

Abstract
Overcoming cisplatin (CDDP) resistance is a major issue in urothelial cancer (UC), in which CDDP‐based chemotherapy is the first‐line treatment. WEE1, a G2/M checkpoint kinase, confers chemoresistance in response to genotoxic agents. However, the efficacy of WEE1 blockade in UC has not been reported. MK‐1775, a WEE1 inhibitor also known as AZD‐1775, blocked proliferation of UC cell lines in a dose‐dependent manner irrespective of TP53 status. MK‐1775 synergized with CDDP to block proliferation, inducing apoptosis and mitotic catastrophe in TP53‐mutant UC cells but not in TP53‐WT cells. Knocking down TP53 in TP53‐WT cells induced synergism of MK‐1775 and CDDP. In UMUC3 cell xenografts and two patient‐derived xenograft lines with MDM2 overexpression, in which the p53/cell cycle pathway was inactivated, AZD‐1775 combined with CDDP suppressed tumor growth inducing both M‐phase entry and apoptosis, whereas AZD‐1775 alone was as effective as the combination in RT4 cell xenografts. Drug susceptibility assay using an ex vivo cancer tissue‐originated spheroid system showed correlations with the in vivo efficacy of AZD‐1775 alone or combined with CDDP. We determined the feasibility of the drug susceptibility assay using spheroids established from UC surgical specimens obtained by transurethral resection. In conclusion, WEE1 is a promising therapeutic target in the treatment of UC, and a highly specific small molecule inhibitor is currently in early phase clinical trials for cancer. Differential antitumor efficacy of WEE1 blockade alone or combined with CDDP could exist according to p53/cell cycle pathway activity, which might be predictable using an ex vivo 3D primary culture system.
Keywords: cell cycle, cisplatin, p53, urothelial cancer, WEE1
WEE1 inhibitor and cisplatin (CDDP) in combination was significantly more effective than the single treatments. Drug susceptibility assay using an ex vivo cancer tissue‐originated spheroid system showed correlations with the in vivo efficacy of AZD‐1775 alone or combined with CDDP.

1. INTRODUCTION
Bladder cancer is a major malignancy, with 430 000 new cases and 165 000 deaths worldwide each year.1 Histopathologically, most bladder cancer is classified as urothelial carcinoma (UC). Although non‐muscle‐invasive bladder cancer can be treated by transurethral resection (TUR) with an excellent survival outcome,2 muscle‐invasive bladder cancer (MIBC) is highly associated with lymph node involvement, distant metastasis, and disease‐specific mortality.
Until recently, the treatment outcomes of patients with MIBC or metastatic UC had not been remarkably improved for decades. For patients with advanced UC, cisplatin (CDDP)‐based chemotherapy is the center of first‐line systemic treatment with an initial response rate ranging from 50% to 70%.2 However, only a subset of patients achieves durable response or disease control because of early acquisition of drug resistance. Thus, sensitization to CDDP or overcoming CDDP resistance has been a major unmet need for effective treatment of UC patients.
WEE1 is a tyrosine protein kinase that regulates G2/M checkpoints in the cell cycle in response to DNA damage.3 Activated WEE1 phosphorylates the cyclin‐dependent kinase 1 (CDC2)‐cyclin B complex, arresting the cell cycle and gaining time for DNA repair. Additionally, WEE1 maintains genomic integrity through regulating histone synthesis in S phase and stabilizing replication forks during the DNA repair process.4, 5 During anticancer treatment, WEE1 is activated following DNA damage by either chemotherapy or irradiation.6 Thus, WEE1 is involved in resistance to such anticancer treatment through its negative regulation of the cell cycle.
MK‐1775 (also known as AZD‐1775 or adavosertib) is a selective WEE1 inhibitor that inhibits the phosphorylation of CDC2.7 Several studies have shown the synergistic effects of MK‐1775 and DNA‐damaging agents such as CDDP8, 9, 10, 11 and gemcitabine.6, 12, 13 Synergistic effects of MK‐1775 with various types of drugs related to cell‐cycle regulators or DNA repair proteins such as the aurora A kinase (AURKA) inhibitor,14 ATR inhibitor,15 and poly(ADP‐ribose) polymerase (PARP) inhibitor16 have been also reported. MK‐1775 as a monotherapy has also shown antitumor effects in multiple cancer cell lines and tumor xenografts.5 Most recently, clinical efficacy and safety of adavosertib in combination with gemcitabine for the treatment of platinum‐resistant or ‐refractory high‐grade serous ovarian cancer has been shown in a double‐blind, randomized, placebo‐controlled phase II trial.17
Several reports have shown that the differential antitumor activity of the WEE1 inhibitor appears to be particularly dependent on TP53 mutation status.9, 10, 11, 14 Given that TP53 is the most frequently altered gene in UC18, 19 and CDDP is the flagship drug for advanced UC, a synergistic antitumor effect of MK‐1775 and CDDP can be expected. However, the antitumor effects of WEE1 blockade, used alone or in combination with other drugs, vary within the same cancer type depending on the genetic background of the tumor.14, 20 Notably, advanced UC is characterized by a broad spectrum of gene alterations18, 19 and several distinct molecular subtypes based on gene expression profiles.21 Although it is clinically important to investigate potential treatment strategies for UC involving WEE1 blockade in combination with CDDP, the efficacy and genetic determinants for treatment with WEE1 inhibitor alone or in combination with CDDP are not yet fully understood.
In this study, we used multiple experimental systems of UC including cell lines, cell‐based and patient‐derived xenografts, and ex vivo 3D primary culture to investigate potential treatment strategies for UC. We found that MK‐1775 in combination with CDDP yields a higher efficacy than MK‐1775 or CDDP monotherapy in TP53‐altered UC, whereas MK‐1775 monotherapy was as effective as MK‐1775 in combination with CDDP in TP53‐WT UC.
2. Materials and Methods
2.1. Cell‐ and patient‐derived xenograft models
Six‐week‐old female BALB/cAJcl‐nu/nu mice were purchased from CLEA Japan. For the cell‐derived xenograft (CDX) model, 3.0 × 106 UMUC3 cells or 5.0 × 106 RT4 cells with 75 μL PBS and an equal volume of Matrigel HC (Corning) were inoculated subcutaneously into the left flank of mice.
For the patient‐derived xenograft (PDX) model, we used two lines of PDX from muscle‐invasive UC specimens according to previously described methods.22 After successful establishment (>80% engraftment rate in three consecutive passages), four developed PDX tumors were divided into 40 fragments and subcutaneously transplanted to the flank of 6‐week‐old CB17/lcr‐crj SCID mice (Charles River). When the tumor volumes reached more than 100 mm3, mice were randomly assigned into five treatment groups (n = 8 per group): (a) vehicle; (b) 4 mg/kg CDDP monotherapy; (c) 60 mg/kg AZD‐1775 monotherapy; (d) 90 mg/kg AZD‐1775 monotherapy; or (e) combination of 4 mg/kg CDDP +60 mg/kg AZD‐1775. The mice were treated for 3 weeks. The doses and schedules were determined based on previous preclinical studies on AZD‐17757, 10, 15 and a clinical trial on AZD1775 in patients with refractory solid tumors (NCT02511795), and fixed in all animal experiments regardless of the aggressiveness of cell lines and PDX.
Cisplatin was dissolved in corn oil and injected intraperitoneally. AZD‐1775 was dissolved in DMSO/0.5% methylcellulose and given by oral gavage on days 2‐4 and 9‐11 in the 60 mg/kg AZD‐1775 monotherapy and combination groups and days 2‐6 and 9‐13 in the 90 mg/kg AZD‐1775 monotherapy group. In the control (vehicle) group, corn oil was injected intraperitoneally on days 1, 8, and 15, and DMSO was given by oral gavage on days 2‐4 and 9‐11.
The tumor size was measured with a caliper and volume (V) was calculated by the following formula: V (mm3) = L × W × W × 0.5, in which L is the largest and W is the orthogonal diameter (mm) of the tumor. All animal experiments were approved by the Kyoto University animal experiment committees (approval number: MedKyo 18 244). All animals used in this study were treated according to the guidelines for animal experimentation of the experimental animal center of Kyoto University.
We expected that the vehicle‐treated control group would show rapid tumor growth. We also predicted approximately 30% of tumor growth inhibition with CDDP treatment and approximately 60% tumor growth inhibition with the combination treatment. When considering comparative analyses on the means of a continuous measurement in the two groups with an effect size of 0.8 and SD of 0.5, at least eight mice in each treatment group (six in the control group) were needed to have the significance level of the test 0.05 and the statistical power of 80% (https://sample‐size.net/). This number was consistent with a previous similar study.10
2.2. Ex vivo study with cancer tissue‐originated spheroids
The cancer tissue‐originated spheroid (CTOS) method23 was used to evaluate treatment efficacy ex vivo. We used human bladder tumor specimens directly obtained by TUR or from PDX tumors. Briefly, 1‐2‐mm tumor tissue fragments were incubated in DMEM/F12 (Gibco) supplemented with 100 U/mL penicillin, 100 U/mL streptomycin (Nacalai Tesque), and 100 μL Liberase DH (Roche) at 37℃ for 1 hour. DNase I (final concentration; 10 μg/mL) was added and samples were digested for another 15 minutes. Samples from 40 μm to 100 μm were separated using two types of cell strainers and then cultured in CTOS medium (9.06 mL DMEM/F‐12 with GlutaMAX [Gibco], 100 U/mL penicillin, 100 U/mL streptomycin, 200 μL StemPro [Gibco], 720 μL of 25% BSA, and 18.2 μL of 55 mM 2‐mercaptoethanol [Wako]) in a 5% CO2 humidified chamber at 37℃ for 24 hours. After confirming spheroids, a single spheroid was plated in a 10‐μL Matrigel HC (Corning) droplet in a 96‐well plate and incubated for 30 minutes. Then CTOS medium was added to each plate before day 1 photographs. After photographs, the wells were divided into four treatment groups: vehicle (DMSO), CDDP, MK‐1775, or the combination at the final concentration of 1 μM CDDP and 0.2 μM MK‐1775. Each treatment was carried out in triplicate. Photographs were taken at day 1 and day 7, and the volume was calculated as follows: V (mm3) = L × W × W × 0.5, in which L is the largest and W is the orthogonal diameter (mm) of the spheroid. The CTOS growth was calculated by dividing the volume measured on day 7 by that measured on day 1. See supporting information for further details on experimental methods.
3. RESULTS
3.1. WEE1 expression in human UC tissues and cells
We first evaluated the expression of WEE1 in human UC tissues using immunohistochemistry (IHC). We used 34 MIBC specimens (pretreatment specimens) (Table S2) obtained from patients that were consequently treated with neoadjuvant platinum‐based systemic chemotherapy and radical cystectomy. As we envisioned that WEE1 inhibition will be used in chemoresistant settings as well as a first‐line or perioperative drug treatment for advanced bladder cancer, we also evaluated postchemotherapy cystectomy specimens from 29 of the 34 patients; we were unable to evaluate specimens from five patients who had no residual viable malignant cells in cystectomy specimens because of complete remission (ypT0).
Approximately 82% (n = 28) of MIBC pretreatment specimens expressed WEE1 (Figure 1A,B), whereas normal urothelial cells did not (data not shown). The majority of postchemotherapy specimens (69%, n = 20) showed WEE1 expression, although it was significantly lower compared with pretreatment specimens (P = .045, χ2 test). Approximately 70% of the MIBC specimens showed detectable p53 in IHC, suggesting that the majority of tumors harbored TP53 mutation, as reported previously.19 There was no statistically significant difference in p53 immunostaining between pretreatment and postchemotherapy settings (74% vs. 76%, P =.26, χ2 test). In UC cell lines, although J82 cells showed an approximately 2‐fold increase of WEE1 mRNA expression following CDDP treatment (Figure S1A), 253J did not show such CDDP‐induced WEE1 expression (Figure S1B). Additionally, WEE1 expression at the protein level did not show CDDP‐induced upregulation, even in J82 cells (Figure S1C).
FIGURE 1.
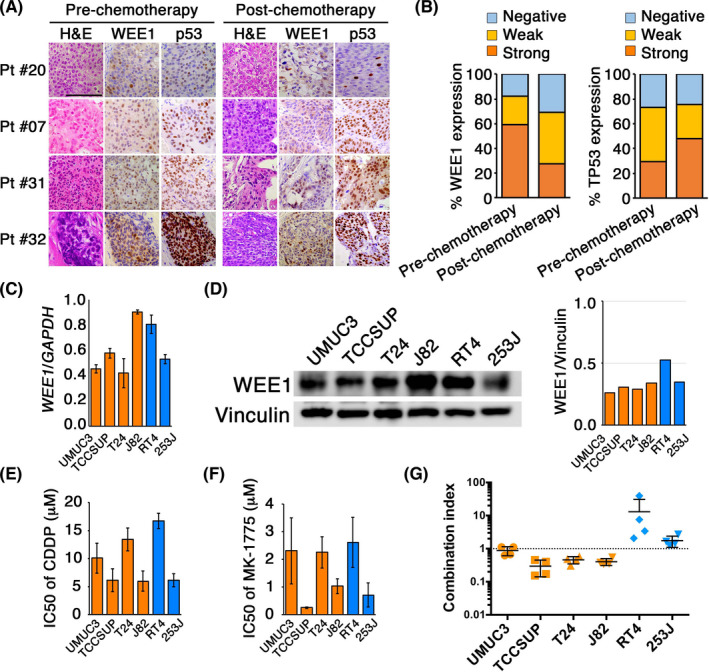
WEE1 expression in human bladder urothelial cancer (UC) tissues and cell lines. A, Muscle‐invasive bladder cancer (MIBC) tissues were evaluated by immunohistochemistry using Abs for WEE1 and p53. Pretreatment tumor tissues were obtained from 34 MIBC patients who consequently received neoadjuvant cisplatin (CDDP)‐based chemotherapy and radical cystectomy. Postchemotherapy tumor tissues were obtained from 29 MIBC patients, excluding five who showed complete response (ypT0) in cystectomy specimens. Representative images of H&E, WEE1, and p53 staining in pre‐ and postchemotherapy tumor specimens from four patients (Pt #20, #07, #31, and #32) are shown. B, Distribution of immunostainings of WEE1 (left) and p53 (right) in pre‐ and postchemotherapy MIBC tissues. C, Expression levels of WEE1 mRNA were evaluated by quantitative RT‐PCR in human UC cell lines with (UMUC3, TCCSUP, T24, and J82 cells; orange bars) or without TP53 mutation (RT4 and 253J cells; blue bars). Values were normalized to GAPDH mRNA levels. D, Western blotting of WEE1 expression in the indicated human UC cell lines. Vinculin served as the loading control. E, F, The IC50 values for (E) CDDP and (F) MK‐1775 as single agents in indicated cells were determined by dose‐effect analyses. Experiments were carried out in triplicate. G, The synergistic effect of CDDP and MK‐1775 was evaluated by combination index (CI) based on WST‐8 assay. CI < 1 indicates synergistic effects, whereas CI = 1 indicates antagonistic effects. Experiments were carried out in triplicate
In terms of correlation between WEE1 and p53 expression, the rate of strongly positive WEE1 increased as p53 immunostaining increased, although the correlation did not reach statistical significance (P =.10, Spearman’s rank correlation test; Figure S1D). There was a statistically significant correlation between WEE1 and p53 expression in postchemotherapy specimens (P =.014, Spearman’s rank correlation test; Figure S1E). These data suggested that WEE1 is expressed in the majority of MIBCs both pre‐ and postchemotherapy and correlated with mutant p53 expression.
We next evaluated WEE1 expression in four human UC cell lines with TP53 alteration (UMUC3, TCCSUP, T24, and J82) and two with wild‐type TP53 (RT4 and 253J). There were no differences in WEE1 mRNA (Figure 1C) and protein (Figure 1D) levels between TP53‐altered and TP53‐WT cell lines. Additionally, no remarkable induction of WEE1 expression was observed following CDDP treatment in TP53‐altered or TP53‐WT cell lines (Figure S1). The six UC cell lines showed dose‐dependent responses to both CDDP and the WEE1 inhibitor MK‐1775 as monotherapy (Figure S2). There were no differences in IC50 values for CDDP (Figure 1E) and MK‐1775 (Figure 1F) between TP53‐altered and TP53‐WT cell lines. However, when the two drugs were given in combination, a synergistic effect was observed in UC cells with TP53 alteration but not in cells with WT TP53 (Figures 1G and S3). These results indicate that CDDP and MK‐1775 show a synergistic effect in UC harboring TP53 mutation.
3.2. Pharmacological and genomic loss of function of WEE1 increased the sensitivity of TP53‐mutant but not TP53‐WT UC cells to CDDP
To further investigate the relationship between TP53 status and the synergistic effect of CDDP and MK‐1775 in UC, we treated the six UC cell lines with CDDP and MK‐1775 alone or in combination. MK‐1775 alone suppressed proliferation by 50%‐75% in all six cell lines (Figures 2A and S4). Cisplatin combined with MK‐1775 showed a significantly greater suppressing effect compared with MK‐1775 alone in TP53‐altered UMUC3, TCCSUP, T24, and J82 cells (P < .05) but not in TP53‐WT RT4 and 253J cells. A similar tendency was observed in colony‐forming assays (Figures 2B and S5).
FIGURE 2.
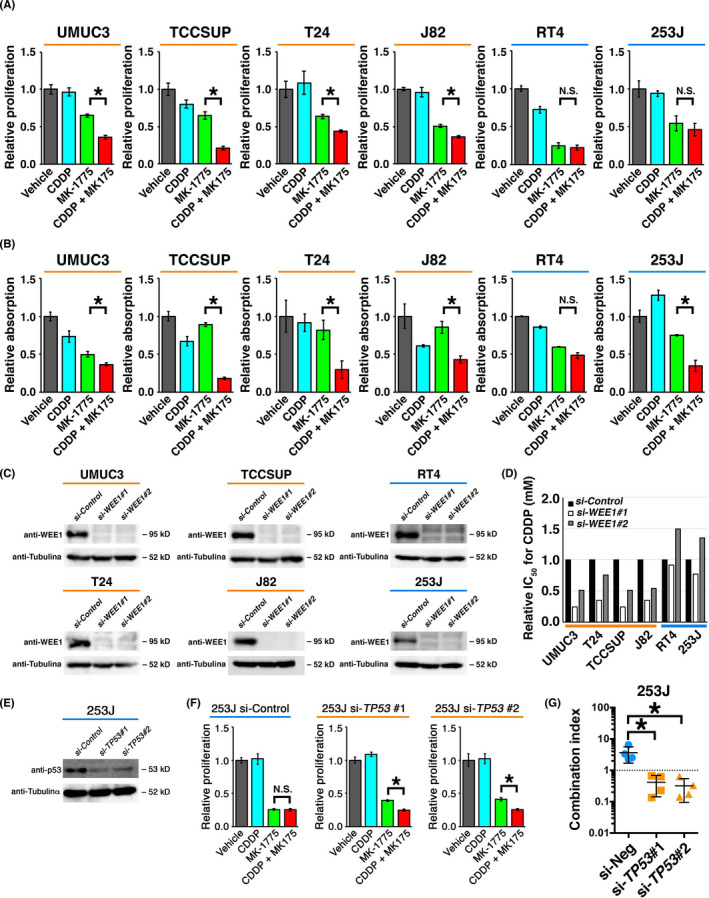
Cisplatin (CDDP) enhanced the inhibitory effect of MK‐1775 on the proliferation of urothelial cancer (UC) cells in a TP53 status‐dependent manner. A, B, Cells were treated with vehicle (DMSO), CDDP, MK‐1775, or CDDP plus MK‐1775. Cell proliferation rate was evaluated by (A) WST‐8 assay after 72 h in triplicate and (B) colony‐forming assay after 7 days. Values were normalized to levels in vehicle‐treated cells. *P < .05. C, Western blot analyses showing the efficiency of siRNA‐mediated knockdown of WEE1 in UC cells 48 h after transfection. D, Chart for relative IC50 values for CDDP. Indicated cells were treated with CDDP for 48 h. Experiments were carried out in triplicate. E, Western blot analysis showing the efficiency of siRNA‐mediated knockdown of TP53 in 253J cells 48 h after transfection. F, Cells were treated as indicated for 72 h and cell proliferation was evaluated by WST‐8 assay. Values were normalized to levels in vehicle‐treated cells. Experiments were carried out in triplicate. *P < .05. G, Charts for combination indices for CDDP and MK‐1775 in cells treated with control siRNA (si‐Neg) or siRNAs for TP53 (si‐TP53#1 and si‐TP53#2). *P < .05. N.S., not significant
To more precisely examine the effect of WEE1 functional loss, WEE1 expression was silenced using two distinct siRNA oligos (Figure 2C). The IC50 of CDDP was lowered by 50%‐75% in TP53‐altered cells with WEE1 knockdown, but not reduced in TP53‐WT cells with WEE1 knockdown (Figure 2D). We next knocked down TP53 in 253J cells (Figure 2E). No significant difference in cell proliferation was observed in 253J cells expressing WT TP53 treated with MK‐1775 alone or in combination with CDDP, but statistically significant differences were observed after TP53 knockdown (approximately 50% reductions; Figures 2F and S6). The combination index was 1 or higher in 253J cells but was significantly decreased to less than 1 by TP53 knockdown (P = .017 for siTP53#1 vs control siRNA [si‐Neg]; P = .015 for siTP53#2 vs si‐Neg; Figures 2G and S7A). These results collectively suggest that WEE1 inhibition alone is somewhat effective in reducing UC cell proliferation irrespective of TP53 status, whereas the synergistic effect with CDDP occurs only in TP53‐mutant UC cells. As WEE1 blockade alone seemed to be effective in retarding the proliferation or tumor growth in TP53‐WT UC cells, we hypothesize that WEE1 inhibition itself confers transient cell‐cycle stoppage by activation of p53 and the subsequent cell‐cycle pathway. When we treated TP53‐WT 253J and RT4 cells with MK‐1775, p53 and downstream p21 were induced (Figure S7B), suggesting that the p53‐p21 axis was activated by WEE1 blockade alone in TP53‐WT UC cells.
3.3. Combined treatment with CDDP and WEE1 blockade induces mitotic catastrophe in TP53‐mutant UC cells
As both CDDP and WEE1 have been implicated in the cell cycle and apoptosis,24 we next investigated the effect of CDDP and MK‐1775 alone or in combination on the cell cycle and apoptosis of UC cells. The proportion of cells in M phase was determined using flow cytometry using Abs against phosphorylated histone H3. Less than 1% of J82 and RT4 control cells were in M phase (Figure 3A,B). J82 cells treated with CDDP and MK‐1775 in combination showed substantial increases of cells in M phase (7%), whereas RT4 cells treated with the combination showed no changes (1%‐2%). Similar results were observed using WEE1 siRNA, with an increase from less than 1.0% to 7% in J82 cells compared with maintaining at 1%‐2% in 253J cells (Figure 3C,D).
FIGURE 3.
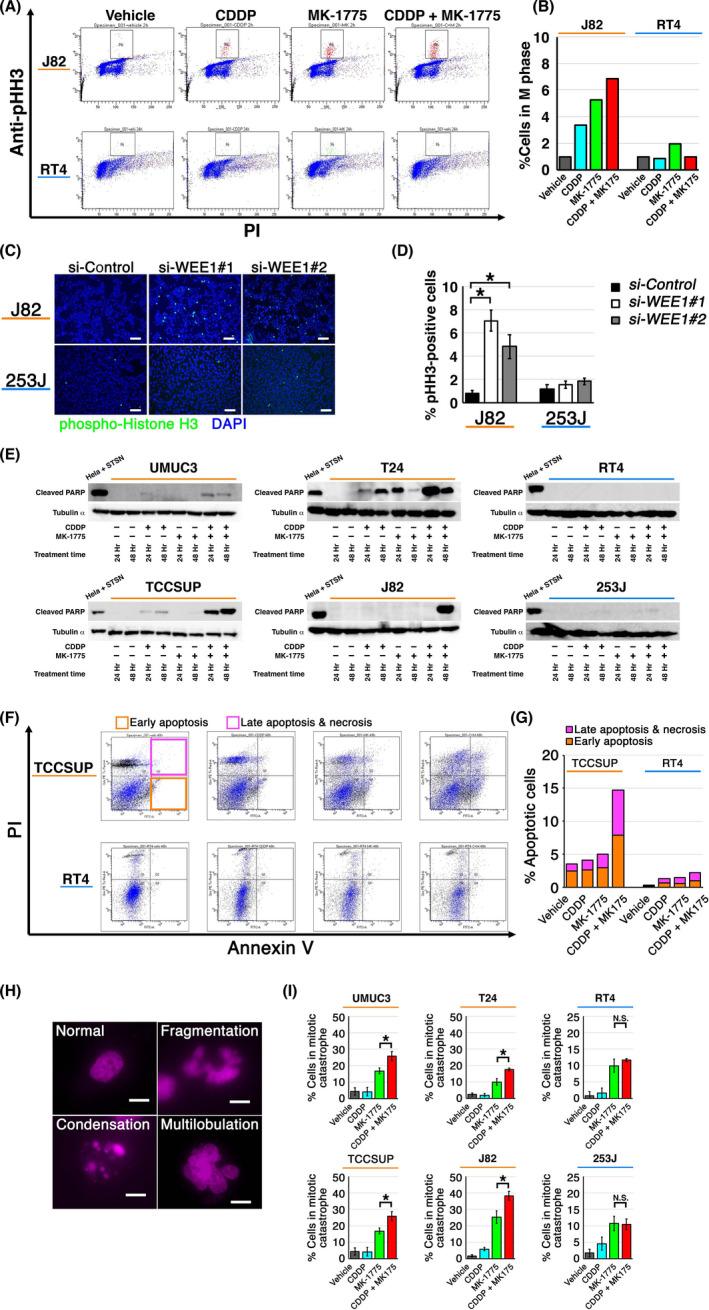
Effect of WEE1 blockade with or without cisplatin (CDDP) on cell cycle and cell death in human urothelial cancer (UC) cells. A, J82 (top) and RT4 (bottom) cells were treated with the indicated drugs for 24 h and then sorted to quantify the M‐phase population defined by phospho‐histone H3 (pHH3)‐high population (gated). B, Proportion of cells in M phase after indicated treatments. Values are normalized to that of vehicle‐treated cells. C, Representative images of immunofluorescence assays carried out using anti‐pHH3 Ab. Cells were transfected with indicated siRNAs and incubated for 48 h. Bar, 100 μm. D, Chart for the proportion of pHH3‐positive cells treated as in (C). Quantitative analysis was performed using five randomly selected microscopic fields. Values were normalized to that for si‐Control. *P < .05. E, UC cell lines were treated as indicated and the expression of cleaved PARP was determined using western blot. HeLa cells treated with 2 μM staurosporine for 4 h served as the positive control. F, TCCSUP (top) and RT4 (bottom) cells were treated with the indicated drugs and then stained with propidium iodide (PI) and annexin V. Cells in early apoptosis and those in late apoptosis or necrosis were gated as indicated. G, Proportion of apoptotic cells treated as in (F). H, Representative fluorescent images of cells in mitotic catastrophe, defined by nuclear fragmentation or multilobulation. Bar, 10 μm. I, Proportion of cells in mitotic catastrophe. Cells were treated with indicated drugs for 24 h in triplicate, fixed and stained with DAPI. Five randomly selected microscopic fields were used for quantification. *P < .05
We next examined the expression of cleaved PARP, a marker of caspase‐dependent apoptosis.25 Cleaved PARP was increasingly detected in TP53‐mutant UC cells treated with CDDP and MK‐1775 in combination but not in TP53‐WT cells (Figure 3E). Consistent with these findings, annexin V assays revealed that TCCSUP cells treated with CDDP and MK‐1775 in combination showed a 3‐fold increase in apoptotic cells whereas RT4 cells showed no changes (Figure 3F,G). Furthermore, nuclear morphology analysis26, 27 revealed significantly increased numbers of cells displaying nuclear fragmentation and multilobulation, which are prominent characteristics of mitotic catastrophe, in TP53‐mutant UC cells treated with CDDP and MK‐1775 in combination (MK‐1775 vs CDDP + MK‐1775: P = .0002 for UMUC3, P = .0003 for T24, P = .029 for TCCSUP, and P = .006 for J82) but not in TP53‐WT cells (MK‐1775 vs CDDP + MK‐1775: P = .139 for RT4, and P = .0902 for 253J; Figure 3H,I). These findings indicate that the combination of CDDP and MK‐1775 induces cell death including apoptosis and mitotic catastrophe in TP53‐mutant UC cells.
3.4. Combined treatment with CDDP and WEE1 blockade increases M‐phase fraction and retards tumor growth of UC CDX and PDX in vivo
We next investigated the efficacy of WEE1 blockade alone or in combination with CDDP in vivo using CDX and PDX. As PDX has several advantages in evaluating drug efficacy in vivo,22 we used two PDX lines established from high‐grade, locally advanced UC of the right renal pelvis (PDX1) and the urinary bladder (PDX2) (Table 1) in addition to CDX derived from UMUC3 (CDX UMUC3) and RT4 (CDX RT4) cells. Both PDXs showed morphological similarity to the original tumor on microscopic observation (Figure S8).
TABLE 1.
Clinicopathologic characteristics of urothelial carcinoma patients with specimens subjected to cancer tissue‐originated spheroid assay
| No. | Age (y) | Sex | Tumor site | T stage | Histological grade |
|---|---|---|---|---|---|
| #01 | 80 | Male | Bladder | T1 | HG |
| #02 | 74 | Male | Bladder | T2 | HG |
| #03 | 71 | Female | Bladder | Ta | HG |
| #04 | 65 | Male | Bladder | Ta + Tis | HG |
| #05 | 89 | Male | Bladder | Ta | LG |
| #06 | 89 | Male | Bladder | T1 | HG |
| #07 | 87 | Female | Left ureter | Ta | HG |
| #08 | 85 | Male | Right renal pelvis | T1 | HG |
| #09a | 76 | Female | Right renal pelvis | T3 | HG |
| #10a | 67 | Male | Bladder | T2 | HG |
Abbreviations: HG, high grade; LG, low grade.
#09 was the source for patient‐derived xenograft (PDX) #01; #10 was the source for PDX #02.
The CDX and PDX model mice were randomized into five treatment groups after tumor engraftment was confirmed: (a) vehicle (control); (b) CDDP (4 mg/kg i.p.); (c) AZD‐1775 (same formula as MK‐1775; 60 mg/kg i.p.); (d) AZD‐1775 (90 mg/kg i.p.); and (e) CDDP (4 mg/kg i.p.) plus AZD‐1775 (60 mg/kg i.p.) (Figure 4A). No regimen induced significant body weight loss (Figure S9) or hematological, renal, or liver function changes (Figure S10) compared with vehicle (control) when given to non‐tumor‐bearing mice.
FIGURE 4.
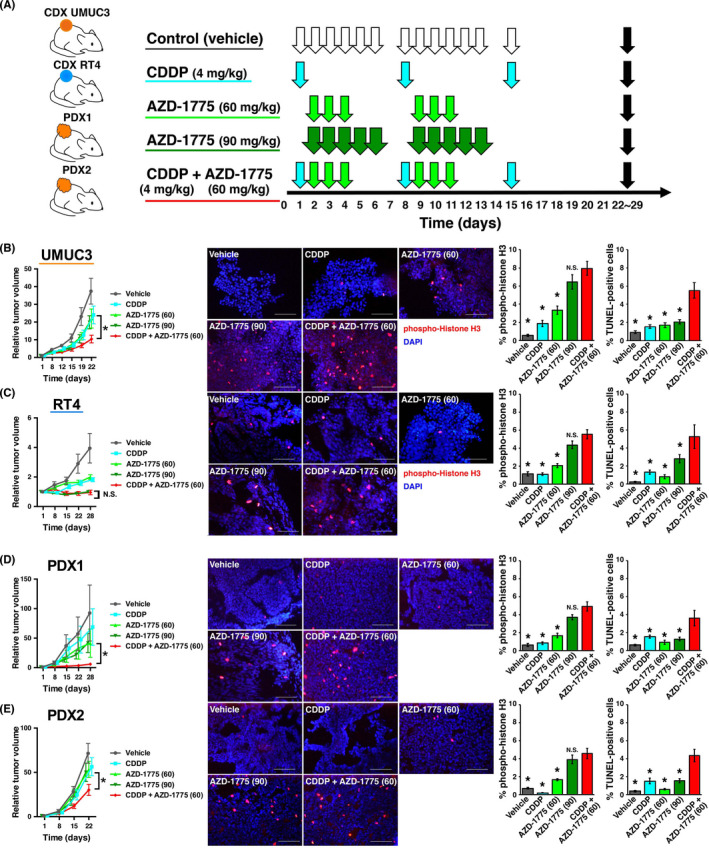
Preclinical modeling of treatment with WEE1 inhibitor alone or in combination with cisplatin (CDDP) using cell‐derived xenografts (CDX) and patient‐derived xenografts (PDX). A, Experimental scheme of preclinical use of AZD‐1775 alone or in combination with CDDP. CDX of UMUC3 cells (CDX UMUC3), CDX of RT4 cells (CDX RT4), and two newly established PDX (PDX1 and PDX2) were implanted into mice. When tumor volumes reached >100 mm3, mice were randomly assigned into five treatment groups (n = 8 per group): control (vehicle, i.p. 6q1w), CDDP alone (4 mg/kg i.p. q1w), AZD‐1775 (same formula as MK‐1775) alone (60 mg/kg i.p. 3q1w), AZD‐1775 alone (90 mg/kg i.p. 5q1w), and CDDP (4 mg/kg i.p. q1w) plus AZD‐1775 (60 mg/kg i.p. 3q1w for two cycles). Tumors were harvested after 3 weeks of treatment (black arrows). B–E, Left panels: tumor growth curves for indicated treatments for (B) CDX UMUC3, (C) CDX RT4, (D) PDX1, and (E) PDX2. Middle panels: representative fluorescent images of tumors harvested after 3 weeks of treatment and immunostained using anti‐phospho‐histone H3 (pHH3). Right panels: proportion of pHH3‐positive and TUNEL‐positive cells quantified from five randomly selected microscopic fields (n = 3 tumors per group). *P < .05 vs CDDP + AZD‐1775 (60 mg)
After two cycles of treatment, tumors on mice treated with CDDP and AZD‐1775 were significantly smaller than tumors from other treatment groups (Figure 4B‐E, left panels), with the exception of CDX RT4 treated with AZD‐1775 alone (90 mg/kg i.p.), which was as effective as the combined treatment (Figure 4C, left panel). In particular, PDX2 did not show growth inhibition or apoptosis by CDDP alone, suggesting that this PDX acts as a CDDP‐resistant model. In this PDX line, combined treatment with CDDP and AZD‐1775 yielded a significant growth inhibition by 2‐fold and increased apoptosis by 3‐fold (Figure 4E).
Tumors that received combined treatment showed the highest proportions of p‐HH3‐positive cells (5%‐8%) in every xenograft line (Figure 4B‐E, middle and right panels), although not significantly different from tumors treated with AZD‐1775 alone (90 mg/kg i.p.). Tumors that received combined treatment showed significantly higher proportions (approximately 2‐fold increases) of TUNEL‐positive cells (from 2%‐3% to 4%‐6%) than other groups in every xenograft line (Figures 4B‐E, right panels, and S11).
These results collectively indicate that high‐dose AZD‐1775 and combined CDDP plus AZD‐1775 are comparable in inducing M phase entry irrespective of TP53 status and suppressing growth of TP53‐WT CDX tumors. These data also suggest that combined CDDP plus AZD‐1775 treatment is the most potent to induce apoptosis and suppress tumor growth in vivo irrespective of TP53 status, although equivalent efficacy can be expected by AZD‐1775 alone in TP53‐WT tumors.
3.5. Efficacy of combined treatment with CDDP and WEE1 blockade in 3D CTOS culture correlates with effects in vivo
Based on the results of drug treatment using the preclinical models, we considered that PDX1 and PDX2 could harbor TP53 alteration. Targeted genomic sequencing of TP53 revealed no mutation (data not shown). However, western blotting and IHC of PDX tumors showed upregulation of MDM2 compared with normal urothelium scarcely expressing it. MDM2 is known as the E3 ligase acting as the pivotal negative regulator of p53 that reportedly showed gene amplification or overexpression in 25% of MIBC19 and protein upregulation in 42%.28 Furthermore, neither PDX1 nor PDX2 showed induction of p53 or p21, a downstream target of p53, following treatment with CDDP and/or AZD‐1775 (Figure 5A,B), which strongly suggested p53/cell cycle pathway inactivation.
FIGURE 5.
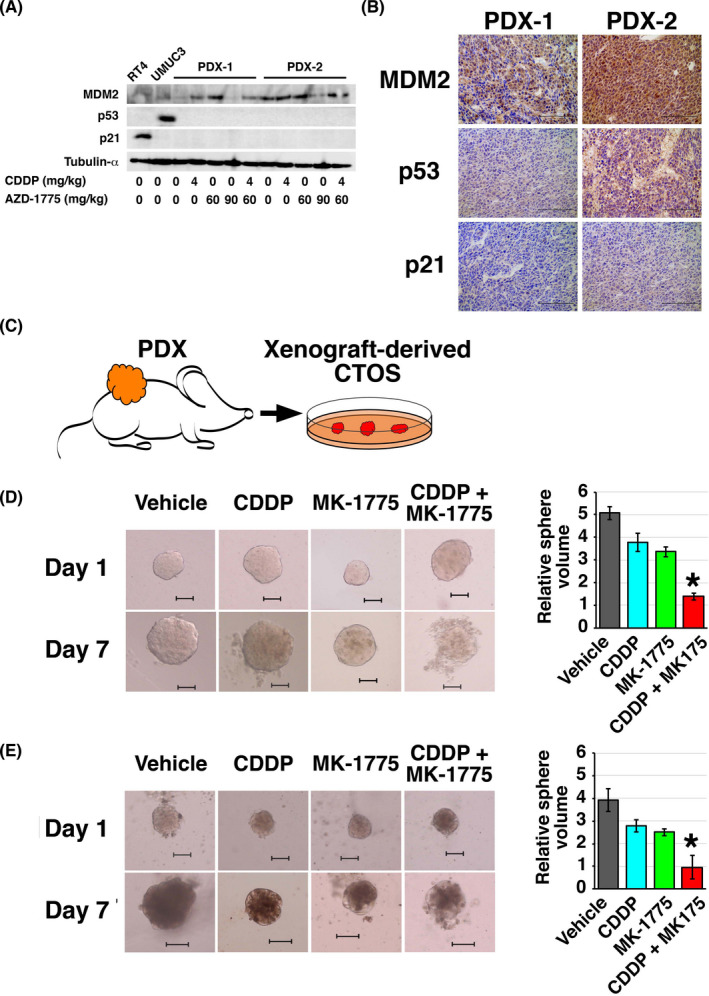
Drug susceptibility assay using an ex vivo patient‐derived xenograft (PDX)‐derived cancer tissue‐originated spheroid (CTOS) culture system. A, MDM2, p53, and p21 expressions were analyzed by western blotting. B, Representative immunohistochemical staining images of the indicated proteins in PDX1 and PDX2. C, Experimental scheme for CTOS culture established from PDX. D, E, Left panels: representative images of CTOS from PDX1 (D) and PDX2 (E) on days 1 and 7 of the indicated treatment. Bar, 100 μm. Right panels: growth of CTOS. CTOS volume was calculated by dividing the volume measured on day 7 with that on day 1. Experiments were carried out in triplicate. *P <.05, CDDP + MK‐1775 vs each of the other groups. CDDP, cisplatin
Our findings indicated that sequencing of TP53 would not accurately predict the efficacy of WEE1 blockade alone or in combination with CDDP but we should look into functional activity of the p53/cell cycle pathway. However, no useful or reliable biomarkers for the p53/cell cycle pathway inactivation have been reported in the clinical setting.29 Therefore, we considered whether a quick primary culture system could predict treatment response. We examined the response of PDX1 and PDX2 to MK‐1775 and/or CDDP using 3D CTOS culture,23 which was reported to predict drug efficacy in vivo30 (Figure 5C). The CTOS culture was successfully established from both PDX1 and PDX2 (Figure 5D,E). MK‐1775 and CDDP in combination was significantly more effective than the single treatments (70%‐80% decrease in sphere volume, Figure 5D,E). Thus, the CTOS primary culture system was shown to be useful to predict the efficacy of WEE1 blockade alone or in combination with CDDP.
3.6. Feasibility of drug susceptibility assessment by direct 3D CTOS culture from human UC surgical specimens
To investigate the feasibility of CTOS culture as a drug susceptibility assay system in the clinical setting, we established CTOSs from eight tumor samples obtained by TUR (Figure 6A, Table 1). Targeting sequencing of TP53 revealed single nucleotide variations of TP53 in three cases (Figure 6B). Drug susceptibility assay using CTOSs showed generally higher efficacy of the combined treatment with MK‐1775 and CDDP compared with MK‐1775 or CDDP alone (Figure 6C) but was not clearly correlated with TP53 status defined by our target sequencing.
FIGURE 6.
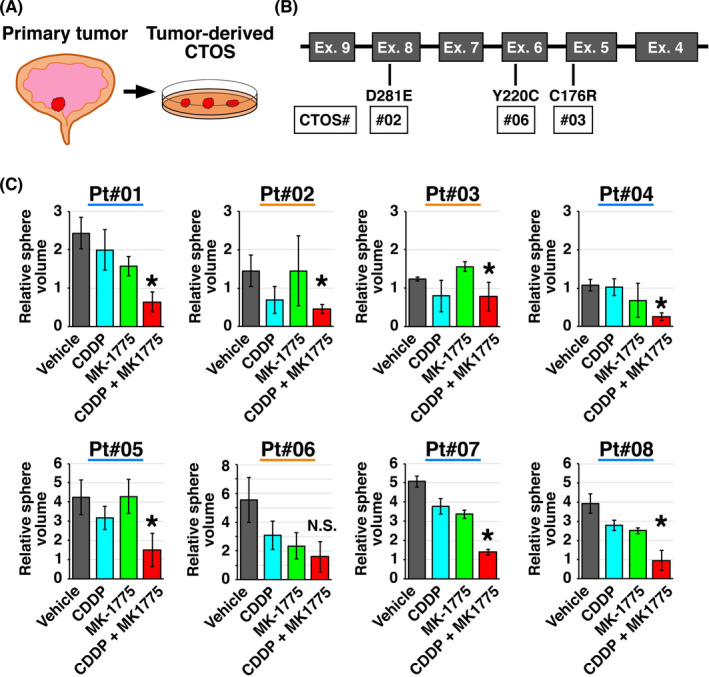
Drug susceptibility tests using tumor‐derived cancer tissue‐originated spheroid (CTOS) models. A, Experimental scheme using CTOS established from surgical specimens obtained by transurethral resection. B, TP53 mutations detected in the original tumors for CTOSs used in the study. C, CTOS from surgical specimens were treated as indicated and growth was calculated by dividing the volume measured on day 7 by that measured on day 1. All experiments were carried out in triplicate. *P <.05, CDDP + MK‐1775 vs MK‐1775 group. CDDP, cisplatin; N.S., not significant; Pt, patient
4. DISCUSSION
The present study has determined the efficacy of WEE1 blockade in UC using multiple in vitro and in vivo models including cell lines, CDX, PDX, and CTOS. Although there has been only limited evidence for the effects of the WEE1 inhibitor in UC, the efficacy of WEE1 blockade has been reported in various other cancers.5, 6, 8, 9, 10, 11, 12, 13, 14, 15, 16, 30 In addition, 56 clinical studies are listed in ClinicalTrials.gov (accessed January 2021) for AZD1775 (MK‐1775), covering a range of cancer types. Some of these trials have been completed and reported promising results with an acceptable toxicity profile.31, 32, 33 Moreover, increasing the tumor shrinkage response by the first‐line chemotherapy is becoming more important in this era when maintenance treatments with checkpoint inhibitors are clinically available.34 Therefore, the present study is timely and clinically relevant. We expect high clinical implementation potential and rapid clinical application for WEE1 inhibitor strategies for UC.
Here we reported the antitumor effect of MK‐1775 as monotherapy in UC. MK‐1775 monotherapy showed equivalent efficacy to the combined treatment using MK‐1775 with CDDP in TP53‐WT UC. This can be explained by p53/cell cycle pathway activation by WT p53 upon immature M phase entry because of WEE1 blockade. These effects could also be attributable to other antitumor functions of MK‐1775, such as DNA‐damaging effects in inducing DNA double‐strand breaks in S phase,5 in addition to the G2/M mechanism. The antitumor efficacy of MK‐1775 alone, but not combined with CDDP, in TP53‐WT tumors is consistent with a previous report.10 However, another report showed conflicting results,9 suggesting the possibility of the presence of other determining factors. Further studies should be undertaken to fully determine the anticancer mechanism of the WEE1 inhibitor alone and in combination with other therapeutic agents and to elucidate predictive biomarkers for the efficacy of the treatments. Until then, the drug susceptibility tests described in the present study could be useful.
We also revealed the synergism of MK‐1775 and CDDP in UC with TP53 alteration. Our findings are clinically relevant, as p53 inactivation is observed in 89% of MIBC cases.19 Additionally, CDDP‐based chemotherapy is frequently used as first‐line systemic therapy for advanced UC. Moreover, our IHC study showed that WEE1 expression was, although not upregulated, maintained even after multiple courses of CDDP‐based chemotherapy and was positively correlated with p53 expression determined by IHC, which could be indicating mutant p53 protein. These observations partly reflect our experimental findings, suggesting that cells with TP53 alteration are more dependent on WEE1. However, caution should be applied in the interpretation of this result because of the limited sample size. Previous studies reported synergistic effects of the WEE1 inhibitor and CDDP using TP53‐deficient or ‐mutant head and neck squamous cell carcinoma (HNSCC) cells.9, 10 These studies focused on isogenic HNSCC cell lines expressing WT or mutant TP53, whereas the present study examined multiple UC cell lines with different backgrounds as well as isogenic cell lines with manipulated TP53 status. These studies and our report together suggest that the synergistic effect of MK‐1775 and CDDP can be achieved in TP53‐altered tumors.
Recent studies have shown that WEE1 is implicated in multidrug resistance, particularly to drugs related to cell cycle modulators, DNA‐damaging agents, or DNA‐repair inhibitors. Consistently, the drug resistance of tumors is associated with increased expression of WEE1 or activation of WEE1 signaling pathways.11, 20 Indeed, several reports have shown the synergistic effect of the WEE1 inhibitor with various anticancer agents in a variety of malignant diseases, such as the AURKA inhibitor alisertib (MLN8237) in HNSCC,14 an ATR inhibitor in diffuse large B‐cell lymphoma,15 a BET inhibitor in non‐small‐cell lung cancer,35 trastuzumab in HER2‐positive breast cancer,36 and avapritinib in gastrointestinal stromal tumors.20 These findings suggest that WEE1 is induced by drug treatment as a process toward resistance. These studies suggest that a synergistic effect with MK‐1775 can be achieved when the cell cycle or DNA damage repair processes in tumor cells are dysregulated by concomitantly administered drugs, which is consistent with the mechanism of action of the WEE1 inhibitor that drives tumor cells to premature mitosis. Therefore, the concomitant inhibition of WEE1 with anticancer agents is a promising strategy to sensitize cancer cells to anticancer treatments and to overcome acquired resistance as well. In particular, CDDP is a strong DNA‐damaging agent and p53 functions as the “gatekeeper” regulating the cell cycle. Therefore, there is a strong rationale for the use of the combined treatment of CDDP and MK‐1775 for UC with p53/cell cycle pathway inactivation.
The lack of a useful disease model has been a barrier in bladder cancer research.37 In this study, in addition to six bladder cancer cell lines and two CDX lines, we also used two PDX lines. Neither of the two PDX lines harbored TP53 alteration but the p53 pathway appeared to be inactivated because the tumors failed to induce p53 or p21 in response to MK‐1775 and/or CDDP. These tumors also showed upregulated expression of MDM2, the E3 ligase of p53 acting as the pivotal negative regulator of p53. Overexpression of MDM2 was reported in approximately 25% of MIBC cases with p53/cell cycle pathway inactivation.19 Additionally, MDM2 activates the AKT/mTOR pathways in a p53‐independent manner.38 There has been a lack of a UC model with p53/cell cycle pathway inactivation by MDM2 overexpression.22, 37 Therefore, our PDX lines are valuable as a UC model of MDM2‐mediated p53 inactivation, particularly considering that previous studies showed conflicting results with regard to the association between MDM2 expression and disease stage and grade in UC.28 In addition to upregulated expression of MDM2, other biological aberrations that cannot be detected by the target sequence of TP53 are known to inactivate the p53/cell‐cycle pathways including loss of CDKN2AARF and homozygous deletion of TP53.36
The present study has shown the potential usefulness of an ex vivo drug susceptibility platform using CTOS of UC. In our two PDX models, the response in CTOS culture correlated with the efficacy of the same treatment in vivo, which is consistent with previous reports from our group30 and others.39 In the eight CTOSs established directly from TUR specimens, combined treatment with CDDP and MK‐1775 showed efficacy in most cases. This may be biased by a higher establishment rate of CTOS in tumors with p53 inactivation, which has been associated with tumor initiation ability or cancer stemness.40, 41 Several investigators have shown the usefulness of organoid‐based platforms for drug screening in various malignancies including lung,42 liver,43 and gynecological cancers.44, 45 Thus, CTOS can be a potent system for drug susceptibility tests that can compensate for the lack of good predictive markers for the efficacy of drugs.
This study has several limitations. The PDX lines used in the present study were not diverse; both appeared to have p53/cell‐cycle pathway inactivation accompanied with upregulation of MDM2 expression despite preservation of WT TP53. Drug susceptibility assays using CTOS included non‐muscle‐invasive tumors to have TP53‐WT tumors, since most of the MIBCs have TP53 alterations. In addition, the outcomes of drug susceptibility assays were not clinically validated because of the unavailability of the WEE1 inhibitor in the clinic.
In conclusion, the present study has revealed that MK‐1775 in combination with CDDP yields higher efficacy than MK‐1775 or CDDP monotherapy in TP53‐altered UC. MK‐1775 monotherapy was as effective as MK‐1775 in combination with CDDP in TP53‐WT UC. Targeting WEE1 is a promising therapeutic strategy in UC, with a highly specific small molecule inhibitor readily available and currently tested in early phase clinical trials. The antitumor efficacy of WEE1 blockade alone or in combination with CDDP could vary according to p53/cell‐cycle pathway activity. The ex vivo 3D primary culture system could be useful for the prediction of treatment efficacy of WEE1‐targeting therapies.
CONFLICT OF INTEREST
T. Kobayashi reports grants from Astra Zeneca during the conduct of the study. No disclosures were reported by the other authors.
Supporting information
Fig S1‐11
Table S1‐2
Supplementary Material
ACKNOWLEDGMENTS
This study was partly supported by grants‐in‐aid from the Ministry of Education and Science (25713055 and 19H03790 to TK, and 26253078 to OO). AZD‐1775 was provided by AstraZeneca. The authors thank Gabrielle White Wolf, PhD, from the Edanz Group for editing a draft of this manuscript. The authors thank the members of the laboratory at the Department of Urology, Kyoto University Graduate School of Medicine for their intellectual support.
Murakami K, Kita Y, Sakatani T, et al. Antitumor effect of WEE1 blockade as monotherapy or in combination with cisplatin in urothelial cancer. Cancer Sci. 2021;112:3669–3681. 10.1111/cas.15051
Funding Information
Ministry of Education and Science, Grant/Award Number: 25713055, 19H03790, 26253078.
REFERENCES
- 1.Antoni S, Ferlay J, Soerjomataram I, Znaor A, Jemal A, Bray F. Bladder cancer incidence and mortality: A global overview and recent trends. Eur Urol. 2017;71:96‐108. [DOI] [PubMed] [Google Scholar]
- 2.Kamat AM, Hahn NM, Efstathiou JA, et al. Bladder cancer. Lancet. 2016;388:2796‐2810. [DOI] [PubMed] [Google Scholar]
- 3.Do K, Doroshow JH, Kummar S. Wee1 kinase as a target for cancer therapy. Cell Cycle. 2013;12:3159‐3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dominguez‐Kelly R, Martin Y, Koundrioukoff S, et al. Wee1 controls genomic stability during replication by regulating the Mus81‐Eme1 endonuclease. J Cell Biol. 2011;194:567‐579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guertin AD, Li J, Liu Y, et al. Preclinical evaluation of the WEE1 inhibitor MK‐1775 as single‐agent anticancer therapy. Mol Cancer Ther. 2013;12:1442‐1452. [DOI] [PubMed] [Google Scholar]
- 6.Bridges KA, Hirai H, Buser CA, et al. MK‐1775, a novel Wee1 kinase inhibitor, radiosensitizes p53‐defective human tumor cells. Clin Cancer Res. 2011;17:5638‐5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hirai H, Iwasawa Y, Okada M, et al. Small‐molecule inhibition of Wee1 kinase by MK‐1775 selectively sensitizes p53‐deficient tumor cells to DNA‐damaging agents. Mol Cancer Ther. 2009;8:2992‐3000. [DOI] [PubMed] [Google Scholar]
- 8.Moser R, Xu C, Kao M, et al. Functional kinomics identifies candidate therapeutic targets in head and neck cancer. Clin Cancer Res. 2014;20:4274‐4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diab A, Kao M, Kehrli K, Kim HY, Sidorova J, Mendez E. Multiple defects sensitize p53‐deficient head and neck cancer cells to the WEE1 kinase inhibition. Mol Cancer Res. 2019;17:1115‐1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Osman AA, Monroe MM, Ortega Alves MV, et al. Wee‐1 kinase inhibition overcomes cisplatin resistance associated with high‐risk TP53 mutations in head and neck cancer through mitotic arrest followed by senescence. Mol Cancer Ther. 2015;14:608‐619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li J, Pan C, Boese AC, et al. DGKA provides platinum resistance in ovarian cancer through activation of c‐JUN‐WEE1 signaling. Clin Cancer Res. 2020;26:3843‐3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aarts M, Sharpe R, Garcia‐Murillas I, et al. Forced mitotic entry of S‐phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov. 2012;2:524‐539. [DOI] [PubMed] [Google Scholar]
- 13.Rajeshkumar NV, De Oliveira E, Ottenhof N, et al. MK‐1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53‐deficient pancreatic cancer xenografts. Clin Cancer Res. 2011;17:2799‐2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee JW, Parameswaran J, Sandoval‐Schaefer T, et al. Combined aurora kinase A (AURKA) and WEE1 inhibition demonstrates synergistic antitumor effect in squamous cell carcinoma of the head and neck. Clin Cancer Res. 2019;25:3430‐3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Young LA, O'Connor LO, de Renty C, et al. Differential activity of ATR and WEE1 inhibitors in a highly sensitive subpopulation of DLBCL linked to replication stress. Cancer Res. 2019;79:3762‐3775. [DOI] [PubMed] [Google Scholar]
- 16.Fang Y, McGrail DJ, Sun C, et al. Sequential therapy with PARP and WEE1 inhibitors minimizes toxicity while maintaining efficacy. Cancer Cell. 2019;35(6):851‐867.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lheureux S, Cristea MC, Bruce JP, et al. Adavosertib plus gemcitabine for platinum‐resistant or platinum‐refractory recurrent ovarian cancer: a double‐blind, randomised, placebo‐controlled, phase 2 trial. Lancet. 2021;397:281‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.The Cancer Genome Atlas Network . Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robertson AG, Kim J, Al‐Ahmadie H, et al. Comprehensive molecular characterization of muscle‐invasive bladder cancer. Cell. 2017;171(540–56):e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ye S, Sharipova D, Kozinova M, et al. Identification of Wee1 as target in combination with avapritinib for Gastrointestinal Stromal Tumor treatment. JCI Insight. 2021;6(2):e143474. 10.1172/jci.insight.143474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kamoun A, de Reynies A, Allory Y, et al. A consensus molecular classification of muscle‐invasive bladder cancer. Eur Urol. 2020;77:420‐433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inoue T, Terada N, Kobayashi T, Ogawa O. Patient‐derived xenografts as in vivo models for research in urological malignancies. Nat Rev Urol. 2017;14:267‐283. [DOI] [PubMed] [Google Scholar]
- 23.Yoshida T, Okuyama H, Endo H, Inoue M. Spheroid cultures of primary urothelial cancer cells: cancer tissue‐originated spheroid (CTOS) method. Methods Mol Biol. 2018;1655:145‐153. [DOI] [PubMed] [Google Scholar]
- 24.Gonzalez VM, Fuertes MA, Alonso C, Perez JM. Is cisplatin‐induced cell death always produced by apoptosis? Mol Pharmacol. 2001;59:657‐663. [DOI] [PubMed] [Google Scholar]
- 25.Whitacre CM, Zborowska E, Willson JK, Berger NA. Detection of poly(ADP‐ribose) polymerase cleavage in response to treatment with topoisomerase I inhibitors: a potential surrogate end point to assess treatment effectiveness. Clin Cancer Res. 1999;5:665‐672. [PubMed] [Google Scholar]
- 26.Swanson PE, Carroll SB, Zhang XF, Mackey MA. Spontaneous premature chromosome condensation, micronucleus formation, and non‐apoptotic cell death in heated HeLa S3 cells. Ultrastructural observations. Am J Pathol. 1995;146:963‐971. [PMC free article] [PubMed] [Google Scholar]
- 27.Takai H, Tominaga K, Motoyama N, et al. Aberrant cell cycle checkpoint function and early embryonic death in Chk1(‐/‐) mice. Genes Dev. 2000;14:1439‐1447. [PMC free article] [PubMed] [Google Scholar]
- 28.Kriegmair MC, Balk M, Wirtz R, et al. Expression of the p53 inhibitors MDM2 and MDM4 as outcome predictor in muscle‐invasive bladder cancer. Anticancer Res. 2016;36:5205‐5213. [DOI] [PubMed] [Google Scholar]
- 29.Yoshida T, Kates M, Fujita K, Bivalacqua TJ, McConkey DJ. Predictive biomarkers for drug response in bladder cancer. Int J Urol. 2019;26:1044‐1053. [DOI] [PubMed] [Google Scholar]
- 30.Kita Y, Hamada A, Saito R, et al. Systematic chemical screening identifies disulfiram as a repurposed drug that enhances sensitivity to cisplatin in bladder cancer: a summary of preclinical studies. Br J Cancer. 2019;121:1027‐1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cuneo KC, Morgan MA, Sahai V, et al. Dose escalation trial of the Wee1 inhibitor adavosertib (AZD1775) in combination with gemcitabine and radiation for patients with locally advanced pancreatic cancer. J Clin Oncol. 2019;37:2643‐2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leijen S, van Geel RM, Pavlick AC, et al. Phase I study evaluating WEE1 inhibitor AZD1775 as monotherapy and in combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. J Clin Oncol. 2016;34:4371‐4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leijen S, van Geel RM, Sonke GS, et al. Phase II study of WEE1 inhibitor AZD1775 plus carboplatin in patients with TP53‐mutated ovarian cancer refractory or resistant to first‐line therapy within 3 months. J Clin Oncol. 2016;34:4354‐4361. [DOI] [PubMed] [Google Scholar]
- 34.Powles T, Park SH, Voog E, et al. Avelumab maintenance therapy for advanced or metastatic urothelial carcinoma. N Engl J Med. 2020;383:1218‐1230. [DOI] [PubMed] [Google Scholar]
- 35.Takashima Y, Kikuchi E, Kikuchi J, et al. Bromodomain and extraterminal domain inhibition synergizes with WEE1‐inhibitor AZD1775 effect by impairing nonhomologous end joining and enhancing DNA damage in nonsmall cell lung cancer. Int J Cancer. 2020;146:1114‐1124. [DOI] [PubMed] [Google Scholar]
- 36.Sand A, Piacsek M, Donohoe DL, et al. WEE1 inhibitor, AZD1775, overcomes trastuzumab resistance by targeting cancer stem‐like properties in HER2‐positive breast cancer. Cancer Lett. 2020;472:119‐131. [DOI] [PubMed] [Google Scholar]
- 37.Kobayashi T, Owczarek TB, McKiernan JM, Abate‐Shen C. Modelling bladder cancer in mice: opportunities and challenges. Nat Rev Cancer. 2015;15:42‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zheng L, Ren JQ, Li H, Kong ZL, Zhu HG. Downregulation of wild‐type p53 protein by HER‐2/neu mediated PI3K pathway activation in human breast cancer cells: its effect on cell proliferation and implication for therapy. Cell Res. 2004;14:497‐506. [DOI] [PubMed] [Google Scholar]
- 39.Kondo J, Ekawa T, Endo H, et al. High‐throughput screening in colorectal cancer tissue‐originated spheroids. Cancer Sci. 2019;110:345‐355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakayama M, Hong CP, Oshima H, Sakai E, Kim SJ, Oshima M. Loss of wild‐type p53 promotes mutant p53‐driven metastasis through acquisition of survival and tumor‐initiating properties. Nat Commun. 2020;11:2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ohtsuka J, Oshima H, Ezawa I, Abe R, Oshima M, Ohki R. Functional loss of p53 cooperates with the in vivo microenvironment to promote malignant progression of gastric cancers. Sci Rep. 2018;8:2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim M, Mun H, Sung CO, et al. Patient‐derived lung cancer organoids as in vitro cancer models for therapeutic screening. Nat Commun. 2019;10:3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Broutier L, Mastrogiovanni G, Verstegen MM, et al. Human primary liver cancer‐derived organoid cultures for disease modeling and drug screening. Nat Med. 2017;23:1424‐1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kopper O, de Witte CJ, Lohmussaar K, et al. An organoid platform for ovarian cancer captures intra‐ and interpatient heterogeneity. Nat Med. 2019;25:838‐849. [DOI] [PubMed] [Google Scholar]
- 45.Maru Y, Tanaka N, Itami M, Hippo Y. Efficient use of patient‐derived organoids as a preclinical model for gynecologic tumors. Gynecol Oncol. 2019;154:189‐198. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐11
Table S1‐2
Supplementary Material