

Abstract
While lung cancer is known to be caused by environmental factors, it has also been shown to have genetic components, and the genetic etiology of lung cancer remains understudied. We previously identified a lung cancer risk locus on 6q23-25 using microsatellite data in families with a history of lung cancer. To further elucidate that signal, we performed targeted sequencing on 9 of our most strongly linked families. Two-point linkage analysis of the sequencing data revealed that the signal was heterogeneous and that different families likely had different risk variants. Three specific haplotypes were shared by some of the families: 6q25.3-26 in families 42 and 44, 6q25.2-25.3 in families 47 and 59, and 6q24.2-25.1 in families 30, 33, and 35. Region-based LOD scores and expression data identified the likely candidate genes for each haplotype overlap: ARID1B at 6q25.3, MAP3K4 at 6q26, and UTRN (6q24.1) and PHACTR2 (6q24.2). Further annotation was used to zero in on potential risk variants in those genes. All four genes are good candidate genes for lung cancer risk, having been linked to either lung cancer specifically or other cancers. However, this is the first time any of these genes has been implicated in germline risk. Functional analysis of these four genes is planned for future work.
Keywords: lung cancer, genetic linkage, family studies, targeted sequencing, 6q
Introduction
The United States is projected to have 228,820 lung cancer cases and 135,720 lung cancer deaths in 2020 (1). Lung cancer is affected by a variety of environmental factors. Tobacco smoke is the most common high-risk exposure (2); occupational hazards such as mining, shipbuilding, radon and asbestos exposure (3) also contribute. Smoking has been found to cause somatic mutations and loss of heterozygosity in oncogenes and tumor suppressors (4-6). Lung cancer risk is directly correlated to the number and duration of tobacco products used (2). Smoking is responsible for 85-90% of lung cancer incidence (7) in European-descent populations. However, about 10-25% of lung cancers occur in nonsmokers (8) and cannot be explained by other environmental factors. Distressingly, the number of lung cancer cases in nonsmokers may be increasing despite stricter laws against smoking (9).
Genetic factors have been shown to increase lung cancer risk. Tokuhata and Lilienfeld found that nonsmoking relatives of smoking lung cancer cases had higher susceptibility risk than nonsmoking relatives of smoking controls (10,11). Multiple studies confirmed a higher risk of lung cancer for individuals with an affected family member even after adjusting for smoking (12-16). Subsequent segregation analyses found the data matched codominant Mendelian inheritance of a rare autosomal genetic risk variant in a subset of families, acting in concert with smoking/polygenic risk factors (12,17-19).
Many recent lung cancer genetic studies have been genome-wide association studies (GWAS). GWAS are population-based and are best equipped to identify association with common, low penetrance variants with a small/moderate effect on lung cancer risk, i.e., the genetic variants that contribute to the polygenic risk of lung cancer. One notable risk locus found by GWAS was 15q25, which contains the neuronal acetylcholine receptor cluster subunits (CHRNA3, CHRNA5, and CHRNB4) (4-6). Multiple GWAS have recently discovered additional genetic loci involved in polygenic risk for lung cancer (20-22).
Family-based linkage studies are designed to find rare and highly penetrant loci that greatly affect disease risk. They are used to search for the autosomal dominant high penetrance major gene component predicted by the segregation analyses described above (12,17-19). Because only a small subset of families with multiple affected individuals are expected to be segregating such a high-penetrance genetic risk variant, linkage study designs concentrate on ascertaining families with a large number of affected individuals, particularly when some of these lung cancer patients exhibit early age at onset of cancer. These types of families are more likely to be segregating the high penetrance risk variants and ascertaining them for the study will increase power to detect the risk variant(s).
The Genetic Epidemiology of Lung Cancer Consortium (GELCC) has recruited patients and their relatives with a strong history of lung cancer from across the US, obtaining archival blood/tissue samples and information such as cancer status and age-at-diagnosis. Initial microsatellite studies using 52 GELCC families identified a familial lung cancer risk locus at 6q23-25 (23). The GELCC collected an additional 93 high risk lung cancer families for an updated study in 2010 and the same region on 6q was found to be genome-wide significant (24). These previous studies used multiallelic microsatellite genotypes, which are sparsely distributed across the genome. The analysis consisted of multipoint linkage, which uses these sparse genotypes to detect long linked haplotypes in a particular region. These regions generally span many potential candidate genes; our selected region on 6q contained over 260 genes. Thus, multipoint linkage is not usually able to identify the causal variant(s) that may be the source of the linkage peak. However, once dense sequence data became available for this region it is possible to use two-point linkage and bioinformatic annotation data to detect the most probable causal variant(s) in each family. In this study, we performed targeted sequencing on the linked 6q region with the 9 of the most highly linked families from the two previous studies to elucidate potential causal genes and variants.
Materials and Methods
Sequencing and Quality Control
Targeted sequencing was performed at the NIH Intramural Sequencing Center (NISC) on 75 individuals from our 9 families most strongly linked to the 6q region from the previous study (23). Illumina technology with a custom Agilent kit was used to capture a 37 Mb region on 6q ranging from 130 Mb to 167 Mb. All participants provided written informed consent. This study was conducted in accordance with the guidelines and tenets of the Declaration of Helsinki. This study was approved by the institutional review boards of the National Human Genome Research Institute, the University of Cincinnati, Karmanos Cancer Institute, Johns Hopkins University, the University of Toledo, and the Mayo Clinic. This study adhered to the tenets of the Declaration of Helsinki and was approved by the institutional review boards of the participating institutions. Quality control was performed using SNP & Variation Suite v7 (Golden Helix, Inc., Bozeman, MT, www.goldenhelix.com) following the best practices of the Genome Analysis Tool Kit (GATK) (25).
An additional 74 unsequenced individuals were included in the dataset to reflect the proper familial relationships needed to connect sections of each pedigree. These were individuals who were reported by family history and medical records but were either unwilling/unable to participate in the study. All unsequenced individuals were coded as having unknown genotypes. They were coded as having an affected phenotype if they were known to have been affected with lung cancer or unknown phenotype otherwise.
Sib-pair (https://genepi.qimr.edu.au/staff/davidD/Sib-pair/Documents/sib-pair.html) was then used to check for Mendelian errors (MEs). Variants with a ME in a single pedigree were removed from that pedigree, while variants with a ME in more than one pedigree were removed from all pedigrees. Sib-pair was also used to calculate allele frequencies for each variant.
The final dataset consisted of 131,760 variants from 149 individuals (75 sequenced subjects and 74 unsequenced subjects needed for pedigree connections) from 9 families. There were 48 affecteds (24 sequenced affecteds); all affected individuals are histologically known to be adenocarcinoma non-small cell lung cancer (NSCLC) affected individuals (with one exception and analyses using this individual as an unknown did not significantly change the results. Detailed information about the samples is presented in Supplementary Table S1.
Parametric Linkage Analyses
We assumed an autosomal dominant inheritance model with a disease allele frequency of 1% and penetrance values of 80% for carriers and 1% for non-carriers. The disease allele frequency and penetrances were chosen in accordance with segregation analyses predicting a risk variant of low frequency and high penetrance (12,17-19). The 1% risk for non-carriers in this model allows for the occurrence of affected family members who do not carry a high-risk susceptibility allele and are affected with lung cancer solely due to their environmental/polygenic risk factors.
Affected individuals were coded as affected while both unaffected individuals and unknown individuals were coded as unknown/missing. This design allowed for the high degree of uncertainty in the relationship between smoking status, age and lung cancer risk, since a young unaffected heavy smoker or an elderly unaffected non-smoker both could have a fairly high risk of being a non-penetrant carrier of the disease allele according to the published segregation analysis models. It also jointly allowed for smoking status to influence the analysis (approximately 95% of the affecteds were smokers), while the small phenocopy rate accounted for heavy smokers affected solely by environmental and polygenic risk factors.
Parametric two-point linkage analysis between each marker and the disease phenotype was performed using TwoPointLods (http://www-genepi.med.utah.edu/~alun/software/). The evidence in favor of linkage at each location was expressed in terms of logarithm of the odds in favor of linkage (LOD scores). When combining data across families we used heterogeneity LOD (HLOD) scores.
We also performed collapsed haplotype pattern (CHP) based linkage analysis. This method is performed by the software SEQLinkage (26) creates short regional haplotypes that correspond to specific genes and intergenic regions (as determined by RefSEQ). The gene-based haplotypes essentially function as multiallelic pseudo-markers (similar to microsatellites) in the two-point linkage analysis. Two-point linkage analysis was performed on the CHP haplotypes using MERLIN (27). This method was primarily used to confirm that the signal identified in the 2004 study using microsatellites was still present. This method did result in the recovery of the initial signal, with 25 total variants with a HLOD score > 3.00 and 114 variants with a HLOD > 2.5 (Supplementary Figure 1). So we were able to recover the initial signal from the 2004 study, but the fact that the signals were spread throughout the entire targeted region meant this method was little help in establishing any causal variants beyond our 2004 microsatellite study.
Annotation and Expression Data
We annotated all variants using wANNOVAR (28), which provided genic location and function and compiled prediction scores by SIFT, PolyPhen and other prediction algorithms. RegulomeDB (29) was used to evaluate regulatory potential for noncoding variants. We used the Genotype-Tissue Expression (GTEx) Project to look at lung tissue expression for candidate genes. GTEx (dbGaP Accession phs000424.v8.p2) is a comprehensive atlas and open database that has expression data from healthy individuals, including 515 lung cancer tissue donors. We used expression within lung tissue as a prioritization factor in deciding which genes would be the best candidate genes.
Region-based LOD Scores
We anticipated the potential for considerable heterogeneity for risk loci across the 9 families, whether from different variants in different genes or different variants within the same gene. The variant-based approach used above would be underpowered in this scenario. Consider the example where three families have rare, high-risk variants within the same gene, but each high-risk variant is at a different position. Under the variant-based approach described above, the high-risk variant in family 1 would be summed with the other families, which under this example all exhibit the homozygous reference allele and thus are uninformative for linkage. This is repeated for families 2 and 3 and the result is three risk variants within the same gene that are underpowered with respect to HLOD score; there is no ability to combine the evidence in favor of linkage across the families within this gene.
To account for this scenario, we developed a region-based LOD score approach. This approach assigned all variants to a given genomic region, which consisted of either a gene proper (including UTRs) or an intergenic region. For each family, the variant with the highest absolute value was taken to represent the region for the family. The chosen regional variants were then added across all families to obtain a cumulative LOD score for the region. Thus, if more than one family shows linkage to a region or gene, the evidence in favor of linkage will be cumulative across the linked families.
Results
Two-point Linkage Analysis across Families
Linkage analysis across the 9 families yielded no genome-wide significant results and 20 genome-wide suggestive results, all of which were noncoding (Supplementary Figure S2A) We use the Lander and Kruglyak suggested threshold of (H)LOD ≥ 3.3 for genome-wide significant and (H)LOD ≥ 1.9 for genome-wide suggestive (30). The highest HLOD score was located at rs9478640, an intergenic variant between TFB1M and NOX3 at 6q25.3. Overall, 16 of the 20 suggestive variants were located at 6q25.2-25.3. The HLOD and LOD scores of every family for all variants can be found in Supplementary Table S2A.
Two-point Linkage Analysis within Families
In complex diseases like lung cancer there is likely locus heterogeneity within these families; different genes and rare variants may be driving the signal at this region in different families. All the families (except family 102) exhibited long strings of linked variants that are likely due to long linked haplotypes across the region. Family 102 exhibited a series of peaks across the targeted region clustered in 6q25.3. The highest LOD score was rs9480313 in the intergenic region between NOX3 and ARID1B (Supplementary Figure S2B).
All other families displayed long, linked haplotypes across at least part of the targeted region. This is expected for family-based linkage, since there are only a small number of meioses that can break up founder haplotypes within each family. These long haplotypes complicate the procedure of finding potential causal variants, since these haplotypes contain many variants with similar LOD scores. We filtered all variants to a MAF < 0.01, since prior segregation and linkage analyses give strong evidence that the predicted high-penetrance risk variants in these families are individually rare. We used a less stringent threshold of MAF < 0.03 for coding variants to ensure that an excellent coding candidate was not missed. When describing the results below, we refer only to the filtered, rare variants (Supplementary Table S2B).
The haplotype in family 12 was uninformative, as it stretched across the entire targeted region (Supplementary Figure S2C). The other seven families showed strings of linked markers, indicating putative haplotypes that only stretched across parts of the targeted 6q region; these haplotypes overlapped between certain families at certain loci. Figure 1 shows the highest linked haplotype for each of the seven families, and the overlap regions. Three main regions of overlap were identified: 6q25.3-26 (approximately 160,523,000 – 161,537,000 bp) in families 42 and 44, 6q25.2-25.3 (approximately 155,720,000 – 158,279,000 bp) in families 47 and 59, and 6q24.2-25.1 (approximately 144,046,000 – 150,507,000 bp) in families 30, 33, and 35.
Figure 1:

Overlapping haplotypes across 6q region. The top linked haplotypes for the seven families displaying long linked haplotypes across the region with each color representing a different family. The color key is family 30 = blue, family 33 = pink, family 35 = orange, family 42 = black, family 44 = green, family 47 = red, family 59 = gray.
Families 42 and 44 had small haplotypes across 6q25.3-26. The highest scoring overall variant in family 42 was the intronic variant rs140997681 in MAP3K4 at 6q26 (LOD = 0.5351 (Figure 2A). Family 44 had higher magnitude LOD scores, with the highest overall score (LOD = 1.294) in a nonsynonymous exonic variant (rs41267809) in LPA at 6q26 (Figure 2B). This SNV is predicted damaging by PolyPhen2 and MutationTaster and is moderately rare (MAF = 0.023) in gnomAD NFE. Family 42 did not have any top LOD scores in LPA while family 44 had a high LOD score (1.287) in MAP3K4 at the intronic SNV rs62435519.
Figure 2:
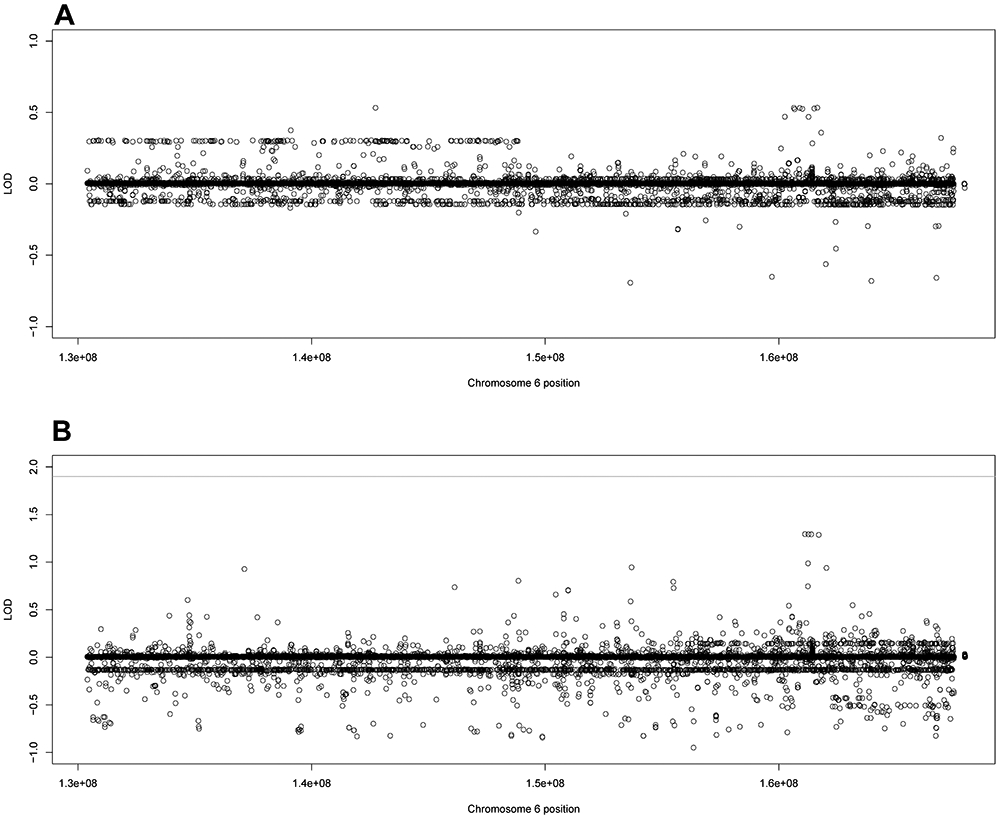
Family specific LOD scores using filtered variants for families 42 and 44. The family specific LOD score for A) family 42 and B) family 44 using the filtered variant set (noncoding variants MAF < 0.01, coding variants MAF < 0.03). The line represents genome-wide suggestive, defined as LOD ≥ 1.9 by Lander and Kruglyak.
Families 47 and 59 shared an overlapping haplotype at 6q25.2-6q25.3. All the variants along the haplotype for both families were intronic or intergenic. Both families had high LOD scores in ARID1B at 6q25.3. In family 47, the second and third overall scoring variants were the intronic variants rs150018283 (LOD = 0.8132) and rs187390535 (LOD = 0.8131) in ARID1B (Figure 3A) and in family 59, the first and second overall scoring variants were the intronic rs180708725 (LOD = 0.7659) and rs185173861 (LOD = 0.7655) in ARID1B (Figure 3B).
Figure 3:
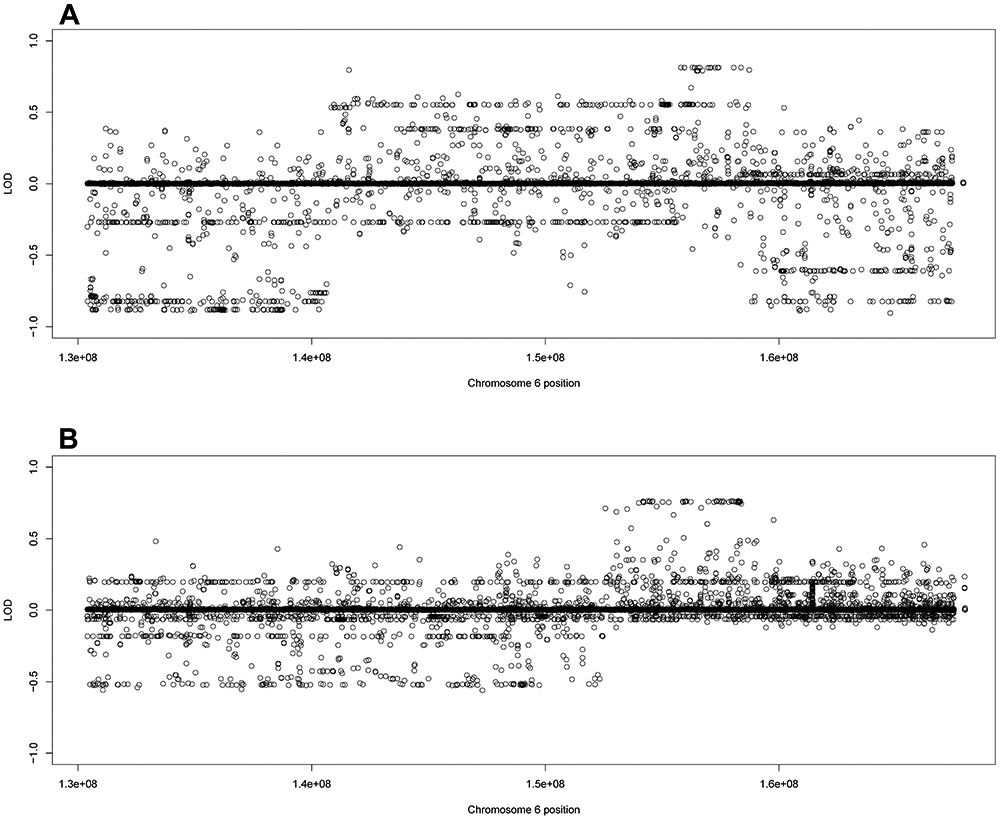
Family specific LOD scores using filtered variants for families 47 and 59. The family specific LOD score for A) family 47 and B) family 59 using the filtered variant set (noncoding variants MAF < 0.01, coding variants MAF < 0.03)
Families 30, 33, and 35 all had long linked haplotypes: 6q24.1-25.1 for family 30, 6q23.3-6q25.3 for family 33, and 6q24.2-6q25.2 for family 35 (Figure 4A-4C). There is significant overlap between all three families at 6q24.2-25.1. All the variants along these haplotypes were noncoding. All three families had (different) variants with top overall LOD score in the genes UTRN and PHACTR2.
Figure 4:
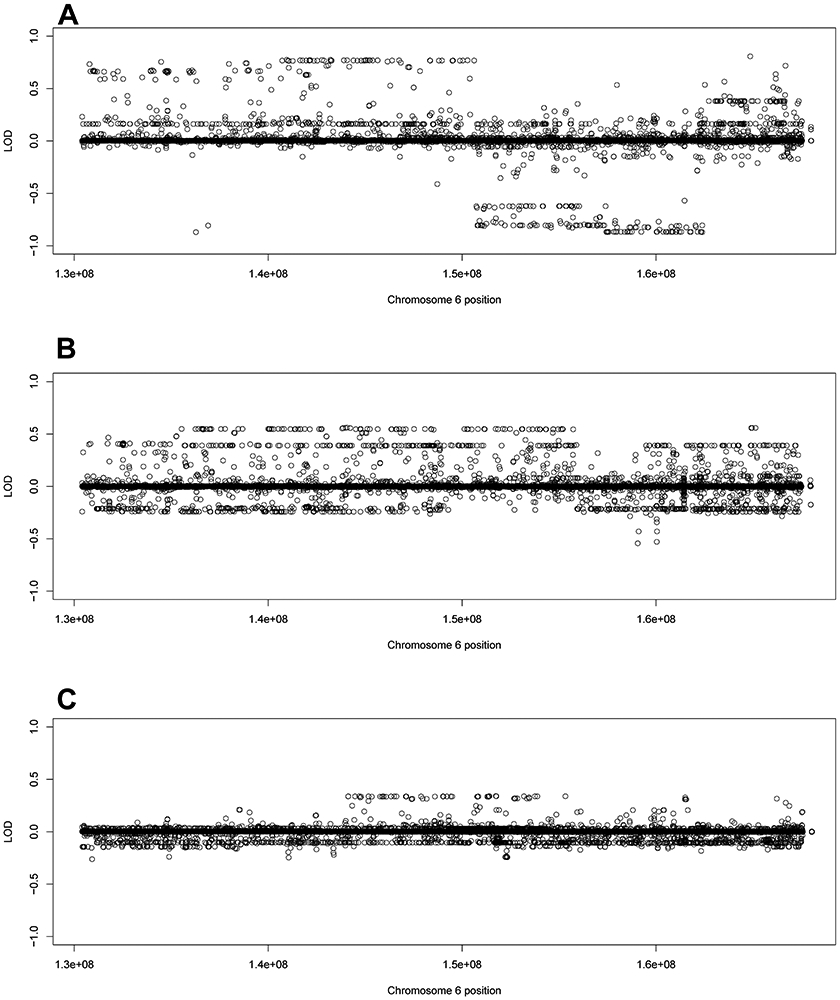
Family specific LOD scores using filtered variants for families 30, 33, and 35. The family specific LOD score for A) family 30, B) family 33, and C) family 35 using the filtered variant set (noncoding variants MAF < 0.01, coding variants MAF < 0.03)
Region-based LOD Scores
In many of the overlapping haplotypes, families shared top LOD scores in the same genes or intergenic regions, just not at the same variant. Working under the hypothesis that different mutations within the same region might cause disease risk by the same or similar functional effect, we performed region-based grouping of the LOD scores using the filtered variants. Cumulative LOD scores were calculated across the families that had overlapping putative haplotypes: families 30, 33, and 35, families 42 and 44, and families 47 and 59. The highest LOD scores for each linked putative haplotype can be found in Supplementary Table S3A, while the LOD scores for all regions can be found in Supplementary Tables S3B-S3D.
The first set of overlapping haplotypes consisted of families 30, 33, and 35 (Figure 5A). PHACTR2 and UTRN had the highest cumulative LOD scores across these three families. (LOD = 1.671 and 1.659 respectively). Three other regions were within 0.02 LOD of the top score, the gene GRM1 (6q24.3), the intergenic region between SAMD5 and SASH1 (6q24.3) and the intergenic region between UTRN and EPM2A (6q24.2).
Figure 5:
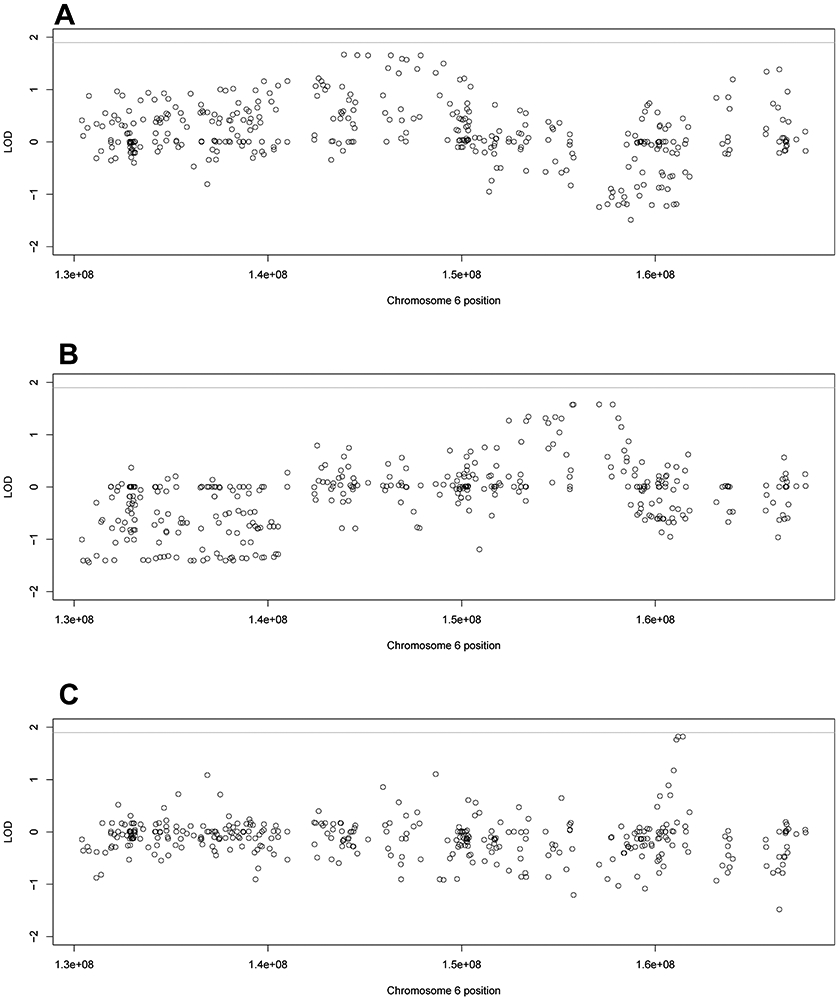
Region-based LOD scores of the three linked haplotypes. The region-based LOD scores for the A) linked haplotype shared by families 30, 33, and 35, B) linked haplotype shared by families 47 and 59, and C) linked haplotype shared by families 42 and 44. The line at 1.9 represents the genome-wide suggestive significance threshold.
Family 47 and 59 also had an overlapping haplotype at 6q25.2-6q25.3 (Figure 5B). There were four regions all within 0.006 of the top LOD score of 1.579; all located clustered together in 6q25.3. The top four regions consisted of three genes – ARID1B, ZDHHC14, and NOX3, and the intergenic region between NOX3 and ARID1B. In all four cases, the LOD scores were split almost equally between the two families, with family 47 having a LOD score of approximately 0.81 and family 59 having a LOD score of approximately 0.76.
The final haplotype consisted of a small region on 6q25.3-26 in families 42 and 44 (Figure 5C). The top two regions were within 0.0001 of each other with LODs of about 1.82; these top regions were the MAP3K4 gene and the intergenic region between PLG and MAP3K4 both at 6q26. The LOD scores in the top regions were higher in family 44 than 42; however, both families had their top overall LOD scores in these regions.
Candidate Gene Expression Data
The region-based analysis provided a total of twelve regions – seven genes and five intergenic regions. We further prioritized the genes by looking at their expression in lung tissue using GTEx. All genes showed at least some baseline expression in the lung except for NOX3 (Table 1). UTRN had the highest level of expression in lung tissue with a median of 32.3 transcripts per million (TPM) followed by MAP3K4 with a median of 17.63 TPM. Every gene had higher expression in tissues other than lung, though three genes – ARID1B, UTRN, and PHACTR2 had ratios greater than 0.5 when compared to the highest tissue value.
Table 1:
Candidate Gene Expression Data
| Gene | Median TPM in Lung | Highest Median TPM | Tissue | Ratio |
|---|---|---|---|---|
| UTRN | 32.30 | 55.35 | Tibial Nerve | 0.58 |
| PHACTR2 | 14.95 | 23.19 | Adrenal Gland | 0.65 |
| GRM1 | 0.06 | 38.35 | Brain | 0.002 |
| NOX3 | 0.00 | 0.05 | Testis | 0.00 |
| ARID1B | 8.95 | 15.67 | Ovary | 0.57 |
| ZDHHC14 | 7.45 | 24.74 | Uterus | 0.30 |
| MAP3K4 | 17.63 | 36.64 | Uterus | 0.48 |
| LPA | 0.02 | 14.98 | Liver | 0.001 |
Legend: The gene expression data from GTEx from the seven candidate genes on the linked haplotypes. Here, the headers represent: Gene = gene, Median TPM in Lung = the median expression level of gene transcripts in lung tissue, measured in transcripts per million (TPM), Highest Median TPM = the highest median expression level of gene transcripts in any tissue, measured in transcripts per million (TPM), Tissue = the tissue location of the highest median TPM for the gene, Ratio = the ratio in median TPM between the highest tissue and lung tissue.
Potential Causal Variants
All the variants with top LOD scores in the candidate genes described above were intronic, with the notable exception of the nonsynonymous exonic rs41267809 in LPA in Family 44. This makes it difficult to determine which variants might be causal. We used the criterion of potentiality for being a transcription factor (TF) binding site via RegulomeDB as a factor to zero in on candidate causal variants. Further, since all variants were rare (MAF < 0.01), the minor allele for these variants was only present in one family; the variants were informative in just one family. Due to the rarity of these SNPs, none of these variants are in LD with any other variant and it is difficult to calculate the founder haplotypes in these families because most of the founders within the main pedigrees are unsequenced. We present our best estimates for potential causal variants along each linked haplotype for each family in Table 2, based on LOD scores, potentiality for TF binding, MAF, and cosegregation of the minor allele with the phenotype.
Table 2:
Candidate Variants for Causality in Candidate Genes
| ID | POS | Family | LOD | Gene | FREQ1 | FREQ2 | TF Binding |
Potential TF Protein |
|---|---|---|---|---|---|---|---|---|
| rs564982701 | 157406633 | 59 | 0.7561 | ARID1B | 0.0168 | 0.0011 | 0.93 | ATF5 CCTCTTCCTTA |
| rs187390535 | 157172983 | 47 | 0.8131 | ARID1B | 0.0143 | 0.0077 | 0.87 | BARHL2 TAAACG |
| rs150018283 | 157322342 | 47 | 0.8132 | ARID1B | 0.0142 | 0.0015 | 1.0 | IRF7 ACTTTCGCTTTCG TRIM63 AGTTTCACTTT |
| rs62435519 | 161536849 | 44 | 1.2865 | MAP3K4 | 0.0164 | 0.0061 | 0.61 | N/A |
| rs140997681 | 161489813 | 42 | 0.5351 | MAP3K4 | 0.0156 | 0.0015 | 0.24 | GABPA ATGACTCAGCA |
| rs186871831 | 144973790 | 30 | 0.7701 | UTRN | 0.0151 | 0.0066 | 0.70 | N/A |
| rs532363235 | 144709986 | 33 | 0.5484 | UTRN | 0.0149 | 0.0023 | 0.59 | N/A |
| rs191491353 | 144670343 | 35 | 0.3277 | UTRN | 0.0171 | 0.0086 | 0.39 | NFKB1 GGGGATTCCCT, NFKB2 GGGGAATCCCC, REL GGGTTTCC |
| rs966382235 | 144001704 | 30 | 0.7693 | PHACTR2 | 0.0153 | 0.0003 | 0.61 | N/A |
| rs79313503 | 143998579 | 33 | 0.5629 | PHACTR2 | 0.0154 | 0.0030 | 0.61 | N/A |
| rs553447284 | 144051910 | 35 | 0.3388 | PHACTR2 | 0.0142 | 0.0002 | 0.58 | N/A |
Legend: The best candidate causal variants for each family in the best candidate gene along the linked haplotype. All variants are intronic. Here, the headers represent: ID = rs ID of the variant, POS = the physical position in base pairs, Family = Family that the minor allele of the variant appears in, LOD = LOD score of the variant from the family in column 3, Gene = the genic location of the variant, FREQ1 = the minor allele frequency of the variant in the founders in all families in our dataset, FREQ2 = the minor allele frequency of the variant in gnomAD NFE, TF binding = the transcription factor binding probability score of the variant as calculated by RegulomeDB, with 1.0 being the highest score and corresponding to a 100% chance of the variant being a TF binding site, Potential TF Protein = Protein that is known to bind to the motif that the SNP is found in (according to RegulomeDB). The binding motif of the protein is provided below the protein name (with the SNP allele bolded).
There were three candidate genes along the linked haplotype at 6q25.3 for families 47 and 59: ARID1B, ZDHHC14, and NOX3. NOX3 is not expressed in the lungs. ARID1B was more highly expressed in lung tissue and has higher LOD scores than ZDHHC14. We identified potential causal variants in ARID1B for both families. In family 47, rs150018283 has a 100% probability of being a TF binding site. It also has the highest LOD score in the gene (0.8132) for this family and the second highest overall LOD score for filtered variants overall, only 0.007 behind the top LOD score. It is extremely rare in non-Finnish Europeans (NFE) according to gnomAD, with a MAF of 0.0015. RegulomeDB predicts this variant to be part of two known binding motifs for IRF7 (antisense strand) and TRIM63 (sense strand). IRF7 is strongly expressed in lung tissue. rs187390535 is also an excellent candidate variant for this family based on a high LOD score (0.8131). It is slightly more common than rs150018283, with a MAF of 0.0077 in gnomAD NFE and has about an 87% chance of being a TF binding site. It is predicted to be in a binding motif of BARHL2, which is not expressed in lung tissue.
For family 59, the best candidate causal variant is rs564982701. It has about a 93% chance of being a TF binding site and has a MAF of 0.0011. It is within 0.0098 of the top LOD score overall and within the gene. It is predicted to be in a binding motif of ATF5, which is expressed in lung tissue.
For all three variants, the minor allele only appears in a single family and nowhere else in the dataset. Family 47 is three-generation pedigree with five affecteds (four sequenced) (Supplementary Figure S3A). The minor alleles of rs150018283 and rs187390535 appear six times – in the four affected individuals and two unknown individuals. One unknown is only 40 and could still develop lung cancer. Further, the one unsequenced affected individual becomes an obligate carrier of both minor alleles based on the genotypes of his wife and son. This means all affecteds carry the minor alleles of rs150018283 and rs187390535.
Family 59 is a four-generation pedigree with four affecteds (two sequenced) (Supplementary Figure S3B). rs564982701 shows good segregation in the family; the minor allele appears in the two sequenced affecteds and two young unknowns (51 and 46 years old). The two unsequenced affecteds also become obligate carriers of the minor allele based on the genotypes of their relatives.
The two candidate genes along the 6q26 haplotype comprising families 42 and 44 were MAP3K4 and LPA. MAP3K4 is probably the better candidate since it has the higher regional LOD scores and is much more highly expressed in lung tissue. There are a few interesting potential candidate genes in MAP3K4, including rs62435519 for family 44. This variant had a LOD score of 1.287, one of the highest overall LOD scores in both the gene and overall for this family. There is a 61% chance the variant is a TF binding site and its MAF is 0.0061 in gnomAD NFE.
rs140997681 is the best candidate variant for family 42; it is the highest LOD score in both the gene and overall in the family (for filtered variants). There is only a 24% chance that the variant is part of a TF binding site; known to be part of a motif for GABPA, a TF known to be expressed in lung tissue. rs140997681 has a MAF of 0.0015 in gnomAD NFE.
The minor alleles of both variants appear only in their respective families and nowhere else in the dataset. Family 42 is a four-generation pedigree with six affecteds (two sequenced) (Supplementary Figure S4A). The rs140997681 minor allele appears four times, in the two sequenced affecteds and two unknowns (one who is only 30 years old). Two of the four unsequenced affecteds become obligate carriers based on relatives’ genotypes.
Family 44 is a four-generation family with seven affecteds (four sequenced) (Supplementary Figure S4B). The minor allele of rs62435519 appears in all four sequenced cases and one unknown and two of the three unsequenced affecteds are obligate carriers of the rare allele.
There were three candidate genes at the 6q24.2 haplotype for families 30, 33, and 35: UTRN, PHACTR2, and GRM1. GRM1 is only expressed at very low levels in the lung. Both UTRN and PHACTR2 are expressed in the lung and are both good candidates. In family 30, the best candidate variants are rs186871831 (UTRN) and rs966382235 (PHACTR2). Both variants have the highest LOD score in the gene and one of the highest LOD scores overall. Both variants have a high probability of being a TF being site, rs186871831 (70%) and rs966382235 (61%) and both are rare in NFE.
The best candidate variants for family 33 are rs532363235 (UTRN) and rs79313503 (PHACTR2). Both are predicted to be TF binding sites - rs532363235 (59% probability) and rs79313503 (61% probability). Both variants are rare in Europeans (MAF > 0.003). rs79313503 in PHACTR2 has the highest overall LOD score (0.5629) in the family and rs532363235 in UTRN is close behind at 0.5484.
For family 35, the best candidate variants are rs191491353 (UTRN) and rs553447284 (PHACTR2). Both variants are within 0.02 of the top LOD score in the family and a MAF < 0.0087 in NFE. rs553447284 has a 58% probability of being a TF binding site while rs191491353 has a 39% chance. rs191491353 is predicted to encompass multiple TF binding motifs: NFKB1, NFKB1, and REL. NFKB1 and REL are expressed in the lung more than any other tissue (excluding EBV-transformed lymphocytes), NFKB2 is also strongly expressed in lung tissue.
The candidate variants show good segregation within the families. Family 30 is three-generation pedigree with four affecteds (two sequenced) (Supplementary Figure S5A). The minor alleles of rs186871831 and rs966382235 appear in the same individuals – two sequenced affecteds and three unknown individuals. The two unsequenced affecteds are obligate carriers of the rare variants.
Family 33 is a three-generation family with five affecteds (one sequenced) (Supplementary Figure S5B). The minor alleles of rs532363235 and rs79313503 appear in the sequenced affected and either five (rs532363235) or four (rs79313503) unknowns. The unknowns are young in age (33, 38, 43, and 47) and three of the four unsequenced affecteds are obligate carriers of the rare alleles.
Family 35 is three-generation family with five affecteds (two sequenced) (Supplementary Figure S5C). The minor alleles of rs191491353 and rs553447284 appear in the two sequenced affecteds and two non-affecteds (Supplementary Figure S5C). Two of the three unsequenced affecteds are obligate carriers of the minor allele.
Discussion
This study used targeted sequencing to further examine a previous linkage peak for familial lung cancer that was identified at 6q23-25. Seven of the nine sequenced families exhibited linked sets of variants indicating linked haplotypes across different parts of the targeted region; three distinct haplotypes emerged at 6q25.3, 6q26, and 6q24.1-24.2. The apparent heterogeneity of this signal was surprising to us, as we had originally hypothesized a single, major gene affecting lung cancer risk in this 6q region in all 9 linked families. However, our findings show clear evidence of high-risk variants in different major genes segregating in different families (i.e., locus heterogeneity). In retrospect, finding multiple major genes in lieu of a single major gene should not have been all that surprising, given that there are multiple genes within this linked region that are a priori excellent candidates for lung cancer susceptibility, so this result is reasonable. Locus heterogeneity has been observed for most high-risk cancer syndromes, such as BRCA1 and BRCA2 for breast cancer. Thus, the only surprising aspect of our findings here was that these candidate lung cancer risk genes are all located close together in this 6q region.
We were able to identify potential candidate genes on each of the haplotypes: ARID1B at 6q25.3, MAP3K4 at 6q26, UTRN at 6q24.1, and PHACTR2 at 6q24.2. We further postulate potential causal variants within these genes, based on enrichment of very rare minor alleles co-segregating strongly with the lung cancer in the families and potentiality for TF binding sites.
Candidate Gene – ARID1B
ARID1B is the candidate gene for the 6q25.3 haplotype comprising families 47 and 59. The gene has the highest region-based LOD score, variants in this gene were among the top overall LOD score in both the families and it has been shown to be expressed in lung tissue. ARID1B encodes a component of the SWI/SNF chromatin remodeling complex which is often mutated in cancer cells and is believed to be a tumor suppressor (31). Decreases in expression of SWI/SNF have been observed in non-small cell lung cancer cells (32). ARID1B has been identified as a potential lung cancer risk gene in a microsatellite study of case/control exome data (33). It has also been shown to be mutated in lung carcinomas (34) and is mutated in other cancers, including pancreatic cancer (35) and neuroblastoma (36).
All variants along the ARID1B haplotype were noncoding, however potential causal variants were identified at rs150018283 and rs187390535 in family 47 and rs564982701 in family 59. These variants had among the highest LOD scores in the family and were all extremely rare in the general European population but highly enriched in affected individuals within the families. Further, these variants were strongly predicted to affect TF binding sites by RegulomeDB. rs150018283 is predicted to be in a binding motif for IRF7 (which is expressed strongly in lung tissue) that mediates pathways that promote bone metastasis in breast cancer (37). rs564982701 is in a possible binding motif for activation transcription factor 5 (ATF5), which expressed in lungs and is known to increase radiation resistance, malignancy, and invasiveness of lung carcinomas (38).
Candidate Gene – MAP3K4
The best candidate gene for the 6q26 haplotype in family 42 and 44 was MAP3K4. MAP3K4 had the highest region-based LOD score and is expressed in lung tissue. Germline truncation mutations in MAP3K4 are associated with both ovarian cancer (39) and Wilms tumor (40), a rare type of kidney cancer that primarily affects children. This is the first time that germline mutations in this gene have been implicated in lung cancer. Somatic mutations have been reported in endometrial tumors (41). MAP3K4 has been shown to affect the epithelial-mesenchymal transition in cancer cells (42). It is interesting to note that both ARID1B and MAP3K4 are associated with chromatin remodeling. Our best potential causal variants in MAP3K4 are rs62435519 (family 44) and rs140997681 (family 42). Both variants have among the highest LOD scores in their corresponding families. They are very rare in the general population and occur only in one family, where they show good co-segregation with affected individuals. Further, rs140997681 is predicted to be in a possible binding motif for GABPA, which has been shown to bind in coordination with a gain-of-function mutant p53 (43).
Candidate Genes – UTRN and PHACTR2
For the 6q24.1-24.2 haplotype in families 30, 33, and 35, there were two good candidate genes. The first was utrophin (UTRN). It had one of the highest region-based LOD scores in these families and was expressed in the lung. Somatic mutations of utrophin have been identified in multiple cancers, including breast cancer, neuroblastoma, and melanoma (44), as well as sporadic endocrine pancreatic tumors (45). RNAi knockdown of UTRN in glioma cells was found to inhibit cell proliferation (46). This is the first time any germline mutations in utrophin have been implicated in lung cancer. While all variants along the linked haplotype in the three families were noncoding variants; good potential causal variants are rs186871831 in family 30, rs532363235 in family 33, and rs191491353 in family 35. These variants were exceedingly rare in the general European population. In this dataset, they appeared only in the corresponding family and showed good segregation with affected individuals. rs186871831 and rs532363235 were predicted to be likely transcription factor binding sites.
The other candidate gene along the 6q24.1-24.2 haplotype was PHACTR2. PHACTR2 had the highest region-based LOD score along the haplotype and is expressed in the lung. It is differentially expressed in lung adenocarcinoma (47), and polymorphisms in the gene have been shown to affect DNA repair capacity, which is a risk factor for lung cancer (48). Long noncoding antisense RNA PHACTR2-AS1 has been shown to promote metastasis in breast cancer (49). Good potential causal variants were identified at rs966382235, rs79313503, and rs553447284. All three variants are rare in the general population but enriched in one family in the dataset, were predicted to be TF binding sites, and showed good segregation with the affected individuals in the family.
Conclusions
We note that there is a lack of power within each of these individual families. Many families had a lack of sequenced affecteds, since many affecteds had aggressive lung cancer and died before any sample could be obtained; we genotyped spouses, parents, and children to reconstruct genotypes. Some of these families have lower information content in this sequencing study than in the original published linkage studies because adequate DNA samples were not available for some affected individuals who were genotyped in the original study. All these factors cause this study to be slightly underpowered. Further, many of the founders of these families are grandparents or great grandparents of the probands who died long before this study began, meaning we have many families with unsequenced founders. This, coupled with the fact we were focusing on rare variants, makes calculation of LD blocks around the potential causal variants difficult. We emphasize that in the case of potential causal variants listed here, they should be treated with some caution. We zeroed in on these variants in each family by looking for enrichment of very rare variants in affected individuals that had high LOD scores. We also used RegulomeDB to predict if any variants might be in TF binding sites. These variants represent our best estimates at causality given our data and the annotation data, but functional analysis is needed to confirm these results.
We also note that our initial 2004 study found an overall genome-wide significant LOD score of 4.26, while none of the LOD scores from this work reached a LOD score of 3.00. This is not surprising given the differences between those two studies. This study had a much higher genomic resolution than that of the 2004 study. Thus, the genome-wide significant signal observed in the 2004 study has not disappeared, rather it has been fragmented into smaller signals that the 2004 study was unable to differentiate. The earlier study used microsatellite markers that covered a wide swath of a chromosome (e.g., there were only 18 microsatellites covering the entirety of chromosome 6). Further, the highest LOD scores were found using long linked haplotypes that spanned three microsatellites across an 18 cM (≈12,216,600 basepair) region. Heterogeneous multipoint linkage within that large region would appear as a single, significant peak with a high LOD score, because the low resolution of the study simply cannot detect the different signals. By contrast, our targeted sequencing study contained thousands of variants within that same region. With that greater resolution, we are able to identify the multiple signals that created that large LOD score in the 2004 microsatellite. This fragmentation of the signal from one large peak to multiple smaller peaks means that no single gene in the region will reach the level of linkage evidence from the multipoint analysis in 2004.
One of the challenges in working with lung cancer is the relationship between lung cancer and smoking. As noted in the Methods, most of the affected individuals in these families were smokers. Our analyses were modeled as affected only, which allowed for the uncertainty of disease allele status in nonsmokers. We also modeled a small phenocopy rate, which controlled any individuals whose cancer status might be caused solely by smoking. This study investigated the targeted sequence of a known familial lung cancer linkage peak on 6q in nine families that were highly aggregated for familial lung cancer. We identified three discrete linked sets of rare variants in putative haplotypes across the region at 6q26, 6q25.3, and 6q24.1-24.2. The best candidate genes at the haplotypes were ARID1B (6q25.3), MAP3K4 (6q26), UTRN (6q24.1) and PHACTR2 (6q24.2). We identified some potential causal variants within these genes. All these variants were rare and enriched in affected individuals and many have a high probability of being transcription factor binding sites. We stress that these variants are speculative for causality at this time and functional assays are needed to confirm any biological significance. These further functional studies are indeed planned on these candidate genes and variants to fully determine each gene’s specific effect on lung cancer.
Supplementary Material
Statement of Significance.
This study identifies four genes associated with lung cancer risk, which could help guide future lung cancer prevention and treatment approaches.
Acknowledgments:
The authors thank all study participants and their families. This work was funded in part by the National Institutes of Health, National Cancer Institute grants U01CA76293 (S.M. Pinney and M.W. Anderson), U01CA243483 (C.I. Amos and S.M. Pinney), P50CA125123 (C.I. Amos), P30CA22453 (A.G. Schwartz), R03CA77118 (P. Yang), R01CA80127 (P. Yang), R01CA84354 (P. Yang), National Institutes of Health, National Institute of Environmental Health Sciences P30ES006096 (S.M. Pinney), and Department of Health and Human Services contracts HHSN26820100007C (D. Mandal), HHSN268201700012C (D. Mandal), and HHSN75N92020C00001 (D. Mandal). C.I. Amos is a Research Scholar of the Cancer Prevention Research Institute of Texas (CPRIT). This research was partially supported by CPRIT grant RR170048. J.E. Bailey-Wilson, A.M. Musolf, B.A. Moiz, C.D. Middlebrooks, and C.L. Simpson were funded in part by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health. P. Yang and M. de Andrade were funded in part by the Mayo Foundation Fund. This work was funded in part by the National Lung Cancer Partnership (now Free to Breathe) Young Investigator Research Grant to C.L. Simpson. The GTEx Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used in this study was obtained from the GTEx portal on April 17, 2020. This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov).
Footnotes
Conflicts of Interest: The authors declare no potential conflicts of interest
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA: a cancer journal for clinicians; 2020;70(1):7–30. [DOI] [PubMed] [Google Scholar]
- 2.Doll R, Peto R, Wheatley K, Gray R, Sutherland I. Mortality in relation to smoking: 40 years' observations on male British doctors. Bmj 1994;309(6959):901–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morgan WK, Seaton A. Occupational lung diseases. Philadelphia: W.B. Saunders; 1984. [Google Scholar]
- 4.Amos CI, Wu X, Broderick P, Gorlov IP, Gu J, Eisen T, et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nature genetics 2008;40(5):616–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hung RJ, McKay JD, Gaborieau V, Boffetta P, Hashibe M, Zaridze D, et al. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature 2008;452(7187):633–7. [DOI] [PubMed] [Google Scholar]
- 6.Thorgeirsson TE, Geller F, Sulem P, Rafnar T, Wiste A, Magnusson KP, et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature 2008;452(7187):638–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flanders WD, Lally CA, Zhu BP, Henley SJ, Thun MJ. Lung cancer mortality in relation to age, duration of smoking, and daily cigarette consumption: results from Cancer Prevention Study II. Cancer research 2003;63(19):6556–62. [PubMed] [Google Scholar]
- 8.Mitchell P, Mok T, Barraclough H, Strizek A, Lew R, van Kooten M. Smoking history as a predictive factor of treatment response in advanced non-small-cell lung cancer: a systematic review. Clinical lung cancer 2012;13(4):239–51. [DOI] [PubMed] [Google Scholar]
- 9.Jenks S Is Lung Cancer Incidence Increasing in Never-Smokers? Journal of the National Cancer Institute 2016;108(1). [DOI] [PubMed] [Google Scholar]
- 10.Tokuhata GK, Lilienfeld AM. Familial aggregation of lung cancer in humans. Journal of the National Cancer Institute 1963;30:289–312. [PubMed] [Google Scholar]
- 11.Tokuhata GK, Lilienfeld AM. Familial aggregation of lung cancer among hospital patients. Public health reports 1963;78:277–83. [PMC free article] [PubMed] [Google Scholar]
- 12.Ooi WL, Elston RC, Chen VW, Bailey-Wilson JE, Rothschild H. Increased familial risk for lung cancer. Journal of the National Cancer Institute 1986;76(2):217–22. [PubMed] [Google Scholar]
- 13.Cannon-Albright LA, Thomas A, Goldgar DE, Gholami K, Rowe K, Jacobsen M, et al. Familiality of cancer in Utah. Cancer research 1994;54(9):2378–85. [PubMed] [Google Scholar]
- 14.Goldgar DE, Easton DF, Cannon-Albright LA, Skolnick MH. Systematic population-based assessment of cancer risk in first-degree relatives of cancer probands. Journal of the National Cancer Institute 1994;86(21):1600–8. [DOI] [PubMed] [Google Scholar]
- 15.Etzel CJ, Amos CI, Spitz MR. Risk for smoking-related cancer among relatives of lung cancer patients. Cancer research 2003;63(23):8531–5. [PubMed] [Google Scholar]
- 16.Cote ML, Kardia SL, Wenzlaff AS, Ruckdeschel JC, Schwartz AG. Risk of lung cancer among white and black relatives of individuals with early-onset lung cancer. Jama 2005;293(24):3036–42. [DOI] [PubMed] [Google Scholar]
- 17.Bailey-Wilson JE, Sellers TA, Elston RC, Evens CC, Rothschild H. Evidence for a major gene effect in early-onset lung cancer. The Journal of the Louisiana State Medical Society : official organ of the Louisiana State Medical Society 1993;145(4):157–62. [PubMed] [Google Scholar]
- 18.Sellers TA, Bailey-Wilson JE, Elston RC, Wilson AF, Elston GZ, Ooi WL, et al. Evidence for mendelian inheritance in the pathogenesis of lung cancer. Journal of the National Cancer Institute 1990;82(15):1272–9. [DOI] [PubMed] [Google Scholar]
- 19.Sellers TA, Bailey-Wilson JE, Potter JD, Rich SS, Rothschild H, Elston RC. Effect of cohort differences in smoking prevalence on models of lung cancer susceptibility. Genetic epidemiology 1992;9(4):261–71. [DOI] [PubMed] [Google Scholar]
- 20.Byun J, Schwartz AG, Lusk C, Wenzlaff AS, de Andrade M, Mandal D, et al. Genome-wide association study of familial lung cancer. Carcinogenesis 2018;39(9):1135–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKay JD, Hung RJ, Han Y, Zong X, Carreras-Torres R, Christiani DC, et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nature genetics 2017;49(7):1126–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bosse Y, Amos CI. A Decade of GWAS Results in Lung Cancer. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology 2018;27(4):363–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bailey-Wilson JE, Amos CI, Pinney SM, Petersen GM, de Andrade M, Wiest JS, et al. A major lung cancer susceptibility locus maps to chromosome 6q23-25. American journal of human genetics 2004;75(3):460–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amos CI, Pinney SM, Li Y, Kupert E, Lee J, de Andrade MA, et al. A susceptibility locus on chromosome 6q greatly increases lung cancer risk among light and never smokers. Cancer research 2010;70(6):2359–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research 2010;20(9):1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang GT, Zhang D, Li B, Dai H, Leal SM. Collapsed haplotype pattern method for linkage analysis of next-generation sequence data. European journal of human genetics : EJHG 2015;23(12):1739–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nature genetics 2002;30(1):97–101. [DOI] [PubMed] [Google Scholar]
- 28.Chang X, Wang K. wANNOVAR: annotating genetic variants for personal genomes via the web. Journal of medical genetics 2012;49(7):433–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome research 2012;22(9):1790–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lander E, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nature genetics 1995;11(3):241–7. [DOI] [PubMed] [Google Scholar]
- 31.Yoshimoto T, Matsubara D, Nakano T, Tamura T, Endo S, Sugiyama Y, et al. Frequent loss of the expression of multiple subunits of the SWI/SNF complex in large cell carcinoma and pleomorphic carcinoma of the lung. Pathology international 2015;65(11):595–602. [DOI] [PubMed] [Google Scholar]
- 32.Naito T, Udagawa H, Umemura S, Sakai T, Zenke Y, Kirita K, et al. Non-small cell lung cancer with loss of expression of the SWI/SNF complex is associated with aggressive clinicopathological features, PD-L1-positive status, and high tumor mutation burden. Lung cancer 2019;138:35–42. [DOI] [PubMed] [Google Scholar]
- 33.Velmurugan KR, Varghese RT, Fonville NC, Garner HR. High-depth, high-accuracy microsatellite genotyping enables precision lung cancer risk classification. Oncogene 2017;36(46):6383–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang HT, Chen SM, Pan LB, Yao J, Ma HT. Loss of function of SWI/SNF chromatin remodeling genes leads to genome instability of human lung cancer. Oncology reports 2015;33(1):283–91. [DOI] [PubMed] [Google Scholar]
- 35.Yang C, Wang Y, Xu W, Liu Z, Zhou S, Zhang M, et al. Genome-wide association study using diversity outcross mice identified candidate genes of pancreatic cancer. Genomics 2019;111(6):1882–88. [DOI] [PubMed] [Google Scholar]
- 36.Lee SH, Kim JS, Zheng S, Huse JT, Bae JS, Lee JW, et al. ARID1B alterations identify aggressive tumors in neuroblastoma. Oncotarget 2017;8(28):45943–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bidwell BN, Slaney CY, Withana NP, Forster S, Cao Y, Loi S, et al. Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nature medicine 2012;18(8):1224–31. [DOI] [PubMed] [Google Scholar]
- 38.Ishihara S, Yasuda M, Ishizu A, Ishikawa M, Shirato H, Haga H. Activating transcription factor 5 enhances radioresistance and malignancy in cancer cells. Oncotarget 2015;6(7):4602–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanchi KL, Johnson KJ, Lu C, McLellan MD, Leiserson MD, Wendl MC, et al. Integrated analysis of germline and somatic variants in ovarian cancer. Nature communications 2014;5:3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gadd S, Huff V, Walz AL, Ooms A, Armstrong AE, Gerhard DS, et al. A Children's Oncology Group and TARGET initiative exploring the genetic landscape of Wilms tumor. Nature genetics 2017;49(10):1487–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Le Gallo M, O'Hara AJ, Rudd ML, Urick ME, Hansen NF, O'Neil NJ, et al. Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nature genetics 2012;44(12):1310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mobley RJ, Raghu D, Duke LD, Abell-Hart K, Zawistowski JS, Lutz K, et al. MAP3K4 Controls the Chromatin Modifier HDAC6 during Trophoblast Stem Cell Epithelial-to-Mesenchymal Transition. Cell reports 2017;18(10):2387–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vaughan CA, Deb SP, Deb S, Windle B. Preferred binding of gain-of-function mutant p53 to bidirectional promoters with coordinated binding of ETS1 and GABPA to multiple binding sites. Oncotarget 2014;5(2):417–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Y, Huang J, Zhao YL, He J, Wang W, Davies KE, et al. UTRN on chromosome 6q24 is mutated in multiple tumors. Oncogene 2007;26(42):6220–8. [DOI] [PubMed] [Google Scholar]
- 45.Barghorn A, Speel EJ, Farspour B, Saremaslani P, Schmid S, Perren A, et al. Putative tumor suppressor loci at 6q22 and 6q23-q24 are involved in the malignant progression of sporadic endocrine pancreatic tumors. The American journal of pathology 2001;158(6):1903–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shen SH, Yu N, Xu H, Liu XY, Tan GW, Wang ZX. Inhibition of Human Glioma Cell Proliferation Caused by Knockdown of Utrophin Using a Lentivirus-Mediated System. Cancer biotherapy & radiopharmaceuticals 2016;31(4):133–8. [DOI] [PubMed] [Google Scholar]
- 47.Li J, Li Z, Zhao S, Song Y, Si L, Wang X. Identification key genes, key miRNAs and key transcription factors of lung adenocarcinoma. Journal of thoracic disease 2020;12(5):1917–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang LE, Gorlova OY, Ying J, Qiao Y, Weng SF, Lee AT, et al. Genome-wide association study reveals novel genetic determinants of DNA repair capacity in lung cancer. Cancer research 2013;73(1):256–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chu W, Zhang X, Qi L, Fu Y, Wang P, Zhao W, et al. The EZH2-PHACTR2-AS1-Ribosome Axis induces Genomic Instability and Promotes Growth and Metastasis in Breast Cancer. Cancer research 2020;80(13):2737–50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.