

Abstract
Pyrosequencing (PSQ) represents the golden standard for MGMT promoter status determination. Binary interpretation of results based on the threshold from the average of several CpGs tested would neglect the existence of the “gray zone”. How to define the gray zone and reclassify patients in this subgroup remains to be elucidated. A consecutive cohort of 312 primary glioblastoma patients were enrolled. CpGs 74‐81 in the promoter region of MGMT were tested by PSQ and the protein expression was assessed by immunohistochemistry (IHC). Receiver operating characteristic curves were constructed to calculate the area under the curves (AUC). Kaplan‐Meier plots were used to estimate the survival rate of patients compared by the log‐rank test. The optimal threshold of each individual CpG differed from 5% to 11%. Patients could be separated into the hypomethylated subgroup (all CpGs tested below the corresponding optimal thresholds, n = 126, 40.4%), hypermethylated subgroup (all CpGs tested above the corresponding optimal thresholds, n = 108, 34.6%), and the gray zone subgroup (remaining patients, n = 78, 25.0%). Patients in the gray zone harbored an intermediate prognosis. The IHC score instead of the average methylation levels could successfully predict the prognosis for the gray zone (AUC for overall survival, 0.653 and 0.519, respectively). Combining PSQ and IHC significantly improved the efficiency of survival prediction (AUC: 0.662, 0.648, and 0.720 for PSQ, IHC, and combined, respectively). Immunohistochemistry is a robust method to predict prognosis for patients in the gray zone defined by PSQ. Combining PSQ and IHC could significantly improve the predictive ability for clinical outcomes.
Keywords: glioblastoma, gray zone, immunohistochemistry, MGMT promoter methylation, pyrosequencing
Pyrosequencing (PSQ) is the golden standard for MGMT promoter status determination for glioblastoma patients but a gray zone does exist by this strategy. Our study revealed that immunohistochemistry (IHC) is a robust method to predict prognosis for patients in the gray zone defined by PSQ. Combining PSQ and IHC could significantly improve the predictive ability for clinical outcomes.

Abbreviations
- AUC
area under the curve
- CI
confidence interval
- DMR
differentially methylated region
- FFPE
formalin‐fixed paraffin‐embedded
- GBM
glioblastoma
- GTR
gross total removal
- IDH
isocitrate dehydrogenase
- IHC
immunohistochemistry
- MGMT
O6‐methylguanine‐DNA methyltransferase
- MMR
mismatch repair
- MS‐MLPA
methylation‐specific multiplex ligation‐dependent probe amplification
- MSP
methylation‐specific PCR
- OS
overall survival
- PFS
progression‐free survival
- PR
partial removal
- PSQ
pyrosequencing
- qMSP
quantitative methylation‐specific PCR
- RANO
Response Assessment in Neurooncology
- ROC
receiver operating characteristic curves
- STR
subtotal removal
- TMZ
temozolomide
1. INTRODUCTION
Glioblastoma is the most common and lethal primary malignant brain tumor.1, 2, 3 The Stupp protocol, including radiation therapy with a concurrent guideline‐recommended daily dose of TMZ and maintenance TMZ adjuvant chemotherapy, was highly recommended to eligible patients after the maximal safe removal of the tumor.4, 5 This protocol profoundly improved quality of life and significantly prolonged survival in a certain cohort of patients, whereas chemoresistance to TMZ in others could lead to failure in yielding therapeutic benefit. Thus, identifying the potentially beneficial subgroup from TMZ is of paramount importance for subsequent clinical decision‐making, accurate survival prediction, and suitable clinical trial enrollment.
Temozolomide could add methyl groups to O6 positions of guanine (O6‐G to O6‐MeG) and result in mispairing of methylated guanine with thymine during replication.6 This change ultimately results in DNA double‐strand breaks and cell death. The MGMT gene encodes for a DNA repair enzyme that could obviate this procedure and consequently confer resistance to TMZ.7 Conversely, methylation of MGMT promoter could silence the gene expression and enhance the chemotherapeutic effect.8, 9 Physicians have reached a consensus that the MGMT promoter status determination should be implemented routinely to predict alkylating agent response. Nevertheless, the best methods and optimal cut‐off definitions for MGMT status determination remain controversial.10
Currently, MSP, including qMSP, and PSQ have been widely used to determine the status of MGMT promoter. Pyrosequencing could provide unparalleled quantitative methylation results of each individual CpG site sequenced, which is highly recommended and further considered as the “gold standard” for MGMT promoter methylation testing.11, 12, 13, 14 However, the status of MGMT promoter should not be arbitrarily dichotomized into methylated or unmethylated by the optimal threshold obtained from average methylation levels of CpGs tested. The intermediate methylation status, termed the gray zone, does exist due to the profound heterogeneity of methylation levels among CpG sites. Reclassifying patients in the gray zone might determine the true beneficial subgroup from TMZ treatment and improve the survival prediction efficiency.
Immunohistochemistry could assess MGMT status at the protein level and directly evaluate gene function.15 Posttranscriptional modifications of MGMT mRNA, impaired MMR system function, and methylation in the gene body could result in discordance between methylation status and protein expression.16 Although interobserver variability and inferior survival prediction efficiency render IHC less reliable for MGMT status determination in some research, results from well‐designed detection strategies were still illuminating.17, 18, 19, 20 In this study, we tried to explore whether IHC could provide useful information to classify patients in the gray zone, and further assist to improve clinical predictive performance for PSQ results.
2. MATERIALS AND METHODS
2.1. Patients
From 1 March 2013 to 15 August 2018, a consecutive cohort of adult newly diagnosed GBM patients were included in this retrospective study. All tissue sections were meticulously reviewed by three senior neuropathologists for a consensus diagnosis based on 2016 WHO classification of brain tumors.21 The IDH1/2 mutant GBMs were excluded to eliminate their influence on the outcome.22 The Stupp protocol was prescribed after surgery with a waiting period of approximately 3‐5 weeks. Patients with multifocal lesions, inadequate follow‐up, insufficient clinical or radiological data, and rejection of Stupp protocol, were excluded (Fig. S1). This study was approved by the institutional review board of Capital Medical University, according to the principles of the Declaration of Helsinki.
2.2. DNA isolation and PSQ testing for MGMT promoter methylation
Pyrosequencing was carried out according to the manufacturer’s instructions. Briefly, genomic DNA was isolated from 10 FFPE sections (5‐8 μm) of tumor tissue with QIAamp DNA FFPE Tissue Kits (Qiagen) and further cleaned and purified. DNA concentrations were ≥30 ng/μl as assessed on a Nanodrop2000 and ≥2 μg of sample was used for bisulfite conversion and PCR. Bisulfite‐treated DNA was amplified and eight CpG sites containing CpG sites 74‐81 in exon 1 of the MGMT promoter region (genomic sequence on chromosome 10 from 131 265 507 to 131 265 544, CGctttgCGtccCGaCGccCGcaggtcctCGCGgtgCG) were tested using MGMT Methylation Detection Kits (Gene Tech). Analyzed sequences were YGTTTTGYGTTTYGAYGTTYGTAGGTTTTYGYGGTGYGTA. Pyrosequencing was undertaken using a PyroMarker Q96 instrument and data were analyzed using PyroMarker Q96 software (Qiagen).
2.3. Molecular information
Molecular status was determined as previously described.23, 24 Briefly, IDH1 R132, IDH2 R172, and TERT C228T/C250T mutation was tested by Sanger sequencing. BRAF V600E, FGFR1, and H3K27M mutation were evaluated by Sanger sequencing for exclusion when required. The 1p/19q status was identified by FISH. The Ki‐67 index, expression of epidermal growth factor receptor, MMP‐9, TP53, vascular endothelial growth factor, and PTEN were detected by IHC. Patients were divided into high (≥30%) or low (<30%) expression groups for further analysis based on IHC results.25
2.4. Immunohistochemical staining for MGMT expression
Formalin‐fixed paraffin‐embedded tissues were sectioned and stained with H&E using the standard protocol. Immunohistochemistry was carried out according to the manufacturer’s instructions and previous reports.14, 17, 19, 20, 26, 27 Five famous neuropathologists (Dehong Lu, Xuanwu Hospital, 45 years of experience in neuropathology; Yueshan Piao, Xuanwu Hospital, 25 years of experience in neuropathology; Xueling Qi, Sanbo Brain Hospital, 26 years of experience in neuropathology; Luo Lin, Beijing Tiantan Hospital, 50 years of experience in neuropathology; and Zifen Gao, Peking University Cancer Hospital, 45 years of experience in cancer pathology) provided valuable guidance and approved our protocol. Briefly, IHC staining was carried out on 4‐μm sections heated for 30 minutes at 60°C using a Leica Bond‐MAX fully automated staining system with the Bond Polymer Refine detection system. Antigen retrieval and dilution were carried out as follows: MGMT clone UMAB56 (OriGene) 1:100 with Epitope Retrieval Solution 2 (pH 9) at 100°C for 15 minutes. The MGMT Ab was optimized using breast tissue as a positive control. The whole images of slices were screened and stored by Leica Biosystem Imaging, and the images were reviewed from Aperio ImageScope (version 12.4.3). Fields with more than 80% tumor cells were selected to calculate IHC scores. Two trained neuropathologists (GHD, 15 years of experience in neuropathology; and WWZ, 5 years of experience in neuropathology) independently evaluated the staining results. An individual patient was screened for at least three isolated fields, and each field must be evaluated with more than 100 qualified tumor cells. Necrotic areas and perivascular zones were excluded. The IHC score was calculated as the mean value of the percentage of MGMT positive cells in all assessed tumor cells from all fields. If the IHC scores were ambiguous (ranging between 20% and 60%, or decided by the two neuropathologists), CD31 clone EP78 (OriGene) 1:200 to exclude endothelial cells, LCA clone 2B11 and PD7/26 (OriGene) 1:200 to exclude lymphoma cells, and CD68 clone KP1 (OriGene) 1:100 staining to excluded microglia and macrophages, were used to generate the final IHC scores (Fig. S2).
2.5. Radiological assessment and follow‐up
Contrast‐enhanced MRI was undertaken on a 3.0 T clinical scanner (Siemens Trio Tim or GE) as previously described. The resection degree was defined according to the following equation: (preoperative tumor volume − postoperative tumor volume) / preoperative tumor volume, as GTR (>98% resection), STR (90%‐98%), and PR (50%‐90%) based on 48‐72 h postoperative MRI. Contrast‐enhanced MRI was meticulously followed with an interval of 8‐12 weeks or if necessary. The definition of tumor progression was based on the RANO and modified RANO criteria.28, 29 Progression‐free survival was defined as the duration from initial surgery to the time of tumor progression, and OS was termed as the duration between the initial surgery and the death, or date of the last follow‐up.
2.6. Statistical analysis
All statistical analyses were carried out with GraphPad Prism 8.0.1 (GraphPad Software, R (version 4.0.3), and R studio (version 1.3.1093). For continuous variables, Student’s t test or one‐way ANOVA was applied and the Mann‐Whitney U test or Kruskal‐Wallis tests for nonparametric data. Receiver operating characteristic curves were constructed to calculate the AUC and the optimal cut‐off value by the Youden index (sensitivity + specificity − 1) (package pROC for R). The patients were stratified based on the median OS (20.0 months). Kaplan‐Meier plots were used to estimate the survival rate of patients, and the differences between curves were compared by the log‐rank test. A Cox proportional hazard regression model was constructed to estimate the hazard ratio for each potential prognostic factor. All tests were two‐sided. P < .05 was considered statistically significant.
3. RESULTS
3.1. Descriptive characteristics
In this retrospective study, a total of 345 de novo adult supratentorial IDH WT GBMs were assessed, of which 33 cases were excluded due to preoperative leptomeningeal dissemination, comorbid visceral carcinoma, differential postoperative management protocols, and a loss to follow‐up (Fig. S1). The mean age at diagnosis was 50.0 years (range, 18‐75 years). Two hundred and six patients (66.0%) had a relatively good status (Karnofsky Performance Status score > 70). All patients accepted open surgery, and the number of patients who achieved GTR, STR, and PR was 165 (52.9%), 127 (40.7%), and 20 (6.4%), respectively. In 167 cases with assessable TERT promoter status, more than half of them (93/167, 55.7%) were mutant (Table 1). The median follow‐up time was 28.0 months, and the median PFS and OS were 9.5 months and 20.0 months, respectively. The number of patients who developed tumor recurrence was 267 (85.6%), and 221 patients (70.8%) have died.
TABLE 1.
Clinical, radiological, and pathological information of 312 glioblastoma patients in this study
| Study cohort n = 312 | n | % |
|---|---|---|
| Gender | ||
| Female | 118 | 37.8 |
| Male | 194 | 62.2 |
| Age at diagnosis (y) | ||
| Mean | 50.0 | – |
| Median | 51.5 | – |
| Range | 18‐75 | – |
| Preoperative KPS score | ||
| >70 | 206 | 66.0 |
| ≤70 | 106 | 34.0 |
| Extent of resection | ||
| GTR | 165 | 52.9 |
| STR | 127 | 40.7 |
| PR | 20 | 6.4 |
| TERT promoter (n = 167) | ||
| Mutant | 93 | 55.7 |
| WT | 74 | 44.3 |
| MGMTp | ||
| Methylated (mean ≥ 12%) | 130 | 41.7 |
| Unmethylated (mean < 12%) | 182 | 58.3 |
| MGMT protein | ||
| <30% | 147 | 47.1 |
| ≥30% | 165 | 52.9 |
| PFS (mo) | ||
| Median | 9.5 | |
| Range | 1.0‐75.0 | |
| OS (mo) | ||
| Median | 20.0 | |
| Range | 1.0‐80.0 | |
Abbreviations: GTR, gross total removal; KPS, Karnofsky Performance Status; OS, overall survival; PFS, progression‐free survival; PR, partial removal; STR, subtotal removal.
3.2. Heterogeneity of methylation levels among CpGs and different cut‐off values for predicting OS
Two genomic regions, DMR1 (CpG sites 25‐50) and DMR2 (CpG sites 73‐90), are strongly concordant with MGMT expression and patient outcome (Figure 1A), and the latter is the critical region for methylation testing.30 Thus, the CpG sites 74‐81 in DMR2 were tested in our study to determine MGMT promoter status. The heterogeneity of methylation levels among CpGs implied their different ability in predicting the outcome (example in Figure 1B). Although all the CpGs could predict survival, each individual CpG site harbored its specific AUC value (range, 0.677‐0.722; Figure 1C and Table 2). Based on the maximal of Youden’s index, the optimal cut‐off values for each individual CpG site varied pronouncedly (range, 5%‐11%; Figure 1D
FIGURE 1.
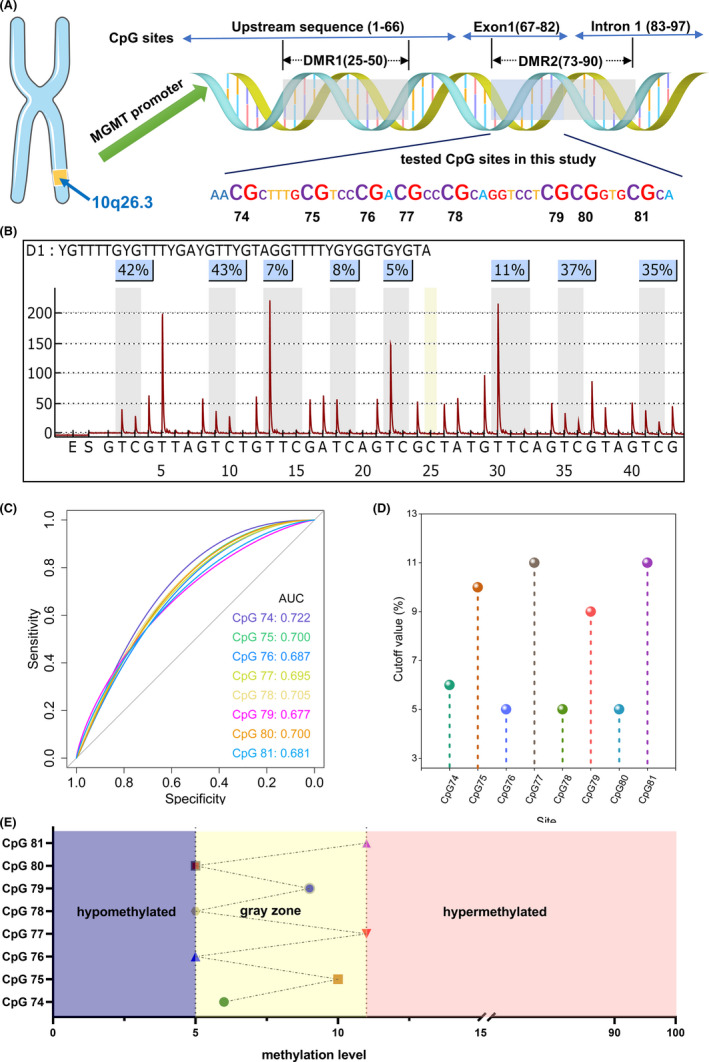
(A) MGMT locates at 10q26.3 and includes 97 CpG sites. Differentially methylated region 1 (DMR1, CpG 25‐50) and DMR2 dominate the gene expression. DMR1 is always methylated when DMR2 is methylated, thus CpGs 74‐81 were tested in our study. (B) A typical example with heterogeneous CpGs (5%‐43%). Although the average methylation level was high (23.5%), the heterogeneity of CpGs was quite prominent. (C) Each individual CpG could predict survival and harbored a specific value of area under the receiver operating characteristic curve. (D) Optimal threshold for each CpG was quite different (5%‐11%). (E) Patients could be separated into the hypomethylated subgroup (methylation levels of all CpGs below the corresponding optimal thresholds, violet), hypermethylated subgroup (methylation levels of all CpG sites above the corresponding optimal thresholds, pink), and the gray zone subgroup (remaining patients, yellow)
TABLE 2.
Pyrosequencing results of individual CpGs and immunohistochemistry (IHC) results for determining MGMT status in glioblastoma
| Site | AUC (95% CI) | Cut‐off value (%) | Sensitivity (%) | Specificity (%) |
|---|---|---|---|---|
| Average | 0.709 (0.652‐0.768) | 12 | 60.1 | 72.2 |
| CpG 74 | 0.722 (0.655‐0.776) | 6 | 70.3 | 66.1 |
| CpG 78 | 0.705 (0.644‐0.763) | 5 | 67.7 | 64.4 |
| CpG 75 | 0.700 (0.642‐0.759) | 10 | 65.2 | 67.2 |
| CpG 80 | 0.700 (0.643‐0.760) | 5 | 76.0 | 60.0 |
| CpG 77 | 0.695 (0.634‐0.757) | 11 | 57.6 | 76.7 |
| CpG 76 | 0.687 (0.626‐0.750) | 5 | 73.4 | 58.9 |
| CpG 81 | 0.681 (0.624‐0.742) | 11 | 60.1 | 71.7 |
| CpG 79 | 0.677 (0.623‐0.744) | 9 | 65.2 | 67.2 |
| IHC | 0.653 (0.527‐0.768) | 30% | 73.4 | 58.9 |
Abbreviations: AUC, area under the receiver operating characteristic curve; CI, confidence interval.
and Table 2). This phenomenon informed us that using a single cut‐off value from averaged results to dichotomize patients into methylated and unmethylated subgroups might be technically insufficient and neglect some crucial information.
3.3. Gray zone defined by PSQ and its intermediate prognosis
Based on the distinct optimal cut‐off values amongst CpGs 74‐81, we further separated patients into three subgroups: the hypomethylated subgroup (methylation levels of all CpGs were below the corresponding optimal cut‐off values, n = 126, 40.4%), hypermethylated subgroup (methylation levels of all CpGs were above the corresponding optimal cut‐off values, n = 108, 34.6%), and the gray zone subgroup (remaining patients, n = 78, 25.0%) (Figures 1E and 2A).
FIGURE 2.
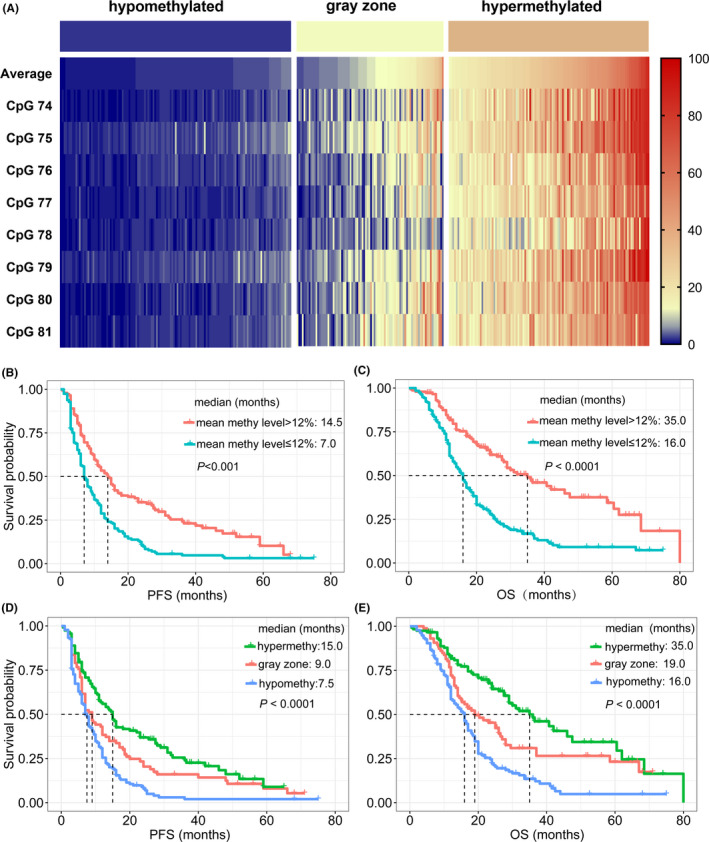
(A) Heatmap for methylation levels amongst CpGs 74‐81 in hypomethylated, gray zone, and hypermethylated groups of glioblastoma patients. (B, C) The optimal threshold (12%) for average methylation levels could effectively predict survival, in terms of (B) progression‐free survival (PFS) and (C) overall survival (OS). (D, E) An intermediate prognosis was observed for patients in the gray zone (PFS in (D) and OS in (E))
Although the optimal threshold of mean methylation levels from CpGs 74‐81 (12%) could effectively predict clinical outcomes (median PFS for ≤12% and >12%: 7.0 vs 14.5 months, P < .001, Figure 2B; median OS: 16.0 vs 35.0 months, P < .001, Figure 2B), refinement of the classification strategy could help us better understand survival significance of MGMT. An intermediate prognosis for gray zone patients was observed in this cohort (median PFS: 7.5, 9.0, and 15.0 months in hypomethylated, gray zone, and hypermethylated groups, respectively; P < .001, Figure 2D; median OS: 16.0, 19.0, and 35.0 months, respectively; P < .001, Figure 2E). This result confirmed that the gray zone was not merely a mathematical product due to heterogeneity amongst CpGs but endowed specific prognostic significance. Determining whether gray zone patients could benefit from TMZ was essential for prognosis assessment and subsequent clinical decision‐making. Other methods for MGMT status evaluation might be beneficial.
3.4. MGMT protein expression identified by IHC and its utility in GBM prognosis stratification
The protein expression of MGMT is the most immediate evidence reflecting gene function. Thus, we further used IHC to evaluate MGMT protein expression levels. The results indicated that the IHC score of MGMT could be a reliable tool to predict OS (AUC 0.656; 95% CI, 0.527‐0.768, P < .001; Figure 3A, brown line), but the efficiency was lower than mean methylation levels obtained from PSQ (AUC 0.709; 95% CI, 0.652‐0.768, P < .001; Figure 3A, red line). Using the median percentage of cells staining for MGMT from IHC as the optimal threshold, patients could be separated into low IHC score (<30%; 147/312, 47.1%) and high IHC score groups (≥30%; 165/312, 52.9%). Patients with low IHC scores showed a median PFS of 13.0 months, whereas patients with high IHC scores showed a median PFS of 7.0 months (log‐rank, P < .001; Figure 3C). Overall survival in patients with low IHC score was also more favorable than the high IHC score group (27.0 vs 15.0 months; log‐rank, P < .001). This result reconfirmed the superiority of PSQ for prognosis stratification and pointed out that IHC also could be applied to assess the status of MGMT as well.
FIGURE 3.
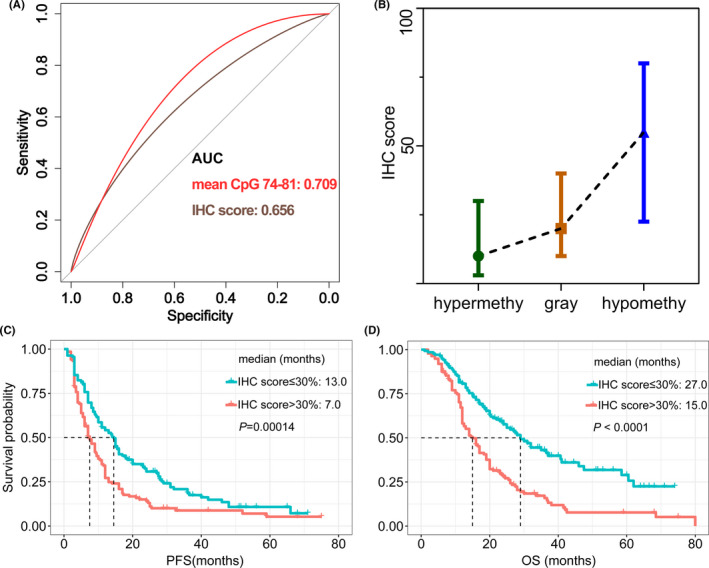
(A) Immunohistochemistry (IHC) score could be used for MGMT status determination to predict overall survival (OS) in glioblastoma patients (area under the receiver operating characteristic curve [AUC] 0.656, brown line), but the efficiency was inferior to pyrosequencing (PSQ) (AUC: 0.709, red line). (B) Differences in IHC scores among PSQ‐defined hypermethylated, gray zone, and hypomethylated groups were significant (mean: 18.0%, 27.5%, and 49.7%, respectively; P < .001). (C, D) Patients with high MGMT expression (IHC score > 30%) showed inferior (C) progression‐free survival (PFS) and (D) overall survival (OS)
The mean IHC scores for the methylated group (mean methylation level >12%) and unmethylated group (mean methylation level ≤12%) were 17.7% and 44.2%, respectively (P < .001). Furthermore, 70.0% of patients in the methylated group (91/130) had low MGMT expression (<30%, IHC‐defined methylated); in the unmethylated group, 65.3% (119/182) of patients showed high MGMT expression (≥30%, IHC‐defined unmethylated). The difference of mean IHC score among hypermethylated, gray zone, hypomethylated groups was also significant (mean: 18.0%, 27.5%, and 49.7%, respectively; P < .001, one‐way ANOVA; Figure 3B). In the hypermethylated group, 76.4% of patients (81/106) had a low IHC score (<30%), and 70.2% (87/124) patients in the hypomethylated group had a higher IHC score (≥30%). In gray zone patients, only 22 patients (mean methylation level >12%, 22/78, 28.2%) were methylated by PSQ, but 56.4% (44/78) had low MGMT expression (<30%). This prominent distinction implied that IHC might play a role in prognosis stratification for patients in the gray zone (Figure 4A).
FIGURE 4.
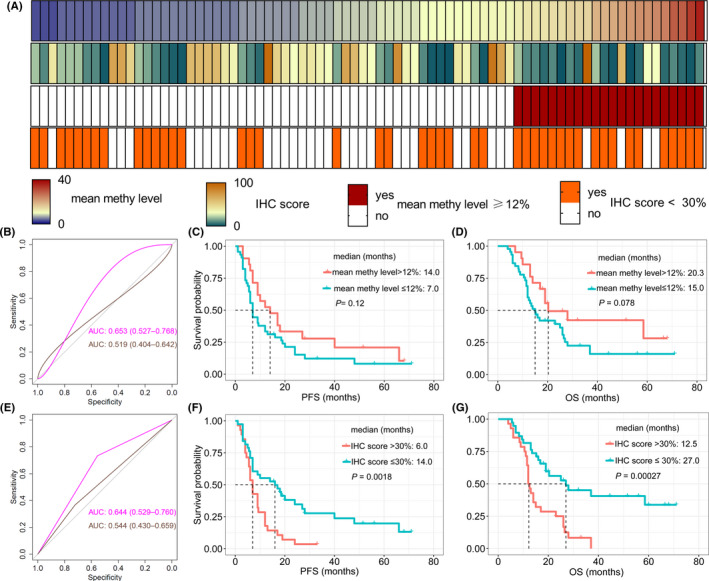
(A) Average methylation levels (first row) and immunohistochemistry (IHC) scores (second row) heatmap for glioblastoma patients in the pyrosequencing (PSQ)‐defined gray zone subgroup. The MGMT status could be divided into methylated or unmethylated based on the thresholds from PSQ (threshold: 12%, third row) and IHC (threshold: 30%, fourth row). (B) IHC scores (pink line) instead of mean methylation levels (brown line) could predict overall survival (OS). (C, D) The threshold of 12% for the average methylation levels failed to assess the prognosis for gray zone, regardless of progression‐free survival (PFS) or OS. (E) The threshold for IHC score (30%) rather than the threshold for average methylation results from PSQ (12%) could effectively evaluate OS for gray zone patients. (F, G) The prognosis difference between high and low IHC scores subgroups for patients in the gray zone was significant (F, PFS; G, OS)
3.5. Immunohistochemistry score of MGMT instead of mean methylation level could predict outcomes of gray zone patients
Patients in the gray zone showed intermediate PFS and OS, thus, separating them into different subgroups based on survival was important for prognosis assessment. The mean methylation levels of CpGs 74‐81 failed to predict survival for patients in the gray zone (AUC 0.519; 95% CI, 0.404‐0.642, P > .05; Figure 4B, brown line). Based on the optimal cut‐off established in the whole cohort (12%), methylation status ascertained by PSQ still could not predict survival (AUC 0.544; 95% CI, 0.430‐0.659, P > .05; Figure 4E, brown line). Neither PFS nor OS was significantly different between methylated and unmethylated patients from the gray zone (median PFS: 14.0 vs 7.0 months, P = .12, Figure 4C; median OS: 20.3 vs 15.0 months, P = .08, Figure 4D).
We also attempted to explore whether IHC score could differentiate outcomes for patients in the gray zone. The result showed that the IHC score was effective (AUC 0.653; 95% CI, 0.528‐0.766, P < .001; Figure 4B, pink line), and using the cut‐off value of 30%, the result of IHC could still be a robust tool to predict OS (AUC 0.644; 95% CI, 0.529‐0.760, P < .001; Figure 4E, pink line). The Kaplan‐Meier analysis showed that the median PFS of low MGMT expression patients from the gray zone was 14.0 months, while the median PFS of high MGMT expression patients was only 6.0 months (P = .0018; Figure 4F). A similar result was observed in terms of OS (median OS: 27.0 vs 12.5 months, P < .001; Figure 4G).
3.6. Combining MGMT promoter PSQ and protein expression to optimize prognosis evaluation
Although the predictive ability of IHC for survival was marginally inferior to PSQ, its specific value in assessing survival for patients in the gray zone enlightened us that IHC might be a perfect supplement to PSQ. Thus, we introduced a new classification strategy to dichotomize gray zone patients into methylated (IHC score <30%) and unmethylated (IHC score ≥30%) groups. In the whole cohort, patients could be divided into refined methylated (hypermethylated + IHC score <30% from gray zone; Figure 5A) and refined unmethylated (hypomethylated + IHC score ≥30% from gray zone; Figure 5A) groups. Comparing mean methylation levels by PSQ (cut‐off value, 12%; AUC 0.662; 95% CI, 0.611‐0.0712, P < .001; Figure 5B, blue line) and protein expression (cut‐off value, 30%; AUC 0.648; 95% CI, 0.591‐0.705, P < .001; Figure 5B, orange line), this novel category strategy substantially improved survival predictive ability (AUC 0.720; 95% CI, 0.667‐0.774, P < .001; Figure 5B, red line). A more favorable prognosis was observed in the refined methylated compared to refined unmethylated group, both PFS (median, 16.0 vs 7.0 months; log‐rank, P < .001; Figure 5C) and OS (35.0 vs 14.0 months; log‐rank, P < .001; Figure 5D). These results, including remarkably improved AUC value and much pronounced prognosis difference in novel category strategy, inspired us to use a combined strategy for survival prediction.
FIGURE 5.
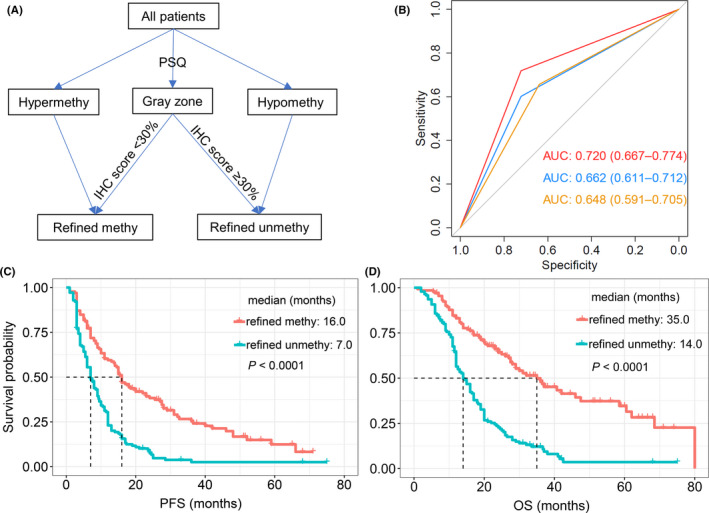
(A) Combining pyrosequencing (PSQ) and immunohistochemistry (IHC) to determine MGMT status in glioblastoma. The refined methylated group included patients from hypermethylated and gray zone groups with low IHC scores, whereas the refined unmethylated group was comprised of patients from unmethylated and gray zone groups with high IHC scores. (B) Compared with PSQ alone (threshold: 12%, blue line) and IHC alone (threshold: 30%, orange line), the combined strategy (red line) significantly improved survival prediction efficiency. (C, D) Prognosis difference for refined methylated and unmethylated patients was more prominent (C, PFS; D, OS). AUC, area under the receiver operating characteristic curve
4. DISCUSSION
In this retrospective study, we defined the gray zone subgroup with PSQ, the gold standard for MGMT promoter status determination. Patients in the gray zone harbored an intermediate prognosis and could be further divided into different subgroups by IHC staining. Combining PSQ and IHC significantly improved outcome predictive efficiency compared with the single threshold from average methylation levels (AUC 0.662‐0.720). Our results indicated IHC could be a reliable supplement to PSQ to predict clinical outcomes in the TMZ era.
Until now, chemotherapy regimens based on TMZ formed the backbone of malignant glioma treatment.31 However, not all patients could derive a survival benefit from this alkylating agent. MGMT could reverse the TMZ‐induced DNA damage and attenuate chemotherapeutic effects. Epigenetic modification of specific CpG islands within the MGMT promoter region could silence the gene transcription and enhance the cytotoxic effect of TMZ. Thus, identifying the status of the MGMT promoter is of paramount importance for predicting therapeutic response and evaluating individual survival. As the most effective biomarker for predicting TMZ response, the status of MGMT determination has reached a consensus amongst neuro‐oncologists and neuropathologists. Although this detection should be implemented into routine clinical practice, the optimal method for this purpose remains controversial.10 Simple MSP, qMSP, PSQ, high‐resolution melt, Infinium Methylation EPIC Bead Chip Array, MS‐MLPA, IHC, quantitative real‐time PCR, and enzymatic assays have been attempted for MGMT status determination, but none is perfect enough for clinical practice.
Methylation‐specific PCR is a PCR‐dependent protocol that tests the conversion of unmethylated cytosines to uracils to generate positive and negative results. This method is insufficient in terms of stability and inconsistent results were observed among replicates.32 Quantitative MSP normalizes the copy number of methylated MGMT to an unmethylated reference gene by standard curves. However, MSP and qMSP could only detect fully methylated or unmethylated sequences through designed primers.33 Other shortcomings of MSP and qMSP include poor reliability in FFPE tissues, only limited tested CpGs (typically a series of three to five CpGs), and failure in reflecting heterogeneous methylation levels among CpGs.34
Pyrosequencing could quantify methylation levels within each CpG site tested and present as a pyrogram PyroMark Q96 (QIAGEN, Hilden, Germany).16 Thus, PSQ could reflect methylation heterogeneity more effectively than MSP‐based protocols, which means PSQ is superior to MSP in precisely assessing epigenetic modification status.35 Nevertheless, the optimal cut‐off value for determining methylated or unmethylated subgroups remains controversial. Each individual CpG site harbors a specific cut‐off value and typically the optimal threshold is obtained from mean methylation levels among CpGs tested.36, 37, 38 Small sample size, the mixture of molecular and histologically distinct tumors, and different commercial kits contribute to the lack of universal criteria for clinical practice. In our study, we confirmed the optimal cut‐off values for CpGs 74‐81 with a large cohort of de novo GBM patients. We further defined a subgroup of patients with intermediate survival as the gray zone by interrogating the heterogeneity of CpGs tested. A careful eye should be cast on the interpretation of results from the gray zone for their potential benefit from TMZ.
How to stratify gray zone patients into distinct subgroups remains an intractable issue to be elucidated. Chai et al developed a novel analytical model to dichotomize patients into methylated or unmethylated groups based on the number of CpGs methylated. The results indicated that the strategy could accurately predict the prognosis for patients in the gray zone and improve clinical predictive performance. However, this study incorporated a substantial number of IDH1/2 mutant and WHO grade III patients, and the positive outcome in this subgroup could lead to analytic bias to some extent.35 Radke et al simply divided IDH WT patients into highly methylated (>20%), unmethylated (<10%), and low methylated (10%‐20% mean methylation) groups, and found an intermediate prognosis for low methylated patients. Significantly improved test precision for survival was observed when MSP results were supplemented to PSQ. Although the definition of highly methylated, unmethylated, and low methylated groups needed some refinement, the combination of MSP and PSQ for MGMT status identification was quite thoughtful and illuminating.39
Immunohistochemistry is the direct assay to assess MGMT status at the protein level focusing on regions of high tumor purity. However, high interobserver variability and non‐neoplastic cell contamination decrease the accuracy and reliability of IHC for MGMT status confirmation.40, 41 Our protocol used an evaluation strategy by interrogating a large number of tumor cells from isolated fields, and the result confirmed that IHC was a reliable detection method, despite the inferior predictive ability compared with PSQ. Notably, IHC could reclassify patients from the gray zone into distinct subgroups based on survival, and further combining PSQ and IHC significantly improved the efficiency and accuracy for TMZ response evaluation and prognosis prediction. Lalezari et al reported patients with tandem promoter methylation and low expression had a much more favorable prognosis than a single method or other combinations.17 Although the IHC results for MGMT status evaluation were not perfect, our research informed us that it could be an ideal complementary test to the current gold standard PSQ.
The status of MGMT promoter is the most important biomarker for predicting TMZ response, but other mechanisms could also play a role in impacting the alkylating agent effect. Posttranscriptional modulation, IDH1/2 mutation, and the CpG island methylator phenotype are well‐elaborated factors correlated with the association between MGMT methylation and patient outcome.42, 43, 44 In addition, TMZ could upregulate MGMT expression activity during GBM recurrence regardless of changes in promoter methylation status, which might link to genomic rearrangements.45 Cytotoxicity of TMZ depends on the robust MMR system. Deficiency in the MMR system was often observed in recurrent tumors and led to TMZ resistance.46, 47 Therefore, predicting TMZ response exclusively dependent on MGMT promoter determination strategies, such as current gold standard PSQ and MSP, is not sufficient. Immunohistochemistry provides alternative insight into MGMT assessment at the protein level in an economical, accessible, and time‐saving manner, which makes it a perfect complement to PSQ.
Limitations in this study do exist. It is retrospective research within a single institution, thus inevitable selective and analytical bias might have some influence on the final results. Other commercial PSQ testing kits for different CpG sites and detection assays including MSP, MS‐MLPA, and Infinium Methylation EPIC BeadChip Array were not applied in this cohort.
In conclusion, IHC is a robust method to predict clinical outcomes for GBM patients in the PSQ defined gray zone. A combination of PSQ and IHC could profoundly improve predictive performance for clinical outcomes.
DISCLOSURE
No conflicts of interest are declared by the authors.
Supporting information
Fig S1
Fig S2
ACKNOWLEDGMENT
The authors sincerely thank the patients and their families for their participation in the present study.
Li M, Dong G, Zhang W, et al. Combining MGMT promoter pyrosequencing and protein expression to optimize prognosis stratification in glioblastoma. Cancer Sci. 2021;112:3699–3710. 10.1111/cas.15024
Funding information
This study was supported by the National Natural Science Foundation of China (Grant Nos. 81571632 and 81771309).
REFERENCES
- 1.Bm A, Tf C. Adult glioblastoma. J Clin Oncol. 2017;35(21):2402‐2409. [DOI] [PubMed] [Google Scholar]
- 2.Lapointe S, Perry A, Butowski NA. Primary brain tumours in adults. The Lancet. 2018;392(10145):432‐446. [DOI] [PubMed] [Google Scholar]
- 3.Aldape K, Brindle KM, Chesler L, et al. Challenges to curing primary brain tumours. Nat Rev Clin Oncol. 2019; 16: 509‐520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stupp R, Mason WP, van den Bent MJ , et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987‐996. [DOI] [PubMed] [Google Scholar]
- 5.Perry JR, Laperriere N, Mason WP. Radiation plus temozolomide in patients with glioblastoma. N Engl J Med. 2017;376(22):2197. [DOI] [PubMed] [Google Scholar]
- 6.Malmström A, Łysiak M, Kristensen B, Hovey E, Henriksson R, Söderkvist P. MGMTDo we really know who has an methylated glioma? Results of an international survey regarding use of analyses for glioma. Neuro‐oncology Pract. 2020;7(1):68‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brigliadori G, Goffredo G, Bartolini D, et al. Influence of intratumor heterogeneity on the predictivity of MGMT gene promoter methylation status in glioblastoma. Front Oncol. 2020;10: 533000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997‐1003. [DOI] [PubMed] [Google Scholar]
- 9.Jonsson P, Lin A, Young R, et al. Genomic correlates of disease progression and treatment response in prospectively characterized gliomas. Clin Cancer Res. 2019;25(18):5537‐5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mansouri A, Hachem LD, Mansouri S, et al. MGMT promoter methylation status testing to guide therapy for glioblastoma: refining the approach based on emerging evidence and current challenges. Neuro Oncol. 2019;21:167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johannessen LE, Brandal P, Myklebust TORÅ, Heim S, Micci F, Panagopoulos I. MGMT gene promoter methylation status – assessment of two pyrosequencing kits and three methylation‐specific PCR methods for their predictive capacity in glioblastomas. Cancer Genom Proteom. 2018;15(6):437‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Carlo E, Gerratana L, De Maglio G, et al. Defining a prognostic score based on O6‐methylguanine‐DNA methyltransferase cut‐off methylation level determined by pyrosequencing in patients with glioblastoma multiforme. J Neurooncol. 2018;140(3):559‐568. [DOI] [PubMed] [Google Scholar]
- 13.Quillien V, Lavenu A, Sanson M, et al. Outcome‐based determination of optimal pyrosequencing assay for MGMT methylation detection in glioblastoma patients. J Neurooncol. 2014;116(3):487‐496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quillien V, Lavenu A, Karayan‐Tapon L, et al. Comparative assessment of 5 methods (methylation‐specific polymerase chain reaction, MethyLight, pyrosequencing, methylation‐sensitive high‐resolution melting, and immunohistochemistry) to analyze O6‐methylguanine‐DNA‐methyltranferase in a series of 100 glioblastoma patients. Cancer. 2012;118(17):4201‐4211. [DOI] [PubMed] [Google Scholar]
- 15.Dahlrot R, Dowsett J, Fosmark S, et al. Prognostic value of O‐6‐methylguanine‐DNA methyltransferase (MGMT) protein expression in glioblastoma excluding nontumour cells from the analysis. Neuropathol Appl Neurobiol. 2018;44(2):172‐184. [DOI] [PubMed] [Google Scholar]
- 16.Butler M, Pongor L, Su Y, et al. MGMT Status as a clinical biomarker in glioblastoma. Trends Cancer. 2020;6(5):380‐391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lalezari S, Chou AP, Tran A, et al. Combined analysis of O6‐methylguanine‐DNA methyltransferase protein expression and promoter methylation provides optimized prognostication of glioblastoma outcome. Neuro Oncol. 2013;15(3):370‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burke E, Grobler M, Elderfield K, et al. Double‐labelling immunohistochemistry for MGMT and a "cocktail" of non‐tumourous elements is a reliable, quick and easy technique for inferring methylation status in glioblastomas and other primary brain tumours. Acta Neuropathol Communicat. 2013;1:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sasai K, Nodagashira M, Nishihara H, et al. Careful exclusion of non‐neoplastic brain components is required for an appropriate evaluation of O6‐methylguanine‐DNA methyltransferase status in glioma: relationship between immunohistochemistry and methylation analysis. Am J Surg Pathol. 2008;32(8):1220‐1227. [DOI] [PubMed] [Google Scholar]
- 20.Nakasu S, Fukami T, Baba K, Matsuda M. Immunohistochemical study for O6‐methylguanine‐DNA methyltransferase in the non‐neoplastic and neoplastic components of gliomas. J Neurooncol. 2004;70(3):333‐340. [DOI] [PubMed] [Google Scholar]
- 21.Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131(6):803‐820. [DOI] [PubMed] [Google Scholar]
- 22.Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765‐773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li M, Ren X, Jiang H, et al. Supratentorial high‐grade astrocytoma with leptomeningeal spread to the fourth ventricle: a lethal dissemination with dismal prognosis. J Neurooncol. 2019;142:253–261. [DOI] [PubMed] [Google Scholar]
- 24.Yang K, Ren X, Tao L, et al. Prognostic implications of epidermal growth factor receptor variant III expression and nuclear translocation in Chinese human gliomas. Chin J Cancer Res. 2019;31(1):188‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang H, Yu K, Li M, et al. Classification of progression patterns in glioblastoma: analysis of predictive factors and clinical implications. Front Oncol. 2020;10: 590648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Christmann M, Nagel G, Horn S, et al. MGMT activity, promoter methylation and immunohistochemistry of pretreatment and recurrent malignant gliomas: a comparative study on astrocytoma and glioblastoma. Int J Cancer. 2010;127(9):2106‐2118. [DOI] [PubMed] [Google Scholar]
- 27.Chinot O, Barrié M, Fuentes S, et al. Correlation between O6‐methylguanine‐DNA methyltransferase and survival in inoperable newly diagnosed glioblastoma patients treated with neoadjuvant temozolomide. J Clin Oncol. 2007;25(12):1470‐1475. [DOI] [PubMed] [Google Scholar]
- 28.Wen PY, Macdonald DR, Reardon DA, et al. Updated response assessment criteria for high‐grade gliomas: response assessment in neuro‐oncology working group. J Clin Oncol. 2010;28(11):1963‐1972. [DOI] [PubMed] [Google Scholar]
- 29.Ellingson BM, Wen PY, Cloughesy TF. Modified criteria for radiographic response assessment in glioblastoma clinical trials. Neurotherapeutics. 2017;14(2):307‐320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malley D, Hamoudi R, Kocialkowski S, Pearson D, Collins V, Ichimura K. A distinct region of the MGMT CpG island critical for transcriptional regulation is preferentially methylated in glioblastoma cells and xenografts. Acta Neuropathol. 2011;121(5):651‐661. [DOI] [PubMed] [Google Scholar]
- 31.Jiang H, Zeng W, Ren X, et al. Super‐early initiation of temozolomide prolongs the survival of glioblastoma patients without gross‐total resection: a retrospective cohort study. J Neurooncol. 2019;144:127–135. [DOI] [PubMed] [Google Scholar]
- 32.Xia D, Reardon D, Bruce J, Lindeman N. The Clinical implications of inconsistently methylated results from glioblastoma MGMT testing by replicate methylation‐specific PCR. J Mol Diagnost. 2016;18(6):864‐871. [DOI] [PubMed] [Google Scholar]
- 33.Vlassenbroeck I, Califice S, Diserens AC, et al. Validation of real‐time methylation‐specific PCR to determine O6‐methylguanine‐DNA methyltransferase gene promoter methylation in glioma. J Mol Diagn. 2008;10(4):332‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Silber JR, Bobola MS, Blank A, Chamberlain MC. O(6)‐methylguanine‐DNA methyltransferase in glioma therapy: promise and problems. Biochim Biophys Acta. 2012;1826(1):71‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chai RC, Liu YQ, Zhang KN, et al. A novel analytical model of MGMT methylation pyrosequencing offers improved predictive performance in patients with gliomas. Mod Pathol. 2019;32(1):4‐15. [DOI] [PubMed] [Google Scholar]
- 36.Yuan G, Niu L, Zhang Y, et al. Defining optimal cutoff value of MGMT promoter methylation by ROC analysis for clinical setting in glioblastoma patients. J Neurooncol. 2017;133(1):193‐201. [DOI] [PubMed] [Google Scholar]
- 37.Zhao H, Wang S, Song C, Zha Y, Li L. The prognostic value of MGMT promoter status by pyrosequencing assay for glioblastoma patients' survival: a meta‐analysis. World J Surg Oncol. 2016;14(1):261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brigliadori G, Foca F, Dall'Agata M, et al. Defining the cutoff value of MGMT gene promoter methylation and its predictive capacity in glioblastoma. J Neurooncol. 2016;128(2):333‐339. [DOI] [PubMed] [Google Scholar]
- 39.Radke J, Koch A, Pritsch F, et al. Predictive MGMT status in a homogeneous cohort of IDH wildtype glioblastoma patients. Acta Neuropathol Communicat. 2019;7(1):89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang L, Li Z, Liu C, et al. Comparative assessment of three methods to analyze MGMT methylation status in a series of 350 gliomas and gangliogliomas. Pathol Res Pract. 2017;213(12):1489‐1493. [DOI] [PubMed] [Google Scholar]
- 41.Schulze Heuling E, Knab F, Radke J, et al. Prognostic relevance of tumor purity and interaction with MGMT methylation in glioblastoma. Mol Cancer Res. 2017;15(5):532‐540. [DOI] [PubMed] [Google Scholar]
- 42.Wick W, Meisner C, Hentschel B, et al. Prognostic or predictive value of MGMT promoter methylation in gliomas depends on IDH1 mutation. Neurology. 2013;81(17):1515‐1522. [DOI] [PubMed] [Google Scholar]
- 43.Mulholland S, Pearson DM, Hamoudi RA, et al. MGMT CpG island is invariably methylated in adult astrocytic and oligodendroglial tumors with IDH1 or IDH2 mutations. Int J Cancer. 2012;131(5):1104‐1113. [DOI] [PubMed] [Google Scholar]
- 44.Yin A, He Y, Etcheverry A, et al. Novel predictive epigenetic signature for temozolomide in non‐G‐CIMP glioblastomas. Clin Epigenet. 2019;11(1):76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oldrini B, Vaquero‐Siguero N, Mu Q, et al. MGMT genomic rearrangements contribute to chemotherapy resistance in gliomas. Nat Commun. 2020;11(1):3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nagel ZD, Kitange GJ, Gupta SK, et al. DNA repair capacity in multiple pathways predicts chemoresistance in glioblastoma multiforme. Cancer Res. 2017;77(1):198‐206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thomas A, Tanaka M, Trepel J, Reinhold WC, Rajapakse VN, Pommier Y. Temozolomide in the era of precision medicine. Cancer Res. 2017;77(4):823‐826. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2