

Abstract
Antimitotic drugs such as vinca alkaloids and taxanes cause mitotic cell death after prolonged mitotic arrest. However, a fraction of cells escape from mitotic arrest by undergoing mitotic slippage, which is related to resistance to antimitotic drugs. Tipping the balance to mitotic cell death thus can be a way to overcome the drug resistance. Here we found that depletion of a mitotic regulator, CHAMP1 (chromosome alignment‐maintaining phosphoprotein, CAMP), accelerates the timing of mitotic cell death after mitotic arrest. Live cell imaging revealed that CHAMP1‐depleted cells died earlier than mock‐treated cells in the presence of antimitotic drugs that resulted in the reduction of cells undergoing mitotic slippage. Depletion CHAMP1 reduces the expression of antiapoptotic Bcl‐2 family proteins, especially Mcl‐1. We found that CHAMP1 maintains Mcl‐1 expression both at protein and mRNA levels independently of the cell cycle. At the protein level, CHAMP1 maintains Mcl‐1 stability by suppressing proteasome‐dependent degradation. Depletion of CHAMP1 reduces cell viability, and exhibits synergistic effects with antimitotic drugs. Our data suggest that CHAMP1 plays a role in the maintenance of Mcl‐1 expression, implying that CHAMP1 can be a target to overcome the resistance to antimitotic drugs.
Keywords: anticancer drug resistance, apoptosis, CHAMP1, Mcl‐1, mitotic cell death
We found that depletion of chromosome alignment‐maintaining phosphoprotein CHAMP1, a mitotic regulator, accelerates the timing of mitotic cell death in the presence of antimitotic drugs. CHAMP1 maintains expression of Mcl‐1, an antiapoptotic Bcl‐2 family protein, both at protein and mRNA levels. CHAMP1 depletion suppresses tumor growth in a xenograft model, and reduces cell viability synergistically with antimitotic drugs, implying that CHAMP1 can be a target to overcome drug resistance.

1. INTRODUCTION
Among chemotherapeutic drugs, antimitotic drugs, such as vinca alkaloids and taxanes, target the mitotic spindle and cause mitotic arrest through the activation of the spindle assembly checkpoint (SAC).1 After prolonged mitotic arrest, cells undergo apoptotic cell death, known as mitotic cell death (Figure 1A). However, a fraction of cells slip into the next cell cycle, called mitotic slippage, which is one of the causes of resistance to antimitotic drugs. Whether cells undergo mitotic cell death or mitotic slippage after prolonged mitotic arrest differs not only between cell lines, but also within a population of the same cell line.2
FIGURE 1.
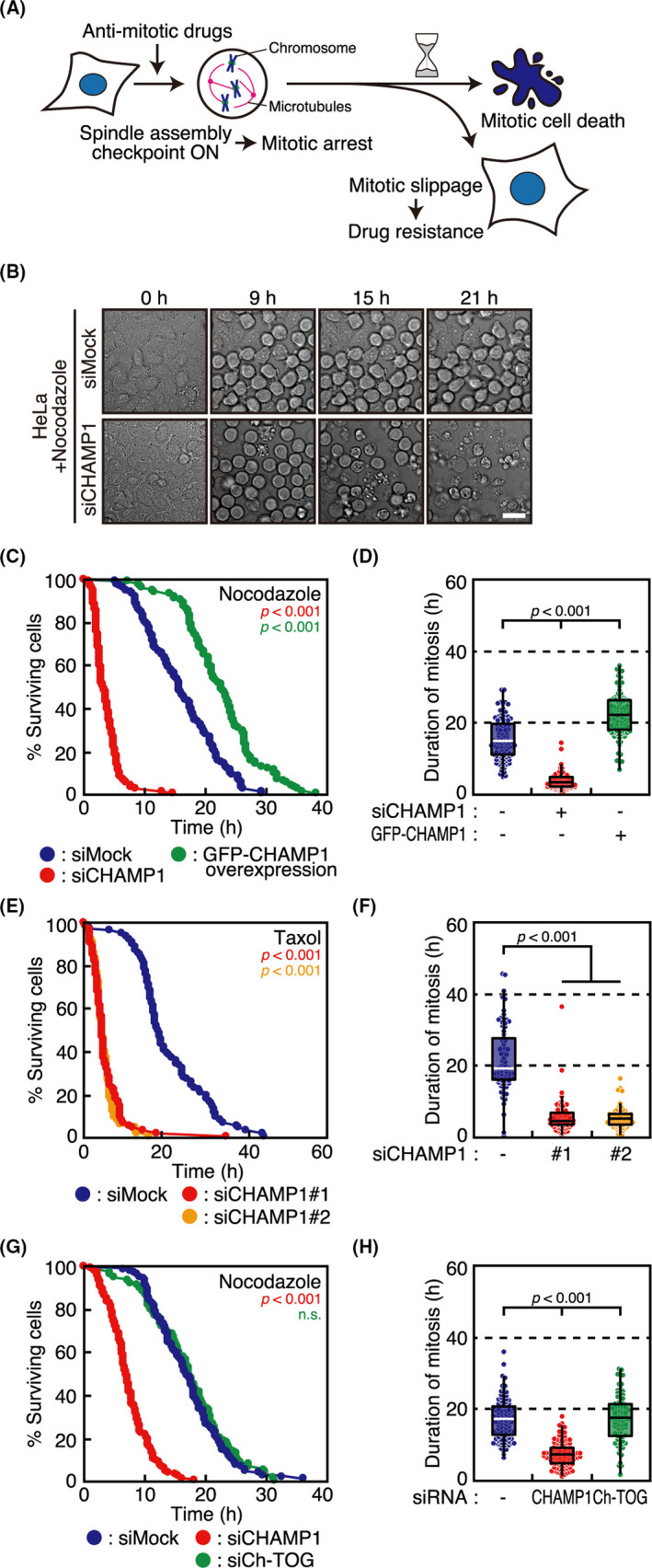
Chromosome alignment‐maintaining phosphoprotein (CHAMP1)‐depleted cells show accelerated mitotic cell death. A, Schematic diagram of the fates of cells treated with antimitotic drugs. B, Phase‐contrast time‐lapse imaging of nocodazole‐treated HeLa cells with or without CHAMP1 depletion. Numbers show elapsed time (h) relative to the start of imaging. Scale bar, 20 µm. C, Survival of HeLa cells treated with mock or CHAMP1 siRNA, or overexpressing GFP‐CHAMP1, after mitotic arrest by nocodazole treatment. Cumulative survival curves are plotted as a function of time after mitotic arrest. P values were obtained using the log‐rank test compared with siMock. D, Duration of mitosis, which was determined as the duration from mitotic cell rounding to cell rupture (mitotic cell death), in nocodazole‐treated HeLa cells shown in (C). In box plots, the bottom and top of the box show the lower and upper quartile values, respectively. The median is indicated with a bar in the box, and the whiskers denote the range within 1.5× size of the box. P values were obtained using the Steel‐Dwass multiple comparison test. E, Survival of HeLa cells treated with mock or CHAMP1 siRNA after mitotic arrest by taxol treatment. Cumulative survival curves are shown as in (C). P values were obtained using the log‐rank test compared with siMock. F, Duration of mitotic arrest before mitotic cell death in cells shown in (E), which is displayed as in (D). P values were obtained using the Steel‐Dwass multiple comparison test. G, Survival of HeLa cells treated with mock, CHAMP1, or ch‐TOG siRNA after mitotic arrest by nocodazole treatment. Cumulative survival curves are shown as in (C). P values were obtained using the log‐rank test compared with siMock. n.s., not statistically significant. H, Duration of mitotic arrest before mitotic cell death in cells shown in (G), which is displayed as in (D). P values were obtained using the Steel‐Dwass multiple comparison test
Mitotic cell death is induced by an intrinsic pathway of apoptosis in which Bcl‐2 family proteins function to initiate mitochondrial outer membrane permeabilization (MOMP).3 Mitochondrial outer membrane permeabilization results in the release of cytochrome c from mitochondria into the cytoplasm that triggers caspase activation. During mitotic arrest, cells are “primed” for death by apoptosis, which means that cells are poised to MOMP by tipping the balance in favor of proapoptotic signal.4 Among the antiapoptotic Bcl‐2 family (Bcl‐2, Mcl‐1, Bcl‐xL, and Bcl‐w),3 Mcl‐1 has a particularly short half‐life, and is considered as a major determinant of the timing of mitotic cell death.5, 6, 7 Mitotic slippage occurs as a result of decrease in the level of cyclin B below the threshold to keep cells in mitosis, which depends on the residual activity of the anaphase‐promoting complex/cyclosome (APC/C) in the presence of the SAC.8 Whether cells undergo mitotic cell death or mitotic slippage is explained by the competing networks model, in which cell fate is determined by the competition between cyclin B degradation and proapoptotic signal accumulation.9, 10
We identified a novel protein, CHAMP1 (chromosome alignment‐maintaining phosphoprotein, CAMP, C13orf8, ZNF828), as a molecule involved in the maintenance of kinetochore‐microtubule attachment.11 An 812 amino acid protein, CHAMP1 contains zinc finger domains at both termini, and localizes to the nucleus in interphase and to chromosomes and the spindle in mitosis. It was identified as a binding protein of Rev7 (Mad2L2),11, 12 and also interacts with heterochromatin protein 1 (HP1) and POGO transposable element with ZNF domain (POGZ).13 It was later found that CHAMP1, as well as POGZ, is one of the genes mutated in individuals with intellectual disability.13, 14, 15 Cells depleted of CHAMP1 show chromosome misalignment and mitotic arrest in a SAC‐dependent manner.11 We have noticed that CHAMP1‐depleted cells die in a short period after the mitotic arrest, and hence we explored its underlying mechanism. We found that CHAMP1 regulates the stability of antiapoptotic Bcl‐2 family molecules, especially Mcl‐1, at both mRNA and protein levels. Depletion of CHAMP1 accelerates the timing of mitotic cell death in cancer cell lines after mitotic arrest, which suppresses the occurrence of mitotic slippage.
2. MATERIALS AND METHODS
2.1. Cell culture, synchronization, and drug treatment
HeLa Kyoto (human cervical carcinoma), A549 (human lung carcinoma; ATCC CCL‐185), U2OS (human osteosarcoma; ATCC HTB‐96), and 293FT (HEK; Thermo Fisher Scientific) cells were grown at 37°C in a 5% CO2 atmosphere in DMEM (Nacalai Tesque), supplemented with 10% FBS. The HeLa Kyoto cell line is a popular subline of HeLa cells, which is suitable for mitotic study due to its slower migration.16 To synchronize cells in G1/S phase, cells were treated with 2 mM thymidine (Wako) for 18 hours. To synchronize cells in M phase, cells were treated with 2 mM thymidine for 18 hours, then treated with 2 μM nocodazole (Merck) after washout of thymidine with PBS (137 mM NaCl, 8.1 mM Na₂HPO₄, 2.68 mM KCl, and 1.47 mM KH₂PO₄) three times. Taxol (Wako), vincristine (Abcam), dimethylenastron (Merck), BI2536 (Axon Medchem), and N‐methyl‐N’‐nitro‐N‐nitrosoguanidine (MNNG) (Wako) were used at 1 μM, 10 nM, 1 μM, and 0.5 μM, respectively, unless otherwise stated. MG132 (Z‐Leu‐Leu‐Leu‐al; Merck), cycloheximide (Nacalai Tesque), and actinomycin D (Wako) were used at 10 μg/mL, 10 μM, and 100 μg/mL, respectively.
2.2. RNA interference and plasmid transfection
The siRNA sequences for targeting CHAMP1 were described previously.11 The siRNA sequences for targeting POGZ and ch‐TOG were 5′‐UAUAAUCUUCAACUACAGAGUCCUC‐3′ and 5′‐UUUAUUUGAAUCAUUGAGUCGACCC‐3′, respectively (Stealth; Thermo Fisher Scientific). RNA duplexes (50 nM) were transfected into cells using Lipofectamine RNAiMAX reagent (Thermo Fisher Scientific). Transfection of the GFP‐CHAMP1 construct was carried out using Lipofectamine 2000 (Thermo Fisher Scientific) as described previously.11
2.3. Western blot analysis
Cells were lysed in lysis buffer (1% NP‐40, 150 mM NaCl, 20 mM Tris‐HCl, pH 7.5, 0.5 mM EGTA, and 0.1 mM DTT) containing cOmplete mini protease inhibitor cocktail tablets (Merck) and phosphatase inhibitor cocktail (Nacalai Tesque). Cell lysates were boiled for 10 minutes with 4× NuPAGE LDS sample buffer (Thermo Fisher Scientific). Proteins were separated using the NuPAGE SDS gel system (Thermo Fisher Scientific), electroblotted onto a PVDF membrane (Amersham Hybond‐P; Cytiva) and subjected to immunodetection using the following primary Abs at 1:1000 dilution; monoclonal mouse antibodies: anti‐α‐tubulin B‐5‐1‐2 (T5168; Merck), anti‐Cyclin B (610219; BD Biosciences), and anti‐Bcl‐2 (sc‐509; Santa Cruz Biotechnology); polyclonal rabbit antibodies: anti‐CHAMP1 (HPA008900; Atlas Antibodies), anti‐Mcl‐1 (4572; Cell Signaling Technology), anti‐Bcl‐xL (2762; Cell Signaling Technology), anti‐MAD2L2 (12683‐1‐AP; Proteintech), and anti‐Bak (3814; Cell Signaling Technology); and monoclonal rabbit antibody: anti‐Bim (2933; Cell Signaling Technology). Blocking and Ab incubations were carried out in 3% nonfat dry milk. Proteins were visualized using HRP‐labelled secondary Abs (1:5000; Santa Cruz Biotechnology) and enhanced chemiluminescence using ECL Prime Western Blotting Detection Reagents (Cytiva), according to the manufacturer’s instructions.
2.4. Immunoprecipitation
Cells were lysed in lysis buffer (1% NP‐40, 150 mM NaCl, 20 mM Tris‐HCl, pH 7.5, and 0.5 mM EGTA) containing cOmplete mini protease inhibitor cocktail tablets (Merck) and phosphatase inhibitor cocktail (Nacalai Tesque), and cleared by centrifugation for 10 minutes at 4°C at 21 130 g. After preclearing with sheep anti‐rabbit IgG conjugated to magnetic beads (Dynabeads M‐280; Thermo Fisher Scientific), the lysates were incubated with anti‐CHAMP1 Ab for 3 h at 4°C, followed by incubation with anti‐rabbit IgG conjugated to magnetic beads for 1 h at 4°C. After washing three times with wash buffer (0.1% NP‐40, 150 mM NaCl, 20 mM Tris‐HCl, pH 7.5, and 0.5 mM EGTA), the beads were subjected to western blotting analysis.
2.5. Live cell imaging
Cells were grown in glass chambers (Thermo Fisher Scientific). One hour before imaging, the medium was changed to prewarmed Leibovitz’s L‐15 medium (Thermo Fisher Scientific) supplemented with 20% FBS and 20 mM HEPES, pH 7.0. Recordings were made in a temperature‐controlled incubator at 37°C, as described previously.11, 17, 18, 19, 20 Z‐series of three sections in 3‐µm increments were captured every 15 minutes. Image stacks were projected. Images were collected with an Olympus IX‐71 inverted microscope controlled by DeltaVision softWoRx (Cytiva) using a ×20 0.75 NA UPlanSApo objective lens (Olympus).
2.6. RNA purification and quantitative RT‐PCR
Total RNA samples were prepared from HeLa cells 48 hours after siRNA transfection using the RNeasy Mini Kit (Qiagen), according to the manufacturer’s instructions. The cDNAs were synthesized from 200 ng total RNA by using ReverTraAce qPCR RT Master Mix with gDNA Remover (Toyobo). Real‐time PCR was carried out using Thunderbird SYBR qPCR Mix (Toyobo) and the following primers: Mcl‐1#1 forward, 5′‐CCAAGGCATGCTTCGGAAAC‐3′ and reverse, 5′‐GGTTCGATGCAGCTTTCTTG‐3′; Mcl‐1#2 forward, 5′‐GAAAGCTGCATCGAACCATT‐3′ and reverse, 5′‐CCAGCTCCTACTCCAGCAAC‐3′; CHAMP#1 forward, 5′‐ACATGCATCCCCAGACAAAT‐3′ and reverse, 5′‐GGAAGAGGAGGGCTTTTCAC‐3′; CHAMP1#2 forward, 5′‐CGTTTGGAGTGTGACCATTG‐3′ and reverse, 5′‐CACCAGCATCCATTTCATCA‐3′; Actin#1 forward, 5′‐AGAGCTACGAGCTGCCTGAC‐3′ and reverse, 5′‐AGCACTGTGTTGGCGTACAG‐3′; and Actin#2 forward, 5′‐CACCATTGGCAATGAGCGGTTC‐3′ and reverse 5′‐AGGTCTTTGCGGATGTCCACGT‐3′.
2.7. Luciferase assay
Cells were transfected with reporter plasmids (pGL2‐Mcl‐1 vector [a gift from Wafik El‐Deiry; Addgene plasmid #19132] or pGL4.25_5× upstream activating sequence [UAS] vector) coding firefly luciferase (Fluc), driver plasmids (pcDNA3.1‐GAL4‐DBD, pcDNA3.1‐CHAMP1‐GAL4‐DBD, or pcDNA3.1‐mFOXO1‐GAL4‐DBD), and phRL‐TK vector (Promega), a normalization plasmid coding Renilla luciferase (Rluc), 6 hours after siRNA transfection, and subjected to the assay after 48 hours. Luciferase assay was undertaken using the Dual‐Glo Luciferase Assay System (Promega), according to the manufacturer’s instructions. Luminescence was measured using the GloMax Explorer System (Promega), and relative luciferase activity was determined by the ratio of Fluc / Rluc activity.
2.8. Cell viability assay
For annexin V and propidium iodide (PI) staining, cells were stained using the ApoAlert Annexin V‐FITC Apoptosis Kit (Takara Bio), according to the manufacturer’s instructions. For cleaved caspase‐3 and cytochrome c immunostaining, cells were fixed with 3% paraformaldehyde in PBS for 10 minutes at 37°C, and permeabilized with 1% Triton X‐100 in PBS for 5 minutes. Fixed cells were incubated with anti‐cleaved caspase‐3 (1:2000, 9664; Cell Signaling Technology) and anti‐cytochrome c (1:2000, NBP2‐24872; NovusBio) Abs for 1 hour, followed by incubation with secondary Abs coupled with Alexa Fluor 488/594 (Thermo Fisher Scientific, 1:3000) for 1 hour. Then cells were observed under an Olympus IX‐83 inverted microscope controlled by cellSens (Olympus) using a ×60 1.42 NA PlanApoN objective lens (Olympus). For the MTT assay, 1 × 103 cells were seeded onto 96‐well culture plates and were analyzed using a Cell Proliferation Kit I (MTT) (Merck), according to the manufacturer’s instructions.
2.9. Mouse xenograft model
Animal experiments conformed to the Regulations for Animal Experiments and Related Activities at Tohoku University, reviewed by the Institutional Laboratory Animal Care and Use Committee of Tohoku University, and approved by the President of Tohoku University (2019AcA‐023). HeLa or A549 cells (5 × 106) were suspended with PBS, and were subcutaneously injected into flanks of 12‐week‐old athymic nude mice (BALB/cAJcl‐Foxn1nu ; CREA Japan). Before injection, mice were anesthetized with isoflurane (Escain; Pfizer). Tumor size was measured every week using digital calipers. Tumor volumes were calculated using the formula v = width2 × length / 2.
2.10. Statistical analysis
The Mann‐Whitney U test was used for comparison of dispersion, and a two‐sided t test was used for comparisons of average. If one of the samples had a fixed value, a significant difference between samples was validated by a one‐sample t test. A two‐sided F test validated the dispersibility of each category before the Student’s t test. If the result of the F test was an unequal variance, a significant difference between samples was validated by a two‐sided Welch’s t test. For comparisons between all groups showing normal distribution, one‐way ANOVA was used with the Tukey‐Kramer post hoc test. The Kolmogorov‐Smirnov test verified the normality of data distribution for each group before the one‐way ANOVA. If the result of the Kolmogorov‐Smirnov test was non‐nominal distribution, the significant differences between all groups were validated by the Kruskal‐Wallis test, which was used with the Steel‐Dwass post hoc test. Repeated‐measures ANOVA was used for comparison of repeated observation. Log‐rank test with the Bonferroni correction was used for comparison of survival curves. All statistical analyses were undertaken with EZR,21 which is a graphical user interface for R (R Core Team, https://www.R‐project.org/).
3. RESULTS
3.1. Depletion of CHAMP1 accelerates the timing of mitotic cell death and suppresses mitotic slippage
We identified CHAMP1 as a molecule involved in the maintenance of kinetochore‐microtubule attachment.11 Cells depleted of CHAMP1 show chromosome misalignment and mitotic arrest in a SAC‐dependent manner. When we observed CHAMP1‐depleted HeLa cells, a cervical cancer‐derived cell line, in live cell imaging, we noticed that cells arrested in mitosis died in a short period (data not shown), suggesting that CHAMP1 is involved in cell survival during mitotic arrest. In this study, we aimed to clarify the effect of CHAMP1 depletion on cells that are treated with antimitotic drugs, and determine whether treatment with antimitotic drugs and simultaneous depletion of CHAMP1 synergistically cause mitotic cell death in various cancer cell lines. First, we treated HeLa cells with a high‐dose of nocodazole, a microtubule‐depolymerizing agent, and observed the cells with or without CHAMP1 depletion in live cell imaging (Figures 1B and S1A, Movie S1). As shown in Figures 1C,D and S1B, the timing from mitotic arrest to mitotic cell death was significantly shorter in CHAMP1‐depleted cells compared to mock‐treated cells. We confirmed the result using two independent siRNAs (Figure S1A,C,D). Depletion of POGZ, a CHAMP1 binding protein that is also involved in chromosome segregation,22 did not accelerate the timing of mitotic cell death (Figure S1C,D). When we overexpressed CHAMP1, cells survived longer than mock‐treated cells after mitotic arrest (Figures 1C,D and S1A,B). The timing of mitotic cell death was also accelerated when cells were treated with taxol, a microtubule stabilizer (Figure 1E,F). To exclude the possibility that the early occurrence of mitotic cell death is related to the role of CHAMP1 in kinetochore‐microtubule attachment, we observed cells depleted of ch‐TOG, which is also involved in kinetochore‐microtubule attachment.23 In contrast to CHAMP1 depletion, ch‐TOG depletion did not accelerate the timing of mitotic cell death, excluding the possibility (Figure 1G,H). These data suggest that CHAMP1 is involved in cell survival after mitotic arrest, which is independent of its role in kinetochore‐microtubule attachment.
After mitotic arrest induced by nocodazole or taxol treatment, the majority of HeLa cells underwent mitotic cell death, and mitotic slippage was not frequently seen (Figure S1B). To examine whether the early occurrence of mitotic cell death by CHAMP1 depletion results in the reduction of mitotic slippage, we examined the effect of CHAMP1 depletion in other cell lines. In A549 cells, a lung cancer‐derived cell line, nearly half of the cells arrested in mitosis after nocodazole treatment showed mitotic slippage (Figures 2A,B and S2A, Movie S2). Intriguingly, CHAMP1 depletion resulted in not only the reduction of time arrested in mitosis before cell death, but also the decrease of mitotic slippage (Figure 2B‐D, Movie S2). This was also the case in U2OS cells, an osteosarcoma‐derived cell line, although the duration of mitotic arrest was shorter compared to A549 cells (Figures 2B‐D and S2A). We also investigated the acceleration of the timing of mitotic cell death by CHAMP1 depletion in response to vincristine, a vinca alkaloid widely used in cancer therapy. In HeLa cells, a small fraction of cells that underwent mitotic slippage disappeared following CHAMP1 depletion, concomitant with the reduced duration of mitosis (Figure 2E‐G). In A549 cells, the majority of cells exhibited mitotic slippage in the presence of vincristine, and survived until the end of the imaging (Figure 2E‐G). When CHAMP1 was depleted, the number of cells that underwent mitotic slippage significantly decreased, and most of the cells died after shortened mitotic arrest (Figure 2E‐G). We further confirmed the acceleration of the timing of mitotic cell death by CHAMP1 depletion using other antimitotic drugs. As shown in Figure S2B‐D, the timing of cell death was significantly accelerated in A549 cells after mitotic arrest induced by dimethylenastron, an inhibitor for a mitotic motor protein, Eg5, or BI2536, an inhibitor for a mitotic kinase, Plk1. These data suggest that CHAMP1 is related to the survival of cells arrested in mitosis, and CHAMP1 depletion accelerates the timing of mitotic cell death and suppresses mitotic slippage in cancer cell lines.
FIGURE 2.

Chromosome alignment‐maintaining phosphoprotein (CHAMP1)‐depleted cells reduce the occurrence of mitotic slippage. A, Phase‐contrast time‐lapse imaging of nocodazole‐treated A549 cells. An example of cells undergoing mitotic cell death (left) or mitotic slippage (right) is shown. Numbers show elapsed time (h:min) relative to the start of imaging. Scale bar, 10 µm. B, Fate profiles of nocodazole‐treated A549 and U2OS cells treated with mock or CHAMP1 siRNA during 60 h of imaging. Individual cells were monitored by phase‐contrast time‐lapse imaging, and cell cycle phases during the time course were indicated by different colors. Cells were categorized into three groups depending on their fates; cell death in interphase, mitotic cell death, and mitotic slippage. The number and percentage of cells that underwent mitotic slippage against the total number of mitotic cells are indicated. C, Survival of nocodazole‐treated A549 and U2OS cells treated with mock or CHAMP1 siRNA shown in (B). Cumulative survival curves are plotted as a function of time after mitotic arrest. P values were obtained using the log‐rank test compared with siMock. D, Duration of mitosis, which was determined as the duration from mitotic cell rounding to cell rupture (in the case of mitotic cell death) or cell flattening (in the case of mitotic slippage), in nocodazole‐treated A549 and U2OS cells shown in (B). In box plots, the bottom and top of the box show the lower and upper quartile values, respectively. The median is indicated with a bar in the box, and the whiskers denote the range within 1.5× size of the box. P value was obtained using the Mann‐Whitney U test. E, Fate profiles of HeLa and A549 cells treated with mock or CHAMP1 siRNA in the presence or absence of vincristine during 60 h of imaging. Individual cells were analyzed as in (B). F, Survival of HeLa and A549 shown in (E). Cumulative survival curves are shown as in (C). 1C P values were obtained using the log‐rank test compared with siMock. G, Duration of mitotic arrest before mitotic cell death or mitotic slippage in HeLa and A549 cells shown in (E), displayed as in (D) 1D. P value was obtained using the Mann‐Whitney U test
3.2. CHAMP1 maintains Mcl‐1 protein stability
To explore the molecular basis for the role of CHAMP1 on cell survival after mitotic arrest, we examined the expression of proteins involved in cell cycle control and apoptosis in nocodazole‐treated HeLa cells. We observed the expression of Bcl‐2 family proteins in cell lysates collected from HeLa cells synchronized by thymidine treatment and arrested in mitosis by nocodazole treatment in immunoblot analysis. In mock‐treated cells, the expression of Mcl‐1 was reduced during the time course (Figure 3A), consistent with the finding that Mcl‐1 protein has a shorter half‐life compared with other Bcl‐2 family proteins and its expression is reduced in accordance with mitotic progression.24, 25, 26, 27, 28 In CHAMP1‐depleted cells, expression of antiapoptotic Bcl‐2 family proteins, Mcl‐1 and Bcl‐2, was reduced compared to mock‐treated cells throughout the time course even before cells were treated with nocodazole (Figure 3A). These data suggest that CHAMP1 maintains the expression of antiapoptotic Bcl‐2 family proteins, which could be related to the role of CHAMP1 in cell survival after mitotic arrest.
FIGURE 3.

Chromosome alignment‐maintaining phosphoprotein (CHAMP1) maintains Mcl‐1 protein stability. A, Western blotting of nocodazole‐treated HeLa cells with or without CHAMP1 depletion. Cells were released from thymidine block, treated with nocodazole after 9 h, and harvested at indicated times. The cell lysates were subjected to immunoblot analysis using Abs as indicated. B, Immunoblot analysis of HeLa cells transfected with mock or CHAMP1 siRNA or overexpressing RNAi‐resistant GFP‐CHAMP1. Cells were treated with 10 μg/mL MG132 or 10 μM cycloheximide (CHX) for 6 h before harvest. The cell lysates were subjected to immunoblot analysis using Abs as indicated. C, Expression of Mcl‐1 protein in HeLa cells treated as in (B). Relative Mcl‐1 protein levels against α‐tubulin are shown by adjusting the level of mock‐treated cells without drug treatment as 1. Error bars represent SD of four independent experiments. P values were obtained using the Tukey‐Kramer multiple comparison test. n.s., not statistically significant
As Mcl‐1 protein degradation is known to induce mitotic cell death by antimitotic drugs,5, 6 we examined whether CHAMP1 is involved in the protein stability of Mcl‐1. When we treated HeLa cells with cycloheximide (CHX), which inhibits protein synthesis, Mcl‐1 protein level was significantly reduced (Figure 3B), consistent with the notion that Mcl‐1 protein is degraded through the ubiquitin‐proteasome pathway.29 In CHAMP1‐depleted cells, Mcl‐1 expression was significantly reduced, which was further reduced following CHX treatment (Figure 3B). Then we treated HeLa cells with MG132, a proteasome inhibitor, to suppress protein degradation. As shown in Figure 3B,C, Mcl‐1 expression was increased in mock‐treated cells, as expected. Importantly, in CHAMP1‐depleted cells, Mcl‐1 expression was markedly increased by MG132 treatment, suggesting that CHAMP1 depletion promotes proteasome‐dependent Mcl‐1 degradation (Figure 3B,C). Expression of RNAi‐resistant GFP‐CHAMP1 rescued the Mcl‐1 expression level, which was further increased by MG132 treatment (Figure 3B,C), showing that reduced Mcl‐1 expression is not an off‐target effect of CHAMP1 RNAi. These data suggest that CHAMP1 suppresses proteasome‐dependent Mcl‐1 degradation. To examine the possibility that the role of CHAMP1 in Mcl‐1 stability is through direct binding to Mcl‐1, we undertook immunoprecipitation assays in HeLa cells. As shown in Figure S3, Mcl‐1 was not detected in CHAMP1 immunoprecipitates from interphase or mitotic phase lysates, suggesting that the role of CHAMP1 on Mcl‐1 stability is not through direct binding of these proteins. We further found that CHAMP1 does not bind to other antiapoptotic Bcl‐2 family proteins, Bcl‐xL and Bcl‐2 (Figure S3).
3.3. CHAMP1 regulates Mcl‐1 mRNA expression
Expression of Mcl‐1 is known to be regulated by various mechanisms both at mRNA and protein levels.30 As Mcl‐1 expression is reduced before cells enter mitosis in CHAMP1‐depleted cells (Figure 3A), we next studied the possibility that CHAMP1 regulates Mcl‐1 expression not only at the protein level, but also at the mRNA level. We compared Mcl‐1 mRNA expression by reverse transcription quantitative real‐time PCR (RT‐qPCR) in asynchronous HeLa cells with or without CHAMP1 depletion. We found that Mcl‐1 mRNA was reduced in CHAMP1‐depleted cells (Figure 4A), which suggests that CHAMP1 regulates Mcl‐1 mRNA expression. To examine whether Mcl‐1 mRNA degradation is promoted in CHAMP1‐depleted cells, we treated HeLa cells with actinomycin D, a transcription inhibitor, and compared Mcl‐1 mRNA levels by RT‐qPCR in HeLa cells with or without CHAMP1 depletion. As shown in Figure 4B, Mcl‐1‐mRNA was reduced to less than 50% in 30 minutes in mock‐treated cells, confirming that Mcl‐1 mRNA has a shorter half‐life.31 In CHAMP1‐depleted cells, Mcl‐1 mRNA was significantly reduced at the beginning of the time course, and was not reduced further by actinomycin D treatment (Figure 4B), suggesting that the reduction of Mcl‐1 mRNA in CHAMP1‐depleted cells is mainly due to reduced mRNA production rather than increased degradation. Then we verified the possibility that CHAMP1 regulates transcription of Mcl‐1 mRNA. First, we examined the activity of Mcl‐1 promoter by a dual‐luciferase reporter assay. A reporter construct containing the 325 bp promoter region upstream of the luciferase cDNA, which contains binding sites for ATF4, CREB, Ets‐1, Elk‐4, SRF, STAT3, and NF‐κB,32 was used to study the Mcl‐1 promoter activity in mock‐ or CHAMP1‐depleted HeLa cells (Figure 4C). As shown in Figure 4D, CHAMP1‐depleted cells showed comparable levels of luciferase activity to mock‐treated cells. Luciferase activity was also not affected by CHAMP1 depletion in a longer construct containing the 3261‐bp promoter region of the Mcl‐1 gene (data not shown). As CHAMP1 is a zinc‐finger protein implicated in transcriptional regulation,33 we further used a luciferase assay to examine the transcriptional activity of CHAMP1. Full‐length CHAMP1 was fused with GAL4 DNA‐binding domain (GAL4‐DBD), and expressed in 293FT cells together with a reporter construct in which the UAS, a GAL4‐binding sequence, was placed upstream of luciferase cDNA (Figure S4A). As shown in Figure S4B, luciferase activity was not increased by CHAMP1‐GAL4‐DBD compared to GAL4‐DBD, showing that transcriptional activation by CHAMP1 was not detected in this experimental setting. Taken together, our data suggest that CHAMP1 is involved in Mcl‐1 mRNA expression, which is probably not through transcriptional regulation.
FIGURE 4.
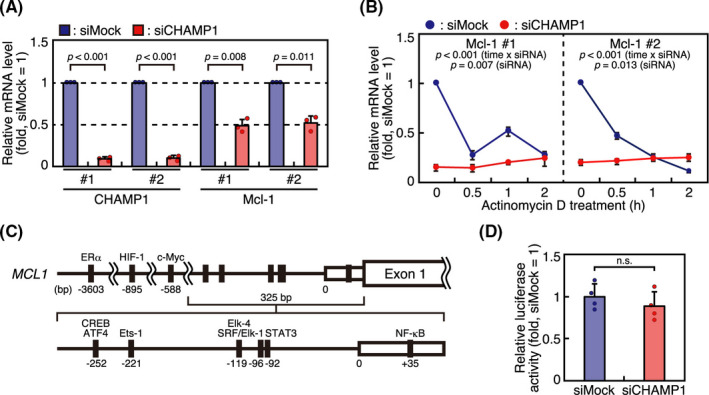
Chromosome alignment‐maintaining phosphoprotein (CHAMP1) depletion leads to reduced expression of Mcl‐1 mRNA, which is not through transcriptional control. A, Reverse transcription quantitative real‐time PCR (RT‐qPCR) analysis of Mcl‐1 mRNA in CHAMP1‐depleted cells. Total RNA extracted from HeLa cells transfected with mock or CHAMP1 siRNA were subjected to RT‐qPCR analysis to determine the relative expression levels of CHAMP1 and Mcl‐1 mRNAs. Two sets of primers were used to detect each mRNA. Relative mRNA levels are shown by adjusting the levels of mock‐treated cells as 1. Error bars represent SD of three independent experiments. P values were obtained using a one‐sample t test. B, Actinomycin D chase experiments to determine Mcl‐1 mRNA stability in HeLa cells transfected with mock or CHAMP1 siRNA. Cells were treated with 100 μg/mL actinomycin D for 30 min, 1 h, and 2 h before extracting RNAs. Two sets of primers were used to detect Mcl‐1 mRNA. Relative mRNA levels are shown by adjusting the level of mock‐treated cells before drug treatment as 1. Error bars represent SD of three independent experiments. P values were obtained using a repeated‐measures ANOVA. C, Schematic diagram of Mcl‐1 gene indicating the 325‐ and 3261‐bp promoter regions used in the reporter assay. Binding sites for transcription factors are shown. D, Luciferase assay of Mcl‐1 promoter activity in CHAMP1‐depleted cells. HeLa cells treated with mock or CHAMP1 siRNA were transfected with pGL2‐Mcl‐1 vector, the 325‐bp Mcl‐1 gene promoter construct, and phRL‐TK vector, an internal control construct. Relative luciferase activity was determined by the ratio of firefly luciferase / Renilla luciferase activity. Error bars represent SD of four independent experiments. P value was obtained using Student’s t test. n.s., not statistically significant
3.4. Depletion of CHAMP1 reduces cell viability synergistically with antimitotic drugs
As Mcl‐1 is downregulated in CHAMP1‐depleted cells before mitotic arrest, CHAMP1 could regulate cell survival irrespective of the cell cycle phase. To verify the possibility, we detected apoptotic cells in asynchronous HeLa cells with or without CHAMP1 depletion by annexin V and PI staining. As shown in Figure S5A, annexin V‐ and PI‐positive cells increased in CHAMP1‐depleted cells compared to mock‐treated cells, suggesting that CHAMP1 depletion facilitates apoptotic cell death throughout the cell cycle. However, the increase of apoptotic cells in interphase was small, which might not be sufficient to explain the marked effect on tumor growth in vivo (see below). The result was confirmed by the increase of cells positive for apoptotic markers cleaved caspase‐3 and cytochrome c (Figure S5B). Consistent with the notion that CHAMP1 maintains cell survival throughout the cell cycle, CHAMP1 depletion accelerated cell death in interphase A549 cells after treatment with MNNG, an alkylating agent that causes DNA damage (Figure 5A). We further examined the role of CHAMP1 in tumor cell viability in xenograft model. When we injected HeLa or A549 cells into nude mice with or without CHAMP1 depletion, tumor growth was significantly suppressed by CHAMP1 depletion (Figure 5B,C).
FIGURE 5.

Chromosome alignment‐maintaining phosphoprotein (CHAMP1) depletion facilitates apoptosis and reduces cell viability. A, Survival of A549 cells treated with mock or CHAMP1 siRNA in the presence or absence of N‐methyl‐N’‐nitro‐N‐nitrosoguanidine (MNNG). Cumulative survival curves are plotted as a function of time after mitotic arrest. P value was obtained using the log‐rank test. B, Representative image of tumors formed by HeLa cells 3 wk after injection in BALB/c nude mice. Mice were injected subcutaneously with 5 × 106 mock (white arrowhead) or CHAMP1‐ (red arrowhead) depleted cells. C, CHAMP1 depletion suppresses tumor growth in a xenograft model. BALB/c nude mice were injected subcutaneously with 5 × 106 mock or CHAMP1‐depleted cells. Tumor size was measured every week, and the volumes of tumors formed by HeLa (left) and A549 cells (right) are shown. Relative tumor volumes are shown by adjusting the levels of 1 wk as 1. Error bars represent SD of three mice. P values were obtained using a repeated‐measures ANOVA. D, Cell viability of HeLa and A549 cells with or without CHAMP1 depletion treated with vincristine, determined by MTT assay at 48 h after drug treatment. Viabilities are shown by adjusting the levels of nontreated siMock cells as 100%. Error bars represent SD of three independent experiments. P values were obtained using a repeated‐measures ANOVA. E, Cell viability of HeLa and A549 cells with or without CHAMP1 depletion treated with taxol, determined by MTT assay at 48 h after drug treatment. Viabilities are shown by adjusting the levels of nontreated siMock cells as 100%. Error bars represent SD of three independent experiments. P values were obtained using a repeated‐measures ANOVA
Finally, we examined the synergistic effect of CHAMP1 depletion and antimitotic drugs. We treated HeLa cells or A549 cells with vincristine or taxol, and determined their viability after 48 hours. As shown in Figure 5D,E, CHAMP1 depletion reduced cell viability even without drug treatment, and synergistically reduced cell viability in the presence of the antimitotic drugs. Overall, our data suggest that CHAMP1 depletion reduces cell viability, and shows synergistic effects with antimitotic drugs.
4. DISCUSSION
Here we report that CHAMP1 depletion accelerates mitotic cell death and suppresses mitotic slippage in cancer cell lines. This function of CHAMP1 for cell survival is independent of the role of CHAMP1 in kinetochore‐microtubule attachment,11 because accelerated mitotic cell death was observed in the absence of kinetochore‐microtubule attachment by treatment with high‐dose nocodazole. Mitotic cell death is driven by the intrinsic apoptosis pathway, where the balance of Bcl‐2 family proteins shifts to prime cells for apoptosis.4 We found that CHAMP1 depletion reduces the expression of antiapoptotic Bcl‐2 family proteins, suggesting that CHAMP1 is involved in the mitochondrial priming for apoptosis. Focusing on Mcl‐1, which plays a critical role in determining the timing of mitotic cell death,5, 6, 7 we found that CHAMP1 maintains Mcl‐1 expression both at protein and mRNA levels even before mitosis. Mcl‐1 protein degradation is regulated by the ubiquitin‐proteasome pathway,29 which is promoted by phosphorylation by kinases such as Jnk, p38, and Cdk1,34 and ubiquitylation by E3 ligases including MULE, APC/C, and FBW‐7/SCF.5, 6, 35, 36 We did not detect interaction between CHAMP1 and antiapoptotic Bcl‐2 family proteins, nor have we identified kinases and E3‐ligases related to the role of CHAMP1 in Mcl‐1 protein stability. Regarding Mcl‐1 mRNA expression, our data suggest that CHAMP1 is not involved in the transcription of Mcl‐1. Mcl‐1 mRNA expression is controlled not only transcriptionally, but also posttranscriptionally, through pre‐mRNA splicing and regulation of mRNA levels by RNA‐binding proteins and regulatory RNAs.30 These complex effects on Mcl‐1 and Bcl‐2 expression is supposed to be indirectly caused by CHAMP1 depletion, which warrants further investigation. Regarding the known interacting proteins for CHAMP1 (Rev7, HP1, and POGZ), involvement in apoptotic regulation has not been reported so far, and we found that POGZ depletion does not accelerate the timing of mitotic cell death in HeLa cells. Whether the role of CHAMP1 on cell survival is related to the pathogenesis of intellectual disability in individuals with CHAMP1 mutations13 is an interesting possibility worth investigating.
To avoid resistance to antimitotic drugs due to mitotic slippage, suppression of mitotic slippage by directly inhibiting the APC/C, not through SAC activation, was proposed.37 However, CHAMP1 depletion suppresses mitotic slippage by accelerating the timing of mitotic cell death. It was reported that the Myc oncogene promotes mitochondrial priming by upregulating BH3‐only proteins and downregulating Bcl‐xL, facilitating mitotic cell death.38 Thus, CHAMP1 and Myc play opposing roles in mitochondrial priming, and CHAMP1 depletion can be a strategy to avoid drug resistance. We found that CHAMP1 depletion suppresses tumor growth in a xenograft model. Furthermore, CHAMP1 depletion reduces cell viability synergistically with antimitotic drugs. Further study on the role of CHAMP1 in cell survival will reveal the way to overcome resistance to chemotherapeutic drugs.
DISCLOSURE
The authors declare no competing financial interests.
Supporting information
Fig S1‐S5
Movie S1
Movie S2
ACKNOWLEDGEMENTS
The authors thank T. Hirota for the HeLa Kyoto cell line, H. Motohashi for pGL2‐X1.4 vector, Wafik El‐Deiry for pGL2‐Mcl‐1 vector, and A. Kubo for pGL4.25_5xUAS vector. We also thank members of the KT laboratory for discussions, and A. Harata for technical assistance. This work was supported by JSPS KAKENHI Grant Numbers 24370078, 24650616, 19K22410; MEXT KAKENHI Grant Numbers, 26116501, 16H01296; P‐DIRECT from AMED; and grants from the Takeda Science Foundation to KT, and JSPS KAKENHI Grant Numbers 20K16295 to KI.
Hino M, Iemura K, Ikeda M, Itoh G, Tanaka K. Chromosome alignment‐maintaining phosphoprotein CHAMP1 plays a role in cell survival through regulating Mcl‐1 expression. Cancer Sci. 2021;112:3711–3721. 10.1111/cas.15018
Funding information
JSPS KAKENHI, Grant/Award Numbers: 24370078, 24650616, 19K22410, 20K16295; MEXT KAKENHI, Grant/Award Numbers 26116501, 16H01296; P‐DIRECT from AMED; Takeda Science Foundation.
REFERENCES
- 1.Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253‐265. [DOI] [PubMed] [Google Scholar]
- 2.Gascoigne KE, Taylor SS. Cancer cells display profound intra‐ and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell. 2008;14:111‐122. [DOI] [PubMed] [Google Scholar]
- 3.Youle RJ, Strasser A. The BCL‐2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47‐59. [DOI] [PubMed] [Google Scholar]
- 4.Pedley R, Gilmore AP. Mitosis and mitochondrial priming for apoptosis. Biol Chem. 2016;397:595‐605. [DOI] [PubMed] [Google Scholar]
- 5.Harley ME, Allan LA, Sanderson HS, Clarke PR. Phosphorylation of Mcl‐1 by CDK1‐cyclin B1 initiates its Cdc20‐dependent destruction during mitotic arrest. EMBO J. 2010;29:2407‐2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wertz IE, Kusam S, Lam C, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471:110‐114. [DOI] [PubMed] [Google Scholar]
- 7.Sloss O, Topham C, Diez M, Taylor S. Mcl‐1 dynamics influence mitotic slippage and death in mitosis. Oncotarget. 2016;7:5176‐5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brito DA, Rieder CL. Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr Biol. 2006;16:1194‐1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang HC, Mitchison TJ, Shi J. Stochastic competition between mechanistically independent slippage and death pathways determines cell fate during mitotic arrest. PLoS One. 2010;5:e15724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Topham CH, Taylor SS. Mitosis and apoptosis: how is the balance set? Curr Opin Cell Biol. 2013;25:780‐785. [DOI] [PubMed] [Google Scholar]
- 11.Itoh G, Kanno S, Uchida KS, et al. CAMP (C13orf8, ZNF828) is a novel regulator of kinetochore‐microtubule attachment. EMBO J. 2011;30:130‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hara K, Taharazako S, Ikeda M, et al. Dynamic feature of mitotic arrest deficient 2‐like protein 2 (MAD2L2) and structural basis for its interaction with chromosome alignment‐maintaining phosphoprotein (CAMP). J Biol Chem. 2017;292:17658‐17667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Isidor B, Kury S, Rosenfeld JA, et al. De novo truncating mutations in the kinetochore‐microtubules attachment gene CHAMP1 cause syndromic intellectual disability. Hum Mutat. 2016;37:354‐358. [DOI] [PubMed] [Google Scholar]
- 14.Hempel M, Cremer K, Ockeloen CW, et al. De novo mutations in CHAMP1 cause intellectual disability with severe speech impairment. Am J Hum Genet. 2015;97:493‐500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tanaka AJ, Cho MT, Retterer K, et al. De novo pathogenic variants in CHAMP1 are associated with global developmental delay, intellectual disability, and dysmorphic facial features. Cold Spring Harb Mol Case Stud. 2016;2:a000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu YJ, Le Berre M, Lautenschlaeger F, et al. Confinement and low adhesion induce fast amoeboid migration of slow mesenchymal cells. Cell. 2015;160:659‐672. [DOI] [PubMed] [Google Scholar]
- 17.Amin MA, Itoh G, Iemura K, Ikeda M, Tanaka K. CLIP‐170 recruits PLK1 to kinetochores during early mitosis for chromosome alignment. J Cell Sci. 2014;127:2818‐2824. [DOI] [PubMed] [Google Scholar]
- 18.Itoh G, Sugino S, Ikeda M, et al. Nucleoporin Nup188 is required for chromosome alignment in mitosis. Cancer Sci. 2013;104:871‐879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ikeda M, Tanaka K. Plk1 bound to Bub1 contributes to spindle assembly checkpoint activity during mitosis. Sci Rep. 2017;7:8794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Itoh G, Ikeda M, Iemura K, et al. Lateral attachment of kinetochores to microtubules is enriched in prometaphase rosette and facilitates chromosome alignment and bi‐orientation establishment. Sci Rep. 2018;8:3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanda Y. Investigation of the freely available easy‐to‐use software 'EZR' for medical statistics. Bone Marrow Transplant. 2013;48:452‐458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nozawa RS, Nagao K, Masuda HT, et al. Human POGZ modulates dissociation of HP1alpha from mitotic chromosome arms through Aurora B activation. Nat Cell Biol. 2010;12:719‐727. [DOI] [PubMed] [Google Scholar]
- 23.Herman JA, Miller MP, Biggins S. chTOG is a conserved mitotic error correction factor. eLife. 2020;9:e61773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akgul C, Moulding DA, White MR, Edwards SW. In vivo localisation and stability of human Mcl‐1 using green fluorescent protein (GFP) fusion proteins. FEBS Lett. 2000;478:72‐76. [DOI] [PubMed] [Google Scholar]
- 25.Craig RW. MCL1 provides a window on the role of the BCL2 family in cell proliferation, differentiation and tumorigenesis. Leukemia. 2002;16:444‐454. [DOI] [PubMed] [Google Scholar]
- 26.Cuconati A, Mukherjee C, Perez D, White E. DNA damage response and MCL‐1 destruction initiate apoptosis in adenovirus‐infected cells. Genes Dev. 2003;17:2922‐2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kozopas KM, Yang T, Buchan HL, Zhou P, Craig RW. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc Natl Acad Sci USA. 1993;90:3516‐3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iglesias‐Serret D, Pique M, Gil J, Pons G, Lopez JM. Transcriptional and translational control of Mcl‐1 during apoptosis. Arch Biochem Biophys. 2003;417:141‐152. [DOI] [PubMed] [Google Scholar]
- 29.Michels J, O'Neill JW, Dallman CL, et al. Mcl‐1 is required for Akata6 B‐lymphoma cell survival and is converted to a cell death molecule by efficient caspase‐mediated cleavage. Oncogene. 2004;23:4818‐4827. [DOI] [PubMed] [Google Scholar]
- 30.Senichkin VV, Streletskaia AY, Gorbunova AS, Zhivotovsky B, Kopeina GS. Saga of Mcl‐1: regulation from transcription to degradation. Cell Death Differ. 2020;27:405‐419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang T, Buchan HL, Townsend KJ, Craig RW. MCL‐1, a member of the BLC‐2 family, is induced rapidly in response to signals for cell differentiation or death, but not to signals for cell proliferation. J Cell Physiol. 1996;166:523‐536. [DOI] [PubMed] [Google Scholar]
- 32.Ricci MS, Kim SH, Ogi K, et al. Reduction of TRAIL‐induced Mcl‐1 and cIAP2 by c‐Myc or sorafenib sensitizes resistant human cancer cells to TRAIL‐induced death. Cancer Cell. 2007;12:66‐80. [DOI] [PubMed] [Google Scholar]
- 33.Lambert SA, Jolma A, Campitelli LF, et al. The human transcription factors. Cell. 2018;175:598‐599. [DOI] [PubMed] [Google Scholar]
- 34.Thomas LW, Lam C, Edwards SW. Mcl‐1; the molecular regulation of protein function. FEBS Lett. 2010;584:2981‐2989. [DOI] [PubMed] [Google Scholar]
- 35.Zhong Q, Gao W, Du F, Wang X. Mule/ARF‐BP1, a BH3‐only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl‐1 and regulates apoptosis. Cell. 2005;121:1085‐1095. [DOI] [PubMed] [Google Scholar]
- 36.Inuzuka H, Shaik S, Onoyama I, et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature. 2011;471:104‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang HC, Shi J, Orth JD, Mitchison TJ. Evidence that mitotic exit is a better cancer therapeutic target than spindle assembly. Cancer Cell. 2009;16:347‐358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Topham C, Tighe A, Ly P, et al. MYC is a major determinant of mitotic cell fate. Cancer Cell. 2015;28:129‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S5
Movie S1
Movie S2