

Abstract
Patients with BRAF‐mutated colorectal cancer (CRC) have a poor prognosis despite recent therapeutic advances such as combination therapy with BRAF, MEK, and epidermal growth factor receptor (EGFR) inhibitors. To identify microRNAs (miRNAs) that can improve the efficacy of BRAF inhibitor dabrafenib (DAB) and MEK inhibitor trametinib (TRA), we screened 240 miRNAs in BRAF‐mutated CRC cells and identified five candidate miRNAs. Overexpression of miR‐193a‐3p, one of the five screened miRNAs, in CRC cells inhibited cell proliferation by inducing apoptosis. Reverse‐phase protein array analysis revealed that proteins with altered phosphorylation induced by miR‐193a‐3p were involved in several oncogenic pathways including MAPK‐related pathways. Furthermore, overexpression of miR‐193a‐3p in BRAF‐mutated cells enhanced the efficacy of DAB and TRA through inhibiting reactivation of MAPK signaling and inducing inhibition of Mcl1. Inhibition of Mcl1 by siRNA or by Mcl1 inhibitor increased the antiproliferative effect of combination therapy with DAB, TRA, and anti‐EGFR antibody cetuximab. Collectively, our study demonstrated the possibility that miR‐193a‐3p acts as a tumor suppressor through regulating multiple proteins involved in oncogenesis and affects cellular sensitivity to MAPK‐related pathway inhibitors such as BRAF inhibitors, MEK inhibitors, and/or anti‐EGFR antibodies. Addition of miR‐193a‐3p and/or modulation of proteins involved in the miR‐193a‐3p–mediated pathway, such as Mcl1, to EGFR/BRAF/MEK inhibition may be a potential therapeutic strategy against BRAF‐mutated CRC.
Keywords: BRAF, colorectal cancer, MAPK signaling, Mcl1, miR‐193a‐3p
We screened 240 miRNAs in BRAF‐mutated colorectal cancer cells and found that miR‐193a‐3p was involved in several oncogenic pathways including MAPK‐related pathways. In addition, we found that overexpression of miR‐193a‐3p in BRAF‐mutated cells enhanced the efficacy of a BRAF inhibitor and a MEK inhibitor through inhibiting reactivation of MAPK signaling and inducing inhibition of Mcl1. Addition of miR‐193a‐3p and/or modulation of proteins involved in the miR‐193a‐3p–mediated pathway, such as Mcl1, to EGFR/BRAF/MEK inhibition is a potential therapeutic strategy against BRAF‐mutated colorectal cancer.
1. INTRODUCTION
The serine/threonine protein kinase BRAF plays an important role in the activation of the RAS‐RAF‐MEK‐ERK signaling pathway.1, 2 Oncogenic BRAF mutations occur in various malignancies including malignant melanoma (50%), thyroid cancer (30%‐50%), and colorectal cancer (CRC, 10%).2, 3, 4 Over 90% of BRAF mutations in CRC are V600E BRAF V600E mutations, which lead to constitutive RAS‐independent upregulation of BRAF kinase activity and promote tumorigenesis.3
Several studies have shown that patients with BRAF‐mutated CRC have a poor prognosis and exhibit poor responsiveness to chemotherapy compared with those with BRAF wild‐type CRC.5, 6, 7, 8 BRAF inhibitor monotherapy, as a targeted therapy, is effective against BRAF‐mutated advanced melanoma.9, 10 In contrast, BRAF inhibitor monotherapy was not found to be effective in advanced CRC patients with a BRAF mutation.11 Resistance to BRAF inhibitors in BRAF‐mutated CRC was shown to be mainly due to reactivation of MAPK signaling; addition of epidermal growth factor receptor (EGFR) inhibition to BRAF inhibitors was found to suppress MAPK reactivation and increase antitumor activity in vitro and in vivo.12, 13 The combination of BRAF inhibitors and anti‐EGFR antibodies or MEK inhibitors has shown promising results in some phase II studies.14, 15, 16 More recently, in the phase III BEACON trial, triplet therapy with BRAF inhibitor encorafenib, MEK inhibitor binimetinib, and anti‐EGFR antibody cetuximab (Cmab) and doublet therapy with encorafenib and Cmab were found to be superior to standard chemotherapy with a combination of FOLFIRI or irinotecan with Cmab for BRAF‐mutated CRC.17 Despite the recent therapeutic advances, clinical outcomes of patients with BRAF‐mutated CRC still remain poor.5, 17 Therefore, development of more effective therapies for BRAF‐mutated CRC is a key imperative.
MicroRNAs (miRNAs), small noncoding RNAs of 19‐25 nucleotides, suppress gene expression by binding to complementary sequences in the 3′‐untranslated regions (UTRs) of the target mRNAs. miRNAs are involved in cancer growth, migration, and sensitivity to anticancer drugs.18, 19, 20 The potential application of miRNAs as diagnostic and prognostic biomarkers in the context of cancer is a current research hotspot.18, 19 Some studies have investigated the alterations of miRNA expressions in BRAF‐mutated CRC.21, 22, 23 We earlier identified 22 dysregulated miRNAs in BRAF‐mutated CRC using genome‐wide miRNA expression analysis.23 However, it is not clear whether miRNAs can affect the chemosensitivity of BRAF‐mutated CRC to the combination therapy of a BRAF inhibitor, a MEK inhibitor, and an anti‐EGFR antibody.
In the present study, we aimed to identify miRNAs that may potentially improve the therapeutic efficacy of BRAF inhibitor dabrafenib (DAB) and MEK inhibitor trametinib (TRA) through screening of miRNA mimics in RKO cells. Moreover, among the screened miRNAs, we identified a novel role of miR‐193a‐3p in oncogenesis and elucidated the previously unknown molecular mechanisms by which miR‐193a‐3p overexpression enhances the therapeutic efficacy of DAB and TRA (DAB/TRA) and DAB/TRA plus Cmab. We demonstrate that the enhanced sensitivity to DAB/TRA induced by miR‐193a‐3p overexpression is mediated at least in part by Mcl1 inhibition and that miR‐193a‐3p overexpression or addition of Mcl1 inhibitor to the DAB/TRA combination or the combination of DAB/TRA plus Cmab could be a novel therapeutic strategy for BRAF‐mutated CRC.
2. MATERIALS AND METHODS
2.1. Reagents
DAB, TRA, Cmab, and S63845 were purchased from LC Laboratories, Cayman Chemical, Merck Serono, and Selleck, respectively.
2.2. Cell lines and cell culture
The human CRC cell lines SW480, RKO, and SW1417 were purchased from the ATCC. COLO320 and LOVO were purchased from RIKEN. LIM1215 was purchased from the ECACC. HT‐29 was provided by Dr Mariadason at the Ludwig Institute for Cancer Research (Heidelberg, Australia). SW480 was cultured in RPMI‐1640 (Sigma‐Aldrich) with 10% FBS, whereas the other six cell lines were grown in DMEM (Sigma‐Aldrich) with 10% FBS.
2.3. Functional screening using miRNA mimic library
Screening of the 240 miRNAs from miR‐1‐3p to miR‐511‐5p included in the human miRIDIAN microRNA Mimic Library version 19 (Dharmacon) was performed. RKO cells were separately transfected with 25 nM of each miRNA mimic using DharmaFECT‐1 Transfection Reagent (Dharmacon) in 96‐well plates. We included miRIDIAN microRNA Mimic Transfection Control with Dy547 (Dharmacon) as a negative control of miRNA mimic (NC‐mimic). At 6 hours after transfection, cells were treated with DAB and TRA or dimethyl sulfoxide (DMSO) vehicle for 48 hours. Cell viability was assessed using Cell Counting Kit‐8 assay (Dojindo).
2.4. Transfection of miR‐193a‐3p mimic and siRNAs
MiRVana miRNA‐193a‐3p mimic (#4464066) and MiRVana miRNA mimic negative control (#4464058) were purchased from Thermo Fisher Scientific. siRNAs used in this study are listed in Table S1. Cells were transfected with 33‐40 nM of miR‐193a‐3p mimic and siRNAs using Lipofectamine 2000 (Thermo Fisher Scientific).
2.5. Cell viability assay
Cell viability was assessed using the Cell Counting Kit‐8 assay according to the manufacturer's protocol. Combination indices were calculated using the CompuSyn software version 1.0 (ComboSyn, Inc).
2.6. Cell cycle analysis
Cells were collected after the treatment, washed in PBS, and fixed overnight with 70% ethanol at 4℃. Cells were incubated with RNase and propidium iodide. The samples were analyzed by FC‐500 (Beckman Coulter).
2.7. Apoptosis assay
Cells were collected after the treatment, and apoptosis was detected using Annexin V‐FITC Apoptosis Detection Kit (Nacalai Tesque) according to the manufacturer's protocol. The measurement was performed by FC‐500.
2.8. Western blot
Western blot analyses were performed following the standard protocol.24 Primary antibodies used in this study are listed in Table S2. The band intensities were quantified by densitometry using ImageJ version 1.52a.25 The expression levels of each protein were normalized by those of internal proteins such as tubulin or β‐actin, as determined by densitometry.
2.9. Reverse‐phase protein array (RPPA) analysis
The Phospho Explorer Antibody Array (Full Moon Biosystems) contains 1318 antibodies. Cell lysates from LIM1215 cells transfected with miR‐193a‐3p mimic and NC‐mimic for 48 hours were used as experimental samples. Experiments were performed by Filgen, Inc. according to the manufacturer's protocol. Each of the antibodies had two replicates, and an average of duplicate signals was calculated as the signal value for each antibody. The signal value for each antibody was normalized by the median signal value for all antibodies. The phosphorylation rate was calculated by dividing the signal value for the phospho‐specific protein antibodies by the signal value for the corresponding total protein–specific antibody for each protein.
2.10. Pathway analysis
The Database for Annotation, Visualization, and Integrated Discovery (DAVID) tool was employed for pathway analyses using altered phosphorylation rates.26, 27 The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways that belong to Environmental Information Processing and Cellular Processes categories were selected in this pathway analysis. A signaling pathway was regarded as significantly changed when it met the screening criteria, P ≤ .1, false discovery rate (FDR) < 0.01, and included at least 10 proteins with fold change (FC) < 0.75 or FC > 1.33.
2.11. Luciferase reporter assay
The 3′‐UTR sequences containing the potential miR‐193a‐3p binding sites of KRAS, ATF2, and MYC genes were cloned into the XhoI and NotI restriction sites of the psiCHECK‐2 vector (Promega). The primer sequences are listed in Table S3. RKO cells were cotransfected with 40 ng of psiCHECK2 construct and 33 nM miR‐193a‐3p mimic or NC‐mimic using Lipofectamine 2000. After 48 hours of transfection, firefly and Renilla luciferase activities were measured by the Dual‐Luciferase Reporter Assay System (Promega). The relative luciferase activity is presented as the ratio of firefly to Renilla luciferase activity and normalized to NC‐mimic.
2.12. Statistical analysis
Statistical analyses were performed with JMP Pro version 14.0. All data except for screening using miRNA mimic library and RPPA analysis were obtained from three or more independent experiments. Results are presented as mean ± standard error of the mean (SEM). Between‐group differences were assessed using Student’s t‐test; P‐values < 0.05 were considered indicative of statistical significance.
3. RESULTS
3.1. Screening of miRNA mimics that serve as sensitizers to BRAF and MEK inhibitors
To screen miRNAs that enhance the sensitivity to DAB/TRA, 240 human miRNA mimics were transfected into the BRAF‐mutated CRC cell line RKO, and the cell viability was assessed using the cell viability assay (Figure 1A). The growth of cells transfected with miRNA mimics or NC‐mimic was measured 48 hours after DAB/TRA treatment. Treatment with DAB 100 nM and TRA 10 nM decreased the viability of RKO cells transfected with NC‐mimic by 30%. Each miRNA was classified as a DAB/TRA sensitizer if its transfection caused a decrease in cell viability by >50% with DAB/TRA treatment but a decrease in cell viability by <20% with DMSO treatment. As a result, five miRNAs were identified as DAB/TRA sensitizers (Figure 1B and Table S4). Out of these five miRNAs, miR‐193a‐3p was found to be downregulated in BRAF‐mutated CRCs in our previous study23; therefore, we focused on miR‐193a‐3p for the subsequent analyses.
FIGURE 1.
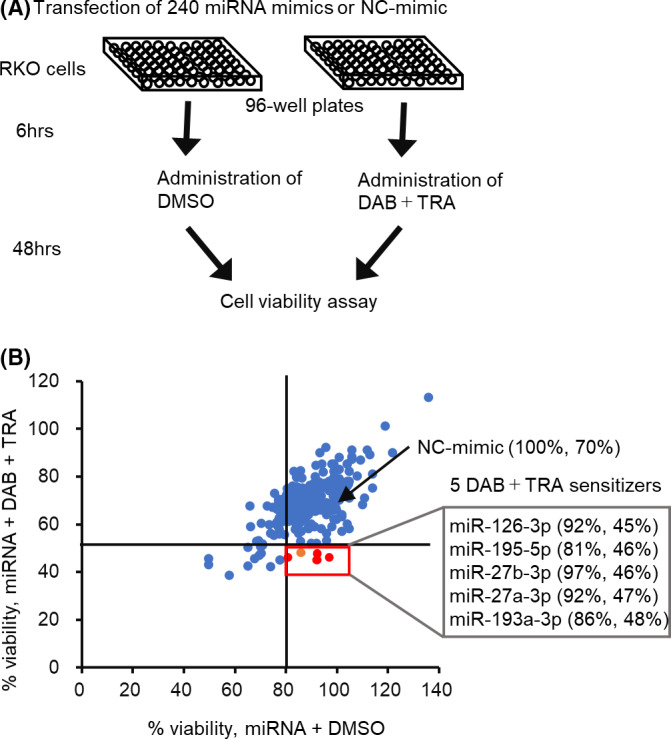
Screening of miRNAs that sensitize RKO cells to dabrafenib and trametinib (DAB/TRA) using miRNA mimic library. A, A schematic representation of the miRNA screening. B, Screening data plotted as percentage of cell viability after transfection of miRNAs or negative control of miRNA mimic (NC‐mimic), in combination with DAB/TRA (y‐axis) vs DMSO (x‐axis). RKO cells were transfected with miRNA mimic or NC‐mimic, and cell viability was determined 48 h after treatment with DAB 100 nM and TRA 10 nM. The percentage of viable cells was calculated by normalization to cells transfected with NC‐mimic and treated with DMSO. The five miRNAs are identified as DAB/TRA sensitizers
3.2. miR‐193a‐3p exerts tumor‐suppressive functions in CRC cell lines
miR‐193a‐3p has been shown to act as a tumor suppressor in various cancers including CRC28, 29, 30, 31, 32, 33; however, the precise molecular mechanisms of miR‐193a‐3p in CRC carcinogenesis are not fully elucidated. To identify the novel roles of miR‐193a‐3p in CRC, we first analyzed miR‐193a‐3p expression level and the effect of miR‐193a‐3p overexpression on CRC cell lines. The mean miR‐193a‐3p expression value tended to be lower in BRAF‐mutated CRC cell lines compared with BRAF wild‐type CRC cell lines (Figure S1). Next, for the further experiments, we used six of 12 cell lines whose miR‐193a‐3p expression level was lower than the mean of the total of 17 cell lines to determine whether miR‐193a‐3p overexpression affects cellular proliferation. miR‐193a‐3p overexpression significantly inhibited cell proliferation by 15‐60% compared with NC‐mimic overexpression in six CRC cell lines (Figure 2A). Although the inhibition rate in the various cell lines varied to some extent, cell growth was inhibited by miR‐193a‐3p overexpression regardless of the cellular genotype of KRAS/BRAF (Table S5). LIM1215 showed the most significant inhibitory effect of miR‐193a‐3p on cell proliferation by 62% ± 7% (Figure 2A). We therefore considered LIM1215 to be a suitable model to characterize the functional role of miR‐193a‐3p in the subsequent analyses. To investigate the mechanism of growth inhibition by miR‐193a‐3p, the effect of this miRNA on cell cycle or induction of apoptosis was analyzed by flow cytometry. miR‐193a‐3p overexpression resulted in a significant increase of subG1 population and decrease of G2/M population compared with NC‐mimic overexpression in LIM1215 and LOVO cells. The G1 and S fractions were slightly decreased by miR‐193a‐3p overexpression in LOVO cells, whereas both fractions were not significantly changed in LIM1215 cells (Figures 2B and S2). In addition, apoptotic cells were significantly increased by miR‐193a‐3p overexpression compared with NC‐mimic overexpression (Figure 2C). These results indicate that miR‐193a‐3p overexpression inhibited the growth of CRC cell lines mainly due to induction of apoptosis.
FIGURE 2.
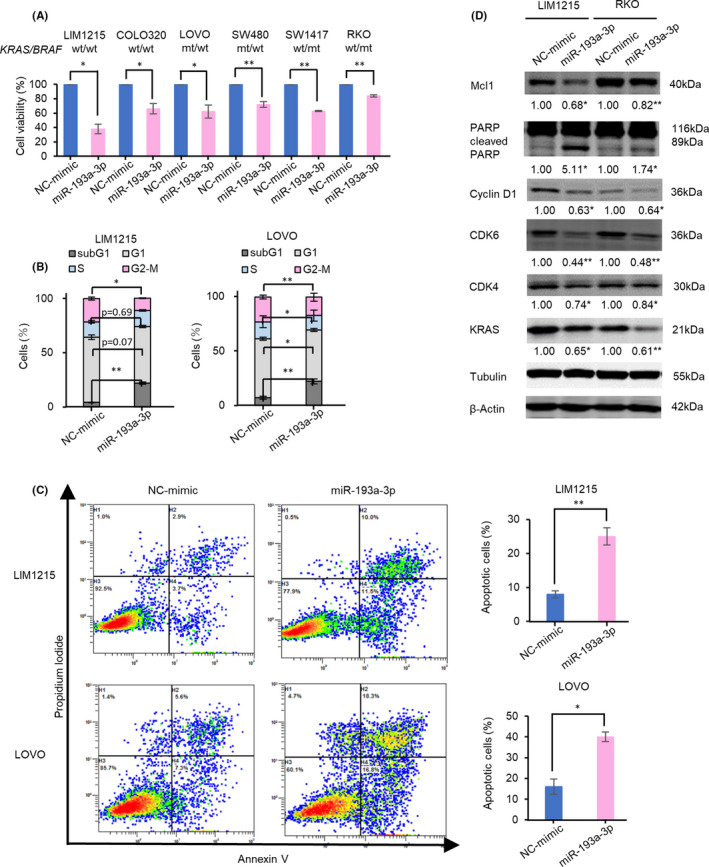
Overexpression of miR‐193a‐3p inhibits proliferation of colorectal cancer (CRC) cells by induction of apoptosis. A, Overexpression of miR‐193a‐3p inhibited cell viability of the CRC cells at 72 h after transfection. B, C, miR‐193a‐3p overexpression increased cells in the subG1 phase and apoptotic cells at 48 h after transfection. D, Western blots show that miR‐193a‐3p overexpression decreased protein expressions of KRAS, Mcl1, Cyclin D1, and CDK6 and increased the expression of cleaved PARP at 48 h after transfection. Numbers below each band represent the expression levels of each protein relative to that of tubulin or β‐actin, as determined by densitometry. Data of miR‐193a‐3p were normalized to that of negative control of miRNA mimic (NC‐mimic). *P < .05; **P < .01
Next, we investigated the protein expression changes to evaluate the molecular mechanisms involved in the induction of apoptosis. We first examined known direct targets of miR‐193a‐3p including KRAS,29, 34, 35, 36, 37 Mcl1,38, 39 Cyclin D1,40, 41, 42, 43, 44 CDK440 and CDK6,40 and cleaved PARP,39 an indirectly dysregulated protein by miR‐193a‐3p expression in various cancers. As shown in Figure 2D, miR‐193a‐3p overexpression reduced the protein expressions of KRAS, Mcl1, Cyclin D1, and CDK6 and increased the protein expression of cleaved PARP, compared with NC‐mimic overexpression in LIM1215 and RKO cells.
These results indicate that overexpression of miR‐193a‐3p inhibited cell proliferation through induction of apoptosis by affecting the expressions of multiple proteins involved in signaling pathway, cell cycle, and apoptotic pathway.
3.3. Characterization of proteins with significant phosphorylation changes induced by miR‐193a‐3p overexpression
To further clarify the molecular mechanisms of miR‐193a‐3p in colorectal carcinogenesis, we performed RPPA analysis to assess the overall signaling changes induced by miR‐193a‐3p overexpression in LIM1215 cells. The Phospho Explorer Antibody Array facilitates detection of both proteins with phosphorylation changes and potential candidate miRNA targets.45, 46 Antibodies with suppressed phosphorylation rates and enhanced phosphorylation rates were identified using a threshold of FC (miR‐193a‐3p/NC‐mimic): FC < 0.75 and FC > 1.33, respectively. The results showed 93 upregulated phosphorylation sites in 68 proteins and 44 downregulated phosphorylation sites in 40 proteins in miR‐193a‐3p overexpression cells relative to NC‐mimic–overexpressing cells (Table S6).
In total, approximately 26% of proteins (106 out of 412) were found to exhibit significant phosphorylation changes caused by miR‐193a‐3p overexpression. To identify the key pathways associated with miR‐193a‐3p, we performed pathway analyses using DAVID, to identify KEGG pathways linked to these differentially expressed proteins. As shown in Table 1 and Figure 3, 14 KEGG pathways were enriched with proteins with phosphorylation change.
TABLE 1.
KEGG pathway analysis of proteins with significant phosphorylation change induced by miR‐193a‐3p overexpression
| KEGG pathway | Number of proteins | Phosphorylation change ratea (%) | P‐value | FDR | Proteins |
|---|---|---|---|---|---|
| AMPK signaling pathway | 14 | 44 | 2.18E‐08 | 2.70E‐05 | FKHRL1/FOXO3A, PDK1, CREB, LKB1, PPAR‐gamma, AKT1, mTOR, AMPK beta1, AKT1S1, ACC1, AMPK1, IRS‐1, SREBP‐1, 4E‐BP1 |
| VEGF signaling pathway | 11 | 42 | 1.44E‐08 | 1.78E‐05 | Caspase 9, FAK, PLCG1, c‐PLA2, PLCG2, Raf1, PKC alpha/beta II, MEK1, VEGFR2, BAD, AKT1 |
| PI3K‐AKT signaling pathway | 34 | 39 | 5.17E‐19 | 6.39E‐16 | Caspase 9, Raf1, PKC alpha/beta II, EGFR, NFkB‐p65, GSK3 beta, p27Kip1, LKB1, mTOR, SYK, ATF2, AMPK1, 4E‐BP1, BAD, IL‐2RA/CD25, FAK, SREBP‐1, FKHRL1/FOXO3A, CD19, MEK1, HSP90 co‐chaperone Cdc37, VEGFR2, PDK1, CREB, Integrin beta‐4, HSP90B, AKT1, p53, IRS‐1, 14‐3‐3 beta/zeta, NFkB‐p105/p50, Myc, S6 Ribosomal Protein, Cyclin D3 |
| ERBB signaling pathway | 19 | 35 | 1.70E‐16 | 2.78E‐13 | c‐Jun, FAK, HER2, Raf1, PKC alpha/beta II, Elk1, EGFR, MEK1, GSK3 beta, p27Kip1, AKT1, mTOR, MKK4/SEK1, PLCG1, PLCG2, STAT5B, 4E‐BP1, BAD, Myc |
| Cell cycle | 13 | 34 | 2.11E‐07 | 2.62E‐04 | SMC1, Chk2, GSK3 beta, Chk1, p27Kip1, p53, CDK1/CDC2, PLK1, 14‐3‐3 beta/zeta, HDAC2, Cyclin B1, Myc, Cyclin D3 |
| Focal adhesion | 22 | 34 | 1.15E‐12 | 1.42E‐09 | c‐Jun, FAK, HER2, Raf1, PKC alpha/beta II, Elk1, EGFR, MEK1, Vinculin, GSK3 beta, Ras‐GRF1, VEGFR2, PDK1, p130Cas, XIAP, VASP, Caveolin‐1, Integrin beta‐4, AKT1, Catenin beta, BAD, Cyclin D3 |
| mTOR signaling pathway | 10 | 33 | 1.30E‐07 | 1.61E‐04 | PKC alpha/beta II, AKT1S1, AMPK1, IRS‐1, 4E‐BP1, PDK1, LKB1, AKT1, S6 Ribosomal Protein, mTOR |
| HIF‐1 signaling pathway | 13 | 33 | 1.16E‐08 | 1.44E‐05 | HER2, PLCG1, PLCG2, PKC alpha/beta II, EGFR, MEK1, NFkB‐p65, 4E‐BP1, NFkB‐p105/p50, p27Kip1, AKT1, S6 Ribosomal Protein, mTOR |
| MAPK signaling pathway | 24 | 32 | 9.43E‐13 | 1.17E‐09 | c‐Jun, MEF2C, Tau, Raf1, MSK1, c‐PLA2, PKC alpha/beta II, EGFR, Elk1, NFkB‐p65, MEK1, PKA CAT, Ras‐GRF1, AKT1, p53, MKP1, MKP2, MKK4/SEK1, Arrestin‐1, FAS, Stathmin 1, ATF2, NFkB‐p105/p50, Myc |
| TNF signaling pathway | 11 | 30 | 3.20E‐06 | 3.96E‐03 | c‐Jun, MKK4/SEK1, FAS, MSK1, NFkB‐p65, ATF2, MEK1, NFkB‐p105/p50, CREB, AKT1, JunD |
| cAMP signaling pathway | 14 | 29 | 5.53E‐06 | 6.84E‐03 | c‐Jun, NMDAR1, Raf1, NFkB‐p65, MEK1, PKA CAT, ATPase, CREB, RyR2, AKT1, PLD1, BAD, NFkB‐p105/p50, DARPP‐32 |
| Rap1 signaling pathway | 15 | 27 | 1.94E‐06 | 2.41E‐03 | NMDAR1, Raf1, PKC alpha/beta II, EGFR, PLC beta3, MEK1, VEGFR2, p130Cas, VASP, LAT, AKT1, Catenin beta, PKD2, PLCG1, PKD1/PKC mu |
| Ras signaling pathway | 18 | 27 | 2.29E‐08 | 2.83E‐05 | NMDAR1, c‐PLA2, Raf1, PKC alpha/beta II, Elk1, EGFR, NFkB‐p65, MEK1, PKA CAT, Ras‐GRF1, VEGFR2, LAT, AKT1, PLCG1, PLCG2, PLD1, BAD, NFkB‐p105/p50 |
| FoxO signaling pathway | 13 | 25 | 4.96E‐07 | 6.13E‐04 | Raf1, EGFR, FKHRL1/FOXO3A, MEK1, PDK1, LKB1, p27Kip1, AKT1, AMPK beta1, PLK1, AMPK1, IRS‐1, CyclinB1 |
Proteins with altered phosphorylation rate (FC < 0.75 or FC > 1.33) were subjected to KEGG pathway analysis.
Abbreviations: FDR, false discovery rate; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Phosphorylation change rate means percentage of proteins with phosphorylation change, which was calculated by dividing proteins with phosphorylation change by all analyzed proteins in each pathway.
FIGURE 3.
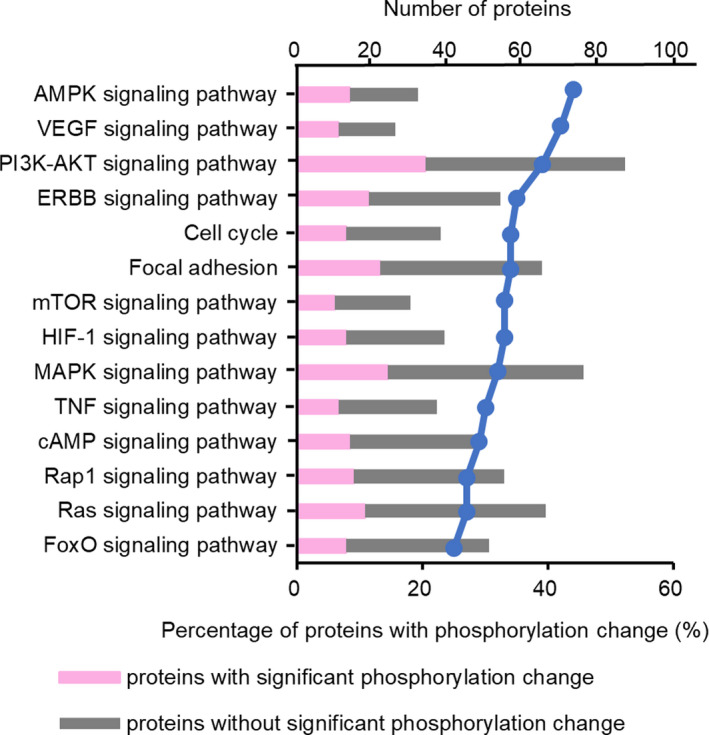
Pathway functional annotations of proteins with altered phosphorylation levels induced by miR‐193a‐3p overexpression. Percentage of proteins with phosphorylation change divided by all analyzed proteins in each pathway is represented by line plot
Our analysis indicated that the proteins with significantly altered phosphorylation levels were intricately involved in various oncogenic pathways such as AMPK, VEGF, PI3K‐AKT, and ERBB signaling pathways. For example, approximately one‐third of the proteins belonged to the PI3K‐AKT pathway and showed altered phosphorylation rates. These results suggest that miR‐193a‐3p is involved in the regulation of several oncogenic pathways.
3.4. Identification and validation of novel target candidates of miR‐193a‐3p using RPPA
In order to detect novel target candidates of miR‐193a‐3p, we next performed RPPA to screen proteins that were downregulated by miR‐193a‐3p overexpression. Proteins with the standardized signal values exceeding the median value of those of all proteins from the cells transfected with NC‐mimic were analyzed. When several total protein–specific antibodies were present on a single protein, the median value of the FCs calculated for each antibody was used for the analysis. Of the 225 proteins that qualified for the aforementioned criteria, 13 proteins (6%) showed reduced expression using FC < 0.70 as the threshold.
To validate the protein expression changes, of the 13 downregulated proteins identified on RPPA, the expression levels of seven proteins associated with cell growth were assessed by Western blot analysis. Overexpression of miR‐193a‐3p in LIM1215 and RKO cells induced a mild but significant reduction in the expressions of ATF2, MYC, and FOXO3A; these results were consistent with the findings of RPPA (Figure 4A,B). This is the first study to demonstrate the regulation of ATF2 and FOXO3A by miR‐193a‐3p.
FIGURE 4.
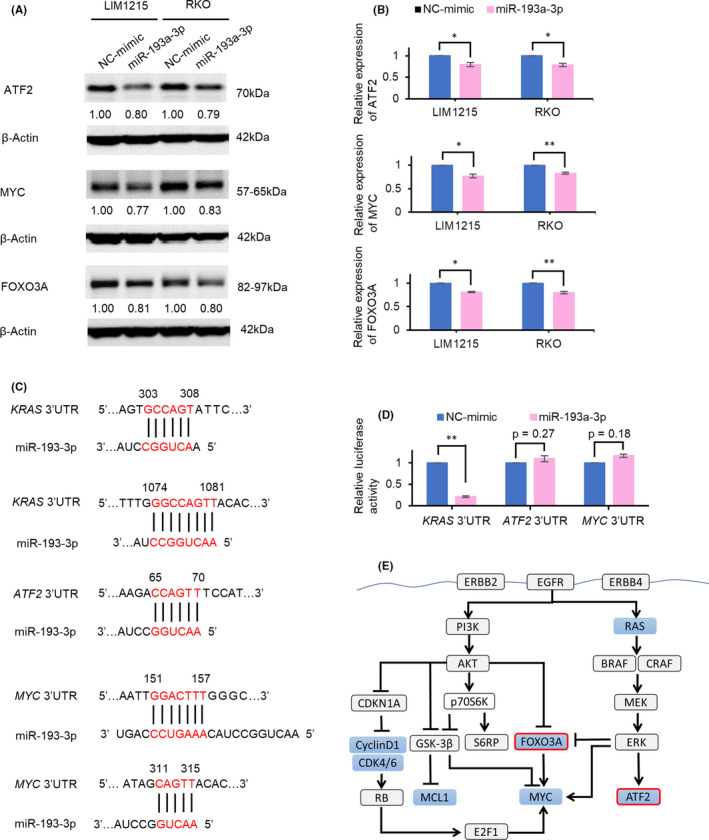
miR‐193a‐3p represses the protein expressions of ATF2, MYC, and FOXO3A. A, B, Western blots show that miR‐193a‐3p overexpression reduced protein expressions of ATF2, MYC, and FOXO3A. Numbers below each band represent the expression levels of each protein relative to that of β‐actin, as determined by densitometry. Relative protein expression was calculated by normalization to negative control of miRNA mimic (NC‐mimic). C, Sequences of the potential miR‐193a‐3p binding sites in the 3′‐UTR of KRAS, ATF2, and MYC. D, The luciferase activity of luciferase reporter with 3′‐UTR of ATF2 and MYC shows no significant difference by miR‐193a‐3p overexpression. E, Potential model for mechanism of tumor suppression of miR‐193a‐3p. miR‐193a‐3p decreases the known targets and decreases ATF2, FOXO3A, and MYC. Proteins downregulated by miR‐193a‐3p overexpression in Western blots are shown in blue. *P < .05; **P < .01
We next performed luciferase reporter assays to verify whether downregulation of ATF2 and MYC (whose expressions in LIM1215 were reduced by >20% after overexpression of miR‐193a‐3p) resulted from direct targeting of miR‐193a‐3p. We used KRAS, a known direct target of miR‐193a‐3p, as a positive control.29, 34, 35, 36, 37 We made a luciferase reporter plasmid incorporating the 3′‐UTRs of KRAS, ATF2, and MYC (Figure 4C). The luciferase activity of 3′‐UTR of ATF2 and MYC showed no significant decrease by miR‐193a‐3p overexpression, whereas that of 3′‐UTR of KRAS was markedly decreased (Figure 4D). The results suggested that ATF2 and MYC are indirectly regulated by miR‐193a‐3p. Collectively, the tumor‐suppressive function of miR‐193a‐3p is likely attributable to the regulation of several oncogenic pathways through indirect modulation of the expressions of ATF2 and MYC, in addition to that of known direct targets such as KRAS,29, 34, 35, 36, 37 Mcl1,38, 39 and Cyclin D140, 41, 42, 43, 44 (Figure 4E).
3.5. Overexpression of miR‐193a‐3p potentiates inhibition of cell proliferation by the combination therapy of DAB/TRA and DAB/TRA plus Cmab
We next tried to validate the effect of overexpression of miR‐193a‐3p, identified as a sensitizer of DAB/TRA, on the efficacy of DAB/TRA, and to clarify the molecular mechanisms of the enhanced drug efficacy. Overexpression of miR‐193a‐3p reduced the growth of RKO cells treated with DAB alone and the combination of DAB/TRA compared with NC‐mimic overexpression (Figure 5A). Because miR‐193a‐3p overexpression remarkably enhanced the inhibitory effect of DAB/TRA on cell growth, we further analyzed the effect of miR‐193a‐3p overexpression on the combination of DAB/TRA.
FIGURE 5.
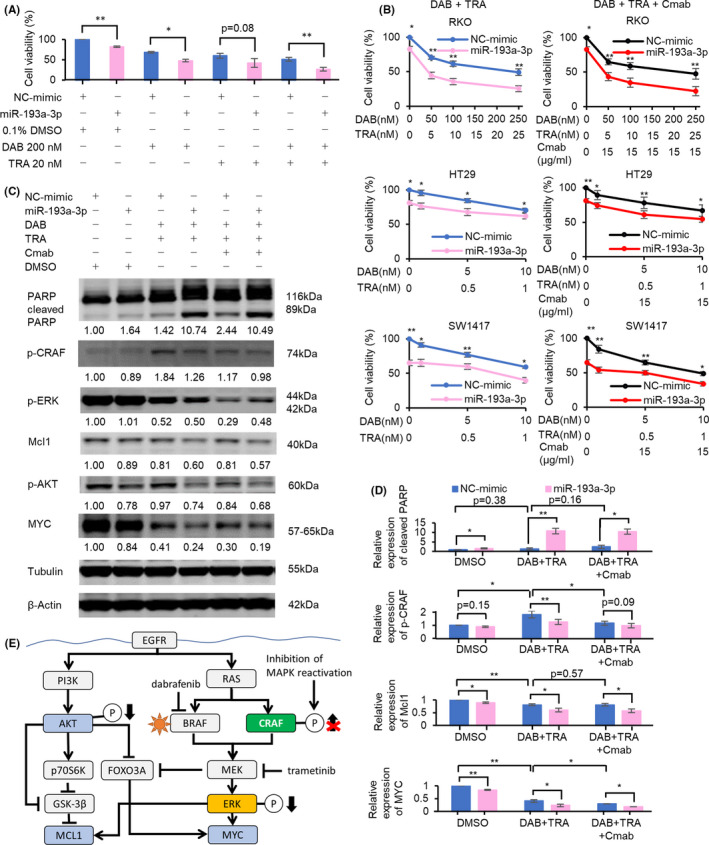
miR‐193a‐3p potentiates the efficacy of dabrafenib and trametinib (DAB/TRA) and DAB/TRA plus Cmab in BRAF‐mutated colorectal cancer (CRC) cells. A, miR‐193a‐3p overexpression enhanced the inhibition of cell proliferation after treatment with DAB alone and DAB/TRA combination for 48 h in RKO cells. B, Inhibition of cell growth after treatment with DAB/TRA and DAB/TRA plus Cmab for 60 h was potentiated by overexpression of miR‐193a‐3p. C, D, Western blots show that miR‐193a‐3p overexpression enhanced the efficacy of DAB/TRA and DAB/TRA plus Cmab for 36 h and suppressed MAPK reactivation in RKO cells. Numbers below each band represent the expression levels of each protein relative to that of tubulin or β‐actin, as determined by densitometry. Relative protein expression was calculated by normalization to negative control of miRNA mimic (NC‐mimic) with DMSO treatment. E, Potential model for miR‐193a‐3p regulation of efficacy of DAB/TRA in BRAF‐mutated CRC. Overexpression of miR‐193a‐3p in addition to DAB/TRA treatment inhibited reactivation of p‐CRAF and intensified reduction of Mcl1, p‐AKT, and MYC. *P < .05; **P < .01
The enhanced inhibitory effect of DAB/TRA in RKO cells with miR‐193a‐3p overexpression was also confirmed in other BRAF‐mutated CRC cell lines, HT29 and SW1417; however, the extent of enhancement in HT29 and SW1417 was modest compared with that in RKO (Figure 5B). Similar results were also obtained with DAB/TRA plus Cmab (Figure 5B). The greater sensitization to DAB/TRA induced by miR‐193a‐3p overexpression in RKO compared with HT29 and SW1417 may be due in part to the lower sensitivity to DAB/TRA and the lower inhibitory effect of miR‐193a‐3p alone on growth. On Western blot analysis, DAB/TRA treatment of RKO cells transfected with NC‐mimic decreased the expression levels of Mcl1, phospho‐ERK (p‐ERK), and MYC, and increased that of phospho‐CRAF (p‐CRAF), probably due to MAPK reactivation.12, 13 Notably, DAB/TRA treatment of cells with miR‐193a‐3p overexpression resulted in marked increase in cleaved PARP and decrease in Mcl1, phospho‐AKT (p‐AKT), and MYC (Figure 5C–E). Reactivation of p‐CRAF induced by DAB/TRA was inhibited by addition of miR‐193a‐3p as well as addition of Cmab to DAB/TRA. Moreover, addition of miR‐193a‐3p to DAB/TRA increased apoptosis and decreased Mcl1 and MYC more obviously than addition of Cmab. Furthermore, miR‐193a‐3p–transfected cells treated with DAB/TRA plus Cmab showed increased expression of cleaved PARP and decreased expressions of Mcl1, p‐AKT, and MYC compared with NC‐mimic–transfected RKO (Figure 5C,D) and SW1417 cells treated with DAB/TRA plus Cmab (Figure S3). These results suggest that the enhanced efficacy of DAB/TRA induced by miR‐193a‐3p overexpression is at least partly attributable to the suppression of MAPK reactivation.
3.6. Silencing of Mcl1 enhanced the efficacy of combination therapy with DAB/TRA and DAB/TRA plus Cmab
We then hypothesized that miR‐193a‐3p affects the response to DAB/TRA via directly or indirectly targeting the molecules involved in oncogenic pathways. To address this issue, we next determined whether silencing of known direct targets of miR‐193a‐3p including KRAS, Mcl1 and Cyclin D1, or MYC, an indirectly dysregulated protein by miR‐193a‐3p expression, can reproduce the effects of miR‐193a‐3p overexpression on the efficacy of DAB/TRA in RKO cells. Western blot analysis showed reduced protein expressions of Mcl1, KRAS, MYC, and Cyclin D1 in cells transfected with si‐Mcl1, si‐KRAS, si‐MYC, and si‐Cyclin D1, respectively, at 48 hours after transfection (Figure 6A). Silencing of Mcl1 and Cyclin D1 enhanced the inhibitory effect of DAB/TRA on cell growth (Figure 6A). Owing to obvious augmentation of growth inhibition after si‐Mcl1 transfection, we further evaluated the effects of Mcl1 inhibition. Enhanced inhibition of cell growth by DAB/TRA in addition to Mcl‐1 silencing was also observed after treatment of SW1417 cells with high doses of DAB/TRA (Figure 6B). These results were also validated after treatment with DAB/TRA plus Cmab in RKO and SW1417 cells (Figure 6B).
FIGURE 6.
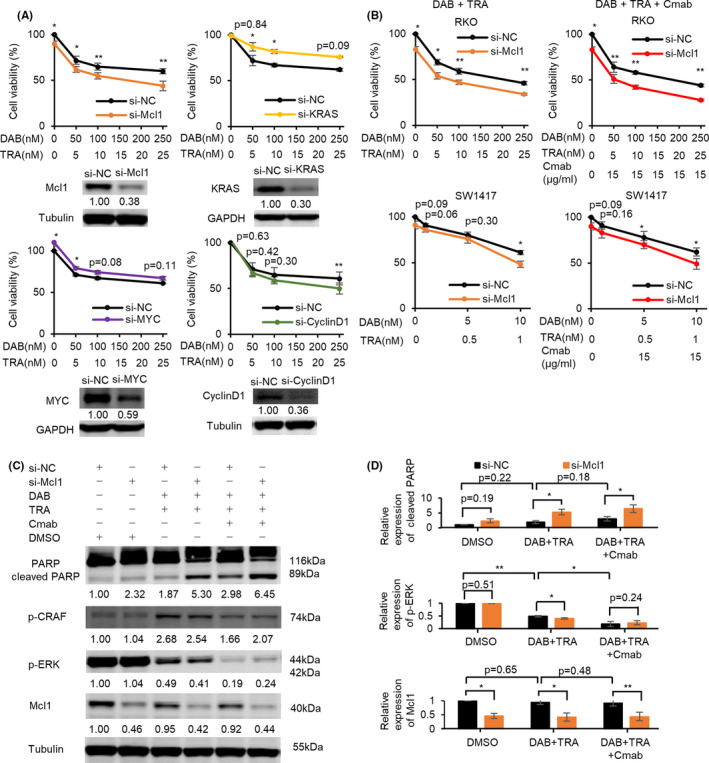
Silencing of Mcl1 enhances the efficacy of dabrafenib and trametinib (DAB/TRA) and DAB/TRA plus Cmab. A, Silencing of Mcl1 and Cyclin D1 promoted growth inhibition by treatment with DAB/TRA for 48 h in RKO cells. B, Inhibition of cell growth by treatment with DAB/TRA and DAB/TRA plus Cmab for 60 h was potentiated by Mcl1 suppression. C, D, Western blots show that Mcl1 suppression enhanced apoptosis induced by treatment with DAB/TRA and DAB/TRA plus Cmab for 36 h in RKO cells. Numbers below each band represent the expression levels of each protein relative to that of tubulin, as determined by densitometry. Relative protein expression was calculated by normalization to si‐negative control (si‐NC) with DMSO treatment. *P < .05; **P < .01
We next assessed the effect of Mcl1 silencing on the expression of proteins involved in apoptotic or MAPK pathways. RKO cells transfected with si‐Mcl1 showed a significant increase in cleaved PARP and a significant decrease in Mcl1 after DAB/TRA treatment compared with RKO cells transfected with si‐negative control (si‐NC) (Figure 6C,D). Reactivation of p‐CRAF by DAB/TRA was inhibited by miR‐193a‐3p overexpression but not by Mcl1 silencing alone (Figures 5C,6C). Moreover, under treatment with DAB/TRA plus Cmab, Mcl1 silencing also induced a further increase in cleaved PARP, but along with persistent suppression of p‐ERK, compared with si‐NC–transfected RKO cells (Figure 6C,D).
3.7. Mcl1 inhibitor S63845 enhanced apoptosis induced by combination therapy with DAB/TRA and DAB/TRA plus Cmab
To validate whether the enhancement of DAB/TRA efficacy by Mcl1 silencing with siRNA was also achieved by Mcl1 inhibition using Mcl1 inhibitors, we next investigated whether Mcl1 inhibitors promote the inhibitory effect of DAB/TRA. Cell viability assay revealed that treatment with the selective Mcl1 inhibitor S63845 1 µM enhanced the inhibitory effect of DAB alone, TRA alone, combination of DAB/TRA, and combination of DAB/TRA plus Cmab on the growth of RKO cells (Figure 7A). The enhanced inhibitory effect of combination therapy with DAB/TRA or DAB/TRA plus Cmab by S63845 was also observed in SW1417 cells (Figure 7B). The interaction between S63845 and DAB/TRA was synergistic as determined by calculation of the combination indices that were lower than 1, and the dose‐dependent cytotoxicity of S63845 was observed in RKO cells (Figure S4A,B and Table S7). In SW1417 cells, a synergistic effect with S63845 was observed at higher doses of DAB/TRA (DAB 10 nM and TRA 1 nM) (Figure S4C). Cotreatment with DAB/TRA and S63845 induced a remarkable increase in apoptosis of RKO cells, compared with treatment with DAB/TRA alone or S63845 alone (Figure 7C). Furthermore, Mcl1 inhibition by S63845 caused more significant increase in apoptosis after cotreatment with DAB/TRA plus Cmab than after cotreatment with DAB/TRA (Figure 7C).
FIGURE 7.
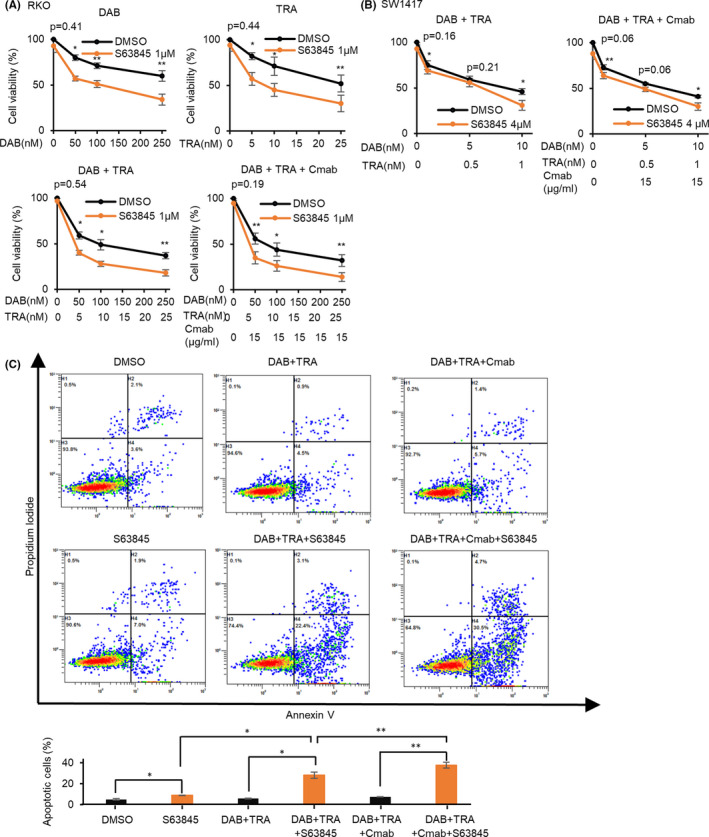
Mcl1 inhibitor S63845 enhances apoptosis induced by treatment with dabrafenib and trametinib (DAB/TRA) and DAB/TRA plus Cmab. A, S63845 enhanced growth inhibition by DAB alone, TRA alone, combination therapy with DAB/TRA, and DAB/TRA plus Cmab for 72 h in RKO cells. B, S63845 also enhanced growth inhibition by treatment with DAB/TRA and DAB/TRA plus Cmab for 72 h in SW1417 cells. C, S63845 further increased apoptosis induced by treatment with DAB/TRA and DAB/TRA plus Cmab for 48 h in RKO cells. *P < .05; **P < .01
Collectively, we demonstrated that similar to miR‐193a‐3p overexpression, suppression of Mcl1 by siRNA or Mcl1 inhibitors further promoted apoptosis and inhibition of cell growth by the combination of DAB/TRA or by DAB/TRA plus Cmab. The enhanced effects of miR‐193a‐3p on the efficacy of treatment with DAB/TRA and/or DAB/TRA plus Cmab are likely to be mediated at least partly by inhibition of Mcl1, which is a known direct target of miR‐193a‐3p.38, 39
4. DISCUSSION
The aims of this study were to identify miRNAs that sensitize BRAF‐mutated CRC cells to BRAF inhibitors and MEK inhibitors and to clarify the function and potential of the screened miRNAs that could lead to novel therapeutic strategies against BRAF‐mutated CRC. Herein, we demonstrated the novel role of miR‐193a‐3p in CRC as follows. First, we identified five miRNAs including miR‐193a‐3p, which sensitized BRAF‐mutated CRC cells to DAB/TRA. Second, we found that miR‐193a‐3p regulates multiple oncogenic pathways in CRC and acts as a tumor suppressor by indirectly downregulating the expressions of ATF2 and MYC, in addition to known direct targets of miR‐193a‐3p. Third, we found that miR‐193a‐3p overexpression enhanced growth inhibition by BRAF and MEK inhibitors by inhibiting the reactivation of MAPK signaling. We examined the effect of silencing the targets of miR‐193a‐3p, including Mcl1, KRAS, and Cyclin D1, or MYC, whose expression is regulated by miR‐193a‐3p. The results showed that silencing of Mcl1 can reproduce the effects of miR‐193a‐3p overexpression on the efficacy of DAB/TRA, so we focused on Mcl1. The enhanced effects of miR‐193a‐3p on the cytotoxicity of the combination of DAB/TRA or DAB/TRA plus Cmab are partially mediated by suppression of Mcl1.
Although some miRNAs exert different functions in different organs, miR‐193a‐3p has consistently been shown to have tumor‐suppressive functions in various carcinomas. Several studies suggest that miR‐193a‐3p acts as a tumor suppressor in CRC by targeting KRAS,28, 29 ERBB4,30, 31 and IL17RD.32 In our study, we also observed reduction of KRAS expression by miR‐193a‐3p overexpression and confirmed that miR‐193a‐3p directly targets the 3′‐UTR of KRAS mRNA, which is consistent with previous reports.28, 29 In addition to KRAS, the following genes have been reported as direct targets of miR‐193a‐3p: Mcl1 in ovarian cancer38 and glioblastoma39; Cyclin D1 in breast,40 prostate,41 gastric,42 thyroid,43 and pancreatic cancers44; and CDK4 and CDK6 in breast cancer.40 To the best of our knowledge, this is the first study to show that miR‐193a‐3p overexpression also decreases the expressions of Mcl1, Cyclin D1, CDK4, and CDK6 proteins in CRC, although whether the regulation by miR‐193a‐3p is via directly targeting the 3’‐UTR of mRNAs also in CRC requires further elucidation.
Two previous studies have investigated the protein expression changes induced by miR‐193a‐3p using RPPA in a breast cancer cell line. Uhlmann et al induced expression of 810 miRNAs and performed RPPA analysis of 26 proteins.47 They demonstrated that miR‐124, miR‐147, and miR‐193a‐3p regulate the proteins involved in EGFR‐related pathways.47 Seviour et al conducted RPPA analysis of 120 proteins after inducing overexpression of 879 miRNAs and demonstrated that miR‐193a‐3p directly targets KRAS.34 In our study, as many as 412 proteins were analyzed with 1318 antibodies. The results revealed that miR‐193a‐3p regulates many oncogenic pathways including EGFR‐related pathways, suggesting that the inhibitory effects on several targets and protein interactions in those signaling pathways altered the expression of multiple proteins.
Recent studies have revealed that miRNAs may serve as prognostic biomarkers of anticancer therapies.18, 19 In our previous study, low expression of miR‐193a‐3p in CRC tissues showed an association with worse progression‐free survival of patients who received anti‐EGFR antibody; this suggested that the miR‐193a‐3p expression status affects the sensitivity to anti‐EGFR antibody.23 In the present study, miR‐193a‐3p was found to regulate the PI3K‐AKT and ERBB‐MAPK pathways; in addition, miR‐193a‐3p overexpression enhanced the inhibitory effect of BRAF and MEK inhibitors and suppressed MAPK reactivation in BRAF‐mutated CRC cell lines. These findings seem to support our hypothesis that miR‐193a‐3p is a key regulator of these signaling pathways and is involved in mechanisms that determine the sensitivity to therapies targeted against these pathways (including anti‐EGFR therapy and/or BRAF/MEK inhibitor therapy). The mechanisms underlying the drug sensitivity may involve, at least partly, the inhibition of known direct target genes by miR‐193a‐3p, ie, KRAS 29, 34, 35, 36, 37 and ERBB4.30, 31 We found that miR‐193a‐3p overexpression suppressed MAPK pathway reactivation explained by p‐CRAF activation under the combination therapy of a BRAF inhibitor and a MEK inhibitor, but did not further suppress MAPK pathway reactivation under the combination therapy of a BRAF inhibitor, a MEK inhibitor, and an anti‐EGFR antibody. This result suggests that miR‐193a‐3p mediates the sensitization of cells to BRAF and MEK inhibitors through inhibiting MAPK pathway reactivation mainly via inhibition of EGFR; however, the precise underlying mechanism is not clear. Inhibition of Mcl1 suppressed cell growth induced by the combination of BRAF inhibitors, MEK inhibitors, and/or anti‐EGFR antibodies, similar to that observed with miR‐193a‐3p overexpression. In contrast, Mcl1 inhibition did not suppress MAPK reactivation by BRAF and MEK inhibitors, which was different from the effect of miR‐193a‐3p overexpression. These results suggest that the effects of miR‐193a‐3p on the increased toxicity of BRAF inhibitors, MEK inhibitors and/or anti‐EGFR antibodies, are mediated partly through suppression of Mcl1, but also through other unknown mechanisms.
A small number of in vitro and in vivo studies have demonstrated the association of Mcl1 with the sensitivity of BRAF V600E–mutated cancers to BRAF inhibitors or MEK inhibitors. Fofaria et al and Sale et al reported suppression of Mcl1 enhanced the antitumor effect of BRAF inhibitor vemurafenib and MEK inhibitor selumetinib against BRAF‐mutated melanoma.48, 49 Kawakami et al showed that the combination of Mcl1 suppression and MEK inhibitor cobimetinib enhanced the antitumor effect in BRAF‐mutated CRC.50 These findings together with our findings support that Mcl1 suppression is a useful strategy to enhance the efficacy of BRAF and MEK inhibitors against BRAF‐mutated cancers. Combination therapy of BRAF inhibitors, MEK inhibitors, and anti‐EGFR antibodies has been introduced in clinical practice for patients with BRAF‐mutated CRC17, 51; however, further development of therapies to potentiate the efficacy of the combination therapy is warranted. Of note, the present study shows for the first time that Mcl1 suppression in combination with BRAF inhibitors, MEK inhibitors, and anti‐EGFR antibodies enhance the antitumor effect in CRC. In vivo studies will be required to evaluate efficacy and safety toward the therapeutic application of miR‐193a‐3p mimic and Mcl1 inhibitors. Further research on new combination therapies such as use of miR‐193a‐3p mimic or Mcl1 inhibitors could lead to the development of novel therapeutic strategies for patients with BRAF‐mutated cancers, including CRC.
In conclusion, we have shown the possibility that miR‐193a‐3p is a key modulator of multiple proteins involved in CRC oncogenesis; in addition, miR‐193a‐3p affects the drug sensitivity to MAPK‐related pathway inhibitors such as BRAF inhibitors, MEK inhibitors, and/or anti‐EGFR antibodies. We propose that the addition of miR‐193a‐3p or Mcl1 inhibitors to combination therapy with BRAF inhibitors, MEK inhibitors, and anti‐EGFR antibodies may be a potential novel treatment strategy for BRAF‐mutated cancers.
DISCLOSURE
C. Ishioka received research funding from MSD, Merck Biopharma, RIKEN Genesis, Ono Pharmaceutical Company. C. Ishioka received research grants from Chugai, Ono, Taiho Pharmaceutical Company. C. Ishioka received honoraria from Chugai, Taiho Pharmaceutical Company. M. Takahashi has received research funding from Ono Pharmaceutical Company.
Supporting information
Figures S1‐S4
Table S1‐S7
ACKNOWLEDGMENTS
We thank Ms Nobuko Saeki and Ms Hiromi Nakano for their technical assistance. We thank Dr John M. Mariadason at Ludwig Institute for Cancer Research for kindly providing the CRC cell lines.
Hiraide S, Takahashi M, Yoshida Y, Yamada H, Komine K, Ishioka C. Tumor suppressor miR‐193a‐3p enhances efficacy of BRAF/MEK inhibitors in BRAF‐mutated colorectal cancer. Cancer Sci. 2021;112:3856–3870. 10.1111/cas.15075
Funding information
This study was supported by the grants‐in‐aid from the Ministry of Education, Science, Sports, and Culture of Japan (18K07993 to M. Takahashi and 19H03508 to C. Ishioka).
Contributor Information
Masanobu Takahashi, Email: masanobu.takahashi.a7@tohoku.ac.jp.
Chikashi Ishioka, Email: chikashi@tohoku.ac.jp.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949‐954. [DOI] [PubMed] [Google Scholar]
- 2.Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med. 2013;19:1401‐1409. [DOI] [PubMed] [Google Scholar]
- 3.Dankner M, Rose AAN, Rajkumar S, Siegel PM, Watson IR. Classifying BRAF alterations in cancer: new rational therapeutic strategies for actionable mutations. Oncogene. 2018;37:3183‐3199. [DOI] [PubMed] [Google Scholar]
- 4.Karoulia Z, Gavathiotis E, Poulikakos PI. New perspectives for targeting RAF kinase in human cancer. Nat Rev Cancer. 2017;17:676‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cremolini C, Loupakis F, Antoniotti C, et al. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first‐line treatment of patients with metastatic colorectal cancer: updated overall survival and molecular subgroup analyses of the open‐label, phase 3 TRIBE study. Lancet Oncol. 2015;16:1306‐1315. [DOI] [PubMed] [Google Scholar]
- 6.Kadowaki S, Kakuta M, Takahashi S, et al. Prognostic value of KRAS and BRAF mutations in curatively resected colorectal cancer. World J Gastroenterol. 2015;21:1275‐1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dienstmann R, Mason MJ, Sinicrope FA, et al. Prediction of overall survival in stage II and III colon cancer beyond TNM system: a retrospective, pooled biomarker study. Ann Oncol. 2017;28:1023‐1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taieb J, Lapeyre‐Prost A, Laurent Puig P, Zaanan A. Exploring the best treatment options for BRAF‐mutant metastatic colon cancer. Br J Cancer. 2019;121:434‐442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hauschild A, Grob J‐J, Demidov LV, et al. Dabrafenib in BRAF‐mutated metastatic melanoma: a multicentre, open‐label, phase 3 randomised controlled trial. Lancet. 2012;380:358‐365. [DOI] [PubMed] [Google Scholar]
- 10.McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAFV600E and BRAFV600K mutation‐positive melanoma (BRIM‐3): extended follow‐up of a phase 3, randomised, open‐label study. Lancet Oncol. 2014;15:323‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kopetz S, Desai J, Chan E, et al. Phase II Pilot Study of vemurafenib in patients with metastatic BRAF‐mutated colorectal cancer. J Clin Oncol. 2015;33:4032‐4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prahallad A, Sun C, Huang S, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100‐103. [DOI] [PubMed] [Google Scholar]
- 13.Corcoran RB, Ebi H, Turke AB, et al. EGFR‐mediated re‐activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2:227‐235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corcoran RB, Atreya CE, Falchook GS, et al. Combined BRAF and MEK inhibition with dabrafenib and trametinib in BRAF V600‐mutant colorectal cancer. J Clin Oncol. 2015;33:4023‐4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kopetz S, McDonough SL, Lenz H‐J, et al. Randomized trial of irinotecan and cetuximab with or without vemurafenib in BRAF‐mutant metastatic colorectal cancer (SWOG S1406). J Clin Oncol. 2017;35(15_suppl):3505–3505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tabernero J, Geel RV, Guren TK, et al. Phase 2 results: Encorafenib (ENCO) and cetuximab (CETUX) with or without alpelisib (ALP) in patients with advanced BRAF‐mutant colorectal cancer (BRAFm CRC). J Clin Oncol. 2016;34:3544‐3544. [Google Scholar]
- 17.Kopetz S, Grothey A, Yaeger R, et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E‐Mutated Colorectal Cancer. N Engl J Med. 2019;381:1632‐1643. [DOI] [PubMed] [Google Scholar]
- 18.Hayes J, Peruzzi PP, Lawler S. MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol Med. 2014;20:460‐469. [DOI] [PubMed] [Google Scholar]
- 19.Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16:203‐222. [DOI] [PubMed] [Google Scholar]
- 20.Magee P, Shi L, Garofalo M. Role of microRNAs in chemoresistance. Ann Transl Med. 2015;3:332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nosho K, Igarashi H, Nojima M, et al. Association of microRNA‐31 with BRAF mutation, colorectal cancer survival and serrated pathway. Carcinogenesis. 2014;35:776‐783. [DOI] [PubMed] [Google Scholar]
- 22.Lundberg IV, Wikberg ML, Ljuslinder I, et al. MicroRNA expression in KRAS‐ and BRAF‐mutated colorectal cancers. Anticancer Res. 2018;38:677‐683. [DOI] [PubMed] [Google Scholar]
- 23.Takahashi H, Takahashi M, Ohnuma S, et al. microRNA‐193a‐3p is specifically down‐regulated and acts as a tumor suppressor in BRAF‐mutated colorectal cancer. BMC Cancer. 2017;17:723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takahashi M, Koi M, Balaguer F, Boland CR, Goel A. MSH3 mediates sensitization of colorectal cancer cells to cisplatin, oxaliplatin, and a poly(ADP‐ribose) polymerase inhibitor. J Biol Chem. 2011;286:12157‐12165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671‐675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44‐57. [DOI] [PubMed] [Google Scholar]
- 27.da Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mamoori A, Wahab R, Islam F, et al. Clinical and biological significance of miR‐193a‐3p targeted KRAS in colorectal cancer pathogenesis. Hum Pathol. 2018;71:145‐156. [DOI] [PubMed] [Google Scholar]
- 29.Zhu Z, Du S, Yin K, et al. Knockdown long noncoding RNA nuclear paraspeckle assembly transcript 1 suppresses colorectal cancer through modulating miR‐193a‐3p/KRAS. Cancer Med. 2019;8:261‐275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu T, Li J, Yan M, et al. MicroRNA‐193a‐3p and ‐5p suppress the metastasis of human non‐small‐cell lung cancer by downregulating the ERBB4/PIK3R3/mTOR/S6K2 signaling pathway. Oncogene. 2015;34:413‐423. [DOI] [PubMed] [Google Scholar]
- 31.Yue B, Cai D, Liu C, Fang C, Yan D. Linc00152 functions as a competing endogenous RNA to confer oxaliplatin resistance and holds prognostic values in colon cancer. Mol Ther. 2016;24:2064‐2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pekow J, Meckel K, Dougherty U, et al. miR‐193a‐3p is a key tumor suppressor in ulcerative colitis‐associated colon cancer and promotes carcinogenesis through upregulation of IL17RD. Clin Cancer Res. 2017;23:5281‐5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grossi I, Salvi A, Abeni E, Marchina E, De Petro G. Biological function of MicroRNA193a‐3p in health and disease. Int J Genomics. 2017;2017:5913195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seviour EG, Sehgal V, Mishra D, et al. Targeting KRas‐dependent tumour growth, circulating tumour cells and metastasis in vivo by clinically significant miR‐193a‐3p. Oncogene. 2017;36:1339‐1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iliopoulos D, Rotem A, Struhl K. Inhibition of miR‐193a expression by Max and RXRalpha activates K‐Ras and PLAU to mediate distinct aspects of cellular transformation. Cancer Res. 2011;71:5144‐5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen K, Liu MX, Mak CS, et al. Methylation‐associated silencing of miR‐193a‐3p promotes ovarian cancer aggressiveness by targeting GRB7 and MAPK/ERK pathways. Theranostics. 2018;8:423‐436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fan Q, Hu X, Zhang H, et al. MiR‐193a‐3p is an Important Tumour Suppressor in Lung Cancer and Directly Targets KRAS. Cell Physiol Biochem. 2017;44:1311‐1324. [DOI] [PubMed] [Google Scholar]
- 38.Nakano H, Yamada Y, Miyazawa T, Yoshida T. Gain‐of‐function microRNA screens identify miR‐193a regulating proliferation and apoptosis in epithelial ovarian cancer cells. Int J Oncol. 2013;42:1875‐1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kwon JE, Kim BY, Kwak SY, Bae IH, Han YH. Ionizing radiation‐inducible microRNA miR‐193a‐3p induces apoptosis by directly targeting Mcl‐1. Apoptosis. 2013;18:896‐909. [DOI] [PubMed] [Google Scholar]
- 40.Hydbring P, Wang Y, Fassl A, et al. Cell‐cycle‐targeting microRNAs as therapeutic tools against refractory cancers. Cancer Cell. 2017;31:576‐590.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu Y, Xu X, Xu X, et al. MicroRNA‐193a‐3p inhibits cell proliferation in prostate cancer by targeting cyclin D1. Oncol Lett. 2017;14:5121‐5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chou NH, Lo YH, Wang KC, Kang CH, Tsai CY, Tsai KW. MiR‐193a‐5p and ‐3p play a distinct role in gastric cancer: miR‐193a‐3p suppresses gastric cancer cell growth by targeting ETS1 and CCND1. Anticancer Res. 2018;38:3309‐3318. [DOI] [PubMed] [Google Scholar]
- 43.Li XJ, Wen R, Wen DY, et al. Downregulation of miR193a3p via targeting cyclin D1 in thyroid cancer. Mol Med Rep. 2020;22:2199‐2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen ZM, Yu Q, Chen G, et al. MiR‐193a‐3p inhibits pancreatic ductal adenocarcinoma cell proliferation by targeting CCND1. Cancer Manag Res. 2019;11:4825‐4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tan X, Liu P, Huang Y, et al. Phosphoproteome analysis of invasion and metastasis‐related factors in pancreatic cancer cells. PLoS One. 2016;11:e0152280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bracamontes CG, Lopez‐Valdez R, Subramani R, et al. The serum protein profile of early parity which induces protection against breast cancer. Oncotarget. 2016;7:82538‐82553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Uhlmann S, Mannsperger H, Zhang JD, et al. Global microRNA level regulation of EGFR‐driven cell‐cycle protein network in breast cancer. Mol Syst Biol. 2012;8:570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fofaria NM, Frederick DT, Sullivan RJ, Flaherty KT, Srivastava SK. Overexpression of Mcl‐1 confers resistance to BRAFV600E inhibitors alone and in combination with MEK1/2 inhibitors in melanoma. Oncotarget. 2015;6:40535‐40556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sale MJ, Minihane E, Monks NR, et al. Targeting melanoma's MCL1 bias unleashes the apoptotic potential of BRAF and ERK1/2 pathway inhibitors. Nat Commun. 2019;10:5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kawakami H, Huang S, Pal K, Dutta SK, Mukhopadhyay D, Sinicrope FA. Mutant BRAF upregulates MCL‐1 to confer apoptosis resistance that is reversed by MCL‐1 antagonism and cobimetinib in colorectal cancer. Mol Cancer Ther. 2016;15:3015‐3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Corcoran RB, Andre T, Atreya CE, et al. Combined BRAF, EGFR, and MEK inhibition in patients with BRAF(V600E)‐mutant colorectal cancer. Cancer Discov. 2018;8:428‐443. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1‐S4
Table S1‐S7
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.