

Abstract
Cell‐free methylated DNA immunoprecipitation and high‐throughput sequencing (cfMeDIP‐seq) is a new bisulfite‐free technique, which can detect the whole‐genome methylation of blood cell‐free DNA (cfDNA). Using this technique, we identified differentially methylated regions (DMR) of cfDNA between lung tumors and normal controls. Based on the top 300 DMR, we built a random forest prediction model, which was able to distinguish malignant lung tumors from normal controls with high sensitivity and specificity of 91.0% and 93.3% (AUROC curve of 0.963). In summary, we reported a non–invasive prediction model that had good ability to distinguish malignant pulmonary nodules.
Keywords: cfDNA methylation, cfMeDIP‐seq, lung cancer, machine learning, pulmonary nodule
This study reported the whole‐genome methylation profiling of plasma cfDNA of patients with pulmonary nodules by cfMeDIP‐seq. A random forest model was used to distinguish pulmonary malignant tumors from normal controls, based on the whole‐genome methylation profiling.

Abbreviations
- cfDNA
cell free DNA
- cfMeDIP‐seq
cell‐free methylated DNA immunoprecipitation and high‐throughput sequencing
- DMR
differentially methylated regions
- LDCT
low‐dose CT
- NGS
next‐generation sequencing
1. INTRODUCTION
Early detection of lung cancer has significant potential for reducing the mortality and improving the survival of patients. Low‐dose CT (LDCT) screening has been widely used in the early detection of lung cancer, especially for those aged >55 years and smokers.1 However, due to the high sensitivity but low specificity of LDCT screening, many non–tumorous pulmonary nodules are also detected. Although CT imaging can estimate the malignant risk of nodules based on the size, lobar location, density, and margin characteristics, approximately 20% of the nodules are misdiagnosed as malignant by CT screening. Several pulmonary nodules are still difficult to distinguish as benign or malignant using CT imaging, especially the ground‐glass nodules that are approximately 1 cm in diameter.2, 3 Therefore, there is an unmet need for new diagnostic options with high sensitivity and specificity to distinguish malignant tumors from benign pulmonary nodules, preferably through non–invasive procedures.
Cell‐free methylated DNA immunoprecipitation and high‐throughput sequencing (cfMeDIP‐seq) is a new bisulfite‐free technique, which can detect the whole‐genome methylation of blood cell‐free DNA (cfDNA). Compared with bisulfite sequencing, this technique can enrich genome‐wide CpG methylated cfDNA, with advantages of low input DNA (<10 ng) and cost efficiency.4, 5 This technique has been used to classify renal cell carcinomas and gliomas in cancer patients and healthy controls with high sensitivity and specificity.6, 7 In this study, based on cfMeDIP‐seq methylation profiling, we built a prediction model to effectively differentiate malignant pulmonary nodules.
2. MATERIALS AND METHODS
2.1. Patients
This study included healthy individuals without pulmonary nodules (n = 7) and patients with lung cancers (tumor size >3 cm, n = 32). Patients with malignant pulmonary nodules were screened as positive for pulmonary nodules (nodule size <3 cm, n = 35) by CT scan. They subsequently underwent surgical resection and were diagnosed histologically as having lung cancer. There were two categories of patients among those with benign pulmonary nodules (n = 23). Some patients were positive for pulmonary nodules, which were diagnosed as benign nodules by CT scan. The other patients were positive for pulmonary nodules based on CT scans; they subsequently underwent surgical resection but were diagnosed histologically as having benign lesions. All selected patients were from the Second Affiliated Hospital of Anhui Medical University (Hefei, Anhui, China) and Shanxi Cancer Hospital (Taiyuan, Shanxi, China). All patients provided written informed consent, and the study was approved by the institutional review board (YX2020‐019) of the Ethics Committee of Anhui Medical University in accordance with all relevant ethical regulations.
2.2. cfMeDIP‐seq and machine learning
The plasma sample collection, cfDNA isolation, library construction and sequencing of cfMeDIP‐seq, peak calling of sequencing data, detection of DMR, and establishment of the random forest prediction model were performed as described in the supplementary materials and methods.
3. RESULTS AND DISCUSSION
We collected plasma cfDNA samples of 30 normal controls (n = 7 healthy individuals without pulmonary nodules and n = 23 benign pulmonary nodules) and 67 lung cancer patients (n = 35 malignant pulmonary nodules with nodule size <3 cm and n = 32 lung cancers with tumor size >3 cm). The clinical information of the healthy controls and patients is shown in Table S1. The patients with malignant pulmonary nodules (6.93 [5.02, 8.96] ng/mL) and lung cancer (6.64 [5.18, 9.24] ng/mL) had significantly higher cfDNA levels than those in patients with benign pulmonary nodules (4.85 [3.55, 7.74] ng/mL) and healthy individuals (3.53 [3.27, 4.43] ng/mL) (Figure 1A). The cfDNA samples were all performed by cfMeDIP‐seq (Figure 1B). After removing the PCR duplication, average sequence reads of 42.3 million (approximately 6G data/sample) were obtained from all samples, 87.6% of which was mapped to the human reference genome. The average number of peaks was 74 335/sample (Table S2). To characterize methylation signatures specific to lung cancer, we compared differentially methylated regions (DMR) between normal and tumor samples. The top 300 significant DMR were identified by limma‐trend test statistic (Figure 1C). The 300 DMR were located in the distal intergenic (43.80%), intron (37.21%), promoter (12.02%), exon (3.49%), 5ʹ UTR (0.78%), 3ʹ UTR (2.33%), and downstream (0.39%) regions (Figure S1). In the top 300 DMR, a total of 49 DMR were associated with the promoter, exon, 5ʹ UTR, 3ʹ UTR or downstream region of 42 genes (Table S3). Using the top 300 DMR, visualization using principal component plots showed clear separation of normal controls and lung cancer patients (Figure 1D).
FIGURE 1.
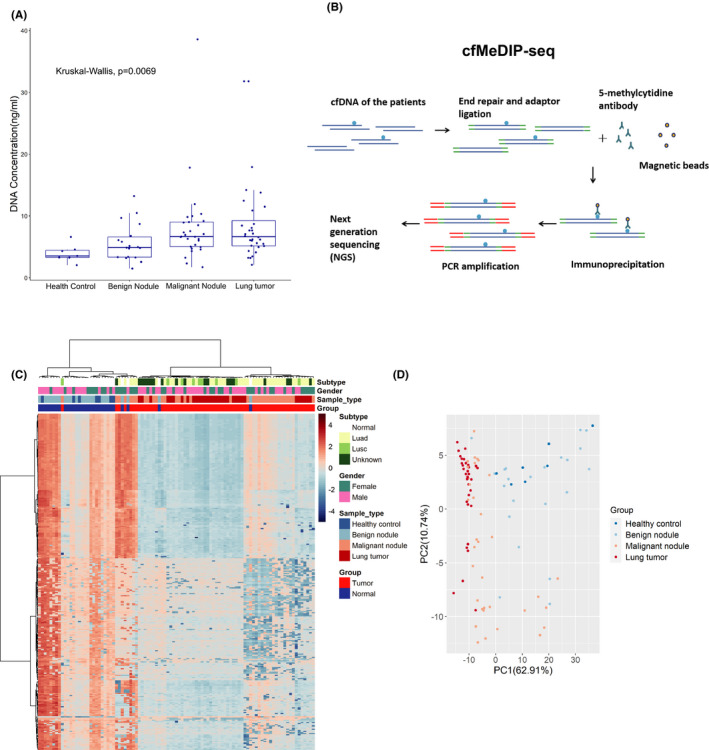
Cell‐free methylated DNA immunoprecipitation and high‐throughput sequencing (cfMeDIP‐seq) identified the differentially methylated regions (DMR) between lung tumors and normal controls. A, Cell‐free DNA (cfDNA) concentrations of patients from different groups. Box plots are displayed with a median center line; box range from the 25th to 75th percentile. B, Workflow of the cfMeDIP‐seq. cfDNA is ligated with sequencing adaptors. Methylated cfDNA fragments are immunoprecipitated with the 5‐methylcytidine antibody, followed by PCR amplification and next‐generation sequencing (NGS). C, Heatmap of the top 300 DMR identified in the plasma cfDNA between 30 normal controls and 67 lung tumor patients. Luad, lung adenocarcinoma; Lusc, lung squamous cell carcinoma. D, The top 300 DMR were used to generate the principal component (PC) plot to classify lung tumors and normal controls
We carried out a cross‐validation to evaluate the ability of cfMeDIP profiles in the prediction of normal and tumor samples. The 30 normal controls and 67 lung tumor patients were randomly split into 80% of controls and cases for a training set and 20% of controls and cases for a test set. Using the training‐set samples, we selected the top 300 DMR to classify control and case samples. Based on the top 300 DMR, we built a random forest model that was used to assign a malignant risk score to the test samples. This process was repeated 100 times (Figure 2A). Across the 100 training‐test sets, at a malignant risk threshold (0.65), all lung cancers (tumor size >3 cm, n = 32) and healthy controls (without pulmonary nodules, n = 7) were predicted as tumors and normal samples, respectively; 29 of 35 malignant pulmonary nodules were predicted as tumors, and 21 of 23 benign pulmonary nodules were predicted as normal samples (Figure 2B). Using the prediction model, we observed the 91.0% sensitivity and 93.3% specificity to distinguish pulmonary malignant tumors from normal controls with a mean AUROC of 0.963 (Figure 2C). Furthermore, as shown in Figure S2, we calculated the mean feature importance of the frequently emerged DMR (emerged more than 50 times in the 100 sets). In the frequently emerged DMR, a total of 34 DMR were associated with the promoter, exon, 5ʹ UTR, 3ʹ UTR or downstream region of 33 genes (Table S4). The 34 DMR were all overlapped with the 49 DMR in Table S3, which suggested their important contributions in distinguishing normal and lung tumor samples.
FIGURE 2.
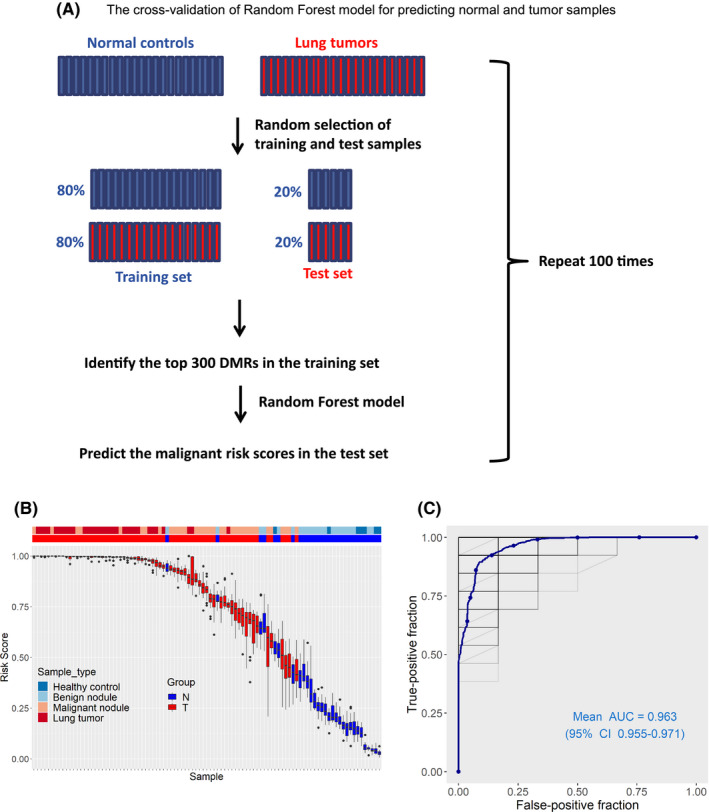
The cross‐validation to evaluate the ability of cell‐free methylated DNA immunoprecipitation and high‐throughput sequencing (cfMeDIP‐seq) profiles in the prediction of normal and tumor samples. A, Workflow of the cross‐validation. The tumor and normal samples were partitioned into 100 independent training and test sets in an 80%‐20% split. Based on the top 300 DMR identified in the training set, a random forest model was used to predict a malignant risk score for the test samples. B, Box plots of the predicted risk scores of individual plasma samples of normal controls (N) and lung tumor patients (T) from 100 randomly selected training‐test sets. Box plots are displayed with a median center line; box range from the 25th to 75th percentile. C, AUROC curve for the cross‐validation generated from 100 randomly selected training‐test sets to evaluate the predictive ability of the model in distinguishing malignant pulmonary nodules from normal controls
Next, we tested the utility of this prediction model for new samples. The pulmonary nodules of 3 patients were suspected of being malignant based on CT scans. Plasma cfDNA samples were analyzed by cfMeDIP‐seq, and then the prediction model was used to predict the benign and malignant nature of pulmonary nodules. The pulmonary nodules were all predicted to be malignant by the random forest model. Finally, all 3 patients underwent surgeries, and these pulmonary nodules were pathologically confirmed to be lung adenocarcinomas (Figure 3).
FIGURE 3.
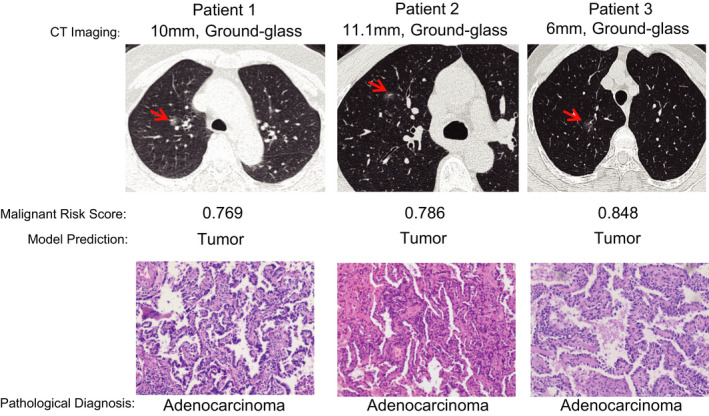
The utility of this random forest prediction model in three independent patients with pulmonary nodules. The CT images, median malignant risk scores, predicted results, and pathological diagnoses are shown
The use of bisulfite modification for analysis of the whole‐genome methylation is limited for plasma cfDNA due to its low abundance. The studies on cfDNA methylation detection for early diagnosis of lung cancer have been restricted to targeted sequencing and locus‐specific PCR with bisulfite‐converted cfDNA.8 Using high‐throughput bisulfite DNA methylation targeted sequencing, Liang et al9 identified nine cancer‐specific methylation markers for differentiating patients with malignant pulmonary nodules, with the sensitivity and specificity of 79.5% and 85.2%. Using bisulfite‐converted methylation‐specific PCR, Chen et al10 demonstrated that a three‐gene combination (CDO1, SOX17, and HOXA7) had sensitivity and specificity of 90% and 71% in distinguishing malignant from benign pulmonary nodules. However, cfMeDIP‐seq also has limitations in its application. cfMeDIP‐seq relies on the methylated DNA fragments (counts of reads), which are immunoprecipitated by the antibody. The batch effect can influence the outcome. The batch effect may derive from different antibody vendor production lots, cfDNA abundance, or NGS sequenced depth.5 In our study, the bioinformatics approach was used to normalize methylation values between batches.
In this study, using a low‐input and bisulfite‐free sequencing method, we reported the first whole‐genome methylation profile of plasma cfDNA of patients with pulmonary nodules and demonstrated accurate classification of lung cancer and normal control. After performing larger prospective validation, this method could transform clinical management by enabling early detection of lung cancer.
DISCLOSURE
JN, HW, and BH are board members of Casgenome Medicine (Hefei). The other authors have no financial/commercial conflicts of interest.
Supporting information
Table S1
Table S2
Table S3
Table S4
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the 100‐Talent Program of the Chinese Academy of Sciences, National Natural Science Foundation of China (Grant Number: 81872438), the Key Program of 13th 5‐year plan of CASHIPS, the Key Research and Development Project of Anhui Province (no. 201904a07020064), the Research Fund of Beijing Cancer Research Institute (no. CAPTRALung2020004), and the open fund of the Key Laboratory of Medical Physics and Technology of Anhui Province (no. LMPT201902).
Qi J, Hong B, Tao R, et al. Prediction model for malignant pulmonary nodules based on cfMeDIP‐seq and machine learning. Cancer Sci. 2021;112:3918–3923. 10.1111/cas.15052
Jian Qi and Bo Hong are co‐first authors.
Contributor Information
Bo Hong, Email: bhong@hmfl.ac.cn.
Hongzhi Wang, Email: wanghz@hfcas.ac.cn.
Dahai Zhao, Email: zhaodahai@ahmu.edu.cn.
Jinfu Nie, Email: nie_jinfu@gibh.ac.cn.
REFERENCES
- 1.National Lung Screening Trial Research T , Aberle DR, Adams AM, et al. Reduced lung‐cancer mortality with low‐dose computed tomographic screening. N Engl J Med. 2011; 365: 395‐409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nasim F, Ost DE. Management of the solitary pulmonary nodule. Curr Opin Pulm Med. 2019;25:344‐353. [DOI] [PubMed] [Google Scholar]
- 3.Kramer BS, Berg CD, Aberle DR, Prorok PC. Lung cancer screening with low‐dose helical CT: results from the National Lung Screening Trial (NLST). J Med Screen. 2011;18:109‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shen SY, Singhania R, Fehringer G, et al. Sensitive tumour detection and classification using plasma cell‐free DNA methylomes. Nature. 2018;563:579‐583. [DOI] [PubMed] [Google Scholar]
- 5.Shen SY, Burgener JM, Bratman SV, De Carvalho DD. Preparation of cfMeDIP‐seq libraries for methylome profiling of plasma cell‐free DNA. Nat Protoc. 2019;14:2749‐2780. [DOI] [PubMed] [Google Scholar]
- 6.Nuzzo PV, Berchuck JE, Korthauer K, et al. Detection of renal cell carcinoma using plasma and urine cell‐free DNA methylomes. Nat Med. 2020;26:1041‐1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nassiri F, Chakravarthy A, Feng S, et al. Detection and discrimination of intracranial tumors using plasma cell‐free DNA methylomes. Nat Med. 2020;26:1044‐1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chabon JJ, Hamilton EG, Kurtz DM, et al. Integrating genomic features for non–invasive early lung cancer detection. Nature. 2020;580:245‐251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liang W, Zhao Y, Huang W, et al. Non–invasive diagnosis of early‐stage lung cancer using high‐throughput targeted DNA methylation sequencing of circulating tumor DNA (ctDNA). Theranostics. 2019;9:2056‐2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen C, Huang X, Yin W, et al. Ultrasensitive DNA hypermethylation detection using plasma for early detection of NSCLC: a study in Chinese patients with very small nodules. Clin Epigenetics. 2020;12:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Table S2
Table S3
Table S4
Supplementary Material