

ABSTRACT
Background
Prenatal alcohol exposure (PAE) is associated with postnatal iron deficiency (ID), which has been shown to exacerbate deficits in growth, cognition, and behavior seen in fetal alcohol spectrum disorders. However, the mechanisms underlying PAE-related ID remain unknown.
Objectives
We aimed to examine biochemical measures of iron homeostasis in the mother, placenta, neonate, and 6.5-month-old infant.
Methods
In a prenatally recruited, prospective longitudinal birth cohort in South Africa, 206 gravidas (126 heavy drinkers and 80 controls) were interviewed regarding alcohol, cigarette, and drug use and diet at 3 prenatal visits. Hemoglobin, ferritin, and soluble transferrin receptor (sTfR) were assayed twice during pregnancy and urinary hepcidin:creatinine was assayed once. Infant ferritin and hemoglobin were measured at 2 weeks and 6.5 months and sTfR was measured at 6.5 months. Histopathological examinations were conducted on 125 placentas and iron transport assays (iron regulatory protein-2, transferrin receptor-1, divalent metal transporter-1, ferroportin-1, and iron concentrations) were conducted on 63.
Results
In multivariable regression models, prenatal drinking frequency (days/week) was related to higher maternal hepcidin and to sequestration of iron into storage at the expense of erythropoiesis in mothers and neonates, as evidenced by a lower hemoglobin (g/dL)-to-log(ferritin) (ug/L) ratio [mothers: raw regression coefficient (β) = −0.21 (95% CI: −0.35 to −0.07); neonates: β = −0.15 (95% CI: −0.24 to −0.06)]. Drinking frequency was also related to decreased placental ferroportin-1:transferrin receptor-1 (β = −0.57 for logged values; 95% CI: −1.03 to −0.10), indicating iron-restricted placental iron transport. At 6.5 months, drinking frequency was associated with lower hemoglobin (β = −0.18; 95% CI: −0.33 to −0.02), and increased prevalences of ID (β = 0.09; 95% CI: 0.02–0.17) and ID anemia (IDA) (β = 0.13; 95% CI: 0.04–0.23). In causal inference analyses, the PAE-related increase in IDA was partially mediated by decreased neonatal hemoglobin:log(ferritin), and the decrease in neonatal hemoglobin:log(ferritin) was partially mediated by decreased maternal hemoglobin:log(ferritin).
Conclusions
In this study, greater PAE was associated with an unfavorable profile of maternal-fetal iron homeostasis, which may play mechanistic roles in PAE-related ID later in infancy.
Keywords: FAS, fetal alcohol syndrome, FASD, fetal alcohol spectrum disorders, prenatal alcohol exposure, ID, iron deficiency, IDA, iron deficiency anemia, placenta
Introduction
Fetal alcohol spectrum disorders (FASD) comprise the most common preventable cause of neurodevelopmental disabilities worldwide, with prevalence estimates of 2.0%–5.0% of the school-age population in the United States and Western Europe and 13.6%–20.9% in South Africa (1, 2). FASD are manifested by growth restriction and a broad range of cognitive and behavioral deficits, and prenatal alcohol exposure (PAE) has been associated with birth defects in almost every organ system (3–8). Fetal alcohol syndrome (FAS), the most severe form of FASD, is characterized by distinctive facial dysmorphology, a small head circumference, and growth restriction (6, 9).
Both animal and human studies have demonstrated PAE-induced alterations in infant iron homeostasis. Miller and colleagues (10) found region-specific, PAE-related alterations in brain iron in rats. In a series of experimental rat models, Smith and colleagues(11–14) demonstrated PAE-related increases in maternal and fetal anemia and hepcidin, decreased fetal brain iron, and increased fetal liver iron. In a prospective, longitudinal human cohort study in Cape Town, South Africa, we found that infants exposed to prenatal binge drinking had 70% greater odds of iron deficiency anemia (IDA) at ages 6–12 months (15). Given the range of long-term neurodevelopmental deficits that result from infant iron deficiency (ID) (16–18), PAE-related alterations in iron homeostasis may have important implications in FASD. ID effects on information processing speed and eyeblink conditioning are remarkably similar to those seen in FASD (18–21). Furthermore, in both our human study (3, 15) and Smith and colleagues’ rat models (11, 14, 22), PAE-related infant ID exacerbated the teratogenic effects of PAE.
As data regarding maternal and fetal iron status were not available, the mechanisms underlying the PAE-related increase in IDA seen in our Cape Town study (15) were unclear and, to our knowledge, remain unknown in FASD literature. PAE-related ID could reflect alcohol-related disruptions in iron availability at any point in the mother-placenta-infant iron chain, including maternal diet, bioavailability of maternal iron, placental iron transport, and alterations in fetal and/or postnatal iron homeostasis/utilization. Chronic heavy alcohol consumption has been associated with hematological alterations and iron overload in studies of nonpregnant adults (23–27). In 1 prospective cohort study in Seattle, ID was more common among the heaviest drinkers (>8 drinks/day) than the rest of the cohort (28), while in 3 South African FASD studies, dietary iron intake was similarly inadequate in both drinkers and nondrinkers (29–31). We and others have demonstrated PAE-related reductions in placental weight and increased maternal vascular underperfusion, both of which may result in decreased iron transfer (5, 32, 33). PAE may also disrupt expression of placental iron transport proteins, as seen in other diseases, such as gestational diabetes (34, 35). Given that the mechanisms underlying fetal alcohol-related infant ID remain unknown, the aims of the present study were to examine the effects of PAE on maternal, fetal, placental, and infant iron homeostasis in a prenatally recruited, prospective, longitudinal cohort designed to examine the developmental effects of PAE (5, 36, 37). The primary outcomes were maternal, neonatal, and 6.5-month ferritin and hemoglobin; 6.5-month ID/IDA; and placental iron transport protein concentrations. Secondary outcomes included red blood cell indices, anemia, and anemia of inflammation. Understanding the mechanisms underlying fetal alcohol-related infant ID has the potential to inform future intervention studies targeting effects of PAE on iron homeostasis.
Methods
Sample
As previously reported, women were recruited from 2 antenatal clinics in Cape Town, South Africa, from 2011–2015 to take part in a prospective, longitudinal cohort study examining the developmental effects of PAE, with eyeblink conditioning as the primary outcome (data not yet published) (19, 29, 36). See Supplemental Figure 1 for a recruitment and retention flow diagram. Alcohol consumption at recruitment was ascertained in “timeline follow-back interviews” (38, 39) used to document alcohol consumption over the preceding 2-week period and a typical 2-week period around the time of conception. Any woman averaging at least 1.0 oz (30 mL) absolute alcohol (AA)/day (≈2 standard drinks) or reporting binge drinking [≥2.0 oz (60 mL) AA/drinking occasion] was invited to participate. Women who abstained or drank only minimally (with no binge episodes) were recruited as controls. Maternal exclusion criteria at recruitment were age <18 years, HIV infection, multiple gestation pregnancy, and pharmacologic treatment for medical conditions. In addition, infants meeting the following exclusion criteria chosen prior to study initiation were excluded from further follow-up due to potential developmental consequences of such conditions: infant major chromosomal anomalies (none), seizures (none), neural tube defects (none), very low birth weight (<1500 g; 1 exposed and 1 control), and extreme prematurity (weeks gestation <32 weeks; 1 exposed and 4 controls). There were 6 drinkers and 4 controls lost to follow-up. Infants with missing data were similar to those seen at 2 weeks and 6.5 months across maternal demographics; alcohol and drug use; weeks gestation at study entry; maternal dietary iron intake, iron supplement use, ferritin, and hemoglobin (Supplemental Table 1). Optimal sample size projections were aimed at collecting placental samples for iron transport protein assays from at least 30 heavy drinkers and 30 controls to achieve 80% power (α = 0.05) to detect intercorrelations between placental transport proteins using effect sizes from prior placental iron studies by coauthor MKG (34, 35). The final sample included 206 mothers (126 drinkers, 80 controls) and 189 infants (118 exposed, 71 controls). Consent was obtained and interviews were conducted in the mother's preferred language (Afrikaans or English). Approval was obtained from the ethics committees at Wayne State University, the University of Cape Town Faculty of Health Sciences, and the Columbia University Irving Medical Center.
Ascertainment of maternal alcohol, cigarette smoking, and drug use
In “timeline follow-back interviews” (38, 39) at recruitment and 4 and 12 weeks thereafter, women were asked about their alcohol use on a day-by-day basis during the previous 2 weeks, with recall linked to specific times of daily activities. Each woman was also asked about her cigarette smoking and drug use [cocaine, methamphetamine, opiates, methaqualone, and marijuana; validated by urine ELISA drug testing (5)]. Summary alcohol measures were constructed by averaging across pregnancy: oz AA/day, oz AA/drinking occasion, and frequency of drinking (1 oz = 30 mL) (19). The predictive validity of the timeline follow-back interview method was supported by consistent relations between PAE and growth and neurobehavior, with similar effect sizes, across longitudinal studies in Detroit and Cape Town (3, 4, 38).
Dietary, hematological, and biochemical iron indicators
Average daily iron and energy intakes were calculated from multipass 24-hour dietary recall interviews administered at each antenatal study visit and processed with FoodFinder3 (South African Medical Research Council) (29). Women were asked whether they had been prescribed iron supplements and how often they took them. Venous blood samples obtained from mothers at recruitment and 12 weeks later and from infants at 2 weeks and 6.5 months postpartum (corrected for prematurity), were analyzed for complete blood count and serum ferritin. Serum soluble transferrin receptor, C-reactive protein (CRP), erythrocyte folate, and serum vitamin B12 concentrations were assessed on maternal and 6.5-month infant samples. ID was defined as 1) serum ferritin <12 μg/L; or 2) in cases with CRP >5 mg/L, elevated soluble transferrin receptor (sTfR; >8.5 mg/L for mothers; >8.3 mg/L for infants at 6.5 months) (40–42). Anemia was defined as hemoglobin < 11.0 g/dL (42). Anemia of inflammation was defined as normocytic [mean corpuscular volume (MCV) ≥80 fL for mothers and ≥71 fL for infants] anemia with ferritin >30 μg/L (43). The hemoglobin:log(ferritin) ratio was calculated to assess the balance of iron in storage compared with iron available for erythropoiesis. The hepcidin:creatinine ratio, which reflects the blood hepcidin concentration, was assayed by competitive ELISA (44) in morning urine samples obtained from the last 91 women recruited (50 drinkers, 41 controls). Further laboratory assay technical details are listed in Supplemental Table 2.
Placenta histopathology examinations and iron transport protein assays
Histopathology examinations, including evaluations for maternal vascular underperfusion using Redline criteria (45), were performed on all available placentas (76 drinkers, 49 controls) stored at 3°C from delivery until examination (median storage = 21.3 hours; IQR, 10.5–46.7) (5). Iron transport protein assays were conducted on snap-frozen, transplacental central cotyledon core samples from placentas collected within 48 hours of delivery (33 drinkers, 30 controls) after excluding mothers with a priori-chosen conditions that lead to placental vascular changes that may affect iron transport [syphilis (1 drinker, 1 control), hypertension (1 control), diabetes (1 control), and methamphetamine use (5 drinkers, 7 controls)]. Infants with missing placental iron transport data were generally similar to those followed at 2 weeks and 6.5 months but were more likely to have had prenatal cigarette exposure (84.6% compared with 66.7%, respectively; P = 0.003; Supplemental Table 1). Protein concentrations of iron regulatory protein-2 (IRP-2), transferrin receptor (TfR-1), divalent metal transporter-1 (DMT-1), and ferroportin-1 (FPN-1) were determined using previously published methods (35, 46). Briefly, after tissue homogenization, 30–40 µg protein separated on SDS-PAGE was transferred onto polyvinylidene difluoride membrane. Membranes were incubated with mouse anti-β-actin antibody and rabbit antibodies against the following: IRP-2, TfR-1, DMT-1, and FPN-1 (see Supplemental Table 2 for antibody specifications). Fluorescent secondary antibodies were applied, and membranes were imaged (Odyssey Infrared Imaging System; LI-COR Biosciences) to determine the intensity of the target protein relative to β-actin (relative optical density). The placental non-heme iron concentration was measured using spectrophotometry on a colorimetric assay of acidified tissue homogenates mixed with a ferrozine/thioglycolic acid chromogen solution that produces a stable colored complex (A590) (47). The ratio of FPN-1 to TfR-1, also known as the placental iron deficiency index, was calculated since it has been shown to be a sensitive measure of iron deficiency of the maternal-fetal unit, with elevated FPN-1:TfR-1 being indicative of maternal iron restriction (48). In normal development, driven by fetal and placental iron demand, IRP-2 increases expression of TfR-1 on the maternal circulation–facing placental surface, which is then internalized via endosomes with subsequent release of iron into the placental cystosol by DMT-1 and transfer of iron to the fetal circulation by FPN-1 (48–51). In iron-replete states, FPN-1, TfR-1, and DMT-1 remain in relative balance. However, when iron availability is restricted on the maternal side, placental TfR-1 and DMT-1 expression are increased and FPN-1 expression is decreased to preserve placental tissue iron (48).
FASD diagnosis
We held diagnostic clinics in 2013 and 2016, in which the infants in our cohort were independently examined for FASD using the Hoyme 2005 criteria (6) by ≥2 members of a team of dysmorphologists led by expert FASD dysmorphologist H.E. Hoyme, MD (52). Examiners were blinded to the PAE history.
Demographics and potential covariates
Women were interviewed regarding their demographic background, including age, gravidity, education, and household food security (53). Weeks gestation at delivery was determined by early pregnancy ultrasound, if available, or by last menstrual period. At the 6.5-month visit, mothers were interviewed regarding the number of weeks the infant was breastfed and whether/when the infant was given formula and other complementary foods.
Statistical analyses
All statistical analyses were 2-sided (α = .05) using SPSS (v.24; IBM), Stata (v.16.1; Statcorp), or SAS (v.9.4, SAS Institute Inc.). All variables were examined for normality of distribution; AA/day and TfR-1, DMT-1, and FPN-1 concentrations and FPN-1:TfR-1 were log-transformed due to skewness (>3.0). Missing data were treated with list-wise deletion. Mixed-model regression analyses with repeated measures were used to examine associations between PAE and serial maternal iron/hematologic outcomes. Linear or logistic regression models were used to examine associations between PAE and maternal hepcidin:creatinine, placental iron transport measures, and infant iron/hematologic outcomes. To compare iron/hematologic outcomes between FASD diagnostic groups, ANCOVA models were used for continuous measures; chi-square analyses were used for categorical measures. Potential covariates for multivariable models examining maternal and placental outcomes included factors known to affect maternal iron status (age, gravidity, cigarette smoking, average daily dietary iron intake, iron supplementation, household food insecurity), energy intake, weeks gestation at visit, and, for red cell indices, erythrocyte folate and serum vitamin B12 concentrations. Potential covariates for infant outcome models included the above factors that affect maternal iron status, as well as prenatal methamphetamine use, gestational hypertension, and age at time of blood draw; for 6.5-month outcome models, weeks breastfeeding, weeks feeding formula, weeks giving complementary foods, and, for red cell indices, erythrocyte folate and serum vitamin B12 concentrations were also considered. Covariates were included in multivariable models if their removal from a model regressing a given outcome on PAE and all potential covariates resulted in a change in the PAE regression coefficient by >10% (54); covariates were chosen for ANCOVA models if their exclusion resulted in a >10% change in the partial eta-squared value. Given the rapid changes in erythrocytes and ferritin that occur in the first few weeks of life (55), age at the time of assessment was included in all neonatal outcome models. Given the potential for collinearity between hepcidin and ferritin and hemoglobin:log(ferritin), which may be driven by common biologic causes, we examined the stability of the relations of PAE to maternal ferritin and hemoglobin:log(ferritin) when adjusting for hepcidin using the method described by Clogg et al. (56). This method estimates whether the change in magnitude of the association between PAE and ferritin or hemoglobin:log(ferritin) induced by adjusting for hepcidin is significant at an α of 0.05.
Effect modification by PAE of associations between placental iron transport measures was examined in regression models relating a placental iron transport outcome to PAE, a placental iron transport measure predictor, and a PAE by placental iron transport measure predictor interaction term. Given the low power of interaction terms in epidemiological studies, effect modification was inferred when the P value for the interaction term was ≤0.10 (57).
To examine the hypothesis that relations of PAE on infant iron measure outcomes are mediated by placental weight, maternal vascular underperfusion, and/or maternal or neonatal iron measures, mediation analyses were conducted based on marginal structural models and the product method (paramed package, Stata) (58, 59). This method allows for estimation of natural direct effects (exposure→outcome, independent of the mediator), natural indirect effects (exposure→outcome, through the mediator), and total effects with exposure-mediator interactions and control for confounders. The marginal structural models employed here allowed for PAE-mediator interactions modeled as drinking frequency increasing from 0 to 3 days/week or heavy drinking (yes or no) and included covariates from the relevant PAE-mediator and mediator-outcome regression models described above. A limitation of this approach is the potential for collider bias, in which the association between the exposure and outcome is distorted by conditioning on a variable on the causal path to the outcome that is causally influenced by the exposure and by additional, possibly unmeasured, confounders. Causal inferences are best supported under the assumptions that the covariates conditioned upon ensure no confounding of 1) the exposure-outcome relation; 2) the exposure-mediator relation; and 3) the mediator-outcome relation. A further important assumption is that the exposure itself does not confound the mediator-outcome relation. Risks of model misspecification, unmeasured/residual confounding, and collider bias are always present in the analysis of observational studies. We therefore view our findings as suggestive of possible mechanistic pathways that warrant investigation in future observational studies involving more extensive data collection or, where possible, experimental settings.
Results
The mothers in the cohort were poorly educated and socioeconomically disadvantaged (Table 1). While 76.1% had attended high school, only 12.2% had graduated. On average, drinkers were 2 years older than controls and had completed 0.7 years less school. Of the drinkers, 61.6% reported low household food security on the USDA food security survey module (53), compared with 40.8% of controls. Few women developed medical problems after recruitment (8.5% hypertension; <5% diabetes or preeclampsia), with no differences between groups, and positive rapid plasma reagin syphilis serology (part of general antenatal screening) was rare, with all receiving prompt treatment and none resulting in congenital syphilis. Drinkers averaged 8.8 standard drinks/occasion on 2.2 days/week around time of conception and 8.6 standard drinks on 1.3 days/week across pregnancy. Binge drinking among drinkers was prevalent, with 93.6% of drinkers averaging at least 4 standard drinks/occasion. Controls abstained from drinking during pregnancy, except for 7 women who reported light drinking (1–3 occasions for 4 women, monthly for 2, once weekly for 1; none of the 7 reported any binge drinking) and 1 woman who drank 4.2 drinks on 1 occasion. Although smoking was prevalent, with drinkers more likely to smoke than controls, the number of cigarettes smoked/day was similar and generally low among both groups, with 82.2% of smokers reporting <0.5 pack/day and 3.1% reporting >1.0 pack/day. Coexposures were prevalent, with 23.0% of heavy drinkers reporting marijuana use and 11.1% reporting methamphetamine use. Most women had full-term pregnancies (87.3% of controls; 88.1% of drinkers). Low birth weight (<2500 g) was more than twice as prevalent among exposed infants. All but 4 mothers breastfed, with most continuing through the 6.5-month follow-up visit, and more than half of the mothers reported supplementing with formula, with no difference between drinkers and controls. There was a trend for controls to have given their infants complementary foods for 2.0 weeks longer on average than drinkers.
TABLE 1.
Maternal and infant sample characteristics
| Controls, n = 80 | Heavy drinkers, n = 126 | All, n = 206 | ||||
|---|---|---|---|---|---|---|
| n | M ± SD or n (%) | n | M ± SD or n (%) | P 1 | M ± SD or n (%) | |
| Maternal characteristics | ||||||
| Maternal age at conception, years | 80 | 25.6 ± 4.8 | 126 | 27.6 ± 6.1 | 0.009 | 26.8 ± 5.7 |
| Gravidity, n live births | 80 | 1.5 ± 1.3 | 126 | 1.8 ± 1.7 | 0.154 | 2.7 ± 1.5 |
| Education, years school completed | 79 | 10.0 ± 1.7 | 126 | 9.3 ± 1.7 | 0.004 | 9.5 ± 1.7 |
| Marital status, n married (%) | 80 | 33 (41.3) | 126 | 35 (27.8) | 0.045 | 68 (33.0) |
| Gestational diabetes, n (%) | 77 | 3 (3.9) | 123 | 5 (4.1) | 0.953 | 8 (4.0) |
| Hypertension, n (%) | 77 | 6 (7.8) | 123 | 11 (8.9) | 0.776 | 17 (8.5) |
| Preeclampsia, n (%) | 77 | 2 (2.6) | 123 | 6 (4.9) | 0.423 | 8 (4.0) |
| Positive rapid plasma reagin, syphilis serology, n (%) | 77 | 1 (1.3) | 124 | 8 (6.5) | 0.086 | 9 (4.5) |
| Food security2 | 76 | — | 120 | — | 0.019 | — |
| High food security, n (%) | — | 36 (47.4) | — | 33 (27.5) | 69 (35.2) | |
| Marginal food security, n (%) | — | 9 (11.8) | — | 13 (10.8) | 22 (11.2) | |
| Low food security, n (%) | — | 11 (14.5) | — | 19 (15.8) | 30 (15.3) | |
| Very low food security, n (%) | — | 20 (26.3) | — | 55 (45.8) | 75 (38.2) | |
| Maternal Alcohol and drug use | ||||||
| Alcohol use at conception: | 80 | — | 126 | — | — | |
| oz AA/day | — | 0.0 ± 0.0 | — | 1.6 ± 1.6 | <0.001 | — |
| oz AA/drinking day | — | 0.1 ± 0.3 | — | 4.4 ± 2.9 | <0.001 | — |
| Drinking days/week | — | 0.1 ± 0.4 | — | 2.2 ± 1.3 | <0.001 | — |
| Alcohol use across pregnancy: | 80 | — | 126 | — | — | |
| oz AA/day | — | 0.0 ± 0.0 | — | 0.9 ± 1.1 | <0.001 | — |
| oz AA/drinking day | — | 0.1 ± 0.4 | — | 4.3 ± 2.3 | <0.001 | — |
| Drinking days/week | — | 0.0 ± 0.1 | — | 1.3 ± 1.0 | <0.001 | — |
| n reporting cigarette smoking (%) | 80 | 55 (68.8) | 126 | 108 (85.7) | 0.004 | 163 (79.1) |
| Cigarettes/day among smokers | — | 6.3 ± 5.9 | — | 6.7 ± 4.1 | 0.618 | 6.5 ± 4.8 |
| n reporting marijuana use (%) | 80 | 8 (10.0) | 126 | 29 (23.0) | 0.018 | 37 (18.0) |
| Days/month marijuana use among users | — | 4.0 ± 4.7 | — | 9.7 ± 9.4 | 0.108 | 8.5 ± 8.8 |
| n reporting methamphetamine use (%) | 80 | 13 (16.3) | 126 | 14 (11.1) | 0.287 | 27 (13.1) |
| Days/month methamphetamine use among users | — | 9.6 ± 8.3 | — | 4.4 ± 5.0 | 0.055 | 6.9 ± 7.2 |
| Infant characteristics | ||||||
| Weeks gestation at delivery | 71 | 39.1 ± 1.8 | 118 | 38.9 ± 1.9 | 0.482 | 39.0 ± 1.8 |
| Preterm delivery (<38 wks), n (%) | — | 9 (12.7) | — | 14 (11.9) | 0.869 | 23 (12.2) |
| Placental weight, g | 48 | 474.7 ± 103.2 | 74 | 415.3 ± 112.1 | 0.004 | 438.6 ± 112.1 |
| Placental maternal vascular underperfusion, n (%) | 48 | 10 (20.8) | 74 | 36 (48.6) | 0.002 | 46 (24.3) |
| Infant sex, n female (%) | 70 | 33 (47.1) | 119 | 65 (54.6) | 0.320 | 98 (51.9) |
| Infant birth weight, g | 70 | 3078.8 ± 534.5 | 118 | 2880.3 ± 568.5 | 0.019 | 2954.2 ± 562.9 |
| Low birth weight (<2500 g), n (%) | — | 8 (11.4) | — | 29 (24.8) | 0.026 | 37 (19.7) |
| Weeks breastfed | 61 | 26.9 ± 7.7 | 103 | 26.5 ± 7.1 | 0.745 | 26.6 ± 7.3 |
| Weeks formula given | 61 | 8.5 ± 10.4 | 103 | 10.4 ± 10.8 | 0.280 | 9.7 ± 10.8 |
| Weeks complementary foods given | 61 | 5.7 ± 7.4 | 103 | 3.7 ± 6.8 | 0.084 | 4.5 ± 7.1 |
1 oz AA = 30 mL AA ≈ 2 standard drinks. Abbreviation: AA, absolute alcohol.
From independent-sample t-tests for continuous outcomes, chi-square tests for binary outcomes, and linear-by-linear association tests for ordinal outcomes.
USDA food security interview (53).
Prenatal alcohol consumption and maternal iron homeostasis
Table 2 presents bivariate comparisons of iron and hematological measures between controls and heavy drinkers. Drinkers and controls reported similar average daily dietary iron intakes (Table 2), with almost all women in both groups reporting an intake below the Dietary Reference Intake estimated average requirement (23 mg/day for women ≤18 years and <22 mg/day for women >18 years) (60). Most women were prescribed prenatal iron/folic acid supplementation (67 mg elemental iron + 5 mg folic acid/day), and >96% in both groups reported taking the supplement on “most days.” No woman had low erythrocyte folate, and low serum vitamin B12 occurred in <20% in both groups.
TABLE 2.
Maternal and infant iron and hematological measures
| Controls, n = 80 | Heavy drinkers, n = 126 | ||||
|---|---|---|---|---|---|
| n | M ± SD or n (%) | n | M ± SD or n (%) | P 1 | |
| Dietary iron intake,2 mg/day | 80 | 12.6 ± 4.9 | 126 | 11.7 ± 4.0 | 0.153 |
| Inadequate intake2 | — | 76 (95.0) | — | 124 (98.4) | 0.210 |
| Iron/folic acid supplementation, n (%) | 78 | 65 (83.3) | 123 | 104 (84.6) | 0.818 |
| n (%) reporting taking supplement on “most days” | — | 63 (96.9) | — | 100 (96.2) | 0.793 |
| Maternal iron indices (study visit 1) | |||||
| Weeks gestation at visit | 79 | 26.3 ± 4.8 | 125 | 23.6 ± 5.8 | <0.001 |
| Ferritin, ug/L | 79 | 15.4 ± 11.6 | 124 | 35.2 ± 36.7 | <0.001 |
| sTfR, mg/L | 76 | 4.2 ± 2.3 | 122 | 3.2 ± 1.7 | 0.001 |
| sTfR:log(ferritin) | 76 | 1.8 ± 1.3 | 121 | 1.2 ± 1.0 | <0.001 |
| C-reactive protein, mg/L | 73 | 5.8 ± 4.2 | 122 | 6.8 ± 7.2 | 0.263 |
| Hemoglobin, g/dL | 79 | 11.0 ± 1.1 | 125 | 11.2 ± 0.9 | 0.082 |
| Anemia3 | 79 | — | 125 | — | 0.029 |
| No anemia, hemoglobin ≥11 g/dL, n (%) | — | 45 (57.0) | — | 78 (62.4) | |
| Moderate, 8.5 ≤ hemoglobin <11 g/dL, n (%) | — | 25 (31.6) | — | 44 (35.2) | |
| Severe, hemoglobin <8.5 g/dL, n (%) | — | 9 (11.4) | — | 3 (2.4) | |
| Mean corpuscular volume, fL | 79 | 87.2 ± 6.7 | 125 | 91.2 ± 7.3 | <0.001 |
| Mean corpuscular hemoglobin, pg | 79 | 29.0 ± 3.0 | 125 | 30.5 ± 2.7 | <0.001 |
| Red cell distribution width, % | 79 | 14.4 ± 1.7 | 125 | 13.8 ± 1.3 | 0.018 |
| Reticulocyte count, % | 79 | 2.5 ± 0.7 | 124 | 2.5 ± 0.7 | 0.793 |
| Hemoglobin:log(ferritin) | 79 | 4.4 ± 0.9 | 124 | 3.8 ± 1.1 | <0.001 |
| Hepcidin, urine; ng/mg creatinine | 42 | 99.2 ± 124.1 | 49 | 156.8 ± 241.5 | 0.166 |
| Iron deficiency4 | 79 | — | 124 | — | 0.001 |
| Iron sufficient, n (%) | — | 41 (51.9) | — | 95 (76.6) | |
| Iron deficiency without anemia, n (%) | — | 15 (19.0) | — | 12 (9.7) | |
| Iron deficiency with anemia, n (%) | — | 23 (29.1) | — | 17 (13.7) | |
| Anemia of inflammation,5n (%) | 79 | 0 (0.0) | 124 | 18 (14.5) | <0.001 |
| Anemia of indeterminate cause, n (%) | 79 | 11 (13.9) | 124 | 30 (24.2 | 0.076 |
| Erythrocyte folate, nmol/L | 79 | 1518.2 ± 592.3 | 125 | 1458.9 ± 619.5 | 0.499 |
| Low erythrocyte folate,6n (%) | — | 0 (0.0) | — | 0 (0.0) | N/A |
| Vitamin B12, pmol/L | 79 | 143.8 ± 90.8 | 124 | 143.5 ± 55.6 | 0.970 |
| Low vitamin B12,7n (%) | — | 13 (16.5) | — | 18 (14.5) | 0.708 |
| Maternal iron indices (study visit 2) | |||||
| Weeks gestation at visit | 46 | 34.6 ± 3.7 | 90 | 32.8 ± 4.2 | 0.015 |
| Ferritin, ug/L | 45 | 10.9 ± 11.0 | 86 | 22.2 ± 25.9 | 0.001 |
| Soluble transferrin receptor, mg/L | 45 | 4.8 ± 2.1 | 87 | 4.3 ± 2.7 | 0.348 |
| sTfR:log(ferritin) | 45 | 2.3 ± 1.9 | 86 | 1.9 ± 1.7 | 0.191 |
| C-reactive protein, mg/L | 45 | 5.9 ± 4.0 | 84 | 7.7 ± 10.4 | 0.252 |
| Hemoglobin, g/dL | 43 | 11.0 ± 1.0 | 87 | 10.9 ± 1.3 | 0.434 |
| Anemia3 | 43 | — | 87 | — | 0.030 |
| No anemia, hemoglobin ≥11 g/dL, n (%) | — | 28 (65.1) | — | 40 (46.0) | |
| Moderate, 8.5 ≤ hemoglobin <11 g/dL, n (%) | — | 13 (30.2) | — | 36 (41.4) | |
| Severe, hemoglobin <8.5 g/dL, n (%) | — | 2 (4.7) | — | 11 (12.6) | |
| Mean corpuscular volume, fL | 43 | 86.3 ± 6.3 | 87 | 88.7 ± 8.2 | 0.092 |
| Mean corpuscular hemoglobin, pg | 43 | 28.7 ± 2.4 | 87 | 29.4 ± 3.2 | 0.242 |
| Red cell distribution width (%) | 43 | 14.1 ± 1.2 | 87 | 14.0 ± 1.4 | 0.760 |
| Reticulocyte count, % | 43 | 2.6 ± 0.6 | 89 | 2.5 ± 0.7 | 0.427 |
| Hemoglobin:log(ferritin) | 43 | 5.0 ± 0.9 | 86 | 4.2 ± 1.1 | <0.001 |
| Iron deficiency4 | 43 | — | 86 | — | 0.057 |
| Iron sufficient, n (%) | — | 10 (23.3) | — | 48 (55.8) | |
| Iron deficiency without anemia, n (%) | — | 21 (48.8) | — | 12 (14.0) | |
| Iron deficiency with anemia, n (%) | — | 12 (27.9) | — | 26 (30.2) | |
| Anemia of inflammation,5n (%) | 43 | 0 (0.0) | 86 | 6 (7.0) | 0.178 |
| Anemia of indeterminate cause, n (%) | 43 | 3 (7.0) | 86 | 20 (23.3) | 0.027 |
| Erythrocyte folate, nmol/L | 44 | 1727.3 ± 694.5 | 88 | 1559.8 ± 618.2 | 0.162 |
| Low erythrocyte folate,6n (%) | — | 0 (0.0) | — | 0 (0.0) | — |
| Vitamin B12, pmol/L | 46 | 131.3 ± 66.6 | 89 | 137.4 ± 51.9 | 0.564 |
| Low vitamin B12,7n (%) | — | 5 (10.9) | — | 3 (3.4) | 0.080 |
| Placental iron transport measures: | |||||
| Iron concentration, ug/g wet tissue | 26 | 145.6 ± 74.2 | 28 | 179.0 ± 103.5 | 0.181 |
| Iron regulatory protein-2, relative OD | 29 | 1.2 ± 0.8 | 27 | 1.3 ± 0.7 | 0.727 |
| Divalent metal transporter-1, OD | 29 | 2.1 ± 2.0 | 28 | 1.8 ± 2.1 | 0.512 |
| TfR-1; OD | 27 | 2.0 ± 2.6 | 24 | 2.5 ± 1.9 | 0.476 |
| FPN-1; OD | 23 | 15.4 ± 34.0 | 25 | 7.6 ± 14.3 | 0.312 |
| FPN-1:TfR-1 | 20 | 24.3 ± 54.3 | 20 | 5.1 ± 7.8 | 0.133 |
| Neonatal iron indices | |||||
| Age at visit, days | 59 | 15.8 ± 8.7 | 99 | 17.8 ± 10.6 | 0.244 |
| Ferritin, ug/L | 58 | 218.8 ± 136.6 | 92 | 243.1 ± 133.2 | 0.282 |
| Hemoglobin, g/dL | 56 | 15.4 ± 2.7 | 91 | 15.1 ± 2.2 | 0.474 |
| Mean corpuscular volume, fL | 55 | 101.3 ± 6.9 | 91 | 102.2 ± 5.8 | 0.426 |
| Mean corpuscular hemoglobin, pg | 55 | 33.4 ± 2.6 | 91 | 34.0 ± 2.1 | 0.115 |
| Red cell distribution width, % | 55 | 16.4 ± 1.1 | 91 | 16.4 ± 0.9 | 0.901 |
| Reticulocyte count, % | 55 | 1.2 ± 0.7 | 90 | 1.2 ± 0.8 | 0.903 |
| 6.5-month iron indices | |||||
| Age at visit, months | 69 | 6.7 ± 0.5 | 113 | 6.7 ± 0.5 | 0.465 |
| Ferritin, ug/L | 69 | 26.1 ± 17.8 | 111 | 26.3 ± 20.2 | 0.950 |
| Soluble transferrin receptor, mg/L | 65 | 6.0 ± 1.8 | 105 | 6.1 ± 2.0 | 0.689 |
| sTfR:log(ferritin) | 65 | 2.2 ± 1.3 | 104 | 2.3 ± 1.4 | 0.713 |
| C-reactive protein, mg/L | 68 | 4.3 ± 6.0 | 109 | 6.5 ± 21.0 | 0.410 |
| Hemoglobin, g/dL | 69 | 10.9 ± 0.9 | 111 | 10.6 ± 1.2 | 0.026 |
| Anemia3 | 69 | — | 111 | — | 0.321 |
| No anemia, hemoglobin ≥11 g/dL, n (%) | — | 35 (50.7) | — | 49 (44.1) | |
| Moderate, 8.5 ≤ hemoglobin <11 g/dL, n (%) | — | 29 (42.0) | — | 46 (41.4) | |
| Severe, hemoglobin <8.5 g/dL, n (%) | — | 5 (7.2) | — | 16 (14.4) | |
| Mean corpuscular volume, fL | 69 | 73.8 ± 4.8 | 111 | 72.7 ± 5.5 | 0.164 |
| Mean corpuscular hemoglobin, pg | 69 | 24.3 ± 2.0 | 111 | 23.8 ± 2.4 | 0.141 |
| Red cell distribution width, % | 69 | 14.9 ± 1.1 | 111 | 15.0 ± 1.4 | 0.475 |
| Reticulocyte count, % | 69 | 2.1 ± 0.8 | 110 | 2.0 ± 0.6 | 0.234 |
| Iron deficiency4 | 69 | — | 109 | — | 0.459 |
| Iron sufficient, n (%) | — | 53 (76.8) | — | 76 (69.7) | |
| Iron deficiency without anemia, n (%) | — | 7 (10.1) | — | 12 (11.0) | |
| Iron deficiency with anemia, n (%) | — | 9 (13.0) | — | 22 (20.2)) | |
| Anemia of inflammation,5n (%) | 69 | 8 (11.6) | 109 | 17 (15.6) | 0.454 |
| Anemia of indeterminate cause, n (%) | 69 | 25 (36.2) | 109 | 38 (34.9) | 0.852 |
| Erythrocyte folate, nmol/L | 68 | 1755.1 ± 685.1 | 111 | 1666.3 ± 631.5 | 0.378 |
| Low erythrocyte folate,6n (%) | — | 0 (0.0) | — | 0 (0.0) | |
| Vitamin B12, pmol/L | 67 | 284.1 ± 167.5 | 112 | 305.1 ± 170.5 | 0.424 |
| Low vitamin B12,7n (%) | — | 14 (20.9) | — | 20 (17.9) | 0.616 |
Abbreviations: FPN-1, ferroportin-1; NA, not applicable; OD, optical density; sTfR, soluble transferrin receptor; TfR-1, transferrin receptor 1.
From independent-sample t-tests for continuous outcomes, chi-square tests for binary outcomes, and linear-by-linear association tests for ordinal outcomes.
From 3 multipass 24-hour recall dietary interviews. Inadequate intake was defined as less than the Dietary Reference Intake estimated average requirement (23 mg/day for women aged ≤ 18 years and <22 mg/day for women >18 years) (60).
As defined by the WHO (42).
Iron deficiency was defined as 1) serum ferritin <12 μg/L; or 2) in cases with C-reactive protein >5 mg/L, elevated sTfR (>8.5 mg/L for mothers; >8.3 mg/L at 6.5 months) (40–42).
Anemia of inflammation was defined as normocytic (mean corpuscular volume ≥80 fL for mothers and ≥71 for infants) anemia with ferritin >30 μg/L (43).
As expected, hemoglobin and ferritin values declined over the course of pregnancy for both drinkers and controls, while the prevalence of anemia and ID increased. Of note, weeks gestation at the study visit was 2–3 weeks earlier on average for drinkers than controls. At recruitment, drinkers had higher ferritin and lower sTfR, sTfR:log(ferritin), and hemoglobin:log(ferritin) than controls, as well as higher MCV and mean corpuscular hemoglobin (MCH) and lower red cell distribution width. Higher ferritin and lower hemoglobin:log(ferritin) among drinkers were still evident 12 weeks later at visit 2, whereas the group differences in sTfR, sTfR:log(ferritin), and red cell indices were no longer evident. There was a lower prevalence of anemia among drinkers than controls at both visits and a trend for higher hemoglobin at visit 1. At both visits, anemia of inflammation was only present among drinkers, and a smaller proportion of drinkers than controls met criteria for ID.
Table 3 presents regression models examining relations between drinking frequency (days/week) to iron and hematological measures. In univariate models, drinking frequency was associated with higher maternal ferritin, lower sTfR and sTfR:log(ferritin), lower hemoglobin:log(ferritin), higher MCV and MCH, a lower prevalence of ID, higher hepcidin, and a greater prevalence of anemia of inflammation. In multivariable models adjusting for covariates, drinking frequency was associated with higher maternal ferritin but was not associated with hemoglobin, sTfR, or sTfR:log(ferritin), a measure of iron stores that is inversely related to total body iron and is less affected by inflammation than sTfR or ferritin alone (64). Drinking frequency was also associated with lower hemoglobin:log(ferritin), higher hepcidin, and an increased prevalence of anemia of inflammation. This constellation of findings suggests a shift of iron into storage over erythropoiesis, possibly due to inflammation. An alcohol-related decrease in ID prevalence was seen, but this finding is difficult to interpret in the setting of potential inflammation, which alters the parameters used for ID diagnosis (43, 65). The associations of PAE with ferritin and hemoglobin:log(ferritin) were statistically dependent on hepcidin; after adjustment for hepcidin, the relation of drinking frequency (days/week) to log(ferritin) (ug/L) decreased significantly, from [anti-log(β) – 1] = 0.30 (all ranges in parentheses are 95% CI's) (0.07–0.57) to [anti-log(β) – 1] = 0.13 (−0.05 to 0.34; coefficient change P = 0.001), and the relation of drinking frequency (days/week) on hemoglobin:log(ferritin) decreased significantly from β = −0.22 (−0.49 to 0.05) to β = −0.06 (−0.32 to 0.19; P = 0.012). Maternal dietary iron intake (mg/day) was associated with higher hemoglobin (g/dL) among control mothers (β = 0.05; 0.00–0.10; P = 0.049) but not drinkers; effect modification of the relation of dietary iron to hemoglobin by alcohol was confirmed in a regression model regressing hemoglobin on dietary iron, heavy drinking (yes/no), and a heavy drinking (yes/no) by dietary iron interaction term (interaction term P = 0.013).
TABLE 3.
Relation of prenatal alcohol exposure to maternal, neonatal, and postnatal infant iron and hematologic outcomes
| Drinking frequency (days/wk) univariate model | Drinking frequency (days/wk) multivariable model1 | Drinking frequency (days/wk) multivariable model1 adjusted for placental weight2 | Drinking frequency (days/wk) multivariable model1 adjusted for maternal vascular underperfusion2 | |
|---|---|---|---|---|
| β (95% CI) | β (95% CI) | β (95% CI) | β (95% CI) | |
| Mothers, n = 126 drinkers, 80 controls | ||||
| Ferritin,3,4,5 % change | 0.286 (0.16–0.42) | 0.216 (0.09–0.34) | — | — |
| sTfR,4,7,8 mg/L | −0.349 (−0.62 to −0.07) | −0.20 (−0.47 to 0.07) | — | — |
| sTfR:log(ferritin)4,7,8 | −0.189 (−0.33 to −0.03) | −0.08 (−0.22 to 0.06) | — | — |
| Hemoglobin,4,5,7,8,10–14 g/dL | 0.05 (−0.08 to 0.18) | 0.02 (−0.02 to 0.05) | — | — |
| Anemia,4,5,13,14 yes = 1, no = 0 | 0.03 (−0.04 to 0.09) | 0.03 (−0.03 to 0.10) | — | — |
| Mean corpuscular volume,4 fL | 2.526 (1.58–3.46) | 2.186 (1.24–3.12) | — | — |
| Mean corpuscular hemoglobin, pg | 0.936 (0.55–1.30) | 0.936 (0.55–1.30) | — | — |
| Red cell distribution width,4,5,7,8,10–14 % | −0.06 (−0.25 to 0.13) | 0.00 (−0.21 to 0.22) | — | — |
| Reticulocyte count,4,5,7,10–14 % | 0.02 (−0.07 to 0.11) | 0.02 (−0.08 to 0.12) | — | — |
| Hemoglobin:log(ferritin)4,5 | −0.296 (−0.41 to −0.16) | −0.2115 (−0.35 to −0.07) | — | — |
| Hepcidin,5,8,10 urine; ng/mg creatinine | 75.9215 (23.77–128.1) | 64.179 (2.37–125.97) | — | — |
| Iron deficiency,4,5,8 yes = 1, no = 0 | −0.116 (−0.16 to −0.05) | −0.079 (−0.13 to −0.01) | — | — |
| Iron deficiency anemia,4,5,7,8,10,11,14,16,17 yes = 1, no = 0 | −0.03 (−0.10 to 0.03) | 0.00 (−0.07 to 0.07) | — | — |
| Anemia of inflammation,5,17 yes = 1, no = 0 | 0.076 (0.04–0.10) | 0.066 (0.02–0.09) | — | — |
| Erythrocyte folate,4,5,10,11 nmol/L | 25.00 (−53.75 to 103.72) | 40.72 (−38.99 to 120.43) | — | — |
| Vitamin B12,4,5,7,11 pmol/L | −3.37 (−11.43 to 4.68) | −5.13 (−13.77 to 3.50) | — | — |
| Placental iron transport, n = 33 exposed, 30 controls | ||||
| Iron concentration,4,5,7,10,11,14,16 ug/g wet tissue | 12.55 (−14.55 to 39.65) | −2.23 (−35.80 to 31.34) | 0.00 (−34.66 to 34.66) | −6.41 (−41.92 to 29.10) |
| Iron regulatory protein-2,3,5,10,11,14,18 % change | −0.09 (−0.36 to 0.14) | −0.14 (−0.43 to 0.12) | −0.07 (−0.35 to 0.16) | −0.14 (−0.45 to 0.12) |
| TfR-1,3,5,7,10,11,14,18 % change | 0.09 (−0.07 to 0.28) | −0.01 (−0.22 to 0.21) | 0.02 (−0.20 to 0.25) | −0.02 (−0.25 to 0.21) |
| Divalent metal transporter-1,3,4,7 % change | −0.05 (−0.22 to 0.11) | −0.08 (−0.27 to 0.08) | −0.06 (−0.22 to 0.09) | −0.09 (−0.28 to 0.08) |
| FPN-1,3,4,5,7,10,11,14,16,18 % change | −0.02 (−0.51 to 0.43) | −0.03 (−0.72 to 0.63) | −0.04 (−0.80 to 0.68) | −0.02 (−0.80 to 0.72) |
| FPN-1:TfR-1,3,4,5,7 % change | −0.4519 (−0.27 to 0.08) | −0.779 (−1.80 to −0.11) | −0.809 (−1.97 to −0.11) | −0.779 (−1.94 to −0.07) |
| Neonates, n = 99 exposed, 59 controls | ||||
| Ferritin,3,20,21 % change | 0.1119 (−0.01 to 0.22) | 0.139 (0.01–0.25) | 0.149 (0.02–0.27) | 0.1219 (0.00–0.25) |
| Hemoglobin,21 g/dL | −0.27 (−0.67 to 0.13) | −0.2919 (−0.60 to 0.03) | −0.3719 (−0.76 to 0.02) | −0.3419 (−0.73 to 0.04) |
| Mean corpuscular volume,21 fL | 0.86 (−0.17 to 1.89) | 0.8219 (−0.13 to 1.76) | 0.33 (−0.70 to 1.37) | 0.83 (−0.23 to 1.90) |
| Mean corpuscular hemoglobin,21 pg | 0.489 (0.10–0.86) | 0.469 (0.10–0.83) | 0.22 (−0.18 to 0.61) | 0.34 (−0.07 to 0.75) |
| Red cell distribution width,5,7,10,18,20,21 % | 0.03 (−0.14 to 0.20) | −0.04 (−0.23 to 0.15) | 0.12 (−0.06 to 0.31) | 0.1619 (−0.01 to 0.34) |
| Reticulocyte count,4,5,7,10,11,16,18,20,21 % | −0.02 (−0.14 to 0.11) | −0.01 (−0.12 to 0.11) | 0.01 (−0.12 to 0.14) | 0.03 (−0.10 to 0.16) |
| Hemoglobin:log(ferritin)5,20,21 | −0.119 (−0.20 to −0.02) | −0.156 (−0.24 to −0.06) | −0.176 (−0.26 to −0.07) | −0.166 (−0.26 to −0.06) |
| 6.5-month-old infants, n = 113 exposed, 69 controls | ||||
| Ferritin,3,5,10,18,20,22 % change | −0.04 (−0.15 to 0.07) | −0.06 (−0.20 to 0.07) | −0.04 (−0.20 to 0.11) | −0.04 (−0.21 to 0.11) |
| sTfR,5,16,18,20–22 mg/L | 0.26 (−0.06 to 0.57) | 0.15 (−0.19 to 0.49) | 0.07 (−0.33 to 0.47) | 0.08 (−0.31 to 0.47) |
| sTfR:log(ferritin)18,20–22 | 0.16 (−0.07 to 0.39) | 0.13 (−0.11 to 0.36) | 0.10 (−0.15 to 0.36) | 0.12 (−0.14 to 0.37) |
| Hemoglobin,20,23 g/dL | −0.199 (−0.34 to −0.03) | −0.189 (−0.33 to −0.02) | −0.249 (−0.44 to −0.05) | −0.229 (−0.40 to −0.03) |
| Anemia5,6,10,11,16,18,20,23 | 0.04 (−0.04 to 0.11) | 0.02 (−0.06 to 0.10) | 0.04 (−0.06 to 0.14) | 0.03 (−0.07 to 0.12) |
| Mean corpuscular volume,4,5,7,10,11,16,18,20,22–25 fL | −0.15 (−0.91 to 0.62) | −0.15 (−1.10 to 0.80) | 0.05 (−1.07 to 0.97) | 0.03 (−0.93 to 0.98) |
| Mean corpuscular hemoglobin,4,5,7,10,11,16,18,20–25 pg | −0.09 (−0.41 to 0.24) | −0.03 (−0.43 to 0.36) | −0.03 (−0.45 to 0.39) | −0.02 (−0.43 to 0.39) |
| Red cell distribution width,5,10,11,16,18,20,22,25,26 % | 0.14 (−0.04 to 0.33) | 0.06 (−0.18 to 0.30) | 0.19 (−0.08 to 0.47) | 0.19 (−0.09 to 0.46) |
| Reticulocyte count,5,7,16,20,22,23 % | −0.07 (−0.17 to 0.03) | −0.06 (−0.18 to 0.07) | −0.10 (−0.24 to 0.05) | −0.09 (−0.24 to 0.06) |
| Hemoglobin:log(ferritin)4,5,7,10,16,18,20–22,25–27 | −0.02 (−0.18 to 0.14) | −0.01 (−0.21 to 0.19) | −0.03 (−0.26 to 0.19) | −0.02 (−0.25 to 0.21) |
| Iron deficiency,5,10,20,22 yes = 1, no = 0 | 0.05 (−0.02 to 0.12) | 0.099 (0.02–0.17) | 0.109 (0.01–0.19) | 0.109 (0.01–0.19) |
| Iron deficiency anemia,5,7,10,16,17,22 yes = 1, no = 0 | 0.0719 (0.00–0.15) | 0.1315 (0.04–0.23) | 0.129 (0.01–0.23) | 0.139 (0.02–0.24) |
| Anemia of inflammation,5,11,17,18,20,21 yes = 1, no = 0 | −0.02 (−0.08 to 0.05) | −0.01 (−0.08 to 0.06) | −0.02 (−0.11 to 0.07) | −0.02 (−0.11 to 0.07) |
| Erythrocyte folate,20,22 nmol/L | −2.26 (−97.27 to 92.75) | −65.98 (−168.15 to 36.19) | −85.20 (−213.38 to 42.98) | −74.11 (−201.52 to 53.30) |
| Vitamin B12,10,20,22 pmol/L | 2.89 (−21.79 to 27.57) | −22.3019 (−48.46 to 3.86) | −21.03 (−48.32 to 6.27) | −20.79 (−48.20 to 6.62) |
β = raw regression coefficients from mixed models with repeated measures for all maternal outcomes, except for hepcidin (measured once) and linear regression models for hepcidin and all placenta and infant outcomes. Abbreviations: FPN-1, ferroportin-1; PAE, prenatal alcohol exposure; sTfR, soluble transferrin receptor; TfR-1, transferrin receptor 1.
The potential covariates for multivariable models examining maternal and placental outcomes included factors known to affect maternal iron status (age, gravidity, cigarette smoking, average daily dietary iron intake, iron supplementation, household food security), energy intake, weeks gestation at visit, and, for red cell indices, erythrocyte folate and serum vitamin B12 concentrations. Potential covariates for infant outcome models included the above maternal factors that affect iron status as well as prenatal methamphetamine use, gestational hypertension, and age at time of blood draw; the 6.5-month outcome models included weeks breastfeeding, weeks feeding formula, weeks giving complementary foods, and, for red cell indices, erythrocyte folate and serum vitamin B12 concentrations were also considered. Covariates were included in multivariable models if their removal from a model regressing a given outcome on PAE and all potential covariates resulted in a change in the PAE regression coefficient by >10%. Given the rapid changes in erythrocytes and ferritin that occur in the first few weeks of life (55), age at time of assessment was included in all neonatal outcome models.
Available for 65 exposed and 41 control infants with neonatal iron measures and 72 exposed and 45 control infants with 6.5-month iron measures.
Values are antilog − 1 of β (95% CI) from models using logged values due to skewness >3.0, signifying % change per each additional day/week of prenatal drinking.
Maternal age was included as a covariate in multivariable models.
Food insecurity was included as a covariate in multivariable models (53).
P ≤ 0.001.
Prenatal cigarettes/d was included as a covariate in multivariable models.
Weeks gestation at visit was included as a covariate in multivariable models.
P ≤ 0.05.
Prenatal iron supplementation was included as a covariate in multivariable models (yes = 1, no = 0).
Dietary iron intake (mg/day) was included as a covariate in multivariable models.
Maternal erythrocyte folate (nmol/L) was included as a covariate in multivariable models.
Maternal serum vitamin B12 (pmol/L) was included as a covariate in multivariable models.
Energy intake (kcal/day) was included as a covariate in multivariable models.
P ≤ 0.01.
Gravidity was included as a covariate in multivariable models.
Those with anemia of indeterminate cause were excluded.
Weeks gestation at delivery was included as a covariate in multivariable models.
P ≤ 0.10.
Days/month prenatal methamphetamine use was included as a covariate in multivariable models.
Infant age at time of blood draw was included as a covariate in multivariable models.
Weeks infant fed formula was included as a covariate in multivariable models.
6.5-month infant serum vitamin B12 (pmol/L) was included as a covariate in multivariable models.
6.5-month infant erythrocyte folate (nmol/L) was included as a covariate in multivariable models.
Weeks infant fed complementary foods was included as a covariate in multivariable models.
Weeks infant breastfed was included as a covariate in multivariable models.
Maternal gestational hypertension was included as a covariate in multivariable models.
Placental development and iron transport proteins
Placentas from heavy drinkers had smaller weights than those from controls (mean difference = −59.4 g; −99.3 to −19.5) and a higher prevalence of placental maternal vascular underperfusion (48.6 compared with 20.8%, respectively; Table 1), both of which may decrease nutrient transfer to the fetus. In multivariable regression models (Table 3), drinking frequency (days/week) was not related to any individual iron transport measure but was associated with lower FPN-1:TfR-1 (β = −0.57; −1.03 to −0.10), indicating iron restriction on the maternal side. Supplemental Table 3 presents interrelations between placental tissue iron and iron transport protein concentrations, stratified by alcohol exposure. The expected associations between IRP-2 and TfR-1 and DMT-1 and TfR-1 were seen among both control and exposed placentas. DMT-1 was associated with FPN-1 among control but not exposed placentas; effect modification by PAE of the relation of DMT-1 to FPN-1 was confirmed by regression models with an exposure (yes/no) by DMT-1 [logged optical density (OD) values] interaction term {Antilog[β(exposure)] − 1 = 2.56 (−0.09 to 12.87); antilog(β[lg(DMT-1)]) − 1 = 5.11 (1.20–15.94); antilog[β(interaction)] − 1 = −2.32 (−11.30 to 0.13); P(interaction) = 0.073}. In regression models including an exposure (yes/no) by placental iron interaction term, placental iron (mg/g tissue) was associated with FPN-1 expression (logged OD values) in the presence of PAE {Antilog[β(exposure)] − 1 = −5.95 (−27.50 to 0.15); antilog[β(iron)] − 1 = −0.01 (−0.02 to 0.00); antilog[β(interaction)] − 1 = 0.01 (0.00–0.02); P(interaction) = 0.051}, consistent with an iron-restricted state, in which lower placental iron is associated with lower FPN-1 expression.
PAE and infant iron status
Ferritin and hemoglobin concentrations were appropriate for the neonatal period (Table 2) (55). Maternal drinking frequency was related to lower neonatal hemoglobin:log(ferritin) and higher MCH in univariate analyses (Table 3), as well as higher ferritin at a trend. In multivariable models, drinking frequency was related to higher neonatal ferritin and lower hemoglobin:log(ferritin), indicating sequestration of iron into storage at the expense of hemoglobin in a pattern similar to that seen in the mothers. Drinking frequency was also related to higher neonatal MCH, as well as trends for higher MCV and lower hemoglobin. Associations at age 2 weeks were not mediated by PAE-related reductions in placental weight or increased maternal vascular perfusion. In causal inference analyses (Figure 1A), the association of PAE to decreased neonatal hemoglobin:log(ferritin) was partially mediated by the relation of PAE to decreased maternal hemoglobin:log(ferritin) [natural indirect effect of PAE (drinkers compared with controls) through maternal hemoglobin:log(ferritin) β = −0.08 (−0.16 to 0.00; P = 0.043); total effects β = −0.21 (−0.40 to 0.03; P = 0.025); proportion mediated = natural indirect/total effects = 38.1%].
FIGURE 1.
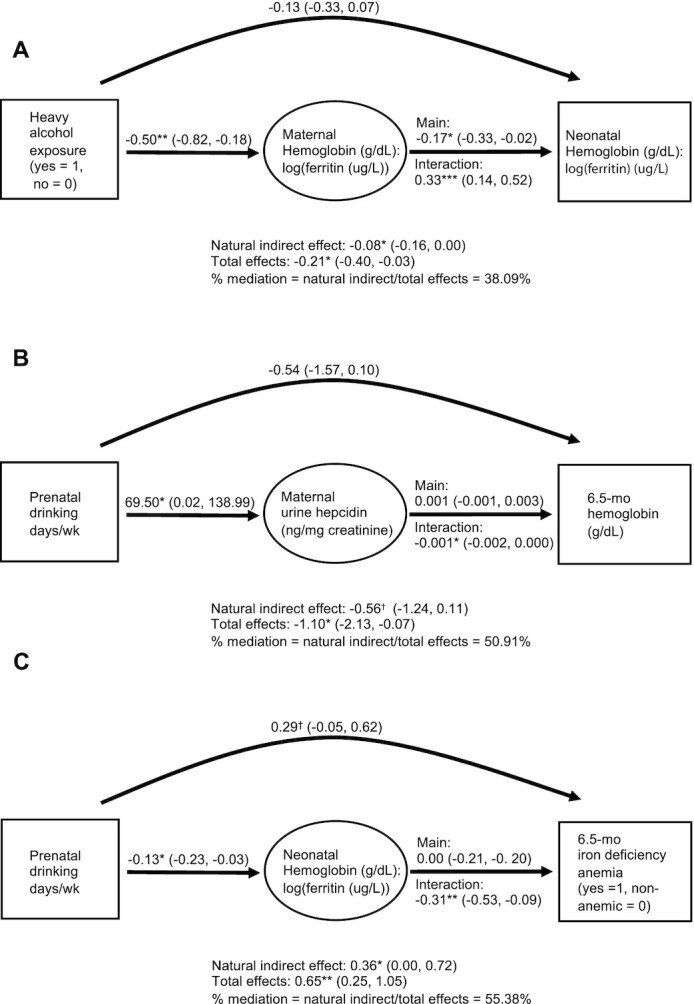
Causal mediation analyses. Directed acyclic graphs demonstrating: (A) maternal hemoglobin-to-log(ferritin) ratio as a mediator of the relation between heavy alcohol exposure and neonatal hemoglobin-to-log(ferritin) ratio. (B) Maternal hepcidin as a mediator of the relation between drinking frequency and 6.5-month infant hemoglobin. (C) Neonatal hemoglobin-to-log(ferritin) ratio as a mediator of the relation between drinking frequency and infant iron deficiency anemia at 6.5 months. Values are β (95% CI) from marginal structural models adjusting for covariates chosen from exposure-mediator and exposure-outcome regression models using 10% change-in-estimate criteria as described in the Methods section. [(A) maternal age, food insecurity (53), prenatal methamphetamine use, and infant age; (B) weeks gestation at time of hepcidin measurement, prenatal iron supplementation, prenatal methamphetamine use, food insecurity, and 6.5-month serum B12; and (C) gravidity, prenatal cigarette smoking, prenatal methamphetamine use, prenatal iron supplementation, food insecurity, infant age, and weeks formula given]. Interaction effects for drinking days/week were modeled as drinking frequency increasing from 0 to 3 days/week. †P ≤ 0.10; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
At 6.5 months, mean hemoglobin was 0.3 g/dL lower among infants born to heavy drinkers than controls (Table 2), and drinking frequency was related to lower hemoglobin in univariate regression models (Table 3). In multivariable models, PAE was associated with lower hemoglobin and a higher risk of ID and IDA. Placental size and maternal vascular underperfusion did not statistically mediate these associations. There was a trend for mediation of the association of drinking frequency to reduced 6.5-month hemoglobin (g/dL) by the PAE-related increase in maternal hepcidin [Figure 1B; natural indirect effect of drinking frequency (days/week) through maternal hepcidin β = −0.56 (−1.24 to 0.11; P = 0.103); total effects β = −1.10 (−2.13, −0.07), P = 0.037; proportion mediated = natural indirect/total effects = 50.9%]. The association between drinking frequency and 6.5-month IDA was partially mediated by the relation of drinking frequency to lower neonatal hemoglobin:log(ferritin) [Figure 1C; natural indirect effect of drinking frequency (days/week) through neonatal hemoglobin:log(ferritin) β = 0.36 (95% CI: 0.00–0.72; P = 0.048); total effects β = 0.65 (95% CI: 0.25–1.05; P = 0.002); proportion mediated = natural indirect/total effects = 55.4%].
In ANCOVA models adjusting for covariates (Table 4), when compared to controls, children later diagnosed with FAS had higher ferritin at 2 weeks and lower ferritin and higher sTfR:log(ferritin) at 6.5 months, indicative of lower iron stores (64). A trend for lower 6.5-month hemoglobin among those with FAS was also seen. Children with FAS or PFAS had higher prevalences of anemia and IDA than nonsyndromal PAE-exposed and control children.
TABLE 4.
Infant iron status measures among 6.5-month FASD diagnostic groups
| Controls | Exposed nonsyndromal | PFAS | FAS | Post hoc FAS vs. controls | |||
|---|---|---|---|---|---|---|---|
| M1 (95% CI) | M1 (95% CI) | M1 (95% CI) | M1 (95% CI) | P 1 | Mean difference1 (95%CI) | P 1 | |
| Neonatal period | n = 57 | n = 73 | n = 6 | n = 19 | — | ||
| Ferritin,2–4 ug/L | 163.0 (147.4–199.3) | 199.3 (180.4–243.7) | 269.4 (163.0–444.9) | 243.7 (180.4–329.3) | 0.108 | 80.7 (5.6–168.5) | 0.046 |
| Hemoglobin,4–7 g/dL | 15.2 (14.7– 15.7) | 15.2 (14.8–15.7) | 13.9 (12.0–15.7) | 15.1 (14.2– 15.9) | 0.581 | −0.1 (−1.1 to 0.9) | 0.856 |
| Hemoglobin:log(ferritin) 3–5 | 3.0 (2.9–3.1) | 2.9 (2.7–3.0) | 2.5 (2.0–3.0) | 2.8 (2.5–3.0) | 0.091 | −0.2 (−0.5 to 0.1) | 0.097 |
| 6.5 months | n = 68 | n = 81 | n = 7 | n = 25 | — | ||
| Ferritin,2–4,6 ug/L | 21.2 (17.2–23.5) | 21.2 (19.1–26.1) | 26.1 (15.4–49.4) | 12.5 (9.0–17.2) | 0.011 | −8.7 (−12.2 to −2.2) | 0.009 |
| sTfR,3,4,6,8–12 mg/L | 6.1 (5.6–6.6) | 5.9 (5.5–6.3) | 6.1 (4.6–7.7) | 6.6 (5.7–7.5) | 0.498 | 0.5 (−0.5 to 1.6) | 0.316 |
| sTfR:log(ferritin) 3,4,6,11,13 | 2.3 (1.9–2.6) | 2.1 (1.8–2.4) | 1.7 (0.6–2.7) | 3.2 (2.6–3.7) | 0.009 | 0.9 (0.2–1.6) | 0.010 |
| Hemoglobin,3–6,9–11,14 g/dL | 10.8 (10.6–11.1) | 10.6 (10.4–10.9) | 10.5 (9.5–11.5) | 10.3 (9.9–10.8) | 0.343 | −0.5 (−1.0 to 0.1) | 0.081 |
| Hemoglobin:log(ferritin) 3–6 | 3.7 (3.5–4.0) | 3.7 (3.4–3.9) | 3.3 (2.2–4.4) | 4.2 (3.7–4.6) | 0.198 | 0.4 (0.1–1.0) | 0.104 |
| Anemia | — | — | — | — | 0.01015 | — | |
| No anemia, hemoglobin ≥11 g/dL | 35 (51.5) | 38 (48.1) | 2 (28.6) | 9 (36.0) | — | ||
| Moderate, 8.5 ≤ hemoglobin <11 g/dL | 28 (41.2) | 34 (43.0) | 3 (42.9) | 9 (36.0) | — | ||
| Severe, hemoglobin <8.5 g/dL | 5 (7.4) | 7 (8.9) | 2 (28.6) | 7 (28.0) | — | ||
| Iron deficiency,16 | — | — | — | — | 0.00215 | — | |
| Iron sufficient | 28 (63.6) | 28 (60.9) | 2 (5.0) | 7 (33.3) | — | ||
| Iron deficiency without anemia | 7 (15.9) | 10 (21.7) | 0 (0.0) | 2 (9.5) | — | ||
| Iron deficiency with anemia | 9 (20.5) | 8 (17.4) | 2 (50.0) | 12 (57.1) | — | ||
| Anemia of inflammation16 | 8 (15.7) | 14 (23.0) | 2 (33.3) | 1(4.5) | 0.35515 | — | |
Abbreviations: FAS, fetal alcohol syndrome; FASD, fetal alcohol spectrum disorders; PFAS, partial fetal alcohol syndrome; sTfR, soluble transferrin receptor.
From ANCOVA models. Potential covariates included factors known to affect maternal iron status (age, gravidity, cigarette smoking, average daily dietary iron intake, iron supplementation, household food insecurity (53)), prenatal methamphetamine use, gestational hypertension, age at time of blood draw; for 6.5-month outcome models, weeks breastfeeding, weeks formula feeding, and weeks giving complementary foods were each considered as well. Covariates were included if their removal from an ANCOVA model including all potential covariates for a given outcome resulted in a change in the partial eta-squared value for FASD diagnostic group by >10%. Given the rapid changes in erythrocytes and ferritin that occur in the first few weeks of life (55), age at time of assessment was included in all neonatal outcome models.
Values are antilog − 1 of estimates from models using logged values due to skewness (>3.0).
Maternal prenatal methamphetamine use (days/mo) was included in ANCOVA models.
Age at time of blood draw was included in ANCOVA models.
Food insecurity was included in ANCOVA models.
Maternal gestational hypertension was included in ANCOVA models.
Prenatal iron supplementation (yes = 1, no = 0) was included in ANCOVA models.
Maternal age was included in ANCOVA models.
Gravidity was included in ANCOVA models.
Dietary iron intake (mg/day) was included in ANCOVA models.
Weeks gestation at delivery was included in ANCOVA models.
Weeks infant breastfed was included in ANCOVA models.
Weeks infant fed formula was included in ANCOVA models.
Prenatal cigarettes/d was included in ANCOVA models.
Linear-by-linear association.
Those with anemia of indeterminate cause were excluded.
Discussion
To our knowledge, this is the first human study to examine the impact of maternal alcohol consumption on iron homeostasis in the mother, placenta, neonate, and postnatal infant. In this prospective longitudinal cohort with serial iron/hematological measures, PAE was associated with a shift of iron into storage over erythropoiesis in both the mother and neonate; iron-restricted placental iron transport; and increased risks of ID and IDA in the infant at age 6.5 months. These associations were independent of a range of known risk factors for maternal and infant anemia and ID and were apparent despite iron supplementation at doses above WHO recommendations. These findings confirm our previous reports of alcohol-related infant IDA and demonstrate effects of alcohol on the mother, placenta, and neonate that appear to underlie effects of PAE on later iron status.
Alcohol consumption was related to higher maternal ferritin, which is indicative of iron in storage, and higher hepcidin, the negative regulatory hormone in iron homeostasis. Elevations in ferritin and hepcidin occur together when higher total body iron induces hepcidin expression or when inflammation increases expression of both ferritin and hepcidin (66, 67). Hepcidin production is profoundly suppressed in pregnancy, but this suppression may be reversed by inflammation (68, 69). If higher total body iron were the underlying cause of alcohol-related increases in maternal ferritin and hepcidin, one would expect an associated increase in hemoglobin and decrease in sTfR:log(ferritin), neither of which were seen. Rather, alcohol was related to decreased hemoglobin-to-log(ferritin) ratio, indicating sequestration of iron into storage at the expense of erythropoiesis and possibly other processes, such as placental iron transfer. This sequestration of iron into storage was statistically dependent on elevations in maternal hepcidin and is consistent with an inflammatory state (68). Alcohol consumption was also related to an increased prevalence of maternal anemia of inflammation. Our finding of an alcohol-related increase in hepcidin is inconsistent with the results of a number of studies that have documented decreased hepcidin production and resultant iron overload among heavy drinkers (26, 27). This discrepancy may reflect a hepcidin response to alcohol that is specific to pregnancy or may be due to our cohort being comprised of younger, episodic binge drinkers, whereas the majority of studies demonstrating alcohol-related iron overload have been in older alcoholics with many years of alcohol abuse and/or alcoholic liver disease. Alcohol has been associated with folate/vitamin B12-independent increases in MCV and MCH, as seen in this study, which are thought to be the result of direct toxic effects of alcohol and its metabolites on erythrocytes (23–25).
In the neonate, a similar PAE-related shift of iron into storage at the expense of hemoglobin production was seen, with increased ferritin, a trend for decreased hemoglobin, and decreased hemoglobin-to-log(ferritin) ratio. The relation of PAE to decreased neonatal hemoglobin-to-log(ferritin) ratio was partially mediated by decreased maternal hemoglobin-to-log(ferritin) ratio. These findings are consistent with those of Smith and colleagues’ (11–13) FASD rat model study findings, in which PAE-related, inflammation-induced increases in maternal and fetal hepcidin were seen, as well as a shift of iron from the fetal brain and red blood cells to liver storage. Heavy alcohol consumption has been associated with a pro-inflammatory state in both humans and animals (13, 70), including PAE-related increases in maternal and fetal IL-6, IL-1β, and TNFα, the chief inflammatory stimulators of hepcidin and ferritin (71). Given the pattern of heavy alcohol consumption on weekends in this cohort, it is not surprising that drinking was not related to CRP, a marker of acute inflammation measured on weekdays; markers of chronic inflammation were not available. Our finding that alcohol consumption was related to a lower prevalence of maternal ID likely reflects limitations in accurately identifying ID in the setting of inflammation among drinkers rather than a true decrease in ID, since ferritin and sTfR (albeit less so) are poor indicators of iron status when inflammation is present (43, 65). Drinking was not associated with maternal sTfR:log(ferritin), a measure of iron stores thought to be less affected by inflammation (64).
The sequestration of iron into storage seen in the neonatal period did not persist through 6.5 months, suggesting that potential PAE-related inflammatory insults were limited to the in utero and early neonatal periods. However, PAE-related disruptions in maternal and fetal iron homeostasis appeared to play important mechanistic roles in postnatal PAE-related ID. At age 6.5 months, PAE was associated with decreased hemoglobin and increases in ID and IDA, and children later diagnosed with FAS/PFAS had lower 6.5-month ferritin, higher sTfR:log(ferritin), and higher prevalences of anemia and ID. The association of PAE to IDA at 6.5 months was partially mediated by the neonatal shift of iron into storage at the expense of hemoglobin, and there was a trend for the relation of PAE to reduced 6.5-month hemoglobin to be partially mediated by increases in maternal hepcidin, though true causation cannot be inferred in this observational study (as noted in Methods).
PAE was associated with lower FPN-1:TfR-1, indicative of insufficient iron on the maternal circulation side of the placenta, when placental TfR-1 is upregulated to enhance absorption of iron from the mother and FPN-1 is downregulated to sequester iron in the placenta, which is needed for cellular function even at the expense of fetal iron stores (48). Restricted iron on the maternal side may have been the result of PAE-related increases in maternal hepcidin and/or decreases in hemoglobin-to-log(ferritin) ratio. Indeed, maternal hepcidin has been shown to decrease placental iron transfer to the fetus (68, 69). The PAE-related reduction in placental size and the increase in maternal vascular underperfusion we previously reported (5) did not mediate relations of PAE to placental FPN-1:TfR-1 or infant iron homeostasis.
This study had limitations common to other longitudinal studies of prenatal exposures. The small sample size, measurement error surrounding exposure estimates and covariates, model misspecification, and residual confounding likely led to unmeasured bias and Type 1 (false positive) and Type 2 (false negative) results. Furthermore, the large numbers of maternal, placental, neonatal, and infant iron/hematological measures examined in this study may have caused false-positive findings (Type 1 errors), and our results should thus be interpreted based on their collective pattern. We have previously demonstrated the predictive validity of the timeline follow-back interview used (38) and a high concordance of maternal drug use reporting and urine testing (5). Although we were unable to ascertain dietary intake on Friday or Saturday, when women were more likely to drink, previous sensitivity analyses showed that even a worst-case scenario, in which women replaced food with alcohol, did not cause clinically significant differences in micronutrient intake (29). Moreover, most women took prenatal iron supplementation at a dose 5.5 times the cohort average for daily dietary iron intake. PAE was very high in this cohort, and potential effects of PAE on iron homeostasis at light-to-moderate levels of PAE need to be studied. Furthermore, our findings may not apply to mother-infant pairs meeting our pre- and postrecruitment exclusionary criteria (e.g., twin pregnancy, extreme prematurity, very low birth weight, infant seizure disorders, etc.).
Given the range of long-term neurodevelopmental deficits that result from ID, our findings of PAE-related iron-restricted placental iron transport, changes in neonatal iron homeostasis, and increased prevalences of postnatal ID and IDA indicate that infants with PAE may warrant screening for ID/IDA in early infancy (16–18). Furthermore, alcohol-related alterations in iron homeostasis may play major roles in the pathogenesis of FASD. PAE-related infant ID has been shown to exacerbate fetal alcohol growth restriction in both rats and humans (3, 11, 15). Since iron is prioritized for hemoglobin synthesis above other tissues, by the time decreases in hemoglobin are seen, tissue-level iron inadequacy in the brain is likely to be well established (72). Given the critical roles of iron in neuronal development and myelination, PAE-related disruptions in iron homeostasis may play mechanistic roles in the teratogenic effects of alcohol. ID effects on growth and neurobehavior have remarkable overlap with those seen in FASD (18–21), and studies are needed to examine whether PAE disruptions in iron homeostasis mediate and/or exacerbate FASD neurodevelopmental deficits in humans. In Smith's FASD rat model studies, maternal iron supplementation ameliorated virtually all effects of PAE on maternal and infant iron homeostasis. Future studies are needed to examine whether pre- and/or postnatal iron supplementation in humans may mitigate PAE-related disruptions in iron homeostasis. However, high and/or intermittent dosing regimens may need to be considered given that the majority of our study participants reported taking prenatal iron supplements and that iron supplements taken daily may increase maternal hepcidin.
Supplementary Material
Acknowledgments
We thank our University of Cape Town and Wayne State University research staff, including Maggie September, Beverley Arendse, Patricia O'Leary, and Patricia Solomon, for their work on subject recruitment and retention; Catherine Lewis, Nadine Lindinger, and Stacey Hall, who performed the measurements of the infants; dietary interviewers Catherine Day, Monika Uys, and Nicola Cooper; Lori Bechard for her computation of dietary data; Marjanne Senekal for her input on dietary data collection and analysis; and Renee Sun, for her assistance with data processing. We thank Elizabeth Leibold for the donation of the iron regulatory protein 2 antibody and the medical technologist Annelie Visser for her assistance with pathology examinations. We thank H. Eugene Hoyme, who examined the children at our 2013 and 2016 fetal alcohol spectrum disorders dysmorphology diagnostic clinics, and Greetje de Jong, Heidre Bezuidenhout, Emma Krzesinski, Prachi Shah, and Vikas Kodali, who assisted him. We also thank Susan Fawcus, Head of the Department of Obstetrics, Mowbray Maternity Hospital; the nursing and records department staff at the Hanover Park and Retreat Midwife Obstetric Units, Mowbray Maternity Hospital, Somerset Hospital, and Groote Schuur Hospital, where the mothers were recruited and the infants were born; and the staff in the National Health Laboratory Service Anatomical Pathology Division at Groote Schuur Hospital. We thank Richard Cook for his consultation regarding the statistical methods. Lastly, we extend our deep appreciation to our Cape Town study participants for their contributions to this study.
Author disclosures: CPD is an Editor for The American Journal of Clinical Nutrition and played no role in the Journal’s evaluation of the manuscript. All other authors report no conflicts of interest.
The authors’ responsibilities were as follows—RCC, SWJ, JLJ, MKG, CPD, and CDM: conceptualized the study design; SWJ, JLJ, RCC, and CDM: oversaw recruitment and assessment of the longitudinal birth cohort, data acquisition and analysis, and interpretation of findings; CDM and EMM: coordinated and supervised data collection; HW: conducted the placental histopathology examinations and assisted with interpretation of results; KME and MKG: performed placenta laboratory assays and assisted with interpretation of results; NCD: coordinated data management and assisted with statistical analyses; RCC: conducted the data analyses and wrote the manuscript; SWJ, JLJ, MKG, and CPD: edited and revised the manuscript; and all authors: read and approved the final manuscript.
Notes
This study was funded by the NIH/National Institute on Alcohol Abuse and Alcoholism (R01AA016781, R21AA022203, K23AA020516; and R01AA027916) and the Lycaki-Young Fund (State of Michigan).
Supplemental Tables 1–3 and Supplemental Figure 1 are available from the “Supplementary data” link in the online posting of the article and from the same link in the online table of contents at https://academic.oup.com/ajcn/.
Abbreviations used: β, raw regression coefficient; CRP, C-reactive protein; DMT-1, divalent metal transporter-1; FAS, fetal alcohol syndrome; FASD, fetal alcohol spectrum disorders; FPN-1, ferroportin-1; ID, iron deficiency; IDA, iron deficiency anemia; IRP-2, iron regulatory protein 2; MCH, mean corpuscular hemoglobin; MCV, mean corpuscular volume; OD, optical density; PAE, prenatal alcohol exposure; sTfR, soluble transferrin receptor; TfR-1, transferrin receptor 1.
Contributor Information
R Colin Carter, Institute of Human Nutrition and Departments of Emergency Medicine and Pediatrics, Columbia University Irving Medical Center, New York, NY, USA.
Michael K Georgieff, Department of Pediatrics, University of Minnesota Medical School, Minneapolis, MN, USA.
Kathleen M Ennis, Department of Pediatrics, University of Minnesota Medical School, Minneapolis, MN, USA.
Neil C Dodge, Department of Psychiatry and Behavioral Neurosciences, Wayne State University School of Medicine, Detroit, MI, USA.
Helen Wainwright, National Health Laboratory Service, Department of Pathology, Groote Schuur Hospital, Cape Town, South Africa.
Ernesta M Meintjes, Department of Human Biology, University of Cape Town Faculty of Health Sciences, Cape Town, South Africa.
Christopher P Duggan, Division of Gastroenterology, Hepatology, and Nutrition, Boston Children's Hospital, Boston, MA, USA.
Christopher D Molteno, Department of Psychiatry and Mental Health, University of Cape Town Faculty of Health Sciences, Cape Town, South Africa.
Joseph L Jacobson, Department of Psychiatry and Behavioral Neurosciences, Wayne State University School of Medicine, Detroit, MI, USA; Department of Human Biology, University of Cape Town Faculty of Health Sciences, Cape Town, South Africa.
Sandra W Jacobson, Department of Psychiatry and Behavioral Neurosciences, Wayne State University School of Medicine, Detroit, MI, USA; Department of Human Biology, University of Cape Town Faculty of Health Sciences, Cape Town, South Africa; Department of Psychiatry and Mental Health, University of Cape Town Faculty of Health Sciences, Cape Town, South Africa.
Data Availability
Deidentified, individual participant data reported in this article (text, tables, figures, and appendices) and the study protocol, statistical analysis plan, and analytic code will be available for sharing to journal editors either before or after publication for checking and to researchers who provide a methodologically sound proposal, as determined by the authors of this article. Proposals from researchers should be directed to SWJ (sandra.jacobson@wayne.edu).Data will be stored in a data warehouse at Wayne State University and transmitted electronically in encrypted form to requestors. Data requestors will need to sign a data access agreement prior to access.
References
- 1.May PA, Baete A, Russo J, Elliott AJ, Blankenship J, Kalberg WO, Buckley D, Brooks M, Hasken J, Abdul-Rahman Oet al. Prevalence and characteristics of fetal alcohol spectrum disorders. Pediatrics. 2014;134(5):855–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.May PA, Chambers CD, Kalberg WO, Zellner J, Feldman H, Buckley D, Kopald D, Hasken JM, Xu R, Honerkamp-Smith Get al. Prevalence of fetal alcohol spectrum disorders in 4 US communities. JAMA. 2018;319(5):474–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carter RC, Jacobson JL, Molteno CD, Jiang H, Meintjes EM, Jacobson SW, Duggan C. Effects of heavy prenatal alcohol exposure and iron deficiency anemia on child growth and body composition through age 9 years. Alcohol Clin Exp Res. 2012;36(11):1973–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carter RC, Jacobson JL, Sokol RJ, Avison MJ, Jacobson SW. Fetal alcohol-related growth restriction from birth through young adulthood and moderating effects of maternal prepregnancy weight. Alcohol Clin Exp Res. 2013;37(3):452–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carter RC, Wainwright H, Molteno CD, Georgieff MK, Dodge NC, Warton F, Meintjes EM, Jacobson JL, Jacobson SW. Alcohol, methamphetamine, and marijuana exposure have distinct effects on the human placenta. Alcohol Clin Exp Res. 2016;40(4):753–64. [DOI] [PubMed] [Google Scholar]
- 6.Hoyme HE, May PA, Kalberg WO, Kodituwakku P, Gossage JP, Trujillo PM, Buckley DG, Miller JH, Aragon AS, Khaole Net al. A practical clinical approach to diagnosis of fetal alcohol spectrum disorders: Clarification of the 1996 Institute of Medicine criteria. Pediatrics. 2005;115(1):39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jacobson SW. Specificity of neurobehavioral outcomes associated with prenatal alcohol exposure. Alcohol Clin Exp Res. 1998;22(2):313–20. [DOI] [PubMed] [Google Scholar]
- 8.Carter RC, Jacobson JL, Molteno CD, Dodge NC, Meintjes EM, Jacobson SW. Fetal alcohol growth restriction and cognitive impairment. Pediatrics. 2016;138(2):e20160775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoyme HE, Kalberg WO, Elliott AJ, Blankenship J, Buckley D, Marais A-S, Manning MA, Robinson LK, Adam MP, Abdul-Rahman Oet al. Updated clinical guidelines for diagnosing fetal alcohol spectrum disorders. Pediatrics. 2016;138(2):e20154256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller MW, Roskams AJ, Connor JR. Iron regulation in the developing rat brain: Effect of in utero ethanol exposure. J Neurochem. 1995;65(1):373–80. [DOI] [PubMed] [Google Scholar]
- 11.Huebner SM, Blohowiak SE, Kling PJ, Smith SM. Prenatal alcohol exposure alters fetal iron distribution and elevates hepatic hepcidin in a rat model of fetal alcohol spectrum disorders. J Nutr. 2016;146(6):(1180–8.). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huebner SM, Helfrich KK, Saini N, Blohowiak SE, Cheng AA, Kling PJ, Smith SM. Dietary iron fortification normalizes fetal hematology, hepcidin, and iron distribution in a rat model of prenatal alcohol exposure. Alcohol Clin Exp Res. 2018;42(6):1022–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saini N, Helfrich KK, Kwan STC, Huebner SM, Abazi J, Flentke GR, Blohowiak SE, Kling PJ, Smith SM. Alcohol's dysregulation of maternal-fetal IL-6 and p-STAT3 is a function of maternal iron status. Alcohol Clin Exp Res. 2019;43(11):2332–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rufer ES, Tran TD, Attridge MM, Andrzejewski ME, Flentke GR, Smith SM. Adequacy of maternal iron status protects against behavioral, neuroanatomical, and growth deficits in fetal alcohol spectrum disorders. PLoS One. 2012;7(10):e47499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carter RC, Jacobson SW, Molteno CD, Jacobson JL. Fetal alcohol exposure, iron-deficiency anemia, and infant growth. Pediatrics. 2007;120(3):559–67. [DOI] [PubMed] [Google Scholar]
- 16.Lozoff B, Beard J, Connor J, Barbara F, Georgieff M, Schallert T. Long-lasting neural and behavioral effects of iron deficiency in infancy. Nutr Rev. 2006;64(5):S34–43.; discussion S72–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carter RC, Jacobson JL, Burden MJ, Armony-Sivan R, Dodge NC, Angelilli ML, Lozoff B, Jacobson SW. Iron deficiency anemia and cognitive function in infancy. Pediatrics. 2010;126(2):e427–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Algarín C, Peirano P, Garrido M, Pizarro F, Lozoff B. Iron deficiency anemia in infancy: long-lasting effects on auditory and visual system functioning. Pediatr Res. 2003;53(2):217–23. [DOI] [PubMed] [Google Scholar]
- 19.Jacobson SW, Stanton ME, Molteno CD, Burden MJ, Fuller DS, Hoyme HE, Robinson LK, Khaole N, Jacobson JL. Impaired eyeblink conditioning in children with fetal alcohol syndrome. Alcohol Clin Exp Res. 2008;32(2):365–72. [DOI] [PubMed] [Google Scholar]
- 20.Jacobson SW, Stanton ME, Dodge NC, Pienaar M, Fuller DS, Molteno CD, Meintjes EM, Hoyme HE, Robinson LK, Khaole Net al. Impaired delay and trace eyeblink conditioning in school-age children with fetal alcohol syndrome. Alcohol Clin Exp Res. 2011;35(2):250–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McEchron MD, Alexander DN, Gilmartin MR, Paronish MD. Perinatal nutritional iron deficiency impairs hippocampus-dependent trace eyeblink conditioning in rats. Dev Neurosci. 2008;30(4):243–54. [DOI] [PubMed] [Google Scholar]
- 22.Huebner SM, Tran TD, Rufer ES, Crump PM, Smith SM. Maternal iron deficiency worsens the associative learning deficits and hippocampal and cerebellar losses in a rat model of fetal alcohol spectrum disorders. Alcohol Clin Exp Res. 2015;39(11):2097–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Niemela O, Parkkila S. Alcoholic macrocytosis–Is there a role for acetaldehyde and adducts?. Addict Biol. 2004;9(1):3–10. [DOI] [PubMed] [Google Scholar]
- 24.Seppa K, Laippala P, Saarni M. Macrocytosis as a consequence of alcohol abuse among patients in general practice. Alcohol Clin Exp Res. 1991;15(5):871–6. [DOI] [PubMed] [Google Scholar]
- 25.Yokoyama A, Yokoyama T, Brooks PJ, Mizukami T, Matsui T, Kimura M, Matsushita S, Higuchi S, Maruyama K. Macrocytosis, macrocytic anemia, and genetic polymorphisms of alcohol dehydrogenase-1B and aldehyde dehydrogenase-2 in Japanese alcoholic men. Alcohol Clin Exp Res. 2014;38(5):1237–46. [DOI] [PubMed] [Google Scholar]
- 26.Dostalikova-Cimburova M, Balusikova K, Kratka K, Chmelikova J, Hejda V, Hnanicek J, Neubauerova J, Vranova J, Kovar J, Horak J. Role of duodenal iron transporters and hepcidin in patients with alcoholic liver disease. J Cell Mol Med. 2014;18(9):1840–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohtake T, Saito H, Hosoki Y, Inoue M, Miyoshi S, Suzuki Y, Fujimoto Y, Kohgo Y. Hepcidin is down-regulated in alcohol loading. Alcohol Clin Exp Res. 2007;31(Suppl 1):S2–8. [DOI] [PubMed] [Google Scholar]
- 28.Streissguth AP, Barr HM, Labbe RF, Smith JR, Darby BL, Smith NJ, Martin DC, Doan RN. Alcohol use and iron status in pregnant women. Alcohol Clin Exp Res. 1983;7(2):7–30. [DOI] [PubMed] [Google Scholar]
- 29.Carter RC, Senekal M, Dodge NC, Bechard L, Meintjes EM, Molteno CD, Duggan C, Jacobson JL, Jacobson SW. Maternal alcohol use and nutrition during pregnancy: diet and anthropometry. Alcohol Clin Exp Res. 2017;41: 2114–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.May PA, Hamrick KJ, Corbin KD, Hasken JM, Marais A-S, Blankenship J, Hoyme HE, Gossage JP. Maternal nutritional status as a contributing factor for the risk of fetal alcohol spectrum disorders. Reprod Toxicol. 2016;59:101–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.May PA, Hamrick KJ, Corbin KD, Hasken JM, Marais A-S, Brooke LE, Blankenship J, Hoyme HE, Gossage JP. Dietary intake, nutrition, and fetal alcohol spectrum disorders in the Western Cape Province of South Africa. Reprod Toxicol. 2014;46:31–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang N, Tikellis G, Sun C, Pezic A, Wang L, Wells JCK, Cochrane J, Ponsonby AL, Dwyer T. The effect of maternal prenatal smoking and alcohol consumption on the placenta-to-birth weight ratio. Placenta. 2014;35(7):437–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tai M, Piskorski A, Kao JC, Hess LA, MdlM S, Gundogan F. Placental morphology in fetal alcohol spectrum disorders. Alcohol Alcohol. 2016;52(2):138–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petry CD, Wobken JD, McKay H, Eaton MA, Seybold VS, Johnson DE, Georgieff MK. Placental transferrin receptor in diabetic pregnancies with increased fetal iron demand. Am J Physiol. 1994;267(4 Pt 1):E507–14. [DOI] [PubMed] [Google Scholar]
- 35.Georgieff MK, Berry SA, Wobken JD, Leibold EA. Increased placental iron regulatory protein-1 expression in diabetic pregnancies complicated by fetal iron deficiency. Placenta. 1999;20(1):87–93. [DOI] [PubMed] [Google Scholar]
- 36.Jacobson SW, Jacobson JL, Molteno CD, Warton CMR, Wintermark P, Hoyme HE, De Jong G, Taylor P, Warton F, Lindinger NMet al. Heavy prenatal alcohol exposure is related to smaller corpus callosum in newborn MRI scans. Alcohol Clin Exp Res. 2017;41(5):965–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carter RC, Chen J, Li Q, Deyssenroth M, Dodge NC, Wainwright HC, Molteno CD, Meintjes EM, Jacobson JL, Jacobson SW. Alcohol-related alterations in placental imprinted gene expression in humans mediate effects of prenatal alcohol exposure on postnatal growth. Alcohol Clin Exp Res. 2018;42:1431–43. [DOI] [PubMed] [Google Scholar]
- 38.Jacobson SW, Chiodo LM, Sokol RJ, Jacobson JL. Validity of maternal report of prenatal alcohol, cocaine, and smoking in relation to neurobehavioral outcome. Pediatrics. 2002;109(5):815–25. [DOI] [PubMed] [Google Scholar]
- 39.Sokol R, Martier S, Ernhart C. Identification of alcohol abuse in the prenatal clinic. In: Chang N, Chao M, eds. NIAAA research monograph 17: early identification of alcohol abuse. Rockville (MD): Alcohol, Drug Abuse, and Mental Health Administration Research; 1983, p. 85–128. [Google Scholar]
- 40.Beard JL. Iron deficiency: assessment during pregnancy and its importance in pregnant adolescents. Am J Clin Nutr. 1994;59(2):502S–10S;. discussion 8S-10S. [DOI] [PubMed] [Google Scholar]
- 41.Carriaga MT, Skikne BS, Finley B, Cutler B, Cook JD. Serum transferrin receptor for the detection of iron deficiency in pregnancy. Am J Clin Nutr. 1991;54(6):1077–81. [DOI] [PubMed] [Google Scholar]
- 42.World Health Organization/United Nations Children's Fund/United Nations University , Iron deficiency anemia: prevention, assessment and control–report of a joint WHO/UNICEF/UNU consultation, World Health Organization, Geneva (Switzerland)1998. [Google Scholar]
- 43.Ganz T. Anemia of inflammation. N Engl J Med. 2019;381(12):1148–57. [DOI] [PubMed] [Google Scholar]
- 44.Ganz T, Olbina G, Girelli D, Nemeth E, Westerman M. Immunoassay for human serum hepcidin. Blood. 2008;112(10):4292–7. [DOI] [PubMed] [Google Scholar]
- 45.Redline RW, Boyd T, Campbell V, Hyde S, Kaplan C, Khong TY, Prashner HR, Waters BL; for the Society for Pediatric Pathology, Perinatal Section, Maternal Vascular Underperfusion Nosology Committee . Maternal vascular underperfusion: nosology and reproducibility of placental reaction patterns. Pediatr Dev Pathol. 2004;7(3):237–9. [DOI] [PubMed] [Google Scholar]
- 46.Ennis K, Felt B, Georgieff MK, Rao R. Early-life iron deficiency alters glucose transporter-1 expression in the adult rodent hippocampus. J Nutr. 2019;149(9):1660–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rebouche CJ, Wilcox CL, Widness JA. Microanalysis of non-heme iron in animal tissues. J Biochem Biophys Methods. 2004;58(3):239–51. [DOI] [PubMed] [Google Scholar]
- 48.Sangkhae V, Fisher AL, Wong S, Koenig MD, Tussing-Humphreys L, Chu A, Lelic M, Ganz T, Nemeth E. Effects of maternal iron status on placental and fetal iron homeostasis. J Clin Invest. 2019;130(2):625–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bradley J, Leibold EA, Harris ZL, Wobken JD, Clarke S, Zumbrennen KB, Eisenstein RS, Georgieff MK. Influence of gestational age and fetal iron status on IRP activity and iron transporter protein expression in third-trimester human placenta. Am J Physiol Regul Integr Comp Physiol. 2004;287(4):R894–901. [DOI] [PubMed] [Google Scholar]
- 50.Cao C, Fleming MD. The placenta: the forgotten essential organ of iron transport. Nutr Rev. 2016;74(7):421–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Georgieff MK, Wobken JK, Welle J, Burdo JR, Connor JR. Identification and localization of divalent metal transporter-1 (DMT-1) in term human placenta. Placenta. 2000;21(8):799–804. [DOI] [PubMed] [Google Scholar]
- 52.Jacobson SW, Hoyme HE, Carter RC, Dodge NC, Molteno CD, Meintjes EM, Jacobson JL. Evolution of the physical phenotype of Fetal Alcohol Spectrum Disorders from childhood through adolescence. Alcohol Clin Exp Res. 2021;45(2):395–408. [DOI] [PubMed] [Google Scholar]
- 53.National Research Council . Food insecurity and hunger in the United States: An assessment of the measure. Washington (DC): The National Academies Press; 2006. [Google Scholar]
- 54.Greenland S, Pearce N. Statistical foundations for model-based adjustments. Annu Rev Public Health. 2015;36:89–108. [DOI] [PubMed] [Google Scholar]
- 55.Siddappa AM, Rao R, Long JD, Widness JA, Georgieff MK. The assessment of newborn iron stores at birth: a review of the literature and standards for ferritin concentrations. Neonatology. 2007;92(2):73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Clogg CC, Petkova E, Haritou A. Statistical methods for comparing regression coefficients between models. Am J Sociol. 1995;100(5):1261–93. [Google Scholar]
- 57.McClelland GH, Judd CM. Statistical difficulties of detecting interactions and moderator effects. Psychol Bull. 1993;114(2):376–90. [DOI] [PubMed] [Google Scholar]
- 58.Pearl J. Direct and indirect effects. In: Breese J, Koller D, ed. The 17th Conference on Uncertainty and Artificial Intelligence. San Francisco (CA): Morgan Kaufmann Publishers Inc; 2001: p. 411–20. [Google Scholar]
- 59.Robins JM, Greenland S. Identifiability and exchangeability for direct and indirect effects. Epidemiology. 1992;3(2):143–55. [DOI] [PubMed] [Google Scholar]
- 60.Institute of Medicine . Iron, Otten JJ, Hellwig JP, Meyers LD, ed. In: Dietary Reference Intakes. Washington (DC): National Academy Press; 2006, p. 328–39. [Google Scholar]
- 61.World Health Organization . Serum and red blood cell folate concentrations for assessing folate status in populations. In: World Health Organization, ed. Vitamin and mineral nutrition information system. Geneva (Switzerland): WHO; 2012. [Internet]. [Accessed 2021 Jan 28]. Available from: http://apps.who.int/iris/bitstream/10665/75584/1/WHO_NMH_NHD_EPG_12.1_eng.pdf. [Google Scholar]
- 62.Sarafoglou K, Rodgers J, Hietala A, Matern D, Bentler K. Expanded newborn screening for detection of vitamin B12 deficiency. JAMA. 2011;305(12):1198–200. [DOI] [PubMed] [Google Scholar]
- 63.Abbassi-Ghanavati M, Greer LG, Cunningham FG. Pregnancy and laboratory studies: a reference table for clinicians. Obstet Gynecol. 2009;114(6):1326–31. [DOI] [PubMed] [Google Scholar]
- 64.Thomas C, Thomas L. Biochemical markers and hematologic indices in the diagnosis of functional iron deficiency. Clin Chem. 2002;48(7):1066–76. [PubMed] [Google Scholar]
- 65.Suchdev PS, Williams AM, Mei Z, Flores-Ayala R, Pasricha SR, Rogers LM, Namaste SM. Assessment of iron status in settings of inflammation: challenges and potential approaches. Am J Clin Nutr. 2017;106(Suppl 6):1626S–33S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ganz T. Hepcidin and iron regulation, 10 years later. Blood. 2011;117(17):4425–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sangkhae V, Nemeth E. Regulation of the iron homeostatic hormone hepcidin. Adv Nutr. 2017;8(1):126–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fisher AL, Nemeth E. Iron homeostasis during pregnancy. Am J Clin Nutr. 2017;106(Suppl 6):1567S–74S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sangkhae V, Fisher AL, Chua KJ, Ruchala P, Ganz T, Nemeth E. Maternal hepcidin determines embryo iron homeostasis in mice. Blood. 2020;136(19):2206–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Miller AM, Horiguchi N, Jeong WI, Radaeva S, Gao B. Molecular mechanisms of alcoholic liver disease: innate immunity and cytokines. Alcohol Clin Exp Res. 2011;35(5):787–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ahluwalia B, Wesley B, Adeyiga O, Smith DM, Da-Silva A, Rajguru S. Alcohol modulates cytokine secretion and synthesis in human fetus: an in vivo and in vitro study. Alcohol. 2000;21(3):207–13. [DOI] [PubMed] [Google Scholar]
- 72.Georgieff MK. Nutrition and the developing brain: nutrient priorities and measurement. Am J Clin Nutr. 2007;85(2):614S–20S. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Deidentified, individual participant data reported in this article (text, tables, figures, and appendices) and the study protocol, statistical analysis plan, and analytic code will be available for sharing to journal editors either before or after publication for checking and to researchers who provide a methodologically sound proposal, as determined by the authors of this article. Proposals from researchers should be directed to SWJ (sandra.jacobson@wayne.edu).Data will be stored in a data warehouse at Wayne State University and transmitted electronically in encrypted form to requestors. Data requestors will need to sign a data access agreement prior to access.