluble extracts and the 5xFAD brain soluble extracts in wild‐type mice demonstrated that Aβ fibrils from both samples transport along the vagus nerve (Fig 6C). HT7 staining showed that fibrillized human Tau from AD patient brain extracts also spread along the vagus nerve, whereas the signal was barely detected with 5xFAD brain extracts (Fig 6D), supporting that the IHC signals are specific and consistent with results with recombinant Aβ and Tau N368 fibrils. Thus, our findings indicate that Aβ and Tau N368 fibrils can spread along the vagus nerve from the gut into the brain.
Colonic injection of recombinant Aβ and Tau N368 fibrils activates C/EBPβ/δ‐secretase axis in the brain
To explore whether recombinant Aβ or Tau N368 fibrils translocated from the colon to the brain induce the activation of C/EBPβ/δ‐secretase pathway, we conducted IF co‐staining on brain sections 6 months after colonic injection. δ‐secretase was evidently augmented in the hippocampus in wild‐type mice by both Aβ and Tau N368 fibrils, and it was tightly co‐localized with fluorescent ATTO 550 signals. Moreover, p‐C/EBPβ, an activated form of the protein, was also robustly elevated by Aβ and Tau N368 fibrils, co‐distributed in the cells stained with ATTO 550. By contrast, no p‐C/EBPβ or δ‐secretase activities were stimulated in the brain by fluorescent dye alone after colonic injection (Fig 7A). To assess the behavioral impact of Aβ and Tau N368 fibril spread into the brain, we performed cognitive behavioral tests. MWM assays showed that wild‐type mice inoculated with pre‐formed fibrils (PFFs) of ATTO 550‐conjugated Aβ or Tau N368 in the colon exhibited impaired memory in the MWM probe trial (Fig 7B). Other measures, including the latency to mount the submerged platform across trials, swim path distances, and swim speed during MWM training, were comparable between groups (Fig 7C–G). The swim speed and swim path distances by these mice remained approximately the same (Fig 7E–G). Therefore, these studies indicate that recombinant Aβ or Tau N368 fibrils propagate from the gut into the brain, where they activate C/EBPβ/δ‐secretase signaling, leading to spatial memory recall deficits in wild‐type mice.
Figure 7. Colonic inoculation of recombinant Aβ PFFs or Tau N368 PFFs activates C/EBPβ/δ‐secretase pathway in WT mice brain.
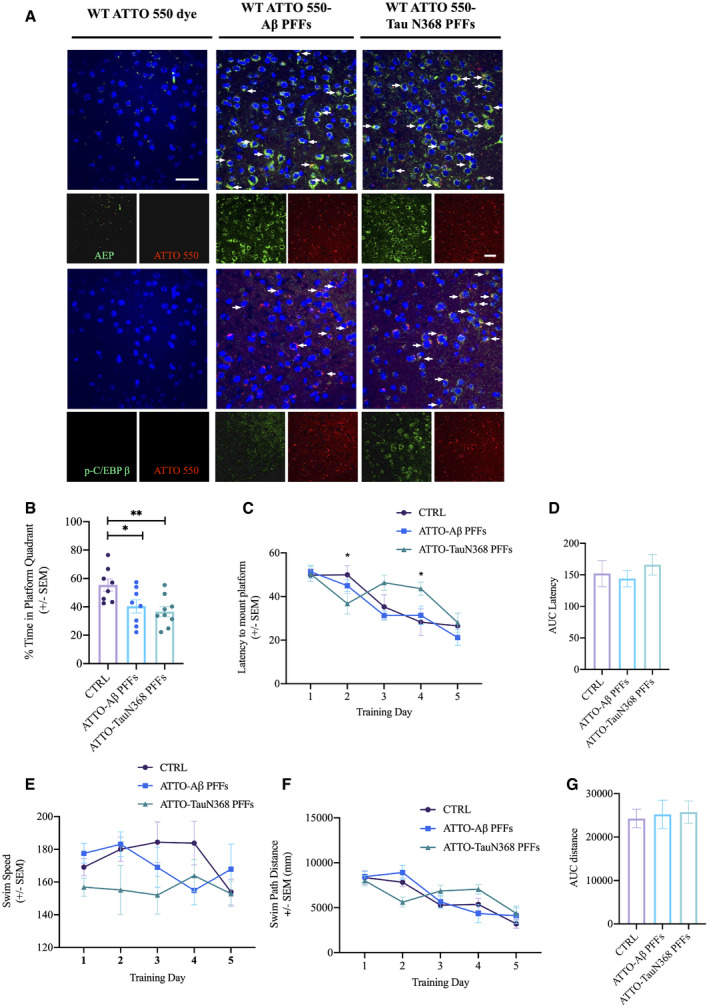
-
AImmunofluorescent staining of AEP and p‐C/EBPβ in cerebral cortex of brains of WT mice 6 months after colonic inoculation with either Aβ PFFs or Tau N368 PFFs. Scale bar: 20 μm. White arrows indicate that AEP and p‐C/EBPβ signals (green) were co‐localized with ATTO 550 signals (red), respectively.
-
B–GMorris water maze test. Data represent the mean ± SEM. Colonic‐injected mice (n = 7 per group) were trained in the water maze over five days. Shown are the percentage of time spent in the target quadrant in the probe trail, *P = 0.0362, **P = 0.0072, unpaired t‐tests (B), Latency to platform, *P = 0.0434, *P = 0.0166, two‐way ANOVA (C), the area under curve of Latency (AUC Latency) (D), Swim Speed (E), Swim Path Distance (F), the area under curve of Swim Path Distance (AUC distance) (G).
Source data are available online for this figure.
Vagotomy blunts AD pathologies in 3xTg mice induced by Colonic inoculated AD brain extracts
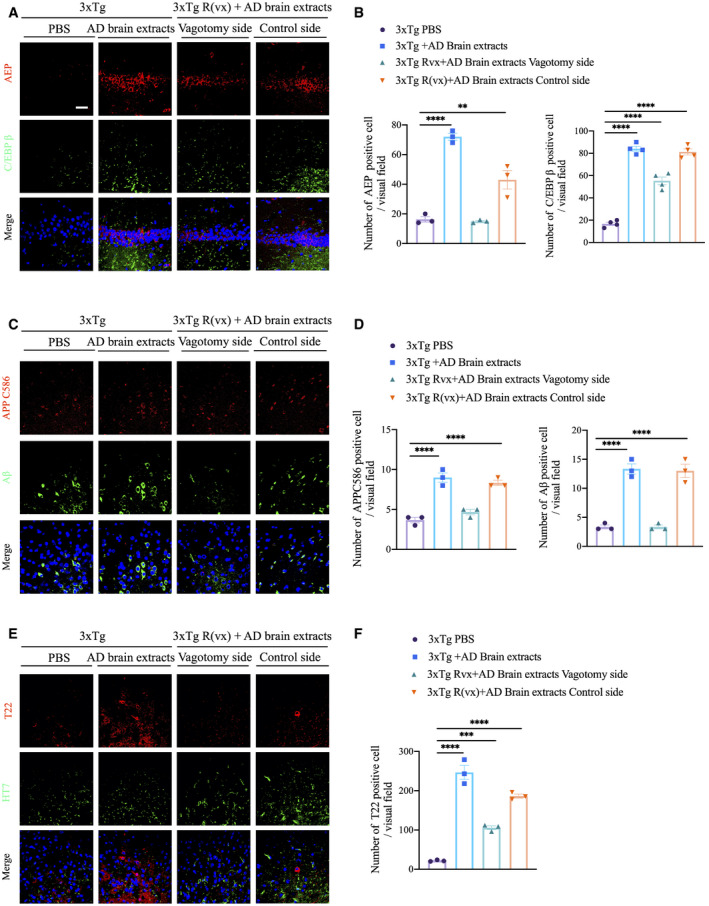
To examine whether Aβ and Tau N368 fibrils derived from AD patient brain extracts can stimulate C/EBPβ/δ‐secretase signaling and accelerate AD pathology onset, we injected soluble AD patients’ brain extracts or PBS control in the colon of 2‐month‐old 3xTg mice. Four months later, we observed that both C/EBPβ and δ‐secretase were prominently escalated in AD brain lysate, and vagotomy significantly diminished these signals compared with the control side (Fig 8A and B). To assess whether active δ‐secretase triggers APP cleavage and leads to Aβ accumulation, we conducted IF co‐staining with anti‐APP C586 and anti‐Aβ and found that more truncated APP C586 co‐localized with enhanced Aβ in AD brain lysate‐injected group versus PBS. Vagotomy notably reduced these activities (Fig 8C and D). Moreover, AD brain lysates potently provoked Tau aggregation, as visualized by conspicuous T22 staining, and δ‐secretase‐cleaved Tau N368 and AT8 immunoreactivity were reduced upon vagotomy (Fig 8E and F). Immunoblotting analysis showed that vagotomy diminished total and p‐C/EBPβ, as well as δ‐secretase. Consequently, APP N585, Tau N368, and p‐Tau AT8 abundance were decreased (Appendix Fig S6A). Enzymatic assays revealed that AD brain extract‐provoked δ‐secretase was repressed by vagotomy surgery (Appendix Fig S6B). Quantitative ELISA revealed that Aβ42 concentrations in soluble fraction, but not guanidine‐HCl fraction, were significantly diminished upon vagotomy treatment (Appendix Fig S6C), consistent with the reduced δ‐secretase activity and APP N585 cleavage by vagotomy in AD brain extract‐injected 3xTg mice. AD brain extract‐injected mice displayed more abundant hippocampal Tau N368 compared with those received PBS, which was also attenuated upon vagotomy (Appendix Fig S7A and B). Neuronal loss, as detected with cleaved caspase‐3 antibody and NeuN staining, was amplified by AD brain lysate administration, and again vagotomy rescued neuronal degeneration (Appendix Fig S7C and D). Thus, vagotomy diminishes multiple AD‐like neuropathologies in the brain elicited by gut inoculation with AD patient brain extracts in young 3xTg mice.
Figure 8. Colonic injection of AD brain extracts activates C/EBPβ/δ‐secretase pathway in 3xTg mice brain.
-
AImmunofluorescent staining of AEP (red) and C/EBPβ (green) in CA1 region from hippocampus of brains from colonic‐injected PBS, colonic‐injected AD brain extracts, and pre‐vagotomy colonic‐injected AD brain extracts 3xTg mice. Scale bar: 20 μm.
-
BQuantitative analysis of AEP‐positive cells and C/EBPβ‐positive cells, respectively. The density of both AEP‐ and C/EBPβ‐positive cells was significantly increased by colonic injection of AD brain extracts and decreased through vagotomy before colonic injection. Representative data of three samples, data are shown as mean ± SEM. **P = 0.016, ****P < 0.0001, one‐way ANOVA.
-
CImmunofluorescent staining of cleaved APPC586 (red) and Aβ (green) in cerebral cortex of brains from colonic‐injected PBS, colonic‐injected AD brain extracts, and pre‐vagotomy colonic‐injected AD brain extracts 3xTg mice.
-
DQuantitative analysis of cleaved APP C586‐positive cells and Aβ‐positive cells, respectively. The density of both cleaved APP C586‐positive cells and Aβ‐positive cells was significantly increased by colonic injection of AD brain extracts and decreased through vagotomy before colonic injection. Representative data of three samples, data are shown as mean ± SEM. ****P < 0.0001, one‐way ANOVA.
-
EImmunofluorescent staining of T22 (red) and HT7 (green) in cerebral cortex of brains from colonic‐injected PBS, colonic‐injected AD brain extracts, and pre‐vagotomy colonic‐injected AD brain extracts 3xTg mice.
-
FQuantitative analysis of T22‐positive cells. The density of T22‐positive cells was significantly increased by colonic injection of AD brain extracts and decreased through vagotomy before colonic injection. Representative data of three samples, data are shown as mean ± SEM. ***P = 0.0007, ****P < 0.0001, one‐way ANOVA.
Discussion
In the current work, we provide compelling evidence that recombinant Aβ or Tau N368 fibrils can be transported from the injected gut along the vagus nerve into the brainstem in wild‐type C57/BL6 mice, where they spread to anatomically connected brain regions including the DMVA, LC, and hippocampus. These exogenous pathogens trigger C/EBPβ/δ‐secretase pathway activation in the hippocampus, leading to cognitive deficits. Noticeably, colonic infusion of AD brain extracts, which encompass both Aβ and Tau N368 fibrils, into the gut of young 3xTg mice also travels along the vagus nerve and activates the C/EBPβ/δ‐secretase pathway, accelerating AD‐like pathologies in the hippocampus. This process depends on the vagus nerve, which when resected can ameliorate the spread of pathology and cognitive impairment. In addition, we show that chronic treatment of 3xTg mice with 1% DSS facilitates C/EBPβ/δ‐secretase signaling activation and AD pathologies in both the gut and the brain, while deletion of either C/EBPβ or δ‐secretase from 3xTg mice abrogated the AD pathologies. Remarkably, vagotomy completely suppressed DSS‐elicited C/EBPβ/δ‐secretase signaling and AD pathologies in both organs of 3xTg mice on the surgically ablated side without affecting the control side, indicating that the vagus nerve mediates AD pathogenesis from the gut into the brain. Although cleaved (N368) and aggregated (T22‐positive) Tau co‐localized with p‐Tau (AT8) in the brain (Figs 3A and 4G), it remains unclear why T22 signals were not co‐localized with AT8 in the gut upon DSS stimulation (Appendix Fig S3C) or in 17‐month‐old 3xTg mice (Fig 1B).
When PD patient brain lysates or recombinant α‐Syn aggregates were infused into the intestinal wall in rats, α‐Syn pathology was propagated via the vagus nerve to brainstem cholinergic neurons (Holmqvist et al, 2014). Similarly, when a viral vector expressing human α‐Syn was injected directly into the vagus nerve in the neck, α‐Syn pathology developed in brainstem neurons and spread to the midbrain and cerebral cortex (Ulusoy et al, 2013). In the current work, we also injected AD patient brain extracts or 5xFAD mouse brain extracts or recombinant Aβ or Tau N368 fibrils into the guts of 3xTg or wild‐type mice and found that both Aβ and Tau fibrils propagated along the vagus nerve into the brain regardless of patient‐derived species or in vitro recombinant strains (Fig 6). These results are consistent with prion‐like seeded aggregation of misfolded abnormal proteinaceous assemblies in neurodegenerative diseases (Jucker & Walker, 2018). One possibility is that vagotomy blunts the transport of proteinaceous aggregates from the GI to the brain in 3xTg mice, thus offering protection from the spread of proteopathic seeds and cognitive dysfunction. Therefore, these findings demonstrate that, like Lew body fibrils in PD, AD‐related aggregates including Aβ and Tau N368 seeds can spread from the gut into the brain, and suggest that chronic gut inflammation contributes to AD pathogenesis.
Accumulating evidence links gut microbiota with neuropsychiatric and neurological diseases, and its relationship with neurodegenerative diseases is particularly intriguing. The intestinal microbiota is a large and diverse collection of microorganisms that play a crucial role in regulating host health. Recently, studies have highlighted that changes in intestinal microbiota contribute to brain dysfunction in various neurological diseases including AD (Seo & Holtzman, 2019). Microbiota compositions are altered in AD patients and animal models, and these changes may increase intestinal permeability and induce inflammation (Garcez et al, 2019). Dysbiosis causes gut inflammation, diarrhea, constipation, visceral hypersensitivity, abdominal pain, dysfunctional metabolic state, and peripheral immune and neuro‐immune communication. Pathogenic gut microbiota are known to upregulate gut and systemic inflammation (due to LPS from pathogenic bacteria and synthesis of pro‐inflammatory cytokines; Daulatzai, 2015). Bacterial endotoxins may play a key role in the inflammatory and pathologic processes associated with amyloidosis and AD (Asti & Gioglio, 2014), as bacterial components, such as endotoxins, have been found within the typical senile plaque lesions of the AD brain. Most recently, it has been proposed that AD may begin in the gut, and is closely related to the imbalance of gut microbiota (Sochocka et al, 2019). For instance, AD pathology in APP/PS1 mice shifts the gut microbiota toward profiles that share features with inflammatory disorders (Bauerl et al, 2018). Furthermore, antibiotic‐induced perturbations in microbial diversity alter amyloid pathology in aged APP/PS1 mice (Minter et al, 2017). Conceivably, due to the gut leakage during aging, the metabolites from gut microbiota elicit chronic gut inflammation, which stimulates C/EBPβ, and Aβ or inflammatory cytokine‐activated transcription factor. Active C/EBPβ strongly upregulates inflammatory cytokines (Straccia et al, 2011) and δ‐secretase expression in the gut, in addition to APP and MAPT (Kfoury & Kapatos, 2009). Consequently, escalated APP and Tau proteins are cleaved by active δ‐secretase, promoting Aβ and NFTs pathologies in the enteric nervous system (Figs 1, 2, 3, 4). We report findings in the colon from AD patients (Fig 1D) and 3xTg mice chronically treated with DSS (Figs 2, 3, 4), which are consistent with the notion that the host bacteria amyloids contribute to misfolding and amyloidogenic diseases such as AD (Schwartz & Boles, 2013; Mezo et al, 2020). The vagus nerve links the viscera with the brain and is an important communication pathway for the gut microbiota to influence brain and behavior. Within the small and large intestine, vagal afferents terminate in the muscle layer as well as in the mucosa. In the muscle layer, vagal afferents form intra‐ganglionic laminar endings and intramuscular arrays, while some vagal fibers synapse onto neurons from the enteric nervous system (Fulling et al, 2019). Unlike other risk factors and genetic causes of AD, neuroinflammation is not typically thought to be causal on its own, but rather a result of one or more AD pathologies or risk factors associated with AD, and serves to increase the severity of the disease by exacerbating β‐amyloid and tau accumulation (Kinney et al, 2018). However, our study shows that local DSS‐triggered gut leakage and inflammation itself robustly activates C/EBPβ/δ‐secretase signaling, which drives Aβ aggregation and Tau fibrillization in the gut (Figs 2, 3, 4 and Appendix Fig S3). These observations are in alignment with the hypothesis that inflammation increases β‐amyloid burden and drives AD pathogenesis (Irwin & Vitiello, 2019). The gut microbiota might be capable of influencing homeostasis by changing the properties of vagus nerve signaling and related neuronal networks (Bonaz et al, 2016).
δ‐secretase‐cleaved Tau N368 fragment is detectable in human AD brains, and it is more prone to fibrillization than full‐length (FL) Tau (Zhang et al, 2014; Leuzy et al, 2019). Tau N368 fibrils have greater neurotoxicity and trans‐synaptic neuronal spreading capacity than FL Tau. α‐Syn N103/Tau N368 fibrils act as seeds to induce phosphorylation and aggregation of endogenous Tau or α‐Syn within neurons, and leads to cell‐to‐cell transmission of pathologies in anatomically interconnected regions after intra‐striatal or colonic inoculation. Tau N368 fibrils or α‐Syn N103/Tau N368 fibrils propagate in the vagus nerve of α‐SNCA mice and wild‐type mice (Ahn et al, 2019). Previous studies demonstrate that both Tau and α‐Syn pathologies spread to distant and synaptically connected regions (Luk et al, 2012; Sanders et al, 2014). Tau displays different conformations in different tauopathies, and they possess various strains with different propagation and prion‐like properties. Moreover, the distinct strains in various tauopathies reveal differential cellular and brain region predispositions (Sanders et al, 2014). Furthermore, some Tau strains can only be propagated in Tau P301S transgenic mice but not wild‐type mice. Hence, we chose to focus Tau N368 instead of FL Tau in the current study. Pathological α‐Syn propagates along major CNS pathways to regions far beyond injection sites and reduces survival with a highly reproducible interval from injection to death in inoculated animals, and inoculation with α‐Syn fibrils assembled from recombinant human α‐Syn induces identical consequences (Holmqvist et al, 2014). Remarkably, colon‐inoculated α‐Syn N103/Tau N368 fibrils propagate much more slowly in SNCA/AEP‐null mice than in α‐SNCA/AEP wild‐type mice, suggesting that δ‐secretase participates in mediating the fibril transportation and spreading of α‐Syn and Tau pathologies (Ahn et al, 2019). In the current study, we also show recombinant fluorescent‐tagged Tau N368 fibrils, Aβ fibrils, and Aβ fibrils from AD brain or 5xFAD brain extracts transport along the vagus nerve in wild‐type mice (Fig 6). Previous studies show that FL Tau fibrils spread from neuron‐to‐neuron only in the context of human α‐Syn or Tau overexpression; they can propagate in α‐SNCA or Tau P301S transgenic mice but not wild‐type mice (Sanders et al, 2014). In order to explore the spread of AD brain extract‐containing fibrils from the gut to the brain and subsequent potential pathological effects by the exogenous fibrils, we employed 3xTg mice and vagotomy surgery (Fig 7; Appendix Fig S6 and S7).
Our findings demonstrate that chronic mild gut inflammation induced by a low concentration (1%) of DSS in the drinking water is sufficient to accelerate the onset of cognitive dysfunction in the 3xTg mouse model of AD. Compared to control 3xTg mice, those subjected to experimental chronic low‐grade gut inflammation displayed robust activation of C/EBPβ/δ‐secretase signaling in both the gut and the brain, associated with exacerbation of Aβ and NFT pathology, neuroinflammation, and synapse loss. As predicted, deletion of C/EBPβ or δ‐secretase from 3xTg mice reduced AD‐like neuropathology in both the colon and the brain of 3xTg/C/EBPβ+/− and 3xTg/AEP−/− (Figs 2 and 3). Moreover, vagotomy in 3xTg also strongly diminished C/EBPβ/δ‐secretase signaling and AD pathologies (Fig 4). These findings show that local gut inflammation can trigger or exacerbate brain Aβ and NFT pathologies and neuronal degeneration by a mechanism involving vagus nerve‐mediated propagation of the AD processes from the gut to the brain. In support of our observations, previous studies have already shown that vagus nerve stimulation (VNS) has a cognitive‐enhancing effect in AD patients (Sjogren et al, 2002; Merrill et al, 2006). Information from the vagus nerve is relayed in the brainstem, where gut vagal afferents synapse onto neurons in the NTS. GI vagal afferents in the NTS are topographically organized, with afferents from the stomach projecting to the medial and gelatinous nucleus, and afferents from the intestines synapsing onto neurons in the medial and commissural nucleus (Fulling et al, 2019). Together, our findings support a model in which Aβ and NFTs pathogenic aggregates can propagate from the gut into the brain via the vagus nerve, and this process is mediated by the C/EBPβ/δ‐secretase pathway. Modulation of gut microbiota through personalized diet or beneficial microbiota intervention to diminish gut inflammation may have utility for the treatment of brain disorders including AD.
Materials and Methods
Mice
3xTg mice were ordered from the Jackson Laboratory (34830). Since some of the homozygous mutations are lethal on pure‐strain backgrounds, C/EBPβ mice were maintained as heterozygotes on two separate strain backgrounds (C57BL/6 and 129 Sv). These two strains were crossed to generate viable F1 hybrid wild‐type and C/EBPβ−/− littermates, which were used for aging studies. AEP‐knockout mice on a mixed C57BL/6 and 129/Ola background were generated as reported. (Shirahama‐Noda et al, 2003) The following animal groups were analyzed: 3xTg, 3xTg/C/EBPβ+/−, and 3xTg/AEP−/−. Both female and male mice were used. Animal care and handling were performed according to NIH animal care guidelines and the Declaration of Helsinki and Emory Medical School guidelines. The protocol was reviewed and approved by the Emory Institutional Animal Care and Use Committee. The animals were randomly allocated to experimental groups. Investigators were blinded to group allocation during the animal experiments. In chronic DSS treatment experiment, 3xTg, 3xTg/C/EBPβ+/− and 3xTg/AEP KO mice were provided 1%DSS in drinking water beginning at 8–9 weeks of age through 24–25 weeks of age; two batches of wild‐type mice were provided 1% DSS in drinking water beginning at 8 weeks of age, and the treatment lasted for 4 weeks and 4 months, respectively. Vagotomy procedure were performed on 3xTg mice and wild‐type mice 4 days before DSS treatment started. Mice were anesthetized with isoflurane, and meloxicam (2 mg/kg) was injected subcutaneously for analgesics. After cervical midline incision, vagus nerve around common carotid artery was isolated and cut on one side.
Antibodies and reagents
δ‐secretase antibody (clone 6E3 and 11B7) was a present from Dr. Colin Watts, University of Dundee. TrkB antibody was from BioVision (Milpitas, CA). Phospho‐TrkB Y816 antibody was raised against [H]‐CKLQNLAKASPV‐pY‐LDILG‐[OH] (a.a.806–822) (EM437 and EM438) as rabbit polyclonal antibody. 4‐HNE antibody was from Abcam (SF, CA). Aβ, AT8, AT100, Tau 5, tubulin, and beta‐actin antibody were from Sigma‐Aldrich (St Louis, MO). CEBP/β (H‐7) antibody was bought from Santa Cruz Biotechnology (Dallas, Texas). Legumain (D6S4H) and p‐C/EBPβ antibodies were bought from Cell Signaling Technology (Boston, MA). Iba‐1 antibodies were bought from FUJIFILM Wako Pure Chemicals Corporation (Richmond, VA). Histostain‐SP kit and Aβ 1‐42 and Aβ 1‐40 ELISA kit were from Invitrogen (Grand Island, NY). TNF‐α, IL‐1β and IL‐6 ELISA kits were from eBioscience (San Diego, CA). All chemicals not included above were purchased from Sigma‐Aldrich.
Western blot analysis
The mice brain tissue was lysed in lysis buffer (50 mM Tris, pH 7.4, 40 mM NaCl, 1 mM EDTA, 0.5% Triton X‐100, 1.5 mM Na3VO4, 50 mM NaF, 10 mM sodium pyrophosphate, 10 mM sodium β‐glycerophosphate, supplemented with protease inhibitors cocktail) and centrifuged for 15 min at 16,000 g. The supernatant was boiled in SDS loading buffer. After SDS–PAGE, the samples were transferred to a nitrocellulose membrane. Western blotting analysis was performed with a variety of antibodies.
Immunostaining
Paraffin‐embedded mouse brain and colon sections went through dewaxing and rehydration process by incubating the slides in xylene first and then immerse them into decreasing percentage of ethanol. The sections were boiled in 10 mM citric acid for 20 min followed by room temperature cooling down for antigen retrieval. Then, sections were treated with 3% H2O2 for 10 min followed by 3 times of wash in PBS and 30 min blocking in 1% RIA‐BSA and 0.3% Triton X‐100 as well as the overnight incubation with p‐TrkB (Y816, homemade, 1:300), 4‐HNE antibody (Abcam, 1:50), and Aβ antibody (Sigma‐Aldrich, 1:500) at 4°C. The signal was developed using Histostain‐SP kit (Invitrogen). To detect the localization of CEBP/β and p‐ CEBP/β, the slides were incubated with CEBP/β antibody (H‐7) (1:100) and p‐CEBP/β antibody (1:200) at 4°C. To detect the localization of AEP, AEP‐derived Tau fragment, and phosphorylated Tau in mouse brain section, the slides were incubated with AEP antibody (11B7) (1:500, from Dr. Colin Watts, University of Dundee), Tau N368 (homemade, 1:1,000), and AT8 (Thermo, MN1020, 1:500) at 4°C. After overnight incubation, the slides were washed three times in PBS and incubated with Texas Red‐conjugated anti‐rabbit IgG or FITC‐conjugated anti‐mouse IgG for 1 h at room temperature. The slides were washed three times in PBS and then covered with a glass cover using mounting solution and examined under a fluorescence microscope (Olympus).
Golgi staining
Mice brains were fixed in 10% formalin for 24 h and then immersed in 3% potassium bichromate for 3 days in the dark. The solution was changed each day. Then, the brains were transferred into 2% silver nitrate solution and incubated for 24 h in the dark. Vibratome sections were cut at 60 μm, air‐dried for 10 min, dehydrated through 95% and 100% ethanol, cleared in xylene, and coverslipped. For measurement of spine density, only spines that emerged perpendicular to the dendritic shaft were counted.
Aβ plaque staining
Amyloid plaques were stained with Thioflavin S. The deparaffinized and hydrated sections were incubated in 0.25% potassium permanganate solution for 20 min, rinsed in distilled water, and incubated in bleaching solution containing 2% oxalic acid and 1% potassium metabisulfite for 2 min. After rinsed in distilled water, the sections were transferred to blocking solution containing 1% sodium hydroxide and 0.9% hydrogen peroxide for 20 min. The sections were incubated for 5 s in 0.25% acidic acid and then washed in distilled water and stained for 5 min with 0.0125% Thioflavin S in 50% ethanol. The sections were washed with 50% ethanol and placed in distilled water. Then, the sections were covered with glass cover using mounting solution.
Aβ ELISA
The mice brains were homogenized in 8X mass of 5 M guanidine‐HCl/50 mM Tris–HCl (pH 8.0) and incubated at room temperature for 3 h. Then, the samples were diluted with cold reaction buffer (phosphate‐buffered saline with 5% BSA and 0.03% Tween 20, supplemented with protease inhibitor cocktail) and centrifuged at 16,000 g for 20 min at 4°C. The supernatant was analyzed by human Aβ40 and Aβ42 ELISA kit according to the manufacturer’s instructions (KHB3481 and KHB3441, respectively, Invitrogen). The Aβ concentrations were determined by comparison with the standard curve.
Th‐T assay
Th‐T stocking solution was prepared by adding 8 mg Th‐T to 10 ml PBS and filtering through a 0.2‐μm syringe filter (Sigma‐Aldrich, Cat# T3516). The stocking solution was diluted into the phosphate buffer (1 ml Th‐T stock to 50 ml buffer) to generate the working solution. Th‐T working solution with PFFs (7 μM) was excited at 440 nm and emitted at 482 nm to measure the fluorescence intensity on the plate reader (BioTek, #251639, Vermont, USA). For the PFF seeding test, different ratios of mixed PFFs/monomer (1:10, 1:35, 1:70 and 1:100) were incubated on the orbital shaker at 37°C for 1 h. The fluorescence values onto plate reader were recorded.
Transmission electron microscopy (TEM)
Electron microscopy images were taken from Tau N368 PFFs. The samples (5 μl) were deposited on Formvar‐coated 400 mesh copper grids, fixed with 0.5% glutaraldehyde, negatively stained with 2% uranyl acetate (Sigma‐Aldrich, Germany), and screened by CM‐10 TEM.
AEP activity assay
Tissue homogenates or cell lysates (10 μg) were incubated in 200 μl reaction buffer (20 mM citric acid, 60 mM Na2HPO4, 1 mM EDTA, 0.1% CHAPS, and 1 mM DTT, pH 5.5) containing 20 μM AEP substrate Z‐Ala‐Ala‐Asn‐AMC (Bachem). AMC released by substrate cleavage was quantified by measuring at 460 nm in a fluorescence plate reader at 37°C in kinetic mode.
Generation of TauN368 PFFs
TauN368 fibrils were prepared in reactions (500 μl per tube) containing 3 mg/ml Tau N368 protein monomers in PFF reaction buffer (50 mM Tris–HCl, pH 7.4, 50 mM NaCl). The protein was incubated for several days at 37 °C, with orbital shaking at 300 rpm until samples appeared cloudy. Usually, the reactions were subjected to 7–9 days shaking for the PFF generation. PFFs were validated by TEM morphology.
Generation of Aβ PFFs
Recombinant Aβ1‐42 HFIP was purchased from rPeptide®. The fibril was prepared through adding 900 μl of deionized water to one vial following 2 s vortex and incubation on ice for 5 mins. Repeated the procedure till 30 mins. Then add 100 μl of fibril‐forming buffer (0.2 M NaPi, 1.5 M NaCl, 0.2% NaN3, pH 7.5) to the vial to make final concentration at 1 mg/ml and vortex. Seal tube and place at 37°C for one week, vortexing daily (15 s each time). Fibrils are stored at −80°C. Fibril formation was validated by Th‐T assay.
Human tissue extracts preparation
Postmortem human AD brain samples and gut biopsy tissues were procured in accordance with institutional guideline and approved by Emory School of Medicine and Capital Medical University Institutional Review Board. Tissue for preparation of extract was derived at autopsy from the temporal cortex of two people who had died of confirmed Alzheimer’s disease provided by Emory ADRC (Goizueta Alzheimer’s Disease Research Center). Neuropathological examination revealed profuse senile plagues and neurofibrillary tangles in the temporal cortex of all two Alzheimer’s subjects and fulfilled the Consortium for the Establishment of a Registry for Alzheimer’s Disease (CERAD) criteria for AD. Tissue samples were fresh‐frozen on dry ice and stored at −80°C. They were homogenized at 10% (W/V) in sterile HBSS, vortexed for 2 min, probe‐sonicated for 3 s, revortexed, and centrifuged at 3,000 g for 5 min to remove tissue debris. The supernatant was recovered and further diluted 1:10 (v/v, in HBSS) to a final concentration of 1% immediately before colonoscopy injection.
Mouse colonoscopy injection
A high‐resolution miniaturized colonoscope system (Coloview Veterinary Endoscope, Karl Storz) was used for colonoscopy injection of PFF solutions (5 μg per mouse). This system consisted of a miniature rigid endoscope (1.9 mm outer diameter), a xenon light source, a triple‐chip high‐resolution charge‐coupled device camera, and an operating sheath with instrument channels (all from Karl Storz). The endoscope with outer operating sheath was inserted into the mid‐descending colon and the injection was performed with the aid of a custom‐made device comprising a needle (OD: 0.2 mm) connected to a small tube fitted with a syringe. Colonoscopy injection procedures were viewed with high‐resolution (1,024 × 768 pixels) live video on a flat‐panel color monitor.
Morris water maze (MWM)
Experimental mice were trained in a round, water‐filled tub (52 inches in diameter) in an environment rich with extra maze cues. An invisible escape platform was located in a fixed spatial location 1 cm below the water surface independent of a subject start position on a particular trial. In this manner, subjects needed to utilize extra maze cues to determine the platform’s location. At the beginning of each trial, the mouse was placed in the water maze with their paws touching the wall from 1 of 4 different starting positions (N, S, E, W). Each subject was given 4 trials/day for 5 consecutive days with a 15‐min intertrial interval. The maximum trial length was 60 s, and if subjects did not reach the platform in the allotted time, they were manually guided to it. Upon reaching the invisible escape platform, subjects were left on it for an additional 5 s to allow for survey of the spatial cues in the environment to guide future navigation to the platform. After each trial, subjects were dried and kept in a dry plastic holding cage filled with paper towels to allow the subjects to dry off. The temperature of the water was monitored every hour so that mice were tested in water that was between 22 and 25°C. Following the 5 days of task acquisition, a probe trial was presented during which time the platform was removed and the percentage of time spent in the quadrant which previously contained the escape platform during task acquisition was measured over 60 s. All trials were analyzed for latency, swim path length, and swim speed by means of MazeScan.
Fear conditioning
The ability to form and retain an association between an aversive experience and environmental cues was tested with a standard fear‐conditioning paradigm that occurred over a period of 3 d. The mouse was placed in the fear‐conditioning apparatus (7 in wide × 7 in deep × 12 in high; Coulbourn) composed of Plexiglas with a metal shock grid floor and was allowed to explore the enclosure for 3 min. Following this habituation period, three conditioned stimulus (CS)–unconditioned stimulus (US) pairings were presented with a 1‐min intertrial interval. The CS was composed of a 20‐s, 85‐dB tone, and the US was composed of a 2‐s 0.5‐mA footshock, which was coterminate with each CS presentation. One minute following the last CS–US presentation, the mouse was returned to its home cage. On day 2, the mouse was presented with a context test during which the subject was placed in the same chamber used during conditioning on day 1, and the amount of freezing was recorded via a camera and software provided by Coulbourn. No shocks were given during the context test. On day 3, a tone test was presented during which the subject was exposed to the CS in a novel compartment. Initially, the animal was allowed to explore the novel context for 2 min. Then, the 85‐db tone was presented for 6 min, and the amount of freezing behavior was recorded.
Statistical analysis
All data are expressed as mean ± SEM from three or more independent experiments, and the level of significance between two groups was assessed with Student’s t‐test. For more than two groups, one‐way ANOVA followed by LSD post hoc test was applied. A value of P < 0.05 was considered to be statistically significant.
Author contributions
KY conceived the project, designed the experiments, analyzed the data, and wrote the manuscript. CC, HW, SSK, and EHA designed and performed most of the experiments. XL prepared primary neurons and assessed with animal experiments. YZ and JJ provided AD patients’ gut biopsy tissues. JJ and AA assisted with data analysis and interpretation and critically read the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Review Process File
Appendix
Source Data for Expanded View and Appendix
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
This work was supported by grants from NIH grants (RF1, AG051538; RF1 AG061175) to K.Y. We thank ADRC (P30 AG066511) at Emory University for human AD patients and healthy control samples. The authors are thankful to Dr. Lary Walker at ADRC, Emory University, and Dr. David Weinshenker at Department of Human Genetics, Emory University, for critical proofreading the manuscript and providing a lot of valuable advices. This study was supported in part by the Rodent Behavioral Core (RBC), which is subsidized by the Emory University School of Medicine and is one of the Emory Integrated Core Facilities. Additional support was provided by the Viral Vector Core of the Emory Neuroscience NINDS Core Facilities (P30NS055077). Further support was provided by the Georgia Clinical & Translational Science Alliance of the National Institutes of Health under Award Number UL1TR002378.
The EMBO Journal (2021) 40: e106320.
Data availability
This study includes no data deposited in external repositories.
References
- Ahn EH, Kang SS, Liu X, Chen G, Zhang Z, Chandrasekharan B, Alam AM, Neish AS, Cao X, Ye K (2019) Initiation of Parkinson's disease from gut to brain by delta‐secretase. Cell Res 30: 70–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asti A, Gioglio L (2014) Can a bacterial endotoxin be a key factor in the kinetics of amyloid fibril formation? J Alzheimers Dis 39: 169–179 [DOI] [PubMed] [Google Scholar]
- Bauerl C, Collado MC, Diaz Cuevas A, Vina J, Perez Martinez G (2018) Shifts in gut microbiota composition in an APP/PSS1 transgenic mouse model of Alzheimer's disease during lifespan. Lett Appl Microbiol 66: 464–471 [DOI] [PubMed] [Google Scholar]
- Bonaz B, Sinniger V, Pellissier S (2016) Anti‐inflammatory properties of the vagus nerve: potential therapeutic implications of vagus nerve stimulation. J Physiol 594: 5781–5790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E (1996) Development of Alzheimer‐related neurofibrillary changes in the neocortex inversely recapitulates cortical myelogenesis. Acta Neuropathol 92: 197–201 [DOI] [PubMed] [Google Scholar]
- Busquets O, Ettcheto M, Pallas M, Beas‐Zarate C, Verdaguer E, Auladell C, Folch J, Camins A (2017) Long‐term exposition to a high fat diet favors the appearance of beta‐amyloid depositions in the brain of C57BL/6J mice. A potential model of sporadic Alzheimer's disease. Mech Ageing Dev 162: 38–45 [DOI] [PubMed] [Google Scholar]
- Cattaneo A, Cattane N, Galluzzi S, Provasi S, Lopizzo N, Festari C, Ferrari C, Guerra UP, Paghera B, Muscio Cet al (2017) Association of brain amyloidosis with pro‐inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol Aging 49: 60–68 [DOI] [PubMed] [Google Scholar]
- Daulatzai MA (2015) Non‐celiac gluten sensitivity triggers gut dysbiosis, neuroinflammation, gut‐brain axis dysfunction, and vulnerability for dementia. CNS Neurol Disord Drug Targets 14: 110–131 [DOI] [PubMed] [Google Scholar]
- de Calignon A, Polydoro M, Suárez‐Calvet M, William C, Adamowicz D, Kopeikina K, Pitstick R, Sahara N, Ashe K, Carlson Get al (2012) Propagation of tau pathology in a model of early Alzheimer's disease. Neuron 73: 685–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejarque‐Ortiz A, Medina MG, Tusell JM, Perez‐Gonzalez AP, Serratosa J, Saura J (2007) Upregulation of CCAAT/enhancer binding protein beta in activated astrocytes and microglia. Glia 55: 178–188 [DOI] [PubMed] [Google Scholar]
- Fulling C, Dinan TG, Cryan JF (2019) Gut microbe to brain signaling: what happens in Vagus. Neuron 101: 998–1002 [DOI] [PubMed] [Google Scholar]
- Garcez ML, Jacobs KR, Guillemin GJ (2019) Microbiota alterations in Alzheimer's disease: involvement of the kynurenine pathway and inflammation. Neurotox Res 36: 424–436 [DOI] [PubMed] [Google Scholar]
- Holmqvist S, Chutna O, Bousset L, Aldrin‐Kirk P, Li W, Bjorklund T, Wang ZY, Roybon L, Melki R, Li JY (2014) Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol 128: 805–820 [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Morris JC, Goate AM (2011) Alzheimer's disease: the challenge of the second century. Sci Transl Med 3: 77sr71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman BT, Van Hoesen GW, Damasio AR, Barnes CL (1984) Alzheimer's disease: cell‐specific pathology isolates the hippocampal formation. Science 225: 1168–1170 [DOI] [PubMed] [Google Scholar]
- Iba M, McBride JD, Guo JL, Zhang B, Trojanowski JQ, Lee VM (2015) Tau pathology spread in PS19 tau transgenic mice following locus coeruleus (LC) injections of synthetic tau fibrils is determined by the LC's afferent and efferent connections. Acta Neuropathol 130: 349–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin MR, Vitiello MV (2019) Implications of sleep disturbance and inflammation for Alzheimer's disease dementia. Lancet Neurol 18: 296–306 [DOI] [PubMed] [Google Scholar]
- Jucker M, Walker LC (2018) Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat Neurosci 21: 1341–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane MD, Lipinski WJ, Callahan MJ, Bian F, Durham RA, Schwarz RD, Roher AE, Walker LC (2000) Evidence for seeding of beta ‐amyloid by intracerebral infusion of Alzheimer brain extracts in beta ‐amyloid precursor protein‐transgenic mice. J Neurosci 20: 3606–3611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauwe JSK, Bailey MH, Ridge PG, Perry R, Wadsworth ME, Hoyt KL, Staley LA, Karch CM, Harari O, Cruchaga Cet al (2014) Genome‐wide association study of CSF levels of 59 alzheimer's disease candidate proteins: significant associations with proteins involved in amyloid processing and inflammation. PLoS Genet 10: e1004758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI (2012) Trans‐cellular propagation of Tau aggregation by fibrillar species. J Biol Chem 287: 19440–19451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kfoury N, Kapatos G (2009) Identification of neuronal target genes for CCAAT/enhancer binding proteins. Mol Cell Neurosci 40: 313–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT (2018) Inflammation as a central mechanism in Alzheimer's disease. Alzheimers Dement 4: 575–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuzy A, Cicognola C, Chiotis K, Saint‐Aubert L, Lemoine L, Andreasen N, Zetterberg H, Ye K, Blennow K, Höglund Ket al (2019) Longitudinal tau and metabolic PET imaging in relation to novel CSF tau measures in Alzheimer's disease. Eur J Nucl Med Mol Imaging 46: 1152–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Strohmeyer R, Liang Z, Lue LF, Rogers J (2004) CCAAT/enhancer binding protein delta (C/EBPdelta) expression and elevation in Alzheimer's disease. Neurobiol Aging 25: 991–999 [DOI] [PubMed] [Google Scholar]
- Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, Duff K (2012) Trans‐synaptic spread of tau pathology in vivo . PLoS One 7: e31302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm VM, Zhang B, O'Brien P, Trojanowski JQ, Lee VM (2012) Intracerebral inoculation of pathological alpha‐synuclein initiates a rapidly progressive neurodegenerative alpha‐synucleinopathy in mice. J Exp Med 209: 975–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukiw WJ (2004) Gene expression profiling in fetal, aged, and Alzheimer hippocampus: a continuum of stress‐related signaling. Neurochem Res 29: 1287–1297 [DOI] [PubMed] [Google Scholar]
- Maesako M, Uemura K, Kubota M, Kuzuya A, Sasaki K, Hayashida N, Asada‐Utsugi M, Watanabe K, Uemura M, Kihara Tet al (2012) Exercise is more effective than diet control in preventing high fat diet‐induced beta‐amyloid deposition and memory deficit in amyloid precursor protein transgenic mice. J Biol Chem 287: 23024–23033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magalini A, Savoldi G, Ferrari F, Garnier M, Ghezzi P, Albertini A, Di Lorenzo D (1995) Role of IL‐1 beta and corticosteroids in the regulation of the C/EBP‐alpha, beta and delta genes in vivo . Cytokine 7: 753–758 [DOI] [PubMed] [Google Scholar]
- Mahley RW (2016) Apolipoprotein E: from cardiovascular disease to neurodegenerative disorders. J Mol Med 94: 739–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques F, Sousa JC, Sousa N, Palha JA (2013) Blood‐brain‐barriers in aging and in Alzheimer's disease. Mol Neurodegener 8: 38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill CA, Jonsson MAG, Minthon L, Ejnell H, Silander H‐S, Blennow K, Karlsson M, Nordlund A, Rolstad S, Warkentin Set al (2006) Vagus nerve stimulation in patients with Alzheimer's disease: Additional follow‐up results of a pilot study through 1 year. J Clin Psychiatry 67: 1171–1178 [DOI] [PubMed] [Google Scholar]
- Meyer‐Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton ALet al (2006) Exogenous induction of cerebral beta‐amyloidogenesis is governed by agent and host. Science 313: 1781–1784 [DOI] [PubMed] [Google Scholar]
- Mezö C, Dokalis N, Mossad O, Staszewski O, Neuber J, Yilmaz B, Schnepf D, de Agüero MG, Ganal‐Vonarburg SC, Macpherson AJet al (2020) Different effects of constitutive and induced microbiota modulation on microglia in a mouse model of Alzheimer's disease. Acta Neuropathol Commun 8: 119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minter MR, Hinterleitner R, Meisel M, Zhang C, Leone V, Zhang X, Oyler‐Castrillo P, Zhang X, Musch MW, Shen Xet al (2017) Antibiotic‐induced perturbations in microbial diversity during post‐natal development alters amyloid pathology in an aged APPSWE/PS1DeltaE9 murine model of Alzheimer's disease. Sci Rep 7: 10411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monson NL, Ireland SJ, Ligocki AJ, Chen D, Rounds WH, Li M, Huebinger RM, Munro Cullum C, Greenberg BM, Stowe AMet al (2014) Elevated CNS inflammation in patients with preclinical Alzheimer's disease. J Cereb Blood Flow Metab 34: 30–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagne A, Barnes S, Sweeney M, Halliday M, Sagare A, Zhao Z, Toga A, Jacobs R, Liu C, Amezcua Let al (2015) Blood‐brain barrier breakdown in the aging human hippocampus. Neuron 85: 296–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olanow CW, Wakeman DR, Kordower JH (2014) Peripheral alpha‐synuclein and Parkinson's disease. Mov Disord 29: 963–966 [DOI] [PubMed] [Google Scholar]
- Papathanasiou A, Nikakis P, Bonakis A, Kilidireas K, Dimitrakopoulos A, Michopoulos S, Kalfakis N, Papageorgiou SG (2014) Rapidly progressive dementia as presenting feature in inflammatory bowel disease. Alzheimer Dis Assoc Disord 28: 294–295 [DOI] [PubMed] [Google Scholar]
- Poli V (1998) The role of C/EBP isoforms in the control of inflammatory and native immunity functions. J Biol Chem 273: 29279–29282 [DOI] [PubMed] [Google Scholar]
- Reichardt F, Chassaing B, Nezami BG, Li GE, Tabatabavakili S, Mwangi S, Uppal K, Liang B, Vijay‐Kumar M, Jones Det al (2017) Western diet induces colonic nitrergic myenteric neuropathy and dysmotility in mice via saturated fatty acid‐ and lipopolysaccharide‐induced TLR4 signalling. J Physiol 595: 1831–1846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotermund C, Truckenmuller FM, Schell H, Kahle PJ (2014) Diet‐induced obesity accelerates the onset of terminal phenotypes in alpha‐synuclein transgenic mice. J Neurochem 131: 848–858 [DOI] [PubMed] [Google Scholar]
- Saksida T, Koprivica I, Vujicic M, Stosic‐Grujicic S, Perovic M, Kanazir S, Stojanovic I (2018) Impaired IL‐17 production in gut‐residing immune cells of 5xFAD mice with Alzheimer's disease pathology. J Alzheimers Dis 61: 619–630 [DOI] [PubMed] [Google Scholar]
- Sanders D, Kaufman S, DeVos S, Sharma A, Mirbaha H, Li A, Barker S, Foley A, Thorpe J, Serpell Let al (2014) Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82: 1271–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz K, Boles BR (2013) Microbial amyloids–functions and interactions within the host. Curr Opin Microbiol 16: 93–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo DO, Holtzman DM (2019) Gut microbiota: from the forgotten organ to a potential key player in the pathology of Alzheimer disease. J Gerontol A Biol Sci Med Sci 75: 1232–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirahama‐Noda K, Yamamoto A, Sugihara K, Hashimoto N, Asano M, Nishimura M, Hara‐Nishimura I (2003) Biosynthetic processing of cathepsins and lysosomal degradation are abolished in asparaginyl endopeptidase‐deficient mice. J Biol Chem 278: 33194–33199 [DOI] [PubMed] [Google Scholar]
- Shoemark DK, Allen SJ (2015) The microbiome and disease: reviewing the links between the oral microbiome, aging, and Alzheimer's disease. J Alzheimers Dis 43: 725–738 [DOI] [PubMed] [Google Scholar]
- Sjogren MJC, Hellstrom PTO, Jonsson MAG, Runnerstam M, C‐son Silander H, Ben‐Menachem E, Sjögren JC, Hellström PTO (2002) Cognition‐enhancing effect of vagus nerve stimulation in patients with Alzheimer's disease: a pilot study. J Clin Psychiatry 63: 972–980 [DOI] [PubMed] [Google Scholar]
- Sochocka M, Donskow‐Lysoniewska K, Diniz BS, Kurpas D, Brzozowska E, Leszek J (2019) The gut microbiome alterations and inflammation‐driven pathogenesis of Alzheimer's disease‐a critical review. Mol Neurobiol 56: 1841–1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straccia M, Gresa‐Arribas N, Dentesano G, Ejarque‐Ortiz A, Tusell JM, Serratosa J, Sola C, Saura J (2011) Pro‐inflammatory gene expression and neurotoxic effects of activated microglia are attenuated by absence of CCAAT/enhancer binding protein beta. J Neuroinflammation 8: 156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohmeyer R, Shelton J, Lougheed C, Breitkopf T (2014) CCAAT‐enhancer binding protein‐beta expression and elevation in Alzheimer's disease and microglial cell cultures. PLoS One 9: e86617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed AK, Boles BR (2014) Fold modulating function: bacterial toxins to functional amyloids. Front Microbiol 5: 401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenfeld SM, Milekic MH, Monti B, Alberini CM (2001) The consolidation of new but not reactivated memory requires hippocampal C/EBPbeta. Nat Neurosci 4: 813–818 [DOI] [PubMed] [Google Scholar]
- Ulusoy A, Rusconi R, Perez‐Revuelta BI, Musgrove RE, Helwig M, Winzen‐Reichert B, Di Monte DA (2013) Caudo‐rostral brain spreading of alpha‐synuclein through vagal connections. EMBO Mol Med 5: 1119–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JM, Dixit S, Saulsberry AC, May JM, Harrison FE (2017) Reversal of high fat diet‐induced obesity improves glucose tolerance, inflammatory response, beta‐amyloid accumulation and cognitive decline in the APP/PSEN1 mouse model of Alzheimer's disease. Neurobiol Dis 100: 87–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LC (2018) Prion‐like mechanisms in Alzheimer disease. Handb Clin Neurol 153: 303–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Liu X, Chen S, Ye K (2018a) Spatiotemporal activation of the C/EBPbeta/delta‐secretase axis regulates the pathogenesis of Alzheimer's disease. Proc Natl Acad Sci USA 115: E12427–E12434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Balaji V, Kaniyappan S, Krüger L, Irsen S, Tepper K, Chandupatla RR, Maetzler W, Schneider A, Mandelkow Eet al (2017) The release and trans‐synaptic transmission of Tau via exosomes. Mol Neurodegener 12: 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZH, Gong K, Liu X, Zhang Z, Sun X, Wei ZZ, Yu SP, Manfredsson FP, Sandoval IM, Johnson PFet al (2018b) C/EBPbeta regulates delta‐secretase expression and mediates pathogenesis in mouse models of Alzheimer's disease. Nat Commun 9: 1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Kang SS, Liu X, Ahn EH, Zhang Z, He L, Iuvone PM, Duong DM, Seyfried NT, Benskey MJet al (2017) Asparagine endopeptidase cleaves alpha‐synuclein and mediates pathologic activities in Parkinson's disease. Nat Struct Mol Biol 24: 632–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Song M, Liu X, Kang SS, Kwon I‐S, Duong DM, Seyfried NT, Hu WT, Liu Z, Wang J‐Zet al (2014) Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer's disease. Nat Med 20: 1254–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Song M, Liu X, Su Kang S, Duong DM, Seyfried NT, Cao X, Cheng L, Sun YE, Ping Yu Set al (2015) Delta‐secretase cleaves amyloid precursor protein and regulates the pathogenesis in Alzheimer's disease. Nat Commun 6: 8762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Dua P, Lukiw WJ (2015) Microbial sources of amyloid and relevance to Amyloidogenesis and Alzheimer's disease (AD). J Alzheimers Dis Parkinsonism 5: 177 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Review Process File
Appendix
Source Data for Expanded View and Appendix
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
This study includes no data deposited in external repositories.