

Abstract
Gut microbiota produce Trimethylamine N-oxide (TMAO) by metabolizing dietary phosphatidylcholine, choline, l-carnitine and betaine. TMAO is implicated in the pathogenesis of chronic kidney disease (CKD), diabetes, obesity and atherosclerosis. We test, whether TMAO augments angiotensin II (Ang II)-induced vasoconstriction and hence promotes Ang II-induced hypertension. Plasma TMAO levels were indeed elevated in hypertensive patients, thus the potential pathways by which TMAO mediates these effects were explored. Ang II (400 ng/kg−1min−1) was chronically infused for 14 days via osmotic minipumps in C57Bl/6 mice. TMAO (1%) or antibiotics were given via drinking water. Vasoconstriction of renal afferent arterioles and mesenteric arteries were assessed by microperfusion and wire myograph, respectively. In Ang II-induced hypertensive mice, TMAO elevated systolic blood pressure and caused vasoconstriction, which was alleviated by antibiotics. TMAO enhanced the Ang II-induced acute pressor responses (12.2 ± 1.9 versus 20.6 ± 1.4 mmHg; P < 0.05) and vasoconstriction (32.3 ± 2.6 versus 55.9 ± 7.0%, P < 0.001). Ang II-induced intracellular Ca2+ release in afferent arterioles (147 ± 7 versus 234 ± 26%; P < 0.001) and mouse vascular smooth muscle cells (VSMC, 123 ± 3 versus 157 ± 9%; P < 0.001) increased by TMAO treatment. Preincubation of VSMC with TMAO activated the PERK/ROS/CaMKII/PLCβ3 pathway. Pharmacological inhibition of PERK, ROS, CaMKII and PLCβ3 impaired the effect of TMAO on Ca2+ release. Thus, TMAO facilitates Ang II-induced vasoconstriction, thereby promoting Ang II-induced hypertension, which involves the PERK/ROS/CaMKII/PLCβ3 axis.
Keywords: Trimethylamine N-oxide, Angiotensin II, Blood pressure, Afferent arteriole, Calcium
Graphical abstract
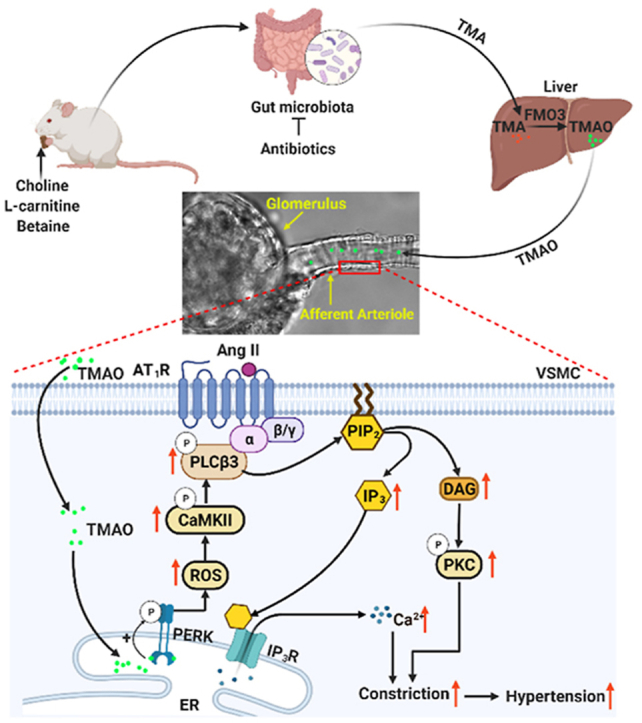
Highlights
-
•
Orally administered TMAO aggravates Ang II-induced hypertension. Antibiotics alleviate Ang II-induced hypertension by reducing TMAO generation.
-
•
High concentrations of TMAO constrict afferent arterioles and mesenteric arteries and increase blood pressure.
-
•
Low concentrations of TMAO enhance Ang II-induced vasoconstriction and acute pressor response via activating PERK/ROS/CaMKII/PLCβ3/Ca2+ pathway.
1. Introduction
More than 30% (1.38 billion) of the global adult population suffer from hypertension [1], a major preventable risk factor for cardiovascular disease (CVD), and the leading cause of death worldwide. A better understanding of the underlying mechanisms causing hypertension is required for preventing and treating high blood pressure [2]. Angiotensin II (Ang II), is a principle component of the renin-angiotensin-aldosterone system, which plays a critical role in hypertension [3]. The kidneys determine ANG II formation and controls plasma volume, making this organ a key players in hypertension. In the kidney, afferent arterioles (Af) constitute the major resistance vessels [4]. By their constriction, Ang II contributes to blood pressure and glomerular filtration rate (GFR) regulation [4,5].
Certain forms of hypertension are linked to gut microbiota [[6], [7], [8]]. For instance, gut microbiota composition differs between normotensive controls and various genetic (SHR and Dahl salt-sensitive) and pharmacological (Ang II and DOCA-salt) hypertension models [[9], [10], [11], [12], [13]]. What is more, Ang II-induced vascular dysfunction and hypertension are mitigated in germ-free (GF) mice (gnotobiotic) [10]. Accordingly, the lower blood pressure and reduced vascular contractility observed in GF rats is restored by introducing microbiota to these GF rats [14]. In the light of these studies, it does not surprise that dietary interventions to alter gut microbiota can modulate blood pressure [13]. Yet, whether gut microbiota affects blood pressure via a certain metabolite, Trimethylamine N-oxide (TMAO), is unknown.
Choline, l-carnitine, phosphatidylcholine and betaine are metabolized by gut microbiota to generate Trimethylamine (TMA), which is rapidly oxidized by hepatic flavin-containing monooxygenase-3 (FMO3) to form TMAO in the liver [15,16]. TMAO may also be obtained directly from foods [16]. Remarkably, plasma TMAO concentration is closely associated with atherosclerosis [15], obesity [17] and chronic kidney disease [18].
Direct targets of TMAO have attracted much attention [19]. Chen et al. demonstrate that TMAO directly binds and activates protein kinase R-like endoplasmic reticulum kinase (PERK) and, thus, promotes metabolic dysfunction [20]. TMAO may also augment platelet intracellular Ca2+ ([Ca2+]i) release via elevation of inositol 1,4,5-triphosphate (IP3) generation [21]. Interestingly, aging and chronic kidney disease (CKD) animal models have higher circulating TMAO levels than their control strains.
TMAO impairs endothelial nitric oxide synthase (eNOS)-derived nitric oxide (NO) production, and reduces endothelium-dependent vasorelaxation of the aorta in response to acetylcholine [22,23]. Whether activation of PERK by TMAO affects Ang II-induced vasoconstriction via enhancing [Ca2+]i release is investigated here, where we test whether a physiological concentration of TMAO augments Ang II–induced intracellular Ca2+ release via activating PERK that enhances vasoconstriction of Af and mesenteric arteries. Furthermore, we explore whether depleting gut microbiota reduces circulating TMAO levels to reduce blood pressure and Ang II-vasoconstriction.
2. Methods
All data to support the findings of this study are available from the corresponding author upon reasonable request. A detailed section of methods and materials is provided in the Data Supplement.
2.1. Animals and experimental model
Adult C57Bl/6 mice, male, 25–28 g, were obtained from SLAC laboratory animal company (Shanghai, China). Animals were housed under climate-controlled conditions with a 12-h light/dark cycle and provided with standard laboratory chow and tap water. All procedures and experimental protocols were approved by the Institute Animal Care and Ethical Committee of Zhejiang University School of Medicine.
Osmotic pumps (Alzet Model 2002, CA, USA) were subcutaneously implanted into mice to deliver either saline or Ang II (400 ng/kg−1min−1) for 2 weeks [24]. TMAO (1%, Trimethylamine N-oxide dihydrate, ACROS Organics, Geel, Belgium) and an antibiotics cocktail (0.5 g/L vancomycin, 1 g/L neomycin sulfate, 1 g/L metronidazole, 1 g/L ampicillin) were administered via drinking water [15,25]. Vancomycin was purchased from Meryer Chemical Technology (Shanghai, China), Neomycin sulfate was purchased from Macklin (Shanghai, China), Metronidazole and Ampicillin were purchased from Aladdin (Shanghai, China). Drinking water was replenished thrice a week. After treating mice with TMAO for 14 days or antibiotics for 7 days, the osmotic pumps were implanted. Systolic blood pressure was measured by non-invasive tail-cuff (Visitech Systems BP-2000, NC, USA) during 14 days of Ang II infusion.
2.2. Human plasma TMAO analysis
The study was approved by the Ethics Committee of the Second Affiliated Hospital of Zhejiang University School of Medicine. Subjects were aged between 18 and 80 years. A total of 69 humans were assigned to the hypertensive or normotensive group. Subjects were allocated to the hypertensive group (37) if they presented with systolic blood pressure >140 mmHg and diastolic blood pressure >90 mmHg, and not under antihypertensive treatment. Subjects will be excluded if they presented with severe hypertension (blood pressure >180/120 mmHg). Healthy individuals were recruited from the physical examination center for the normotensive group (32) at the same time.
Only one freeze-thaw cycle was allowed for all samples in the process. Plasma TMAO levels and SOD activity were assessed with the Human TMAO assay Kit (Jianglai Institute of Biotechnology, Shanghai, China) and Total Superoxide Dismutase Assay Kit (Beyotime Biotechnology, Shanghai, China), following the manufacturer's instructions.
2.3. Renal afferent arteriolar microperfusion
Isolation and microperfusion procedures have been described before [[26], [27], [28], [29]]. In brief, mice were anesthetized with isoflurane and kidneys were removed and sliced along the corticomedullary axis. Slices were placed in ice-cold DMEM with or without 250 μM TMAO and dissected under a stereomicroscope. A single Af with attached glomerulus was isolated and then transferred to a temperature-controlled chamber on the stage of an inverted microscope, and perfused using a micromanipulator system with concentric holding and perfusion pipettes. Once the Af was successfully perfused, 100 mM KCl was added into the bath solution to assess vascular function and then washout. The perfusion pressure was maintained at 60 mmHg. The chamber was perfused with 37 °C DMEM to maintain the temperature. Only one Af was used from each mouse and 5–8 mice were used for each group and all the experiments were finished within 2 h after the mice were killed. The dose-response curve of Af to Ang II was obtained after 1 h equilibration. TMAO and/or PERK inhibitor GSK2606414 (2 μM), ROS scavenger Tempol (100 μM), CaMKII inhibitor KN-93 (1 μM), PLC inhibitor U73122 (10 μM), AT1 inhibitor ZD7155 (0.1 μM) or AT2 inhibitor PD123319 (10 μM) were added to bath solution before equilibration period and existed during the whole experiment. All inhibitors and scavenger used for microperfusion were purchased from MedChemExpress (NJ, USA).
2.4. Mesenteric arterial reactivity
Reactivity of mouse mesenteric arteries were studied by using a wire myograph, as previously described [28]. Second branch mesenteric artery from adult male C57Bl/6 mice were administered saline, Ang II, TMAO or antibiotics, were cleaned of fat and connective tissue and then cut into rings (2 mm in length) in ice-cold Krebs-Henseleit solution. The arteries were mounted to the wire myograph system and resting tension was set according to the manufacturer's protocol. Then the mesenteric artery was equilibrated for 60 min at 37 °C before experiments. Subsequently, the mesenteric artery was challenged three times with a high-potassium solution (KPSS, PSS with 80 mM KCl) to verify vascular reactivity and then washed out. Next, mesenteric arteries were pretreated with or without PLC inhibitor U μM) for 30 min. Cumulative dose response curves of mesenteric arteries to Ang II and phenylephrine (PE) were obtained.
2.5. Pressor response to acute administration of Ang II
Mice were anesthetized with inhaled isoflurane placed on a temperature-controlled operating table to maintain body temperature at 37.0 ± 1.0 °C, as described previously [27]. Then, the right carotid artery and left jugular vein were isolated. A catheter (PE-10) was inserted into the right carotid artery for blood pressure measurement with a PowerLab (ADInstruments, CO, USA) and another catheter was inserted into the left jugular vein for administration of TMAO, Tempol, KN-93 or Ang II dissolved by saline. TMAO, Tempol and KN-93 were given 30 min before Ang II treatment.
2.6. Statistical analyses
Statistical analyses were performed using Prism 8. One-way ANOVA or two-way ANOVA followed by Dunnett's post hoc test or Turkey's post hoc test, respectively (when multiple groups were compared). 2-tailed Student's t-test was performed when only two groups were compared. Data are presented as a mean ± SEM, and a P < 0.05 is considered statistically significant.
3. Results
3.1. TMAO aggravates Ang II–induced hypertension and impairs renal GFR
In mice, there were no significant differences in systolic blood pressure (SBP) among all groups at baseline. TMAO or antibiotics treatment had marginal effects on SBP. In the Ang II–infused mice, hypertension was aggravated by treatment with TMAO, but ameliorated by the supplementation of antibiotics. SBP was measured by tail-cuff (Fig. 1A). GFR gradually decreased from 239 ± 14 μl/min at baseline to 193 ± 6 μl/min at month two and 158 ± 16 μl/min at month six in mice treated with TMAO alone (Fig. 1B). To explore a relationship between circulating TMAO and Ang II-induced hypertension, we measured plasma TMAO and Ang II levels in all groups of mice. In Ang II infused mice, plasma Ang II level was higher than that in saline infused mice (Fig. S1A). In TMAO treated mice, plasma TMAO level was higher than that in control mice. Treatment of mice with antibiotics significantly reduced plasma TMAO levels (Fig. S1B). However, hepatic flavin-containing monooxygenase 3 (FMO3) expression was not significantly different among all groups (Fig. S1C). In mice treated with TMAO for six months, serum Blood Urea Nitrogen (BUN) and renal fibrosis were upregulated, but serum creatinine has no obvious change (Fig. S2). Overall, these data indicate that TMAO promotes Ang II-induced hypertension, impairs renal GFR, and leads to renal injury. Knockdown of endogenous gut microbiota by antibiotic cocktail reduces TMAO generation, which ameliorates Ang II-induced hypertension.
Fig. 1.

Chronic administration of TMAO aggravates angiotensin II (Ang II)-induced hypertension and impairs renal glomerular filtration rate (GFR). (A) Systolic blood pressure (SBP) in mice delivered saline, Ang II (400 ng/kg−1min−1), TMAO (1%) and/or an antibiotic cocktail (n = 4–7; *P < 0.05 vs Control, #P < 0.05 vs Ang II). (B) Time course of glomerular filtration rate (GFR) in mice treated with TMAO (n = 5–6; *P < 0.05 vs 0). Statistical differences were calculated by two-way ANOVA followed by Dunnett's post hoc test (A), one-way ANOVA followed by Turkey's post hoc test (B).
3.2. TMAO exacerbates Chronic Ang II infusion induced vascular dysfunction in afferent arterioles and mesenteric arteries
Vasoconstriction in the mesenteric artery or afferent arteriole (Af) were assessed by myograph or microperfusion, respectively. In TMAO-treated mice, vasoconstrictions to Ang II were significantly enhanced in Af (Fig. 2B) and mesenteric arteries (Fig. 2C) compared with vehicle-treated mice. Interestingly, in Ang II-infused mice treated with TMAO, vasoconstrictions to Ang II were also enhanced in mesenteric artery and Af when compared to Ang II-infused mice treated with vehicle. Chronic Ang II infusion did not affect the vasoconstriction to Ang II in the mesenteric artery from vehicle-treated mice, but in Af from the same mice, Ang II-induced vasoconstriction was enhanced by chronic Ang II infusion. In addition, when Ang II-infused mice were treated with antibiotics, the enhancement of Ang II-induced vasoconstriction in Af was inhibited. Pretreatment with phospholipase C (PLC) inhibitor U73122 completely blocked the Ang II-induced vasoconstriction in mesenteric artery in all groups of mice (Fig. 2 D). There was no difference in phenylephrine (PE)-induced vasoconstriction in mesenteric artery in all groups of mice (Fig. 2E). The vasoconstriction of mesenteric artery in response to PE was partially (≈30%) inhibited by U73122 (Fig. 2F). Taken together, these results indicate that chronic administration of TMAO via drinking water enhances Ang II-induced vasoconstriction via PLC.
Fig. 2.

Chronic administration of TMAO augments Ang II-induced vasoconstriction of mesenteric artery and afferent arterioles via activation of phospholipase C (PLC). Vasoconstriction in afferent arteriole (Af) or mesenteric artery were assessed by microperfusion or wire myograph. (A) Representative images of the isolated perfused Af before (Basal) and during administration of 10−9 and 10−6 mol/L Ang II. Concentration-response curves to Ang II of Af (B) or mesenteric artery (C) from TMAO, Antibiotics or vehicle-treated mice infused with saline or Ang II (n = 4–14; *P < 0.05 vs Control, #P < 0.05 vs Ang II, $P < 0.05 vs Ang II+ Antibiotics). (D) Mesenteric artery response to Ang II in the presence of PLC inhibitor U73122 (n = 8). (E) Mesenteric artery contraction response to phenylephrine (PE). (F) Effect of U73122 on PE induced vasoconstriction of mesenteric artery (n = 6). Statistical differences were calculated by two-way ANOVA followed by Dunnett's post hoc test (B–F).
3.3. Acute administration of TMAO increases the pressor response to Ang II and AT1R-Dependent vessel contraction
To determine the direct effect of TMAO on blood pressure and vascular tone, we used different concentrations of TMAO to treat mice or blood vessel. A PE-10 catheter was inserted into the right carotid artery, and the blood pressure changes were recorded under anesthesia. Low concentrations of TMAO do not affect blood pressure, however, high concentrations of TMAO directly increase blood pressure (Fig. 3A). To examine the effect of TMAO on vascular tone, we used isolated renal Af and mesenteric artery. Low concentrations of TMAO do not affect vascular tone, however, high concentrations of TMAO directly contract Af and mesenteric artery (Fig. 3B and C). In another experiment, 5 min after intubation, Ang II (250 ng/kg, 2 μl/g) was first applied intravenously. There is no difference in acute pressor responses to Ang II between the two groups of mice during the first application. Ten minutes after intubation, TMAO (0.3 mg/kg, 2 μl/g) or saline was administered intravenously, the pressor response to the second application of Ang II was significantly increased as compared with saline group (12.2 ± 1.9 vs 20.6 ± 1.4 mmHg; P < 0.05 versus NaCl). The interval between the two intravenous administrations of Ang II was 25 min (Fig. 3D). Af pretreatment with 250 μM TMAO for 1 h significantly increased maximum vasoconstriction in response to 0.1 μM Ang II (Fig. 3E). Ang II type 1 receptor (AT1R) antagonist ZD7155 blocked the effect of TMAO on Ang II-induced constriction in Af (Fig. S3A). Conversely, Ang II type 2 receptor (AT2R) antagonist PD123319 did not significantly influence TMAO-induced increase in vasoconstriction to Ang II (Fig. S3B). These findings suggest that the pressor response to Ang II is enhanced by TMAO, accompanied by the augmentation of Ang II-induced vasoconstriction of Af via AT1R.
Fig. 3.

Acute administration of TMAO increases pressor response and vasoconstriction to Ang II. (A) Direct effect of TMAO on SBP (n = 4–5; *P < 0.05 vs NaCl). (B–C) Direct effect of TMAO on vascular tone in Af and mesenteric artery (n = 4–5; *P < 0.05 vs 0). (D) The changes of SBP during two intravenous bolus injections of 0.25 μg/kg Ang II. Five minutes after the first injection of Ang II, TMAO or saline was administered (n = 6–7; *P < 0.05 vs NaCl). (E) The changes of Ang II caused vasoconstriction in the absence or presence of TMAO (n = 6–7, *P < 0.05 vs Control). Statistical differences were calculated by two-way ANOVA followed by Dunnett's post hoc test (A, D and E), one-way ANOVA followed by Turkey's post hoc test (B–C). 1st, First Application; 2nd, Second Application.
3.4. TMAO activates the PERK/elF2α/chop/ERO1-α pathway
Since TMAO binds and activates Protein Kinase R-like Endoplasmic Reticulum Kinase (PERK) [20], we tested whether activation of PERK by TMAO is involved in the observed elevation of Ang II-induced vasoconstriction. Treatment with 250 μM TMAO for 1 h enhanced phosphorylation of PERK in Af detected by immunofluorescence (Fig. 4A). Immunohistochemical revealed that the expression of p-PERK was also enhanced in Af from mice treated with TMAO for six months (Fig. S4).
Fig. 4.

TMAO activates the PERK/elF2α/Chop/ERO1-α pathway. (A) Effect of 1 h preincubation with TMAO (250 μM) on the phosphorylation of PERK in Af. (B–C) The levels of p-PERK, PERK, p-elF2α, elF2α, CHOP and ERO1-α were detected by western blotting after incubation of mouse vascular smooth muscle cell (VSMC) with various concentrations of TMAO for 24 h or a fixed concentration of TMAO (100 μM) for different time periods (n = 3; *P < 0.05 vs 0). Statistical differences were calculated by one-way ANOVA followed by Turkey's post hoc test (B–C).
In addition, VSMC were treated with various concentrations of TMAO (0.025–10 mM) for 24 h. The phosphorylation levels of PERK and its downstream target eIF2α were enhanced by TMAO. The total protein levels of CHOP and ERO1-α were also augmented by TMAO (Fig. 4B). A time-dependent increase in p-PERK, p-eIF2α, CHOP and ERO1-α levels were observed when VSMC were treated with 100 μM TMAO for various durations (Fig. 4C).
3.5. Overproduction of ROS contributes to the potentiating effects of TMAO on Ang II response
It is well known that activation of PERK/eIF2α/CHOP/ERO1-α pathway results in overproduction of ROS [30,31], which can effectively facilitate Ang II-induced hypertension and vasoconstriction [5]. Therefore, we next assessed the role of ROS in the TMAO-evoked enhancement of Ang II-induced vasoconstriction and pressor response. ROS scavenger Tempol blocked TMAO induced strengthening of the pressor response to an acute administration of Ang II. Right carotid artery was catheterized to detect blood pressure under anesthesia (Fig. 5A). Tempol blocked the TMAO-evoked enhancement of Ang II-induced vasoconstriction in Af (Fig. 5B). Treatment with TMAO (250 μM, 1 h) or CCT020312 (a selective PERK activator; 10 μM, 1 h), increased cytoplasmic and mitochondrial superoxide generation in Af (Fig. 5C and D). In separate sets of experiments, cytoplasmic and mitochondrial superoxide levels increased by incubation of VSMC with TMAO (100 μM, 24 h). These effects were inhibited by PERK inhibitor GSK2606414 (2 μM, 24 h) and mitochondria ROS scavenger MitoTempo (10 μM, 24 h). However, cytoplasmic ROS scavenger Tempol (100 μM, 24 h) only blocked the TMAO effects on cytoplasmic superoxide. We also observed that Ang II alone upregulated cytoplasmic and mitochondrial superoxide levels; these effects were further enhanced by pre-incubation of VSMC with TMAO (Fig. 5E). The oxidative stress markers, including SOD activity, Catalase activity, MDA and superoxide levels in kidney tissue from mice treated with TMAO for six months were assessed by commercial kits or DHE staining, respectively. The results showed that TMAO promotes oxidative stress in kidney (Figs. S5 and S6). These data demonstrate that activation of PERK by TMAO greatly enhances ROS generation, which augments Ang II-induced vasoconstriction and its pressor response.
Fig. 5.

Reactive oxygen species (ROS) are involved in regulating the effect of TMAO on Ang II-induced pressor responses and vasoconstriction. The changes of the Ang II-induced pressor response (A) and vasoconstriction (B) in the presence of cytoplasmic ROS scavenger Tempol or Tempol+TMAO (n = 4; *P < 0.05 vs NaCl+Tempol or Control+Tempol). (C–D) Afs were incubated with TMAO (250 μM) or CCT020312 (10 μM) for 1 h and then cytoplasmic superoxide or mitochondrial superoxide were measured by staining with DHE or MitoSOX, respectively (n = 3–4; *P < 0.05 vs Control). (E) Changes in cytoplasmic superoxide and mitochondrial superoxide levels in VSMC incubated with TMAO, PERK inhibitor GSK, mitochondrial ROS scavenger MitoTempo, cytoplasmic ROS scavenger Tempol or Ang II (n = 6–18; *P < 0.05 vs Control, $P < 0.05 vs Control+Tempol, #P < 0.05 vs Control+Ang II). Statistical differences were calculated by two-way ANOVA followed by Dunnett's post hoc test (A–B), one-way ANOVA followed by Turkey's post hoc test (C–E). 1st, First Application; 2nd, Second Application; GSK, GSK2606414; CCT, CCT020312.
3.6. Activation of calmodulin-dependent protein kinase II (CaMKII) by ROS is involved in the potentiating effects of TMAO on Ang II responses
Excessive ROS oxidizes CaMKII at Met-281/282 (Ox-CaMKII) and phosphorylates CaMKII (p-CaMKII) at Thr-286, which enhances the activity of CaMKII [32,33]. Inhibition of CaMKII activity reduces endothelin-1 and Ang II-induced vasoconstriction [34,35]. To determine whether CaMKII is involved in TMAO-induced Ang II response strengthening, an inhibitor of CaMKII, KN-93, was used. KN-93 blocked TMAO enhanced vasoconstriction and the pressor response to Ang II. Blood pressure was continuously recorded under anesthesia by right carotid arterial cannula (Fig. 6A and B). Moreover, oxidation and phosphorylation of CaMKII (Ox-CaMKII and p-CaMKII) were increased in VSMC treated with TMAO (Fig. 6C). VSMC exposed to 100 μM TMAO for various durations also enhanced levels of Ox-CaMKII and p-CaMKII (Fig. 6D). GSK2606414, Tempol and KN-93 blocked the TMAO-induced enhancement of Ox-CaMKII and p-CaMKII expression (Fig. S7). Taken together, these data suggest that TMAO enhanced Ang II response is mediated via CaMKII, which can be activated by excessive ROS generation.
Fig. 6.

Calmodulin-dependent protein kinase II (CaMKII) is involved in regulating the effect of TMAO on Ang II-induced pressor response and vasoconstriction. (A–B) The changes of Ang II-induced pressor response and vasoconstriction in the continuous presence of CaMKII inhibitor KN-93 or KN-93 + TMAO (n = 4). (C–D) The levels of Ox-CaMKII, p-CaMKII and CaMKII were detected by western blotting after incubation of VSMC with various concentrations of TMAO for 24 h or a fixed concentration of TMAO (100 μM) for different time periods (n = 3; *P < 0.05 vs 0). Statistical differences were determined by two-way ANOVA followed by Dunnett's post hoc test (A–B), one-way ANOVA followed by Turkey's post hoc test (C–D). 1st, First Application; 2nd, Second Application; GSK, GSK2606414.
3.7. Phosphorylation and activation of PLCβ3 by CaMKII in the potentiating effects of TMAO on Ang II response
CaMKII phosphorylates and activates phospholipase C β3 (PLCβ3) at Ser-537 [36,37]. In human platelets, pre-incubation with TMAO augments inositol-1,4,5-trisphosphate (IP3) generation, which is produced by various PLC isoforms and triggers intracellular Ca2+ release [21]. There was no significant effect of TMAO on Ang II-induced vasoconstriction when Af was pretreated with PLC inhibitor U73122 (Fig. 7A). In VSMC, Ang II-induced PLC activation and IP3 formation were augmented by TMAO treatment (Fig. 7B and C). Furthermore, TMAO progressively increased the phosphorylation levels of PLCβ3 (p-PLCβ3) and PKC-pan (p-PKC-pan) in a dose- and time-dependent manner (Fig. 7D and E). GSK2606414, Tempol, KN-93 and U73122 inhibited TMAO-induced enhancement of p-PLCβ3 expression (Fig. S8). Thus, TMAO activates PLCβ3 through the PERK/ROS/CaMKII pathway. Activated PLCβ3 mediates Ang II-induced vasoconstriction of Af.
Fig. 7.

Activation of the PLCβ3/IP3/Ca2+pathway by TMAO promotes Ang II-induced vasoconstriction. (A) Effect of U73122 treatment on Ang II-induced vasoconstriction in the absence or presence of TMAO (n = 4; *P < 0.05 vs Control+U73122). (B–C) Effect of TMAO on Ang II-induced PLC activation and IP3 formation in VSMC (n = 6–12; *P < 0.05 vs Control, #P < 0.05 vs Control+Ang II). (D–E) The levels of p-PLCβ3 and p-PKC were detected by western blotting after incubation of VSMC with various concentrations of TMAO for 24 h or a fixed concentration of TMAO for different time periods (n = 3; *P < 0.05 vs 0). Statistical differences were calculated by two-way ANOVA followed by Dunnett's post hoc test (A), one-way ANOVA followed by Turkey's post hoc test (B–E).
3.8. TMAO promotes Ang II-Triggered intracellular Ca2+ release
Fig. 8A shows representative images of Ang II-triggered intracellular Ca2+ changes in isolated perfused Af. Pre-incubation with TMAO (250 μM) for 1 h significantly increased Ang II-induced release of Ca2+ in Af, which was blocked by U73122 and Tempol. Similar results were observed in VSMC treated with TMAO. GSK2606414, Tempol, KN-93, U73122 and BAPTA-AM (intracellular Ca2+ chelator) blocked TMAO-evoked augmentation of Ang II-induced Ca2+ mobilization in VSMC (Fig. 8B). Notably, TMAO enhanced Ang II-triggered intracellular Ca2+ release through PERK/ROS/CaMKII/PLCβ3 axis, which was further conformed in mice treated with TMAO for six months (Figs. S9 and S10).
Fig. 8.

TMAO promotes Ang II-evoked intracellular Ca2+ release. (A) Representative images of the isolated perfused Afs loaded with fluo-4 AM for measurement of intracellular Ca2+. In the presence of TMAO, Ang II triggered intracellular Ca2+ release was significantly increased, which was blocked by U73122 and Tempol (n = 3–5; *P < 0.05 vs Control). (B) Representative images and summarized data showing that TMAO enhances intracellular Ca2+ release of VSMC in response to Ang II (0.1 μM), which was impaired by GSK, Tempol, KN-93, U73122 and BAPTA-AM (n = 4–8; *P < 0.05 vs Control, #P < 0.05 vs TMAO). Statistical differences were calculated by two-way ANOVA followed by Dunnett's post hoc test (A), one-way ANOVA followed by Turkey's post hoc test (B). GSK, GSK2606414.
3.9. SBP, SOD activity, creatinine and BUN are correlated with plasma TMAO concentrations in normotensive and hypertensive adults
In hypertensive patients, plasma TMAO (Fig. 9A), SOD activity (Fig. 9C) and creatinine (Fig. 9E) were increased compared with normotensive controls. Hypertension did not affect plasma BUN levels (Fig. 9G). Linear regression analysis indicated that SBP positivity correlated with plasma TMAO concentration (Fig. 9B). Plasma SOD activity negatively correlated with TMAO concentration (Fig. 9D). Plasma creatinine and BUN positivity correlated with TMAO concentration (Fig. 9F and H). Collectively, these data illustrate that TMAO may be involved in the pathogenesis of hypertension and renal injury via activating of oxidative stress.
Fig. 9.

Plasma TMAO is correlated with SBP, SOD activity, creatinine and BUN in normotensive and hypertensive adults. Plasma levels of TMAO (A), SOD activity (C), creatinine (E) and BUN (G) in normotensive (n = 32) or hypertensive patients (n = 37) (*P < 0.05 vs Normotensive). Correlation analysis of plasma TMAO with SBP (B), SOD activity (D), creatinine (F) and BUN (H). Statistical differences were calculated by the paired T-test (A, C, E and G), The correlation between 2 parameters was evaluated by Pearson correlation test (B, D, F and H).
4. Discussion
Major findings of the present study are that the gut microbiota metabolite TMAO directly constricts arterioles, sensitizes blood pressure responses to Ang II and lowers GFR. TMAO does so by activating PERK and its downstream ROS/CaMKII/PLCβ3/Ca2+ pathway, which enhances Ang II-induced vasoconstriction. Depleting gut microbiota by antibiotics reduced TMAO generation, thereby alleviating Ang II-induced hypertension. In hypertensive patients, plasma levels of TMAO correlated positively with the blood pressure.
As a bioactive metabolite of gut microbiota, TMAO plays a critical role in the progression of many diseases, including chronic kidney disease [18], obesity [17], atherosclerosis [15], diabetes [38] and aging [39]. An elevation in circulating TMAO concentration is commonly observed in these disease states. For instance, TMAO plasma concentration is positively correlated with blood pressure and negatively associated with estimated GFR [38]. The present study demonstrates that TMAO aggravates Ang II-induced hypertension in mice and reduces GFR, which is consistent with the prior clinical report. The antibiotics treatment procedure successfully reduced TMAO levels, as shown before [21,40] and blunted Ang II-induced hypertension in mice, indicating that decreasing TMAO levels may be a potential therapeutic approach for the treatment of hypertension.
Several studies underscore the microbiome's role in maintaining hypertension. Karbach et al. report that Ang II-induced hypertension is alleviated in Germ-Free mice [10]. Moreover, high-fiber diet and acetate supplementation blunt DOCA-salt-induced hypertension by modifying gut microbiota [13]. In agreement with the latter, the anti-inflammatory antibiotic, minocycline, blunts Ang II-induced hypertension, again by altering gut microbiota composition [41]. The present study adds to our understanding by putting forward TMAO as a potential link between gut microbiota and hypertension.
Although many studies show an inverse association between plasma TMAO levels and eGFR in CKD [18,42], type 2 diabetic [38] and heart failure [43], the present study is the first to show that TMAO decreases GFR. Resistance of afferent and mesenteric arterioles play an important role in controlling blood pressure [5,24,44]. It is the afferent arterioles that regulate GFR [4,45]. High circulating TMAO levels impair endothelial nitric oxide synthase (eNOS)-derived NO production and reduce endothelium-dependent vasorelaxation of the aorta in response to acetylcholine [22,23]. We show that chronic administration of TMAO enhances Ang II contractions in Af and mesenteric arteries, which is confirmed by the whole animal experiments. Af and mesenteric arteries may therefore be seen as targets of TMAO with regard to Ang II-induced hypertension and GFR.
Although a large number of studies reveal that various intracellular signaling pathways are affected by TMAO [21,46,47], the receptor for TMAO remains enigmatic. Several studies indicate that TMAO acts as a small-molecule protein chaperone [48] to stabilize proteins, which consequently reduces PERK activity and endoplasmic reticulum (ER) stress [49]. PERK is an ER stress kinase. Inositol-requiring protein 1α (IRE1α), activating transcription factor 6 (ATF6) and PERK are three unfolded protein response (UPR) branches [50]. Under normal conditions, the protein chaperone HSPA5 (also known as GRP78 or BiP) binds and inactivates PERK. When perturbed (e.g., by calcium depletion or lipid overload), the unfolded proteins accumulate and recruit HSPA5, which activates PERK [51]. Recently, Chen et al. demonstrated that pathological concentrations of TMAO (0.05 mM) bind and activate PERK causing metabolic dysfunction [20]. TMAO binds to the luminal domain of PERK and disrupts the suppressive interaction between HSPA5 and luminal domain of PERK, which may account for the activation of PERK. Unexpectedly, TMAO binds and activates PERK because, as mentioned above, TMAO reduces PERK activity and ER stress by acting as a protein chaperone [48,49]. Different TMAO concentrations can induce opposite effects of TMAO on PERK. In the study by Chen et al., pathophysiological concentration of TMAO (0.05 mM) increases p-PERK, which is reduced by pharmacological TMAO (300 mM) treatment [20]. In several other studies, pharmacological concentrations (100–2000 mM) of TMAO decrease p-PERK [49]. On the other hand, ER-stress and PERK are involved in vasoconstriction and blood pressure regulation [52,53]. Our results are consistent with these findings. Therefore, we conclude that activation of PERK by pathophysiological concentration of TMAO can account for the augmentation of Ang II-induced vasoconstriction.
ROS production may mediate TMAO's enhancement of Ang II vasoconstriction. Activation of PERK causes excessive ROS production through induction of ER oxidoreductase 1-α (ERO1-α) [30,31,54]. ERO1-α is a eukaryotic flavin adenine nucleotide-containing enzyme located in the ER that produces H2O2, which can increase the ROS burden of cells. ER-stress activates PERK/EIF2α/CHOP signaling pathway, which upregulates ERO1-α expression [30,31,54]. Consequently, overexpression of ERO1-α leads to excessive ROS generation. We observed that TMAO stimulates the PERK/EIF2α/CHOP/ERO1-α signaling pathway causing excessive ROS generation. Since ROS play an important role in regulating Ang II-induced vasoconstriction [5,24], we suggest that PERK activation by TMAO enhances ROS generation, which contributes to the augmentation of Ang II-induced vasoconstriction.
CaMKII (a redox-sensitive enzyme) may be a key player downstream of PERK/ERO1-α/ROS axis to enhance Ang II vasoconstriction. Excessive ROS production activates CaMKII by oxidation of methionine 281/282 (Ox-CaMKII) [33], but CaMKII can also be activated by phosphorylation at threonine 286 by excessive ROS [32,55]. The present study shows that TMAO enhances CaMKII activity via the proposed PERK/ERO1-α/ROS axis. Interestingly, CaMKII inhibition blunts Ang II-induced hypertension [56] and agonists-stimulated vasoconstriction [34,57], which is in line with our conclusion.
It is well known that Ang II elicits vascular contraction by activating AT1R/PLC/IP3/Ca2+ pathway. In human platelets, pre-incubation with TMAO augments inositol-1,4,5-trisphosphate (IP3) generation, which is produced by various PLC isoforms and triggers intracellular Ca2+ release [21]. CaMKII phosphorylates and activates phospholipase C β3 (PLCβ3) at Ser-537 [36,37]. Our results suggest that TMAO activates PLCβ3 via PERK/ROS/CaMKII. We therefore conclude that TMAO enhances Ang II-induced vasoconstriction and hypertension via PERK/ROS/CaMKII/PLCβ3.
Several in vivo studies have documented that inhibition of the PERK/eIF2α/Chop pathway decreases arterial blood pressure in animal models of hypertension [53,58,59]. Intraperitoneal injection of PERK inhibitor GSK2606414 ameliorates vascular remodeling [60], which is a major pathophysiological event in hypertension [61]. Deletion of Chop in Ang II-infused mice impairs U-46619-induced vasoconstriction. This can explain why hypertension is ameliorated by knockout of Chop in Ang II-infused mice [59]. On the other hand, Chop deletion may reduce ROS as verified by animal studies [31,62]. Oral administration of GSK2606414 attenuates the overproduction of mitochondrial ROS in T-cell via inhibition of ERO1α, which is also observed in T-cell-specific PERK knockout mice [63]. Of note, multiple animal studies demonstrate that ROS is closely associated with the development of hypertension [[64], [65], [66]]. Among them, Kawada et al. and Dikalova et al. report that treatment of Ang II-infused mice with Tempol or mitoTempo reduce the Ang II action on blood pressure, respectively [65,66]. In addition, Tempol and mitoTempol also attenuate enhanced vasoconstriction in animal models of diabetes [67] and hypertension [65]. Numerous in vivo studies have demonstrated that excess ROS activate CaMKII [32,33]. Meanwhile, CaMKII also can be effectively inhibited in vivo by Tempol [68]. Overexpression of CaMKII exacerbates Ang II-induced hypertension [69]. In Ang II-infused rats, CaMKII inhibitor KN-93 blunts Ang II-elevated blood pressure [56]. Remarkably, disruption of CaMKII in mice inhibits Ang II-induced vasoconstriction [35]. Chronic administration of KN-93 inhibits Ang II, NE and ET-1 induced vasoconstriction in diabetic mice [70].
Several lines of evidence indicate that CaMKII phosphorylates and activates PLCβ336, 37. It is well-known that PLCβ mediates Ang II-induced vasoconstriction and plays a major role in regulating blood pressure [3]. Aortic ligation-induced hypertensive rats administrated with U73122 show reduced mean arterial pressure [71]. Taken together, there is ample in vivo evidence confirming that hypertension is promoted by the PERK/ROS/CaMKII/PLCβ3 pathway. The inhibitor's ex vivo effects may be extrapolated to our in vivo findings.
5. Conclusion
TMAO promotes Ang II-induced hypertension through Ang II-induced vasoconstriction that is mediated by the PERK/ROS/CaMKII/PLCβ3/Ca2+ axis. These findings provide new insights into the pathogenesis of hypertension and identify TMAO as a potential therapeutic target and biomarker.
Declaration of competing interest
The authors declare that they have no competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by grants to Qichun Wei from National Nature Science Foundation of China (81572952), to En Yin Lai, Andreas Patzak and Pontus B. Persson from Deutsche Forschungsgemeinschaft Project 394046635-SFB 1365, En Yin Lai is a Mercator Fellow supported by the German Research Foundation Project 394046635-SFB 1365, and to Zhihua Zheng from Shenzhen Science and technology Innovation Committee Founding 2019 (33) and 2018 (327), and to Chun Tang by the medical scientific research foundation of Guangdong province (A2018071) and the Guangdong basic and applied basic research foundation (2019A1515110488).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2021.102115.
Contributor Information
En Yin Lai, Email: laienyin@zju.edu.cn.
Qichun Wei, Email: Qichun_Wei@zju.edu.cn.
Zhihua Zheng, Email: zhzhihua@mail.sysu.edu.cn.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Mills K.T., Bundy J.D., Kelly T.N., Reed J.E., Kearney P.M., Reynolds K., Chen J., He J. Global disparities of hypertension prevalence and control: a systematic analysis of population-based studies from 90 countries. Circulation. 2016;134:441–450. doi: 10.1161/CIRCULATIONAHA.115.018912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mills K.T., Stefanescu A., He J. The global epidemiology of hypertension. Nat. Rev. Nephrol. 2020;16:223–237. doi: 10.1038/s41581-019-0244-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Forrester S.J., Booz G.W., Sigmund C.D., Coffman T.M., Kawai T., Rizzo V., Scalia R., Eguchi S. Angiotensin ii signal transduction: an update on mechanisms of physiology and pathophysiology. Physiol. Rev. 2018;98:1627–1738. doi: 10.1152/physrev.00038.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carlstrom M., Wilcox C.S., Arendshorst W.J. Renal autoregulation in health and disease. Physiol. Rev. 2015;95:405–511. doi: 10.1152/physrev.00042.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carlstrom M., Lai E.Y., Ma Z., Steege A., Patzak A., Eriksson U.J., Lundberg J.O., Wilcox C.S., Persson A.E. Superoxide dismutase 1 limits renal microvascular remodeling and attenuates arteriole and blood pressure responses to angiotensin ii via modulation of nitric oxide bioavailability. Hypertension. 2010;56:907–913. doi: 10.1161/HYPERTENSIONAHA.110.159301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marques F.Z., Mackay C.R., Kaye D.M. Beyond gut feelings: how the gut microbiota regulates blood pressure. Nat. Rev. Cardiol. 2018;15:20–32. doi: 10.1038/nrcardio.2017.120. [DOI] [PubMed] [Google Scholar]
- 7.Sharma R.K., Yang T., Oliveira A.C., Lobaton G.O., Aquino V., Kim S., Richards E.M., Pepine C.J., Sumners C., Raizada M.K. Microglial cells impact gut microbiota and gut pathology in angiotensin ii-induced hypertension. Circ. Res. 2019;124:727–736. doi: 10.1161/CIRCRESAHA.118.313882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jama H.A., Kaye D.M., Marques F.Z. The gut microbiota and blood pressure in experimental models. Curr. Opin. Nephrol. Hypertens. 2019;28:97–104. doi: 10.1097/MNH.0000000000000476. [DOI] [PubMed] [Google Scholar]
- 9.Adnan S., Nelson J.W., Ajami N.J., Venna V.R., Petrosino J.F., Bryan R.M., Jr., Durgan D.J. Alterations in the gut microbiota can elicit hypertension in rats. Physiol. Genom. 2017;49:96–104. doi: 10.1152/physiolgenomics.00081.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karbach S.H., Schonfelder T., Brandao I., Wilms E., Hormann N., Jackel S., Schuler R., Finger S., Knorr M., Lagrange J., Brandt M., Waisman A., Kossmann S., Schafer K., Munzel T., Reinhardt C., Wenzel P. Gut microbiota promote angiotensin ii-induced arterial hypertension and vascular dysfunction. J. Am. Heart Assoc. 2016;5 doi: 10.1161/JAHA.116.003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li J., Zhao F., Wang Y., Chen J., Tao J., Tian G., Wu S., Liu W., Cui Q., Geng B., Zhang W., Weldon R., Auguste K., Yang L., Liu X., Chen L., Yang X., Zhu B., Cai J. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome. 2017;5:14. doi: 10.1186/s40168-016-0222-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Santisteban M.M., Qi Y., Zubcevic J., Kim S., Yang T., Shenoy V., Cole-Jeffrey C.T., Lobaton G.O., Stewart D.C., Rubiano A., Simmons C.S., Garcia-Pereira F., Johnson R.D., Pepine C.J., Raizada M.K. Hypertension-linked pathophysiological alterations in the gut. Circ. Res. 2017;120:312–323. doi: 10.1161/CIRCRESAHA.116.309006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marques F.Z., Nelson E., Chu P.Y., Horlock D., Fiedler A., Ziemann M., Tan J.K., Kuruppu S., Rajapakse N.W., El-Osta A., Mackay C.R., Kaye D.M. High-fiber diet and acetate supplementation change the gut microbiota and prevent the development of hypertension and heart failure in hypertensive mice. Circulation. 2017;135:964–977. doi: 10.1161/CIRCULATIONAHA.116.024545. [DOI] [PubMed] [Google Scholar]
- 14.Joe B., McCarthy C.G., Edwards J.M., Cheng X., Chakraborty S., Yang T., Golonka R.M., Mell B., Yeo J.Y., Bearss N.R., Furtado J., Saha P., Yeoh B.S., Vijay-Kumar M., Wenceslau C.F. Microbiota introduced to germ-free rats restores vascular contractility and blood pressure. Hypertension. 2020;76:1847–1855. doi: 10.1161/HYPERTENSIONAHA.120.15939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koeth R.A., Wang Z., Levison B.S., Buffa J.A., Org E., Sheehy B.T., Britt E.B., Fu X., Wu Y., Li L., Smith J.D., DiDonato J.A., Chen J., Li H., Wu G.D., Lewis J.D., Warrier M., Brown J.M., Krauss R.M., Tang W.H., Bushman F.D., Lusis A.J., Hazen S.L. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013;19:576–585. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeisel S.H., Warrier M. Trimethylamine n-oxide, the microbiome, and heart and kidney disease. Annu. Rev. Nutr. 2017;37:157–181. doi: 10.1146/annurev-nutr-071816-064732. [DOI] [PubMed] [Google Scholar]
- 17.Schugar R.C., Shih D.M., Warrier M., Helsley R.N., Burrows A., Ferguson D., Brown A.L., Gromovsky A.D., Heine M., Chatterjee A., Li L., Li X.S., Wang Z., Willard B., Meng Y., Kim H., Che N., Pan C., Lee R.G., Crooke R.M., Graham M.J., Morton R.E., Langefeld C.D., Das S.K., Rudel L.L., Zein N., McCullough A.J., Dasarathy S., Tang W.H.W., Erokwu B.O., Flask C.A., Laakso M., Civelek M., Naga Prasad S.V., Heeren J., Lusis A.J., Hazen S.L., Brown J.M. The tmao-producing enzyme flavin-containing monooxygenase 3 regulates obesity and the beiging of white adipose tissue. Cell Rep. 2017;19:2451–2461. doi: 10.1016/j.celrep.2017.05.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang W.H., Wang Z., Kennedy D.J., Wu Y., Buffa J.A., Agatisa-Boyle B., Li X.S., Levison B.S., Hazen S.L. Gut microbiota-dependent trimethylamine n-oxide (tmao) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ. Res. 2015;116:448–455. doi: 10.1161/CIRCRESAHA.116.305360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Subramaniam S., Fletcher C. Trimethylamine n-oxide: breathe new life. Br. J. Pharmacol. 2018;175:1344–1353. doi: 10.1111/bph.13959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen S., Henderson A., Petriello M.C., Romano K.A., Gearing M., Miao J., Schell M., Sandoval-Espinola W.J., Tao J., Sha B., Graham M., Crooke R., Kleinridders A., Balskus E.P., Rey F.E., Morris A.J., Biddinger S.B. Trimethylamine n-oxide binds and activates perk to promote metabolic dysfunction. Cell Metabol. 2019;30:1141–1151. doi: 10.1016/j.cmet.2019.08.021. e1145. [DOI] [PubMed] [Google Scholar]
- 21.Zhu W., Gregory J.C., Org E., Buffa J.A., Gupta N., Wang Z., Li L., Fu X., Wu Y., Mehrabian M., Sartor R.B., McIntyre T.M., Silverstein R.L., Tang W.H.W., DiDonato J.A., Brown J.M., Lusis A.J., Hazen S.L. Gut microbial metabolite tmao enhances platelet hyperreactivity and thrombosis risk. Cell. 2016;165:111–124. doi: 10.1016/j.cell.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ke Y., Li D., Zhao M., Liu C., Liu J., Zeng A., Shi X., Cheng S., Pan B., Zheng L., Hong H. Gut flora-dependent metabolite trimethylamine-n-oxide accelerates endothelial cell senescence and vascular aging through oxidative stress. Free Radic. Biol. Med. 2018;116:88–100. doi: 10.1016/j.freeradbiomed.2018.01.007. [DOI] [PubMed] [Google Scholar]
- 23.Li T., Gua C., Wu B., Chen Y. Increased circulating trimethylamine n-oxide contributes to endothelial dysfunction in a rat model of chronic kidney disease. Biochem. Biophys. Res. Commun. 2018;495:2071–2077. doi: 10.1016/j.bbrc.2017.12.069. [DOI] [PubMed] [Google Scholar]
- 24.Lai E.Y., Solis G., Luo Z., Carlstrom M., Sandberg K., Holland S., Wellstein A., Welch W.J., Wilcox C.S. P47(phox) is required for afferent arteriolar contractile responses to angiotensin ii and perfusion pressure in mice. Hypertension. 2012;59:415–420. doi: 10.1161/HYPERTENSIONAHA.111.184291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shikata F., Shimada K., Sato H., Ikedo T., Kuwabara A., Furukawa H., Korai M., Kotoda M., Yokosuka K., Makino H., Ziegler E.A., Kudo D., Lawton M.T., Hashimoto T. Potential influences of gut microbiota on the formation of intracranial aneurysm. Hypertension. 2019;73:491–496. doi: 10.1161/HYPERTENSIONAHA.118.11804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lai E.Y., Martinka P., Fahling M., Mrowka R., Steege A., Gericke A., Sendeski M., Persson P.B., Persson A.E., Patzak A. Adenosine restores angiotensin ii-induced contractions by receptor-independent enhancement of calcium sensitivity in renal arterioles. Circ. Res. 2006;99:1117–1124. doi: 10.1161/01.RES.0000249530.85542.d4. [DOI] [PubMed] [Google Scholar]
- 27.Jiang S., Wang X., Wei J., Zhang G., Zhang J., Xie P., Xu L., Wang L., Zhao L., Li L., Wilcox C.S., Chen J., Lai E.Y., Liu R. Nahco3 dilates mouse afferent arteriole via na(+)/hco3(-) cotransporters nbcs. Hypertension. 2019;74:1104–1112. doi: 10.1161/HYPERTENSIONAHA.119.13235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu N., Wang Q., Jiang S., Wang Q., Hu W., Zhou S., Zhao L., Xie L., Chen J., Wellstein A., Lai E.Y. Fenofibrate improves vascular endothelial function and contractility in diabetic mice. Redox Biol. 2019;20:87–97. doi: 10.1016/j.redox.2018.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu W.P., Jiang S., Liao Y.X., Li J.H., Dong F., Guo J., Wang X.H., Fei L.Y., Cui Y., Ren X.Q., Xu N., Zhao L., Chen L.M., Zheng Y.L., Li L.L., Patzak A., Persson P.B., Zheng Z.H., Lai E.Y. High phosphate impairs arterial endothelial function through ampk-related pathways in mouse resistance arteries. Acta Physiol. 2021;231 doi: 10.1111/apha.13595. [DOI] [PubMed] [Google Scholar]
- 30.Qi J., Zhou N., Li L., Mo S., Zhou Y., Deng Y., Chen T., Shan C., Chen Q., Lu B. Ciclopirox activates perk-dependent endoplasmic reticulum stress to drive cell death in colorectal cancer. Cell Death Dis. 2020;11:582. doi: 10.1038/s41419-020-02779-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song B., Scheuner D., Ron D., Pennathur S., Kaufman R.J. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J. Clin. Invest. 2008;118:3378–3389. doi: 10.1172/JCI34587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang X., An N., Zhong C., Guan M., Jiang Y., Li X., Zhang H., Wang L., Ruan Y., Gao Y., Liu N., Shang H., Xing Y. Enhanced cardiomyocyte reactive oxygen species signaling promotes ibrutinib-induced atrial fibrillation. Redox Biol. 2020;30:101432. doi: 10.1016/j.redox.2020.101432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Erickson J.R., Joiner M.L., Guan X., Kutschke W., Yang J., Oddis C.V., Bartlett R.K., Lowe J.S., O'Donnell S.E., Aykin-Burns N., Zimmerman M.C., Zimmerman K., Ham A.J., Weiss R.M., Spitz D.R., Shea M.A., Colbran R.J., Mohler P.J., Anderson M.E. A dynamic pathway for calcium-independent activation of camkii by methionine oxidation. Cell. 2008;133:462–474. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benter I.F., Yousif M.H., Canatan H., Akhtar S. Inhibition of ca2+/calmodulin-dependent protein kinase ii, ras-gtpase and 20-hydroxyeicosatetraenoic acid attenuates the development of diabetes-induced vascular dysfunction in the rat carotid artery. Pharmacol. Res. 2005;52:252–257. doi: 10.1016/j.phrs.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 35.Prasad A.M., Nuno D.W., Koval O.M., Ketsawatsomkron P., Li W., Li H., Shen F.Y., Joiner M.L., Kutschke W., Weiss R.M., Sigmund C.D., Anderson M.E., Lamping K.G., Grumbach I.M. Differential control of calcium homeostasis and vascular reactivity by ca2+/calmodulin-dependent kinase ii. Hypertension. 2013;62:434–441. doi: 10.1161/HYPERTENSIONAHA.113.01508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yue C.P., Sanborn B.M. Kn-93 inhibition of g protein signaling is independent of the ability of ca2+/calmodulin-dependent protein kinase ii to phosphorylate phospholipase c beta(3) on (537)-ser. Mol. Cell. Endocrinol. 2001;175:149–156. doi: 10.1016/s0303-7207(01)00383-5. [DOI] [PubMed] [Google Scholar]
- 37.Hatziapostolou M., Koukos G., Polytarchou C., Kottakis F., Serebrennikova O., Kuliopulos A., Tsichlis P.N. Tumor progression locus 2 mediates signal-induced increases in cytoplasmic calcium and cell migration. Sci. Signal. 2011;4:ra55. doi: 10.1126/scisignal.2002006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winther S.A., Ollgaard J.C., Tofte N., Tarnow L., Wang Z., Ahluwalia T.S., Jorsal A., Theilade S., Parving H.H., Hansen T.W., Hazen S.L., Pedersen O., Rossing P. Utility of plasma concentration of trimethylamine n-oxide in predicting cardiovascular and renal complications in individuals with type 1 diabetes. Diabetes Care. 2019;42:1512–1520. doi: 10.2337/dc19-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li D., Ke Y., Zhan R., Liu C., Zhao M., Zeng A., Shi X., Ji L., Cheng S., Pan B., Zheng L., Hong H. Trimethylamine-n-oxide promotes brain aging and cognitive impairment in mice. Aging Cell. 2018;17 doi: 10.1111/acel.12768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang X., Li Y., Yang P., Liu X., Lu L., Chen Y., Zhong X., Li Z., Liu H., Ou C., Yan J., Chen M. Trimethylamine-n-oxide promotes vascular calcification through activation of nlrp3 (nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3) inflammasome and nf-kappab (nuclear factor kappab) signals. Arterioscler. Thromb. Vasc. Biol. 2020;40:751–765. doi: 10.1161/ATVBAHA.119.313414. [DOI] [PubMed] [Google Scholar]
- 41.Yang T., Santisteban M.M., Rodriguez V., Li E., Ahmari N., Carvajal J.M., Zadeh M., Gong M.H., Qi Y.F., Zubcevic J., Sahay B., Pepine C.J., Raizada M.K., Mohamadzadeh M. Gut dysbiosis is linked to hypertension. Hypertension. 2015;65:1331–1340. doi: 10.1161/HYPERTENSIONAHA.115.05315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stubbs J.R., House J.A., Ocque A.J., Zhang S., Johnson C., Kimber C., Schmidt K., Gupta A., Wetmore J.B., Nolin T.D., Spertus J.A., Yu A.S. Serum trimethylamine-n-oxide is elevated in ckd and correlates with coronary atherosclerosis burden. J. Am. Soc. Nephrol. 2016;27:305–313. doi: 10.1681/ASN.2014111063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang W.H., Wang Z., Fan Y., Levison B., Hazen J.E., Donahue L.M., Wu Y., Hazen S.L. Prognostic value of elevated levels of intestinal microbe-generated metabolite trimethylamine-n-oxide in patients with heart failure: refining the gut hypothesis. J. Am. Coll. Cardiol. 2014;64:1908–1914. doi: 10.1016/j.jacc.2014.02.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chuaiphichai S., Rashbrook V.S., Hale A.B., Trelfa L., Patel J., McNeill E., Lygate C.A., Channon K.M., Douglas G. Endothelial cell tetrahydrobiopterin modulates sensitivity to ang (angiotensin) ii-induced vascular remodeling, blood pressure, and abdominal aortic aneurysm. Hypertension. 2018;72:128–138. doi: 10.1161/HYPERTENSIONAHA.118.11144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wei J., Zhang J., Jiang S., Wang L., Persson A.E.G., Liu R. High-protein diet-induced glomerular hyperfiltration is dependent on neuronal nitric oxide synthase beta in the macula densa via tubuloglomerular feedback response. Hypertension. 2019;74:864–871. doi: 10.1161/HYPERTENSIONAHA.119.13077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen M.L., Zhu X.H., Ran L., Lang H.D., Yi L., Mi M.T. Trimethylamine-n-oxide induces vascular inflammation by activating the nlrp3 inflammasome through the sirt3-sod2-mtros signaling pathway. J. Am. Heart Assoc. 2017;6 doi: 10.1161/JAHA.117.006347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seldin M.M., Meng Y., Qi H., Zhu W., Wang Z., Hazen S.L., Lusis A.J., Shih D.M. Trimethylamine n-oxide promotes vascular inflammation through signaling of mitogen-activated protein kinase and nuclear factor-kappab. J. Am. Heart Assoc. 2016;5 doi: 10.1161/JAHA.115.002767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mello C.C., Barrick D. Measuring the stability of partly folded proteins using tmao. Protein Sci. 2003;12:1522–1529. doi: 10.1110/ps.0372903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Achard C.S., Laybutt D.R. Lipid-induced endoplasmic reticulum stress in liver cells results in two distinct outcomes: adaptation with enhanced insulin signaling or insulin resistance. Endocrinology. 2012;153:2164–2177. doi: 10.1210/en.2011-1881. [DOI] [PubMed] [Google Scholar]
- 50.Hetz C., Zhang K., Kaufman R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020;21:421–438. doi: 10.1038/s41580-020-0250-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rutkowski D.T., Kaufman R.J. That which does not kill me makes me stronger: adapting to chronic er stress. Trends Biochem. Sci. 2007;32:469–476. doi: 10.1016/j.tibs.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 52.Hu X.Q., Song R., Romero M., Dasgupta C., Min J., Hatcher D., Xiao D., Blood A., Wilson S.M., Zhang L. Gestational hypoxia inhibits pregnancy-induced upregulation of ca(2+) sparks and spontaneous transient outward currents in uterine arteries via heightened endoplasmic reticulum/oxidative stress. Hypertension. 2020;76:930–942. doi: 10.1161/HYPERTENSIONAHA.120.15235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liang B., Wang S., Wang Q., Zhang W., Viollet B., Zhu Y., Zou M.H. Aberrant endoplasmic reticulum stress in vascular smooth muscle increases vascular contractility and blood pressure in mice deficient of amp-activated protein kinase-alpha2 in vivo. Arterioscler. Thromb. Vasc. Biol. 2013;33:595–604. doi: 10.1161/ATVBAHA.112.300606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Z., Yin F., Xu J., Zhang T., Wang G., Mao M., Wang Z., Sun W., Han J., Yang M., Jiang Y., Hua Y., Cai Z. Cyt997(lexibulin) induces apoptosis and autophagy through the activation of mutually reinforced er stress and ros in osteosarcoma. J. Exp. Clin. Canc. Res. 2019;38 doi: 10.1186/s13046-019-1047-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Haskew-Layton R.E., Mongin A.A., Kimelberg H.K. Hydrogen peroxide potentiates volume-sensitive excitatory amino acid release via a mechanism involving ca2+/calmodulin-dependent protein kinase ii. J. Biol. Chem. 2005;280:3548–3554. doi: 10.1074/jbc.M409803200. [DOI] [PubMed] [Google Scholar]
- 56.Muthalif M.M., Karzoun N.A., Benter I.F., Gaber L., Ljuca F., Uddin M.R., Khandekar Z., Estes A., Malik K.U. Functional significance of activation of calcium/calmodulin-dependent protein kinase ii in angiotensin ii--induced vascular hyperplasia and hypertension. Hypertension. 2002;39:704–709. doi: 10.1161/hy0202.103823. [DOI] [PubMed] [Google Scholar]
- 57.Waldsee R., Ahnstedt H., Eftekhari S., Edvinsson L. Involvement of calcium-calmodulin-dependent protein kinase ii in endothelin receptor expression in rat cerebral arteries. Am. J. Physiol. Heart Circ. Physiol. 2010;298:H823–H832. doi: 10.1152/ajpheart.00759.2009. [DOI] [PubMed] [Google Scholar]
- 58.Han S., Bal N.B., Sadi G., Usanmaz S.E., Tuglu M.M., Uludag M.O., Demirel-Yilmaz E. Inhibition of endoplasmic reticulum stress protected doca-salt hypertension-induced vascular dysfunction. Vasc. Pharmacol. 2019;113:38–46. doi: 10.1016/j.vph.2018.11.004. [DOI] [PubMed] [Google Scholar]
- 59.Kassan M., Ait-Aissa K., Radwan E., Mali V., Haddox S., Gabani M., Zhang W., Belmadani S., Irani K., Trebak M., Matrougui K. Essential role of smooth muscle stim1 in hypertension and cardiovascular dysfunction. Arterioscler. Thromb. Vasc. Biol. 2016;36:1900–1909. doi: 10.1161/ATVBAHA.116.307869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shimizu T., Higashijima Y., Kanki Y., Nakaki R., Kawamura T., Urade Y., Wada Y. Perk inhibition attenuates vascular remodeling in pulmonary arterial hypertension caused by bmpr2 mutation. Sci. Signal. 2021:14. doi: 10.1126/scisignal.abb3616. [DOI] [PubMed] [Google Scholar]
- 61.Wang L., Zhao X.C., Cui W., Ma Y.Q., Ren H.L., Zhou X., Fassett J., Yang Y.Z., Chen Y., Xia Y.L., Du J., Li H.H. Genetic and pharmacologic inhibition of the chemokine receptor cxcr2 prevents experimental hypertension and vascular dysfunction. Circulation. 2016;134:1353–1368. doi: 10.1161/CIRCULATIONAHA.115.020754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shi Z., Diao D., Zhao Y., Luo Y., Li Y., Liu D., Zhang K., Qiu Y., Yu L., Song Z., Ju Z. C/ebp homologous protein deficiency enhances hematopoietic stem cell function via reducing atf3/ros-induced cell apoptosis. Aging Cell. 2021;20 doi: 10.1111/acel.13382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hurst K.E., Lawrence K.A., Essman M.T., Walton Z.J., Leddy L.R., Thaxton J.E. Endoplasmic reticulum stress contributes to mitochondrial exhaustion of cd8(+) t cells. Canc. Immunol. Res. 2019;7:476–486. doi: 10.1158/2326-6066.CIR-18-0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen Z., Wu C., Liu Y., Li H., Zhu Y., Huang C., Lin H., Qiao Q., Huang M., Zhu Q., Wang L. Elabela attenuates deoxycorticosterone acetate/salt-induced hypertension and renal injury by inhibition of nadph oxidase/ros/nlrp3 inflammasome pathway. Cell Death Dis. 2020;11:698. doi: 10.1038/s41419-020-02912-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kawada N., Imai E., Karber A., Welch W.J., Wilcox C.S. A mouse model of angiotensin ii slow pressor response: role of oxidative stress. J. Am. Soc. Nephrol. 2002;13:2860–2868. doi: 10.1097/01.asn.0000035087.11758.ed. [DOI] [PubMed] [Google Scholar]
- 66.Dikalova A.E., Itani H.A., Nazarewicz R.R., McMaster W.G., Flynn C.R., Uzhachenko R., Fessel J.P., Gamboa J.L., Harrison D.G., Dikalov S.I. Sirt3 impairment and sod2 hyperacetylation in vascular oxidative stress and hypertension. Circ. Res. 2017;121:564–574. doi: 10.1161/CIRCRESAHA.117.310933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xiong X., Lu W., Qin X., Luo Q., Zhou W. Downregulation of the glp-1/creb/adiponectin pathway is partially responsible for diabetes-induced dysregulated vascular tone and vsmc dysfunction. Biomed. Pharmacother. 2020;127:110218. doi: 10.1016/j.biopha.2020.110218. [DOI] [PubMed] [Google Scholar]
- 68.Sepulveda M., Gonano L.A., Viotti M., Morell M., Blanco P., Lopez Alarcon M., Peroba Ramos I., Bastos Carvalho A., Medei E., Vila Petroff M. Calcium/calmodulin protein kinase ii-dependent ryanodine receptor phosphorylation mediates cardiac contractile dysfunction associated with sepsis. Crit. Care Med. 2017;45:e399–e408. doi: 10.1097/CCM.0000000000002101. [DOI] [PubMed] [Google Scholar]
- 69.Basu U., Case A.J., Liu J., Tian J., Li Y.L., Zimmerman M.C. Redox-sensitive calcium/calmodulin-dependent protein kinase iialpha in angiotensin ii intra-neuronal signaling and hypertension. Redox Biol. 2019;27:101230. doi: 10.1016/j.redox.2019.101230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yousif M.H., Benter I.F., Akhtar S. Inhibition of calcium/calmodulin-dependent protein kinase ii normalizes diabetes-induced abnormal vascular reactivity in the rat perfused mesenteric vascular bed. Auton. Autacoid. Pharmacol. 2003;23:27–33. doi: 10.1046/j.1474-8673.2003.00282.x. [DOI] [PubMed] [Google Scholar]
- 71.Wu J., Zhang C., Liu C., Zhang A., Li A., Zhang J., Zhang Y. Aortic constriction induces hypertension and cardiac hypertrophy via (pro)renin receptor activation and the plcbeta3 signaling pathway. Mol. Med. Rep. 2019;19:573–580. doi: 10.3892/mmr.2018.9653. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.