

Abstract
The present study adapted an existing high content imaging-based high-throughput phenotypic profiling (HTPP) assay known as “Cell Painting” for bioactivity screening of environmental chemicals. This assay uses a combination of fluorescent probes to label a variety of organelles and measures a large number of phenotypic features at the single cell level in order to detect chemical-induced changes in cell morphology. First, a small set of candidate phenotypic reference chemicals (n = 14) known to produce changes in the cellular morphology of U-2 OS cells were identified and screened at multiple time points in concentration-response format. Many of these chemicals produced distinct cellular phenotypes that were qualitatively similar to those previously described in the literature. A novel workflow for phenotypic feature extraction, concentration-response modeling and determination of in vitro thresholds for chemical bioactivity was developed. Subsequently, a set of 462 chemicals from the ToxCast library were screened in concentration-response mode. Bioactivity thresholds were calculated and converted to administered equivalent doses (AEDs) using reverse dosimetry. AEDs were then compared to effect values from mammalian toxicity studies. In many instances (68%), the HTPP-derived AEDs were either more conservative than or comparable to the in vivo effect values. Overall, we conclude that the HTPP assay can be used as an efficient, cost-effective and reproducible screening method for characterizing the biological activity and potency of environmental chemicals for potential use in in vitro-based safety assessments.
Keywords: Cell Painting, high content imaging, high-throughput profiling, ToxCast
Introduction
Traditionally, chemical safety assessments have relied on data from toxicity studies performed in animal models or human epidemiological data for hazard identification and dose-response characterization. However, such data are only available for a small percentage of chemicals in commerce or found in the environment (R. Judson et al., 2009). New, high-throughput approaches are needed to obtain hazard information and overcome this gap in knowledge. New approach methodologies (NAMs) are defined as any technology, methodology, approach or combination of approaches that can be used to provide information on chemical hazard or other aspects of chemical risk assessment, that avoids the use of intact animals (USEPA, 2018). Increasingly, members of the scientific community, including some organizations with regulatory authority, have proposed the use of NAMs for generation of hazard information and accelerating the pace of chemical risk assessment (ECHA, 2016; Kavlock et al., 2018; Thomas et al., 2019; USEPA, 2018, 2019). Over the last decade, the United States Environmental Protection Agency (USEPA) National Center for Computational Toxicology has sponsored and/or participated in two research programs, Toxicity Forecaster (ToxCast) and the Tox21 partnership, aimed at screening and characterizing the bioactivity of thousands of chemicals in high-throughput format in several hundred in vitro high-throughput screening (HTS) assays and generating NAMs data for potential use in regulatory decision making contexts (Thomas et al., 2019). A variety of frameworks for use of NAMs data (including ToxCast and Tox21 screening data) in regulatory decision-making contexts such as chemical prioritization and mechanistic prediction have been proposed (Blackwell et al., 2017; Canada, 2016; R. S. Judson et al., 2015; Shah, Liu, Judson, Thomas, & Patlewicz, 2016; Thomas et al., 2013) but are not yet in widespread usage.
Previously, the ToxCast and Tox21 screening programs employed a heterogeneous set of assay technologies, including mainly targeted assays to characterize chemical bioactivity, but also zebrafish, high-content imaging, and cell-based assays with multiple readouts of transcription factor expression or activity. Targeted assays are designed to measure interactions with or effects on one particular protein or pathway at a time (i.e. activity of chemical X at receptor Y). While the ToxCast approach to date has yielded chemical bioactivity data for a variety of target proteins and pathways, only a portion of the biological targets and pathways expressed in the human body have been investigated. As an alternative approach to targeted screening, the recently released Next Generation Blueprint for Computational Toxicology at the USEPA (i.e. EPA CompTox Blueprint) (Thomas et. al., 2019) advocates the use of broad-based high-content profiling assays as an initial approach for characterizing the biological activity of chemicals in human-derived culture models, with a particular emphasis on identifying the threshold concentration(s) that markedly perturb biological processes within a cell. As opposed to targeted assays, profiling assays collect highly-multiplexed measurements of many different features or endpoints and evaluate the global responses of intact cells to chemicals (Caicedo, Singh, & Carpenter, 2016). This approach allows for characterization of cellular effects that may arise from the specific interaction of a chemical at a discrete molecular target or more generalized cellular stress responses arising from promiscuous chemical activity at multiple molecular targets. The highly-multiplexed outputs from profiling assays can also be used to generate response profiles that can be used to facilitate linkage of chemical bioactivity to downstream molecular targets or cellular and tissue level effects. In addition, the EPA CompTox Blueprint envisions the use of profiling assays that can be applied to many different human cell types that express different complements of molecular targets and signaling pathways that could be perturbed by environmental chemicals. Two of the assay types USEPA is currently considering that meet the criteria for profiling-based bioactivity screening are high-throughput transcriptomics (Harrill et al., 2019; Thomas et al., 2019) and high content imaging-based high-throughput-phenotypic profiling (HTPP), the latter of which is the focus of the present work.
Imaging-based phenotypic profiling has been successfully used in a variety of applications. In drug discovery, phenotypic profiling has been used to identify mode of actions, investigate toxicity and reduce the size of molecular libraries while maintaining diversity in biological activities (Wawer, Jaramillo, et al., 2014; Wawer, Li, et al., 2014). In functional genomics, phenotypic profiling has been used to investigate and compare phenotypes of gene knockouts (Caicedo et al., 2016; Rohban et al., 2017). However, to the best of our knowledge, phenotypic profiling has not been applied on a large scale for screening of environmental chemicals for hazard identification, characterization and determination of in vitro potency thresholds for chemical bioactivity. In contrast, a general framework for in-vitro-to-in-vivo extrapolation (IVIVE) of in vitro potency estimates from HTS data has been established for potential use in chemical safety assessment and risk-based decision making (Shin et al., 2015; Sipes et al., 2017; Wetmore et al., 2015). However, the utility of imaging-based phenotypic profiling data in these types of applications has not been extensively evaluated.
In the present study, we adapted an existing phenotypic profiling assay (“Cell Painting”, (Bray et al., 2016)) to be compatible with in-house microfluidics capabilities for 384-well culture format, chemical exposures and fluorescent cytochemistry in order to facilitate concentration-response screening of several hundred environmental chemicals. In this assay, human-derived cells were labeled with multiple fluorescent probes to visualize various subcellular organelles and structural features. High content image analysis workflows were used to measure hundreds of morphological features at the level of the individual cell (i.e. shape of the cells, intensity, texture and distribution of fluorescent labels, etc.). The resultant data were then used to calculate well-level summary values, perform high-throughput concentration-response modeling and generate phenotypic response profiles. First, we identified and screened a set of candidate phenotypic reference chemicals for use as plate-based controls for evaluating HTPP assay performance during large-scale screening studies and identified an optimal exposure duration for HTPP screening. Second, we screened a set of 462 environmental chemicals in the U-2 OS cell model and derived in vitro potency estimates for bioactivity of all active chemicals. In addition, we demonstrated the technical reproducibility of the HTPP assay in concentration-response screening mode using the previously identified phenotypic reference chemicals. Next, we used reverse dosimetry to calculate administered equivalent doses (AEDs) corresponding to the thresholds for chemical bioactivity and compared those values to in vivo effect values from mammalian toxicity studies. For many chemicals the HTPP-derived AEDs were comparable to or lower than the corresponding in vivo effect values, indicating that the HTPP assay could be used to generate putative hazard values for use in chemical safety assessments. Overall, HTPP AEDs tended to be slightly higher than AEDs determined from ToxCast data as well as much higher (and closer to in vivo effect doses) than corresponding threshold of toxicological concern (TTC) values. Finally, we used chemotyping to identify groups of structurally-related chemicals where the HTPP assay did not provide a conservative estimate of bioactivity in comparison to the in vivo studies. We conclude that the HTPP assay is suitable for use as a high-throughput profiling assay for screening of environmental chemicals and when combined with IVIVE can provide conservative or comparable potency estimates as compared to in vivo mammalian data for many chemicals.
Materials and Methods
Materials & Test Chemicals
U-2 OS human osteosarcoma cells (HTB-96®, Lot: 64048673) were purchased from ATCC (Manassas, VA). Dulbecco’s Modified Eagle Medium (DMEM), conical tubes and culture flasks of various sizes (T25, T75, T225) were purchased from Corning (Corning, NY). Heat-inactivated fetal bovine serum (HI-FBS) was purchased from Sigma-Aldrich (St. Louis, MO). 10X Gibco® penicillin-streptomycin-glutamine (PSG), TrypLE Select, 0.4% trypan blue solution, Countess® cell counting chamber slides and MicroAmp® optical adhesive film were purchased from ThermoFisher Scientific (Waltham, MA). Solutions of 16% paraformaldehyde (PFA) were purchased from Electron Microscopy Sciences (Hatfield, PA). CellCarrier-384 Ultra microplates were purchased from PerkinElmer (Waltham, MA). Echo qualified 384-well polypropylene (384PP) and 384-well low dead volume (384LDV) plates were purchased from LabCyte (San Jose, CA). Sourcing information for fluorescent labeling reagents for the cell viability (CV) and HTPP assay are listed in Table 1.
Table 1:
List of fluorescent labels used in the present study.
| Assay | Organelle / purpose | Label | Live-label? | Excitation / Emission wavelength1 [nm] | Provider | Cat-No | Stock concentration | In well concentra tion |
|---|---|---|---|---|---|---|---|---|
| CV | Nucleus | Hoechst-33342 | Y | 405 / 435–480 | Invitrogen | H3570 | 16.2 mM in H2O | 7.7 μM |
| Cytotoxicity | PI | Y | 561 / 570–630 | Invitrogen | P3566 | 1.5 mM in H2O | 3.6 μM | |
| CP | Nucleus | Hoechst-33342 | N | 405 / 435–480 | Invitrogen | H3570 | 16.2 mM in H2O | 6.75 μM |
| Nucleoli / RNA | SYTO14 | N | 488 / 500–550 | Invitrogen | S7576 | 5 mM in DMSO | 3 μM | |
| ER | Concanavalin A-488 | N | Invitrogen | C11252 | 1 mg/ml in 0.1 M NaHCO3 pH 8.37 with 2 mM NaN3 | 100 μg/ml | ||
| Actin skeleton | Alexa Fluor™ 568 Phalloidin | N | 561 / 570–630 | Invitrogen | A12380 | 6.6 μM in methanol | 41.2 nM | |
| Golgi + plasma membrane | Wheat Germ Agglutinin, Alexa Fluor™ 555 Conjugate | N | Invitrogen | W32464 | 1 mg/ml in PBS | 5 μg/ml | ||
| Mitochondria | MitoTracker DeepRed | Y | 640 / 650–760 | Invitrogen | M22426 | 1 mM in DMSO | 475 nM |
Excitation / emission wavelengths refer to the Opera Phenix microscope.
Concentration-response screening was performed on two sets of chemicals in the present study. The first set of chemicals (i.e. Reference Set) was evaluated in order to identify a group of phenotypic reference chemicals to use as plate-based controls for evaluation of assay performance during large-scale HTPP screening. It included 14 candidate phenotypic reference chemicals identified as having produced distinct morphological phenotypes in U-2 OS cells using the Cell Painting assay (Gustafsdottir et al., 2013), two chemicals that were not expected to produce changes in the morphology of U-2 OS cells at concentrations up to and including 100 μM, and two cytotoxic chemicals (Table 2, https://comptox.epa.gov/dashboard/chemical_lists/HTPP2019_REFSET). The second chemical set (i.e. Screening Set) consisted of 462 unique chemicals from the ToxCast inventory (Table S1, https://comptox.epa.gov/dashboard/chemical_lists/HTPP2019_SCREEN), including 448 chemicals from the Accelerating the Pace of Chemical Risk Assessment (APCRA) retrospective chemical list (Paul-Friedman et al., 2019). A subset of 16 chemicals within the Screening Set were screened in duplicate.
Table 2:
Reference Set.
| Group | Chemical Name | CAS # | DTXSID1 | Reported Phenotype (Gustafsdottir et al. 2013) | Vendor | Cat No | Stockcone [mM] | ||
|---|---|---|---|---|---|---|---|---|---|
| Min | max | ||||||||
| reference chemical | Amperozide | 75558-90-6 | DTXSID6048416 | Toroid nuclei | Santa-Cruz | sc-203512 | 20 | 0.100 | 100 |
| Berberine Chloride | 633-65-8 | DTXSID8024602 | Redistribution of mitochondria | Sigma-Aldrich | B3251 | 20 | 0.100 | 100 | |
| Ca-074-Me | 147859-80-1 | DTXSID50881386 | Bright, abundant Golgi staining | Sigma-Aldrich | C5857 | 20 | 0.010 | 10 | |
| Etoposide | 33419-42-0 | DTXSID5023035 | Large, flat nucleoli | Sigma-Aldrich | E1383 | 20 | 0.010 | 10 | |
| Fenbendazole | 43210-67-9 | DTXSID0040672 | Giant, multi-nucleated cells | Sigma-Aldrich | F5396 | 20 | 0.002 | 2 | |
| Fluphenazine | 69-23-8 | DTXSID2023068 | Enhanced Golgi staining and some cells with fused nucleoli | Sigma-Aldrich | F4765 | 20 | 0.100 | 100 | |
| Latrunculin B | 76343-94-7 | DTXSID10881387 | Actin breaks | Sigma-Aldrich | L5288 | 20 | 0.010 | 10 | |
| Metochlopramide | 364-62-5 | DTXSID6045169 | Enhanced Golgi staining and some cells with fused nucleoli | Sigma-Aldrich | M0763 | 20 | 0.100 | 100 | |
| 5-Nitro-2-(3-phenylpropylaminojbenzoic acid | 107254-86-4 | DTXSID90147978 | Redistribution of ER to one side of the nucleus | Sigma-Aldrich | N4779 | 20 | 0.100 | 100 | |
| Oxibendazole | 20559-55-1 | DTXSID5045625 | Large, multi-nucleated cells with fused nucleoli | Sigma-Aldrich | 03132 | 4 | 0.002 | 2 | |
| Rapamycin | 53123-88-9 | DTXSID5023582 | Reduced nucleolar size | Sigma-Aldrich | R0395 | 20 | 0.010 | 10 | |
| Rotenone 2 | 83-79-4 | DTXSID6021248 | Extensive mitochondrial fission | Sigma-Aldrich | R8875 | 20 | 0.001 | 1 | |
| Taxol | 33069-62-4 | DTXSID9023413 | Large, multi-nucleated cells with fused nucleoli | Sigma-Aldrich | T7402 | 5 | 0.100 | 100 | |
| Tetrandrine | 518-34-3 | DTXSID10178062 | Abundant ER | Sigma-Aldrich | T2695 | 5 | 0.100 | 100 | |
| negative control | Saccharin | 81-07-2 | DTXSID5021251 | Sigma-Aldrich | 240931 | 20 | 0.000 | 0.05 | |
| Sorbitol | 50-70-4 | DTXSID5023588 | Sigma-Aldrich | S1876 | 20 | 0.025 | 25 | ||
| cytotoxic control | lonomycin | 56092-81-0 | DTXSID2040521 | Sigma-Aldrich | I9657 | 6 | 30 | ||
| Staurosporine | 62996-74-1 | DTXSID6041131 | Sigma-Aldrich | S5921 | 0.2 | 1 |
DTXSID are unique substance identifiers associated with ToxCast chemicals, as referenced on the CompTox Chemicals Dashboard (Williams et al., 2017)
rotenone was tested in lieu of beta-dihydrorotenone which was tested and described in Gustafsdottir et al. (2013)
Cell Culture
Upon receipt from ATCC, U-2 OS cells were temporarily stored in vapor phase liquid nitrogen, thawed and cultured in U-2 OS growth media (DMEM + 10% HI-FBS + 1X PSG). When U-2 OS cells reached > 80% confluence, they were rinsed gently with 1X phosphate-buffered saline (PBS) and passaged via incubation with 1X TrypLE Select for 8 minutes. The TrypLE was deactivated via addition of 3X volume of U-2 OS growth media and the cell suspension was transferred to fresh 15 or 50 mL conical tubes, centrifuged (5 min @ 250g), resuspended in U-2 OS growth media and seeded in fresh T25, T75 or T225 Corning® culture flasks for continued expansion. Cells were maintained at 37°C in a humidified incubator with 5% CO2 atmosphere. U-2 OS cells were expanded through several consecutive passages as described and cryopreserved at internal passage number 6 (P6) using the manufacturer’s recommended cryopreservation media.
For each replicate within each experiment, a single vial of P6 U-2 OS cells was thawed, cultured in U-2 OS growth media and expanded through one to three consecutive passages in the manner described until a sufficient cell mass was generated for multiwell plating. At each passage, a Countess™ II automated cell counter (ThermoFisher Scientific) was used to quantify the total number of cells and percentage of live cells using trypan blue dye exclusion according to manufacturer’s protocol. All experiments were performed with U-2 OS cells between P7 and P9. Cells were seeded in CellCarrier-384 Ultra microplates at a density of 400 live cells / well. Total culture volume per well was 40 μL. Cells were placed in a humidified incubator with 5% CO2 atmosphere and allowed to recover for 24 hours prior to chemical treatment (Fig. 1A).
Fig. 1: Experimental Strategy Overview.
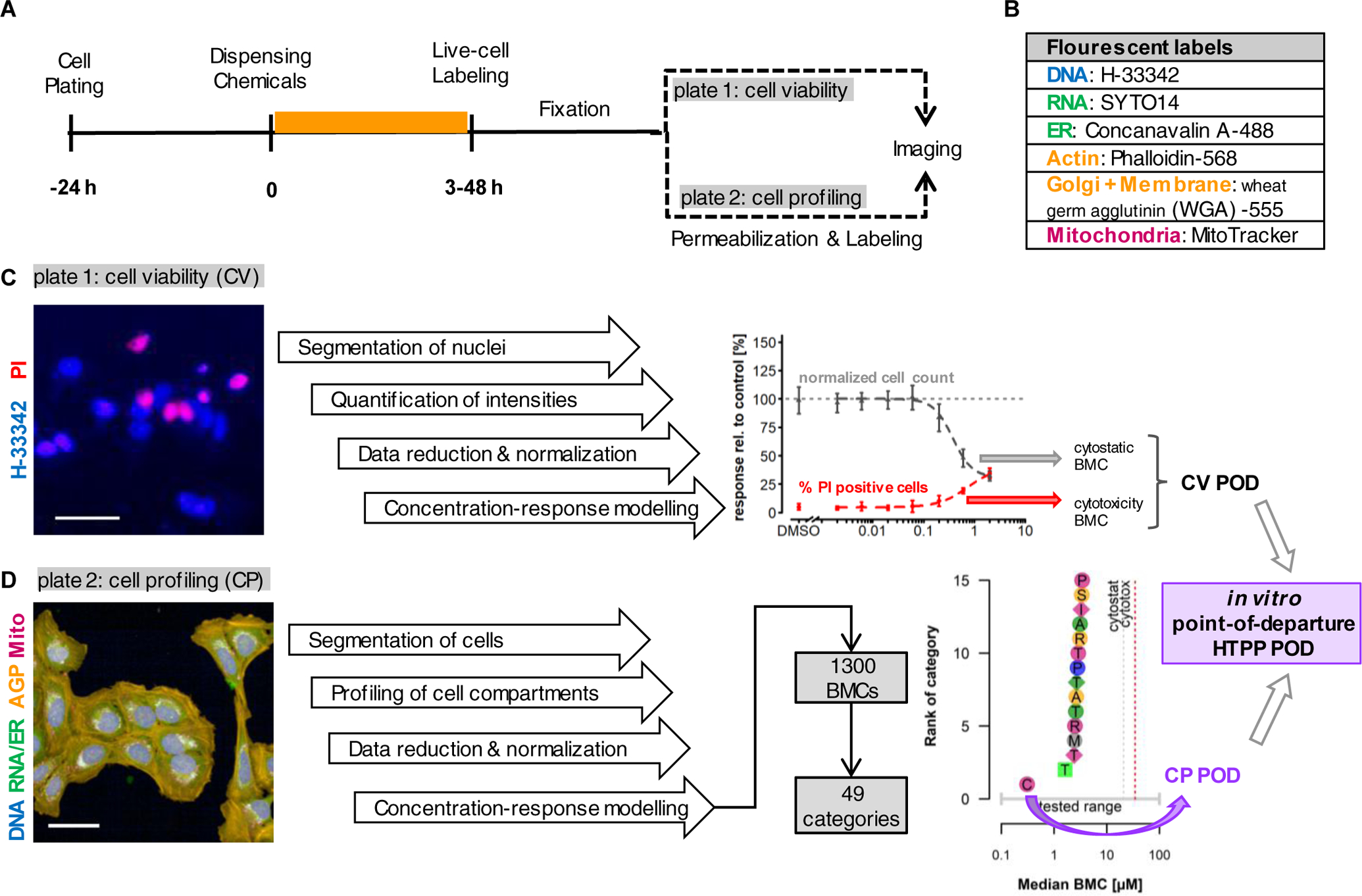
(A) Diagram illustrating the timing of laboratory workflows for parallel assays, i.e. cell viability (CV) and cell profiling (CP). U-2 OS cells were plated in 384-well format and allowed to recover for 24 h prior to dispensing of test chemicals. Cells were then incubated with test chemicals for various exposure durations (3 – 48 h) for the different phases of experimentation. Thirty minutes prior to sampling, live-cell labeling reagents were applied to each assay plate. Cells were then sampled via fixation with PFA and thoroughly rinsed. For CV plates, cells were imaged with no further sample processing. For CP, cells were further permeabilized and labeled with additional fluorescent probes. (B) List of the fluorescent probes used in the CP assay and associated organelles. Note that in some instances multiple organelles were labeled and imaged in the same fluorescent channel. (C) Overview of data analysis steps for the CV assay. Endpoints were derived from analysis of cells labeled with H-33342 and PI as shown in the representative image. Two endpoints were modeled: 1) normalized cell count (nCC) and 2) percent of PI-positive cells. BMCs were calculated for each as described in Methods. The minimum of the cytostatic and cytotoxic BMCs was defined as the CV POD. (D) Overview of data analysis steps for the CP assay. Phenotypic features were derived from analysis of cells labeled with the combination of fluorescent probes in panel B as shown in the representative image. A total of 1300 features were calculated at the cell level and used to calculate well-level data for concentration-response modeling. Results were aggregated into categories as described in Methods. The CP POD was defined as the median BMC of the most sensitive category. The HTPP POD was defined as the minimum of CV POD and CP POD.
Chemical Treatment
Chemicals in the Reference Set were solubilized in dimethylsulfoxide (DMSO) at concentrations ranging from 4 to 20 mM (Table 2) to create chemical stock solutions and stored at −20°C. Prior to dose plate preparation, reference chemical stocks were thawed and pipetted into an Echo qualified 384-well polypropylene (384PP) plate. For the Screening Set, DMSO-solubilized chemical stock solutions were received frozen in 384PP format from the US EPA ToxCast chemical inventory management contractor (EvoTec, Princeton, NJ) and stored at −80°C prior to dose plate preparation. During dose plate preparation, dilution series of reference chemicals and test chemicals were prepared in Echo qualified 384-well low dead volume (384LDV) plates using an Echo 550 acoustic dispenser (LabCyte, San Jose, CA). Varying volumes of reference or test chemical stocks were dispensed into consecutive vertical wells of the 384LDV plates and the wells were backfilled with varying volumes DMSO using a Certus FLEX Microdispenser (LEAP Technologies, Morrisville, NC) equipped with a 0.10 / 0.03 Gyger microvalve. Dose plates were then sealed with an adhesive aluminum film and stored at −80°C until time of use. The dose plates were prepared at 200x of the final concentrations tested in each study.
Chemicals in the Reference Set were tested at seven concentrations (approximate half-log spacing) with three technical replicates per plate. The concentration range tested for each chemical in the Reference Set is listed in Table 2. The two cytotoxicity control chemicals were tested at one concentration (1 μM staurosporine, 30 μM ionomycin) with six technical replicates per plate. Experiments with the Reference Set were repeated using three independent cultures (i.e. biological replicates) established on different days.
Chemicals in the Screening Set were tested at eight concentrations (approximate half-log spacing) with one technical replicate per plate. The concentration range tested for each chemical in the Screening Set is listed in Table S1. Dilution series of four phenotypic reference chemicals identified from testing the Reference Set (e.g. berberine chloride, Ca-074-Me, etoposide, rapamycin) were included on each plate. In addition, a dilution series for staurosporine was included on each plate as a cytotoxicity control. Experiments with the Screening Set were repeated using four independent cultures (i.e. biological replicates) established on different days. A diagram of an example dose plate layout for the Screening Set is provided in Figure S1.
At 24 hours post-plating, 200 nL of 200x dosing solutions were transferred to the assay plates using a LabCyte Echo 550 acoustic dispenser. The final concentration of DMSO in test wells was 0.5%. Well coordinates on each assay plate were uniquely randomized with respect to treatment so that any potential edge-well effects were distributed in an unbiased manner across all possible treatment conditions. Assay plates were placed back in a humidified incubator immediately after dosing.
Cell Viability (CV) Assay
To evaluate cytotoxic and cytostatic chemical effects, wells were live-labeled with a combination of Hoechst-33342 (H-33342) and propidium iodide (PI) (Fig. 1C). A labeling solution of H-33342 and PI was prepared in U-2 OS growth media and 2 μL of labeling solution was dispensed into each assay well using a Certus FLEX micro dispenser equipped with a 0.10 / 0.03 Gyger microvalve. The concentration of each fluorescent probe in the labeling solution and the final concentration in assay wells are listed in Table 1. Plates were placed in a humidified incubator for 30 minutes. Plates were then fixed by direct addition of 12 μL of 16% PFA solution using a MultiFlo FX Microplate Dispenser with a 1 μL peri-pump cassette. After 10 min incubation at room temperature protected from light, plates were rinsed four times with 1X PBS. The final wash remained in the wells as a storage buffer. Plates were then sealed with optical adhesive tape and stored at 4°C. Prior to image acquisition, plates were removed from 4°C storage and allowed to equilibrate to room temperature.
Fluorescent images of H-33342 and PI labeled cells were acquired using an Opera Phenix High Content Screening System (Perkin Elmer) and the Harmony software (v4.8, Perkin Elmer). Excitation and emission wavelengths for each fluorescent probe are listed in Table 1. For experiments involving the Reference Set, images were acquired using a 5x objective and one field per well (2536 μm × 2536 μm). For experiments involving the Screening Set, images were acquired using a 10x objective and four unique fields per well, covering a total of 2590 μm × 2590 μm. There was no marked difference in the performance of the CV assay when comparing results from the 5X and 10X imaging objectives (data not shown).
CV Image Processing and Data Analysis
Image processing was performed using PerkinElmer Harmony® software as outlined in Fig S2 and succinctly illustrated in Fig 1. Briefly, images were prefiltered to exclude regions with bright artifacts (i.e. dust, etc.) in the H-33342 channel, followed by nuclei segmentation in the H-33342 channel. Valid nuclei were selected based on H-33342 intensity and size criteria. The mean intensity in the PI channel was then measured for each valid nucleus. Cell-level data for each plate were then exported for downstream analysis.
For data analysis, R statistical software was used (v3.4.1), as outlined in Fig S3. Briefly, for each plate, valid objects were defined based on nuclear area and roundness. Cells were classified as PI-positive if their mean intensity was above the 95th-percentile derived from vehicle control cells. Two well-level endpoints were calculated: 1) normalized cell count (nCC), the number of H-33342 labeled nuclei relative to solvent control conditions and 2) percentage of PI-positive cells.
For each chemical and both endpoints, well-level data from all technical and biological replicates were combined for use in benchmark concentration (BMC) modelling (Fig S4). Concentration-response data was modelled using concentration-response functions from the R package tcpl (v1.4.3) (Filer, Kothiya, Setzer, Judson, & Martin, 2017). Cytotoxicity data (PI) was fit to three functions: constant, Hill and Gain-Loss function, whereas cell count was only modeled with the first two functions. The best-fitting model was selected based on best Akaike information criterion (AIC). If the best-fitting model was Hill or Gain-Loss, a BMC was estimated. For cytotoxicity, the benchmark response (BMR) was set at 3 times the normalized median absolute deviation (nMAD, with nMAD = 1.4826*MAD), which corresponds approximately to 3 SD. For cell count, the effective concentration 50 (EC50) was calculated. The in vitro point-of-departure (POD) for the cell viability assay (CV POD) was defined as the minimum of BMCPI and BMCnCC and consequently, the no observable effect concentration (NOEC) and lowest observable effect concentration (LOEC) are defined as the highest tested concentration below and lowest tested concentration above CV POD, respectively.
Cell Profiling (CP) Assay
Phenotypic profiling of cells in response to chemical treatments was performed using the ‘Cell Painting’ method as described by Bray et al. (2016), with modifications. A variety of organelles (nucleus, nucleoli, endoplasmic reticulum (ER), actin cytoskeleton, golgi apparatus and plasma membrane (AGP), mitochondria) were labeled using fluorescent probes or fluorophore-conjugated small molecules as listed in Table 1 and illustrated in Fig. 1B. First, a labeling solution of MitoTracker Deep Red was prepared in U-2 OS growth media and 2 μL of labeling solution was dispensed into each assay well using a Certus FLEX micro dispenser equipped with a 0.10 / 0.30 Gyger microvalve. Plates were placed in a humidified incubator for 30 minutes, followed by fixation and washing as described above for the CV assay. Cells were then permeabilized with 0.1% Triton X-100, incubated for 30 minutes at room temperature protected from light and rinsed twice with 1X PBS. Wells were then drained to a residual volume of 10 μL of 1X PBS. A solution containing the remainder of the labeling reagents listed in Table 1 was prepared in 1X PBS with 1% BSA and dispensed (2 μL / well) using a Certus FLEX micro dispenser and 0.10 / 0.30 Gyger microvalve. The concentration for each fluorescent probe in the labeling solution and the final concentration of label in the assay wells are listed in Table 1. After 30 min incubation at room temperature protected from light, plates were rinsed four times with 1X PBS. The final wash remained in the wells as a storage buffer. Plates were sealed with optical adhesive tape and stored at 4°C. Immediately prior to image acquisition, plates were removed from the 4°C storage and allowed to equilibrate to room temperature.
Fluorescent images of labeled cells were acquired using an Opera Phenix High Content Screening System and Harmony® software (v4.8). Five images were acquired using four different excitation wavelengths: 405 nm (DNA), 488 nm (RNA, z-offset 1), 488 nm (ER, z-offset 2), 561 nm (actin, Golgi, plasma membrane) (AGP), 640 nm (mitochondria) (Mito). Images were acquired using a 20x water immersion objective in confocal mode using 2×2 pixel binning. The z-offsets for each channel were optimized by examining randomly selected wells across multiple plates with the goal of acquiring sharp, “in focus” images for the cell compartment or organelle of interest. Optimized z-offsets remained constant throughout the experiments. Exposure times and laser power settings were optimized using vehicle-treated wells with the goal of achieving average pixel intensities in the lowest quartile of the intensity range for the 16-bit large format sCMOS cameras included in the Opera Phenix HCS instrument (e.g. < 15000). Typically, 9 unique fields per well were acquired, covering a total of 1908 μm × 1908 μm per well.
CP Image Processing, Feature Extraction and Data Analysis
Image processing was performed using Harmony® software, as described in Fig. S5 and illustrated in Fig. S6. Briefly, nuclei were segmented in the H-33342 channel, followed by segmentation of the cells in the ER channel using positional information from the nuclei as a seed. Cells touching the image border or with low intensity in the ER channel were excluded. For valid objects, additional cell compartments were defined: membrane, cytoplasm and the perinuclear space (i.e. “ring”). The perinuclear space compartment overlays a portion of the cytoplasm where the ER and Golgi are typically localized but does not extend fully to the outer cell membrane. Next, a variety of features were calculated using different modules within the Harmony® software. Feature types include intensity, texture, localization of fluorescent signal (SCARP morphology) as well as shape and position of the cells. Modules measuring the intensity of fluorescence labeling were run for all meaningful combinations of channel x compartment (e.g. DNA and RNA intensity is only meaningful in the nucleus, and ER only in the cytoplasm/ring whereas membrane intensity was only measured for the AGP channel, see Fig. S7). Overall, a total of 1300 features were measured for each cell. Typically, approximately 300 cells were analyzed per well. Cell-level data for each plate were then exported for downstream analysis. The full list of features output using this approach is listed in Table S2.
For each plate, data reduction was performed using R statistical software, as outlined in Fig. S8 and illustrated in Fig. 1D. Valid objects were defined based on the range of nucleus and cell area parameters, effectively excluding non-cellular debris (i.e. dust, etc) from downstream analysis. For each endpoint, data were normalized to vehicle control using median absolute deviation (MAD) normalization (Bray et al., 2016). Well-level feature data were calculated as the median of normalized cell-level data within each well. Wells with < 50-cells were excluded from further analysis. Well-level data were further standardized by scaling to the SD of solvent control wells (typically 24 wells/plate). Thus, the standardized values represent the number of standard deviations a response is above or below the mean of DMSO controls. Standardized values > +1 or < −1 are considered markedly different from control.
Prior to BMC modelling of CP data, highly cytotoxic treatments were removed by excluding all tested concentrations above the CV LOEC (Fig. S9A). For each endpoint, well-level data from all technical and biological replicates were combined for BMC modelling using the command line version of BMDExpress (v2.2.180) (Fig. S10). Endpoints were only modeled if they had at least one concentration with a | mean response | > 1. Four functions were fit to the data: first- and second-degree polynomial (linear, Poly2), power and Hill. The better fitting polynomial model was identified using a nested χ2-test. The model with the lowest AIC across the best-fitting polynomial model, hill and power models was then selected as the winning model. The BMR was set at +/− 1, corresponding to 1 SD from vehicle control. Only features with a BMC below the highest tested concentration were considered to be responsive to chemical treatment and were retained for downstream analysis.
To determine an overall in vitro POD for the CP assay (CP POD), each of the 1300 features were grouped by channel then module then compartment into 49 categories (Table S2, Fig. S7). For example, the category “Mito_Texture_Ring” contains texture features measured in the ring compartment in the fluorescent channel used to visualize mitochondria. In order for a category to be considered affected by chemical treatment, ≥ 30% of the features contained within the category must have been responsive. The median BMC for each category was calculated from the feature-level BMC for all responsive features contained within the category. Categories were then ranked in ascending order based on the median BMC and the most sensitive category (i.e. the category with the lowest potency values) was defined as the CP POD (Fig. 1D).
IVIVE & Comparison to In Vivo Data
The HTPP POD was defined as the minimum of the CP POD and CV POD. The HTPP PODs were then converted to administered equivalent doses (HTPP AED) using the httk R package (v. 1.9.2) (Pearce, Setzer, Strope, Wambaugh, & Sipes, 2017). Using reverse dosimetry, AEDs were estimated that would have resulted in a steady state plasma concentration equivalent to the HTPP POD. The httk package uses chemical-specific information on in vitro clearance in human hepatocytes and human plasma protein binding (Wetmore et al., 2012). Plasma concentrations are estimated for humans based on a 3-compartment model assuming 100% bioavailability and restrictive clearance. Inter-individual variability in a population was taken into account via Monte Carlo simulation whereby pharmacokinetic parameters were varied (Ring, Pearce, Setzer, Wetmore, & Wambaugh, 2017; Wetmore et al., 2012) as previously described (Paul-Friedman et al., 2019) to calculate the 5th, 50th (median) and 95th percentile of the population distribution.
This HTPP AED was then compared to publicly available in vivo effect values from the USEPA Toxicity Value Database (ToxValDB, development version 7) (Williams et al., 2017) (https://comptox.epa.gov/dashboard/). Selection of in vivo effect values was performed as described (Paul-Friedman et al., 2019). Briefly, only mammalian studies using an oral exposure route with units of mg/kg-bw or mg/kg-bw/day, or units that could be converted to mg/kg-bw/day values such as parts per million or parts per billion in the diet or mg/kg in the diet, were used. Various study types (i.e., subchronic, chronic) in various mammalian species (i.e. rat, mouse, rabbit, dog) were included. Only the following effect value types were included: no observable or no observable adverse effect levels (NOEL, NOAEL) or lowest observable or lowest observable adverse effect levels (LOEL, LOAEL). The in vivo effect values were then summarized by calculating the 5th percentile (PODtrad) using the R function quantile from the base package with option “type=2”. This means, that for compounds with less than 20 studies, the in vivo effect value is shifted towards the most sensitive study. A total of 442 chemicals had in vivo effect values.
From these 442 chemicals, 1 chemical did not have high-throughput toxicokinetics (HTTK) information available and 21 chemicals lacked an HTPP POD, leaving a total of 420 chemicals from the screening set for calculation of an HTPP AED. The HTPP AED and the PODtrad for each of these chemicals were compared and chemicals were grouped relative to whether the HTPP AED range (i.e. 5th to 95th percentile) was above or below the PODtrad or whether the PODtrad was within the HTPP AED range. The HTPP AED was considered to be conservative for chemicals in the “below” category and comparable to in vivo if the PODtrad value was within the HTPP AED range. The HTPP AED was not considered to be conservative or comparable to in vivo for chemicals in the “above” category; meaning that the PODtrad was lower than the lower bound of the AED confidence interval.
For comparison, published data from two NAMs were also compared to the PODtrad: 1) TTC values (Kroes, Kleiner, & Renwick, 2005) and 2) ToxCast assay data, both as reported in Paul-Friedman et al. (2019). In this study, ToxCast potency values were log-transformed, then the in vitro POD was defined as the 5th percentile of the distribution (using the R function quantile from the base package with option “type=7”) and converted to AEDs as described above. ToxCast data was available for 426 chemicals, while 413 chemicals had a TTC value.
Results
Qualitative Reproducibility of Chemical-Induced Cellular Phenotypes
For this study, the ‘Cell Painting’ assay (Bray et al., 2016; Gustafsdottir et al., 2013) was adapted as described above. The first phase of the study was aimed at identification of phenotypic reference chemicals for use as plate-based controls for evaluating assay performance during HTS. A set of 14 candidate phenotypic reference chemicals, with narrative descriptions of the phenotypes produced in U-2 OS cells during single concentration screening studies (Gustafsdottir et al., 2013) was identified (i.e. Reference Set, Table 2). The chemicals were screened in seven-point concentration-response in the same cell type (U-2 OS) and exposure duration (48 h) as used in previous studies (Gustafsdottir et al., 2013). For the majority of the 14 chemicals, visual phenotypes were consistent with observations from previous studies. Berberine chloride affected mitochondria localization (Fig. 2A,B) and Ca-074-Me produced bright, abundant labeling of the Golgi (Fig. 2C,D). Etoposide had potent effects in various organelles and many different features, driven by an overall increase in nuclear and cellular size (i.e. cell swelling) (Fig. 2E,F). Rapamycin had subtle effects on a variety of features driven by changes in nucleus and cell shape, including reduction in nucleolar size (Fig. 2 G,H). In comparison to Gustafsdottir et al. (2013), the ER phenotypes reported for tetrandrine and 5-Nitro-2-(3-phenylpropylamino)benzoic acid were not observed in the present study.
Fig. 2: Qualitative and Quantitative Comparison of Reference Chemical Phenotypes.
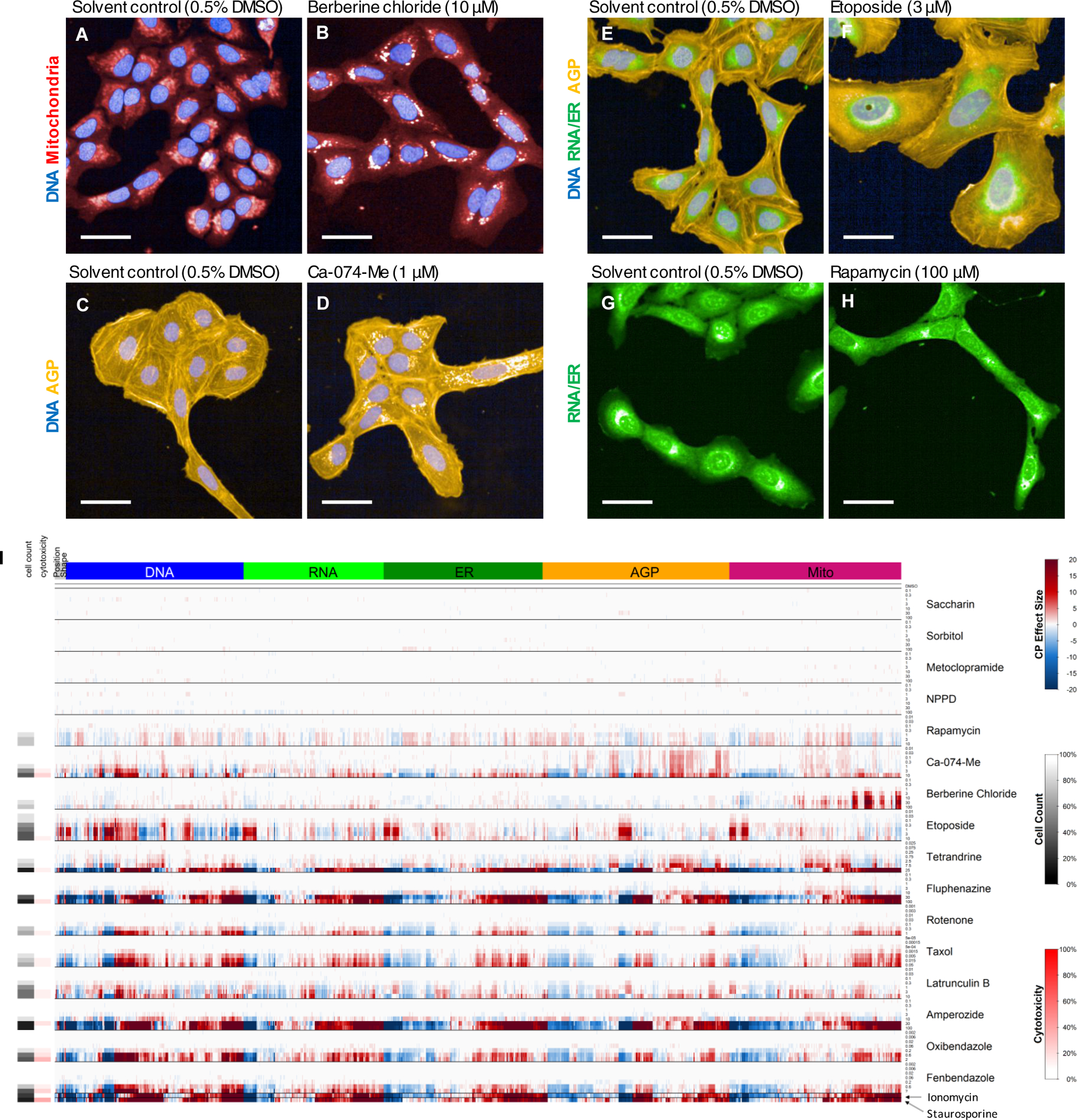
U-2 OS cells were treated with test chemicals for 48 h before sampling and imaging. (A-H) Representative images of reference chemicals and corresponding solvent controls. Only selected channels are shown for each chemical. Scale bar = 50 μm. Distinct effects on cellular morphology were observed for each chemical. (I) Quantitative summary of phenotypic effects for the Reference Set. Well-level feature data were normalized and scaled per plate and then averaged. The columns of the heatmap correspond to the 1300 phenotypic features, organized by fluorescent channel. Colors represent the magnitude of increase or decrease in a measured feature with respect to DMSO control. Gray and red tiles to the far left of the heatmap correspond to cell count (nCC) and percent PI-positive cells, respectively. Rows correspond to individual concentrations of the test chemicals. Data for each chemical is separated by horizontal black lines and test concentrations are in ascending order from top to bottom. Chemicals with weak phenotypic effects are at the top of the heatmap. Chemicals with marked phenotypic effects below the threshold for cytotoxicity are in the middle of the heatmap. Chemicals where phenotypic effects are observed at or near the threshold for cytotoxicity are at the bottom of the heatmap. The distinct phenotypic effects observed qualitatively in panels A-H are recapitulated in quantitative visualization of the CP data. Data were derived from ~300 cells/well across 3 technical replicate wells and 3 biological replicates (n = 9 wells total).
The feature profiles for each chemical in the Reference Set, including two negative controls and two cytotoxic chemicals, are visualized in Fig. 2I. For the two negative controls (saccharin and sorbitol), very few features were changed from control at concentrations up to and including 100 μM. In contrast, the two cytotoxic control compounds (staurosporine and ionomycin) had strong effects on many phenotypic features and resulted in a feature profile very different from the control. This “cytotoxic profile” was also observed at higher concentrations of several candidate reference chemicals (i.e. taxol, amperozide, fluphenazine). To avoid confounding of concentration-response modeling of phenotypic features, highly cytotoxic concentrations were removed from further analysis. Of note, twelve of fourteen chemicals in the Reference Set produced marked phenotypic changes, often at concentrations well below cytotoxicity. For example, the observed qualitative phenotypes for berberine chloride and Ca-074-Me (Fig. 2A–D) were confirmed quantitatively, with strong effects observed in the mitochondria and AGP channels, respectively, at concentrations below the threshold for cytotoxicity. Likewise, the pronounced qualitative phenotype of etoposide (i.e. cell swelling) (Fig. 2E,F) and the subtle qualitative phenotype of rapamycin (Fig. 2G,H) were recapitulated quantitatively at concentrations below the threshold for cytotoxicity. Overall, previously published descriptions of chemical-induced cellular phenotypes were reasonably reproduced in the present study.
Concentration-Response Modelling of Profiling Data
The next step in the present study was to devise a workflow for concentration-response modeling of well-level phenotypic profiling data and estimation of potency thresholds for chemical bioactivity. Fig. 3A visualizes the results of feature-level concentration-response modeling for four phenotypic reference chemicals (berberine chloride, Ca-074-Me, etoposide, rapamycin). Each point in the plot is a phenotypic feature represented by the potency (i.e. BMC) and effect size (normalized magnitude, Fig. S9B) from the concentration-response modeling results, coded by channel, cellular compartment and feature type. It is immediately obvious that the profile of phenotypic effects differs substantially across these four chemicals. Berberine chloride had robust effects on mitochondria compactness and intensity. For Ca-074-Me, the most pronounced effects were on texture and compactness of the AGP channel, in the ring region in particular, reflective of pronounced Golgi labeling. This effect occurs at low concentrations (< 0.1 μM), whereas at increasing concentrations, changes in the DNA channel emerge. Etoposide and rapamycin both produced a variety of effects, with the DNA channel being the most affected for etoposide. Effect sizes for rapamycin were smaller than for the other compounds and only ranged up to 5 times the noise level (compared to ~20 times for some other chemicals). For each chemical, phenotypic effects were observed in the absence of cytotoxicity or well below the threshold for cytotoxicity.
Figure 3. Concentration-Response Modeling Results and Identification of CP POD for Four Reference Chemicals.

(A) Potency-magnitude plots for four reference chemicals. Concentration-response modeling of feature data from Fig. 2B was performed and feature-level BMCs calculated as described in Methods. The normalized magnitude (y-axis) was defined as the largest effect size observed at a non-cytotoxic concentration (see Fig. S9B for details). Points on each plot represent the BMC and normalized magnitude for a feature, coded by fluorescent channel (color), compartment (shape) and feature type (letter). The gray shaded area is at −1 < magnitude < 1 and represents the threshold for a marked response from control for scaled feature data. The onset of cytotoxicity and cytostatic effects are marked by red and gray vertical dotted lines, respectively. Absence of vertical lines indicates that cytotoxicity or cytostatic effects were not observed within the concentration range tested. Distinct profiles of phenotypic changes are observed for each reference chemical and effects are observed well below the threshold for cytotoxicity. (B) Accumulation plots for exemplary reference chemicals. The feature-level BMCs were grouped into 49 categories. Categories where > 30% of the constituent features were concentration-responsive were ranked in ascending order according to the median BMC of the category. The 25 most sensitive categories for each chemical are displayed in the accumulation plot and coded as described in panel A. For each chemical, the most sensitive categories are the leftward most points on the plot. The most sensitive category is defined as the CP POD (see Figure 1).
Due to a lack of foreknowledge regarding what types of phenotypic features would be of greatest importance or most sensitive for detecting the bioactivity of structurally-diverse environmental chemicals, the authors’ guiding principle for design of the phenotypic profiling assay was to measure an abundance of features (n = 1,300 / cell), many of which were likely to be correlated (or anti-correlated) with one another. In fact, there is correlation/anti-correlation among features, as some features measure different transformations of the same image (i.e. SER features in the texture modules; compactness and texture within a channel). For these reasons, and to avoid the influence of rare, spurious curve fits on in vitro potency estimates, features were grouped in 49 categories for the purpose of defining a POD for the CP assay. Categories were defined based on channel, compartment and feature type (e.g. intensity of the ring compartment in the AGP channel, Table S2, Fig. S7). For each category the median BMC of the affected endpoints was determined. A category was considered affected if ≥ 30% of the endpoints had a BMC. Fig. 3B illustrates the result of the category analysis using accumulation plots. Each category is rank-ordered according to the BMC of the category. These plots convey similar information as the potency-magnitude plots and are useful for identifying which categories of features are most sensitive to the effects of chemicals. For berberine chloride, categories associated with mitochondrial morphology were the most sensitive. Likewise, for Ca-074-Me, categories associated with both mitochondrial and AGP effects were the most sensitive, followed by effects on DNA at higher concentrations approaching the threshold for cytotoxicity. For etoposide and rapamycin, the range of potencies for affected categories was much narrower. However, for etoposide it was apparent that categories related to the area of nuclei and cells (“Shape/Position” categories) were strongly affected at low concentrations. For rapamycin, the most sensitive categories were the radial distribution of AGP labeling throughout the cell/cytoplasm; however, many more feature categories were affected within half a log10-unit of potency, meaning that a variety of morphological features of the U-2 OS cells were affected simultaneously at the same test concentration.
The four chemicals displayed in Fig. 3 (i.e. berberine chloride, Ca-074-Me, etoposide and rapamycin) were selected as phenotypic reference chemicals for use in evaluating CP assay performance in HTS applications in U-2 OS cells as detailed below.
Temporal Characterization of HTPP POD Estimates
The initial experiments described above, as well as in Gustafsdottir et al. (2013), were performed using a 48 h exposure duration. In the next phase of this study, we characterized the temporal progression of phenotypic changes in the Reference Set at several time points up to and including 48 h (i.e. 3, 6, 12, 24 and 48 h). We hypothesized that reducing the exposure duration could be beneficial for distinguishing the primary (i.e. most immediate) phenotypic effects of chemicals, from secondary effects that may occur with longer exposure durations. Of note, cytostatic effects are more pronounced at longer exposure times due to the high proliferation rate of the U-2 OS cells. Comparison of PODs obtained from different sampling times demonstrated that PODs decrease over time but remain remarkably comparable between 12–48 h of exposure (within one order of magnitude of the POD at 48 h) (Fig. 4A), even as the number of affected features increased over time for most chemicals (data not shown). For example, for berberine chloride, phenotypic changes were apparent at 6 h and it appeared that the magnitude of the effects on mitochondrial morphology increased with time (Fig. 4B). Secondary effects (e.g. changes in DNA morphology) were apparent at 48 h.
Figure 4. Temporal Characterization of Phenotypic Profiles and CP POD.
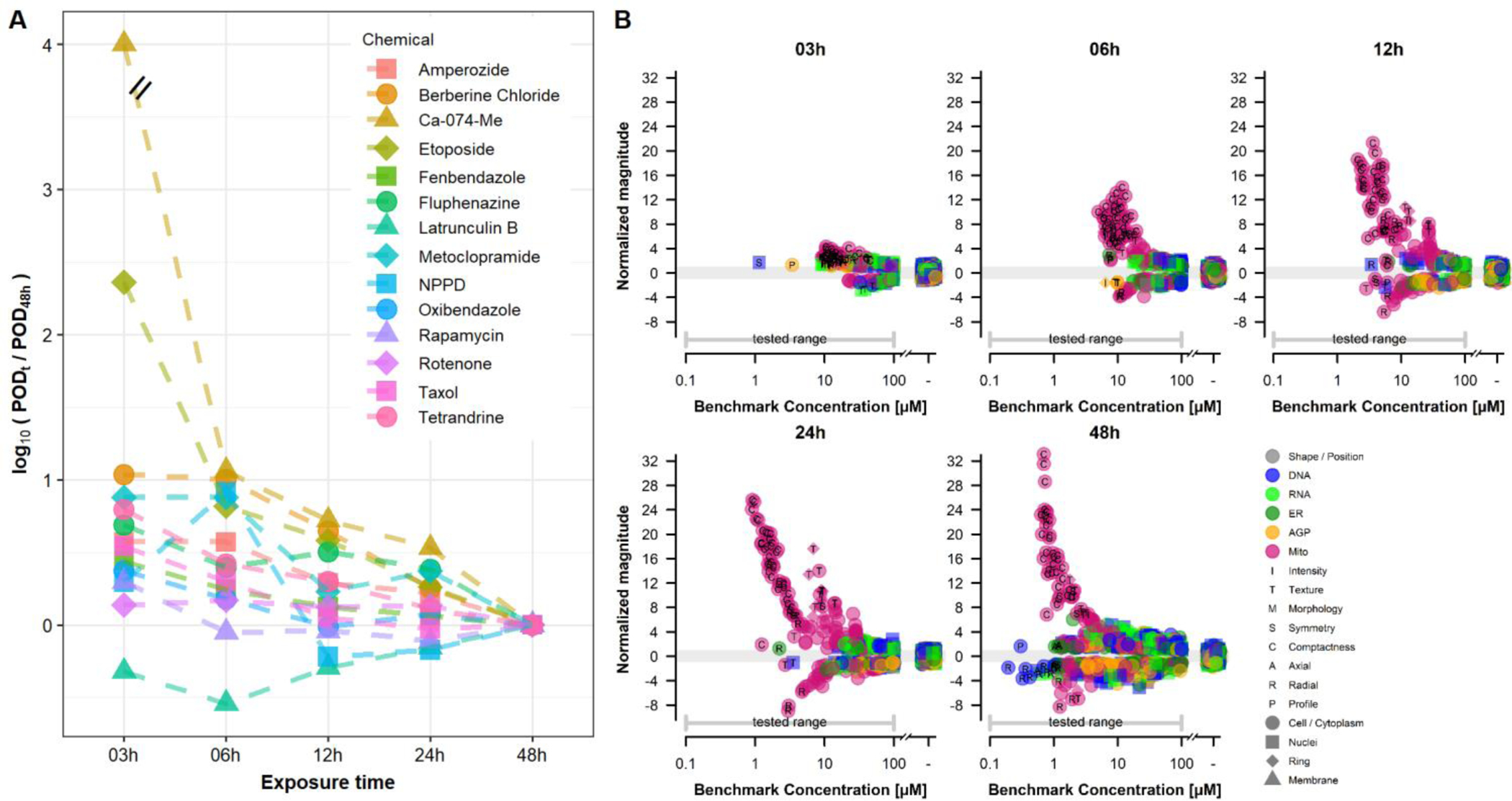
U-2 OS cells were treated with chemicals in the Reference Set for 3, 6, 12, 24 or 48 h prior to sampling and imaging. (A) CP PODs were calculated for each of 14 phenotypic reference chemicals for each exposure duration as described in the Methods. The ratio of the CP POD at 48 hours versus the CP POD at shorter exposure durations was calculated. For four chemical x exposure time combinations, there was no CP POD at the maximal tested concentration. The maximal tested concentration + ½ log10 was used as a surrogate. For most chemicals, the CP POD was stable (i.e. did not vary more than one order of magnitude) between 12 and 48 h exposure durations. (B) Potency-magnitude plots for an exemplary reference chemical, berberine chloride, at different exposure durations. Concentration-response modeling of feature data was performed, and feature-level BMCs calculated as described in Methods. The normalized magnitude (y-axis) was defined as the largest effect size observed at a non-cytotoxic concentration (see Fig. S9B for details). Points on each plot represent the BMC and normalized magnitude for a feature, coded by channel (color), compartment (shape) and feature type (letter). The gray shaded area is at −1 < magnitude < 1 and represents the threshold for a marked response from control for scaled feature data. Note that the magnitude of effects on mitochondrial morphology increases with longer exposure durations. Data was derived from ~300 cells/well across 3 technical replicate wells and 3 biological replicates (n = 9 wells total).
As the phenotypic profiles seemed to more clearly delineate primary effects at 24 h vs 48 h, but the POD changed little, 24 h was chosen as the exposure duration for HTS applications in U-2 OS cells as detailed below.
Evaluation of CV and CP Assay Performance
Concentration-response screening of the Screening Set was performed under similar conditions as those used for concentration-response screening of the Reference Set, with few exceptions. The exposure duration was reduced to 24 h and chemicals were screened using one technical replicate per chemical x concentration across four biological replicates (i.e. independent cultures of the same passage number). The experimental design included a concentration-response of the four phenotypic reference chemicals, as well as staurosporine (i.e. cell viability control) on each assay plate. The overall experimental design across the four biological replicates, as well the dose plate design and schema for randomizing treatments within assay plates is illustrated in Fig. S1. In brief, there were twelve dose plates in the screening study, each of which was used to dose one assay plate within each independent biological replicate culture. CV and CP assay performance were evaluated by aggregating well-level results from the cell viability control chemical and phenotypic reference chemicals, respectively, across biological replicates for each plate group (i.e. the four assay plates associated with each individual dose plate).
Assay performance results for the CV assay are shown in Fig. 5A–5C. Concentration-response curves for the percent of PI-positive cells (Fig. 5A) and nCC (Fig. 5B) were similar (nearly superimposable) across plate groups. In addition, the BMC for cytostatic effects of staurosporine did not differ more than one half order of magnitude across plate groups (Fig. 5C). These data indicate that the results of the CV assay were highly reproducible across plate groups.
Figure 5. Evaluation of CV and CP Assay Performance.
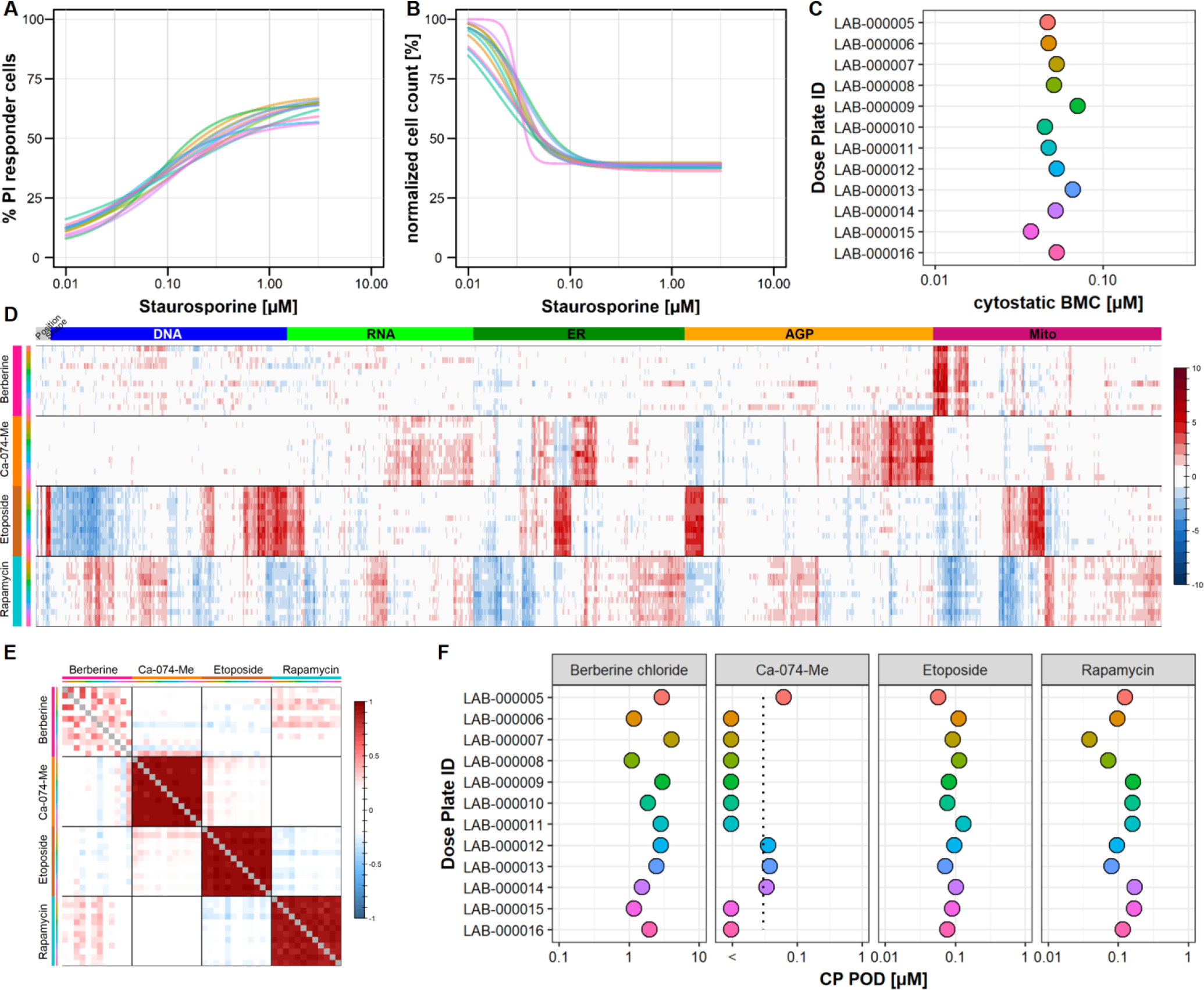
(A-C) Reproducibility of CV assay. A six-point concentration-response of staurosporine was included on every CV assay plate (“t” design, Fig. S1A) in order to evaluate assay performance. Each curve in the left (% PI-positive cells) and middle (nCC) panels represents data pooled within a plate group (i.e. across four biological replicates). Concentration-response curves were fit using the tcpl package as described in Methods. The concentration-response curves for these two endpoints were similar across plate groups (nearly superimposable) indicating a high degree of assay reproducibility. Cytostatic BMCs for staurosporine were also highly reproducible across plate groups, varying by less than one half order of magnitude. (D-F) Reproducibility of CP profiles. A six-point concentration-response of four reference chemicals was included on every CP assay plate (“t” design, Fig. S1A). In the heatmap (panel D) one concentration per chemical that produced marked phenotypic effects is shown: Berberine chloride (10 μM), Ca-074-Me (0.3 μM), Etoposide (0.3 μM), Rapamycin (3 μM). Well-level feature data was normalized and scaled per plate and then averaged within plate group. The columns of the heatmap correspond to the 1300 features, organized by fluorescent channel. Tile colors represent the magnitude of increase or decrease in a measured feature with respect to DMSO control. Each row represents data from a plate group. Data for each chemical is separated by horizontal black lines and labeled according to the multicolored tiles to the left of the heatmap. Reproducible phenotypic profiles were observed for each reference chemical across plate groups. (E) Correlation of CP profiles. Data was normalized and scaled per plate and then averaged within plate group. Normalized area under the curve (nAUC) was computed for each endpoint. The nAUC is defined as the summed mean magnitude across all non-cytotoxic concentrations (see Fig. S9B for details). Each row/column represents data from a plate group and the profiles were compared to each other using Spearman correlation coefficient (red = correlation, blue = anti-correlation). Reference chemical profiles display a high degree of positive correlation across plate groups and low correlation with each other. (F) Reproducibility of CP PODs. Data was normalized and scaled per plate, aggregated within plate group and used for concentration-response modeling with BMDExpress. The CP POD is defined as the median concentration of the most sensitive category that had ≥ 30% affected endpoints. The vertical dotted line indicates the lowest tested concentration. If the POD was below the tested range, the POD was set as half an order of magnitude below the lowest tested concentration. For 3 of 4 reference chemicals, CP PODs were highly reproducible across plate groups, varying by less than one half order of magnitude. For the remaining chemical (Ca-074-Me) CP PODs were mostly below the tested concentration range.
Assay performance results for the CP assay are shown in Fig. 5D–5F. The heatmap in Fig. 5D visualizes the phenotypic effects of each of the four reference chemicals across plate groups. Overall, a broad range of endpoints was affected by at least one of the reference chemicals. Qualitatively, the phenotypic profiles for each of the reference chemicals were remarkably similar across plate groups, but quite dissimilar from one another. The distinct difference in effects between berberine chloride and Ca-074-Me on mitochondrial and Golgi morphology were clearly visible. Likewise, the constellation of many features affected by etoposide and rapamycin were similar across plate groups but did not resemble one another. Correlation of profiles across plate groups was measured by a Spearman correlation performed on the normalized area under the curve (nAUC, see Fig. S9B) of each feature (Fig. 5E). For three chemicals, profiles across plate groups were strongly correlated (Ca-074-Me 0.94, etoposide 0.94, rapamycin 0.89 on average). Berberine chloride was the only compound with poor correlation (mean 0.22), probably due to the fact than few (100–300) endpoints were affected across a limited concentration range. Finally, for three of the reference chemicals, the CP POD varied less than 1 order of magnitude across plate groups (Fig. 5F). For Ca-074-Me, the calculated CP POD was below the concentration range tested in some instances and approximated the lower end of the tested concentration range in other instances. Overall, these data indicate that the results of the CP assay, in terms of phenotypic profiles and test chemical potency, were highly reproducible across plate groups.
The screen also included 16 chemicals run in duplicate. For most chemicals, the number of affected features as well as the CP POD were consistent between the duplicates (Fig. S11). However, there were two chemicals that had a large difference in the number of affected features across the duplicates, but remarkably consistent CP PODs (2,4-dinitrophenol, oryzalin). Three chemicals produced hits in only one of the duplicates. All three of those chemicals (2-anisidine, dimethylarsinic acid, 2,6-dimethylanaline) had < 200 affected features in each duplicate and category summation failed to yield a CP POD for one of the duplicates. For the remaining chemicals, the two PODs were within one order of magnitude. For screening purposes, the three chemicals with mismatching duplicate results were treated as active in the CP assay.
Summary of Screening Results
Overall, after accounting for the presence of duplicates, a total of 462 unique chemicals were screened. A total of 441/462 chemicals (95%) were active in the CP assay, whereas only 123/462 chemicals (27%) were positive in the CV assay (Fig. 6A). Of note, all CV hits were also CP hits. This was expected, as an induction of cytotoxicity or a decrease in cell count should also be accompanied by some type of phenotypic change in the cells.
Figure 6. HTPP Screening Summary.

(A) Comparison of hits for the CV and CP assays. A total of 462 unique chemicals were screened. The Venn diagram indicates the number of chemicals that were CV hits (left), CP hits (right), both (union) or negative in the HTPP assay. (B) Histogram of the number of affected categories across all positive chemicals (n = 441). (C) Scatterplot of the CP POD (log10, μM) versus the ratio of the median category potency to the CP POD (defined as the most sensitive category potency). Each point represents a chemical that was positive in the HTPP assay. Color coding represents the number of categories that were affected by a chemical. (D) Accumulation plots for exemplary chemicals labeled in panel C. The feature-level BMCs were grouped into 49 categories. Categories where ≥ 30% of the constituent features were concentration-responsive were ranked in ascending order according to the median BMC of the category. Only the 20 most potent categories are shown. The onset of cytotoxicity and cytostatic effects are marked by red and gray vertical dotted lines, respectively. Absence of vertical lines indicates that cytotoxicity or cytostatic effects were not observed within the concentration range tested.
Twenty-one chemicals were negative in the CP (and CV) assays, i.e. had no category with enough affected features to derive a HTPP POD (Fig. 6A). The negative chemicals and their reported use are listed in Fig S12. This group comprised three therapeutics, four chemicals used as food additives / fragrance / flavoring agents, six herbicides, two other pesticides and six chemicals used in manufacturing and industrial settings.
In order for a chemical to be identified as a hit in the CP assay, it must have produced effects in 30% of the features of at least one phenotypic category and may have potentially produced effects in all (n = 49) phenotypic categories. The histogram in Fig. 6B illustrates the broad range in the number of phenotypic categories affected by chemicals in the Screening Set. Many of the chemicals identified as hits (~34%) produced effects on 10 or less phenotypic categories, whereas a similar percentage of chemicals produced effects on 40 or more phenotypic categories.
Additional information regarding the biological activity and potential organelle-specific effects of test chemicals can be gleaned by comparing the CP POD (i.e. the potency value for the most sensitive category) to the median potency of all categories affected by a particular chemical (Fig. 6C). Purple points in Figure 6C represent chemicals that affect few categories (< 10). For such chemicals, the identity of the affected categories tended to be from the same organelle / fluorescent channel. Examples include farglitazar, atrazine and acenaphthylene (Fig. 6D, top row). Chemicals in the top half of Figure 6C, generally affected many categories (>10), but the difference between the CP POD and the median category potency was relatively large (≥ 1 on the log10-μM scale). Examples include endosulfan, pyrimethamine and 2-methy-4,6-dinitrophenol (Fig. 6D, second row). Chemicals in the middle part of Fig. 6C also tended to affect many categories (> 10), but nearly all phenotypic categories were affected at approximately the same potency. For some chemicals, these effects were observed at or near the threshold for cytotoxicity. Examples include triphenyltin hydroxide, thiram and disulfiram (Fig. 6D, third row). However, there were also chemicals that affected many categories at the same potency in the absence of cytotoxicity. Examples include cladribine and cycloheximide (Fig. 6D, bottom row). Overall, review of phenotypic profiling data in this manner can help distinguish chemicals that may be producing effects on a particular organelle from chemicals that produce a more generalized or global cellular response.
Comparison of Screening Results to In Vivo Effect Values
In order to compare the bioactivity potency estimates obtained from the HTPP screen to in vivo effect values, the HTPP PODs were used to predict HTPP AEDs using reverse dosimetry. The HTPP AEDs were then compared to the lower 5th percentile of effect values (PODtrad) assembled from various mammalian toxicity studies that used oral gavage as the route of administration. In extreme cases, the difference between the HTPP AED and the PODtrad was 5 orders of magnitude. However, the distribution of the HTPP AEDs was centered around the PODtrad with a median difference of 0.2 (Fig. 7A). Accordingly, 50% and 81% of the HTPP AEDs were within 1 and 2 orders of magnitudes from the PODtrad, respectively. For comparison, potency estimates from two other NAMs were included: TTC and ToxCast (Paul-Friedman et al., 2019). Whereas the TTC approach was on average roughly 4 orders of magnitude below the PODtrad, AEDs calculated from the ToxCast assays fell in between the two approaches and were roughly 1 order of magnitude below the PODtrad.
Figure 7. Comparison of HTPP Assay Results to In Vivo Effect Values and published NAM Results.
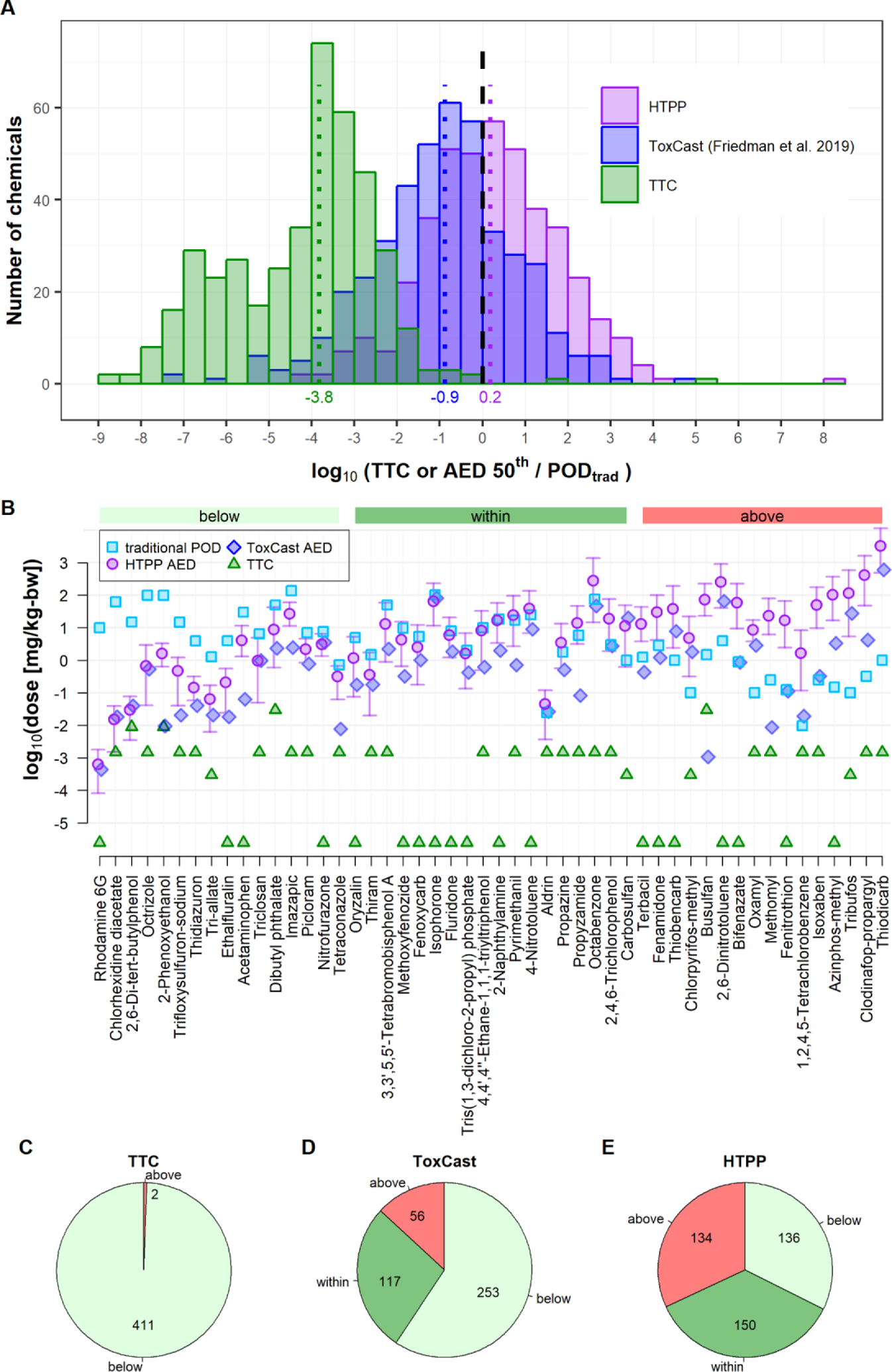
(A) Comparison of different alternative approaches to the PODtrad. HTPP PODs were used to extrapolate an administered equivalent dose (HTPP AED) that would correspond to a plasma concentration equivalent to the HTPP POD in a human population. The median HTPP AED (HTPP AED 50th) was then compared to the 5th percentile (PODtrad) derived from a collection of in vivo mammalian effect values from a variety of study types and test species that used an oral route of administration. Similarly, PODs from the ToxCast assay suite were calculated from published literature (Paul-Friedman et al., 2019), extrapolated to AEDs and compared to PODtrad. For comparison, TTCs from the same study were added as well. Vertical dotted lines and numbers below indicate the median of the distribution for each NAM. The vertical dashed line indicates the unity line. The histogram comprises only chemicals that had available HTTK and in vivo data (ToxCast n=426, TTC n=413, HTPP n=420 (only HTPP hits)).
(B) IVIVE results for exemplary chemicals. The PODtrad values (lightblue squares) are compared to HTPP AED results. Monte Carlo simulation was used during reverse dosimetry analysis to account for physiological diversity in an average human population. The median and 5th to 95th confidence interval are shown for HTPP AED (purple circle and error bars). For ToxCast, only the median AED is displayed (darkblue diamonds). Chemicals were sorted according to the difference between the HTPP AED distribution and the PODtrad and grouped into “below”, “within” and “above” categories as described in Methods.
(C) Tally of the number of chemicals grouped in the “above”, “within” and “below” categories. Chemicals in the former group had AEDs that overpredicted the dose associated with in vivo effects. Chemicals in the two latter groups provided either a comparable or conservative surrogate for the PODtrad, respectively. Since no dose range exists for the TTC approach, the chemicals were grouped into “above” and “below” only.
For reverse dosimetry, the httk R package (Pearce, Setzer, Strope, et al., 2017) uses a Monte Carlo approach to vary pharmacokinetic parameters representative of inter-individual variability in a population in order to compute an AED range (5th – 95th percentile), as illustrated for HTPP AEDs in Fig. 7B. The PODtrad and distributions around the AEDs were used to bin chemicals into three groups: (1) “below” - chemicals where the AED interval is less than the PODtrad (2) “within” - chemicals where the PODtrad lies within the AED interval, and (3) “above” - chemicals where the AED interval is above the PODtrad (Fig. 7B–7E). This grouping approach was used to compare HTPP AEDs and ToxCast AEDs, respectively, to the corresponding PODtrad for each chemical. In contrast, TTCs provide a point estimate for toxicity. Therefore, for TTCs, chemicals were binned into two groups: (1) “below” – TTC was below the PODtrad and (2) “above” – TTC value was above PODtrad. As expected from Fig. 7A, the TTC approach provided a conservative estimate for 411/413 (99.5%) of chemicals (Fig. 7C). AEDs based on ToxCast data were conservative (i.e. “below” group) for 59% (253/426) and comparable to PODtrad (i.e. “within” group) for 27% (117/426) of cases (Fig. 7D). HTPP AEDs were evenly distributed among the three groups and led to a conservative or comparable surrogate for 68% (285/420) of tested chemicals.
For 21 chemicals in the “above” group of the HTPP AED comparison, the lower bound of the HTPP AED was > 2 orders of magnitude above the PODtrad (i.e. HTPP AED overestimated the dose associated with in vivo effects) (Fig. S13A). The majority of chemicals (15/21) were insecticides or nematicides and most of them target acetylcholinesterase. Moreover, there were two herbicides and two therapeutics, the latter of which targets the estrogen and thyroid systems. To exclude the possibility that the large difference was due to an outlier in the in vivo data set, comparison to the individual effect values demonstrated that the HTPP AED was often less sensitive than the least sensitive in vivo effect value (Fig. S13B).
For 20 chemicals in the “below” group of the HTPP AED comparison, the upper bound of the HTPP AED was > 2 orders of magnitude below the PODtrad (i.e. HTPP AED would have underestimated the doses associated with in vivo effects) (Fig. S14A). These chemicals were diverse and included a number of herbicides, pesticides, drugs, food additives and industrial chemicals. A common characteristic of these chemicals was that they had fewer in vivo effect values available in ToxValDB, often less than ten. A statistical analysis of the entire chemical set revealed that chemicals where the predicted HTPP AED was lower than the PODtrad had significantly fewer in vivo observations from which to determine the PODtrad (Fig. S14B).
Overall, while for a subset of chemicals the HTPP AED was several orders of magnitude away from the PODtrad, 50% and 81% of the HTPP AEDs were within 1 and 2 orders of magnitudes, respectively. Moreover, the median HTPP AED was less conservative but closer to the in vivo effect value than estimates for two other NAMs.
Discussion
In support of NAM-based chemical safety assessment, this study used an imaging-based HTPP assay to estimate bioactive doses as part of a broad-based initial screening strategy for environmental chemicals. The present study adapts an existing high content imaging-based HTPP assay (Bray et al. 2016) for bioactivity screening of environmental chemicals. In the first phase of this study, we identified a set of candidate phenotypic reference chemicals (i.e. Reference Set) from the literature for potential use as plate-based controls for assessing assay performance and screened them in concentration-response mode in U-2 OS cells. U-2 OS cells were used in the present work to facilitate comparison to previously published studies (Bray et al., 2016; Gustafsdottir et al., 2013; Wawer, Li, et al., 2014). We also developed a novel image analysis protocol for measurement of phenotypic features at the single cell level, developed the computational pipelines required for modeling and analysis of this high-dimensional data and developed a schema for calculating the in vitro threshold for chemical bioactivity (HTPP POD) using a phenotypic categorization approach (Fig. 1). For most of the Reference Set, we were able to demonstrate reproducible cellular phenotypes as compared to previous studies (Gustafsdottir et al., 2013). We also evaluated the temporal progression of phenotypic effects and stability of in vitro bioactivity values as a function of exposure duration in order to identify a suitable exposure duration (24 h) for screening applications. In the second phase of this study, we performed concentration-response screening of several hundred chemicals from the ToxCast collection (i.e. Screening Set). We demonstrated a high degree of reproducibility in phenotypic profiles and in vitro potency estimates derived from the HTPP assay using the aforementioned phenotypic reference chemicals. We also compared HTPP AEDs calculated from in vitro bioactivity thresholds to in vivo effect values using IVIVE. In many instances (68%), the HTPP AEDs were either more conservative or comparable to the PODtrad. Overall, we conclude that the HTPP assay can be used as an efficient, cost-effective and reproducible screening method for characterizing the biological activity and potency of environmental chemicals for potential use in NAMs-based safety assessments.
The EPA CompTox Blueprint (Thomas et al., 2019) envisions use of broad-based high-content profiling assays that are tractable across many different human-derived cell types as a means for generating NAMs data for potential use in chemical safety assessment. Imaging-based phenotypic profiling is one such assay. There are many different combinations of fluorescent protein markers and molecular probes that may be used in phenotypic profiling assays (Gustafsdottir et al., 2013; Ljosa et al., 2013; Loo, Wu, & Altschuler, 2007). For the present study, we employed a combination of non-antibody-based markers of organelles and sub-cellular structures as described in the “Cell Painting” assay developed by Gustafsdottir et al. (2013) and described in Bray et al. (2016). In previously published studies, the Cell Painting assay has been used primarily in either single-concentration chemical screening mode (Wawer, Jaramillo, et al., 2014; Wawer, Li, et al., 2014) or to investigate the effects of genetic perturbations (i.e. protein knockdown or overexpression) on cellular phenotypes (Rohban et al. 2017). Previous studies comparing Cell Painting profiles to HTS data have demonstrated that this method can detect bioactive small molecules that have selective activity at discrete molecular targets as well as those that have promiscuous biological activity (Wawer et al. 2014a) and that chemicals with similar biological activity produce similar phenotypic profiles (Gustafsdottir et al. 2013; Wawer et al. 2014b). In addition, HTS screening has demonstrated that chemicals in the ToxCast collection (which is enriched for environmental chemicals) have a spectrum of biological response profiles ranging from specific activity at a discrete molecular target to promiscuous activity at multiple molecular targets (Kleinstreuer et al., 2014; Richard et al., 2016; Sipes et al., 2013). Therefore, it was hypothesized that the combination of organelle markers from the Cell Painting assay would provide a broad-based approach for detecting and profiling the biological activity of environmental chemicals within the ToxCast collection (as well as other environmental chemicals of interest) that have either selective or promiscuous biological activity. To our knowledge, we are the first to apply the Cell Painting assay to large-scale screening of environmental chemicals in concentration-response mode.
There are several aspects of the present work that differ from previous Cell Painting studies. From an imaging standpoint, we used single z-plane confocal microscopy that provides greater optical resolution of cellular microarchitecture as compared to widefield fluorescent microscopy conducted at the same magnification. From an image analysis standpoint, we developed a novel set of feature measurements that are amenable for characterizing chemical effects on organelle morphology in different compartments of the cell (i.e. nucleus, cytoplasm, perinuclear space, outer membrane). One of the recognized challenges associated with image-based profiling studies is deciding which morphological features are important for detecting differences in the phenotypes of treated cells as compared to control conditions (Caicedo et al. 2016). In targeted HTS, the researcher typically has a priori knowledge regarding which changes in cellular phenotype, and associated quantitative features, are of interest for characterizing effects on the biological process being studied. For instance, in a targeted screen for genotoxic chemicals, one may choose to focus on measurements of nucleus morphology and DNA-damage marker proteins (Persson et al., 2014; Takeiri et al., 2019). However, in unbiased profiling studies in which there is no a priori knowledge regarding the phenotypes a diverse set of chemicals may produce (or the molecular mechanisms underlying them), restricting or limiting the number of morphological features measured may negatively impact the sensitivity of the assay for detecting chemical bioactivity. Thus, the guiding principle for development of the feature set used in the present study was to measure an abundance of features across many cellular compartments and domains of feature space (i.e. texture, intensity, distribution, etc.). Of note, a subset of the phenotypic features measured in the present study are comparable to those generated by the Cell Profiler software package employed in previous Cell Painting studies (Bray et al., 2016). However, some feature types measured by CellProfiler such as correlation / colocalization of intensity across fluorescent channels are not included in the present study. Reanalysis of images using the CellProfiler approach could be used to provide complementary information to the feature set defined here.
Many of the features measured in the present study have some degree of correlation or anti-correlation. The measurement of correlated features in phenotypic profiling studies is one of the recognized challenges associated with this type of assay (Caicedo et al., 2016). Development of methods for dimensionality reduction and effective mapping of morphological features to schema that facilitate interpretation of biological effects are both areas of active research (Caicedo et al., 2017; Caicedo et al., 2016). Our current solution to these challenges in terms of defining the threshold for chemical bioactivity and facilitating biological interpretation of effects is category-based aggregation of feature-level potency values as illustrated in Fig. 1. This concept was adapted from transcriptomics profiling studies that sequentially used concentration-response modeling of gene level expression values and mapping to gene sets and/or biological pathways to determine potency thresholds for chemical bioactivity (Thomas et al., 2007; Thomas et al., 2012). Mapping to gene sets / pathways allowed for putative identification of perturbed gene networks that may be associated with chemical bioactivity or toxicity with the most sensitive category providing a transcriptional POD (Harrill et al., 2019). Likewise, the phenotypic categorization approach described in the present study can be used to identify an in vitro threshold for chemical bioactivity corresponding to the most sensitive affected phenotypic category (i.e. HTPP POD). A strength of this category-based approach is that it facilitates biological interpretation of the feature-level profiling data, while reducing the impact of correlated features on the HTPP POD, and aids in identification of cellular effects that may be associated with chemical bioactivity or toxicity. However, a limitation of the current category-based approach is the requirement for >= 30% of the features within a category to be concentration-responsive in order for a category to be identified as affected by a test chemical. This criterion might favor identification of phenotypes that are associated with changes in many correlated features within a category, while phenotypes that are associated with changes in only a few non-correlated features (potentially across different categories) might be overlooked. Moreover, the current approach does not account for correlation across feature categories; indeed, future research to understand the relatedness of the features and their corresponding error could be used to reduce the dimensionality of these data using established methods (Caicedo et al., 2017; Haggard et al., 2018) and provide more information regarding identification and biological interpretation of feature profiles.
Interpretation of HTPP data faces some of the same challenges as interpretation of in vivo toxicologic pathology data. Namely, while a change in the morphology of a cell (or tissue) in response to increasing concentration (or dose) of a chemical is indicative of biological activity, the molecular mechanism underlying the morphological effect may not be immediately obvious from the quantitative (or qualitative) observational data. Take for instance berberine chloride, a chemical known to decrease mitochondrial respiration through inhibition of mitochondrial respiratory complex I (Zhang & Ye, 2012). The HTPP data for this chemical identifies mitochondrial compactness as the most sensitive phenotypic category (Fig. 3 & 4). From this cellular phenotype, one could reasonably hypothesize that mitochondrial function is impacted by berberine chloride, but one could not discern the molecular target site from the HTPP information alone. Information from other sources, such as orthogonal molecular assays or mapping of phenotypic profiles to a reference database containing profiles of well-characterized chemicals with known molecular mechanisms-of-action would be needed. This same logic would hold true for other types of phenotypic effects observed in various organelles evaluated in the HTPP assay. Overall, while the phenotypic categorization approach can readily provide information on chemical bioactivity thresholds and provide insight into cellular functions that may be perturbed by a chemical, additional data sources would be needed to make inferences regarding molecular targets underlying the cellular phenotypic effects.
The HTPP phenotypic categorization analysis assigns a potency value to each category where ≥30% of the constituent features are affected by a chemical. We noted that the distribution and rank order of these category-level potency values varied from chemical-to-chemical. For some chemicals, only a few phenotypic categories were affected within the concentration range tested and the affected categories tended to be associated with the same organelle (Fig 6D, top row). This pattern was indicative of an organelle-specific effect that does not progress to more generalized effects within the concentration range tested. For other chemicals, many phenotypic categories were affected within the concentration range tested, but the potency values for the most sensitive categories were left-shifted compared to the median potency across all categories and the most sensitive categories tended to be associated with the same organelle (Fig 6D, second row). The left-shifted phenotypic category potencies may be indicative of specific effects on a particular organelle occurring at or near the threshold for chemical bioactivity in the absence of other widespread effects in the cell. In the case of endosulfan, it appears that more phenotypic categories are affected as the threshold for cytotoxicity is approached.
For still other chemicals, many features and associated phenotypic categories were affected by a chemical and the potency estimates for each phenotypic category were nearly equivalent, resulting in a near “vertical stacking” of potency values when visualized as an accumulation plot (Fig. 6D, bottom two rows). The pattern is indicative of a simultaneous onset of effects across many organelles at approximately the same test concentration. In some cases, this phenomenon was observed at or near the threshold for cytotoxicity. Examples include chemicals that are associated with promiscuous bioactivity in HTS screening assays (Sipes et al., 2013) such as organometallics (e.g. triphenyltin hydroxide) and dimethyldithiocarbamates (e.g. thiram, disulfiram) (Fig. 6D, third row). Also of note, we observed a positive correlation between the number of phenotypic features affected by a chemical in the HTPP assay and the hit rate within the ToxCast assay set (Fig S15A). The reason for this association is unclear but suggests that chemicals with promiscuous bioactivity as determined from HTS screening assay batteries tend to produce a greater number of phenotypic effects in HTPP assay.
The “vertical stacking” phenomenon was sometimes observed in the complete absence of cytotoxicity. Examples include the reference chemicals etoposide and rapamycin (Fig. 3B) as well as several chemicals from the Screening Set such as cladribine and cycloheximide (Fig. 6D, bottom row). These four chemicals have well-characterized molecular mechanisms of action that could be impacted in many different types of human cells. Etoposide is a chemotherapeutic that inhibits DNA-topoisomerase II (Wu et al., 2011), rapamycin is an immunosuppressant that inhibits mTOR / FKBP12 (Balgi et al., 2009), cladribine is a chemotherapeutic adenosine analogue that produces DNA strand breakage (Seto, Carrera, Kubota, Wasson, & Carson, 1985) and cycloheximide is a protein synthesis inhibitor (Obrig, Culp, McKeehan, & Hardesty, 1971). Prolonged in vitro exposure to any of these chemicals should eventually result in cytotoxicity. However, in the present study, cytotoxicity was not observed at 24 h in U-2 OS cells within the concentration range tested. From the rank order of phenotypic category potencies, it is difficult to discern if one organelle is more sensitive than another to these chemicals. Even so, each of these chemicals produced a distinct profile of feature-level effects in terms of the identity, directionality and effect magnitude (Fig. S16). This indicates that the molecular activities of these chemicals manifest as a distinct signature, or fingerprint, of cellular effects. In the pharmaceutical space, these types of phenotypic fingerprints have been used for data-driven grouping and categorization of chemicals based on known or suspected molecular mechanisms-of-action (Gustafsdottir et al., 2013; Wawer, Jaramillo, et al., 2014). It is possible that HTPP phenotypic fingerprints could also be used in conjunction with chemical descriptor information to facilitate read-across predictions in the environmental chemical space (Shah et al., 2016). This is a topic that will be investigated in future work from our group. To summarize, we conclude that the phenotypic categorization approach can be used to putatively identify chemicals that have specific effects on a particular organelle at the threshold for bioactivity and distinguish them from chemicals that appear to act through a promiscuous molecular mechanism of action.
A necessary step in development of an HTS assay is identification of reference chemicals (i.e. positive controls) that can be used to track assay performance and evaluate assay reproducibility (Buchser et al., 2012). For targeted HTS assays, selection of reference chemicals can be based on prior knowledge of biological activity at a molecular target of interest and confirmed by measuring a defined biological response following reference chemical treatment (e.g., nuclear receptor translocation in response to a ligand) (Mackowiak et al., 2019). Identification of reference chemicals for high-throughput profiling assays is more complicated given the highly multiplexed nature of the measured feature set and lack of prior knowledge regarding what cellular phenotypes may be produced in response to chemicals. In the present study, we identified 14 candidate phenotypic reference chemicals (Table 2) from previous Cell Painting studies where the cellular phenotype was described in a narrative fashion based on results from single concentration screening (Gustafsdottir et al. 2013). We then evaluated these chemicals in U-2 OS cells in concentration-response mode. Many of the candidate reference chemicals produced effects on cellular morphology at non-cytotoxic concentrations (Fig. 2I). In particular, berberine chloride, Ca-074-Me, etoposide and rapamycin produced phenotypic responses that were qualitatively similar to those described in previous studies (Fig. 2I, Fig. 3) at concentrations well below the threshold for cytotoxicity. These four chemicals were selected as reference chemicals for the screening portion of this study and used to evaluate assay performance. During screening, the profile of phenotypic effects produced by the four reference chemicals was qualitatively and quantitively similar across plates in terms of the subset of phenotypic features affected by each chemical (Fig. 5D), the magnitude and direction of observed effects (Fig. 5D) and the CP POD values derived using the phenotypic categorization approach (Fig. 5F). The phenotypic profiles produced by the four reference chemicals were also quite different from one another (Fig. 5E). In fact, a majority of the phenotypic features measured in this study were affected by at least one of the reference chemicals. Therefore, the combined use of these four chemicals allows for comprehensive evaluation of assay performance across a large portion of the measured feature space. The consistency of the CP POD values also indicated that the HTPP assay provided reproducible estimates of the threshold for chemical bioactivity for multiple chemicals. From these data, we conclude that the HTPP assay yields reproducible results in terms of both profile characteristics and chemical bioactivity threshold estimates.
In an effort to determine an optimal exposure duration for chemical screening, the temporal progression of phenotypic effects was characterized using the Reference Set. We hypothesized that reducing the exposure duration from 48 h could be beneficial for distinguishing the primary (i.e. most immediate) phenotypic effects of chemicals from secondary effects that may occur with longer exposure durations. We evaluated the consistency of CP POD values as a function of exposure duration as well as the number of phenotypic features affected and the relative magnitude of the response at each time point. We observed that for most chemicals, the CP POD values tended to decrease as a function of exposure duration and were similar across the 12, 24 or 48 h exposure durations (i.e. did not vary by more than 1 order of magnitude) (Fig. 4A). In addition, for most chemicals, the number of affected features and the number of affected phenotypic categories increased with increasing exposure duration (data not shown). For many chemicals in the Reference Set, features that were affected at shorter exposure durations continued to be affected at longer exposure durations, but with larger effect magnitudes. Fig. 4 illustrates this phenomenon using berberine chloride as an example chemical. Low magnitude effects on phenotypic features associated with mitochondrial compactness are observed beginning at 3 hours post-exposure and increase in magnitude up to 24–48 hours post-exposure. These data suggest that, for many chemicals, affected features with the greatest response magnitude at later time points (24–48 h) may be representative of the primary (i.e. earliest onset) phenotypic effects produced by a chemical downstream of a molecular target interaction. Based on these data, we selected 24 h as the exposure duration for the screening portion of this study.
In the latter portion of the present study, we performed concentration-response screening of 462 chemicals. The Screening Set mostly consisted of chemicals identified for use in a retrospective case study comparing PODs derived from various NAMs to PODs derived from traditional in vivo effect studies conducted as part of the multinational APCRA collaboration (Kavlock et al., 2018; Paul-Friedman et al., 2019). The case study chemicals were selected based on the availability of previously-generated NAMs screening data, HTTK information, in vivo effect values, and exposure predictions. As a result, the Screening Set was enriched for bioactive chemicals, such as pesticidal active ingredients, which may partially explain the high hit rate (95%) observed in the present study. Another contributing factor to the high hit rate is that the data analysis pipeline was tailored for high sensitivity, which bears the risk of false positive results. Of the 16 duplicated compounds, three were positive in only one of the duplicates (i.e. discordant hit-calls). Each of these three chemicals affected less than 200 endpoints and very few categories.
Only 21 chemicals were inactive in the HTPP assay and included four food additives (saccharin, methyleugenol, ethylparaben, alpha-isomethylionone), two common analgesics (aspirin, ibuprofen), an anticonvulsant (5,5-diphenylhydantoin), six herbicides (chloramben, imazethapyr, MCPA, metribuzin, vernolate, propoxycarbazone-sodium) and several other chemicals of varying use categories (thiamethoxam, anilazine, 2-methoxy-5-nitroaniline, dipropylene glycol monomethyl ether, acenaphthene, diisopropyl methylphosphonate, 2,6-diethylaniline, diallyl phthalate) (Fig. S12). The reason why these chemicals were inactive in the HTPP assay is unclear, but may be due to lack of molecular target expression in U-2 OS cells, biological activity at molecular target(s) that does not manifest as a change in cell morphology, physicochemical properties that limit in vitro bioavailability or amenability to in vitro screening, and/or a requirement for metabolic activation or general lack of effects on human biology at concentrations up to and including the highest concentration screened in this study. Of note, the chemicals inactive in the HTPP assay also had low hit rates in the ToxCast assay set (Fig. S15B). It is important to note that the HTPP assay can be applied to many different human-derived cell models, and that the hit rate and hit call determinations reported in the present study are specific to the U-2 OS cell model.
To compare in vitro results to in vivo effect values, extrapolation of an administered equivalent dose (AED) in mg/kg-bw/day units is necessary. When the HTPP POD values from the present study were used to calculate AEDs, 80% of the chemicals had AEDs within two orders of magnitude of the corresponding PODtrad. The distribution of the log10 ratio of the PODtrad to HTPP-derived AEDs (i.e. HTPP AED 50th) was centered near zero with a median value of 0.2.
In contrast to the HTPP results, the distribution of the log10 ratio of PODtrad to ToxCast-derived AEDs was centered near −1 with a median value of −0.9, whereas the TTC distribution was centered near −4. Both HTPP and ToxCast provided closer approximations of actual in vivo effect doses as compared to TTC. Overall, this indicates that the ToxCast suite of assays was more sensitive for detecting the bioactivity of chemicals in the Screening Set as compared to the HTPP assay. The reason for this difference in sensitivity is unclear but may be due to differences in the number of molecular targets expressed or represented in U-2 OS cells and the number of molecular targets represented in the ToxCast assay suite, respectively. Alternatively, the use of cell free assays and heterologous reporter systems within the ToxCast assay suite may result in lower estimates of chemical potency than an assay designed to evaluate effects on morphology in an intact cell. Additional analyses would be required to reach a definitive conclusion regarding the comparative sensitivity of the HTPP and ToxCast approaches.
In total, 95% of the Screening Set was active in the HTPP assay (Fig. 6A). Roughly two-thirds (68%) of the active chemicals resulted in an HTPP AED that was either conservative or comparable to the corresponding in vivo potency. Roughly one-third (32 %) of the active chemicals resulted in HTPP AEDs that over-predicted the in vivo dose that would correspond to an effect. There are many factors and uncertainties associated with this IVIVE analysis that may contribute to underprediction or overprediction of in vivo effect doses by HTPP AEDs. One such factor is the biology of U-2 OS cells in comparison to the biology of an intact animal. For example, several of the chemicals in the Screening Set with HTPP AEDs that overpredicted the in vivo effect dose (or that were negative in the HTPP assay) are known to target proteins that are lowly-expressed or not expressed in U-2 OS cells. Examples include the potent estrogen receptor agonist diethylstibestrol and the thyroperoxidase inhibitor 6-propyl-2-thiouracil (Fig. S13). Neither isoform of these proteins are highly expressed in the U-2 OS cell model (https://www.proteinatlas.org/ENSG00000091831-ESR1/cell, https://www.proteinatlas.org/ENSG00000140009-ESR2/cell, https://www.proteinatlas.org/ENSG00000137857-DUOX1/cell, https://www.proteinatlas.org/ENSG00000140279-DUOX2/cell). Therefore, the molecular biological activity underlying the HTPP phenotypes produced by these chemicals was likely associated with “off-target” effects in U-2 OS cells whereas the most sensitive in vivo effect values are likely to be associated with “on-target” biological activity in target tissue(s) in the intact animal that express these proteins. The relative difference in target expression may have contributed to the discrepancy in the HTPP AED and PODtrad. Furthermore, manual review of the group of chemicals with HTPP AEDs that overpredicted the in vivo effect dose revealed a large number of organophosphate (OP) and carbamate cholinesterase inhibitors (Fig. S13). Chemotype enrichment analysis confirmed and anchored this observation to ToxPrint structural features (Yang et al., 2015) (Fig S17). Whereas U-2 OS cells express acetylcholinesterase (https://www.proteinatlas.org/ENSG00000087085-ACHE/cell), these cells generally lack the metabolizing enzymes necessary to convert many of the OP and carbamates to their active metabolite forms and inhibit acetylcholinesterase. Therefore, overprediction of the in vivo effect dose of OP and carbamate chemicals in a U-2 OS cell model would be expected and consistent with IVIVE analysis of other in vitro data sets that lack metabolic competence, such as the ToxCast assay suite (Paul-Friedman et al., 2019; Thomas et al., 2019). Masking of chemicals that contain one or more of the OP or carbamate-associated chemotypes enriched in the over-predicting chemical group resulted in an increase in the percentage of chemicals with either conservative or comparable HTPP AED values from 68% to 77%. We conclude that chemotype analysis can be used to independently identify structural features or confirm anecdotal observations of chemical influences producing poor IVIVE predictivity in a quantitative manner.
The PODtrad used herein were derived from a compilation of different study types in a variety of species. In contrast, U-2 OS cells are a human-derived cell model. Therefore, it is possible that interspecies differences in sensitivity to certain chemicals may also contribute to discrepancies between HTPP AEDs and the PODtrad. We also observed that chemicals where the HTPP AED underpredicted the dose associated with in vivo effects tended to have fewer records available in ToxValDB for use in calculation of the PODtrad (Fig. S14). We hypothesize that fewer records may have corresponded to a lower amount of supporting information and may be associated with more uncertainty in the PODtrad, which may in turn contribute to discrepancies in the IVIVE analysis. Lastly, there are technical uncertainties associated with the HTTK assumptions that may contribute to discrepancies in the IVIVE analysis. In the HTTK modeling, chemical-specific experimental values for first-order metabolic clearance and plasma protein binding are used and population variability is incorporated for liver blood flow and kidney clearance rate (Pearce, Setzer, Davis, & Wambaugh, 2017; Wetmore et al., 2012). The HTTK model used assumes 100% gut absorption and does not take into account chemical-specific bioavailability via the oral route. Therefore, for chemicals with low gut absorption, the HTPP AED may underpredict the in vivo effect dose. For example, only 3% of FD&C Yellow 6 (CAS 2783-94-0) is absorbed upon oral gavage (IARC, 1975). Taking this into account for the reverse dosimetry would shift the HTPP AED for this chemical higher by approximately 1.5 orders of magnitude and more closely approximate the PODtrad. Overall, despite the many uncertainties in the IVIVE analysis, the HTPP AED was more conservative or comparable to the PODtrad for a majority (68%) of chemicals.
The results of this analysis are specific to U-2 OS cells; thus, screening the same set of chemicals in a different cell model may result in differences in the phenotypic profile, hit call determination and potency estimate for any particular chemical due to varying complements of genes (i.e. molecular targets, xenobiotic metabolizing enzymes, etc.) that different cells express and the morphological appearance that different cell types assume (i.e. large vs. small cytoplasm, isolated vs. tight cell packing, prominent vs. inconspicuous cytoarchitecture) when cultured in multiwell format. Therefore, it is possible that combining HTPP data from U-2 OS cells with HTPP data from other cell types, as well as HTS or profiling data from other assay formats, may increase the sensitivity or improve the predictivity of IVIVE analyses. As mentioned above, this “cell line battery” approach for hazard evaluation using high-throughput profiling assays is a component of the EPA CompTox Blueprint (Thomas et al., 2019). A future aim of our group is to extend the HTPP assay to other mammalian cell types for the purpose of bioactivity screening of environmental chemicals. Another future aim is to explore how to integrate HTPP assay data with other NAMs data such as ToxCast, high-throughput transcriptomics and in vitro functional assays.
In summary, we conclude that the HTPP assay is an efficient, cost-effective and reproducible screening method for assessing the potential hazard of environmental chemicals. The resultant data could be used in several ways to accelerate the pace of chemical safety assessment, including provision of bioactivity profiles and potency information for NAMs-based safety assessments and guiding prioritization of chemicals for further testing. Future work will involve investigating the utility of phenotypic profiles for grouping and read-across of environmental chemicals.
Supplementary Material
Highlights:
HTPP assay performance was demonstrated with 14 reference chemicals.
Multi-concentration screening of 462 ToxCast chemicals was performed using HTPP.
A novel workflow was used to predict a bioactive concentration based on HTPP.
Administered equivalent doses (AEDs) based on bioactive concentration was derived.
HTPP AED was less than or similar to in vivo effect values for 68% of chemicals.
Acknowledgements
The authors would like to thank Drs. Scott Auerbach from the National Toxicology Program and Derik Haggard from the USEPA National Center for Computational Toxicology for assistance implementing the command-line version of BMDExpress 2.0 used in this manuscript. The authors would also like to thank Terri Fairley, Daniel Hallinger and Sandra Roberts for operations support activities during conduct of this research. Finally, the authors would like to thank Drs. Richard Judson, Timothy Shafer, Megan Culbreth and Maureen Gwinn for their insightful comments during review of this manuscript.
Funding Information
The U.S. Environmental Protection Agency through its Office of Research and Development provided funding for this research.
Abbreviations
- AED
Administered Equivalent Dose
- AGP
Actin Cytoskeleton, Golgi Apparatus and Plasma Membrane
- AIC
Akaike Information Criterion
- APCRA
Accelerating the Pace of Chemical Risk Assessment
- BSA
Bovine serum albumin
- BMC
Benchmark concentration
- BMR
Benchmark response
- CV
Cell Viability
- DMSO
Dimethylsulfoxide
- EC50
Effective concentration 50
- ER
Endoplasmic Reticulum
- HI-FBS
Heat Inactivated Fetal Bovine Serum
- HTPP
High-Throughput Phenotypic Profiling
- HTPP POD
High-Throughput Phenotypic Profiling Point-of-Departure
- HTS
High-Throughput Screening
- HTTK
High-throughput toxicokinetics
- IVIVE
In vitro-to-in vivo extrapolation
- LOAEL
Lowest observable adverse effect level
- LOEC
Lowest observable effect concentration
- LOEL
Lowest observable effect level
- MAD
Median absolute deviation
- Mito
mitochondria
- NAM
New Approach Methodology
- nAUC
Normalized area under the curve
- nCC
normalized cell count
- nMAD
Normalized median absolute deviation
- NOAEL
No observable adverse effect level
- NOEC
No observable effect concentration
- NOEL
No observable effect level
- OP
Organophosphate
- PBS
Phosphate-buffered saline
- PFA
Paraformaldehyde
- PI
Propidium iodide
- POD
Point-of-departure
- PODtrad
traditional point-of-departure
- PSG
Penicillin-streptomycin-glutamine
- SCARP
Symmetry-compactness-axial-radial-profile
- ToxValDB
Toxicity Value Database
- USEPA
United States Environmental Protection Agency
Footnotes
Conflict of Interest
The authors declare no conflicts of interest regarding this manuscript. The U.S. Environmental Protection Agency has provided administrative review and has approved this paper for publication. The views expressed in this paper are those of the authors and do not necessarily reflect the view or policies of the U.S. Environmental Protection Agency. Reference to commercial products or services does not constitute endorsement.
Supplementary Information
A supplementary document is available at the following online resource: https://doi.org/10.1016/j.taap.2019.114876. Additionally, supplementary data and code is accessible at ftp://newftp.epa.gov/COMPTOX/CCTE_Publication_Data/BCTD_Publication_Data/Nyffeler/Bioactivity_screening_of_environmental_chemicals.
Bibliography
- Balgi AD, Fonseca BD, Donohue E, Tsang TC, Lajoie P, Proud CG, … Roberge M (2009). Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling. PLoS One, 4(9), e7124. doi: 10.1371/journal.pone.0007124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell BR, Ankley GT, Corsi SR, DeCicco LA, Houck KA, Judson RS, … Villeneuve DL (2017). An “EAR” on Environmental Surveillance and Monitoring: A Case Study on the Use of Exposure-Activity Ratios (EARs) to Prioritize Sites, Chemicals, and Bioactivities of Concern in Great Lakes Waters. Environ Sci Technol, 51(15), 8713–8724. doi: 10.1021/acs.est.7b01613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray MA, Singh S, Han H, Davis CT, Borgeson B, Hartland C, … Carpenter AE (2016). Cell Painting, a high-content image-based assay for morphological profiling using multiplexed fluorescent dyes. Nat Protoc, 11(9), 1757–1774. doi: 10.1038/nprot.2016.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchser W, Collins M, Garyantes T, Guha R, Haney S, Lemmon V, … Trask OJ (2012). Assay Development Guidelines for Image-Based High Content Screening, High content Analysis and High Content Imaging. In Sittampalam GS, Grossman A, & Brimacombe K (Eds.), Assay Guidance Manual. Bethesda, MD: Eli Lilly & Company and the National Center for Advancing Translational Sciences. [PubMed] [Google Scholar]
- Caicedo JC, Cooper S, Heigwer F, Warchal S, Qiu P, Molnar C, … Carpenter AE (2017). Data-analysis strategies for image-based cell profiling. Nat Methods, 14(9), 849–863. doi: 10.1038/nmeth.4397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caicedo JC, Singh S, & Carpenter AE (2016). Applications in image-based profiling of perturbations. Curr Opin Biotechnol, 39, 134–142. doi: 10.1016/j.copbio.2016.04.003 [DOI] [PubMed] [Google Scholar]
- Canada H (2016). Integrating New Approach Methodologies within the CMP: Identifying Priorities for Risk Assessment, Existing Substances Risk Assessment Program. http://www.ec.gc.ca/ese-ees/172614CE-D9F6-43DA-A528-B8175438210F/Objectives%20Paper%20November%202016_EN.pdf
- ECHA. (2016). New Approach Methodologies in Regulatory Science.
- Filer DL, Kothiya P, Setzer RW, Judson RS, & Martin MT (2017). tcpl: the ToxCast pipeline for high-throughput screening data. Bioinformatics, 33(4), 618–620. doi: 10.1093/bioinformatics/btw680 [DOI] [PubMed] [Google Scholar]
- Gustafsdottir SM, Ljosa V, Sokolnicki KL, Anthony Wilson J, Walpita D, Kemp MM, … Shamji AF (2013). Multiplex cytological profiling assay to measure diverse cellular states. PLoS One, 8(12), e80999. doi: 10.1371/journal.pone.0080999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggard DE, Karmaus AL, Martin MT, Judson RS, Setzer RW, & Paul Friedman K (2018). High-Throughput H295R Steroidogenesis Assay: Utility as an Alternative and a Statistical Approach to Characterize Effects on Steroidogenesis. Toxicol Sci, 162(2), 509–534. doi: 10.1093/toxsci/kfx274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrill J, Shah I, Setzer RW, Haggard D, Auerbach S, Judson R, & Thomas RS (2019). Considerations for strategic use of high-throughput transcriptomics chemical screening data in regulatory decisions. Current Opinion in Toxicology, 15, 64–75. doi: 10.1016/j.cotox.2019.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- IARC. (1975). IARC monographs on the evaluation of the carcinogenic risk of chemicals to man: some aromatic azo compounds. IARC Monogr Eval Carcinog Risk Chem Man, 8. [Google Scholar]
- Judson R, Richard A, Dix DJ, Houck K, Martin M, Kavlock R, … Smith E (2009). The toxicity data landscape for environmental chemicals. Environ Health Perspect, 117(5), 685–695. doi: 10.1289/ehp.0800168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson RS, Magpantay FM, Chickarmane V, Haskell C, Tania N, Taylor J, … Thomas RS (2015). Integrated Model of Chemical Perturbations of a Biological Pathway Using 18 In Vitro High-Throughput Screening Assays for the Estrogen Receptor. Toxicol Sci, 148(1), 137–154. doi: 10.1093/toxsci/kfv168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavlock RJ, Bahadori T, Barton-Maclaren TS, Gwinn MR, Rasenberg M, & Thomas RS (2018). Accelerating the Pace of Chemical Risk Assessment. Chem Res Toxicol, 31(5), 287–290. doi: 10.1021/acs.chemrestox.7b00339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstreuer NC, Yang J, Berg EL, Knudsen TB, Richard AM, Martin MT, … Houck KA (2014). Phenotypic screening of the ToxCast chemical library to classify toxic and therapeutic mechanisms. Nat Biotechnol, 32(6), 583–591. doi: 10.1038/nbt.2914 [DOI] [PubMed] [Google Scholar]
- Kroes R, Kleiner J, & Renwick A (2005). The threshold of toxicological concern concept in risk assessment. Toxicol Sci, 86(2), 226–230. doi: 10.1093/toxsci/kfi169 [DOI] [PubMed] [Google Scholar]
- Ljosa V, Caie PD, ter Horst R, Sokolnicki KL, Jenkins EL, Daya S, … Carpenter AE (2013). Comparison of Methods for Image-Based Profiling of Cellular Morphological Responses to Small-Molecule Treatment. Journal of Biomolecular Screening, 18(10), 1321–1329. doi: 10.1177/1087057113503553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo LH, Wu LF, & Altschuler SJ (2007). Image-based multivariate profiling of drug responses from single cells. Nat Methods, 4(5), 445–453. doi: 10.1038/nmeth1032 [DOI] [PubMed] [Google Scholar]
- Mackowiak B, Li L, Lynch C, Ziman A, Heyward S, Xia M, & Wang H (2019). High-content analysis of constitutive androstane receptor (CAR) translocation identifies mosapride citrate as a CAR agonist that represses gluconeogenesis. Biochem Pharmacol, 168, 224–236. doi: 10.1016/j.bcp.2019.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obrig TG, Culp WJ, McKeehan WL, & Hardesty B (1971). The mechanism by which cycloheximide and related glutarimide antibiotics inhibit peptide synthesis on reticulocyte ribosomes. J Biol Chem, 246(1), 174–181. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/5541758 [PubMed] [Google Scholar]
- Paul-Friedman K, Gagne M, Loo LH, Karamertzanis P, Netzeva T, Sobanski T, … Thomas RS (2019). Examining the utility of in vitro bioactivity as a conservative point of departure: a case study. Toxicol Sci. [Google Scholar]
- Pearce RG, Setzer RW, Davis JL, & Wambaugh JF (2017). Evaluation and calibration of high-throughput predictions of chemical distribution to tissues. J Pharmacokinet Pharmacodyn, 44(6), 549–565. doi: 10.1007/s10928-017-9548-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce RG, Setzer RW, Strope CL, Wambaugh JF, & Sipes NS (2017). httk: R Package for High-Throughput Toxicokinetics. J Stat Softw, 79(4), 1–26. doi: 10.18637/jss.v079.i04 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson M, Loye AF, Jacquet M, Mow NS, Thougaard AV, Mow T, & Hornberg JJ (2014). High-content analysis/screening for predictive toxicology: application to hepatotoxicity and genotoxicity. Basic Clin Pharmacol Toxicol, 115(1), 18–23. doi: 10.1111/bcpt.12200 [DOI] [PubMed] [Google Scholar]
- Richard AM, Judson RS, Houck KA, Grulke CM, Volarath P, Thillainadarajah I, … Thomas RS (2016). ToxCast Chemical Landscape: Paving the Road to 21st Century Toxicology. Chem Res Toxicol, 29(8), 1225–1251. doi: 10.1021/acs.chemrestox.6b00135 [DOI] [PubMed] [Google Scholar]
- Ring CL, Pearce RG, Setzer RW, Wetmore BA, & Wambaugh JF (2017). Identifying populations sensitive to environmental chemicals by simulating toxicokinetic variability. Environ Int, 106, 105–118. doi: 10.1016/j.envint.2017.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohban MH, Singh S, Wu X, Berthet JB, Bray MA, Shrestha Y, … Carpenter AE (2017). Systematic morphological profiling of human gene and allele function via Cell Painting. Elife, 6. doi: 10.7554/eLife.24060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto S, Carrera CJ, Kubota M, Wasson DB, & Carson DA (1985). Mechanism of deoxyadenosine and 2-chlorodeoxyadenosine toxicity to nondividing human lymphocytes. J Clin Invest, 75(2), 377–383. doi: 10.1172/JCI111710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah I, Liu J, Judson RS, Thomas RS, & Patlewicz G (2016). Systematically evaluating read-across prediction and performance using a local validity approach characterized by chemical structure and bioactivity information. Regul Toxicol Pharmacol, 79, 12–24. doi: 10.1016/j.yrtph.2016.05.008 [DOI] [PubMed] [Google Scholar]
- Shin HM, Ernstoff A, Arnot JA, Wetmore BA, Csiszar SA, Fantke P, … Bennett DH (2015). Risk-Based High-Throughput Chemical Screening and Prioritization using Exposure Models and in Vitro Bioactivity Assays. Environ Sci Technol, 49(11), 6760–6771. doi: 10.1021/acs.est.5b00498 [DOI] [PubMed] [Google Scholar]
- Sipes NS, Martin MT, Kothiya P, Reif DM, Judson RS, Richard AM, … Knudsen TB (2013). Profiling 976 ToxCast chemicals across 331 enzymatic and receptor signaling assays. Chem Res Toxicol, 26(6), 878–895. doi: 10.1021/tx400021f [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipes NS, Wambaugh JF, Pearce R, Auerbach SS, Wetmore BA, Hsieh JH, … Ferguson SS (2017). An Intuitive Approach for Predicting Potential Human Health Risk with the Tox21 10k Library. Environ Sci Technol, 51(18), 10786–10796. doi: 10.1021/acs.est.7b00650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeiri A, Matsuzaki K, Motoyama S, Yano M, Harada A, Katoh C, … Mishima M (2019). High-content imaging analyses of gammaH2AX-foci and micronuclei in TK6 cells elucidated genotoxicity of chemicals and their clastogenic/aneugenic mode of action. Genes Environ, 41, 4. doi: 10.1186/s41021-019-0117-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas RS, Allen BC, Nong A, Yang L, Bermudez E, Clewell HJ 3rd, & Andersen ME (2007). A method to integrate benchmark dose estimates with genomic data to assess the functional effects of chemical exposure. Toxicol Sci, 98(1), 240–248. doi: 10.1093/toxsci/kfm092 [DOI] [PubMed] [Google Scholar]
- Thomas RS, Bahadori T, Buckley TJ, Cowden J, Deisenroth C, Dionisio KL, … Williams AJ (2019). The Next Generation Blueprint of Computational Toxicology at the U.S. Environmental Protection Agency. Toxicol Sci, 169(2), 317–332. doi: 10.1093/toxsci/kfz058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas RS, Clewell HJ 3rd, Allen BC, Yang L, Healy E, & Andersen ME (2012). Integrating pathway-based transcriptomic data into quantitative chemical risk assessment: a five chemical case study. Mutat Res, 746(2), 135–143. doi: 10.1016/j.mrgentox.2012.01.007 [DOI] [PubMed] [Google Scholar]
- Thomas RS, Philbert MA, Auerbach SS, Wetmore BA, Devito MJ, Cote I, … Nong A (2013). Incorporating new technologies into toxicity testing and risk assessment: moving from 21st century vision to a data-driven framework. Toxicol Sci, 136(1), 4–18. doi: 10.1093/toxsci/kft178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- USEPA. (2018). Strategic Plan to Promote the Development and Implementation of Alternative Test Methods Within the TSCA Program.
- USEPA. (2019). Directive to Prioritize Efforts to Reduce Animal Testing. Retrieved from https://www.epa.gov/sites/production/files/2019-09/documents/image2019-09-09-231249.pdf website:
- Wawer MJ, Jaramillo DE, Dancik V, Fass DM, Haggarty SJ, Shamji AF, … Clemons PA (2014). Automated Structure-Activity Relationship Mining: Connecting Chemical Structure to Biological Profiles. J Biomol Screen, 19(5), 738–748. doi: 10.1177/1087057114530783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wawer MJ, Li K, Gustafsdottir SM, Ljosa V, Bodycombe NE, Marton MA, … Clemons PA (2014). Toward performance-diverse small-molecule libraries for cell-based phenotypic screening using multiplexed high-dimensional profiling. Proc Natl Acad Sci U S A, 111(30), 10911–10916. doi: 10.1073/pnas.1410933111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetmore BA, Wambaugh JF, Allen B, Ferguson SS, Sochaski MA, Setzer RW, … Andersen ME (2015). Incorporating High-Throughput Exposure Predictions With Dosimetry-Adjusted In Vitro Bioactivity to Inform Chemical Toxicity Testing. Toxicol Sci, 148(1), 121–136. doi: 10.1093/toxsci/kfv171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetmore BA, Wambaugh JF, Ferguson SS, Sochaski MA, Rotroff DM, Freeman K, … Thomas RS (2012). Integration of dosimetry, exposure, and high-throughput screening data in chemical toxicity assessment. Toxicol Sci, 125(1), 157–174. doi: 10.1093/toxsci/kfr254 [DOI] [PubMed] [Google Scholar]
- Williams AJ, Grulke CM, Edwards J, McEachran AD, Mansouri K, Baker NC, … Richard AM (2017). The CompTox Chemistry Dashboard: a community data resource for environmental chemistry. J Cheminform, 9(1), 61. doi: 10.1186/s13321-017-0247-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu WB, Ou JB, Huang ZH, Chen SB, Ou TM, Tan JH, … Huang ZS (2011). Synthesis and evaluation of mansonone F derivatives as topoisomerase inhibitors. Eur J Med Chem, 46(8), 3339–3347. doi: 10.1016/j.ejmech.2011.04.059 [DOI] [PubMed] [Google Scholar]
- Yang CH, Tarkhov A, Marusczyk J, Bienfait B, Gasteiger J, Kleinoeder T, … Rathman J (2015). New Publicly Available Chemical Query Language, CSRML, To Support Chemotype Representations for Application to Data Mining and Modeling. Journal of Chemical Information and Modeling, 55(3), 510–528. doi: 10.1021/ci500667v [DOI] [PubMed] [Google Scholar]
- Zhang Y, & Ye J (2012). Mitochondrial inhibitor as a new class of insulin sensitizer. Acta Pharm Sin B, 2(4), 341–349. doi: 10.1016/j.apsb.2012.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.