

Abstract
Acute myeloid leukemia (AML) is hierarchically organized by self‐renewing leukemic stem cells (LSCs). LSCs originate from hematopoietic stem cells (HSCs) by acquiring multistep leukemogenic events. To specifically eradicate LSCs, while keeping normal HSCs intact, the discrimination of LSCs from HSCs is important. We have identified T‐cell immunoglobulin and mucin‐domain containing‐3 (TIM‐3) as an LSC‐specific surface molecule in human myeloid malignancies and demonstrated its essential function in maintaining the self‐renewal ability of LSCs. TIM‐3 has been intensively investigated as a “coinhibitory” or “immune checkpoint” molecule of T cells. However, little is known about its distinct function in T cells and myeloid malignancies. In this review, we discuss the structure of TIM‐3 and its function in normal blood cells and LSCs, emphasizing the specific signaling pathways involved, as well as the therapeutic applications of TIM‐3 molecules in human myeloid malignancies.
Keywords: acute myeloid leukemia, immune checkpoint inhibitor, leukemic stem cells, T‐cell immunity, TIM‐3
Abbreviations
- ADC
antibody‐drug conjugate
- AML
acute myeloid leukemia
- BAT3
HLA‐B–associated transcript 3
- CAR
chimeric antigen receptor
- CEACAM1
carcinoembryonic antigen–related cell adhesion molecule 1
- CTLA‐4
cytotoxic T lymphocyte–associated protein 4
- DCs
dendritic cells
- HCC
hepatocellular carcinoma
- HIF1a
hypoxia‐inducible factor 1‐alpha
- HMA
hypomethylating agent
- HMGB1
high‐mobility group box 1
- HSCs
hematopoietic stem cells
- IFN‐γ
interferon‐γ
- LSCs
leukemic stem cells
- mTOR
mammalian target of rapamycin
- NK
natural killer
- PAG1
phosphoprotein associated with glycosphingolipid‐enriched microdomains1
- PD‐1
programmed cell death 1
- PtdSer
phosphatidylserine
- SH2
Src homology 2
- SPTCL
subcutaneous panniculitis‐like T‐cell lymphoma
- TCR
T‐cell receptor
- TIM‐3
T‐cell immunoglobulin and mucin‐domain containing‐3
- TNF‐a
tumor necrosis factor alpha
- Treg
regulatory T
- VEGF
vascular endothelial growth factor
1. INTRODUCTION
Acute myeloid leukemia (AML) originates from self‐renewing leukemic stem cells (LSCs), and purified LSCs can repopulate human AML in immunodeficient mice after xenogeneic transplantation.1, 2 LSCs possess high self‐renewal ability and can propagate leukemia in vivo. Therefore, eradication of LSCs is important to achieve a “cure” for AML. In humans, AML LSCs were originally identified in the CD34+CD38− fraction, whose phenotype is analogous to normal hematopoietic stem cells (HSCs). Recent advances in research on immunodeficient mice enable us to isolate LSCs out of the CD34+CD38− fraction;3 however, the quiescent LSCs harboring high self‐renewal ability are concentrated within this fraction.4 LSCs originate from multipotent self‐renewing preleukemic HSCs, in which somatic mutations or genetic events required for leukemia progression are accumulated.5, 6, 7, 8, 9 Thus, the discrimination between HSCs and LSCs within the identical CD34+CD38− fraction is important to identify molecular mechanisms that LSCs, but not HSCs, specifically depend upon. Furthermore, identification of LSC‐specific markers can promote the development of novel therapeutic approaches that selectively kill LSCs, while keeping normal HSCs intact.
We have intensively investigated the differences between normal HSCs and LSCs using primary AML samples, and identified T‐cell immunoglobulin and mucin‐domain containing‐3 (TIM‐3) as an ideal surface molecule that clearly distinguishes LSCs from normal HSCs.10, 11 We also demonstrated that TIM‐3 is a functional signaling molecule required for the self‐renewal ability of LSCs in humans.12 TIM‐3 has been thoroughly studied as a “coinhibitory” or “immune checkpoint” molecule of T cells like programmed cell death 1 (PD‐1) and cytotoxic T‐lymphocyte–associated protein 4 (CTLA‐4), and therefore, blockade of TIM‐3 is currently investigated in clinical trials as a novel therapeutic strategy for cancers beyond PD‐1 and CTLA‐4 blockade.13 A unique molecular feature of TIM‐3, however, is the lack of known inhibitory signaling motifs; thus, TIM‐3 can potentially act as a costimulatory molecule in a cellular context–dependent manner.
In this review, we discuss the structure of TIM‐3 and its function in normal blood cells and AML cells, emphasizing the distinct signaling pathways in normal and malignant cells. We also discuss the therapeutic applications of TIM‐3 molecules in human myeloid malignancies.
2. IDENTIFICATION OF TIM‐3 AS AN LSC‐SPECIFIC SURFACE MOLECULE
LSC‐specific surface molecules have been intensively investigated in the last decade. Initial studies identified CD33, CD44, and CD123 as surface molecules exhibiting higher expression in LSCs than in normal HSCs and other normal tissues, and monoclonal antibodies targeting CD33 and CD123 were developed for the treatment of AML. The Antibody‐drug conjugates (ADCs) targeting CD33, such as gemutuzumab ozogamicin, are now clinically available in the treatment of AML, and ADCs targeting CD123 have been investigated in clinical studies. Such ADCs exhibits prominent anti‐AML effects; however, these antibodies also impair CD33 or CD123‐expressing normal HSCs, and hematological toxicities represent the major adverse effect of these ADCs. This warrants the identification of more LSC‐specific surface molecules and the development of novel antibody‐based therapeutic strategies in AML.14
To identify the molecules exclusively expressed in human LSCs, we conducted a comprehensive gene expression analysis of purified CD34+CD38− HSCs and LSCs. Based on transcriptome analysis, we found that HAVCR2 was highly expressed in the vast majority of LSCs, but not in HSCs. HAVCR2 encodes TIM‐3 in humans; therefore, we evaluated TIM‐3 expression in HSCs as well as LSCs. CD34+CD38− LSCs universally express TIM‐3 irrespective of AML subtypes, whereas CD34+CD38− HSCs completely lack this expression (Figure 1). Importantly, not only CD34+CD38− AML cells but also CD34+CD38+ AML cells expressed TIM‐3 at a high level, whereas differentiated CD34− AML cells showed lower expression of TIM‐3 in the same patients (Figure 1). As LSCs harboring leukemia reconstitution potential can be found within more differentiated CD34+CD38+ or CD34− AML fractions, we tested whether TIM‐3 expression could mark functional LSCs. TIM‐3+ AML cells efficiently reconstituted human AML in immunodeficient mice, whereas TIM‐3− AML cells did not.10, 15 Thus, self‐renewing LSCs exclusively reside within TIM‐3+ AML cells irrespective of the conventional CD34+ immunophenotype.
FIGURE 1.
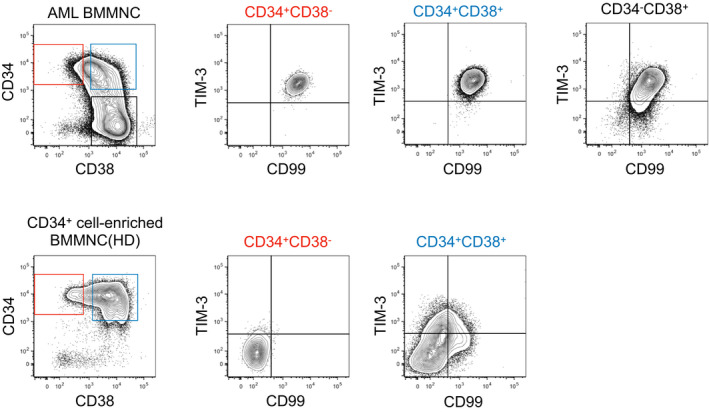
Representative FACS analysis of T‐cell immunoglobulin and mucin domain containing‐3 (TIM‐3) expression in bone marrow mononuclear cells from an acute myeloid leukemia (AML) patient (upper panels) and a healthy donor (HD) (lower panels). Of note, TIM‐3 expression clearly discriminates leukemic stem cells (LSCs) from normal hematopoietic stem cells (HSCs) within the same CD34+CD38− fraction
3. STRUCTURE OF TIM‐3 AND ITS LIGANDS
TIM‐3 was first identified as a 60‐kDa surface molecule expressed on interferon‐γ (IFN‐γ)–producing CD4+ and CD8+ T cells in mice.16 However, many other types of blood cells, including monocytes/dendritic cells (DCs),17 mast cells,18 regulatory T cells (Treg),19 and natural killer (NK) cells20 have been shown to express TIM‐3. TIM‐3 belongs to the TIM family molecules encoded by HAVCR1, HAVCR2, and TIMD4 genes for TIM‐1, TIM‐3, and TIM‐4, respectively. The structure of TIM‐3 consists of an N‐terminal immunoglobulin variable domain (IgV domain), a mucin domain, a transmembrane domain, and a cytoplasmic tail. Figure 2 shows TIM‐3 structure and its ligands. Four distinct TIM‐3 ligands have been identified: galectin‐9, phosphatidylserine (PtdSer), high‐mobility group box 1 (HMGB1), and carcinoembryonic antigen–related cell adhesion molecule 1 (CEACAM1). TIM‐3 has conserved tyrosine residues and a Src homology 2 (SH2) binding motif in its cytoplasmic tail (Figure 2, right panel).
FIGURE 2.
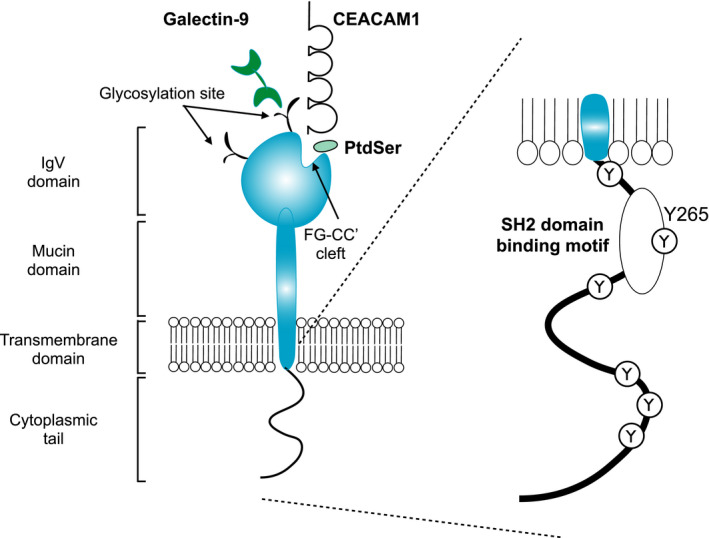
Molecular structure of human T‐cell immunoglobulin and mucin domain containing‐3 (TIM‐3) and its ligands. Galectin‐9, phosphatidylserine (PtdSer), high‐mobility group box 1 (HMGB1), and carcinoembryonic antigen–related cell adhesion molecule 1 (CEACAM1) have been identified as TIM‐3 ligands. Galectin‐9 binds to TIM‐3 via the glycosylation sites of the IgV domain, whereas ligation of PtdSer and CEACAM1 can occur in the FG‐CC cleft. The cytoplasmic tail of TIM‐3 has one Src homology 2 (SH2) binding motif and six tyrosine residues
The first identified TIM‐3 ligand was galectin‐9, an S‐type lectin, which is highly expressed and secreted by various hematopoietic cells. Galectin‐9 has two distinct carbohydrate recognition domains and binds to carbohydrate motifs on the IgV domain of the TIM‐3 molecule. Galectin‐9 ligation to TIM‐3 induces apoptosis of Th1 cells and inhibits their IFN‐γ production.21 TIM‐3 is a heavily glycosylated protein harboring several N‐ or O‐glycosylation sites. It has been shown that galectin‐9 binds to nonglycosylated TIM‐3 with nanomolar affinity (Kd = 2.8 × 10−8M),22 and this binding affinity can be strengthened in the case of the galectin‐9 ligation to glycosylated TIM‐3, which can induce major conformational changes of the TIM‐3/galectin‐9 protein complex.23
PtdSer was identified as a common ligand for all TIM family members based on the crystal structure analysis of TIM‐1, TIM‐3, and TIM‐4.24, 25 PtdSer binds to the pocket framed by the FG and CC’ loops in the IgV domain of TIM‐3,25 and it is postulated that TIM‐3 is crucial for the phagocytosis of apoptotic cells and cross‐presentation by TIM‐3+ DCs.26
HMGB1, known as an alarmin, was identified as a TIM‐3 ligand in DCs; TIM‐3+ tumor‐infiltrating DCs attenuated the nucleic acid–mediated innate immune response through interaction with TIM‐3 and HMGB1.27 In contrast to other TIM‐3 ligands, the binding site of HMGB1 has not been determined; however, HMGB1 binds to TIM‐3 independent of other ligands such as galectin‐9 and PtdSer, suggesting a different binding mechanism from galectin‐9 and PtdSer.27
The most recently identified TIM‐3 ligand is CEACAM1, and it is considered to bind to the cleft framed by the FG and CC’ loops of TIM‐3, similar to PtdSer.28 CEACAM1 is expressed in some hematopoietic cells, epithelial, and endothelial cells and controls immune responses in a contact‐dependent manner.29
Identification of multiple ligands and their suppressive effects on T‐cell immunity prompted the analysis of TIM‐3 molecular structures, especially for the ligand‐binding portions. Binding of PtdSer and CEACAM1 to TIM‐3 does not hamper galectin‐9 ligation to TIM‐3 in mice; the binding sites of these ligands are localized on opposite sides of TIM‐3 IgV domain. A recent study, however, demonstrated significant conformational differences between human and mouse IgV domains of TIM‐3, including the presence of potential glycosylation sites (Asn124) of human TIM‐3 near the framed cleft.30 This suggests the possibility that galectin‐9 ligation to human TIM‐3 can occur adjacent to the PtdSer and CEACAM1 binding sites. Consistently, a single anti‐human TIM‐3 monoclonal antibody (M6903) has been shown to block the binding of three ligands (galectin‐9, PtdSer, and CEACAM1) to TIM‐3.31 Further studies would help us understand the complex interaction of TIM‐3 and its ligands.
4. TIM‐3 SIGNALING AND FUNCTION IN T CELLS
Interestingly, TIM‐3 lacks the conventional inhibitory signaling motifs despite its inhibitory function in T‐cell immunity. Although the detailed intracellular signaling pathway has not been fully elucidated, it has been shown that the Src family kinase proto‐oncogene tyrosine‐protein kinase Fyn (FYN) and HLA‐B–associated transcript 3 (BAT3) can interact with the cytoplasmic tyrosine residues of TIM‐3.32, 33 In the ligand‐unbound form of TIM‐3, BAT3 binds to the TIM‐3 cytoplasmic tail and recruits lymphocyte‐specific protein tyrosine kinase (LCK)34 to maintain T‐cell function (Figure 3A). Thus, ligand‐unbound TIM‐3 can be permissive to T‐cell activation. In contrast, galectin‐9 and/or CEACAM1 ligation to the IgV domain of TIM‐3 triggers the phosphorylation of specific tyrosine residues by (ITK) IL2 Inducible T Cell Kinase,28, 35 resulting in BAT3 release. Subsequently, FYN can be recruited to the same region (Figure 3B). Thus, BAT3 and FYN potentially compete for binding to the cytoplasmic tail of TIM‐3.36 FYN has been shown to suppress T‐cell receptor (TCR) signaling via interaction with phosphoproteins associated with glycosphingolipid‐enriched microdomains1 (PAG1).37 Therefore, FYN‐recruitment induced by TIM‐3 ligands represents one of the possible mechanisms of TIM‐3–mediated inhibitory signaling in T cells. Furthermore, TIM‐3 is found at the immunological synapse where it colocalizes with receptor phosphatases CD45 and CD148, and blocking TIM‐3 has been shown to enhance functional immunological synapse formation in human T cells.34 Thus, TIM‐3 exerts its suppressive effect on T‐cell immunity through multiple molecular mechanisms (Figure 3B).
FIGURE 3.
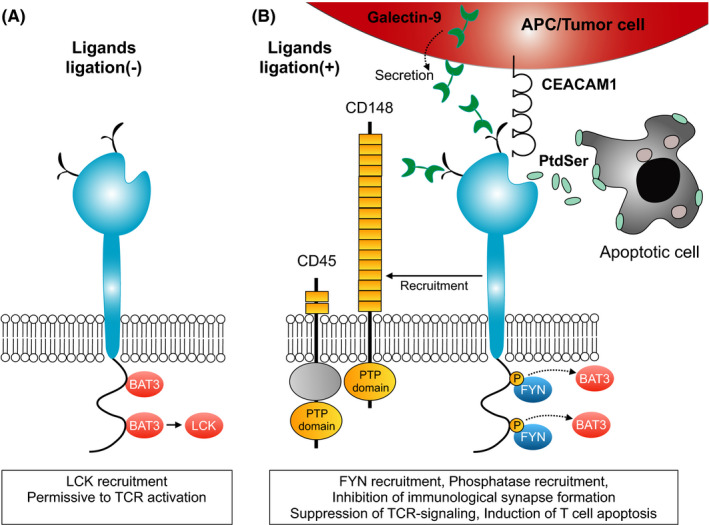
T‐cell immunoglobulin and mucin domain containing‐3 (TIM‐3) signaling in normal T cells. A, In the ligand‐unbound form of TIM‐3, HLA‐B–associated transcript 3 (BAT3) binds to the cytoplasmic tail of TIM‐3 and recruits LCK for maintaining T‐cell receptor (TCR) signaling. B, Ligands binding to the TIM‐3 IgV domain result in the phosphorylation of tyrosine residues in the cytoplasmic tail, leading to the recruitment of FYN instead of BAT3. Furthermore, ligand‐bound TIM‐3 recruits specific phosphatases such as CD45 and CD148 to TIM‐3. These molecules coordinately suppress TCR signaling
Recently, two studies revealed that germline loss‐of‐function mutations in HAVCR2 are associated with the development of subcutaneous panniculitis‐like T‐cell lymphoma (SPTCL).38, 39 Mutations in the IgV domain of TIM‐3 (Tyr82Cys and Ile97Met) result in misfolding of TIM‐3 and the loss of surface TIM‐3 expression in T cells and monocytes, leading to excessive production of proinflammatory molecules because of the absence of the inhibitory function of TIM‐3.38 A dysregulated inflammatory status can promote the development of SPTCL. Furthermore, the high frequency of homozygous and compound Tyr82Cys mutations in SPTCL patients (11 out of 13) indicates the significant role of HAVCR2 mutations in SPTCL development.39 These studies confirm the inhibitory function of TIM‐3 in normal human hematopoiesis.
The suppressive function of TIM‐3 as a “coinhibitory” or “immune checkpoint” molecule has been investigated in the cancer research field,40 because TIM‐3 marks the most dysfunctional T‐cell subset among tumor‐infiltrating CD8+PD‐1+ T cells.41, 42, 43 Consistent with observations that TIM‐3 expression could define the exhausted T cells, antibody blockade of both PD1‐ and TIM‐3 exhibited a synergistic effect against tumor growth in mouse models,41 and improved the tumor antigen‐specific human CD8+ T‐cell response in vitro.42, 43 In addition to the suppressive role in CD8+‐exhausted T cells, recent studies have argued the significance of TIM‐3 in tumor immunity by modifying Treg function; the majority of tumor‐infiltrating human Treg cells were positive for TIM‐3 expression.19, 44 Accordingly, antibody blockade of TIM‐3 suppressed the expression of Treg effector molecules45 and attenuated tumor growth46 in tumor‐bearing mice.
5. TIM‐3 IS A FUNCTIONAL MOLECULE IN AML LSCS
As discussed above, the inhibitory function of TIM‐3 in T cells has been widely accepted. The question is why self‐renewing AML LSCs require TIM‐3 in a cell‐intrinsic manner, as shown in the initial studies.10, 15 To address this issue, we investigated the function of TIM‐3 in AML and found that AML cells secrete galectin‐9 in an autocrine manner. This unique TIM‐3/galectin‐9 autocrine loop enhances the self‐renewal ability of LSCs by inducing β‐catenin accumulation in LSCs.12 Accumulation of β‐catenin plays a pivotal role in maintaining LSCs in murine AML models47 and is associated with the poor prognosis of AML,48 representing the β‐catenin pathway as a potential therapeutic target for AML.47 In humans, genes related to the β‐catenin degradation pathways are frequently mutated in many types of cancers. For example, APC is mutated in the majority of colorectal cancers,49, 50 and CTNNB1 is frequently mutated in endometrial carcinoma50 and hepatocellular carcinoma (HCC).50 CTNNB1 mutations attenuate β‐catenin degradation, leading to the aberrant accumulation of β‐catenin to maintain cancer stemness.51 AML cells, however, do not have canonical Wnt pathway–related mutations,50 suggesting that activation of β‐catenin in AML is mutation independent.52 The TIM‐3/galectin‐9 autocrine stimulatory loop represents a unique molecular mechanism that AML cells universally depend upon to activate the β‐catenin pathway independent of specific mutations. Furthermore, a recent study has shown that β‐catenin induces expression of multiple immune checkpoint molecules, including TIM‐3, through its binding to gene loci in LSCs of murine AML models.53 Thus, TIM‐3 expression itself might be augmented by the TIM‐3/galectin‐9 autocrine stimulatory loop in human LSCs.
In addition to the β‐catenin pathway activation, accumulating evidence supports the leukemia‐propagating function of TIM‐3 in AML. TIM‐3 signaling has been shown to trigger the mammalian target of rapamycin (mTOR) activation via PI3K activation, leading to hypoxia‐inducible factor 1‐alpha (HIF1α) accumulation22 and the secretion of tumor necrosis factor alpha (TNF‐α) and vascular endothelial growth factor (VEGF) in primary AML cells.54 Furthermore, galectin‐9 secreted from AML cells could suppress the function of TIM‐3–expressing immune cells such as NK cells, suggesting that the TIM‐3/galectin‐9 secretory pathway is involved in the immune escape of human AML cells.23 Thus, TIM‐3/galectin‐9 interaction has dual effects in AML: it promotes AML propagation in a cell‐intrinsic manner, while suppressing the immune surveillance activity against AML. In addition to galectin‐9, CEACAM1, and PtdSer, other ligands of TIM‐3 expressed in the tumor microenvironment and apoptotic cells might be involved in the dual effect of TIM‐3 signaling in human myeloid malignancies (Figure 4).
FIGURE 4.
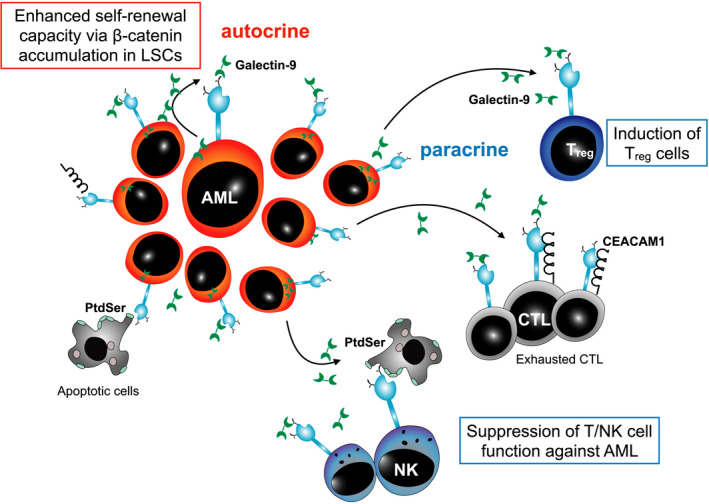
T‐cell immunoglobulin and mucin domain containing‐3 TIM‐3 signaling in human myeloid malignancies. Acute myeloid leukemia (AML) cells secrete galectin‐9 in an autocrine manner. Ligation of galectin‐9 to TIM‐3 can enhance the self‐renewal ability of TIM‐3–expressing AML/LSCs via β‐catenin accumulation. Thus, AML/leukemic stem cells (LSCs) utilize TIM‐3 signaling to propagate leukemia. Strikingly, the identical TIM‐3/galectin‐9 interaction suppressed the immune surveillance activity against AML through the induction of Treg and suppression of T/natural killer (NK) cell function. Other TIM‐3 ligands, such as phosphatidylserine (PtdSer) and carcinoembryonic antigen–related cell adhesion molecule 1 (CEACAM1), might be involved in this dual effect of TIM‐3 signaling in human myeloid malignancies
The question is how TIM‐3 signaling causes opposite effects in TIM‐3+ AML cells and TIM‐3+ T/NK cells. The function and signaling molecules of TIM‐3 have been mainly investigated in T cells, and little is known about the signaling molecules involved in TIM‐3 signaling in AML.
TIM‐3 itself lacks tyrosine kinase activity and requires binding of activated Src family kinases to its cytoplasmic tail; thus, the signal transduction of TIM‐3 mainly depends on the type of Src family kinases, which are coupled with ligand‐stimulated TIM‐3. In T cells, FYN and LCK can interact with TIM‐3 and contribute to its signal transduction; however, we found that AML cells lacked LCK expression and expressed FYN at a lower level than T cells. We, therefore, hypothesized that distinct Src family kinases could be involved in TIM‐3 signal transduction in human AML and found that a Src family kinase, which is expressed in LSCs at a high level but not in normal HSCs, could bind to the TIM‐3 cytoplasmic tail and is activated following galectin‐9 ligation. Specific inhibition of this Src family kinase canceled the β‐catenin accumulation induced by TIM‐3 and galectin‐9 interaction in AML (Sakoda and Kikushige, 2021, unpublished data). Thus, the downstream molecules involved in TIM‐3 signaling are quite different between T cells and AML cells. Consistent with its pivotal role in regulating LSC properties, TIM‐3 represents a stably expressed LSC‐specific surface molecule even in relapsed AML cases.55
Intriguingly, the ectopic expression of TIM‐3 has been identified in several types of human cancers, including HCC,56 breast cancer,57 cervical cancer,58 and renal cell carcinoma.59, 60 Tumor cell–intrinsic TIM‐3 exerts protumoral activity via the NF‐κB/IL‐6/STAT3 axis in HCC,56 and breast cancer cells utilize the TIM‐3/galectin‐9 pathway to facilitate an immune escape from antitumor immunity.57 Thus, TIM‐3 contributes to tumor propagation, suggesting the pleotropic function of TIM‐3 beyond a “coinhibitory” or “immune checkpoint” molecule. Further studies would help us to understand how such different types of cancer can universally utilize TIM‐3 molecules in tumor progression.
6. CLINICAL ASPECTS OF TIM‐3 IN MYELOID MALIGNANCIES
We developed an anti‐human TIM‐3 monoclonal antibody harboring cytotoxic effects against human TIM‐3–expressing cells, and confirmed that this antibody treatment effectively killed LSCs without affecting normal human hematopoiesis in vivo.10 By targeting the TIM‐3 and galectin‐9 interaction, the blocking antibody also attenuated the function of LSCs in vivo.12 In addition to the therapeutic approach using monoclonal antibodies, a recent study reported that the bispecific chimeric antigen receptor (CAR) T cells, targeting CD13 and TIM‐3, efficiently eradicated human AML cells with reduced cytotoxicity against normal hematopoiesis in vivo.61 These preclinical studies using monoclonal antibodies and CAR T cells provide a rationale for the development of therapeutic strategies directly targeting TIM‐3+ LSCs in myeloid malignancies.
The anti‐human TIM‐3 monoclonal antibody has been developed as an “immune checkpoint” inhibitor so far, and the first‐in‐human phase I/II studies have been initiated to test the safety and efficacy of these antibodies in cancer treatment.13 Many of these anti‐human TIM‐3 monoclonal antibodies have been tested in combination with other immune checkpoint inhibitors such as anti‐PD‐1/PD‐L1 monoclonal antibodies.36 Sabatolimab (MBG453), an anti‐human TIM‐3 humanized IgG4 monoclonal antibody with an S228P mutation, has been tested for the treatment of myeloid malignancies, including AML, high‐risk myelodysplastic syndromes, and chronic myelomonocytic leukemia. The combination therapy of sabatolimab and hypomethylating agents (HMAs) has exhibited preliminary encouraging efficacy and safety findings in clinical trials (NCT03066648 for phase I and NCT03946670 for phase II).62 Based on these preceding studies, a phase III multicenter, double‐blind, two‐arm parallel‐group, randomized, placebo‐controlled study of MBG453 added to azacitidine, started in 2020 (NCT04266301). In addition to myeloid malignancies, recent studies identified TIM‐3 expression on primary Hodgkin lymphoma cells63 and adult T‐cell leukemia/lymphoma cells,64 and further studies are required to establish therapeutic strategies targeting TIM‐3 in human blood cancers.
7. CONCLUSION
TIM‐3 has been investigated as a “coinhibitory” molecule in normal hematopoiesis, and recent studies describing the loss‐of‐function germline mutations in HAVCR2 have confirmed this inhibitory function. In contrast, accumulating evidence has shown that human myeloid malignancies employ TIM‐3 signaling to propagate leukemia and escape from the immune surveillance system. Furthermore, several types of cancers have been shown to ectopically express TIM‐3 and utilize the protumoral function of TIM‐3. Thus, TIM‐3 exerts a pleotropic function in a cellular context–dependent manner, an inhibitory function in T cells, and a protumoral effect in malignant cells. Blockade of TIM‐3 signaling, therefore, represents a promising therapeutic approach to eradicate tumor and tumor‐initiating cells by directly inhibiting protumoral TIM‐3 signaling in a cell‐intrinsic manner as well as recovering the antitumor immunity of T cells. Further studies are warranted to clarify how such a structurally simple TIM‐3 molecule exerts pleotropic effects. Elucidation of its unique function in normal and malignant cells can facilitate the future development of highly effective treatment strategies against a variety of cancers by targeting TIM‐3.
CONFLICT OF INTEREST
The authors have the following financial relationships to disclose: patent royalties/gain from Novartis International AG and research funds under contract from Astellas Pharm and Gilead Sciences.
ACKNOWLEDGMENTS
This study was supported in part by a Grant‐in‐Aid for Scientific Research (B) (to YK No. 19109659) and Grant‐in‐Aid for Challenging Exploratory Research (to YK No. 20269344) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan. This study was also supported in part by a research grant from the Princess Takamatsu Cancer Research Fund.
Kikushige Y. TIM‐3 in normal and malignant hematopoiesis: Structure, function, and signaling pathways. Cancer Sci. 2021;112:3419–3426. 10.1111/cas.15042
REFERENCES
- 1.Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645‐648. [DOI] [PubMed] [Google Scholar]
- 2.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730‐737. [DOI] [PubMed] [Google Scholar]
- 3.Goyama S, Wunderlich M, Mulloy JC. Xenograft models for normal and malignant stem cells. Blood. 2015;125:2630‐2640. [DOI] [PubMed] [Google Scholar]
- 4.Ishikawa F, Yoshida S, Saito Y, et al. Chemotherapy‐resistant human AML stem cells home to and engraft within the bone‐marrow endosteal region. Nat Biotechnol. 2007;25:1315‐1321. [DOI] [PubMed] [Google Scholar]
- 5.Miyamoto T, Nagafuji K, Akashi K, et al. Persistence of multipotent progenitors expressing AML1/ETO transcripts in long‐term remission patients with t(8;21) acute myelogenous leukemia. Blood. 1996;87:4789‐4796. [PubMed] [Google Scholar]
- 6.Miyamoto T, Weissman IL, Akashi K. AML1/ETO‐expressing nonleukemic stem cells in acute myelogenous leukemia with 8;21 chromosomal translocation. Proc Natl Acad Sci USA. 2000;97:7521‐7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jan M, Snyder TM, Corces‐Zimmerman MR, et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med. 2012;4:149ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shlush LI, Zandi S, Mitchell A, et al. Identification of pre‐leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506:328‐333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shima T, Miyamoto T, Kikushige Y, et al. The ordered acquisition of Class II and Class I mutations directs formation of human t(8;21) acute myelogenous leukemia stem cell. Exp Hematol. 2014;42(11):955‐965.e5. [DOI] [PubMed] [Google Scholar]
- 10.Kikushige Y, Shima T, Takayanagi S‐I, et al. TIM‐3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell. 2010;7:708‐717. [DOI] [PubMed] [Google Scholar]
- 11.Kikushige Y, Akashi K. TIM‐3 as a therapeutic target for malignant stem cells in acute myelogenous leukemia. Ann N Y Acad Sci. 2012;1266:118‐123. [DOI] [PubMed] [Google Scholar]
- 12.Kikushige Y, Miyamoto T, Yuda J, et al. A TIM‐3/Gal‐9 autocrine stimulatory loop drives self‐renewal of human myeloid leukemia stem cells and leukemic progression. Cell Stem Cell. 2015;17:341‐352. [DOI] [PubMed] [Google Scholar]
- 13.Qin S, Xu L, Yi M, Yu S, Wu K, Luo S. Novel immune checkpoint targets: moving beyond PD‐1 and CTLA‐4. Mol Cancer. 2019;18:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kikushige Y, Miyamoto T. TIM‐3 as a novel therapeutic target for eradicating acute myelogenous leukemia stem cells. Int J Hematol. 2013;98:627‐633. [DOI] [PubMed] [Google Scholar]
- 15.Jan M, Chao MP, Cha AC, et al. Prospective separation of normal and leukemic stem cells based on differential expression of TIM3, a human acute myeloid leukemia stem cell marker. Proc Natl Acad Sci USA. 2011;108:5009‐5014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Monney L, Sabatos CA, Gaglia JL, et al. Th1‐specific cell surface protein Tim‐3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002;415:536‐541. [DOI] [PubMed] [Google Scholar]
- 17.Anderson AC, Anderson DE, Bregoli L, et al. Promotion of tissue inflammation by the immune receptor Tim‐3 expressed on innate immune cells. Science. 2007;318:1141‐1143. [DOI] [PubMed] [Google Scholar]
- 18.Phong BL, Avery L, Sumpter TL, et al. Tim‐3 enhances FcepsilonRI‐proximal signaling to modulate mast cell activation. J Exp Med. 2015;212:2289‐2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao X, Zhu Y, Li G, et al. TIM‐3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PLoS One. 2012;7:e30676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ndhlovu LC, Lopez‐Vergès S, Barbour JD, et al. Tim‐3 marks human natural killer cell maturation and suppresses cell‐mediated cytotoxicity. Blood. 2012;119:3734‐3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu C, Anderson AC, Schubart A, et al. The Tim‐3 ligand galectin‐9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6:1245‐1252. [DOI] [PubMed] [Google Scholar]
- 22.Prokhorov A, Gibbs BF, Bardelli M, et al. The immune receptor Tim‐3 mediates activation of PI3 kinase/mTOR and HIF‐1 pathways in human myeloid leukaemia cells. Int J Biochem Cell Biol. 2015;59:11‐20. [DOI] [PubMed] [Google Scholar]
- 23.Gonçalves Silva I, Yasinska IM, Sakhnevych SS, et al. The Tim‐3‐galectin‐9 secretory pathway is involved in the immune escape of human acute myeloid leukemia cells. EBioMedicine. 2017;22:44‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kobayashi N, Karisola P, Peña‐Cruz V, et al. TIM‐1 and TIM‐4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity. 2007;27:927‐940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeKruyff RH, Bu X, Ballesteros A, et al. T cell/transmembrane, Ig, and mucin‐3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J Immunol. 2010;184:1918‐1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakayama M, Akiba H, Takeda K, et al. Tim‐3 mediates phagocytosis of apoptotic cells and cross‐presentation. Blood. 2009;113:3821‐3830. [DOI] [PubMed] [Google Scholar]
- 27.Chiba S, Baghdadi M, Akiba H, et al. Tumor‐infiltrating DCs suppress nucleic acid‐mediated innate immune responses through interactions between the receptor TIM‐3 and the alarmin HMGB1. Nat Immunol. 2012;13:832‐842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang Y‐H, Zhu C, Kondo Y, et al. CEACAM1 regulates TIM‐3‐mediated tolerance and exhaustion. Nature. 2015;517:386‐390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gray‐Owen SD, Blumberg RS. CEACAM1: contact‐dependent control of immunity. Nat Rev Immunol. 2006;6:433‐446. [DOI] [PubMed] [Google Scholar]
- 30.Gandhi AK, Kim WM, Sun Z‐Y, et al. High resolution X‐ray and NMR structural study of human T‐cell immunoglobulin and mucin domain containing protein‐3. Sci Rep. 2018;8:17512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang D, Jiang F, Zaynagetdinov R, et al. Identification and characterization of M6903, an antagonistic anti‐TIM‐3 monoclonal antibody. Oncoimmunology. 2020;9:1744921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee J, Su EW, Zhu C, et al. Phosphotyrosine‐dependent coupling of Tim‐3 to T‐cell receptor signaling pathways. Mol Cell Biol. 2011;31:3963‐3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rangachari M, Zhu C, Sakuishi K, et al. Bat3 promotes T cell responses and autoimmunity by repressing Tim‐3‐mediated cell death and exhaustion. Nat Med. 2012;18:1394‐1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clayton KL, Haaland MS, Douglas‐Vail MB, et al. T cell Ig and mucin domain‐containing protein 3 is recruited to the immune synapse, disrupts stable synapse formation, and associates with receptor phosphatases. J Immunol. 2014;192:782‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van de Weyer PS, Muehlfeit M, Klose C, Bonventre JV, Walz G, Kuehn EW. A highly conserved tyrosine of Tim‐3 is phosphorylated upon stimulation by its ligand galectin‐9. Biochem Biophys Res Commun. 2006;351:571‐576. [DOI] [PubMed] [Google Scholar]
- 36.Wolf Y, Anderson AC, Kuchroo VK. TIM3 comes of age as an inhibitory receptor. Nat Rev Immunol. 2020;20:173‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davidson D, Schraven B, Veillette A. PAG‐associated FynT regulates calcium signaling and promotes anergy in T lymphocytes. Mol Cell Biol. 2007;27:1960‐1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gayden T, Sepulveda FE, Khuong‐Quang D‐A, et al. Germline HAVCR2 mutations altering TIM‐3 characterize subcutaneous panniculitis‐like T cell lymphomas with hemophagocytic lymphohistiocytic syndrome. Nat Genet. 2018;50:1650‐1657. [DOI] [PubMed] [Google Scholar]
- 39.Polprasert C, Takeuchi Y, Kakiuchi N, et al. Frequent germline mutations of HAVCR2 in sporadic subcutaneous panniculitis‐like T‐cell lymphoma. Blood Adv. 2019;3:588‐595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Acharya N, Sabatos‐Peyton C, Anderson AC. Tim‐3 finds its place in the cancer immunotherapy landscape. J Immunother Cancer. 2020;8:e000911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim‐3 and PD‐1 pathways to reverse T cell exhaustion and restore anti‐tumor immunity. J Exp Med. 2010;207:2187‐2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fourcade J, Sun Z, Pagliano O, et al. PD‐1 and Tim‐3 regulate the expansion of tumor antigen‐specific CD8(+) T cells induced by melanoma vaccines. Cancer Res. 2014;74:1045‐1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fourcade J, Sun Z, Benallaoua M, et al. Upregulation of Tim‐3 and PD‐1 expression is associated with tumor antigen‐specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. 2010;207:2175‐2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yan J, Zhang Y, Zhang JP, Liang J, Li L, Zheng L. Tim‐3 expression defines regulatory T cells in human tumors. PLoS One. 2013;8:e58006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sakuishi K, Ngiow SF, Sullivan JM, et al. TIM3(+)FOXP3(+) regulatory T cells are tissue‐specific promoters of T‐cell dysfunction in cancer. Oncoimmunology. 2013;2:e23849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu J‐F, Wu L, Yang L‐L, et al. Blockade of TIM3 relieves immunosuppression through reducing regulatory T cells in head and neck cancer. J Exp Clin Cancer Res. 2018;37:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y, Krivtsov AV, Sinha AU, et al. The Wnt/beta‐catenin pathway is required for the development of leukemia stem cells in AML. Science. 2010;327:1650‐1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ysebaert L, Chicanne G, Demur C, et al. Expression of beta‐catenin by acute myeloid leukemia cells predicts enhanced clonogenic capacities and poor prognosis. Leukemia. 2006;20:1211‐1216. [DOI] [PubMed] [Google Scholar]
- 49.Aghabozorgi AS, Bahreyni A, Soleimani A, et al. Role of adenomatous polyposis coli (APC) gene mutations in the pathogenesis of colorectal cancer; current status and perspectives. Biochimie. 2019;157:64‐71. [DOI] [PubMed] [Google Scholar]
- 50.Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333‐339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fodde R, Brabletz T. Wnt/beta‐catenin signaling in cancer stemness and malignant behavior. Curr Opin Cell Biol. 2007;19:150‐158. [DOI] [PubMed] [Google Scholar]
- 52.Lane SW, Wang YJ, Lo Celso C, et al. Differential niche and Wnt requirements during acute myeloid leukemia progression. Blood. 2011;118:2849‐2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perry JM, Tao F, Roy A, et al. Overcoming Wnt‐beta‐catenin dependent anticancer therapy resistance in leukaemia stem cells. Nat Cell Biol. 2020;22:689‐700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Silva IG, Gibbs BF, Bardelli M, Varani L, Sumbayev VV. Differential expression and biochemical activity of the immune receptor Tim‐3 in healthy and malignant human myeloid cells. Oncotarget. 2015;6:33823‐33833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Haubner S, Perna F, Köhnke T, et al. Coexpression profile of leukemic stem cell markers for combinatorial targeted therapy in AML. Leukemia. 2019;33(1):64‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang H, Song Y, Yang H, et al. Tumor cell‐intrinsic Tim‐3 promotes liver cancer via NF‐kappaB/IL‐6/STAT3 axis. Oncogene. 2018;37:2456‐2468. [DOI] [PubMed] [Google Scholar]
- 57.Yasinska IM, Sakhnevych SS, Pavlova L, et al. The Tim‐3‐Galectin‐9 pathway and its regulatory mechanisms in human breast cancer. Front Immunol. 2019;10:1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cao Y, Zhou X, Huang X, et al. Tim‐3 expression in cervical cancer promotes tumor metastasis. PLoS One. 2013;8:e53834. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 59.Komohara Y, Morita T, Annan DA, et al. The coordinated actions of TIM‐3 on cancer and myeloid cells in the regulation of tumorigenicity and clinical prognosis in clear cell renal cell carcinomas. Cancer Immunol Res. 2015;3:999‐1007. [DOI] [PubMed] [Google Scholar]
- 60.Kato R, Jinnouchi N, Tuyukubo T, et al. TIM3 expression on tumor cells predicts response to anti‐PD‐1 therapy for renal cancer. Transl Oncol. 2020;14:100918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.He X, Feng Z, Ma J, et al. Bispecific and split CAR T cells targeting CD13 and TIM3 eradicate acute myeloid leukemia. Blood. 2020;135:713‐723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zeidan AM, Komrokji RS, Brunner AM. TIM‐3 pathway dysregulation and targeting in cancer. Expert Rev Anticancer Ther. 2021;21:523‐534. [DOI] [PubMed] [Google Scholar]
- 63.El Halabi L, Adam J, Gravelle P, et al. Expression of the immune checkpoint regulators LAG‐3 and TIM‐3 in classical Hodgkin lymphoma. Clin Lymphoma Myeloma Leuk. 2021;21(4):257‐266.e3. [DOI] [PubMed] [Google Scholar]
- 64.Horlad H, Ohnishi K, Ma C, et al. TIM‐3 expression in lymphoma cells predicts chemoresistance in patients with adult T‐cell leukemia/lymphoma. Oncol Lett. 2016;12:1519‐1524. [DOI] [PMC free article] [PubMed] [Google Scholar]