

Abstract
Elastic fibers provide recoil to tissues that undergo repeated deformation, such as blood vessels, lungs and skin. Composed of elastin and its accessory proteins, the fibers are produced within a restricted developmental window and are stable for decades. Their eventual breakdown is associated with a loss of tissue resiliency and aging. Rare alteration of the elastin (ELN) gene produces disease by impacting protein dosage (supravalvar aortic stenosis, Williams Beuren syndrome and Williams Beuren region duplication syndrome) and protein function (autosomal dominant cutis laxa). This review highlights aspects of the elastin molecule and its assembly process that contribute to human disease and also discusses potential therapies aimed at treating diseases of elastin insufficiency.
Keywords: Elastin, supravalvar aortic stenosis, autosomal dominant cutis laxa, Williams Beuren syndrome, Williams Beuren duplication syndrome, emphysema
1. Introduction
Elastic fibers are essential elements of the extracellular matrix (ECM), providing recoil to a broad range of tissues. Elastin is the main component of elastic fibers. It is formed through multimerization and crosslinking of tropoelastin monomers in the presence of elastic fiber proteins including fibrillins, fibulins, and lysyl oxidases. Once deposited in the ECM, elastin is exceedingly stable, with a half-life of approximately 70 years [1]. Genetic alterations that impact either the quantity or quality of elastin deposited have the potential to impact the function of elastic tissues. This review aims to describe the medical conditions caused by rare variation in the ELN gene. To do so, it first introduces the elastin protein and highlights protein domains that contribute to the efficient assembly and appropriate function of the molecule. In the next section, the review provides a comprehensive report of the genetic variants that cause rare elastin mediated disease, while outlining the mechanism by which the various mutation types cause disease. Third, the review discusses the specific disease phenotypes produced by the different mutation mechanisms and finally it examines current and potential future treatment strategies for the care of affected individuals.
2. Elastin domain structure and expression
The human elastin gene contains 34 exons in its longest transcript (NM_0012789391.1, ENST00000358929.8). The canonical transcript, however, has 33 exons with exon 22 typically spliced out (NM_000501.3, ENST00000252034.11). The exons encode a series of repeated pairs of hydrophobic and crosslinking domains (Figure 1) [2, 3]. Crosslinking exons are important for binding tropoelastin monomers to one another through the formation of desmosine, isodesmosine, and lysinonorleucine crosslinks [4–6], while the hydrophobic regions are essential for generating the entropic forces that contribute to elastic recoil [7–11].
Figure 1. Elastin domain structure.

Pictured is the domain structure of the canonical form of human tropoelastin (NM_000501.3). Exon 22 is not included as it is commonly spliced out and is not part of this transcript. Domains 34 and 35 are also absent as they are not present in the human gene (but are present in other species). Domain 36 is encoded by human exon 34. Domains are scaled to the size of the exon and are color coded as noted in the key to depict domain type.
Small-angle X-ray and neutron scattering studies performed on un-crosslinked tropoelastin show that the monomer takes on boot-like configuration with an N-terminal asymmetric coil structure at the top and a protruding C-terminal foot domain at the end. In between are the spur regions and flexible bridge regions linking the two [12, 13]. Although the domain structure is repetitive, three regions deserve special discussion for their impact on elastic fiber assembly and disease.
First, exon 30, a hydrophobic exon, is believed to mediate the initial association of tropoelastin monomers with one another, in a process referred to as microassembly [14, 15]. Exon 30 of elastin contains a repeated (GGLG(V/A)) sequence that was shown by fourier transform infrared spectroscopy to form anti-parallel beta sheets [16]. With the addition of transmission electron microscopy and atomic force microscopy, Tamburro et al found that fibers were formed from self-association and alignment of primary cross beta structures formed by the exon 30 sequences. Glycine-rich hydrophobic sequences such as this are found in many molecules that form amyloid-like aggregates [16]. Although many regions in tropoelastin are hydrophobic in nature, exon 30 displays a relative paucity of proline residues. It is this lack of proline within the hydrophobic region that increases its ability to form amyloid structures [17]. In in vitro cells systems, when exon 30 was deleted from the elastin cDNA in a bovine assembly system, multimerization and assembly of elastin by cells was reduced [18]. More recent experiments using multiple tropoelastin constructs bearing genetic variations in exon 30 sequence upheld a role for exon 30 in guiding elastic fiber formation and also revealed the region’s contribution to viscoelastic and tensile properties of the final crosslinked polymer[19]. Consequently, human mutations causing the loss of this region are expected to have decreased matrix accumulation of elastin. Miao et al also showed that single point variants within this exon have the potential to affect the mechanical properties of the resulting monomer, potentially affecting its ability to assemble [20].
The exon 16–17 region has also been implicated as a possible assembly domain. Like the exon 30 studies, cDNA constructs of human tropoelastin carrying an exon16–17 deletion failed to deposit elastic fibers when transfected into pigmented epithelial cells [21]. Mutant protein was able to bind fibrillin-1 and fibulin-5 but showed an increased coacervation temperature suggesting decreased association between mutant tropoelastin monomers.
The final region of interest lies in the terminal exon of the human ELN gene. Although the 34th consecutive exon in the human gene, it is referred to as domain 36 in much of the literature. The numbering convention is used because the majority of species have two additional ELN exons, exons 34 and 35, that were evolutionarily lost in humans and higher primates [22]. This domain contains a group of basic amino acids (KxxxRKRK) that are important for cell surface heparin binding. Such interactions are thought to assist in growth/stabilization of the growing elastin multimer and potentially to aid in the release of tropoelastin from chaperones/binding proteins that assist in assembly [23–25]. When exon 36 was deleted in bovine cDNA constructs, the resulting elastin proteins were secreted and deposited in the extracellular space but showed reduced numbers of desmosine crosslinks [18, 26], potentially due to the absent interaction with other relevant matrix associated proteins. As such, mutations affecting the extreme C-terminus may affect the quality of the elastin produced.
Elastin production is highly regulated in a temporal and tissue specific manner. The ELN promoter contains purported TGF-β, insulin and glucocorticoid responsive elements [27–30], but the regulation of elastin protein production is thought to occur mainly through the binding of microRNA (miR) regulatory elements both in the 3’ UTR and in coding sequences of ELN [31, 32]. It is suggested that high expression of miRs-29 and -15 is responsible for posttranscriptional repression of elastin in the adult aorta [32]. Binding of the miRs causes the degradation of ELN RNA. Tissue specific alternative splicing resulting in different tropoelastin isoforms have been identified and may play a role in the diversity of disease manifestations [20, 33–38]. In addition, deposition of elastin into the ECM is known to be impacted by the relative quantity of other ECM glycoproteins (e.g. versican, fibrillins, fibulins, MFAPs, etc) [18, 39–46].
2.1. Rare variation in the ELN gene:
Most described mutations within the ELN gene cause one of two OMIM-designated diseases, supravalvar aortic stenosis (SVAS-MIM #185500) and autosomal dominant cutis laxa (ADCL-MIM#123700). More than 100 pathogenic or presumed pathogenic variants have been described in ELN to date in the literature, in the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar) and in the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk).
Variants causing the SVAS phenotype are generally expected to cause a decrease in total elastin. Reported variants include missense mutations affecting the initiation methionine, non-sense changes leading to a premature stop codon prior to exon 30, and insertion/deletion changes that produce frameshift transcripts expected to be destroyed by non-sense mediated decay (Figure 2 and Supplemental Table 1).
Figure 2. Elastin gene structure and pathogenic variants to date.
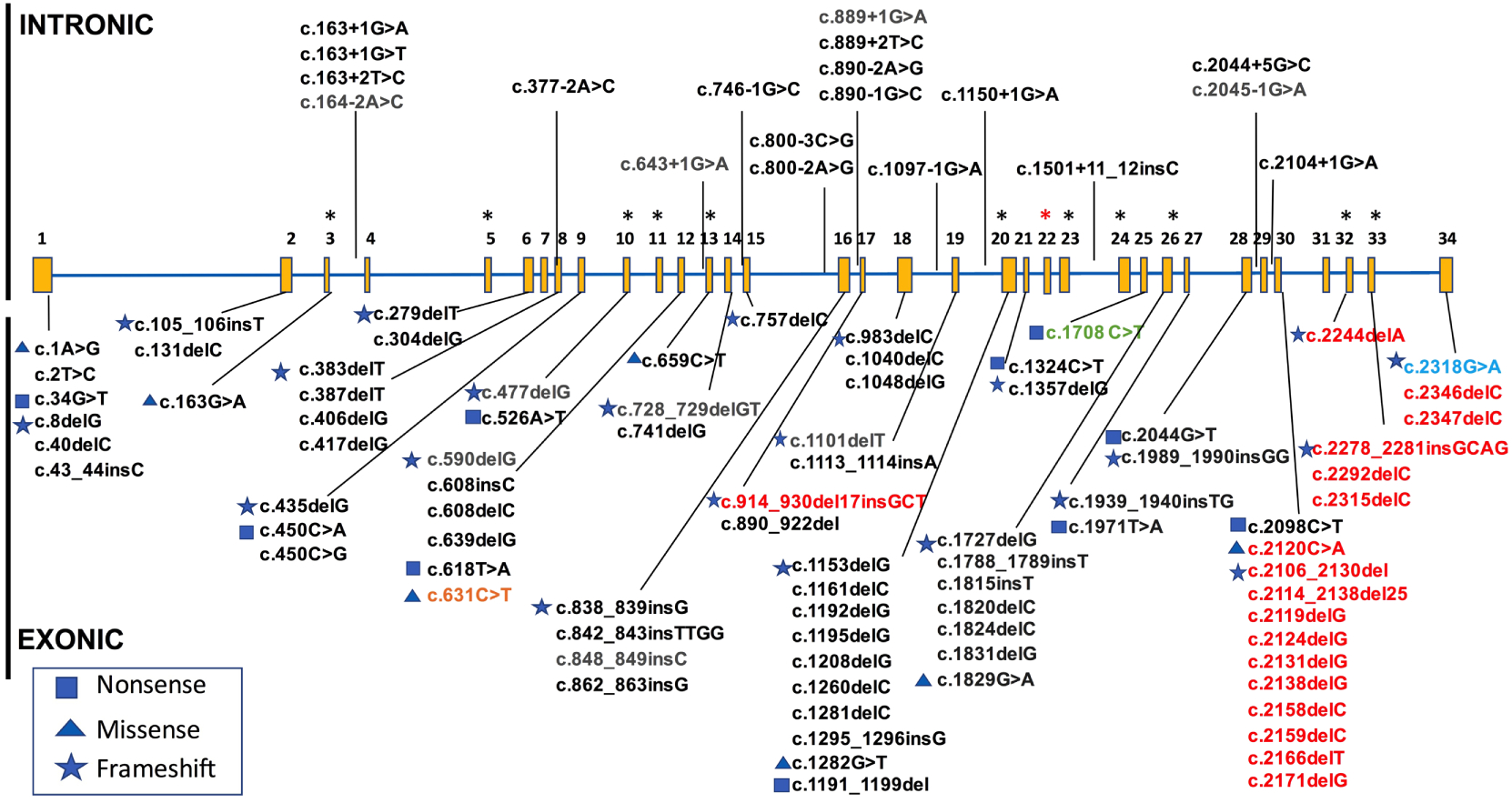
Known exonic and intronic variants curated from published case reports, ClinVar and HGMD. Variants resulting in SVAS (black), ADCL (red), COPD (light blue), and pathogenic/likely pathogenic variants as classified by a clinical laboratory and submitted to ClinVar, but without phenotype available (gray). Green font denotes variant in exon 25 that has been described in individuals with features of SVAS as well as one individual with possible features of ADCL (for full details refer to text) and orange depicts a patient with proposed autosomal recessive cutis laxa. All intronic variants to date have been described as causing SVAS, whereas exonic variants can cause either SVAS or ADCL. Most ADCL-causing variants are located in the C-terminal domain exons 30–34, with few exceptions. Alternatively spliced exons are shown (asterisk), as well as commonly spliced exon 22 (red asterisk). Lines point to intron/exon where variants are located, not specific mutation site.
In addition, splice site mutations are also reported to cause the SVAS phenotype (19 of the 104 reported variants are thought to impact splicing). Because all elastin exons are in the same frame, exon skipping has the potential to produce an in frame, although shorter, protein product. Miao et al showed that changing the domain structure of tropoelastin by adding and subtracting exons from the molecule affected both its hydrodynamic radius and tensile mechanical properties, suggesting that exon skipping may impact the function of the protein product [20]. However, it is not clear whether in most cases, the mutant protein is made. For one variant, two groups evaluated the same gene change, c.800-3C>G, in skin fibroblasts from different patients. This variant causes a change in intron 15, affecting the splice site. Wachi et al found stable expression of a message missing exons 16 and 17 in the mutant fibroblasts [21], while Urban et al noted that the variant led to the generation of a cryptic splice that ultimately led to frameshift and reduced expression [47]. Unfortunately, because of the primers chosen by each team, the groups did not identify the transcript shown by the other laboratory. The group that found stable expression of the mutant Δ16–17 mRNA followed those studies with in vitro work using an expression system that showed exon 16–17 to be important for tropoelastin self-association. An additional study by Park et al showed that the loss of 11 in frame amino acids from exon 17 also led to the SVAS phenotype [48], potentially due to dysfunctional assembly. However, the authors were unable to find an aberrant protein by western.
As for the remaining splice variants, most of the references do not contain functional studies to determine the impact of the gene change. Given the number of such variants, it would seem more likely that the majority would either extend into an intron where a premature stop is encoded or serve to generate cryptic splices leading to aberrant message. In each case, the expectation is that the transcripts will largely be destroyed by nonsense mediated decay. It is possible, however, that like the Δ16–17 story described above, some of the abnormal splices generate shorter variants that delete additional assembly domains. More work is needed to determine this empirically.
In addition to these single base pair/short variations, a translocation [49], a deletion that removes the C-terminus [50], a deletion of exons 2–28 [51] or loss of the entire ELN gene [52] can cause SVAS. These findings suggest that loss of one functional copy of the elastin gene and decreased elastin protein production may contribute to the SVAS phenotype.
The most common genetic alterations affecting the elastin gene, however, are large deletions that remove one copy of ELN in addition to the neighboring 25–27 genes as part of the recurrent microdeletion disorder Williams-Beuren syndrome (WBS)[53–55]. Patients with WBS have vascular features similar to SVAS, but have additional phenotypes owing to the loss of other genes in the deletion [56–61]. Patients with WBS/SVAS mutations deposit less total elastin but the elastic fibers typically appear normal, if a bit less organized [62, 63]. These findings, together with phenotyping data from murine models outlined below suggest that the SVAS phenotype is caused by elastin haploinsufficiency.
A few of the reported SVAS mutations deviate from the above convention. These include missense mutations at amino acids p.Ala55Thr (exon 3), p.Pro220Leu (exon 13), p.Gly610Gln (exon 26), and p.Ala707Asp (exon 30) that cause an SVAS phenotype. The p.Ala55Thr variant occurs as the last basepair in exon 3 and likely contributes to aberrant splicing, as variation in the next two nucleotides in the intron also produce the SVAS phenotype. Both exon 13 and 26 are alternatively spliced and it is possible that increased splicing occurs in the setting of these variants that prevents the accumulation of mutant protein. Dosage could be affected if tissue or timing specific requirements for particular isoforms were critical. The p.Ala707Asp variant is of particular interest due to its location in the putative exon 30 microassembly domain [18], a region also potentially involved in posttranscriptional regulation of message stability [31, 32]. The finding of missense variation at these locations suggests a change to the structure of the tropoelastin monomer that inhibits multimerization or stability of the tropoelastin transcript that leads to functional haploinsufficiency.
By contrast, ADCL is predominantly caused by frameshift mutations in exons 30–34 (Figure 2 and Supplemental Table 1). Elastic fibers deposited by these individuals are abnormal and display fiber fragmentation along with reduced deposition [64, 65]. In general, mutant mRNA in ADCL patients is stable and in some, protein containing the frameshifted alleles has been detected in the matrix using an antibody to the frame-shifted product [64], suggesting a dominant negative mechanism of disease. In addition, Callewaert et al suggested that ADCL variants generate tropoelastin monomers that are aberrantly folded and induce endoplasmic reticulum stress and apoptosis [64]. Of note, due to alternative splicing of exon 32, ADCL mutations in this domain may be phenotypically less severe [64]. Tissues where exon 32 is typically spliced out, for example, would be expected to show no phenotypic consequences of the genetic alteration.
Reported variants not fitting the above mutational prescription for ADCL include a frameshift mutation (c.914_930del17insGCT; p.Ala305Glyfs) that was reported in exon 17 by a clinical lab in ClinVar. The indication for the test was reported to be cutis laxa but no additional clinical details are available. Care should be taken in interpretation of this variant. One additional variant, c.1708C>T (c.1621C>T in [66] which uses NM_000501.3 rather than the full length transcript) has been reported to cause ADCL like features in one patient and SVAS-like features in others [66–68]. The variant changes in the last base pair of exon 25. In the ADCL-like proband and his apparently very mildly affected father, the variant induced in frame splicing out of exon 25 in a fraction of the skin derived RNA. While the proband had quantitatively normal amounts of ELN message (suggesting equal parts WT and splice form), the father had reduced message (suggesting haploinsufficiency). Interestingly, in the transcripts where splicing of exon 25 does not occur, a new stop codon is generated by the exon 25 mutation and the first two base pairs in exon 26, leading to p.Arg570Ter. The child’s skin fibroblasts showed ADCL-like features of scant and small fragmented elastic fibers while the father’s findings were subtler. Consequently, it appears that some with this genotype express a predominantly haploinsufficiency mediated SVAS phenotype (the father in this case (albeit with decreased penetrance) and SVAS cases from [68, 69]), while others (the proband in this case) have increased exon 25 splicing and the retention of aberrant protein leading to ADCL. Exon 25 has been implicated in interchain crosslinking [70]. Of note, this proband had additional features such as agenesis of the corpus collosum and seizures, features not typically seen with ADCL. Sequencing was only performed for ELN, ARX, and FBLN5 and FBLN4. Other genetic changes could be responsible for the unique phenotypic presentation of this proband.
Of note, a homozygous missense mutation in exon 12, p.Pro211Ser, was reported to cause a mild form of autosomal recessive cutis laxa in two related consanguineous families [71]. The phenotype was more severe in a family member who carried biallelic ELN variants and an additional variant in the FBLN5 gene. The carrier parents were clinically unaffected in all cases except one mother who possessed a single ELN p.Pro211Ser mutation and the FBLN5 variant—she possessed mild features of cutis laxa. These findings may suggest that more minor missense changes in non-critical portions of the ELN gene may have limited phenotypic effects when present in isolation. However, when the burden of ELN or other cutis laxa gene variants increases, a clinical phenotype may be appreciated due to the cumulative effect of the total variant load. However, because this testing was done using a candidate gene approach (only ELN and FBLN5 were sequenced), re-evaluation using current technologies (exome or whole genome sequencing) may provide an alternative explanation for the disease features in these patients.
In addition to the SVAS and ADCL phenotypes, the rare ELN point mutation c.2318G>A resulting in p.Gly773Asp (exon 34) was identified in a large pedigree with severe early onset chronic obstructive pulmonary disease (COPD) [72, 73]. This mutation results in a non-conservative amino acid change in a position that is conserved across species, and is predicted to be probably damaging by in silico testing. This variant was identified in one further proband following screening of ~1300 individuals genotyped through the Boston Early-Onset COPD Study and the National Emphysema Treatment Trial but other causative variants in ELN have not been identified in the numbers evaluated [72].
The conditions above discuss the impact of reduced or aberrant elastin, but a small number of individuals are known to possess increased elastin gene dosage due to a duplication event of the WBS region. Patients with this condition have three copies of the ELN gene and mild cardiovascular phenotypes including aortic dilation. Because the vascular phenotype of the WBS duplication patients appears inverse to the stenotic phenotype of the deletion patients, scientists have speculated that increased production of elastin from the 3rd allele leads to subtle changes in aortic mechanics that produce to dilation over time [74].
3. Disease manifestations
Individuals with genetic changes affecting the elastin gene have phenotypes in a range of elastic tissues, although features vary between genetic changes affecting gene dosage and those leading to dominant negative production of aberrant protein.
3.1. Elastin Insufficiency (WBS and SVAS)
3.1.1. Vessel disease- Arteriopathy
In the vasculature, elastic lamellae are organized circumferentially in the media and are layered between sheets of smooth muscle cells. Studies performed on arteries from elastin insufficient mice and humans show increased numbers of smooth muscle and elastic lamellae (Figure 3A and B); each layer, however, has decreased elastin content [63, 69]. These changes impact the biomechanical properties of the vessel wall, leading to a vessel with a smaller lumen, thicker wall and decreased compliance [75, 76]. Eln−/− mice show total disorganization of the smooth muscle layers and obliteration of the luminal space by cells, a process reported to be independent of blood flow, hemodynamic stress, endothelial damage, thrombosis, inflammation, or fibrosis [77], suggesting that the presence of elastin in the extracellular matrix and cell proliferation/orientation are tightly linked [62, 78–80]. Eln+/− mice showed ~50% reduction in Eln mRNA and had ~25–35% more elastic lamellae and smooth muscle in their arteries. Adult hemizygous mice have higher systolic blood pressure, increased arterial stiffness and smaller caliber vessels, with longer segmental length than WT littermates, [69, 75, 76]. Tortuosity is noted in multiple tissue beds ([81] and Figure 3D and E). Gene dosage seems to be important as transgenic mice expressing human elastin from a bacterial artificial chromosome containing the human ELN gene on a Eln −/− null background (hELN BAC; mEln−/−) deposit ~35% of normal elastin content and show higher blood pressure than Eln+/− mice and the ascending aorta is increasingly thickened with more poorly organized lamellae than Eln+/− mice (Figure 3 C). Vessel caliber is markedly reduced and some decrease in longevity was noted [82].
Figure 3. Elastin insufficiency mediated vascular disease.

Images of aorta taken from WT (A), Eln+/− (B), and mEln−/−; hBAC-ELN (C) mice and stained with Verhoeff Van Gieson stain. Full scale bar = 50 μm. Elastin is black with the vessel lumen to the left. Vessels were fixed with a flow rate of 1.5ml/min. Note the thicker wall and increased number of elastic lamellae as Eln dosage decreases. The next two panels are microCT images of left kidneys from WT (D) and Eln+/− (E) mice. Latex containing a radio-opaque dye (Microfil, Flow Tech, Inc. USA) was injected into the left ventricle of the mice and the arteries were back-perfused until the contrast reached the capillary phase of the kidneys. After hardening, kidneys were removed and imaged. Tracing was done in Analyze software (AnalyzeDirect, USA) to highlight the vessels. Note the smooth arteries in the WT mice and the tortuosity of the Eln+/− vessels. Panels F and G are CT angiograms of patients with Williams Beuren syndrome. Both images show SVAS (arrow) in different orientations. Photo credit for A-C to Angela Troia, for D and E to Russell Knutsen and Dr. Delong Liu and to Dr. Shabana Shahanavaz and for F and G.
In humans, the prototypical defect associated with elastin insufficiency is focal stenosis of the large elastic arteries (Figure 3F and G). This narrowing can occur in any elastic artery, but is most frequently described in supravalvar aorta and branch pulmonary arteries [83, 84]. Vascular disease varies from person to person with ~30% of individuals requiring surgical intervention for their stenosis and ~20% having no appreciable stenosis. In addition to the aortic and pulmonary disease, narrowing and anatomical abnormalities of more distal vasculature have been described. Coronary artery abnormalities are also reported in SVAS and WBS and may contribute to sudden death in this population [83, 85–88]. Strokes have been reported in individuals with WBS [89–91], and with isolated SVAS [92], both in the presence and absence of cerebral artery stenosis or aneurysms. A small study performing magnetic resonance angiography in patients with WBS vs. controls found no noticeable stenosis in cerebral arteries. However, they did note that the posterior communicating segment of the anterior cerebral artery was longer in patients with WBS [93]. In addition to focal stenosis, patients with elastin insufficiency have globally narrow vasculature, even in locations without focal stenosis. Recent studies suggest that deficient circumferential growth during development may play a role in this generalized arteriopathy [94]. The vessels show increased stiffness [95, 96]. This stiffness may increase risk of negative cardiovascular outcomes [97–99].
It is important to note that none of the mouse models develop the pathognomonic hourglass appearing SVAS seen in many human patients. Instead, the mouse models show more homogeneous long segment wall thickening and narrowing, with the mEln−/−; hBAC ELN mouse revealing the most profound arch narrowing. Consequently, although endothelial damage and hemodynamic stress may not be necessary for the increased smooth muscle layers seen in both humans and mice, the focal aortic stenosis that typically worsens postnatally in humans may depend on additional mechanical, genetic or environmental factors that are currently incompletely understood.
Hypertension is an important health problem in individuals with elastin insufficiency. Renal artery stenosis may contribute to hypertension in this population [83, 100–103]. However, hypertension often occurs independently of renal artery stenosis in WBS/SVAS. Studies in humans and mice have shown that hypertension is less common in WBS patients with larger deletions that include the NCF1 gene [96, 104–106]. NCF1 encodes p47phox and is a component of the NADPH oxidase complex. The finding that patients with reduced capacity to generate oxidative stress are protected from hypertension suggests that increased reactive oxygen species, generated in response to altered mechanical stresses may be important in the pathology of hypertension in elastin insufficiency.
3.1.2. Lungs
In the lungs, elastin is located within the arteries and elastic cartilages but is also present at the tips of alveolar septa. During expiration, the diaphragm relaxes, allowing the elastic recoil of the lungs to move air out of the air spaces. Lungs of Eln+/− pups displayed a 50% reduction in tropoelastin, significantly fewer microvessels, including lung capillaries, and two-fold increase in collagen-1 and lysyl oxidase [107]. No difference in alveolar size and number has been found [107, 108]. The hELN BAC+ mEln −/− mice, however, have ~65% decrease in elastin level, and present with congenital emphysema characterized by enlarged thoracic cavities, large distended lungs and massively dilated airspaces on microscopy [82]. The lungs in Eln −/− mice demonstrated arrest of development at the level of the distal airways. These mice had dilated distal air sacs and attenuated septae demonstrated even before alveologenesis [109].
Respiratory symptoms in human patients with ELN insufficiency are similarly mild and may include cough, dyspnea and wheezing. Spirometry in 16 young adults with WBS was largely normal [110]. Although uncommon, early onset emphysema has been described in several WBS patients [110–112]. It is unclear whether the severe disease presented in these studies is as a result of their elastin insufficiency alone or if they, in fact, have variations in additional modifiers genes that contribute to the severe phenotype. Additional studies in older individuals with elastin insufficiency are needed to more completely understand the impact on lung physiology. Studies in Eln+/− mice exposed to cigarette smoke suggest that environmental toxins such as smoke are particularly damaging in the face of elastin insufficiency and should be avoided in affected individuals [108]. The differences in phenotypes found in mice and humans may represent the species specific variations in elastin content in tissues [113].
3.1.3. Skin & Integument
Several skin and integument findings have been described in the WBS patient population including, soft skin, dry skin, wrinkles, and loose periorbital connective tissue [114–117]. Elastin insufficiency results in decreased diameter of oxytalan fibers and mature elastic fibers in the skin [118]. Likewise, Urban and colleagues found that individuals with WBS had reduced deposition of amorphous elastin when viewed under electron micrograph despite having a similar distribution of the elastic network when compared to controls [119]. Biomechanical skin properties studied in WBS individuals revealed diminished skin viscoelasticity relative to controls [116].
3.1.4. Gastrointestinal
Feeding difficulties and gastrointestinal complaints have been frequently described in individuals with WBS. The most common findings include chronic abdominal pain, gastroesophageal reflux, vomiting, chronic constipation, and rectal prolapse. It is unclear to what extent pain and motility issues can be attributed to vascular and connective tissue complications of the disease versus changes in innervation owing to the deletion of genes other than ELN in the WBS critical region. Diverticulosis and diverticulitis are common in adults with WBS [114, 120–122]. Although there are data to suggest increased elastin in the longitudinal smooth muscle of colonic samples with diverticular disease, there is little information about the effect of elastin haploinsufficiency in the intestinal wall [123].
3.1.5. Genitourinary
Like the other organs discussed, the bladder also undergoes repeated cycles of stretch and recoil. The smooth muscle there deposits relatively high amounts of elastin and Eln+/− mice revealed decreased bladder compliance and capacity, and increase in contractility [124].
In patients with elastin insufficiency, lower urinary tract symptoms such as urgency, frequency, incontinence and enuresis are common [125–127]. Bladder diverticula, undescended testis, retractile testis, and inguinal hernias are also seen [125, 126, 128–130]. Some of these symptoms may be attributed to neurologic and developmental factors in WBS, but the role of elastin insufficiency should be considered given its relatively high expression in the bladder wall.
3.1.6. Voice
Elastin is found in human vocal cord lamina propia (VCLP) as oxytalan and elaunin in the superficial layer, and as mature elastic fibers in the deeper layers [131, 132], making up 9% of total protein present in the VCLP [132]. Elastin plays an important role in vocal cord vibration, and biomechanics of voice quality. Hoarse voice is a common finding in individuals with WBS [121, 133–135]. The voice quality of individuals with SVAS/WBS has been found to be rough, hoarse, and to have a lower pitch when compared to normal controls [136]. Histological evaluation of vocal cord on autopsy specimen of a WBS patient revealed decreased elastin when compared to an age-matched control [135]. Mice heterozygous for deletions in the Eln gene showed decreased elastin within the vocal folds when compared to Eln WT mice [137].
3.1.7. Hearing
Audiological findings in individuals with WBS include sensorineural hearing loss (SNHL), conductive hearing loss and hyperacusis [114, 138, 139]. Increased sensitivity to sound has been reported in up to 95% of individuals with WBS [138] and is associated with high-frequency hearing loss, similar to noise-induced hearing loss [140, 141]. 60–70% of school-aged-children with WBS display mild to moderate high frequency hearing loss or mixed hearing loss [139, 141]. The pattern of hearing loss is progressive with up to 92% of adults with WBS having some hearing loss [114, 140]. Hearing loss phenotype and hyperacusis were reported in two related individuals with non-syndromic SVAS, suggesting that elastin haploinsufficiency may contribute to the pathogenesis of SNHL in WBS [139]. Conductive hearing loss is related to excessive cerumen buildup [114, 139], and middle ear pathology [140]. Distortion product otoacustic emissions findings were suggestive of subclinical cochlear pathology even in children with WBS with normal behavioral hearing threshold [140].
Different mechanisms for hearing loss related to elastin insufficiency have been proposed including poor cochlear perfusion secondary to vascular stenosis, increased rigidity of the basilar membrane, dysregulation of cochlear cell proliferation and disruption of middle ear mechanics [139, 140, 142]. Hyperacusis may be caused by deficiency of acoustic reflex from auditory nerve dysfunction [141]. More recently, LIMK1 haploinsufficiency has been proposed to be responsible for the auditory phenotype in WBS [143]. Further research into the role of elastin in warranted.
3.2. Dominant negative mediated elastic fiber disease (autosomal dominant cutis laxa)
3.2.1. Skin
Skin findings are the major clinical feature of ADCL, with loose, redundant, inelastic skin and appearance of premature aging being present in 100% of affected individuals. Electron micrographs of skin from affected individuals show disorganized elastin with abnormal branching and fragmentation of elastic fibers [64, 65].
Individuals with ADCL have normal wound healing despite abnormal and decreased dermal elastin. Transgenic mice expressing human tropoelastin with a single nucleotide deletion in exon 30 (c.2012delG) previously described in patients with ADCL, hBACCL, in addition to mELN+/+ showed accumulation of the mutant protein within the skin and skin laxity, requiring half as much force to be displaced when compared with WT and hemizygous mice [36]. Biomechanical testing in skin of individuals with various types of cutis laxa, including ADCL, confirmed that reduced force was necessary to displace patient skin, and showed abnormal and prolonged recoil [144].
3.2.2. Vessel Disease
ADCL was initially thought to be a mild disease with findings limited to the skin [145]. However, multiple individuals have been described with aortopathy, valvulopathies and significant pulmonary disease. Aortic root dilation has been described in 30–50% of patients with ADCL [64, 146]. Aortic dilation can range from mild to severe aneurysms or aortic rupture [147]. Cardiac valve abnormalities, namely bicuspid aortic valve, aortic regurgitation, mitral regurgitation and mitral valve prolapse are common in affected individuals [64, 146, 148]. Studies with hBACCL; mEln+/+ mice showed limited incorporation of the mutant protein into the aorta and no effect on vessel compliance. Combined with the skin findings presented above, these data suggest that tissue specific alternative splicing may occur that modifies the phenotypic presentation in different organs [36].
3.2.3. Lung
A recent literature review states 28% of individuals with ELN variants resulting in ADCL have pulmonary findings [148]. Prior reports state that approximately 35% of patients with ADCL have severe emphysema [64, 146, 149]. Transgenic mice expressing a 25-nucleotide deletion in exon 30 (c.2114_2138del25) as described in a family with ADCL, in the presence of mEln+/+ developed severe emphysema and had increased mortality [150]. Both this and the Sugitani et al study which followed [36] showed the co-polymerization of mutant with WT elastin as well as the numerous imaging studies that show abnormal elastic fibers[64, 65], underscore the claim that ADCL is caused by a dominant negative mechanism.
3.2.4. Genitourinary
Genital prolapse has been described in at least 2 individuals with ADCL caused by ELN variants [146, 149]. Biopsy from cardinal and uterosacral ligaments in a patient with cutis laxa and genital prolapse revealed decreased elastin and increased collagen type VI, suggesting a role for extracellular matrix components in the stability of the pelvic floor [151].
3.3. Increased ELN gene dosage
In contrast to individuals with elastin haploinsufficiency, individuals with three copies of the ELN gene as a result of the Williams Beuren duplication syndrome have an increased rate of aortic dilation, ranging from mild to moderate, and mostly affecting the ascending aorta [74, 152–154]. Two individuals, a mother-son kinship harboring a 2.53Mb duplication that included the WBS region in its entirety, along with 18 additional genes, have required surgical intervention for aortic dilation/aneurysm [154]. In another cohort, possessing a triplication of just the ELN and neighboring LIMK1 gene, 10/11 affected family members exhibited ascending aortic aneurysm [155]. Patent ductus arteriosus and atrial septal defects have also been described in individuals with 7q11.23 microduplication [74, 153, 156]. The ductus arteriosus is a vessel that connects the aorta and pulmonary artery in fetal life. This vessel should normally close after birth to accommodate postnatal circulation [157]. The anatomical closure of the ductus arteriosus depends on vascular smooth muscle cell proliferation, migration and patterning [157–159]. Because elastin haploinsufficiency has been associated with increased smooth muscle cell proliferation [62, 69, 76], one could speculate that elastin overexpression could decrease smooth muscle cell proliferation or alter smooth muscle contractile phenotype, hindering the physiologic closure of the ductus arteriosus. Human studies have not presented expression or tissue based data to confirm or refute this hypothesis. However, work in animal models highlights the importance of elastin/smooth muscle cell interaction to maintain the ductus arteriosis [160–162]. It is possible that overexpression of other genes in the WBS region influence ductus closure such as Baz1b, a gene implicated in neural crest cell function[163].
A single patient with WBS duplication has been reported to have supravalvular aortic stenosis with post-stenotic dilation on echocardiogram, along with several other congenital defects not usually described as part of this syndrome. This child was the product of consanguineous parents which raises concern for additional genetic disorder [164]. Cutis marmorata, a condition with increased vascular predominance on the skin’s surface, has been described in the WBS duplication patient population, but there has been no description of biomechanical properties of the skin in individuals with WBS duplication [153, 165, 166]. No lung findings have been reported to date.
As in the haploinsufficiency model, overexpression of elastin produces a less severe phenotype in mice. In a transgenic mouse expressing human elastin in addition to normal mouse elastin (hELN+ BAC, mEln+/+), overexpression of WT human elastin resulted in no detectable lung disease [150]. Vascular changes were minimal [150] and there was no change in physiological parameters, blood pressure, vessel wall remodeling or compliance [82]. These differences could represent relative differences in gene expression, location, or timing between humans and mice. Alternatively, it could suggest that the cardiovascular phenotypes in the WBS duplication are arising from a different gene. However, data from the ELN-LIMK1 triplication family presented above would seem to limit the gene dosage effects to ELN or LIMK1.
4. Treatment
Genetic changes in elastin cause multi-organ dysfunction and intervention is warranted to improve outcomes. Current treatment for individuals with elastin defects is largely symptomatic. Individuals with elastin insufficiency are monitored regularly for evidence of arterial stenosis and hypertension, as well as the other health concerns described above [84, 167, 168]. If identified hemodynamically significant, stenosis is treated with catheter based dilation or surgical repair. Hypertension is managed with anti-hypertension medications, however limited information is available regarding the best choice of antihypertensive for this population [169, 170]. Research studies are only beginning to assess the role for arterial stiffness and aberrant flow generated by narrow caliber and tortuous vessels over the course of a lifetime [96, 171].
Similarly, patients with ADCL are monitored for cardiac, lung, and urinary symptoms [147, 172]. Emphysema and aortic dilation are managed with the same techniques used for non-cutis laxa induced forms of these conditions. Some individuals with ADCL may choose to undergo plastic surgery to correct the appearance of loose skin [173, 174]. However, results may not be permanent. In both SVAS and cutis laxa, avoidance of environmental toxins such as smoking is recommended [110, 175]. Cutis laxa patients are advised to avoid sun bathing, which can damage the skin.
Currently, there are no FDA approved treatments aimed at the molecular cause of these conditions. Investigational treatments in elastin insufficiency have focused on two targets: increasing elastin production and decreasing smooth muscle proliferation.
4.1. Increased elastin production
The ability to increase elastin deposition is particularly intriguing. Due to its long half-life, an effective treatment may only need to be administered over a short therapeutic window to produce life-long beneficial effects. To improve elastin production, two potential therapies have been tested in mice or tissues: 1) inhibition of microRNA (miR) 29 and 2) KATP channel openers.
As described previously, elastin protein production is controlled largely by the rate and degree of binding of several miRs to the elastin transcript. The miR 29 family, in particular, has been a particularly interesting candidate as it has fourteen binding sites within the ELN exons and 3’ untranslated region [32]. Binding of miRs to a mRNA causes translational repression and mRNA degradation. Consequently, the loss of a miR might be expected to increase its usual target’s mRNA translation and protein production. Given this finding, Zhang et al chose to treat cells and engineered vessels from patients with elastin insufficiency with miR 29 inhibitors [176]. They showed that when cells were treated with miR 29 mimics, ELN transcript levels decreased, while treatment with miR inhibitors increased ELN expression levels above the untreated control levels and resulted in increased elastin in the ECM [176]. Challenges for the clinical use of these reagents include delivery and specificity. The miR 29 family, like most miR’s binds multiple mRNAs [177]. In addition to elastin, miR 29 family members have been shown to regulate fibronectin, laminin, integrin-B1, multiple collagens and matrix metalloproteinase-2 and have been implicated heavily in fibrosis [177–179]. Consequently, use of anti-miRs as therapeutics requires the ability to tune the miR to mRNAs of interest and the ability to deliver the medication to a specific location in the body so as to avoid inappropriate production of ECM in tissues where it is not needed.
Minoxidil, a KATP channel opener and known vasodilator has been shown to increase elastin deposition [180–182]. Postnatal treatment of rats with lower levels of endogenous elastin and Eln+/− mice led to increased accumulation of elastin in the vasculature of those animals [171, 180]. Collagen content, as measured by hydroxyproline, however, was unchanged [171]. The medication also decreased blood pressure, increased lumen diameter, normalized pulse wave velocity and improved blood flow to end organs including the brain [171]. Cell work with this drug decreased proliferation of currently dividing cells [183], but evaluation of treated animals showed generally thicker vessel walls owing to increased matrix and relaxation of smooth muscle cells [171]. Treatment with other KATP openers (diazoxide and pinacidil) had similar effects [180] and subsequent work evaluating mechanisms for how KATP activators lead to increased elastin deposition points to increased calcium influx and Erk1/2 phosphorylation [184, 185]. More recent RNAseq experiments combined with distensibility data suggest that rather than minoxidil playing a specific role in inducing elastin production, it may instead cause outward remodeling and production of an overall larger vessel[171]. Elastin is deposited, but at a rate only modestly higher than other proteins in the vessel wall. However, because the lumen size does increase, this allows for improvement in blood flow to end organs, a potentially useful by-product. Minoxidil too, has side effects due to KATP channel expression in non-vascular tissues. Side effects in non-WBS/SVAS patients include hirsutism, cardiomegaly and edema. However, such changes were limited in the Eln+/− mouse treatment studies and changes in vascular diameter induced by minoxidil in treated Eln+/− vessels persisted at some level for at least 1 month after treatment was discontinued suggesting that the drug’s role is not limited to simple vasodilation but that it has the capacity to “clamp the vessel in the relaxed condition” as previously suggested [186] and that chronic/continuous treatment may not be necessary. Like the miRs, use of KATP channel openers would be optimized by the identification of similar molecules with activity only in smooth muscle cells to limit side effects. Neither drug has been tested for its effect on existing focal stenosis.
4.2. Inhibition of smooth muscle proliferation
Because stenosis is the most obvious and life-limiting pathology associated with elastin insufficiency, the other major approach to treatment is through inhibition of smooth muscle proliferation. Work by multiple investigators has outlined the dramatic, primarily postnatal, increase in vascular smooth muscle numbers that occurs in elastin insufficient arteries [76, 79, 187, 188]. Evidence of smooth muscle cell proliferation was present as early as E15.5 in Eln−/− mice but happened closer to E18.0 in Eln+/−. To inhibit hyperproliferation, investigators have administered mammalian target of rapamycin (mTOR) inhibitors and integrin β3 blockers to mice.
In the mTOR experiments [189], investigators noted increased mTOR signaling in arterial smooth muscle cells of elastin deficient (Eln−/−) mice. They then administered rapamycin, an mTOR inhibitor, to pregnant dams starting at E16.5. Pups showed decreased smooth muscle cell proliferation. Eln−/− revealed reduced obstruction but did not live longer. Eln+/− pups had reduced lamellar number relative to untreated mice and preserved vascular growth. However, in both Eln+/− and Eln−/− somatic growth was reduced. Subsequent work showed reduced collagen accumulation and stiffness in rapalog treated Eln−/−; hBAC ELN mice [78]. In these experiments, treatment did not improve arterial narrowing per se but did alter the mechanosensor response to elastin insufficiency, highlighting the role of integrin and cell matrix interactions in the pathology of elastin insufficiency.
In the integrin study, Misra et al showed increased β3 integrin expression in Eln+/− aortas and in tissue taken from patients with WBS. To test whether inhibition of β3 integrin might serve as a useful therapeutic in elastin insufficiency, Eln mutants were raised in a β3 null genetic background. Eln+/−; Itgb3−/− or Itgb3+/− mice showed decreased smooth muscle proliferation, improvement in smooth muscle cell alignment and retention of lumen size [79]. Eln−/−; Itgb3−/− mice lived longer (from approximately p2 in the Itgb3+/+ mice to p4 in the Itgb3−/− or Itgb3+/−). Similarly, prenatal administration of the β3 blocking drug, cilengitide, led to less muscular arteries and reduced stenosis.
For both the mTor and the cilengitide experiments, the drugs were given early in development and led to the prevention of smooth muscle proliferation and therefore would be predicted to prevent stenosis. The side effect profiles for these drugs are significant though and did not preserve life more than two days. Additional studies are needed to determine whether these drugs would be of benefit after the appearance of stenosis and whether chronic management with the medications would be needed to avoid the development of stenosis or if treatment during a sensitive period would be sufficient to prevent long term disease.
Conclusion:
Genetic alterations in the elastin gene cause disease in a variety of tissues with consequences ranging from mild to life threatening. Additional studies are needed to understand the mechanism by which both gene dosage and dominant negative effects of the protein cause disease and how to treat them. While this review focused on genetic forms of elastin mediated disease, the findings in these rare conditions are surely to impact the management of more common diseases such as vascular stiffness, skin changes and emphysema related to chronic damage to elastic fibers. Consequently, the treatments outlined here, once optimized, may be important for the treatment of a variety of age related illnesses.
Supplementary Material
Highlights.
Rare ELN gene mutations affect vascular, lung, skin and genitourinary tissues.
Elastin quantity (SVAS, WBS duplication) and quality (ADCL) changes cause disease.
ELN variants predict important elastin assembly domains.
Potential therapies may target elastin production and inhibition of proliferation.
Acknowledgements:
The authors would like to acknowledge Ms. Angela Troia, Mr. Russell Knutsen, Dr. Delong Liu, and Dr. Shabana Shahanavaz who donated images for this study. The de-identified images shown here were collected under NIH 16-H-0074 and we acknowledge the many families who participate in this study. Images in this review utilized the Pathology core and the Animal Imaging core of the National Heart, Lung and Blood Institute of the NIH.
Funding:
This work was supported by the National Institutes of Health [grant numbers HL006210, HL006212].
Abbreviations used
- ADCL
autosomal dominant cutis laxa
- BAC
bacterial artificial chromosome
- COPD
chronic obstructive pulmonary disease
- ER
endoplasmic reticulum
- ERK
extracellular signal–regulated kinases
- hELN
human elastin gene
- HGMD
Human Gene Mutation Database
- Itgb3
Integrin beta-3
- mEln
mouse elastin gene
- miR
microRNA
- mTOR
mechanistic target of rapamycin
- SVAS
supravalvar aortic stenosis
- TGFβ
transforming growth factor β
- UTR
untranslated region
- VCLP
vocal cord lamina propria
- WBS
Williams Beuren Syndrome
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- [1].Shapiro SD, Endicott SK, Province MA, Pierce JA, Campbell EJ, Marked longevity of human lung parenchymal elastic fibers deduced from prevalence of D-aspartate and nuclear weapons-related radiocarbon, J Clin Invest 87(5) (1991) 1828–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Foster JA, Gray WR, Franzblau C, Isolation and characterization of crosslinked peptides from elastin, Biochimica et biophysica acta 303(2) (1973) 363–9. [DOI] [PubMed] [Google Scholar]
- [3].Sandberg LB, Zeikus RD, Coltrain IM, Tropoelastin purification from copper-deficient swine: a simplified method, Biochimica et biophysica acta 236(3) (1971) 542–5. [DOI] [PubMed] [Google Scholar]
- [4].Lent R, Franzblau C, Studies on the reduction of bovine elastin: evidence for the presence of delta-6,7-dehydrolysinonorleucine, Biochemical and biophysical research communications 26(1) (1967) 43–50. [DOI] [PubMed] [Google Scholar]
- [5].Partridge SM, Elsden DF, Thomas J, Constitution of the cross-linkages in elastin, Nature 197 (1963) 1297–8. [DOI] [PubMed] [Google Scholar]
- [6].Lent RW, Smith B, Salcedo LL, Faris B, Franzblau C, Studies on the reduction of elastin. II. Evidence for the presence of alpha-aminoadipic acid delta-semialdehyde and its aldol condensation product, Biochemistry 8(7) (1969) 2837–45. [DOI] [PubMed] [Google Scholar]
- [7].Bochicchio B, Tamburro AM, Polyproline II structure in proteins: identification by chiroptical spectroscopies, stability, and functions, Chirality 14(10) (2002) 782–92. [DOI] [PubMed] [Google Scholar]
- [8].Dyksterhuis LB, Carter EA, Mithieux SM, Weiss AS, Tropoelastin as a thermodynamically unfolded premolten globule protein: The effect of trimethylamine N-oxide on structure and coacervation, Arch Biochem Biophys 487(2) (2009) 79–84. [DOI] [PubMed] [Google Scholar]
- [9].Hoeve CA, Flory PJ, The elastic properties of elastin, Biopolymers 13(4) (1974) 677–86. [DOI] [PubMed] [Google Scholar]
- [10].Martino M, Coviello A, Tamburro AM, Synthesis and structural characterization of poly(LGGVG), an elastin-like polypeptide, International journal of biological macromolecules 27(1) (2000) 59–64. [DOI] [PubMed] [Google Scholar]
- [11].Urry DW, Long MM, Conformations of the repeat peptides of elastin in solution: an application of proton and carbon-13 magnetic resonance to the determination of polypeptide secondary structure, CRC critical reviews in biochemistry 4(1) (1976) 1–45. [DOI] [PubMed] [Google Scholar]
- [12].Baldock C, Oberhauser AF, Ma L, Lammie D, Siegler V, Mithieux SM, Tu Y, Chow JY, Suleman F, Malfois M, Rogers S, Guo L, Irving TC, Wess TJ, Weiss AS, Shape of tropoelastin, the highly extensible protein that controls human tissue elasticity, Proceedings of the National Academy of Sciences of the United States of America 108(11) (2011) 4322–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yeo GC, Tarakanova A, Baldock C, Wise SG, Buehler MJ, Weiss AS, Subtle balance of tropoelastin molecular shape and flexibility regulates dynamics and hierarchical assembly, Science advances 2(2) (2016) e1501145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kozel BA, Ciliberto CH, Mecham RP, Deposition of tropoelastin into the extracellular matrix requires a competent elastic fiber scaffold but not live cells, Matrix Biol 23(1) (2004) 23–34. [DOI] [PubMed] [Google Scholar]
- [15].Kozel BA, Rongish BJ, Czirok A, Zach J, Little CD, Davis EC, Knutsen RH, Wagenseil JE, Levy MA, Mecham RP, Elastic fiber formation: a dynamic view of extracellular matrix assembly using timer reporters, J Cell Physiol 207(1) (2006) 87–96. [DOI] [PubMed] [Google Scholar]
- [16].Tamburro AM, Pepe A, Bochicchio B, Quaglino D, Ronchetti IP, Supramolecular amyloid-like assembly of the polypeptide sequence coded by exon 30 of human tropoelastin, J Biol Chem 280(4) (2005) 2682–90. [DOI] [PubMed] [Google Scholar]
- [17].Muiznieks LD, Reichheld SE, Sitarz EE, Miao M, Keeley FW, Proline-poor hydrophobic domains modulate the assembly and material properties of polymeric elastin, Biopolymers 103(10) (2015) 563–73. [DOI] [PubMed] [Google Scholar]
- [18].Kozel BA, Wachi H, Davis EC, Mecham RP, Domains in tropoelastin that mediate elastin deposition in vitro and in vivo, J Biol Chem 278(20) (2003) 18491–8. [DOI] [PubMed] [Google Scholar]
- [19].Muiznieks LD, Miao M, Sitarz EE, Keeley FW, Contribution of domain 30 of tropoelastin to elastic fiber formation and material elasticity, Biopolymers 105(5) (2016) 267–75. [DOI] [PubMed] [Google Scholar]
- [20].Miao M, Reichheld SE, Muiznieks LD, Sitarz EE, Sharpe S, Keeley FW, Single nucleotide polymorphisms and domain/splice variants modulate assembly and elastomeric properties of human elastin. Implications for tissue specificity and durability of elastic tissue, Biopolymers 107(5) (2017). [DOI] [PubMed] [Google Scholar]
- [21].Wachi H, Sato F, Nakazawa J, Nonaka R, Szabo Z, Urban Z, Yasunaga T, Maeda I, Okamoto K, Starcher BC, Li DY, Mecham RP, Seyama Y, Domains 16 and 17 of tropoelastin in elastic fibre formation, Biochem J 402(1) (2007) 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Szabo Z, Levi-Minzi SA, Christiano AM, Struminger C, Stoneking M, Batzer MA, Boyd CD, Sequential loss of two neighboring exons of the tropoelastin gene during primate evolution, J Mol Evol 49(5) (1999) 664–71. [DOI] [PubMed] [Google Scholar]
- [23].Broekelmann TJ, Kozel BA, Ishibashi H, Werneck CC, Keeley FW, Zhang L, Mecham RP, Tropoelastin interacts with cell-surface glycosaminoglycans via its COOH-terminal domain, J Biol Chem 280(49) (2005) 40939–47. [DOI] [PubMed] [Google Scholar]
- [24].Privitera S, Prody CA, Callahan JW, Hinek A, The 67-kDa enzymatically inactive alternatively spliced variant of beta-galactosidase is identical to the elastin/laminin-binding protein, J Biol Chem 273(11) (1998) 6319–26. [DOI] [PubMed] [Google Scholar]
- [25].Gheduzzi D, Guerra D, Bochicchio B, Pepe A, Tamburro AM, Quaglino D, Mithieux S, Weiss AS, Pasquali Ronchetti I, Heparan sulphate interacts with tropoelastin, with some tropoelastin peptides and is present in human dermis elastic fibers, Matrix Biol 24(1) (2005) 15–25. [DOI] [PubMed] [Google Scholar]
- [26].Hsiao H, Stone PJ, Toselli P, Rosenbloom J, Franzblau C, Schreiber BM, The role of the carboxy terminus of tropoelastin in its assembly into the elastic fiber, Connective tissue research 40(2) (1999) 83–95. [DOI] [PubMed] [Google Scholar]
- [27].Broekelmann TJ, Ciliberto CH, Shifren A, Mecham RP, Modification and functional inactivation of the tropoelastin carboxy-terminal domain in cross-linked elastin, Matrix Biol 27(7) (2008) 631–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Del Monaco M, Covello SP, Kennedy SH, Gilinger G, Litwack G, Uitto J, Identification of novel glucocorticoid-response elements in human elastin promoter and demonstration of nucleotide sequence specificity of the receptor binding, The Journal of investigative dermatology 108(6) (1997) 938–42. [DOI] [PubMed] [Google Scholar]
- [29].Marigo V, Volpin D, Vitale G, Bressan GM, Identification of a TGF-beta responsive element in the human elastin promoter, Biochemical and biophysical research communications 199(2) (1994) 1049–56. [DOI] [PubMed] [Google Scholar]
- [30].Shi J, Wang A, Sen S, Wang Y, Kim HJ, Mitts TF, Hinek A, Insulin induces production of new elastin in cultures of human aortic smooth muscle cells, The American journal of pathology 180(2) (2012) 715–26. [DOI] [PubMed] [Google Scholar]
- [31].Zhang M, Pierce RA, Wachi H, Mecham RP, Parks WC, An open reading frame element mediates posttranscriptional regulation of tropoelastin and responsiveness to transforming growth factor beta1, Mol Cell Biol 19(11) (1999) 7314–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ott CE, Grunhagen J, Jager M, Horbelt D, Schwill S, Kallenbach K, Guo G, Manke T, Knaus P, Mundlos S, Robinson PN, MicroRNAs differentially expressed in postnatal aortic development downregulate elastin via 3’ UTR and coding-sequence binding sites, PLoS One 6(1) (2011) e16250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Barrineau LL, Rich CB, Przybyla A, Foster JA, Differential expression of aortic and lung elastin genes during chick embryogenesis, Developmental biology 87(1) (1981) 46–51. [DOI] [PubMed] [Google Scholar]
- [34].Heim RA, Pierce RA, Deak SB, Riley DJ, Boyd CD, Stolle CA, Alternative splicing of rat tropoelastin mRNA is tissue-specific and developmentally regulated, Matrix (Stuttgart, Germany) 11(5) (1991) 359–66. [DOI] [PubMed] [Google Scholar]
- [35].Indik Z, Yeh H, Ornstein-Goldstein N, Sheppard P, Anderson N, Rosenbloom JC, Peltonen L, Rosenbloom J, Alternative splicing of human elastin mRNA indicated by sequence analysis of cloned genomic and complementary DNA, Proceedings of the National Academy of Sciences of the United States of America 84(16) (1987) 5680–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sugitani H, Hirano E, Knutsen RH, Shifren A, Wagenseil JE, Ciliberto C, Kozel BA, Urban Z, Davis EC, Broekelmann TJ, Mecham RP, Alternative splicing and tissue-specific elastin misassembly act as biological modifiers of human elastin gene frameshift mutations associated with dominant cutis laxa, J Biol Chem 287(26) (2012) 22055–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wrenn DS, Parks WC, Whitehouse LA, Crouch EC, Kucich U, Rosenbloom J, Mecham RP, Identification of multiple tropoelastins secreted by bovine cells, J Biol Chem 262(5) (1987) 2244–9. [PubMed] [Google Scholar]
- [38].Yeh H, Anderson N, Ornstein-Goldstein N, Bashir MM, Rosenbloom JC, Abrams W, Indik Z, Yoon K, Parks W, Mecham R, et al. , Structure of the bovine elastin gene and S1 nuclease analysis of alternative splicing of elastin mRNA in the bovine nuchal ligament, Biochemistry 28(6) (1989) 2365–70. [DOI] [PubMed] [Google Scholar]
- [39].Keire PA, Bressler SL, Mulvihill ER, Starcher BC, Kang I, Wight TN, Inhibition of versican expression by siRNA facilitates tropoelastin synthesis and elastic fiber formation by human SK-LMS-1 leiomyosarcoma smooth muscle cells in vitro and in vivo, Matrix Biol. 50(Supplement C) (2016) 67–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Pilecki B, Holm AT, Schlosser A, Moeller JB, Wohl AP, Zuk AV, Heumuller SE, Wallis R, Moestrup SK, Sengle G, Holmskov U, Sorensen GL, Characterization of Microfibrillar-associated Protein 4 (MFAP4) as a Tropoelastin- and Fibrillin-binding Protein Involved in Elastic Fiber Formation, J Biol Chem 291(3) (2016) 1103–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yanagisawa H, Schluterman MK, Brekken RA, Fibulin-5, an integrin-binding matricellular protein: its function in development and disease, J Cell Commun Signal (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Nonaka R, Onoue S, Wachi H, Sato F, Urban Z, Starcher BC, Seyama Y, DANCE/fibulin-5 promotes elastic fiber formation in a tropoelastin isoform-dependent manner, Clin Biochem 42(7–8) (2009) 713–21. [DOI] [PubMed] [Google Scholar]
- [43].Choudhury R, McGovern A, Ridley C, Cain SA, Baldwin A, Wang MC, Guo C, Mironov A Jr., Drymoussi Z, Trump D, Shuttleworth A, Baldock C, Kielty CM, Differential regulation of elastic fiber formation by fibulin-4 and −5, J Biol Chem 284(36) (2009) 24553–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lemaire R, Bayle J, Mecham RP, Lafyatis R, Microfibril-associated MAGP-2 stimulates elastic fiber assembly, J Biol Chem 282(1) (2007) 800–8. [DOI] [PubMed] [Google Scholar]
- [45].Nakamura T, Lozano PR, Ikeda Y, Iwanaga Y, Hinek A, Minamisawa S, Cheng CF, Kobuke K, Dalton N, Takada Y, Tashiro K, Ross J Jr, Honjo T, Chien KR, Fibulin-5/DANCE is essential for elastogenesis in vivo, Nature 415(6868) (2002) 171–5. [DOI] [PubMed] [Google Scholar]
- [46].Trask TM, Trask BC, Ritty TM, Abrams WR, Rosenbloom J, Mecham RP, Interaction of tropoelastin with the amino-terminal domains of fibrillin-1 and fibrillin-2 suggests a role for the fibrillins in elastic fiber assembly, J Biol Chem 275(32) (2000) 24400–6. [DOI] [PubMed] [Google Scholar]
- [47].Urban Z, Michels VV, Thibodeau SN, Donis-Keller H, Csiszar K, Boyd CD, Supravalvular aortic stenosis: a splice site mutation within the elastin gene results in reduced expression of two aberrantly spliced transcripts, Hum Genet 104(2) (1999) 135–42. [DOI] [PubMed] [Google Scholar]
- [48].Park S, Seo EJ, Yoo HW, Kim Y, Novel mutations in the human elastin gene (ELN) causing isolated supravalvular aortic stenosis, Int J Mol Med 18(2) (2006) 329–32. [PubMed] [Google Scholar]
- [49].Curran ME, Atkinson DL, Ewart AK, Morris CA, Leppert MF, Keating MT, The elastin gene is disrupted by a translocation associated with supravalvular aortic stenosis, Cell 73(1) (1993) 159–68. [DOI] [PubMed] [Google Scholar]
- [50].Ewart AK, Jin W, Atkinson D, Morris CA, Keating MT, Supravalvular aortic stenosis associated with a deletion disrupting the elastin gene, J Clin Invest 93(3) (1994) 1071–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Olson TM, Michels VV, Urban Z, Csiszar K, Christiano AM, Driscoll DJ, Feldt RH, Boyd CD, Thibodeau SN, A 30 kb deletion within the elastin gene results in familial supravalvular aortic stenosis, Hum Mol Genet 4(9) (1995) 1677–9. [DOI] [PubMed] [Google Scholar]
- [52].Fryssira H, Palmer R, Hallidie-Smith KA, Taylor J, Donnai D, Reardon W, Fluorescent in situ hybridisation (FISH) for hemizygous deletion at the elastin locus in patients with isolated supravalvular aortic stenosis, J Med Genet 34(4) (1997) 306–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Stromme P, Bjornstad PG, Ramstad K, Prevalence estimation of Williams syndrome, J Child Neurol 17(4) (2002) 269–71. [DOI] [PubMed] [Google Scholar]
- [54].Perez Jurado LA, Peoples R, Kaplan P, Hamel BC, Francke U, Molecular definition of the chromosome 7 deletion in Williams syndrome and parent-of-origin effects on growth, Am J Hum Genet 59(4) (1996) 781–92. [PMC free article] [PubMed] [Google Scholar]
- [55].Robinson WP, Waslynka J, Bernasconi F, Wang M, Clark S, Kotzot D, Schinzel A, Delineation of 7q11.2 deletions associated with Williams-Beuren syndrome and mapping of a repetitive sequence to within and to either side of the common deletion, Genomics 34(1) (1996) 17–23. [DOI] [PubMed] [Google Scholar]
- [56].Antonell A, Del Campo M, Magano LF, Kaufmann L, de la Iglesia JM, Gallastegui F, Flores R, Schweigmann U, Fauth C, Kotzot D, Perez-Jurado LA, Partial 7q11.23 deletions further implicate GTF2I and GTF2IRD1 as the main genes responsible for the Williams-Beuren syndrome neurocognitive profile, J Med Genet 47(5) (2010) 312–20. [DOI] [PubMed] [Google Scholar]
- [57].Ferrero GB, Howald C, Micale L, Biamino E, Augello B, Fusco C, Turturo MG, Forzano S, Reymond A, Merla G, An atypical 7q11.23 deletion in a normal IQ Williams-Beuren syndrome patient, Eur J Hum Genet 18(1) (2010) 33–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Fusco C, Micale L, Augello B, Teresa Pellico M, Menghini D, Alfieri P, Cristina Digilio M, Mandriani B, Carella M, Palumbo O, Vicari S, Merla G, Smaller and larger deletions of the Williams Beuren syndrome region implicate genes involved in mild facial phenotype, epilepsy and autistic traits, Eur J Hum Genet 22(1) (2014) 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Francke U, Williams-Beuren syndrome: genes and mechanisms, Hum Mol Genet 8(10) (1999) 1947–54. [DOI] [PubMed] [Google Scholar]
- [60].Franke Y, Peoples RJ, Francke U, Identification of GTF2IRD1, a putative transcription factor within the Williams-Beuren syndrome deletion at 7q11.23, Cytogenetics and cell genetics 86(3–4) (1999) 296–304. [DOI] [PubMed] [Google Scholar]
- [61].Wang YK, Sporle R, Paperna T, Schughart K, Francke U, Characterization and expression pattern of the frizzled gene Fzd9, the mouse homolog of FZD9 which is deleted in Williams-Beuren syndrome, Genomics 57(2) (1999) 235–48. [DOI] [PubMed] [Google Scholar]
- [62].Urban Z, Riazi S, Seidl TL, Katahira J, Smoot LB, Chitayat D, Boyd CD, Hinek A, Connection between elastin haploinsufficiency and increased cell proliferation in patients with supravalvular aortic stenosis and Williams-Beuren syndrome, Am J Hum Genet 71(1) (2002) 30–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Dridi SM, Foucault Bertaud A, Igondjo Tchen S, Senni K, Ejeil AL, Pellat B, Lyonnet S, Bonnet D, Charpiot P, Godeau G, Vascular wall remodeling in patients with supravalvular aortic stenosis and Williams Beuren syndrome, J Vasc Res 42(3) (2005) 190–201. [DOI] [PubMed] [Google Scholar]
- [64].Callewaert B, Renard M, Hucthagowder V, Albrecht B, Hausser I, Blair E, Dias C, Albino A, Wachi H, Sato F, Mecham RP, Loeys B, Coucke PJ, De Paepe A, Urban Z, New insights into the pathogenesis of autosomal-dominant cutis laxa with report of five ELN mutations, Hum Mutat 32(4) (2011) 445–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Tassabehji M, Metcalfe K, Hurst J, Ashcroft GS, Kielty C, Wilmot C, Donnai D, Read AP, Jones CJ, An elastin gene mutation producing abnormal tropoelastin and abnormal elastic fibres in a patient with autosomal dominant cutis laxa, Hum Mol Genet 7(6) (1998) 1021–8. [DOI] [PubMed] [Google Scholar]
- [66].Graul-Neumann LM, Hausser I, Essayie M, Rauch A, Kraus C, Highly variable cutis laxa resulting from a dominant splicing mutation of the elastin gene, Am J Med Genet A 146A(8) (2008) 977–83. [DOI] [PubMed] [Google Scholar]
- [67].Li DY, Toland AE, Boak BB, Atkinson DL, Ensing GJ, Morris CA, Keating MT, Elastin point mutations cause an obstructive vascular disease, supravalvular aortic stenosis, Hum Mol Genet 6(7) (1997) 1021–8. [DOI] [PubMed] [Google Scholar]
- [68].Metcalfe K, Rucka AK, Smoot L, Hofstadler G, Tuzler G, McKeown P, Siu V, Rauch A, Dean J, Dennis N, Ellis I, Reardon W, Cytrynbaum C, Osborne L, Yates JR, Read AP, Donnai D, Tassabehji M, Elastin: mutational spectrum in supravalvular aortic stenosis, Eur J Hum Genet 8(12) (2000) 955–63. [DOI] [PubMed] [Google Scholar]
- [69].Li DY, Faury G, Taylor DG, Davis EC, Boyle WA, Mecham RP, Stenzel P, Boak B, Keating MT, Novel arterial pathology in mice and humans hemizygous for elastin, J Clin Invest 102(10) (1998) 1783–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Brown-Augsburger P, Tisdale C, Broekelmann T, Sloan C, Mecham RP, Identification of an elastin cross-linking domain that joins three peptide chains. Possible role in nucleated assembly, J Biol Chem 270(30) (1995) 17778–83. [DOI] [PubMed] [Google Scholar]
- [71].Megarbane H, Florence J, Sass JO, Schwonbeck S, Foglio M, de Cid R, Cure S, Saker S, Megarbane A, Fischer J, An autosomal-recessive form of cutis laxa is due to homozygous elastin mutations, and the phenotype may be modified by a heterozygous fibulin 5 polymorphism, The Journal of investigative dermatology 129(7) (2009) 1650–5. [DOI] [PubMed] [Google Scholar]
- [72].Cho MH, Ciulla DM, Klanderman BJ, Hersh CP, Litonjua AA, Sparrow D, Raby BA, Silverman EK, Analysis of exonic elastin variants in severe, early-onset chronic obstructive pulmonary disease, Am J Respir Cell Mol Biol 40(6) (2009) 751–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Kelleher CM, Silverman EK, Broekelmann T, Litonjua AA, Hernandez M, Sylvia JS, Stoler J, Reilly JJ, Chapman HA, Speizer FE, Weiss ST, Mecham RP, Raby BA, A functional mutation in the terminal exon of elastin in severe, early-onset chronic obstructive pulmonary disease, Am J Respir Cell Mol Biol 33(4) (2005) 355–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Parrott A, James J, Goldenberg P, Hinton RB, Miller E, Shikany A, Aylsworth AS, Kaiser-Rogers K, Ferns SJ, Lalani SR, Ware SM, Aortopathy in the 7q11.23 microduplication syndrome, Am J Med Genet A 167A(2) (2015) 363–70. [DOI] [PubMed] [Google Scholar]
- [75].Faury G, Pezet M, Knutsen RH, Boyle WA, Heximer SP, McLean SE, Minkes RK, Blumer KJ, Kovacs A, Kelly DP, Li DY, Starcher B, Mecham RP, Developmental adaptation of the mouse cardiovascular system to elastin haploinsufficiency, J Clin Invest 112(9) (2003) 1419–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Wagenseil JE, Nerurkar NL, Knutsen RH, Okamoto RJ, Li DY, Mecham RP, Effects of elastin haploinsufficiency on the mechanical behavior of mouse arteries, Am J Physiol Heart Circ Physiol 289(3) (2005) H1209–17. [DOI] [PubMed] [Google Scholar]
- [77].Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, Eichwald E, Keating MT, Elastin is an essential determinant of arterial morphogenesis, Nature 393(6682) (1998) 276–280. [DOI] [PubMed] [Google Scholar]
- [78].Jiao Y, Li G, Li Q, Ali R, Qin L, Li W, Qyang Y, Greif DM, Geirsson A, Humphrey JD, Tellides G, mTOR (Mechanistic Target of Rapamycin) Inhibition Decreases Mechanosignaling, Collagen Accumulation, and Stiffening of the Thoracic Aorta in Elastin-Deficient Mice, Arterioscler Thromb Vasc Biol 37(9) (2017) 1657–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Misra A, Sheikh AQ, Kumar A, Luo J, Zhang J, Hinton RB, Smoot L, Kaplan P, Urban Z, Qyang Y, Tellides G, Greif DM, Integrin beta3 inhibition is a therapeutic strategy for supravalvular aortic stenosis, The Journal of experimental medicine 213(3) (2016) 451–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Karnik SK, Brooke BS, Bayes-Genis A, Sorensen L, Wythe JD, Schwartz RS, Keating MT, Li DY, A critical role for elastin signaling in vascular morphogenesis and disease, Development 130(2) (2003) 411–23. [DOI] [PubMed] [Google Scholar]
- [81].Wagenseil JE, Ciliberto CH, Knutsen RH, Levy MA, Kovacs A, Mecham RP, Reduced vessel elasticity alters cardiovascular structure and function in newborn mice, Circ Res 104(10) (2009) 1217–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Hirano E, Knutsen RH, Sugitani H, Ciliberto CH, Mecham RP, Functional rescue of elastin insufficiency in mice by the human elastin gene: implications for mouse models of human disease, Circulation research 101(5) (2007) 523–31. [DOI] [PubMed] [Google Scholar]
- [83].Collins RT 2nd, Cardiovascular disease in Williams syndrome, Circulation 127(21) (2013) 2125–34. [DOI] [PubMed] [Google Scholar]
- [84].Pober BR, Johnson M, Urban Z, Mechanisms and treatment of cardiovascular disease in Williams-Beuren syndrome, J Clin Invest 118(5) (2008) 1606–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Kim YM, Yoo SJ, Choi JY, Kim SH, Bae EJ, Lee YT, Natural course of supravalvar aortic stenosis and peripheral pulmonary arterial stenosis in Williams’ syndrome, Cardiology in the young 9(1) (1999) 37–41. [DOI] [PubMed] [Google Scholar]
- [86].Pieles GE, Ofoe V, Morgan GJ, Severe left main coronary artery stenosis with abnormal branching pattern in a patient with mild supravalvar aortic stenosis and Williams-Beuren syndrome, Congenital heart disease 9(3) (2014) E85–9. [DOI] [PubMed] [Google Scholar]
- [87].Szaflik K, Kazmierczak P, Moll JJ, Moll JA, Severe Congenital Obstruction of the Left Main Coronary Artery Coexisting With Supravalvular Aortic Stenosis in Williams Syndrome: A Dangerous Association, World J Pediatr Congenit Heart Surg 7(2) (2016) 216–9. [DOI] [PubMed] [Google Scholar]
- [88].Voges I, Franklin RC, Wage R, Babu-Narayan SV, Fatal severe coronary artery stenosis in Williams syndrome: decision making using late gadolinium enhancement cardiovascular MRI, Cardiology in the young 27(7) (2017) 1398–1401. [DOI] [PubMed] [Google Scholar]
- [89].Ardinger RH Jr., Goertz KK, Mattioli LF, Cerebrovascular stenoses with cerebral infarction in a child with Williams syndrome, Am J Med Genet 51(3) (1994) 200–2. [DOI] [PubMed] [Google Scholar]
- [90].Putman CM, Chaloupka JC, Eklund JE, Fulbright RK, Multifocal intracranial occlusive vasculopathy resulting in stroke: an unusual manifestation of Williams syndrome, AJNR. American journal of neuroradiology 16(7) (1995) 1536–8. [PMC free article] [PubMed] [Google Scholar]
- [91].Wollack JB, Kaifer M, LaMonte MP, Rothman M, Stroke in Williams Syndrome, Stroke 27(1) (1996) 143–146. [DOI] [PubMed] [Google Scholar]
- [92].Jelsig AM, Urban Z, Hucthagowder V, Nissen H, Ousager LB, Novel ELN mutation in a family with supravalvular aortic stenosis and intracranial aneurysm, Eur J Med Genet 60(2) (2017) 110–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Wint DP, Butman JA, Masdeu JC, Meyer-Lindenberg A, Mervis CB, Sarpal D, Morris CA, Berman KF, Intracranial arteries in individuals with the elastin gene hemideletion of Williams syndrome, AJNR. American journal of neuroradiology 35(1) (2014) 90–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Jiao Y, Li G, Korneva A, Caulk AW, Qin L, Bersi MR, Li Q, Li W, Mecham RP, Humphrey JD, Tellides G, Deficient Circumferential Growth Is the Primary Determinant of Aortic Obstruction Attributable to Partial Elastin Deficiency, Arteriosclerosis, thrombosis, and vascular biology 37(5) (2017) 930–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Bassareo PP, Mercuro G, Increased arterial stiffness in children with Williams syndrome and normal blood pressure, Blood Press Monit 15(5) (2010) 257–61. [DOI] [PubMed] [Google Scholar]
- [96].Kozel BA, Danback JR, Waxler JL, Knutsen RH, de Las Fuentes L, Reusz GS, Kis E, Bhatt AB, Pober BR, Williams syndrome predisposes to vascular stiffness modified by antihypertensive use and copy number changes in NCF1, Hypertension 63(1) (2014) 74–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Chirinos JA, Zambrano JP, Chakko S, Veerani A, Schob A, Willens HJ, Perez G, Mendez AJ, Aortic pressure augmentation predicts adverse cardiovascular events in patients with established coronary artery disease, Hypertension 45(5) (2005) 980–5. [DOI] [PubMed] [Google Scholar]
- [98].Coutinho T, Turner ST, Kullo IJ, Aortic pulse wave velocity is associated with measures of subclinical target organ damage, JACC Cardiovasc Imaging 4(7) (2011) 754–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Franklin SS, Beyond blood pressure: Arterial stiffness as a new biomarker of cardiovascular disease, J Am Soc Hypertens 2(3) (2008) 140–51. [DOI] [PubMed] [Google Scholar]
- [100].Bouchireb K, Boyer O, Bonnet D, Brunelle F, Decramer S, Landthaler G, Liutkus A, Niaudet P, Salomon R, Clinical features and management of arterial hypertension in children with Williams-Beuren syndrome, Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association 25(2) (2010) 434–8. [DOI] [PubMed] [Google Scholar]
- [101].Ingelfinger JR, Newburger JW, Spectrum of renal anomalies in patients with Williams syndrome, The Journal of pediatrics 119(5) (1991) 771–3. [DOI] [PubMed] [Google Scholar]
- [102].Pober BR, Lacro RV, Rice C, Mandell V, Teele RL, Renal findings in 40 individuals with Williams syndrome, Am J Med Genet 46(3) (1993) 271–4. [DOI] [PubMed] [Google Scholar]
- [103].Rose C, Wessel A, Pankau R, Partsch CJ, Bursch J, Anomalies of the abdominal aorta in Williams-Beuren syndrome--another cause of arterial hypertension, Eur J Pediatr 160(11) (2001) 655–8. [DOI] [PubMed] [Google Scholar]
- [104].Del Campo M, Antonell A, Magano LF, Munoz FJ, Flores R, Bayes M, Perez Jurado LA, Hemizygosity at the NCF1 gene in patients with Williams-Beuren syndrome decreases their risk of hypertension, Am J Hum Genet 78(4) (2006) 533–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Kozel BA, Knutsen RH, Ye L, Ciliberto CH, Broekelmann TJ, Mecham RP, Genetic modifiers of cardiovascular phenotype caused by elastin haploinsufficiency act by extrinsic noncomplementation, J Biol Chem 286(52) (2011) 44926–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Campuzano V, Segura-Puimedon M, Terrado V, Sanchez-Rodriguez C, Coustets M, Menacho-Marquez M, Nevado J, Bustelo XR, Francke U, Perez-Jurado LA, Reduction of NADPH-oxidase activity ameliorates the cardiovascular phenotype in a mouse model of Williams-Beuren Syndrome, PLoS Genet 8(2) (2012) e1002458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Hilgendorff A, Parai K, Ertsey R, Navarro E, Jain N, Carandang F, Peterson J, Mokres L, Milla C, Preuss S, Alcazar MA, Khan S, Masumi J, Ferreira-Tojais N, Mujahid S, Starcher B, Rabinovitch M, Bland R, Lung matrix and vascular remodeling in mechanically ventilated elastin haploinsufficient newborn mice, American journal of physiology. Lung cellular and molecular physiology 308(5) (2015) L464–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Shifren A, Durmowicz AG, Knutsen RH, Hirano E, Mecham RP, Elastin protein levels are a vital modifier affecting normal lung development and susceptibility to emphysema, Am J Physiol Lung Cell Mol Physiol 292(3) (2007) L778–87. [DOI] [PubMed] [Google Scholar]
- [109].Wendel DP, Taylor DG, Albertine KH, Keating MT, Li DY, Impaired distal airway development in mice lacking elastin, Am J Respir Cell Mol Biol 23(3) (2000) 320–6. [DOI] [PubMed] [Google Scholar]
- [110].Wan ES, Pober BR, Washko GR, Raby BA, Silverman EK, Pulmonary function and emphysema in Williams-Beuren syndrome, Am J Med Genet A 152A(3) (2010) 653–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Louw JJ, Verleden G, Gewillig M, Devriendt K, Haploinsufficiency of elastin gene may lead to familial cardiopathy and pulmonary emphysema, American journal of medical genetics. Part A158a(8) (2012) 2053–4. [DOI] [PubMed] [Google Scholar]
- [112].Wojcik MH, Carmichael N, Bieber FR, Wiener DC, Madan R, Pober BR, Raby BA, A new diagnosis of Williams-Beuren syndrome in a 49-year-old man with severe bullous emphysema, Am J Med Genet A 173(8) (2017) 2235–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Takubo Y, Hirai T, Muro S, Kogishi K, Hosokawa M, Mishima M, Age-associated changes in elastin and collagen content and the proportion of types I and III collagen in the lungs of mice, Experimental Gerontology 34(3) (1999) 353–364. [DOI] [PubMed] [Google Scholar]
- [114].Cherniske EM, Carpenter TO, Klaiman C, Young E, Bregman J, Insogna K, Schultz RT, Pober BR, Multisystem study of 20 older adults with Williams syndrome, Am J Med Genet A 131(3) (2004) 255–64. [DOI] [PubMed] [Google Scholar]
- [115].Ewart AK, Morris CA, Atkinson D, Jin W, Sternes K, Spallone P, Stock AD, Leppert M, Keating MT, Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome, Nat Genet 5(1) (1993) 11–6. [DOI] [PubMed] [Google Scholar]
- [116].Kozel BA, Bayliss SJ, Berk DR, Waxler JL, Knutsen RH, Danback JR, Pober BR, Skin findings in Williams syndrome, American journal of medical genetics. Part A164a(9) (2014) 2217–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Morris CA, Demsey SA, Leonard CO, Dilts C, Blackburn BL, Natural history of Williams syndrome: physical characteristics, J Pediatr 113(2) (1988) 318–26. [DOI] [PubMed] [Google Scholar]
- [118].Dridi SM, Ghomrasseni S, Bonnet D, Aggoun Y, Vabres P, Bodemer C, Lyonnet S, de Prost Y, Fraitag S, Pellat B, Sidi D, Godeau G, Skin elastic fibers in Williams syndrome, Am J Med Genet 87(2) (1999) 134–8. [PubMed] [Google Scholar]
- [119].Urban Z, Peyrol S, Plauchu H, Zabot MT, Lebwohl M, Schilling K, Green M, Boyd CD, Csiszar K, Elastin gene deletions in Williams syndrome patients result in altered deposition of elastic fibers in skin and a subclinical dermal phenotype, Pediatr Dermatol 17(1) (2000) 12–20. [DOI] [PubMed] [Google Scholar]
- [120].Bedeschi MF, Bianchi V, Colli AM, Natacci F, Cereda A, Milani D, Maitz S, Lalatta F, Selicorni A, Clinical follow-up of young adults affected by Williams syndrome: experience of 45 Italian patients, American journal of medical genetics. Part A155a(2) (2011) 353–9. [DOI] [PubMed] [Google Scholar]
- [121].Morris CA, Mervis CB, Williams syndrome and related disorders, Annu Rev Genomics Hum Genet 1 (2000) 461–84. [DOI] [PubMed] [Google Scholar]
- [122].Stagi S, Lapi E, Chiarelli F, de Martino M, Incidence of diverticular disease and complicated diverticular disease in young patients with Williams syndrome, Pediatric surgery international 26(9) (2010) 943–4. [DOI] [PubMed] [Google Scholar]
- [123].Golder M, Burleigh DE, Ghali L, Feakins RM, Lunniss PJ, Williams NS, Navsaria HA, Longitudinal muscle shows abnormal relaxation responses to nitric oxide and contains altered levels of NOS1 and elastin in uncomplicated diverticular disease, Colorectal disease : the official journal of the Association of Coloproctology of Great Britain and Ireland 9(3) (2007) 218–28. [DOI] [PubMed] [Google Scholar]
- [124].Jiang X, Luttrell I, Li DY, Yang CC, Chitaley K, Altered bladder function in elastin-deficient mice at baseline and in response to partial bladder outlet obstruction, BJU international 110(3) (2012) 413–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Sammour ZM, de Bessa J Jr., Hisano M, Bruschini H, Kim CA, Srougi M, Gomes CM, Lower urinary tract symptoms in children and adolescents with Williams-Beuren syndrome, J. Pediatr. Urol 13(2) (2017) 203.e1–203.e6. [DOI] [PubMed] [Google Scholar]
- [126].Schulman SL, Zderic S, Kaplan P, Increased prevalence of urinary symptoms and voiding dysfunction in Williams syndrome, J Pediatr 129(3) (1996) 466–9. [DOI] [PubMed] [Google Scholar]
- [127].von Gontard A, Niemczyk J, Borggrefe-Moussavian S, Wagner C, Curfs L, Equit M, Incontinence in children, adolescents and adults with Williams syndrome, Neurourology and Urodynamics 35(8) (2016) 1000–1005. [DOI] [PubMed] [Google Scholar]
- [128].Babbitt DP, Dobbs J, Boedecker RA, Multiple bladder diverticula in Williams “Elfin-Facies” syndrome, Pediatric radiology 8(1) (1979) 29–31. [DOI] [PubMed] [Google Scholar]
- [129].Morris CA, Leonard CO, Dilts C, Demsey SA, Adults with Williams syndrome, Am J Med Genet Suppl 6 (1990) 102–7. [DOI] [PubMed] [Google Scholar]
- [130].Sammour ZM, Gomes CM, Duarte RJ, Trigo-Rocha FE, Srougi M, Voiding dysfunction and the Williams-Beuren syndrome: a clinical and urodynamic investigation, J Urol 175(4) (2006) 1472–6. [DOI] [PubMed] [Google Scholar]
- [131].Gray SD, Titze IR, Alipour F, Hammond TH, Biomechanical and histologic observations of vocal fold fibrous proteins, The Annals of otology, rhinology, and laryngology 109(1) (2000) 77–85. [DOI] [PubMed] [Google Scholar]
- [132].Hahn MS, Kobler JB, Starcher BC, Zeitels SM, Langer R, Quantitative and comparative studies of the vocal fold extracellular matrix. I: Elastic fibers and hyaluronic acid, The Annals of otology, rhinology, and laryngology 115(2) (2006) 156–64. [DOI] [PubMed] [Google Scholar]
- [133].Preus M, The Williams syndrome: objective definition and diagnosis, Clin Genet 25(5) (1984) 422–8. [DOI] [PubMed] [Google Scholar]
- [134].Stewart FJ, Dalzell M, McReid M, Cinnamond MJ, Bilateral vocal cord paralysis in Williams syndrome, Clin Genet 44(3) (1993) 164–5. [DOI] [PubMed] [Google Scholar]
- [135].Vaux KK, Wojtczak H, Benirschke K, Jones KL, Vocal cord abnormalities in Williams syndrome: a further manifestation of elastin deficiency, American journal of medical genetics. Part A119a(3) (2003) 302–4. [DOI] [PubMed] [Google Scholar]
- [136].Watts CR, Awan SN, Marler JA, An investigation of voice quality in individuals with inherited elastin gene abnormalities, Clinical linguistics & phonetics 22(3) (2008) 199–213. [DOI] [PubMed] [Google Scholar]
- [137].Watts CR, Knutsen RH, Ciliberto C, Mecham RP, Evidence for heterozygous abnormalities of the elastin gene (ELN) affecting the quantity of vocal fold elastic fibers: a pilot study, Journal of voice : official journal of the Voice Foundation 25(2) (2011) e85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Klein AJ, Armstrong BL, Greer MK, Brown FR 3rd, Hyperacusis and otitis media in individuals with Williams syndrome, J. Speech Hear. Disord 55(2) (1990) 339–44. [DOI] [PubMed] [Google Scholar]
- [139].Marler JA, Elfenbein JL, Ryals BM, Urban Z, Netzloff ML, Sensorineural hearing loss in children and adults with Williams syndrome, Am J Med Genet A 138(4) (2005) 318–27. [DOI] [PubMed] [Google Scholar]
- [140].Marler JA, Sitcovsky JL, Mervis CB, Kistler DJ, Wightman FL, Auditory Function and Hearing Loss in Children and Adults with Williams Syndrome: Cochlear Impairment in Individuals with Otherwise Normal Hearing, American journal of medical genetics. Part C, Seminars in medical genetics154C(2) (2010) 249–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Gothelf D, Farber N, Raveh E, Apter A, Attias J, Hyperacusis in Williams syndrome: characteristics and associated neuroaudiologic abnormalities, Neurology 66(3) (2006) 390–5. [DOI] [PubMed] [Google Scholar]
- [142].Barozzi S, Soi D, Comiotto E, Borghi A, Gavioli C, Spreafico E, Gagliardi C, Selicorni A, Forti S, Ambrosetti U, Cesarani A, Brambilla D, Audiological findings in Williams syndrome: a study of 69 patients, American journal of medical genetics. Part A158A(4) (2012) 759–71. [DOI] [PubMed] [Google Scholar]
- [143].Matsumoto N, Kitani R, Kalinec F, Linking LIMK1 deficiency to hyperacusis and progressive hearing loss in individuals with Williams syndrome, Commun. Integr. Biol 4(2) (2011) 208–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Kozel BA, Su CT, Danback JR, Minster RL, Madan-Khetarpal S, McConnell JS, Mac Neal MK, Levine KL, Wilson RC, Sciurba FC, Urban Z, Biomechanical properties of the skin in cutis laxa, The Journal of investigative dermatology 134(11) (2014) 2836–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Beighton P, The dominant and recessive forms of cutis laxa, J Med Genet 9(2) (1972) 216–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Hadj-Rabia S, Callewaert BL, Bourrat E, Kempers M, Plomp AS, Layet V, Bartholdi D, Renard M, Backer JD, Malfait F, Vanakker OM, Coucke PJ, De Paepe AM, Bodemer C, Twenty patients including 7 probands with autosomal dominant cutis laxa confirm clinical and molecular homogeneity, Orphanet Journal of Rare Diseases 8 (2013) 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Szabo Z, Crepeau MW, Mitchell AL, Stephan MJ, Puntel RA, Yin Loke K, Kirk RC, Urban Z, Aortic aneurysmal disease and cutis laxa caused by defects in the elastin gene, J Med Genet 43(3) (2006) 255–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Duz MB, Kirat E, Coucke PJ, Koparir E, Gezdirici A, Paepe A, Callewaert B, Seven M, A novel case of autosomal dominant cutis laxa in a consanguineous family: report and literature review, Clinical dysmorphology 26(3) (2017) 142–147. [DOI] [PubMed] [Google Scholar]
- [149].Urban Z, Gao J, Pope FM, Davis EC, Autosomal dominant cutis laxa with severe lung disease: synthesis and matrix deposition of mutant tropoelastin, The Journal of investigative dermatology 124(6) (2005) 1193–9. [DOI] [PubMed] [Google Scholar]
- [150].Hu Q, Shifren A, Sens C, Choi J, Szabo Z, Starcher BC, Knutsen RH, Shipley JM, Davis EC, Mecham RP, Urban Z, Mechanisms of emphysema in autosomal dominant cutis laxa, Matrix Biol 29(7) (2010) 621–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Paladini D, Di Spiezio Sardo A, Mandato VD, Guerra G, Bifulco G, Mauriello S, Nappi C, Association of cutis laxa and genital prolapse: a case report, Int Urogynecol J Pelvic Floor Dysfunct 18(11) (2007) 1367–70. [DOI] [PubMed] [Google Scholar]
- [152].Earhart BA, Williams ME, Zamora I, Randolph LM, Votava-Smith JK, Marcy SN, Phenotype of 7q11.23 duplication: A family clinical series, Am J Med Genet A 173(1) (2017) 114–119. [DOI] [PubMed] [Google Scholar]
- [153].Morris CA, Mervis CB, Paciorkowski AP, Abdul-Rahman O, Dugan SL, Rope AF, Bader P, Hendon LG, Velleman SL, Klein-Tasman BP, Osborne LR, 7q11.23 Duplication syndrome: Physical characteristics and natural history, Am J Med Genet A 167A(12) (2015) 2916–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Zarate YA, Lepard T, Sellars E, Kaylor JA, Alfaro MP, Sailey C, Schaefer GB, Collins RT 2nd, Cardiovascular and genitourinary anomalies in patients with duplications within the Williams syndrome critical region: phenotypic expansion and review of the literature, Am J Med Genet A 164A(8) (2014) 1998–2002. [DOI] [PubMed] [Google Scholar]
- [155].Guemann AS, Andrieux J, Petit F, Halimi E, Bouquillon S, Manouvrier-Hanu S, Van De Kamp J, Boileau C, Hanna N, Jondeau G, Vaksmann G, Houfflin-Debarge V, Holder-Espinasse M, ELN gene triplication responsible for familial supravalvular aortic aneurysm, Cardiol Young 25(4) (2015) 712–7. [DOI] [PubMed] [Google Scholar]
- [156].Van der Aa N, Rooms L, Vandeweyer G, van den Ende J, Reyniers E, Fichera M, Romano C, Delle Chiaie B, Mortier G, Menten B, Destree A, Maystadt I, Mannik K, Kurg A, Reimand T, McMullan D, Oley C, Brueton L, Bongers EM, van Bon BW, Pfund R, Jacquemont S, Ferrarini A, Martinet D, Schrander-Stumpel C, Stegmann AP, Frints SG, de Vries BB, Ceulemans B, Kooy RF, Fourteen new cases contribute to the characterization of the 7q11.23 microduplication syndrome, Eur J Med Genet 52(2–3) (2009) 94–100. [DOI] [PubMed] [Google Scholar]
- [157].Bokenkamp R, DeRuiter MC, van Munsteren C, Gittenberger-de Groot AC, Insights into the pathogenesis and genetic background of patency of the ductus arteriosus, Neonatology 98(1) (2010) 6–17. [DOI] [PubMed] [Google Scholar]
- [158].Coceani F, Baragatti B, Mechanisms for ductus arteriosus closure, Seminars in perinatology 36(2) (2012) 92–7. [DOI] [PubMed] [Google Scholar]
- [159].Wu JR, Yeh JL, Liou SF, Dai ZK, Wu BN, Hsu JH, Gamma-secretase Inhibitor Prevents Proliferation and Migration of Ductus Arteriosus Smooth Muscle Cells through the Notch3-HES1/2/5 Pathway, International journal of biological sciences 12(9) (2016) 1063–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Hinek A, Boyle J, Rabinovitch M, Vascular smooth muscle cell detachment from elastin and migration through elastic laminae is promoted by chondroitin sulfate-induced “shedding” of the 67-kDa cell surface elastin binding protein, Exp Cell Res 203(2) (1992) 344–53. [DOI] [PubMed] [Google Scholar]
- [161].Hinek A, Mecham RP, Keeley F, Rabinovitch M, Impaired elastin fiber assembly related to reduced 67-kD elastin-binding protein in fetal lamb ductus arteriosus and in cultured aortic smooth muscle cells treated with chondroitin sulfate, J Clin Invest 88(6) (1991) 2083–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Hinek A, Rabinovitch M, The ductus arteriosus migratory smooth muscle cell phenotype processes tropoelastin to a 52-kDa product associated with impaired assembly of elastic laminae, J Biol Chem 268(2) (1993) 1405–13. [PubMed] [Google Scholar]
- [163].Cus R, Maurus D, Kuhl M, Cloning and developmental expression of WSTF during Xenopus laevis embryogenesis, Gene Expr Patterns 6(4) (2006) 340–6. [DOI] [PubMed] [Google Scholar]
- [164].Orellana C, Bernabeu J, Monfort S, Rosello M, Oltra S, Ferrer I, Quiroga R, Martinez-Garay I, Martinez F, Duplication of the Williams-Beuren critical region: case report and further delineation of the phenotypic spectrum, J Med Genet 45(3) (2008) 187–9. [DOI] [PubMed] [Google Scholar]
- [165].Abbas E, Cox DM, Smith T, Butler MG, The 7q11.23 Microduplication Syndrome: A Clinical Report with Review of Literature, Journal of pediatric genetics 5(3) (2016) 129–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Merla G, Brunetti-Pierri N, Micale L, Fusco C, Copy number variants at Williams-Beuren syndrome 7q11.23 region, Human genetics 128(1) (2010) 3–26. [DOI] [PubMed] [Google Scholar]
- [167].Pober BR, Morris CA, Diagnosis and management of medical problems in adults with Williams-Beuren syndrome, Am J Med Genet C Semin Med Genet 145C(3) (2007) 280–90. [DOI] [PubMed] [Google Scholar]
- [168].Pober BR, Williams-Beuren syndrome N Engl J Med 362(3) (2010) 239–52. [DOI] [PubMed] [Google Scholar]
- [169].Walton JR, Martens MA, Pober BR, The proceedings of the 15th professional conference on Williams Syndrome, Am J Med Genet A 173(5) (2017) 1159–1171. [DOI] [PubMed] [Google Scholar]
- [170].Halabi CM, Broekelmann TJ, Knutsen RH, Ye L, Mecham RP, Kozel BA, Chronic antihypertensive treatment improves pulse pressure but not large artery mechanics in a mouse model of congenital vascular stiffness, Am J Physiol Heart Circ Physiol 309(5) (2015) H1008–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Knutsen RH, Beeman SC, Broekelmann TJ, Liu D, Tsang KM, Kovacs A, Ye L, Danback JR, Watson A, Wardlaw A, Wagenseil JE, Garbow JR, Shoykhet M, Kozel BA, Minoxidil improves vascular compliance, restores cerebral blood flow and alters extracellular matrix gene expression in a model of chronic vascular stiffness Am J Physiol Heart Circ Physiol Accepted (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Hadj-Rabia S, Callewaert BL, Bourrat E, Kempers M, Plomp AS, Layet V, Bartholdi D, Renard M, De Backer J, Malfait F, Vanakker OM, Coucke PJ, De Paepe AM, Bodemer C, Twenty patients including 7 probands with autosomal dominant cutis laxa confirm clinical and molecular homogeneity, Orphanet journal of rare diseases 8 (2013) 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Nahas FX, Sterman S, Gemperli R, Ferreira MC, The role of plastic surgery in congenital cutis laxa: a 10-year follow-up, Plast Reconstr Surg 104(4) (1999) 1174–8; discussion 1179. [PubMed] [Google Scholar]
- [174].Beighton P, Bull JC, Edgerton MT, Plastic surgery in cutis laxa, Br J Plast Surg 23(3) (1970) 285–90. [DOI] [PubMed] [Google Scholar]
- [175].Corbett E, Glaisyer H, Chan C, Madden B, Khaghani A, Yacoub M, Congenital cutis laxa with a dominant inheritance and early onset emphysema, Thorax 49(8) (1994) 836–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Zhang P, Huang A, Ferruzzi J, Mecham RP, Starcher BC, Tellides G, Humphrey JD, Giordano FJ, Niklason LE, Sessa WC, Inhibition of microRNA-29 enhances elastin levels in cells haploinsufficient for elastin and in bioengineered vessels--brief report, Arterioscler Thromb Vasc Biol 32(3) (2012) 756–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Sassi Y, Avramopoulos P, Ramanujam D, Gruter L, Werfel S, Giosele S, Brunner AD, Esfandyari D, Papadopoulou AS, De Strooper B, Hubner N, Kumarswamy R, Thum T, Yin X, Mayr M, Laggerbauer B, Engelhardt S, Cardiac myocyte miR-29 promotes pathological remodeling of the heart by activating Wnt signaling, Nat Commun 8(1) (2017) 1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Cushing L, Kuang P, Lu J, The role of miR-29 in pulmonary fibrosis, Biochem Cell Biol 93(2) (2015) 109–18. [DOI] [PubMed] [Google Scholar]
- [179].Deng Z, He Y, Yang X, Shi H, Shi A, Lu L, He L, MicroRNA-29: A Crucial Player in Fibrotic Disease, Mol Diagn Ther 21(3) (2017) 285–294. [DOI] [PubMed] [Google Scholar]
- [180].Slove S, Lannoy M, Behmoaras J, Pezet M, Sloboda N, Lacolley P, Escoubet B, Bujan J, Jacob MP, Potassium channel openers increase aortic elastic fiber formation and reverse the genetically determined elastin deficit in the BN rat, Hypertension 62(4) (2013) 794–801. [DOI] [PubMed] [Google Scholar]
- [181].Coquand-Gandit M, Jacob MP, Fhayli W, Romero B, Georgieva M, Bouillot S, Esteve E, Andrieu JP, Brasseur S, Bouyon S, Garcia-Honduvilla N, Huber P, Bujan J, Atanasova M, Faury G, Chronic Treatment with Minoxidil Induces Elastic Fiber Neosynthesis and Functional Improvement in the Aorta of Aged Mice, Rejuvenation Res 20(3) (2017) 218–230. [DOI] [PubMed] [Google Scholar]
- [182].Tsoporis J, Keeley FW, Lee RM, Leenen FH, Arterial vasodilation and vascular connective tissue changes in spontaneously hypertensive rats, J Cardiovasc Pharmacol 31(6) (1998) 960–2. [DOI] [PubMed] [Google Scholar]
- [183].Hayashi A, Suzuki T, Wachi H, Tajima S, Nishikawa T, Murad S, Pinnell SR, Minoxidil stimulates elastin expression in aortic smooth muscle cells, Arch Biochem Biophys 315(1) (1994) 137–41. [DOI] [PubMed] [Google Scholar]
- [184].Lannoy M, Slove S, Louedec L, Choqueux C, Journe C, Michel JB, Jacob MP, Inhibition of ERK1/2 phosphorylation: a new strategy to stimulate elastogenesis in the aorta, Hypertension 64(2) (2014) 423–30. [DOI] [PubMed] [Google Scholar]
- [185].Tokimitsu I, Tajima S, Inhibition of elastin synthesis by high potassium salt is mediated by Ca2+ influx in cultured smooth muscle cells in vitro: reciprocal effects of K+ on elastin and collagen synthesis, Journal of biochemistry 115(3) (1994) 536–9. [DOI] [PubMed] [Google Scholar]
- [186].Quast U, Guillon JM, Cavero I, Cellular pharmacology of potassium channel openers in vascular smooth muscle, Cardiovasc Res 28(6) (1994) 805–10. [DOI] [PubMed] [Google Scholar]
- [187].Wagenseil JE, Ciliberto CH, Knutsen RH, Levy MA, Kovacs A, Mecham RP, The importance of elastin to aortic development in mice, Am J Physiol Heart Circ Physiol 299(2) (2010) H257–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, Eichwald E, Keating MT, Elastin is an essential determinant of arterial morphogenesis, Nature 393(6682) (1998) 276–80. [DOI] [PubMed] [Google Scholar]
- [189].Li W, Li Q, Qin L, Ali R, Qyang Y, Tassabehji M, Pober BR, Sessa WC, Giordano FJ, Tellides G, Rapamycin inhibits smooth muscle cell proliferation and obstructive arteriopathy attributable to elastin deficiency, Arterioscler Thromb Vasc Biol 33(5) (2013) 1028–35. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.